

SUMMARY
Radish, Raphanus sativus L., is an important root crop that is cultivated worldwide. Owing to its evolutionary proximity to Arabidopsis thaliana, radish can be used as a model root crop in research on the molecular basis of agronomic traits. Pithiness is a significant defect that reduces the production of radish with commercial value; however, traditional breeding to eliminate this trait has thus far been unsuccessful. Here, we performed transcriptomics and genotype‐by‐sequencing (GBS)‐based quantitative trait locus (QTL) analyses of radish inbred lines to understand the molecular basis of pithiness in radish roots. The transcriptome data indicated that pithiness likely stems from the response to oxidative stress, leading to cell death of the xylem parenchyma during the root‐thickening process. Subsequently, we narrowed down a list of candidates responsible for pithiness near a major QTL and found polymorphisms in a radish homologue of Arabidopsis ANAC013 (RsNAC013), an endoplasmic reticulum bound NAC transcription factor that is targeted to the nucleus to mediate the mitochondrial retrograde signal. We analysed the effects of polymorphisms in RsNAC013 using Arabidopsis transgenic lines overexpressing RsNAC013 alleles as well as in radish inbred lines bearing these alleles. This analysis indicated that non‐synonymous variations within the coding sequence result in different levels of RsNAC013 activities, thereby providing a genetic condition for root pithiness. The elevated oxidative stress or hypoxia that activates RsNAC013 for mitochondrial signalling enhances this process. Collectively, this study serves as an exemplary case of translational research taking advantage of the extensive information available from a model organism.
Keywords: Raphanus sativus, radish root, pithiness, oxidative stress, cell death, transcriptomics, quantitative trait locus analysis, NAC013, genotype‐by‐sequencing, root crop
Significance Statement
This study is an exemplary translational research employing extensive information available from a model organism. To find the molecular basis of root pithiness, a significant defect reducing the commercial value of radish taproots, we performed transcriptomics and QTL analyses, compared these results with regulatory networks in Arabidopsis, and thereby found that the pithiness is mediated by NAC013, a transcription factor activating the oxidative stress response that leads to cell death in radish taproots.
INTRODUCTION
Radish (Raphanus sativus L.), a Brassicaceae species closely related to Arabidopsis thaliana (hereafter Arabidopsis) is commonly cultivated for its edible taproots and fresh sprouts. Radish together with cassava (Manihot esculenta), sweet potato (Ipomoea batatas), sugar beet (Beta vulgaris) and carrot (Daucus carota) are economically important root crops grown worldwide. Radish roots can grow incredibly fast, and reach up to 2 m in length and 30 cm in diameter within a period of 4 months (Yamagishi, 2017).
Radish root pithiness (also known as sponginess) is an agriculturally undesirable trait in breeding programmes because it turns crisp and juicy textures to puffy and dry textures. Previous morphological and physiological studies suggest that radish root pithiness is associated with the death of xylem parenchyma cells (Magendans, 1991; Marcelis, 1997; Takano, 1963). During this process, dry and spongy pith or hollow cavities are formed and become filled with air within the rapidly elongating part of the root. Radish root pithiness has been shown to be largely influenced by genetic programming but is, however, strongly affected by several environmental conditions, including light intensity, plant density and nutrients (Marcelis, 1997; Tanaka and Nagatomo, 2017). Despite the significance of this issue with regard to radish production, very little information pertaining to the molecular mechanism underlying radish root pithiness is available.
Phenomena similar to pithiness were also reported in other plants, including aerenchyma formation in maize (Buckner et al., 1998) and rice roots (Yamauchi et al., 2017), pithiness in sorghum stems (Fujimoto et al., 2018), and hypocotyl lysigenous aerenchyma in Arabidopsis (Muhlenbock et al., 2007). The formation of pithiness or aerenchyma involves several highly connected processes, including secondary cell wall biosynthesis, programmed cell death (PCD) and lignin deposition. Several transcription factors (TFs) have been found to act as master regulators of these processes, and some common causal pathways and proteins that are directly responsible for pithiness or aerenchyma have been identified in different species. Among these, PCD and the PCD‐executing enzymes are the most common downstream causes of pithiness‐like phenotypes. PCD is promoted by several TFs, including NAC DOMAIN‐CONTAINING PROTEINS (NAC) directly through PCD‐executing genes or indirectly through intermediate pathways such as the production of reactive oxygen species (ROS). For example, in Arabidopsis, ANAC046 and ANAC087 mediate PCD in the xylem and lateral root cap (Huysmans et al., 2018), while ANAC053 is involved in ROS‐mediated PCD (Lee et al., 2012, 2014). In sorghum, SbNAC074 activates the PCD of stem pith parenchyma cells through cell autolytic enzymes (Fujimoto et al., 2018). A number of PCD‐executing enzymes, including A. thaliana BIFUNCTIONAL NUCLEASE I (AtBFN1), RIBONUCLEASE 3 (AtRNS3), EXITUS1 (AtEXI1) and PUTATIVE ASPARTIC PROTEINASE A3 (AtPASPA3), were reported to be common downstream targets of different master switches in the aforementioned developmental and stress‐induced PCD (Daneva et al., 2016; Van Durme and Nowack, 2016; Fujimoto et al., 2018; Huysmans et al., 2018; Olvera‐Carrillo et al., 2015). Pithiness in radish root involves the death of xylem parenchyma cells, suggesting the involvement of PCD‐executing enzymes. Therefore, the identification of the environmental factors that induce the formation of pithiness, the regulators that activate the PCD cascade, and possible intermediate pathways would allow us to control this unfavourable trait and to enhance future breeding programmes.
Here, we provide several lines of evidence showing that radish root pithiness is related to an oxidative stress response pathway that leads to the death of xylem parenchyma cells in the root pith. We identified a major quantitative trait locus (QTL) for the radish root pithiness phenotype, and demonstrated that a TF, R. sativus (Rs)NAC013, located within the QTL interval and its downstream pathways may be involved in this process through ROS detoxification and antioxidation. Using Arabidopsis transgenic lines overexpressing two alleles of RsNAC013, it was found that the allele associated with the pithiness line generates a RsNAC013 variant more active in regulating a direct downstream target gene than that from the non‐pithiness line. By triggering the oxidative stress response in radish roots, we confirmed the involvement of RsNAC013‐related pathways in the formation of root pithiness. On the basis of our results here, it is likely that radish root pithiness is caused by oxidative stress, which mainly occurs toward the end of the root‐thickening stage. Our data can be used as a starting platform for further investigations of means by which to improve radish taproot quality through breeding programmes.
RESULTS AND DISCUSSION
Exploration of environmentally dependent pithiness trait using a transcriptome analysis
Among the radish recombinant inbred collection at the National Institute of Horticultural and Herbal Science (NIHHS) of the Republic of Korea, we identified a radish line, WonKyo10040ho (WK40), with vigorous growth and the root pithiness phenotype. In this line, we observed two types of root defects, sponginess and cavitation, depending on the growth conditions (Figures S1 and S2). Given that both may stem from the same process, hereafter we refer to them collectively as root pithiness in this paper. These defects typically appear in the pith of rapidly growing taproots between 6 and 10 weeks after planting (WAP) in field conditions (Figure 1a). Consistent with a previous report (Marcelis, 1997), we also noted that pithiness phenotypes occur stochastically (Figure S3), suggesting that pithiness is influenced not only by genetic factors but also by environmental factor(s). For example, WK40 plants grown in a greenhouse never showed pithiness (i.e. until 10 WAP in our condition), whereas those in the NIHHS field showed either sponginess or cavities at varying frequencies.
Figure 1.
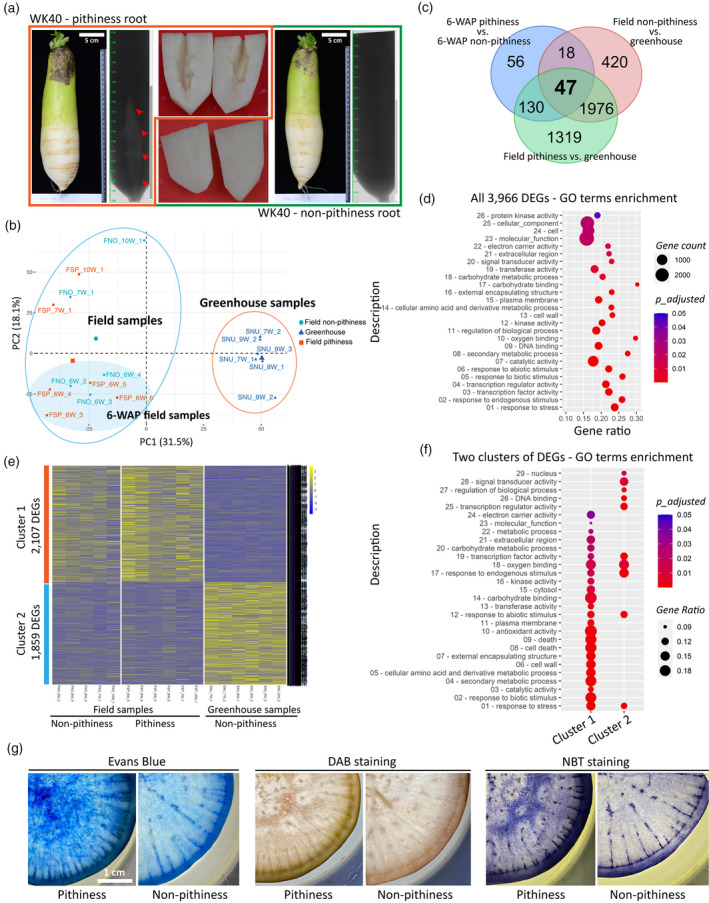
Comparative root transcriptome analysis of radish pithiness line WK40 grown under two different conditions.
(a) Representative images of root samples with and without pithiness, their micro‐computer tomography (CT) scan profiles, and the dissected lower elongated parts used for RNA extraction. For visualization, roots grown in the field collected at 10 WAP (weeks after planting) were used.
(b) A principal component analysis (PCA) of 17 RNA‐seq samples based on the top 5000 most variable genes.
(c) A comparison of differentially expressed genes [DEGs; false discovery rate (FDR) ≤ 0.05, |fold change| ≥ 2] identified in this study. The comparisons were made between field‐collected 6‐WAP pithiness and non‐pithiness samples, field‐collected pithiness samples and greenhouse samples, and between field‐collected non‐pithiness and greenhouse samples.
(d) Gene ontology (GO) term enrichment analysis (FDR ≤ 0.05) of the total 3966 DEGs from panel (c).
(e) K‐means clustering of all DEGs from panel (c) into two clusters of 2107 and 1859 DEGs, respectively. Gene‐wise normalized fragments per kilobase of exon per million mapped fragments (FPKM) values were used for the heatmap. FNO and FSP denote field‐collected samples with and without pithiness, respectively. SNU denotes samples collected from the greenhouse. W represents WAP in the sample names.
(f) GO term enrichment (FDR ≤ 0.05) of the two clusters identified in panel (e).
(g) Evans Blue staining for cell death, 3,3‐diaminobenzidine (DAB) and nitro blue tetrazolium (NBT) staining for hydrogen peroxide (H2O2) and superoxide (O2−) accumulation, respectively, in 12‐WAP samples with and without the pithiness phenotype.
As one step toward learning about genes and pathways ultimately to explain root pithiness, we phenotypically and genotypically analysed a total of 17 root samples collected from 6 to 10 WAP of line WK40 grown under both field and greenhouse conditions. To assess root pithiness accurately, each whole root was initially subjected to non‐destructive micro‐computer tomography (CT) scanning, after which the bottom part (excluding the outer layer, containing mostly xylem tissue where pithiness typically occurs) was excised for RNA extraction (Figure 1a; Movie S1). The micro‐CT results indicated that all six root samples collected from the greenhouse did not show the pithiness phenotype, whereas six out of 11 samples collected from the field exhibited visible pithiness phenotype (Figure S3). We therefore sequenced all 17 RNA samples to obtain over 477 million high‐quality clean RNA‐seq paired‐end reads (Table S1; EXPERIMENTAL PROCEDURES), and then used these for mapping to a total of 50 032 radish reference CDS sequences (Hoang et al., 2020; Jeong et al., 2016). The expression levels (FPKM) of all expressed transcripts are provided in Table S2, together with their Arabidopsis homologous gene IDs and the common gene names. A principal component analysis (PCA) based upon the 5000 most variable genes showed that the field‐collected samples were clustered separately from those collected from the greenhouse by PC1 (Figure 1b). Despite showing different phenotypes, field‐collected samples with and without the pithiness phenotype were not clearly separated by either of the two PCs. This suggests that at the transcriptional level, gene expression profiles were more similar among samples grown under the same conditions regardless of whether or not they showed the pithiness phenotype.
We identified 3966 differentially expressed genes [DEGs; false discovery rate (FDR) ≤ 0.05; |fold‐change| ≥ 2] by comparing three groups of samples, referred to here as the field non‐pithiness, field pithiness and greenhouse non‐pithiness groups (Table S3); and validated their expression levels by quantitative reverse transcriptase‐polymerase chain reaction (qRT‐PCR) on a set of 12 selected DEGs (r = 0.9, P < 0.0001; Table S4). Consistent with the PCA analysis, only 251 DEGs were identified in a comparison between field non‐pithiness and field pithiness, whereas 2461 and 3472 DEGs were identified by comparing each of the two groups with the greenhouse non‐pithiness samples, respectively (Figure 1c). A gene ontology (GO) enrichment analysis (FDR ≤ 0.05) of the total DEGs revealed 110 significantly over‐represented biological process terms, 16 molecular function terms and two cellular component terms (Table S5). Due to the large number of GO terms, we then focused on GOSlim categorizations differentially enriched in the DEGs to obtain a broad overview of the GO content. This indicated that response to stress (GO:0006950), cell wall (GO:0005618) and carbohydrate metabolic process (GO:0005975) were among the most significantly enriched terms (Figure 1d; Table S6). Consistent with these findings, a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis identified four enriched pathways (FDR ≤ 0.05; Table S7) in this case: the metabolic pathways (ath01100, 276 genes), biosynthesis of secondary metabolites (ath01110, 153 genes), the MAPK signalling pathway – plant (ath04016, 28 genes; Figure S4), and starch and sucrose metabolism (ath00500, 26 genes; Figure S5). Several genes in the MAPK signalling pathway are known to be associated with plant responses to oxidative stress, ROS homeostasis and hydrogen peroxide (H2O2)‐mediated cell death (Kovtun et al., 2000; Pitzschke and Hirt, 2006). Genes related to starch and sucrose metabolism could be part of the cell wall biosynthesis and carbohydrate metabolism processes, two processes that were enriched in the GOSlim analysis.
We then conducted K‐means clustering (Saeed et al., 2003) of the DEGs to obtain two major clusters based on their expression patterns. Cluster 1 consisted of 2107 upregulated DEGs in the field‐collected samples, while cluster 2 consisted of 1859 upregulated DEGs in the greenhouse‐collected samples (Figure 1e; Table S8). There were more enriched GO terms identified in cluster 1 than in cluster 2 (Figure 1f; Table S8). Cell death (GO:0008219) and antioxidant activity (GO:0016209) were among those significantly enriched in the upregulated genes in the field samples, of which the pithiness phenotype was prevalently observed. GO term antioxidant activity is part of cellular oxidant detoxification (GO:0098869), and in this group we found several DEGs that were annotated as sub‐terms, specifically superoxide dismutase activity (GO:0004784), peroxidase activity (GO:0004601), L‐ascorbate peroxidase activity (GO:0016688) and the ROS metabolic process (GO:0072593). A KEGG pathway analysis revealed phenylpropanoid biosynthesis (ath00940) and glycolysis/gluconeogenesis (ath00010) as two additional enriched pathways in cluster 1 (Figure S6), while no significantly enriched pathways were found in cluster 2 (Table S9). Collectively, our transcriptome data suggest that cell death, oxidative stress response and cell wall biosynthesis could be among those processes that are responsible for root pithiness formation. Indeed, using Evans Blue, 3,3‐diaminobenzidine (DAB) and nitro blue tetrazolium (NBT) staining as respective indicators of cell death and ROS accumulation (H2O2 and superoxide, O2 −), it was confirmed that, compared with those without any clear sign of pithiness, roots with pithiness were more apt to fix the dye and show a large stained area (Figures 1g and S7).
Pithiness in radish root is associated with the death of xylem parenchyma cells
Given that our transcriptome and histological data strongly supported the previous notion that radish root pithiness is associated with the death of xylem parenchyma cells (Magendans, 1991; Marcelis, 1997; Takano, 1963), we analysed the expression pattern of genes belonging to the cluster 1 DEGs (upregulated in the field samples) that are known to be involved in PCD. To this end, we compared Arabidopsis homologous gene IDs (to the radish cluster 1 DEGs) with a core set of 98 conserved developmentally induced PCD (dPCD) marker genes that were previously identified in Arabidopsis xylem tissues and the lateral root cap (Olvera‐Carrillo et al., 2015), thereby identifying 19 common dPCD genes (Figure 2a; Table S10). Considering that radish pithiness is also influenced by environmental factors, we also compared our data against a set of 84 environmentally induced PCD (ePCD) marker genes (Olvera‐Carrillo et al., 2015). These ePCD genes were upregulated at least twofold according to the results of several shared biotic, osmotic and genotoxic stress experiments. Here, we found another 13 ePCD genes that were common with our cluster 1 DEGs, with only one gene being commonly shared with the dPCD gene set (Figure 2a; Table S11). In total, 21 and 18 radish homologous genes were identified for the aforementioned 19 dPCD and 13 ePCD Arabidopsis genes, respectively, and their expression levels in our transcriptome data are shown in Figure 2b. A previous study indicated that distinct gene sets are involved in dPCD and ePCD, and most dPCD genes are not upregulated in ePCD‐induced conditions (Olvera‐Carrillo et al., 2015). We detected marker genes in both conditions (19/98 dPCD and 13/84 ePCD genes) that were differentially expressed, suggesting that both cell death processes occur in parallel during the formation of radish root pithiness.
Figure 2.
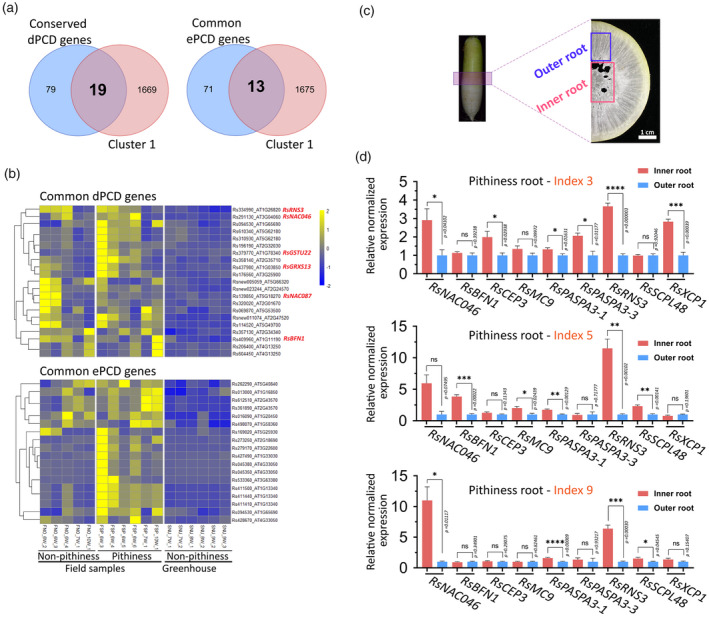
Pithiness in radish root is associated with the death of xylem parenchyma cells.
(a) Venn diagrams showing the common developmental and environmental programmed cell death (dPCD and ePCD) genes, respectively, between differentially expressed genes (DEGs) in cluster 1 in radish and PCD genes identified in the Arabidopsis study. Arabidopsis homologous gene IDs to the radish cluster 1 DEGs were used.
(b) Expression levels of radish homologous genes to common dPCD and ePCD genes (identified in panel a) in the radish transcriptome data. Gene‐wise normalized fragments per kilobase of exon per million mapped fragments (FPKM) values were used for the heatmaps. W represents WAP (weeks after planting) in the sample names.
(c) A cross‐section illustrating the tissues collected for spatial expression analyses of dPCD markers in radish outer and inner root pith tissues.
(d) Spatial expression levels of nine selected dPCD genes in the inner and outer root tissues as analysed by quantitative reverse transcriptase‐polymerase chain reaction (qRT‐PCR). Three 9‐WAP roots from pithiness line WK40 growing under the field condition (Figure 4a) were used. The pithiness index was scored for each root based on the index system in Figure 3a. For each sample, at least three technical replicates were run, with the results analysed by Student’s t‐tests using the outer tissue sample as a control. Expression values are quoted as the mean ± SEM. Asterisks; statistical significance based on Student’s t‐tests, ****P ≤ 0.0001, ***P ≤ 0.001, **P ≤ 0.01, *P ≤ 0.05 and non‐significant (ns) P > 0.05.
Our results revealed several well‐characterized dPCD regulators and executers in the common dPCD DEGs, including a RsNAC046, a RsNAC087, a RsBFN1, a RsRNS3, a GLUTAREDOXIN FAMILY PROTEIN (RsGRXS13) and a GLUTATHIONE S‐TRANSFERASE TAU 22 (RsGSTU22; Figure 2b). In Arabidopsis, ANAC046 and ANAC087 have been shown to be highly expressed in the xylem tracheary element cells and in the lateral root cap, and to mediate PCD (via the nuclease BFN1 and ribonuclease RNS3; Huysmans et al., 2018). AtBFN1 and AtRNS3 are known to be dPCD executers related to the cell content degradation process (Fujimoto et al., 2018; Olvera‐Carrillo et al., 2015), while AtGRXS13 and AtGSTU22 are involved in the response to photo‐oxidative stress through O2 ‐ radical detoxification (Laporte et al., 2012; Mueller et al., 2008). Because our transcriptome data were derived from whole‐root RNA samples, genes that are differentially expressed in a tissue‐specific manner (i.e. expressed highly in the outer root or in inner pith tissues) may not have been well captured. We therefore examined more closely the spatial expression patterns of three selected genes from the aforementioned dPCD DEGs and six other well‐known PCD marker genes (Fujimoto et al., 2018; Olvera‐Carrillo et al., 2015) in three 9‐WAP roots of line WK40 undergoing pithiness formation at different levels in the field condition. The expression levels of the nine genes, RsNAC046, RsBFN1, CYSTEINE ENDOPEPTIDASE 3 (RsCEP3), METACASPASE 9 (RsMC9), two RsPASPA3s, RsRNS3, SERINE CARBOXYPEPTIDASE‐LIKE 48 (RsSCPL48) and XYLEM CYSTEINE PEPTIDASE 1 (RsXCP1) were measured in two regions, the outer xylem and the inner pith (including the tissue part showing pithiness) of each root (Figure 2c). In general, the results suggest that these genes were expressed either at higher levels in the inner root compared with the outer root (P ≤ 0.05), or at the same level between the two root regions. Moreover, all nine selected PCD genes were expressed at a significantly higher level in the inner tissue in at least one out of the three roots (Figure 2d; Table S12), indicating that these genes were both spatially and temporally differentially expressed during pithiness formation. Taken together, our results strongly suggest that genes responsible for PCD execution are induced in the xylem parenchyma cells of radish roots undergoing pithiness formation.
QTL analyses point to a transcriptional switch responsive to oxidative stress for radish root pithiness
To identify the genomic regions associated with radish root pithiness, we generated an F2 population from a cross between the pithiness inbred line WK40 and the non‐pithiness inbred line WonKyo10029ho (WK29; Figure 3a), and subjected it to a QTL analysis. It is noteworthy that F1 individuals typically showed no pithiness phenotype, suggesting that non‐dominant genetic factor(s) contribute to this phenotype. After growing 92 F2 individuals in the NIHHS field for 9 WAP, we harvested the root and scored the pithiness of vertically cut taproots (Figure 3a; also see Experimental Procedures and Table S23 for the phenotyping procedure and data). For genotyping, we employed the genotyping‐by‐sequencing (GBS) method with the EcoRI‐MseI restriction enzyme cut and subsequent sequencing nearby the cut sites to retrieve genetic markers (Poland et al., 2012). A GBS library of the F2 progenies and the two parental lines was sequenced, which resulted in more than 216 million clean single‐end reads with indices (Table S13 and Experimental Procedures). After mapping against the radish genome v2.20 (GenBank: GCA_002197605.1), a total of 17 054 raw single‐nucleotide polymorphisms (SNPs) were called, of which 2702 were biparental SNPs, with 224 SNPs found to segregate at a 1:2:1 ratio in the F2 individuals (Table S14). Through a genome‐wide linkage analysis of these 224 SNPs with the pithiness scores, the QTL analysis resulted in only one major peak via Bonferroni correction (α = 0.05) of chromosome 1 surrounding position 17.8 Mb (locus S1_17807256; LOD = 6.49, hereafter referred to as qPITH‐1; Figures 3b and S8).
Figure 3.
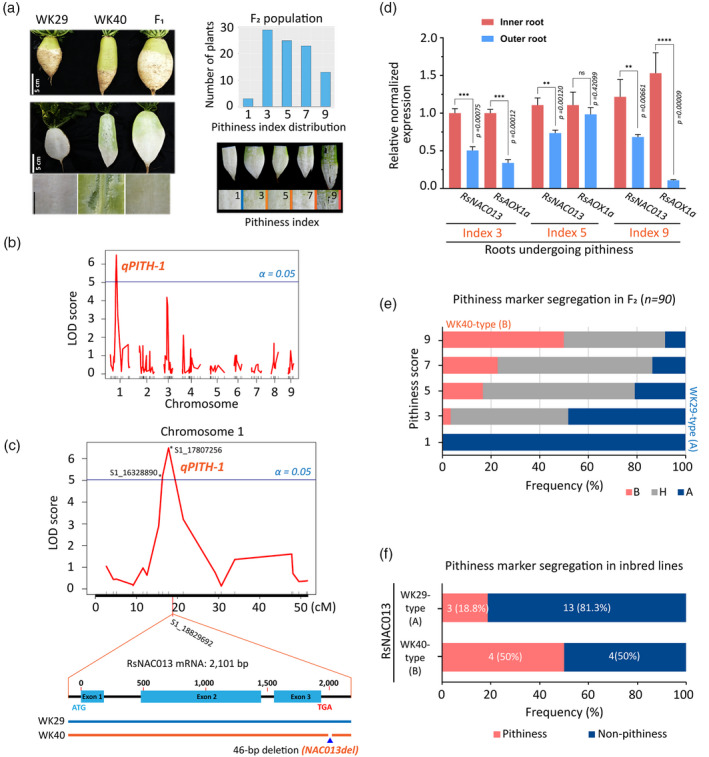
Identifying quantitative trait locus (QTL) for radish root pithiness.
(a) Left, parental inbred lines (WK29 and WK40), F1 and F2 progenies used for the QTL analysis in this study. Samples were collected at 9 WAP (weeks after planting). Right, root pithiness index used for phenotyping and determining the distribution of the pithiness index among the F2 individuals.
(b) The logarithm of odds (LOD) score plot of nine radish chromosomes according to a genome scan with composite interval mapping (CIM) method using R/qtl package and the identification of a major QTL (qPITH‐1; locus S1_17807256; α = 0.05) on chromosome 1. The plot was generated using 224 single‐nucleotide polymorphisms (SNPs) and the five‐point pithiness index data. Radish genome v2.20 (GenBank: GCA_002197605.1) was used. The blue line indicates the 5% significant LOD threshold = 5.03.
(c) A CIM result of chromosome 1 showing the major qPITH‐1 QTL for the radish root pithiness phenotype, and the location of candidate gene RsNAC013 with a 46‐bp deletion (NAC013del marker) in line WK40 compared with line WK29.
(d) Expression levels of RsNAC013 and its direct target RsAOX1a in inner and outer root samples as analysed by quantitative reverse transcriptase‐polymerase chain reaction (qRT‐PCR). Three 9‐WAP roots from pithiness line WK40 growing under the field condition (Figure 4a) were used. The pithiness index was scored for each root based on the index system in (a). Sample collection is illustrated in Figure 2c. For each sample, at least three technical replicates were run, with the results analysed by Student’s t‐tests using the outer tissue sample as a control. Expression values are quoted as the mean ± SEM. Asterisks; statistical significance based on Student’s t‐test, ****P ≤ 0.0001, ***P ≤ 0.001, **P ≤ 0.01, *P ≤ 0.05 and non‐significant (ns) P > 0.05.
(e) Correlation of the two pithiness markers (NAC013del and qPITH‐1 markers) and pithiness score among 90 F2 samples. The segregation of the two markers among the F2 samples based on PCR results and genotype‐by‐sequencing (GBS) data was 100% consistent (n = 90). A allele denotes WK29 type, while B and H denote WK40 and heterozygous types, respectively.
(f) A significant under‐representation of WK40‐type allele of RsNAC013 (NAC013del) among 24 radish inbred lines.
As our transcriptome analysis suggested that ‘cell death’ and the ‘oxidative stress response’ are related to pithiness, we searched for protein coding genes and TFs that are annotated for these terms located within an extended 8‐Mb interval (11–19 Mb, up‐ and downstream of the major qPITH‐1 QTL; Table S15). To this end, we initially compared a total of 1400 radish genes that significantly hit this interval (e‐value = 0, High Scoring Pair length from 350 to 4751 bp) with the 3966 pithiness DEGs identified in our transcriptome analysis. This revealed 97 genes commonly shared between the two datasets, including seven TF genes (Table S15). Among these, we focused on a gene that encodes a TF homologous to Arabidopsis ANAC013 (AT1G32870), located at locus S1_18829692, approximately 1 Mb downstream of the qPITH‐1 QTL marker (Figure 3c). ANAC013, also known as NAC WITH TRANSMEMBRANE MOTIF1‐LIKE (NTL) 1 (Kim et al., 2007), is a membrane‐bound TF that functions during mitochondrial retrograde regulation (MRR) and thereby triggers an increased oxidative stress response in Arabidopsis (De Clercq et al., 2013). The expression of ANAC013 is induced by several factors including UV‐B and R irradiation (Safrany et al., 2008) and chemicals causing mitochondrial dysfunction conditions (e.g. antimycin A or rotenone; De Clercq et al., 2013). Radish NAC013 (Rs333230) and its direct target ALTERNATIVE OXIDASE1a (RsAOX1a), a mitochondrial dysfunction stimulon (MDS) gene (De Clercq et al., 2013), were upregulated in the field‐pithiness samples (Table S16). Additionally, the expressions levels of RsNAC013 and RsAOX1a were generally significantly higher (P ≤ 0.05) in the inner tissue as compared with the outer tissue of three 9‐WAP roots of line WK40 undergoing pithiness in the field condition (Figure 3d; Table S16).
By mapping the whole‐genome sequencing data of the two parental lines to the RsNAC013 genomic sequence, we found five SNPs within the coding sequences (Figure S9a; Table S17). Furthermore, the 3′‐UTR regions showed significant variations between lines WK40 and WK29, including a 46‐bp deletion in line WK40 and several SNPs/small INDELs, while the 5′‐UTR sequences were identical (Figure S9a,b). All of these differences were validated by the Sanger sequencing of CDS and genomic DNA (Table S18). Among the five SNPs within the CDS sequences, two were found to be non‐synonymous, while the other three were synonymous (Figure S9c,d). Furthermore, the segregation of the 46‐bp deletion in the 3′‐UTR of RsNAC013 (termed as NAC013del) among the F2 samples based on PCR results was 100% consistent (n = 90) with the aforementioned GBS qPITH‐1 QTL marker on chromosome 1 (Figure S9e; Table S19). Genotyping results of these markers indicated that all non‐pithiness F2s (pithiness index score = 1) had the WK29‐type allele (A), while approximately 85% of those with pithiness (index score > 5) were heterozygous (H) or homozygous (B) for the WK40‐type allele (Figure 3e).
We also sought to determine if the NAC013del marker can explain the pithiness phenotype in other recombinant inbred lines. A collection of 22 inbred lines as well as WK29 and WK40 were planted in the NIHHS field, and the presence of pithiness and RsNAC013 genotypes was examined (Figure 3f; Table S19). In this analysis, we found a significant under‐representation of the WK40‐type allele of RsNAC013 (8 out of 24 inbred lines examined). This under‐representation may stem from the negative selection of inbred lines with the pithiness phenotype. Consistent with the F2 analyses, 50% of lines with the WK40‐type allele showed pithiness (four out of eight), while only 18.8% of lines with the WK29‐type allele did so (three out of 16). Together, these results indicate that the sequence variations and expression changes found in RsNAC013 are strongly associated with the root pithiness phenotype.
RsNAC013 pathway is upregulated in roots with pithiness phenotype
To gain a deeper understanding of the potential role of RsNAC013 during the formation of radish root pithiness, we measured its expression level together with those of genes related to oxidative pathways and dPCD in 9‐WAP roots collected from the pithiness line WK40 and non‐pithiness line WK29 (as a control) grown in the NIHHS field (Figure 4a). Among the three WK40 individuals collected, one showed very mild pithiness (index 3), while the other two exhibited clear pithiness phenotype (index of 5 and 9). The WK29 sample, as expected, did not show any signs of pithiness (index 1). In total, 66 genes were selected based on previous Arabidopsis studies of ANAC013 (De Clercq et al., 2013; Ng et al., 2013), ROS production and scavenging (Orman‐Ligeza et al., 2016; Sagi and Fluhr, 2006), and dPCD (Fujimoto et al., 2018; Olvera‐Carrillo et al., 2015). These genes formed an interconnected co‐expression network with the ANAC013 direct target, AtAOX1a, being the central node (Figure S10). Interestingly, among 61 Arabidopsis genes homologous to the 66 selected radish genes, 24 genes (approximately 39%), including ANAC013 and AtAOX1a, were upregulated in ANAC017 overexpression lines compared with the wild‐type (Meng et al., 2019; Table S20). This could be expected, as ANAC013 is functionally homologous to and modulated by ANAC017 (De Clercq et al., 2013); accordingly, their target genes and pathways may partially overlap. To determine the whole‐root expression level, RNAs were collected from four different areas (top, middle outer, middle inner and bottom) in each root and equally pooled into a single sample for analysis (Figure S11a). The expression levels of 66 genes categorized based on their functions, regulators of AtAOX1a, ROS‐related, ANAC013 downstream and PCD‐related genes are presented in Figure 4b and Table S20.
Figure 4.
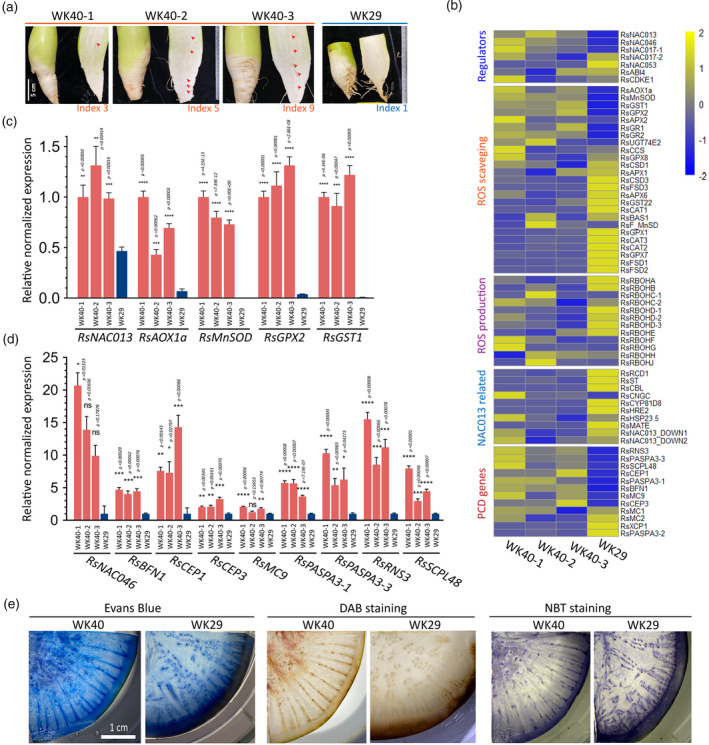
Expression survey of genes involved in the RsNAC013 pathway, oxidative stress response and programmed cell death (PCD).
(a) Root samples used for the gene expression analysis by quantitative reverse transcriptase‐polymerase chain reaction (qRT‐PCR), for which the results are presented in panel (b). Samples were collected at 9 WAP (weeks after planting) from plants grown in the field condition. Three samples from pithiness line WK40 showed different levels of pithiness. One sample collected from non‐pithiness line WK29 was included as a control sample. The pithiness index was scored for each root based on the index system in Figure 3a. NAC013‐related genes refer to those reported to be modulated by ANAC013 in Arabidopsis. For visualization, gene‐wise normalized average expression values from three technical replicates in qRT‐PCR were used for the heatmaps.
(c, d) The high induction of genes related to oxidative stress response, cell detoxification (c) and cell death (d) in samples undergoing pithiness formation in line WK40 compared with that of line WK29. For qRT‐PCR, RNA were collected from four different areas (top, middle outer, middle inner and bottom) in each root and pooled into a single sample for analysis. For each pooled sample, at least three technical replicates were run, with the results analysed by Student’s t‐tests compared with samples from line WK29. Expression values are quoted as the mean ± SEM. Asterisks; statistical significance based on Student’s t‐tests, ****P ≤ 0.0001, ***P ≤ 0.001, **P ≤ 0.01, *P ≤ 0.05 and non‐significant (ns) P > 0.05.
(e) Histological analysis of samples from the two parental lines at 12 WAP using Evans Blue staining for cell death, DAB (3,3‐diaminobenzidine) staining for hydrogen peroxide (H2O2) accumulation and NBT (nitro blue tetrazolium) staining for superoxide (O2−) accumulation.
In this analysis, we found strikingly high upregulation (P ≤ 0.05) in the line WK40 samples compared with WK29 for several genes related to the ANAC013 pathway, including RsNAC013 (2.1–2.8‐fold), RsAOX1a (6.3–14.7‐fold), MANGANESE SUPEROXIDE DISMUTASE 1 (RsMnSOD/MSD1, from 3838‐ to 4820.8‐fold), GLUTATHIONE PEROXIDASE 2 (RsGPX2, 21–31.1‐fold) and GLUTATHIONE S‐TRANSFERASE 1 (RsGST1, 121–162‐fold; Figure 4c). AtAOX1a, AtMnSOD, AtGPX2 and AtGST1 take part in cell detoxification and are normally induced by a high level of ROS accumulation. AtAOX1a plays a role in lowering mitochondrial ROS formation and maintaining ROS equilibrium in plant cells (Giraud et al., 2008), and can be activated by several factors, including inhibitors of the mitochondrial electron transport chain (e.g. antimycin A or rotenone), nutrient availability (e.g. phosphate, nitrate) or mitochondrial genetic defects (De Clercq et al., 2013; Giraud et al., 2008; Pham et al., 2018). The upregulation of RsAOX1a indicates that the radish roots undergoing pithiness exhibited high stress response, with a shift toward the alternative oxidative pathway from normal oxidative respiration. Because the AtAOX1a gene is modulated directly by ANAC013 via a mitochondrial dysfunction motif (a cis‐regulatory element) on its promoter region (De Clercq et al., 2013), the upregulation of the corresponding homologous genes in radish line WK40 may indicate their involvement in root pithiness. The expression levels of other radish homologous genes to those also known to directly influence AtAOX1a, including RsNAC017, RsABI4 and RsCDKE1 (Ng et al., 2013), were induced at a low level or inconsistent among the pithiness samples, suggesting that they may not be responsible for the induction of RsAOX1a in this case. AtMnSOD, a key component of the plant antioxidant defence system, catalyses the destruction of O2 ‐ to H2O2 and O2 (Bowler et al., 2011); AtGPX2 and AtGST1, in turn, take part in the conversion process of H2O2 to H2O (Bela et al., 2015; Dixon and Edwards, 2010). The high induction of these genes is in line with the enriched GO terms ‘antioxidant activity’/‘cellular oxidant detoxification’ found in the upregulated DEGs of the field‐collected samples and the higher levels of H2O2 and O2 − accumulation in roots of line WK40 undergoing pithiness compared with WK29 as revealed by DAB and NBT staining (Figures 4e and S12).
It is important to note here that, in our data, two TFs that mediate the MRR signalling pathway, RsNAC013 and RsNAC017, were expressed higher in roots undergoing pithiness of line WK40 compared with that of line WK29 (2.1–2.8‐fold and 1.2–1.6‐fold, respectively, P ≤ 0.05). In contrast, the expression of the inhibitor of the MRR signalling pathway, RADICAL‐INDUCED CELL DEATH1 (RsRCD1; Shapiguzov et al., 2019), was higher in WK29 root compared with WK40 (>2‐fold, P ≤ 0.05; Figure 4b; Table S20). More recently, Shapiguzov et al. (2020) found that the increased expression of genes in MRR pathways is linked to hypoxic response in Arabidopsis, probably due to the alteration of O2 availability by MDS gene products. The authors identified a total of 19 genes commonly upregulated in rcd1 mutant and wild‐type plants treated with antimycin A or hypoxia. We found 8 out of 19 genes overlapped with our DEG list (including genes with more than one copy in the radish genome, totaling 13 Rs genes). Notably, these included HYPOXIA RESPONSIVE UNIVERSAL STRESS PROTEIN 1 (RsHRU1), RsWRKY25 and several MDS genes (De Clercq et al., 2013) RsAOX1a, SULFOTRANSFERASE 1 (RsSOT12/ST) and CYTOCHROME P450 81D8 (RsCYP81D8). The expression of the 13 Rs DEGs together with other radish genes homologous to common 19 genes in the MRR signalling pathway that linked to hypoxia are shown in Figure S11b. Most of the 13 Rs DEGs, except one, were upregulated in the field samples, especially in pithiness roots, compared with the greenhouse samples. These data strongly confirm that the MRR signalling pathway was involved in radish root pithiness formation.
Among nine NADPH oxidase RESPIRATORY BURST OXIDASE HOMOLOG (RBOH) genes (Orman‐Ligeza et al., 2016), most (except RsRBOHF and RsRBOHG) were either downregulated in WK40 or not significantly differentially expressed between the two lines (Figure 4b). RsRBOHF was upregulated in one WK40 roots (2.7‐fold, P ≤ 0.05), while RsRBOHG was only upregulated in one root (2.3‐fold, P ≤ 0.05). Moreover, only two RsRBOHD genes were identified as DEGs in our transcriptome analysis between WK40 samples from different conditions. This suggest that these genes were either not responsible for ROS production or were induced very early in this case. In relation to cell death, nine selected PCD genes (RsNAC046, RsBFN1, RsRNS3, RsCEP1, RsCEP3, RsMC9, RsPASPA3s and RsSCPL48) were upregulated in the samples from line WK40 (Figure 4d), consistent with the stronger Evans Blue staining of this line compared with WK29 (Figures 4e and S12). Our results collectively support that oxidative stress responses and PCD are part of pithiness formation in radish root, and RsNAC013 acts upstream of these processes.
Artificial induction of oxidative stress enhances the root pithiness
The oxidative stress response and PCD can sequentially coordinate with each other under a given stress condition. For example, in Arabidopsis, heat‐stress‐induced H2O2 activates ANAC053 (NTL4) and NTL4‐mediated signals in turn promote PCD (Lee et al., 2014). Moreover, as a major stress sensor, any disturbance in mitochondrial signalling pathway may lead to PCD activation (Burke et al., 2020). We thus asked whether artificial induction of these two processes can enhance the formation of radish root pithiness. To this end, we treated WK40 plants grown in the greenhouse with 300 mm of exogenous H2O2 to trigger oxidative stress and subsequently analysed the phenotypic changes. We chose 7‐WAP (early) and 10‐WAP (late) samples to start the stress experiment, as pithiness formation is expected to start later in the greenhouse condition compared with that in the field. The expression levels of RsNAC013 and RsAOX1a, as oxidative stress markers, were indeed upregulated (P ≤ 0.05) after 1 day of the H2O2 treatment in the treated group line WK40 in comparison with the non‐treated lines WK40 and WK29 (Figure 5a; Tables S21 and S22, for two independent replicates). This is in line with the findings of a previous study on Arabidopsis (De Clercq et al., 2013), in which an H2O2 treatment induced the expression of AtAOX1a and MDS genes, which are directly mediated by ANAC013. To capture the long‐term phenotypic changes induced by the application of exogenous H2O2, roots were collected and examined at 12 WAP (for the early‐treated plants, for a total of 5 weeks of treatment) and 14 WAP (for the late‐treated plants, for a total of 4 weeks of treatment). At these stages, the expression of RsNAC013 was similar (P > 0.05) to that in the non‐treated WK40 and WK29 plants (Figure 5b,c; Table S21). The expression of RsAOX1a, on the other hand, remained high in both 12‐ and 14‐WAP treated plants, whereas it gradually increased from 12 WAP to 14 WAP in the non‐treated WK40 plants. The results suggest that the induction of these genes occurs earlier in treated plants compared with non‐treated plants of line WK40. This also indicates that RsNAC013 and RsAOX1a may be naturally induced in line WK40 toward the end of the root‐thickening stage (in the presence of inducing environmental factors), whereas they remain lowly expressed in the non‐pithiness line WK29.
Figure 5.
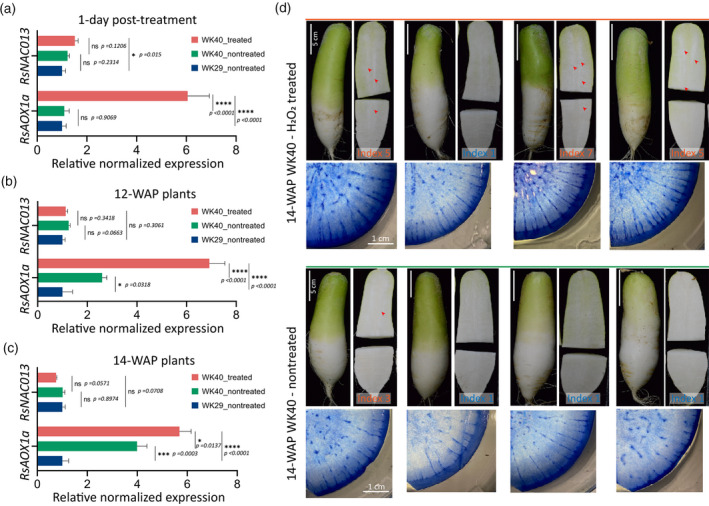
Radish oxidative stress experiment.
(a–c) Expression levels of RsNAC013 and RsAOX1a in radish roots collected at 1 day after the hydrogen peroxide (300 mm H2O2) treatment and at 12 WAP (weeks after planting) and 14 WAP (corresponding to early and late long‐term H2O2 treatments). For the 12‐WAP and 14‐WAP samples, the treatment started at 7 WAP and 10 WAP, respectively, and was repeated once a week until 1 week before collection. RNAs were collected from seven roots (12 WAP) and four roots (for other stages), and equally pooled into a single sample for each treatment for quantitative reverse transcriptase‐polymerase chain reaction (qRT‐PCR) analysis. Four technical replicates were run for each pooled sample, with the results analysed by one‐way anovas (Fisher’s LSD post hoc tests). Expression values are quoted as the mean ± SEM, normalized against line WK29 data. Asterisks; statistical significance, ****P ≤ 0.0001, ***P ≤ 0.001, **P ≤ 0.01, *P ≤ 0.05 and non‐significant (ns) P > 0.05.
(d) Whole roots, longitudinal cross‐sections and Evans blue staining of the 14‐WAP samples from the H2O2 treatment experiment in panel (c). Only root samples from the pithiness line WK40 (treated and non‐treated) are shown. None of the non‐treated samples from the non‐pithiness line WK29 exhibited the pithiness phenotype, the corresponding cross‐sections are presented in Figure S14. The pithiness index was scored for each root based on the index system in Figure 3a.
Among the seven early‐treated plants collected at 12 WAP, two showed pithiness phenotype (one with clear spongy tissue and one with a very mild level of pithiness, index score of 5 and 3, respectively), whereas none of the seven non‐treated plants showed clear signs of pithiness (Figure S13). However, at 14 WAP, we observed three out of four late‐treated plants that exhibited a clear pithiness phenotype, from an intermediate to an almost severe level (index score of 5 and 7), in contrast to one out of four non‐treated plants that showed a very mild level of pithiness (index of 3; Figure 5d). None of the 12‐ and 14‐WAP non‐treated plants from line WK29 included in the experiment showed a sign of pithiness (Figure S14).
Non‐synonymous changes enhancing RsNAC013 activity provide a genetic condition for the root pithiness
Our investigation pointed RsNAC013 as a strong candidate associated with root pithiness phenotype. Because there are sequence variations within the 3′‐UTR region and CDS of RsNAC013 between two parental lines, we investigated whether these polymorphisms affect RsNAC013 function. We asked if the differences in the 3′‐UTR region influence the transcription, mRNA stability or translation. To address this question, we generated Arabidopsis transgenic lines expressing nlsGFP under GL2 promoter and attached the 3′‐UTR of RsNAC013 from either WK29 or WK40 after CDS of nlsGFP. We reasoned that if the 3′‐UTR of RsNAC013 has any role, then we would see the different levels of nlsGFP signal. However, we did not find any difference in the nlsGFP signal between pGL2::nlsGFP:3′UTRWK29 and pGL2::nlsGFP:3′UTRWK40 (Figure 6a,b), indicating that the difference in the 3′‐UTR of RsNAC013 does not lead to a functional difference.
Figure 6.
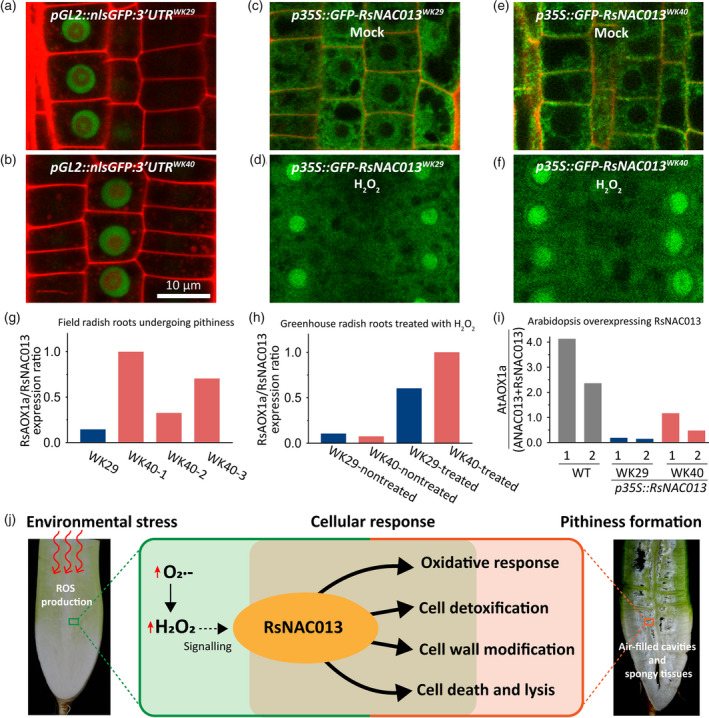
Analysis of transcriptional activity of two RsNAC013 variants in radish inbred lines and Arabidopsis overexpression lines.
(a–f) Confocal microscopy images of root epidermis in the Arabidopsis pGL2::nlsGFP:3′UTRWK29 (a), pGL2::nlsGFP:3′UTRWK40 (b), p35S::GFP‐RsNAC013WK29 mock (c) and H2O2 treatment (d), and p35S::GFP‐RsNAC013WK40 mock (e) and H2O2 treatment (f).
(g) RsAOX1a/RsNAC013 expression ratio in radish root samples collected from the field condition. Expression data from three 9‐WAP (weeks after planting) roots from pithiness line WK40 growing under the field condition (Figure 4a; Table S22) were used. Three samples from line WK40 showed different levels of pithiness, while sample from the non‐pithiness line WK29 did not show pithiness.
(h) RsAOX1a/RsNAC013 expression ratio in 8‐WAP radish root samples grown in greenhouse condition treated with H2O2 for 1 day. All samples showed no pithiness phenotype at the time collected. RNAs were collected from pooled samples of four roots for each treatment. At least four technical replicates were run for each sample. A same pattern was also observed in an independent biological replicate in Figure 5a and Table S22. The expression levels used in panels (g) and (h) were obtained by quantitative reverse transcriptase‐polymerase chain reaction (qRT‐PCR).
(i) AtAOX1a/(ANAC013 + RsNAC013) expression ratio in Arabidopsis transgenic lines overexpressing RsNAC013 CDS sequences from lines WK29 and WK40 (p35S::RsNAC013WK29 and p35S::RsNAC013WK40 , respectively). The expression level of each gene was measured by Droplet Digital PCR (ddPCR; Table S22). Arabidopsis thaliana (Col‐0) was used as a background wild‐type (WT). Two independent plants from each line were used.
(j) A model of transcriptional regulation of the oxidative stress response and programmed cell death (PCD) during root pithiness formation in radish. During root pithiness formation at the thickening stage, a high reactive oxygen species (ROS) accumulation is activated by stress factors, which in turn sends signals to trigger the expression of genes related to the oxidative stress response, cell detoxification, cell wall biosynthesis/modification and PCD.
Next, we tested whether the differences in the CDS (including two amino acid changes) affect the protein function. RsNAC013 contains a NAC domain in the N‐terminus and a transmembrane motif (TM) in its C‐terminus (Figure S9d). It is known that ANAC013 and ANAC017 are localized in the endoplasmic reticulum, released to the nucleus upon proteolytic cleavage, and then activate downstream target genes (De Clercq et al., 2013; Ng et al., 2013). Given this information, we examined whether the differences in the two alleles of RsNAC013, RsNAC013WK29 and RsNAC013WK40 affect subcellular localization, proteolytic cleavage or TF activity. For that, we generated Arabidopsis transgenic lines overexpressing GFP‐tagged RsNAC013 (p35S::GFP‐RsNAC013WK29 and p35S::GFP‐RsNAC013WK40 ), and observed that both RsNAC013WK29 and RsNAC013WK40 were localized in the ER and translocated to the nucleus after H2O2 treatment (Figure 6c–f). This result suggests that RsNAC013 is post‐translationally processed in the same manner as ANAC013, and RsNAC013WK29 and RsNAC013WK40 alleles do not have any difference in the subcellular localization and proteolytic cleavage.
To explore the possibility that RsNAC013WK29 and RsNAC013WK40 have different activities as TF, we used RsAOX1a expression as a readout of RsNAC013 activity in radish lines. We expected that if the two variants of RsNAC013 exhibit differential biological activities, then the relative expression of RsAOX1a against RsNAC013 in WK29 and WK40 would be different. Indeed, in our qRT‐PCR analyses, the RsAOX1a/RsNAC013 expression ratio was higher in 9‐WAP field‐collected WK40 roots undergoing pithiness than in WK29 roots (Figure 6g; Table S22). We also measured RsAOX1a/RsNAC013 in storage roots when H2O2 was treated for 1 day to WK29 and WK40 plants grown for 8 WAP in the greenhouse. In this stress experiment, RsNAC013 was induced to a comparable level in both lines. By contrast, the induction of RsAOX1a was significantly higher in WK40 than WK29 (Table S22). Considering that the RsAOX1a/RsNAC013 expression ratio is not much different in WK40 and WK29 without H2O2 treatment, the activity of RsNAC013WK40 seems further enhanced when H2O2 is applied (Figure 6h). These results suggest that RsNAC013WK40 may be more active than RsNAC013WK29.
To confirm this finding in a heterologous system, we examined the activities of RsNAC013WK29 and RsNAC013WK40 in Arabidopsis using p35S::GFP‐RsNAC013WK29 and p35S::GFP‐RsNAC013WK40 lines. ANAC013 activates its own expression as well as AtAOX1a (De Clercq et al., 2013), thus RsNAC013 in Arabidopsis could activate not only AtAOX1a but also ANAC013. This autoregulatory activity of NAC013 means that the induction of AtAOX1a in these transgenic lines is a result of combined activities of both RsNAC013 and ANAC013. Using Droplet Digital PCR (ddPCR), we measured the absolute expression level of RsNAC013, ANAC013 and AtAOX1a, and then calculated the relative expression of AtAOX1a against the summed expression of RsNAC013 and ANAC013 (Figure 6i; Table S22). We found a clear difference in AtAOX1a/(RsNAC013 + ANAC013) expression ratio among the wild‐type, p35S::GFP‐RsNAC013WK29 and p35S::GFP‐RsNAC013WK40 lines. Overall, this expression ratio was noticeably lower in p35S::GFP‐RsNAC013WK29 (4–8%) and p35S::GFP‐RsNAC013WK40 (12–50%) compared with that in the wild‐type. Our result indicates that both RsNAC013 variants expressed in Arabidopsis are less active than endogenous ANAC013. More importantly, however, it confirms that RsNAC013WK40 is a more potent transcriptional activator than RsNAC013WK29, suggesting that non‐synonymous substitutions in RsNAC013 of WK40 line led to the pithiness phenotype.
CONCLUSION
Root pithiness, observed during the later stage of the storage root‐thickening process in radish, is the formation of spongy textured regions and/or empty cavities likely via the death of xylem parenchyma cells. Farmers discard the radish harvest if they find pithiness in the roots because these roots no longer have any commercial value. Though breeders have attempted to exclude the pithiness trait using traditional breeding programmes, this effort has proven challenging because the pithiness phenotype appears to be influenced by complex interactions between genotypes and environmental components.
This study aims to investigate a molecular basis and an environmental cue triggering radish root pithiness. Combining transcriptome data, QTL mapping based on the GBS of an F2 population between two radish inbred lines, and gene function analysis using Arabidopsis overexpression lines, we propose that radish root pithiness is caused by transcriptional switch(es) that activate oxidative stress response pathways, followed by PCD. During the formation of root pithiness at the thickening stage, ROS accumulated in response to stresses sends out signals to trigger the expression of genes related to the oxidative stress response, cell detoxification, cell wall biosynthesis/modification and PCD (Figure 6j). We identified a major QTL that includes RsNAC013 as a potential switch for the aforementioned pathways. The data and approaches presented in this investigation will be useful to those who seek to improve radish crop quality through molecular breeding programmes.
EXPERIMENTAL PROCEDURES
Plant materials and growth conditions
Two radish inbred lines WK29 and WK40 and the corresponding F1 and 92 F2 progenies were obtained from the National Institute of Horticultural and Herbal Science (NIHHS) of the Republic of Korea. For samples collected from the field, radishes were grown using the common agronomic practices at the NIHHS in Wanju (35′83″N/127′03″E), Korea. For the experiments in the greenhouse (Seoul National University, Korea), each radish seed was germinated in a pot (20 × 20 × 20 cm3) filled with soil (Nongwoo Bio, Suwon, Gyeonggi, Korea) and growth at 22°C in long‐day conditions (16 h light/8 h dark periods; 156 µmol m−2 sec−1) before sample collection. Each plant was given 1 L of water with 1 ml of a Hyponex nutrient solution 6‐10‐5 (Hyponex, Osaka, Japan) weekly.
Radish root pithiness phenotyping
A phenotypic analysis of radish root pithiness was carried out for the F2 population and the parental lines grown in the field condition using an index system ranging from 1 (non‐pithiness), 3 (partial dryness of the tissue), 5 (intermediate, spongy and small cavities), 7 (spongy tissues and medium cavities) to 9 (severe pithiness phenotype; Figure 3a). Roots were longitudinally sectioned into two halves with a kitchen knife and scored by a panel of judges from the NIHHS (Table S23). The five‐point pithiness index data were subsequently utilized for R/qtl mapping. For transcriptome samples, roots were phenotyped using a micro‐CT scanning system SkyScan 1276 (Bruker, Billerica, MA, USA) before being processed for RNA extraction.
RNA and DNA extraction and sequencing
For total RNA extraction, the lower elongated part of the root was excised, frozen in liquid nitrogen, and pulverized into a fine powder in cryo‐jars using a Retsch TissueLyser (Retsch, Haan, Germany) with a frequency of 25/S for 1 min and 30 sec. An amount of approximately 100 mg of the frozen pulverized sample powder was used for RNA extraction using a RNeasy® Plant Mini Kit (Qiagen, Valencia, CA, USA) with an on‐column DNase digestion step to remove DNA. The RNA quality, integrity and quantity were determined by a Nanovue spectrophotometer (GE Healthcare, South Plainfield, NJ, USA) and by a 2100 Agilent Bioanalyser with a plant RNA NanoChip assay (Agilent Technologies, Santa Clara, CA, USA). For RNA‐seq, samples for which RIN = 9.6 and above were used for library preparation (Table S24).
For the extraction of the total genomic DNA, each leaf sample was collected, frozen in liquid nitrogen and ground to a fine powder using metal beads in an Eppendorf tube before extraction. An amount of approximately 100 mg of the powder sample was used for DNA extraction via the CTAB method (Clarke, 2009) with modifications for radish root tissues, or with a Qiagen DNeasy Plant Mini Kit (Qiagen). DNA quality and quantity levels were assessed using a Nanovue spectrophotometer (GE Healthcare), a PicoGreen assay at NICEM (Seoul National University, Korea) and by resolving on 1% agarose gel with SYBR safe (Invitrogen, Waltham, MA, USA) staining.
Transcriptome sequencing and data analysis
An amount of approximately 1 µg of each of 17 root RNA samples was used for indexed‐library preparation (average insert size of > 300 bp), employing the TruSeq stranded method with a Ribo‐Zero Plant Library Prep Kit as preparation for the total RNA library (Illumina, San Diego, CA, USA). Samples were prepared and sequenced using an Illumina HiSeq4000 instrument to obtain 100‐bp paired‐end read data at Macrogen (Seoul, Korea). Read quality before and after trimming was assessed by FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Adapter sequences, low‐quality and duplicate reads were trimmed using Trimmomatic (Bolger et al., 2014) with the following parameters: ‘ILLUMINACLIP: 2:30:10 SLIDINGWINDOW:4:15 LEADING:5 TRAILING:5 MINLEN:50’.
To estimate transcript abundance, high‐quality clean RNA‐Seq reads were aligned to a reference gene set of 50 032 radish CDS sequences (Hoang et al., 2020; Jeong et al., 2016) using BOWTIE v2.3.5 (Langmead and Salzberg, 2012) with default settings. The mapping files were sorted by SAMTOOLS‐1.3.1 (Li et al., 2009) and subjected to RSEM v1.2.31 (Li and Dewey, 2011) for transcript abundance quantification. The expression level was normalized to as fragments per kilobase of the feature sequence per million fragments mapped (FPKM). The DESeq2 package (Love et al., 2014) was used to identify the DEGs between the sample groups with a FDR‐adjusted P‐value of ≤ 0.05 and a |fold change| ≥ 2. Within two groups (pithiness and non‐pithiness) of the field‐collected samples, when comparing the combined data from the 6‐, 7‐ and 10‐WAP samples, we did not obtain any DEGs; therefore, only the 6‐WAP samples were used. For comparisons between the field and greenhouse conditions, samples from all three time points were used. The DEGs were combined and clustered by MeV software v4.9.0 using a K‐means approach (Saeed et al., 2003) for further analyses. Finally, we utilized qRT‐PCR to validate the expression levels of 12 selected DEGs that were part of our expression survey of genes related to RsNAC013, oxidative stress and PCD pathways.
GO and KEGG pathway analysis of DEGs
The GO term and KEGG pathway enrichment analyses were done via a hypergeometric distribution test on a comparison against the whole reference gene set as the background. GO and GOSlim categories were analysed with the BINGO package (Maere et al., 2005) in Cytoscape v3.8.2 (www.cytoscape.org) using the Arabidopsis homologous gene IDs of the DEGs. Only enriched GO terms with an FDR‐adjusted P‐value of ≤ 0.05 were utilized to look for over‐represented cellular components, functions and processes in the DEGs. KEGG pathway enrichment was conducted using the KEGG Orthology‐Based Annotation System (KOBAS) v3.0 (Xie et al., 2011).
Whole‐genome sequencing of parental lines and genome‐wide SNP detection
Amounts of approximately 3 µg of each DNA sample were used for library preparation and sequenced with the Illumina HiSeq 4000 platform at Macrogen to obtain 100‐bp paired‐end sequencing read data. Read quality before and after trimming was assessed by FastQC. Adapter sequences, low‐quality and duplicate reads were trimmed using Trimmomatic (Bolger et al., 2014) with the following parameters: ‘ILLUMINACLIP:TruSeq3‐PE.fa:2:30:7:1:true LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20; MINLEN:75’. About 57 million clean PE reads (Phred Q score ≥ 30) were obtained for each parent (Table S25). For downstream analyses, clean reads were mapped against the radish genome v2.20 (GenBank: GCA_002197605.1) using both CLC Genomics Workbench v9.0 (CLC‐GWB, CLC Bio‐Qiagen, Aarhus, Denmark) and BWA (Burrows‐Wheeler Aligner). For SNP variant calling and analysis, the results from BWA mapping were processed using the same pipeline used for the F2 GBS data (see ‘GBS of a radish F2 population’) with the options for paired‐end reads. The SNP data were added to the GBS data for those positions that were detected in the F2 samples but missing in the parental lines.
GBS of a radish F2 population
In total, 92 F2 individuals and two parental lines (WK29 and WK40) were genotyped using a modified protocol of the original GBS approach (Poland et al., 2012) employing two enzymes, EcoRI and MseI (New England Biolabs, Ipswich, MA, USA). In brief, amounts of 400 ng of each sample were double‐digested by the two enzymes in CutSmart buffer at 37°C for 2 h, followed by an enzyme inactivation step at 85°C for 10 min. The digested fragments were used for ligation at 22°C for 2 h, followed with an inactivation step at 65°C for 20 min. A set of 94 unique barcodes in the EcoRI adapter sequences (Table S26) was adapted from Park et al. (2019). To minimize the amplification of the same‐ended fragments during the PCR, we generated a Y‐shaped MseI adapter so that the fragments cut by both enzymes were preferentially amplified (see Figure S15 for a scheme). Amounts of 5 ng of each ligated DNA sample were then pooled to form a single tube, and subjected to size selection using the Select‐a‐Size DNA Clean & Concentrator Kit (Zymo Research, Orange, CA, USA) to retain fragments in a size range of 150–700 bp. The size‐selected multiplexed library was then amplified using Phusion DNA polymerase (New England Biolabs) for 25 cycles. The PCR product was cleaned using a QIAGEN Quick PCR Cleanup Kit and sequenced using an Illumina HiSeq4000 instrument to obtain 100‐bp single‐end read data at Macrogen. Two prepared libraries (with and without the clean‐up step) were sequenced, with the results showing a high correlation between reads obtained from them for each sample. Accordingly, we combined reads from each sample for downstream analyses.
SNP detection and QTL analysis
The SNP detection and QTL mapping were done followed the protocol described in Fan et al. (2017) with modifications. Briefly, clean reads (Phred Q score ≥ 25, length ≥ 75 bp) were imported by demultiplexing reads with ‘import’ implemented in the SHORE package (http://1001genomes.org) with the option of perfect matches of the barcode sequences and mapped to the radish reference genome v2.20 (GenBank: GCA_002197605.1) using BWA (Li and Durbin, 2009). Mapped reads were converted into bam files using SAMTOOLS (Li et al., 2009), and sorted and indexed using PICARD tools (http://broadinstitute.github.io/picard). Subsequently, we used the GATK package (McKenna et al., 2010) to create the targets for realignment. SNPs were identified from individual mapped reads, and filtered using BCFTOOLS (Danecek and McCarthy, 2017) with a minor allele frequency of > 0.01. The VCF file from BCFTOOLS was then subjected to TASSEL v5.2.48 (Bradbury et al., 2007) to filter out those SNPs that were genotyped in less than 50 samples using the filter tool. For those SNP positions that were detected in the F2 samples but missing in the parental samples, the available base calls from whole‐genome sequencing data were added. The processed SNP data then were converted into the ABH genotype format using TASSEL software based on the information from the two parental lines, WK29‐type allele (as A), WK40‐type allele (as B) and heterozygous (as H). A Chi‐square test was used to retain only SNPs that segregate into a 1:2:1 ratio in the F2 individuals for further analysis. QTL mapping was conducted using the R/qtl package v1.46‐2 (Broman et al., 2003) in R (http://www.R‐project.org) using the pithiness index score phenotypic data and the genotypic data (processed ABH‐format SNPs). First, the conditional genotype probabilities were calculated using the function ‘calc.genoprob’ with error.prob = 0.0001 and map.function = c("kosambi"). Missing data were imputed by simulation with the function ‘sim.geno’. To identify the QTL regions, a genome scan with composite interval mapping (CIM) method was then implemented with the function ‘cim’ using the phenotypic dataset and the multiple regression algorithm. The genome‐wide significant logarithm of odds (LOD) threshold (P = 0.05) was determined by ‘cim’ with a permutation test of 1000 repeats.
Validation of GBS allele‐specific markers
Validation of the qPITH‐1 QTL marker (locus S1_17807256) on chromosome 1 was done using the allele‐specific SNP/INDEL marker approach as described in Kim et al. (2016). In brief, a common and two allele‐specific primers were designed. The SNP was determined by the last base at the 3′‐prime end of the specific primers. To increase the primer specificity, a single artificial mismatch was introduced into the third base from the SNP position at the 3′‐prime for each primer. Sequences were visualized and manipulated in CLC‐GWB, and allele‐specific primers were designed using NCBI Primer BLAST (Ye et al., 2012). Gradient PCR (55–65°C) was then conducted, and the optimal annealing temperature (62°C) was determined for both SNP‐specific primers, at which each of the allele‐specific primers (to lines WK29 and WK40) binds only to its respective allele. The heterozygous genotype was determined by two identically sized allele‐specific bands from both lines (Figure S9e). PCR was run with the following conditions: 95°C for 3 min, followed by 35 cycles (95°C for 30 sec, optimal temperature for 30 sec, and 72°C for 40 sec). For validation of the 46‐bp deletion marker in the 3′‐UTR of RsNAC013 (NAC013del), a pair of primers was defined to amplify a 258‐bp fragment from line WK29, which encompasses a 46‐bp deletion and other small INDELs in line WK40, resulting in a smaller product of 204 bp. All differences were confirmed by Sanger sequencing. The heterozygote showed two allele‐specific bands 54 bp apart. PCR was run under conditions identical to those used for SNP validation with the annealing temperature set in this case to 58°C. The RsACTIN2/7 gene primers were used to evaluate the DNA template loading. A list of the primers used for marker validation and cloning is provided in Table S27.
Generating Arabidopsis transgenic lines
RsNAC013 CDS and 3′‐UTR sequences of WK29 and WK40 were amplified by a two‐step PCR and cloned into pDONR P2R_P3 by a BP reaction. p35S::GFP‐RsNAC013WK29 , p35S::GFP‐RsNAC013WK40 , pGL2::nlsGFP:3′UTRWK29 and pGL2::nlsGFP:3′UTRWK40 were constructed into dpGreen‐BarT by Multisite Gateway LR recombination, as described previously in Lee et al. (2006). All clones in the binary vector were transformed into Agrobacterium GV3101 with pSOUP for Arabidopsis transformation by floral dipping. Arabidopsis thaliana ecotype Columbia (Col‐0) was used as a background wild‐type. Every transgenic line containing a target transgene was selected with a 2000‐fold diluted Basta (Bayer Crop Science, Monheim am Rhein, Germany) solution on soil. Plants were grown under 16‐h light/8‐h dark and 125 μmol m−2 sec−1. Primers for cloning are listed in Table S27.
Arabidopsis genes expression using ddPCR
About 80 mg tissues pooled from three–four leaves of 6‐week‐old Arabidopsis plants was collected from each genotype. The total RNAs were isolated using RNeasy® Plant Mini Kit (Qiagen), and 1 μg of total RNAs was used for RT with SuperScript III reverse transcriptase (Invitrogen). Droplet generation, PCR, quantification and data analysis were performed as previously described in Kim et al. (2020). The PCR steps were performed with the following cycling conditions: one cycle at 95°C for 3 min, 40 cycles of 95°C for 30 sec, 55°C for 45 sec, 60°C for 1 min, and followed by one cycle of 4°C for 5 min and 90°C for 5 min. All primers for ddPCR are presented in Table S28.
Confocal microscopy
To visualize the GFP protein, 5‐days after transferring Arabidopsis seedlings were stained with 10 µg ml−1 of a propidium iodide (PI) solution (Life Technologies, Carlsbad, CA, USA) and imaged with a confocal microscope (Carl Zeiss LSM700, Germany). Excitation/detection windows are 488 nm/500–530 nm for GFP and 555 nm/591–636 nm for PI. For H2O2 and mock treatments, 20 mm H2O2 and distilled water were sprayed on the root and imaged after 1 h.
Candidate gene search and sequencing of the identified QTL for root pithiness
The sequence corresponding to the major qPITH‐1 QTL was extracted from the radish genome and subjected to a blast search against all radish protein coding genes (Hoang et al., 2020; Jeong et al., 2016) using NCBI BLASTn with ‘‐evalue’ set to 1e‐10. The matched genes were filtered (e‐value = 0) and subsequently annotated by Mercator v4 (https://www.plabipd.de/portal/web/guest/mercator4) against the Arabidopsis TAIR Release 10 with the default settings to look for genes related to cell death and oxidative stress. Candidate gene–gene interactions and co‐expression network were analysed using the STRING database (https://string‐db.org/) for detailed functions. Sanger sequencing of candidate genes was performed at NICEM, Seoul National University, Seoul, Korea.
Radish gene expression analysis by qRT‐PCR
For each RNA sample, a 20 µl mixture of 1 µg total RNA, 1 µl oligo(dT)16 (10 pm µl−1), 1 µl dNTP (10 mm), 4 µl 5 × first‐strand buffer, 1 µl dithiothreitol (100 mm), 1 µl SuperScript III reverse transcriptase (200 U µl−1; Invitrogen) and RNase‐free water was prepared. This mixture was then used for a reverse transcription reaction at 50°C for 1 h, followed by inactivation of the enzyme at 70°C for 15 min. The resulting cDNA library was diluted by 25‐fold and mixed with iQTM SYBER Green Supermix (Bio‐Rad, Hercules, CA, USA) for qRT‐PCR experiments on a BioRad CFX96 Real‐Time PCR detection system. The PCR was run with the following settings: 3 min of denaturation at 95°C followed by 48 cycles of 95°C for 30 sec, 57°C for 10 sec, and 72°C for 30 sec. A list of gene‐specific primer sequences designed by NCBI Primer‐BLAST (Ye et al., 2012) is presented in Table S28. The RsACTIN2/7 gene (Xu et al., 2012) was used as internal control to normalize the expression level of each target gene. For each sample, data from at least three technical replicates were obtained and analysed by Student’s t‐tests in Bio‐Rad CFX Maestro software 1.0 and by one‐way anovas (Fisher’s LSD post hoc test) in GraphPad PRISM v.9.0.0.
Plant oxidative stress experiments
To determine a suitable concentration of H2O2 for the treatment, we initially conducted tests on 6‐WAP radishes from line WK40 using concentrations ranging from 0 to 1000 mm (Figure S16). After 5 days of treatment, the plants treated with 500 and 1000 mm were either wilted or dead. Those plants treated with 100 and 200 mm were found to be affected by the stress treatment but continued growing normally. We therefore chose 300 mm as the concentration for our stress experiments. Radish plants (one plant per pot, arranged randomly with a minimum of four plants per treatment) were grown in a greenhouse, as described earlier, for 7 WAP and subjected to a H2O2 treatment. Two time points, 7 WAP (early treatment, collected at 12 WAP) and 10 WAP (late treatment, collected at 14 WAP) were selected. At each time point, three treatments were set up, referred to here as WK40 treated (H2O2), WK40 non‐treated (water, control group), and WK29 non‐treated (water, control group). For the treated plants, H2O2 at 30% (Sigma‐Aldrich, Darmstadt, Germany) was diluted in water to 300 mm (for a final volume of 1 L) and added to the soil once a week, until 1 week before harvesting. As it has been reported that fertilizer can influence the formation of pithiness (Tanaka and Nagatomo, 2017), we stopped adding fertilizer to all plants upon the stress experiment. A gene expression analysis by qRT‐PCR was conducted 1 day after the addition of H2O2 (short‐term) and at 12 WAP and 14 WAP (long‐term), while histological changes were examined at 12 and 14 WAP.
Histological analysis
For cell death staining using Evans Blue, approximately 1.5‐mm‐thick radish root sections were cut using a slicer and stained in a 1% Evans Blue solution (w/v) overnight at room temperature (RT; 22–25°C), after which they were de‐stained in 100% ethanol. For H2O2 detection, root cross‐sections were treated with 0.1% (w/v) DAB (in Tris‐HCl pH 5) for at least 8 h in the dark at RT, washed with ethanol, and then with water. To visualize superoxide anion (O‐ 2) accumulation, sections were stained with NBT (0.05% in phosphate‐buffered saline buffer, pH 6.1) for 8 h in the dark at RT, washed with ethanol, and then washed with water. Samples were viewed under a light microscope (Leica, Wetzlar, Germany) and photos were taken using an iPhone 11 camera (12 megapixels; Apple).
Quantification and statistical analysis
All statistical analyses, unless otherwise stated, were performed using Microsoft Excel, R program with RStudio software (https://www.rstudio.com) and GraphPad PRISM v.9.0.0. Venn diagrams were drawn using online tools (http://bioinformatics.psb.ugent.be/webtools/Venn). Plots and graphs were generated with RStudio using ggplot2 and pheatmap packages, GraphPad and Excel. All analyses in CLC‐Genomics Workbench and the Linux environment were conducted on local servers hosted by the Plant Systems Genetics lab at Seoul National University and the Institute for Basic Science (IBS), Korea.
AUTHOR CONTRIBUTIONS
J‐YL and SP conceived the project; J‐YL supervised the experiments and data analysis; SP provided the radish parental and F2 materials; NVH performed the experiments, analysed the data and prepared the figures; CP contributed to the collection of field samples, generation of Arabidopsis transgenic lines, ddPCR experiment and confocal images; HS contributed to the collection of field samples and to GBS marker validation; SK and EC contributed to the R/qtl analysis; BK contributed to the GBS experiment design and provided the GBS adapter sequences; NVH and J‐YL wrote the article with contributions by all authors.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Supporting information
Figure S1. Radish experimental set‐up. Plants grown in the field (upper) and greenhouse (lower), at around 8 weeks after planting (WAP).
Figure S2. Different types and levels of root pithiness (spongy tissues) and cavitation (air‐filled cavities) observed in our experiment.
Figure S3. Summary of 17 samples used for the RNA‐seq experiment. (a) Pithiness and non‐pithiness samples collected from the field condition. (b) Non‐pithiness samples collected from the greenhouse condition. All roots were collected, and examined by a micro‐CT scanner before processed. The lower part of each root was retained for RNA extraction. FNO and FSP denote non‐pithiness and pithiness samples collected from the field, while SNU denotes non‐pithiness samples collected from greenhouse condition. W represents WAP in the sample names.
Figure S4. MAPK signalling pathway – plant (ath04016). Red boxes indicate DEGs mapped against the pathway. Map was generated by KEGG PATHWAY Database (https://www.genome.jp/kegg/) using output from KEGG Orthology‐Based Annotation System (KOBAS) v3.0 (Xie et al., 2011).
Figure S5. Starch and sucrose metabolism (ath00500). Red boxes indicate DEGs mapped against the pathway. Map was generated by KEGG PATHWAY Database (https://www.genome.jp/kegg/) using output from KEGG Orthology‐Based Annotation System (KOBAS) v3.0 (Xie et al., 2011).
Figure S6. Phenylpropanoid biosynthesis (ath00940, upper panel) and glycolysis/gluconeogenesis (ath00010, lower panel) pathways. These pathways were found in cluster 1 DEGs. Red boxes indicate DEGs mapped against the pathway. Map was generated by KEGG PATHWAY Database (https://www.genome.jp/kegg/) using output from KEGG Orthology‐Based Annotation System (KOBAS) v3.0 (Xie et al., 2011).
Figure S7. Additional histological images of pithiness and non‐pithiness samples, related to Figure 1g. Evans Blue staining for cell death (a); DAB and NBT staining for hydrogen peroxide (H2O2) (b) and superoxide (O2−) (c) accumulation, respectively, in samples with and without pithiness phenotype. 12‐WAP root samples were used. Root sections were stained and viewed under a light microscopy (Leica). Photos were taken by an iPhone 11 camera (12 megapixels; Apple).
Figure S8. Data statistics summary of the R/qtl analysis of 224 final markers. Radish genome v2.20 (GenBank: GCA_002197605.1) was used.
Figure S9. Identification and validation of variations in RsNAC013 sequences. (a) Mapping of whole‐genome sequencing data of two parental lines (WK29 and WK40) against RsNAC013 sequences identified several SNPs and deletions, including a 46‐bp deletion in the 3′‐UTR of RsNAC013 in line WK40 (red arrow), which were used as pithiness markers (termed as NAC013del). Mapping was done in CLC Genomics Workbench with a length fraction 0.9 and similarity fraction 0.9. (b) Variations in full‐length RsNAC013 sequences from the two parental lines obtained from Sanger sequencing (Table S18). Primers for validation (see panel ‘e’) of the NAC013del marker were highlighted, positions 1909..2166 in line WK29, which encompasses the deletion and other small INDELs in line WK40. (c) Variations of RsNAC013 coding sequences identified between the two parental lines, including five SNPs and their Sanger sequencing validation. (d) Hydrophobicity of RsNAC013 proteins from the two lines, showing a potential region (blue arrow) that may influence the protein function. TM denotes the transmembrane motif. Hydrophobicity plot was generated using the online tool at http://best.bio.jhu.edu/HePCaT/run.pl, based on Kyte & Doolittle parameters. (e) A representative image of marker validation of NAC013del marker and qPITH‐1 QTL marker (locus S1_17807256, related to results in Figure 3e,f). PCR were run using DNA from the two parental lines. Primers for qPITH‐1 QTL included a common primer and two allele‐specific primers for lines WK29 and WK40, respectively. Primers for RsNAC013 deletion were designed to amplify the genomic regions shown in panel (b). The product size was 258 and 204 bp, for WK29 and WK40 lines, respectively. RsACTIN2/7 primers were used to check the loading DNA.
Figure S10. A co‐expression gene network of selected genes used in expression analysis (related to Figure 4). Genes related to ANAC013‐AtAOX1a, ROS production and scavenging in the oxidative pathway and PCD were used. The network was constructed by the STRING database (https://string‐db.org). Genes (nodes) are connected by edges, based on a default setting of evidence.
Figure S11. Schematics of the root RNA sample collection for qRT‐PCR and the expression of MDS genes that linked to hypoxia in our radish transcriptome data. (a) Schematics of the root RNA samples collected for expression analysis. RNAs were collected separately from four areas, top, bottom, inner and outer of each root. For whole‐root expression analysis, all four RNA samples were equally mixed to form one single sample for qRT‐PCR analysis. For spatial expression analysis, inner and outer root RNA samples were used. (b) Expression levels of MDS genes in the mitochondrial signalling pathway that are also linked to hypoxic responses. FNO and FSP denote field‐collected samples with and without pithiness, respectively. SNU denotes samples collected from the greenhouse. DEG genes are marked with ‘DEG’ in their names.
Figure S12. Additional histological images of root samples from the two inbred lines WK29 and WK40, related to Figure 4e. Evans Blue staining for cell death (a); DAB and NBT staining of hydrogen peroxide (H2O2) (b) and superoxide (O2−) (c) accumulation, respectively, in samples from lines WK29 and WK40; 12‐WAP root samples were used. Sample sections were stained and viewed under a light microscopy (Leica). Photos were taken by an iPhone 11 camera (12 megapixels, Apple).
Figure S13. Root collection from 12‐WAP plants treated with H2O2. T and N denote treated and non‐treated, respectively.
Figure S14. Line WK29 roots collected at 12 WAP and 14 WAP. All samples showed no pithiness phenotype. Roots of 12 WAP were dissected, examined but not photographed. N in sample names denotes non‐treated.
Figure S15. Adapter construct and primer design for the GBS library used in this study. A unique barcode was inserted into the EcoRI adapter sequence. To minimize the amplification of the same‐ended fragments (MseI – MseI) during PCR, we generated a Y‐shaped MseI adapter and an overhanging EcoRI adapter so that the fragments cut by both enzymes were preferentially amplified. Primers for PCR amplification are provided below the construct scheme.
Figure S16. Determination of the H2O2 concentration for the radish oxidative stress experiment. 6‐WAP radishes of line WK40 were used for this experiment. Plants were treated with hydrogen peroxide (H2O2) of concentration ranging from 0 to 1000 mm in the greenhouse condition. (a) Plants at 2 days after treatment. (b) Plants at 5 days after treatment.
Table S1. Summary statistics of samples and RNA‐seq sequencing reads.
Table S2. Radish gene IDs and corresponding Arabidopsis homologous gene IDs and their expression in the transcriptome data.
Table S3. List of 3966 DEGs identified from three comparisons between the selected samples and their expression values (FPKM).
Table S4. qRT‐PCR validation of RNA‐seq results using selected genes related to oxidative stress and cell death.
Table S5. GO enrichment analysis of 3966 DEGs by BINGO (FDR‐adjusted P‐value ≤ 0.05).
Table S6. GOSlim analysis of 3966 DEGs by BINGO (FDR‐adjusted P‐value ≤ 0.05).
Table S7. KEGG metabolic pathway analysis of 3966 DEGs by KOBAS.
Table S8. List of DEGs in two major clusters and their GOSlim analyses.
Table S9. KEGG metabolic pathway analysis of two DEG clusters by KOBAS.
Table S10. Common dPCD genes between radish DEGs and the core PCD genes in Arabidopsis (Olvera‐Carrillo et al., 2015).
Table S11. Common ePCD genes between radish DEGs and the core PCD genes in Arabidopsis (Olvera‐Carrillo et al., 2015).
Table S12. Expression levels of selected PCD genes in the inner and outer root tissues of radish roots undergoing pithiness formation measured by qRT‐PCR.
Table S13. Sequencing statistic summary of the GBS library for the QTL analysis.
Table S14. List of 224 SNPs that segregate at a 1:2:1 ratio in the F2 individuals.
Table S15. Gene search of the major QTL peak on chromosome 1.
Table S16. Expression levels of RsNAC013 and its direct target RsAOX1a in the transcriptome and qRT‐PCR data.
Table S17. Variations in the RsNAC013 sequence between the two radish lines WK40 and WK29.
Table S18. gDNA and CDS sequences of RsNAC013 obtained from Sanger sequencing.
Table S19. Correlation between qPITH‐1 QTL (S1_17807256) marker and RsNAC013 46‐bp deletion marker in the F2 samples.
Table S20. Expression levels of selected genes related to the RsNAC013 pathway as measured by qRT‐PCR in radish roots.
Table S21. Expression analysis of RsNAC013 and RsAOX1a in radish roots treated with H2O2 compared with non‐treated control samples by qRT‐PCR.
Table S22. Analysis of transcriptional activity of two variants of RsNAC013.
Table S23. Pithiness index of 92 F2 individuals used for GBS in this study.
Table S24. Concentration, RIN values and rRNA ratios of 17 samples used for RNA‐seq in this study.
Table S25. Whole‐genome sequencing data summary of the two parental lines used in this study.
Table S26. EcoRI adapter sequences used in this study.
Table S27. A list of primers used in SNP validation and RsNAC013 gene cloning.
Table S28. A list of primers used in gene expression analysis.
Video S1. Micro‐CT scanning of a root showing pithiness.
ACKNOWLEDGEMENTS
The authors would like to thank all of the Lee lab members, past and present, for assisting with this work at various stages. This work is supported by the National Research Foundation of Korea (NRF‐2018R1A5A1023599 and 2021R1A2C3006061), and by the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, and Forestry (IPET) through the Golden Seed Project (213006‐05‐5‐SBK30), funded by the Ministry of Agriculture, Food and Rural Affairs (MAFRA), the Ministry of Oceans and Fisheries (MOF), the Rural Development Administration (RDA) and Korea Forest Services (KFS) to J.‐Y.L. N.V.H. was supported by the Brain Korea 21 Plus Program.
DATA AVAILABILITY STATEMENT
The accession numbers for the 17 RNA‐seq datasets and raw GBS read data reported in this paper are available at GenBank: PRJNA682541 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA682541) and PRJNA682792 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA682792), respectively. All other relevant data supporting the findings here are included within the article and supplementary files.
REFERENCES
- Bela, K. , Horvath, E. , Galle, A. , Szabados, L. , Tari, I. & Csiszar, J. (2015) Plant glutathione peroxidases: emerging role of the antioxidant enzymes in plant development and stress responses. Journal of Plant Physiology, 176, 192–201. [DOI] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. & Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowler, C. , Van Camp, W. , Van Montagu, M. , Inzé, D. & Asada, K. (2011) Superoxide dismutase in plants. Critical Reviews in Plant Sciences, 13, 199–218. [Google Scholar]
- Bradbury, P.J. , Zhang, Z. , Kroon, D.E. , Casstevens, T.M. , Ramdoss, Y. & Buckler, E.S. (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics, 23, 2633–2635. [DOI] [PubMed] [Google Scholar]
- Broman, K.W. , Wu, H. , Sen, S. & Churchill, G.A. (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics, 19, 889–890. [DOI] [PubMed] [Google Scholar]
- Buckner, B. , Janick‐Buckner, D. , Gray, J. & Johal, G.S. (1998) Cell‐death mechanisms in maize. Trends in Plant Science, 3, 218–223. [Google Scholar]
- Burke, R. , Schwarze, J. , Sherwood, O.L. , Jnaid, Y. , McCabe, P.F. & Kacprzyk, J. (2020) Stressed to death: the role of transcription factors in plant programmed cell death induced by abiotic and biotic stimuli. Frontiers in Plant Science, 11, 1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, J.D. (2009) Cetyltrimethyl ammonium bromide (CTAB) DNA miniprep for plant DNA isolation. Cold Spring Harbor Protocols, 2009(3). 10.1101/pdb.prot5177 [DOI] [PubMed] [Google Scholar]
- Danecek, P. & McCarthy, S.A. (2017) BCFtools/csq: haplotype‐aware variant consequences. Bioinformatics, 33, 2037–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneva, A. , Gao, Z. , Van Durme, M. & Nowack, M.K. (2016) Functions and regulation of programmed cell death in plant development. Annual Review of Cell and Developmental Biology, 32, 441–468. [DOI] [PubMed] [Google Scholar]
- De Clercq, I. , Vermeirssen, V. , Van Aken, O. , Vandepoele, K. , Murcha, M.W. , Law, S.R. et al. (2013) The membrane‐bound NAC transcription factor ANAC013 functions in mitochondrial retrograde regulation of the oxidative stress response in Arabidopsis. The Plant Cell, 25, 3472–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon, D.P. & Edwards, R. (2010) Glutathione transferases. The Arabidopsis Book, 8, e0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, L. , Chae, E. , Gust, A.A. & Nurnberger, T. (2017) Isolation of novel MAMP‐like activities and identification of cognate pattern recognition receptors in Arabidopsis thaliana using next‐generation sequencing (NGS)‐based mapping. Current Protocols in Plant Biology, 2, 173–189. [DOI] [PubMed] [Google Scholar]
- Fujimoto, M. , Sazuka, T. , Oda, Y. , Kawahigashi, H. , Wu, J. , Takanashi, H. et al. (2018) Transcriptional switch for programmed cell death in pith parenchyma of sorghum stems. Proceedings of the National Academy of Sciences United States of America, 115, E8783–E8792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud, E. , Ho, L.H. , Clifton, R. , Carroll, A. , Estavillo, G. , Tan, Y.F. et al. (2008) The absence of ALTERNATIVE OXIDASE1a in Arabidopsis results in acute sensitivity to combined light and drought stress. Plant Physiology, 147, 595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang, N.V. , Choe, G. , Zheng, Y. , Aliaga Fandino, A.C. , Sung, I. , Hur, J. et al. (2020) Identification of conserved gene‐regulatory networks that integrate environmental sensing and growth in the root cambium. Current Biology, 30, 2887–2900 e2887. [DOI] [PubMed] [Google Scholar]
- Huysmans, M. , Buono, R.A. , Skorzinski, N. , Radio, M.C. , De Winter, F. , Parizot, B. et al. (2018) NAC transcription factors ANAC087 and ANAC046 control distinct aspects of programmed cell death in the Arabidopsis columella and lateral root cap. The Plant Cell, 30, 2197–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong, Y.M. , Kim, N. , Ahn, B.O. , Oh, M. , Chung, W.H. , Chung, H. et al. (2016) Elucidating the triplicated ancestral genome structure of radish based on chromosome‐level comparison with the Brassica genomes. Theoretical and Applied Genetics, 129, 1357–1372. [DOI] [PubMed] [Google Scholar]
- Kim, H. , Zhou, J. , Kumar, D. , Jang, G. , Ryu, K.H. , Sebastian, J. et al. (2020) SHORTROOT‐mediated intercellular signals coordinate phloem development in Arabidopsis roots. The Plant Cell, 32, 1519–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.Y. , Kim, S.G. , Kim, Y.S. , Seo, P.J. , Bae, M. , Yoon, H.K. et al. (2007) Exploring membrane‐associated NAC transcription factors in Arabidopsis: implications for membrane biology in genome regulation. Nucleic Acids Research, 35, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.R. , Ramos, J. , Ashikari, M. , Virk, P.S. , Torres, E.A. , Nissila, E. et al. (2016) Development and validation of allele‐specific SNP/indel markers for eight yield‐enhancing genes using whole‐genome sequencing strategy to increase yield potential of rice, Oryza sativa L. Rice, 9, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun, Y. , Chiu, W.L. , Tena, G. & Sheen, J. (2000) Functional analysis of oxidative stress‐activated mitogen‐activated protein kinase cascade in plants. Proceedings of the National Academy of Sciences United States of America, 97, 2940–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. & Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte, D. , Olate, E. , Salinas, P. , Salazar, M. , Jordana, X. & Holuigue, L. (2012) Glutaredoxin GRXS13 plays a key role in protection against photooxidative stress in Arabidopsis. Journal of Experimental Botany, 63, 503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.Y. , Colinas, J. , Wang, J.Y. , Mace, D. , Ohler, U. & Benfey, P.N. (2006) Transcriptional and posttranscriptional regulation of transcription factor expression in Arabidopsis roots. Proceedings of the National Academy of Sciences United States of America, 103, 6055–6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , Lee, H.J. , Huh, S.U. , Paek, K.H. , Ha, J.H. & Park, C.M. (2014) The Arabidopsis NAC transcription factor NTL4 participates in a positive feedback loop that induces programmed cell death under heat stress conditions. Plant Science, 227, 76–83. [DOI] [PubMed] [Google Scholar]
- Lee, S. , Seo, P.J. , Lee, H.J. & Park, C.M. (2012) A NAC transcription factor NTL4 promotes reactive oxygen species production during drought‐induced leaf senescence in Arabidopsis. The Plant Journal, 70, 831–844. [DOI] [PubMed] [Google Scholar]
- Li, B. & Dewey, C.N. (2011) RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics, 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. & Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. & Durbin, R. ; Genome Project Data Processing Subgroup . (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. & Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere, S. , Heymans, K. & Kuiper, M. (2005) BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics, 21, 3448–3449. [DOI] [PubMed] [Google Scholar]
- Magendans, J.F.C. (1991) Elongation and contraction of the plant axis and development of spongy tissues in the radish tuber (Raphanus sativus L. cv. Saxa Nova). Wageningen: Wageningen Agricultural University papers. [Google Scholar]
- Marcelis, L. (1997) Pithiness and growth of radish tubers as affected by irradiance and plant density. Annals of Botany, 79, 397–402. [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. et al. (2010) The genome analysis toolkit: a mapreduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, X. , Li, L. , De Clercq, I. , Narsai, R. , Xu, Y. , Hartmann, A. et al. (2019) ANAC017 coordinates organellar functions and stress responses by reprogramming retrograde signaling. Plant Physiology, 180, 634–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, S. , Hilbert, B. , Dueckershoff, K. , Roitsch, T. , Krischke, M. , Mueller, M.J. et al. (2008) General detoxification and stress responses are mediated by oxidized lipids through TGA transcription factors in Arabidopsis. The Plant Cell, 20, 768–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlenbock, P. , Plaszczyca, M. , Plaszczyca, M. , Mellerowicz, E. & Karpinski, S. (2007) Lysigenous aerenchyma formation in Arabidopsis is controlled by LESION SIMULATING DISEASE1. The Plant Cell, 19, 3819–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, S. , Ivanova, A. , Duncan, O. , Law, S.R. , Van Aken, O. , De Clercq, I. et al. (2013) A membrane‐bound NAC transcription factor, ANAC017, mediates mitochondrial retrograde signaling in Arabidopsis. The Plant Cell, 25, 3450–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olvera‐Carrillo, Y. , Van Bel, M. , Van Hautegem, T. , Fendrych, M. , Huysmans, M. , Simaskova, M. et al. (2015) A conserved core of programmed cell death indicator genes discriminates developmentally and environmentally induced programmed cell death in plants. Plant Physiology, 169, 2684–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orman‐Ligeza, B. , Parizot, B. , de Rycke, R. , Fernandez, A. , Himschoot, E. , Van Breusegem, F. et al. (2016) RBOH‐mediated ROS production facilitates lateral root emergence in Arabidopsis. Development, 143, 3328–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, M. , Lee, J.H. , Han, K. , Jang, S. , Han, J. , Lim, J.H. et al. (2019) A major QTL and candidate genes for capsaicinoid biosynthesis in the pericarp of Capsicum chinense revealed using QTL‐seq and RNA‐seq. Theoretical and Applied Genetics, 132, 515–529. [DOI] [PubMed] [Google Scholar]
- Pham, H.M. , Kebede, H. , Ritchie, G. , Trolinder, N. & Wright, R.J. (2018) Alternative oxidase (AOX) over‐expression improves cell expansion and elongation in cotton seedling exposed to cool temperatures. Theoretical and Applied Genetics, 131, 2287–2298. [DOI] [PubMed] [Google Scholar]
- Pitzschke, A. & Hirt, H. (2006) Mitogen‐activated protein kinases and reactive oxygen species signaling in plants. Plant Physiology, 141, 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland, J.A. , Brown, P.J. , Sorrells, M.E. & Jannink, J.L. (2012) Development of high‐density genetic maps for barley and wheat using a novel two‐enzyme genotyping‐by‐sequencing approach. PLoS One, 7, e32253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed, A.I. , Sharov, V. , White, J. , Li, J. , Liang, W. , Bhagabati, N. et al. (2003) TM4: a free, open‐source system for microarray data management and analysis. BioTechniques, 34, 374–378. [DOI] [PubMed] [Google Scholar]
- Safrany, J. , Haasz, V. , Mate, Z. , Ciolfi, A. , Feher, B. , Oravecz, A. et al. (2008) Identification of a novel cis‐regulatory element for UV‐B‐induced transcription in Arabidopsis. The Plant Journal, 54, 402–414. [DOI] [PubMed] [Google Scholar]
- Sagi, M. & Fluhr, R. (2006) Production of reactive oxygen species by plant NADPH oxidases. Plant Physiology, 141, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiguzov, A. , Nikkanen, L. , Fitzpatrick, D. , Vainonen, J.P. , Gossens, R. , Alseekh, S. et al. (2020) Dissecting the interaction of photosynthetic electron transfer with mitochondrial signalling and hypoxic response in the Arabidopsis rcd1 mutant. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 375, 20190413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiguzov, A. , Vainonen, J.P. , Hunter, K. , Tossavainen, H. , Tiwari, A. , Jarvi, S. et al. (2019) Arabidopsis RCD1 coordinates chloroplast and mitochondrial functions through interaction with ANAC transcription factors. Elife, 8, e43284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano, T. (1963) Studies on the pithiness of radish. I. Engei Gakkai Zasshi, 32, 114–120. [Google Scholar]
- Tanaka, Y. & Nagatomo, M. (2017) Effects of fertilizer application scheme and planting distance on cavitation and pithiness of ‘Sakurajima Daikon’ (Raphanus sativus L.). Horticultural Research (Japan), 16, 435–441. [Google Scholar]
- Van Durme, M. & Nowack, M.K. (2016) Mechanisms of developmentally controlled cell death in plants. Current Opinion in Plant Biology, 29, 29–37. [DOI] [PubMed] [Google Scholar]
- Xie, C. , Mao, X. , Huang, J. , Ding, Y. , Wu, J. , Dong, S. et al. (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Research, 39, W316–W322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y. , Zhu, X. , Gong, Y. , Xu, L. , Wang, Y. & Liu, L. (2012) Evaluation of reference genes for gene expression studies in radish (Raphanus sativus L.) using quantitative real‐time PCR. Biochemical and Biophysical Research Communications, 424, 398–403. [DOI] [PubMed] [Google Scholar]
- Yamagishi, H. (2017) Speciation and diversification of radish. In: Nishio, T. and Kitashiba, H. (Eds.) The radish genome. Cham: Springer International Publishing, pp. 11–30. [Google Scholar]
- Yamauchi, T. , Yoshioka, M. , Fukazawa, A. , Mori, H. , Nishizawa, N.K. , Tsutsumi, N. et al. (2017) An NADPH oxidase RBOH functions in rice roots during lysigenous aerenchyma formation under oxygen‐deficient conditions. The Plant Cell, 29, 775–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, J. , Coulouris, G. , Zaretskaya, I. , Cutcutache, I. , Rozen, S. & Madden, T.L. (2012) Primer‐BLAST: a tool to design target‐specific primers for polymerase chain reaction. BMC Bioinformatics, 13, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Radish experimental set‐up. Plants grown in the field (upper) and greenhouse (lower), at around 8 weeks after planting (WAP).
Figure S2. Different types and levels of root pithiness (spongy tissues) and cavitation (air‐filled cavities) observed in our experiment.
Figure S3. Summary of 17 samples used for the RNA‐seq experiment. (a) Pithiness and non‐pithiness samples collected from the field condition. (b) Non‐pithiness samples collected from the greenhouse condition. All roots were collected, and examined by a micro‐CT scanner before processed. The lower part of each root was retained for RNA extraction. FNO and FSP denote non‐pithiness and pithiness samples collected from the field, while SNU denotes non‐pithiness samples collected from greenhouse condition. W represents WAP in the sample names.
Figure S4. MAPK signalling pathway – plant (ath04016). Red boxes indicate DEGs mapped against the pathway. Map was generated by KEGG PATHWAY Database (https://www.genome.jp/kegg/) using output from KEGG Orthology‐Based Annotation System (KOBAS) v3.0 (Xie et al., 2011).
Figure S5. Starch and sucrose metabolism (ath00500). Red boxes indicate DEGs mapped against the pathway. Map was generated by KEGG PATHWAY Database (https://www.genome.jp/kegg/) using output from KEGG Orthology‐Based Annotation System (KOBAS) v3.0 (Xie et al., 2011).
Figure S6. Phenylpropanoid biosynthesis (ath00940, upper panel) and glycolysis/gluconeogenesis (ath00010, lower panel) pathways. These pathways were found in cluster 1 DEGs. Red boxes indicate DEGs mapped against the pathway. Map was generated by KEGG PATHWAY Database (https://www.genome.jp/kegg/) using output from KEGG Orthology‐Based Annotation System (KOBAS) v3.0 (Xie et al., 2011).
Figure S7. Additional histological images of pithiness and non‐pithiness samples, related to Figure 1g. Evans Blue staining for cell death (a); DAB and NBT staining for hydrogen peroxide (H2O2) (b) and superoxide (O2−) (c) accumulation, respectively, in samples with and without pithiness phenotype. 12‐WAP root samples were used. Root sections were stained and viewed under a light microscopy (Leica). Photos were taken by an iPhone 11 camera (12 megapixels; Apple).
Figure S8. Data statistics summary of the R/qtl analysis of 224 final markers. Radish genome v2.20 (GenBank: GCA_002197605.1) was used.
Figure S9. Identification and validation of variations in RsNAC013 sequences. (a) Mapping of whole‐genome sequencing data of two parental lines (WK29 and WK40) against RsNAC013 sequences identified several SNPs and deletions, including a 46‐bp deletion in the 3′‐UTR of RsNAC013 in line WK40 (red arrow), which were used as pithiness markers (termed as NAC013del). Mapping was done in CLC Genomics Workbench with a length fraction 0.9 and similarity fraction 0.9. (b) Variations in full‐length RsNAC013 sequences from the two parental lines obtained from Sanger sequencing (Table S18). Primers for validation (see panel ‘e’) of the NAC013del marker were highlighted, positions 1909..2166 in line WK29, which encompasses the deletion and other small INDELs in line WK40. (c) Variations of RsNAC013 coding sequences identified between the two parental lines, including five SNPs and their Sanger sequencing validation. (d) Hydrophobicity of RsNAC013 proteins from the two lines, showing a potential region (blue arrow) that may influence the protein function. TM denotes the transmembrane motif. Hydrophobicity plot was generated using the online tool at http://best.bio.jhu.edu/HePCaT/run.pl, based on Kyte & Doolittle parameters. (e) A representative image of marker validation of NAC013del marker and qPITH‐1 QTL marker (locus S1_17807256, related to results in Figure 3e,f). PCR were run using DNA from the two parental lines. Primers for qPITH‐1 QTL included a common primer and two allele‐specific primers for lines WK29 and WK40, respectively. Primers for RsNAC013 deletion were designed to amplify the genomic regions shown in panel (b). The product size was 258 and 204 bp, for WK29 and WK40 lines, respectively. RsACTIN2/7 primers were used to check the loading DNA.
Figure S10. A co‐expression gene network of selected genes used in expression analysis (related to Figure 4). Genes related to ANAC013‐AtAOX1a, ROS production and scavenging in the oxidative pathway and PCD were used. The network was constructed by the STRING database (https://string‐db.org). Genes (nodes) are connected by edges, based on a default setting of evidence.
Figure S11. Schematics of the root RNA sample collection for qRT‐PCR and the expression of MDS genes that linked to hypoxia in our radish transcriptome data. (a) Schematics of the root RNA samples collected for expression analysis. RNAs were collected separately from four areas, top, bottom, inner and outer of each root. For whole‐root expression analysis, all four RNA samples were equally mixed to form one single sample for qRT‐PCR analysis. For spatial expression analysis, inner and outer root RNA samples were used. (b) Expression levels of MDS genes in the mitochondrial signalling pathway that are also linked to hypoxic responses. FNO and FSP denote field‐collected samples with and without pithiness, respectively. SNU denotes samples collected from the greenhouse. DEG genes are marked with ‘DEG’ in their names.
Figure S12. Additional histological images of root samples from the two inbred lines WK29 and WK40, related to Figure 4e. Evans Blue staining for cell death (a); DAB and NBT staining of hydrogen peroxide (H2O2) (b) and superoxide (O2−) (c) accumulation, respectively, in samples from lines WK29 and WK40; 12‐WAP root samples were used. Sample sections were stained and viewed under a light microscopy (Leica). Photos were taken by an iPhone 11 camera (12 megapixels, Apple).
Figure S13. Root collection from 12‐WAP plants treated with H2O2. T and N denote treated and non‐treated, respectively.
Figure S14. Line WK29 roots collected at 12 WAP and 14 WAP. All samples showed no pithiness phenotype. Roots of 12 WAP were dissected, examined but not photographed. N in sample names denotes non‐treated.
Figure S15. Adapter construct and primer design for the GBS library used in this study. A unique barcode was inserted into the EcoRI adapter sequence. To minimize the amplification of the same‐ended fragments (MseI – MseI) during PCR, we generated a Y‐shaped MseI adapter and an overhanging EcoRI adapter so that the fragments cut by both enzymes were preferentially amplified. Primers for PCR amplification are provided below the construct scheme.
Figure S16. Determination of the H2O2 concentration for the radish oxidative stress experiment. 6‐WAP radishes of line WK40 were used for this experiment. Plants were treated with hydrogen peroxide (H2O2) of concentration ranging from 0 to 1000 mm in the greenhouse condition. (a) Plants at 2 days after treatment. (b) Plants at 5 days after treatment.
Table S1. Summary statistics of samples and RNA‐seq sequencing reads.
Table S2. Radish gene IDs and corresponding Arabidopsis homologous gene IDs and their expression in the transcriptome data.
Table S3. List of 3966 DEGs identified from three comparisons between the selected samples and their expression values (FPKM).
Table S4. qRT‐PCR validation of RNA‐seq results using selected genes related to oxidative stress and cell death.
Table S5. GO enrichment analysis of 3966 DEGs by BINGO (FDR‐adjusted P‐value ≤ 0.05).
Table S6. GOSlim analysis of 3966 DEGs by BINGO (FDR‐adjusted P‐value ≤ 0.05).
Table S7. KEGG metabolic pathway analysis of 3966 DEGs by KOBAS.
Table S8. List of DEGs in two major clusters and their GOSlim analyses.
Table S9. KEGG metabolic pathway analysis of two DEG clusters by KOBAS.
Table S10. Common dPCD genes between radish DEGs and the core PCD genes in Arabidopsis (Olvera‐Carrillo et al., 2015).
Table S11. Common ePCD genes between radish DEGs and the core PCD genes in Arabidopsis (Olvera‐Carrillo et al., 2015).
Table S12. Expression levels of selected PCD genes in the inner and outer root tissues of radish roots undergoing pithiness formation measured by qRT‐PCR.
Table S13. Sequencing statistic summary of the GBS library for the QTL analysis.
Table S14. List of 224 SNPs that segregate at a 1:2:1 ratio in the F2 individuals.
Table S15. Gene search of the major QTL peak on chromosome 1.
Table S16. Expression levels of RsNAC013 and its direct target RsAOX1a in the transcriptome and qRT‐PCR data.
Table S17. Variations in the RsNAC013 sequence between the two radish lines WK40 and WK29.
Table S18. gDNA and CDS sequences of RsNAC013 obtained from Sanger sequencing.
Table S19. Correlation between qPITH‐1 QTL (S1_17807256) marker and RsNAC013 46‐bp deletion marker in the F2 samples.
Table S20. Expression levels of selected genes related to the RsNAC013 pathway as measured by qRT‐PCR in radish roots.
Table S21. Expression analysis of RsNAC013 and RsAOX1a in radish roots treated with H2O2 compared with non‐treated control samples by qRT‐PCR.
Table S22. Analysis of transcriptional activity of two variants of RsNAC013.
Table S23. Pithiness index of 92 F2 individuals used for GBS in this study.
Table S24. Concentration, RIN values and rRNA ratios of 17 samples used for RNA‐seq in this study.
Table S25. Whole‐genome sequencing data summary of the two parental lines used in this study.
Table S26. EcoRI adapter sequences used in this study.
Table S27. A list of primers used in SNP validation and RsNAC013 gene cloning.
Table S28. A list of primers used in gene expression analysis.
Video S1. Micro‐CT scanning of a root showing pithiness.
Data Availability Statement
The accession numbers for the 17 RNA‐seq datasets and raw GBS read data reported in this paper are available at GenBank: PRJNA682541 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA682541) and PRJNA682792 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA682792), respectively. All other relevant data supporting the findings here are included within the article and supplementary files.