

Abstract
Comparison of different membrane anchor motifs for the surface display of a protein of interest (passenger) is crucial for achieving the best possible performance. However, generating genetic fusions of the passenger to various membrane anchors is time‐consuming. We herein employ a recently developed modular display system, in which the membrane anchor and the passenger are expressed separately and assembled in situ via SpyCatcher and SpyTag interaction, to readily combine a model passenger cytochrome P450 BM3 (BM3) with four different membrane anchors (Lpp‐OmpA, PgsA, INP and AIDA‐I). This approach has the significant advantage that passengers and membrane anchors can be freely combined in a modular fashion without the need to generate direct genetic fusion constructs in each case. We demonstrate that the membrane anchors impact not only cell growth and membrane integrity, but also the BM3 surface display capacity and whole‐cell biocatalytic activity. The previously used Lpp‐OmpA as well as PgsA were found to be efficient for the display of BM3 via SpyCatcher/SpyTag interaction. Our strategy can be transferred to other user‐defined anchor and passenger combinations and could thus be used for acceleration and improvement of various applications involving cell surface display.
Keywords: biocatalysis, cytochrome P450 BM3, membrane proteins, SpyCatcher/SpyTag, surface display
Highway to optimized surface display: A modular surface display system, employing the SpyCatcher/SpyTag bioconjugation system, is used for rapid comparison of the membrane anchors Lpp‐OmpA, PgsA, INP and AIDA‐I with respect to their performance for surface display of the model passenger cytochrome P450 BM3. The described combinatorial screening method helps to minimize the workload required for identifying the most suitable membrane anchor for a given application.
Introduction
The presentation of heterologous proteins on the surface of microorganisms is a widely used strategy for many applications in biotechnology ranging from protein library screening,[ 1 , 2 ] live vaccine development, [3] biosensors [4] or the production of high‐performance whole‐cell biocatalysts.[ 4 , 5 ] Due to its ability to produce recombinant proteins in high yields and the ease of genetic manipulation, the gram‐negative bacterium Escherichia coli is a frequently used microorganism for this purpose. The conventional approach to transport a protein of interest (passenger) across the inner and outer membrane onto the E. coli cell surface involves its genetic fusion to a membrane anchor. A variety of different membrane anchors has been described for the display of passengers on E. coli.[ 4 , 5 , 6 ] However, issues such as unfavorable domain interactions, hybrid protein misfolding, degradation or loss of functionality are intrinsic for translational fusion systems and can possibly affect surface display capacity or passenger functionality. Thus, many studies compare genetic fusions to different membrane anchors to enable efficient display of the target passenger. [7] However, considering the high workload required to construct various genetic fusions, a faster approach for screening of different membrane anchors would be desirable.
In contrast to direct genetic fusion approaches, so called post‐translational bioconjugation methods, such as the SpyCatcher/SpyTag[ 8 , 9 ] technology, can be employed to combine protein components in a modular fashion, allowing for the easy and fast generation and comparison of various protein conjugates. Therefore, protein components are expressed with SpyCatcher (SC, 113 amino acids) and SpyTag (ST, 13 amino acids) domains respectively and are spontaneously coupled by formation of a covalent isopeptide bond between a lysine and an aspartic acid residue on the SC and ST domains. This technology has found diverse applications ranging from the assembly of multiple enzymes into defined scaffolds or hydrogels [10] over the immobilization of proteins or living cells onto particles [11] to modular vaccine assembly [12] or the generation of self‐assembled antibody like devices. [13]
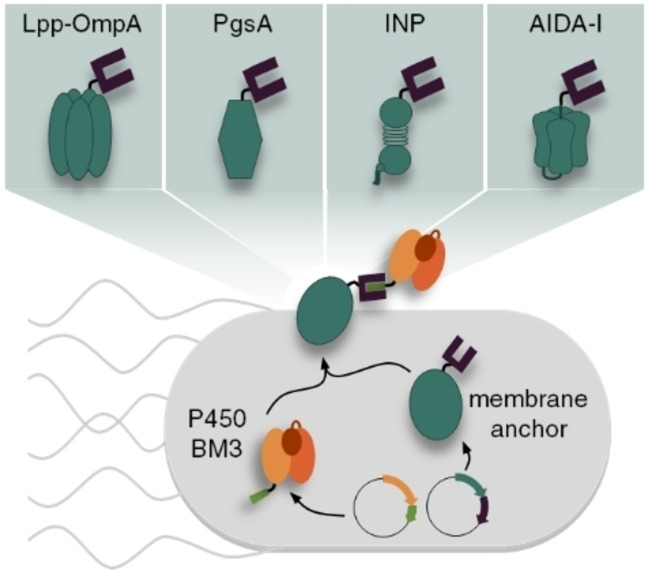
Recently, we employed the SC/ST technology to develop a novel modular surface display method in which the membrane anchor and the passenger are expressed separately and assembled by post‐translational coupling of genetically fused SC and ST domains, thereby mediating the transport to the cell surface (Figure 1A). [14] This novel approach, herein referred to as SC/ST surface display (S3D) system, provided several advantages over the conventional direct genetic fusion. First, it enabled the display of a functional variant of the bulky (119 kDa) heme and diflavin containing cytochrome P450 BM3 monooxygenase (BM3) from Bacillus megaterium (CYP102A1, EC 1.14.14.1) [15] using the well‐established membrane anchor Lpp‐OmpA. With this, a previously described size limitation of this membrane anchor for passengers up to a maximum of 70 kDa [16] has been overcome. Second, in contrast to the conventional display system based on a direct genetic fusion, the S3D system allows the placement of the covalent coupling site at the N‐terminus, C‐terminus and also into an internal loop, depending on the individual needs of the passenger. [14] The potential of the S3D system was recently further highlighted by Kajiwara et al., who employed the system in yeast cells with the goal of enabling in vivo continuous mutagenesis of the separated passenger protein without introducing mutations into the membrane anchor. [17]
Figure 1.

Modular surface display of a BM3 passenger employing different membrane anchors. (A) The membrane anchor is provided with a SC and the BM3 passenger with a matching ST binding unit. Both parts are expressed separately from individual plasmids, post‐translationally assembled through the specific, covalent SC/ST interaction and the complex is finally installed in the E. coli cell envelope via the membrane anchor. (B) Due to the modularity of the system, the different membrane anchors Lpp‐OmpA (I), PgsA (II), INPNC (III) and AIDA‐I (IV) can be compared regarding their performance for surface display of a BM3‐ST passenger. (C) Schematic representation of genetic constructs for the different membrane anchors fused to SC domains via flexible GGGGS linkers. CRD: central repeat domain; ctxB SP: signal peptide of cholera toxin B subunit.
In addition to these features, the modular expression can be utilized to readily combine a passenger with a variety of membrane anchors without the need to construct each individual genetic fusion. While in our initial description of the S3D system we used only one membrane anchor (Lpp‐OmpA) as a proof‐of‐concept, [14] we here report for the first time the transfer of our system to three other established membrane anchors, thus demonstrating the combinatorial screening of membrane anchors. To this end, the performance of the established membrane anchors PgsA, [18] INPNC [19] and AIDA‐I [20] was compared with the Lpp‐OmpA anchor regarding the effect on cell growth and membrane integrity, as well as the surface display capacity and whole‐cell biocatalytic activity using a variant of the challenging enzyme BM3 as a model passenger (Figure 1B). The hybrid membrane anchor Lpp‐OmpA (Figure 1B, I) has been used for stable membrane anchoring of various functional enzymes fused to its C‐terminus with high efficiency.[ 21 , 22 ] However, we and other research groups observed a strong impact on cell viability after overexpression of Lpp‐OmpA.[ 14 , 23 ] For some applications, such as the production of reusable whole‐cell biocatalysts, the employment of other membrane anchors might therefore be beneficial. For example, the membrane anchor PgsA (Figure 1B, II), a membrane integrated protein from Bacillus subtilis involved in poly‐γ‐glutamate synthesis, [24] has been used in E. coli for heterologous surface display of various active enzymes without reports on reduced cell viability.[ 18 , 25 ] However, to the best of our knowledge, PgsA has so far not been used for surface display of passenger proteins larger than 77 kDa [18] such as the BM3 model passenger used in this study. Another well‐established membrane anchor is the ice nucleation protein (INP) (Figure 1B, III) from Pseudomonas syringae comprising of three domains. [26] The hydrophobic N‐terminal domain is anchored to the outer membrane via a glycosylphosphatidylinositol (GPI) anchor and the highly hydrophilic C‐terminal domain is exposed to the surrounding medium. The so called central repeat domain (CRD) consists of a series of repeats which are not required for export to the cell surface and can therefore be used as spacer units to control the distance between the passenger protein and the cell surface. Several constructs such as the full‐length INP as well as different truncated forms lacking the CRD, or consisting of only the N‐terminal domain have been used for successful surface display of active enzymes without affecting the cell viability.[ 2 , 19 , 26 , 27 , 28 ] As the fourth example in this work, we chose the adhesin involved in diffuse adherence‐I (AIDA‐I) (Figure 1B, IV), which belongs to the family of autotransporters. [20] This protein contains a C‐terminal autotransporter structure forming a porin‐like β‐barrel channel on the outer membrane to facilitate the transport of a N‐terminal passenger domain towards the cell surface. By exchanging the native passenger domain against the protein of interest, many enzymes were successfully displayed on E. coli.[ 29 , 30 ] The INP membrane anchor as well as the AIDA‐I anchor are both notably effective for the surface presentation of particularly large or cofactor containing enzymes and have been used successfully for surface display of BM3 using the conventional direct genetic fusion approach.[ 28 , 31 ] Comparison to the S3D system in terms of display capacity is therefore particularly interesting for both anchors.
In order to employ the membrane anchors for the modular S3D system each anchor was provided with a SC domain using (glycine)4‐serine sequences (GGGGS) as a flexible linker between the gene‐encoding sequences. The design of each genetic construct is shown in Figure 1C. Lpp‐OmpA (Figure 1C, I) comprised of the signal peptide and the first 9 N‐terminal amino acids of the E. coli lipoprotein (Lpp) and the transmembrane domain (amino acids 46–159) from outer membrane protein A (OmpA). [21] The SC domain was fused to the surface exposed C‐terminus. PgsA (Figure 1C, II) was used as full length protein [18] with a C‐terminal fusion to the SC domain. For the INP anchor (Figure 1C, III) the N‐ and C‐terminal domains of inaK, a member of the INP family, were used as previously reported [19] with the CRD almost completely deleted (INPNC, the first 32 and the last 48 amino acids of the CRD were remained) and the SC domain was fused to the C‐terminus. The AIDA‐I fusion protein (Figure 1C, IV) was designed to comprise the signal peptide of cholera toxin B subunit (ctxB SP) and the SC domain fused to the N‐terminus of the AIDA‐I sequence, which consisted of a native linker region and the pore forming β‐barrel. [20] The amino acid sequences of all functional proteins are given in the Supporting Information.
Results and Discussion
Expression of the different membrane anchors as SC‐fusion proteins
Initially we examined whether the genetic fusions of SC to the different membrane anchors were successfully expressed in E. coli and whether the expression affected the cell growth or the integrity of the cell membrane (Figure 2). As a reference we analyzed E. coli cells expressing SC intracellularly without genetic fusion to a membrane anchor. Cells were transformed with plasmids encoding for SC alone or SC fused to the different membrane anchors and protein expression was analyzed 20 h post induction after separating the total protein fractions via Coomassie stained SDS‐PAGE (Figure 2A). For the intracellularly expressed SC as well as for each fusion of SC with the different membrane anchors, a distinct protein band was visible that corresponded to the expected calculated molecular weight, indicating successful expression of all fusion proteins. However, it should be noted that expression of the PgsA‐SC fusion protein appeared to be moderately lower than for the other fusion proteins. We had previously observed reduced cell viability after expression of Lpp‐OmpA‐SC [14] and similar effects were reported in several other studies for different membrane anchors.[ 23 , 32 ] Therefore, we also investigated on the cell growth and membrane permeability of the cells. We determined the cell density of all cultures after 20 h of induction (Figure 2B). Compared to the reference cells expressing SC intracellularly, cells expressing Lpp‐OmpA‐SC, PgsA‐SC or SC‐AIDA‐I exhibited significantly reduced cell growth, with PgsA‐SC and SC‐AIDA‐I expression showing a milder impact on cell growth than expression of Lpp‐OmpA‐SC. In contrast, cells expressing INPNC‐SC showed even stronger cell growth than the reference cells. Since it has been reported that reduced growth rates and membrane permeability can be correlated, we analyzed the membrane integrity (Figure 2C) and incubated the cells with the DNA intercalating dye propidium iodide (PI) which can only penetrate into cells with permeabilized membranes (see Figure S1). Cells expressing the SC intracellularly only exhibited very little PI fluorescence, indicating that the expression of SC alone does not affect the membrane integrity. Other than that, the reduced growth observed for cells expressing Lpp‐OmpA‐SC, PgsA‐SC and SC‐AIDA‐I seems to be associated with an increased outer membrane permeability in all cases, as revealed by the strong PI signals determined for these cells. Again cells expressing PgsA‐SC or SC‐AIDA‐I exhibited less severe alterations in membrane permeability then cells expressing Lpp‐OmpA‐SC, which is in line with the observation concerning the growth (Figure 2B). Reducing the incubation time to 4 h after induction of protein expression did not prevent the observed membrane permeabilization (Figure S2). Cells expressing INPNC‐SC did not show any increased PI fluorescence, suggesting that this membrane anchor does not impact the integrity of the cell membrane. Expression of the anchors Lpp‐OmpA, PgsA and AIDA‐I in the absence of a genetically fused SC domain also resulted in reduced cell growth and increased membrane permeability (Figure S3), suggesting that these deficiencies occur independently of the passenger domain. Negative effects on cell integrity and viability are frequently reported upon overexpression of membrane proteins in E. coli.[ 14 , 23 , 32 , 33 ] Studies regarding the physiology underlying this toxicity suggest that the overexpressed membrane proteins may overload the E. coli translocation machinery, thereby preventing the transport of endogenic membrane proteins required for the maintenance of proper cellular function. However, the mechanisms are not yet fully understood and several factors might be involved.[ 33 , 34 ]
Figure 2.

Protein expression, growth and membrane permeability of E. coli cells expressing SC fused to different membrane anchors. Cells expressing the SC intracellularly or as genetic fusion to the membrane anchors Lpp‐OmpA, PgsA, INPNC or AIDA‐I were analyzed 20 h after of induction of protein expression. (A) Total protein fractions separated by Coomassie stained SDS‐PAGE (15 % (w/v) acrylamide). The expected molecular weight (Mw) of each protein as well as the molecular weight of marker bands is given in kDa. The bands of the detected proteins are outlined in green. (B) Density of cell cultures determined by measuring the OD600. (C) Permeability of the cell membrane determined by incubation with the membrane impermeable DNA intercalating dye propidium iodide (PI). A high PI fluorescence signal is related to increased membrane permeability. Details about the staining procedure are given in Figure S1. Error bars were obtained from at least two independent experiments.
Surface exposure and functionality of the SC domains
Apart from integrity and viability of the cells, the surface exposure and functionality of the SC domains fused to the different membrane anchors is important for the application of the S3D system. In order to analyze these properties, we incubated the cells with a purified mRFP‐ST‐his6 fusion protein for covalent binding to functional SC (Figure 3A, I). Although the mRFP‐ST‐his6 probe should not penetrate into intact cells and should therefore only bind to surface exposed SC domains, the observed alterations in membrane permeability (see Figure 2C) might enable the mRFP‐ST‐his6 probe to bind to cytosolic SC domains. To exclude effects due to increased membrane permeability and to ensure specific detection of surface presented SC/ST conjugates we employed double staining with the Alexa488 anti‐his6 antibody (Figure 3A, II). We had previously demonstrated that the antibody is unable to penetrate into cells even after permeabilization caused by the overexpression of a membrane anchor. [14] This was further verified by analysis of control cells expressing cytosolic mRFP‐ST‐his6, as well as the membrane anchor Lpp‐OmpA, which caused the highest degree of membrane permeabilization (see Figure 2C). Staining of these cells with the Alexa488 anti‐his6 antibody resulted in no detectable Alexa488 fluorescence as revealed by fluorescence microscopy (Figure S4). These results demonstrate that the antibody is unable to penetrate into the cells regardless of the membrane permeabilization and that our double‐staining procedure therefore allows for specific detection of surface exposed SC/ST conjugates. The staining procedure was applied to cells expressing SC intracellularly or as genetic fusion to the membrane anchors and cells were analyzed using fluorescence microscopy (Figure 3B). As expected, cells expressing the SC intracellularly did not show any mRFP or Alexa488 fluorescence. Surprisingly, cells expressing INPNC‐SC also showed almost no mRFP or Alexa488 fluorescence. Thus, although the fusion protein was successfully expressed (see Figure 2A), it does not appear to be presented properly on the cell surface. In contrast, we were able to detect mRFP as well as Alexa488 fluorescence for cells expressing Lpp‐OmpA‐SC, PgsA‐SC or SC‐AIDA‐I, suggesting that the SC is functional and indeed presented on the E. coli cell surface. To compare the SC surface display capacity with the different anchors, the Alexa488 fluorescence was quantified using a microplate reader (Figure 3C). Accordingly, the display capacity was highest using the Lpp‐OmpA membrane anchor and slightly lower using the AIDA‐I anchor. However, both anchors achieved a 6‐fold and 4‐fold higher display capacity than the PgsA membrane anchor, respectively. This might be related to the observed lower expression rate of PgsA‐SC compared to Lpp‐OmpA‐SC and SC‐AIDA‐I (see Figure 2A).
Figure 3.

Investigation on functionality and surface exposure of SC fused to different membrane anchors. E. coli cells expressing the SC intracellularly or as genetic fusion to the membrane anchors Lpp‐OmpA, PgsA, INPNC or AIDA‐I were analyzed 20 h after induction of protein expression. (A) Cells were incubated with a purified mRFP‐ST‐his6 protein that covalently binds to functional SC and surface localization was confirmed using a membrane‐impermeable Alexa488 conjugated anti‐his6 antibody. The stained cells were analyzed using fluorescence microscopy (B) and the Alexa488 fluorescence of the different cell suspensions was quantified using a microplate reader in order to compare the display capacities (C). Scale bars correspond to 10 μm. Error bars were obtained from at least two independent experiments. Please note that the aberrant shape of cells expressing SC intracellularly could be due to the use of the pET‐EXPn1 vector as discussed in more detail in Figure S5.
Surface display of a BM3 model passenger
Next, we investigated the ability of the different SC modified membrane anchors to display a co‐translated BM3‐ST passenger enzyme on the E. coli cell surface. To this end, cells were transformed with the plasmid encoding for BM3‐ST as well as plasmids encoding for the different membrane anchors fused to SC. We verified formation of covalent SC/ST conjugates by western blot analysis and observed covalent coupling to BM3‐ST for all SC modified membrane anchors Lpp‐OmpA, PgsA, INPNC and AIDA‐I (Figure S6). In contrast, we observed no conjugation when the respective membrane anchors were either fused to ST or to an inactivated SCEQ, in which formation of the covalent isopeptide bond is prevented by the mutation E77Q [8] (Figure S6). We then investigated on the surface exposure of BM3‐ST conjugated to the SC modified membrane anchors (Figure 4A, S3D system). For comparison, direct genetic fusions of BM3 to the membrane anchors were encoded on a single plasmid and also transformed into E. coli cells (Figure 4A, conventional system). As a control we used cells expressing the BM3‐ST intracellularly without co‐expression of a membrane anchor (Figure 4A, no anchor). The cell growth, analyzed 20 h after induction of protein expression (Figure S7), was similar to that observed in cells expressing the anchor‐SC fusions alone (see Figure 2B). Thus, co‐expression of the BM3‐ST passenger does not seem to further affect cell growth. Moreover, we did not observe any difference in growth behavior when cells presented BM3 using the S3D system or the conventional system (Figure S7). The surface exposure of the BM3 passenger was analyzed by immunofluorescence staining using a DyLight488 conjugated anti‐Myc antibody, which bound to a terminal Myc epitope tag on the BM3 (for details on genetic constructs see Table S1). As previously reported [14] and confirmed in Figure S4, such antibodies bind exclusively to surface presented epitopes even after cell permeabilization by overexpression of membrane anchors and are therefore well suited to reliably confirm the surface exposure of BM3. The DyLight488 fluorescence of the cells was analyzed using fluorescence microscopy (Figure S8) and the display capacity was compared by quantification of the DyLight488 fluorescence with the aid of a microplate reader (Figure 4B). As expected, no fluorescence was detected for control cells expressing no membrane anchor. In case of the Lpp‐OmpA membrane anchor, high immunofluorescence for the S3D system but only very little signal using the conventional system indicated that BM3 could only be displayed using Lpp‐OmpA when employing the S3D system, thereby confirming our previously reported results, in which we found that the S3D system is superior to the conventional system due to absent expression of the Lpp‐OmpA‐BM3 fusion protein. [14] Also, for the PgsA membrane anchor immunofluorescence could only be detected when the BM3 passenger was presented employing the S3D system. SDS‐PAGE and western blot analysis (Figure S9) revealed that this is again related to absent expression of the PgsA‐BM3 fusion protein in the conventional system. These results confirm that the S3D approach can indeed be applied to other membrane anchors to provide advantages over the conventional direct genetic fusion, presumably by avoiding possible unfavorable domain interactions that might occur when the bulky BM3 passenger is directly fused to the membrane anchors. However, the display capacity using PgsA for S3D was significantly lower when compared to the Lpp‐OmpA membrane anchor. This might be related to the lower expression rate (see Figure 2A) and the resulting lower surface coverage (see Figure 3B and C) of PgsA‐SC. Using the INPNC membrane anchor we could not detect any immunofluorescence and thus no BM3 surface presentation with the S3D system, which was to be expected since we observed that the INPNC‐SC fusion was not presented properly on the cell surface (see Figure 3B and C). However, we were surprised that we could also not detect any immunofluorescence using the conventional display system, since successful surface display of BM3 directly fused to INPNC has been reported earlier. [28] Since SDS‐PAGE and western blot indicated successful coupling of INPNC‐SC to BM3‐ST in the S3D system (Figure S6 and S9) as well as successful expression of INPNC‐BM3 in the conventional system (Figure S9), this must be related to unsuccessful translocation of INPNC to the cell surface. Employing the S3D system with the AIDA‐I membrane anchor did not achieve efficient BM3 surface presentation as revealed by the only low immunofluorescence signal for this sample, although covalent coupling of SC‐AIDA‐I to BM3‐ST was confirmed by western blot (Figure S6 and S9). We assume that this might be related to the translocation mechanism of AIDA‐I, which belongs to the family of autotransporters. Although the translocation mechanisms of this family is not yet fully understood, it is assumed that the C‐terminal part of the autotransporter is folded first to form a porin‐like β‐barrel channel in the outer membrane through which the N‐terminal passenger is translocated in a mostly unfolded state. It has been reported that passengers with the propensity to rapidly fold into a complex structure inside the cell may be blocked in translocation.[ 29 , 35 ] Likewise, in‐cell conjugation of SC‐AIDA‐I to the co‐expressed and already folded BM3‐ST might prevent successful translocation to the E. coli cell surface, rendering the AIDA‐I anchor unsuitable for use with the S3D system. In contrast, we were able to detect high immunofluorescence and therefore confirmed successful surface presentation of BM3 by conventional direct genetic fusion to AIDA‐I, which has also been reported previously. [31]
Figure 4.

Surface display of BM3. (A) Control cells expressing BM3‐ST intracellularly (grey) were compared to cells displaying BM3 with the S3D system (light blue) or with the conventional system (dark blue) employing different membrane anchors. (B) Surface display capacity as revealed by immunofluorescence staining of the BM3 passenger. (C) Schematic representation of BM3 whole‐cell biocatalytic activity assay. Cells were incubated with membrane‐impermeable substrate (1) and BM3 catalyzed conversion to intermediate (2) followed by spontaneous decomposition to fluorescent product (3) which was monitored using a microplate reader. Note that in this study a BM3 variant carrying mutations A74G and F78V was used, which exhibits the highest reported conversion rate for substrate (1). [15] The assay results are illustrated in (D). Error bars were obtained from at least two independent experiments.
Apart from the successful surface display, retaining the functionality of the passenger is crucial. To this end, the BM3 whole‐cell biocatalytic activity was determined by incubating the cells with the fluorogenic surrogate substrate 12‐(4‐trifluoromethylcoumarin‐7‐yloxy)dodecanoic acid (1) and the cofactor NADPH (Figure 4C). BM3 catalyzed conversion of (1) leads to formation of the unstable hemiacetal intermediate (2) which spontaneously decomposes to release the fluorophore 7‐hydroxy‐4‐trifluoromethylcoumarin (3). Formation of product (3) was followed by fluorescence read‐out using a microplate reader (Figure 4D). No product formation could be detected for control cells expressing intracellular BM3, since substrate (1) cannot penetrate the intact membrane of cells. In general BM3 whole‐cell activity correlated strongly with previously determined BM3 display capacity (compare Figure 4B), confirming that successfully displayed BM3 passengers retained enzymatic activity and that the corresponding cells could be used as whole‐cell biocatalysts for membrane‐impermeable substrates. However, uncorrelated to the observed BM3 display capacities, we could detect strong BM3 whole‐cell activity when we employed the AIDA‐I membrane anchor for the S3D system, performing even slightly better than the conventional AIDA‐I display system. Since the BM3 passenger has been shown not to be presented on the cell surface using AIDA‐I for S3D (see Figure 4B), this is likely due to the fact that expression of SC‐AIDA‐I leads to an increased membrane permeability (see Figure 2C), allowing substrate (1) to penetrate into the cell and be converted by intracellularly expressed BM3‐ST. Likewise, the strong BM3 whole‐cell activity observed for cells employing Lpp‐OmpA or PgsA for S3D, might not only originate from surface presented but also partially from intracellular BM3, since expression of both anchors also caused increased membrane permeability (see Figure 2C). This example demonstrates that whole‐cell activity assays using membrane‐impermeable substrates are not completely conclusive to prove the passenger localization on the cell surface and should solely be considered as evidence for the preservation of passenger functionality and to demonstrate the cells usability as whole‐cell biocatalysts. Instead, as conducted in this study, surface presentation should always be confirmed by using by immunofluorescence staining.
Conclusion
In this work we expanded our recently developed, modular S3D system to different membrane anchors in order to establish a toolbox of SC modified membrane anchors that can be readily compared for surface display of a given ST modified passenger without the need to construct each individual genetic fusion. To this end, we analyzed E. coli strains expressing fusion proteins consisting of SC and the well‐established membrane anchors Lpp‐OmpA, PgsA, INPNC or AIDA‐I in terms of protein expression, cell growth and membrane integrity as well as surface exposure of the SC domain and subsequently employed the strains for surface display of a co‐expressed ST modified BM3 passenger enzyme via the S3D system. The display capacities and BM3 whole‐cell biocatalytic activities were compared to cells displaying the BM3 passenger by conventional direct genetic fusion to each membrane anchor. In our experiments we were not able to detect any surface presentation of BM3 using a literature described INPNC [19] membrane anchor regardless of whether the S3D or the conventional system was used. Since a variety of INP variants have been reported,[ 2 , 36 ] the simplicity of the S3D system can be exploited to identify other INP constructs which enable efficient surface presentation. In contrast, by using the AIDA‐I membrane anchor we were able to display BM3 by conventional direct genetic fusion as described previously. [31] However, display of BM3 using AIDA‐I with the S3D system was not efficient, even though SC‐AIDA‐I alone was confirmed to be presented on the cell surface and covalent bond formation to co‐expressed BM3‐ST could be demonstrated. This might be related to the translocation mechanism of AIDA‐I, which requires the passenger to be in a mostly unfolded state for translocation through its β‐barrel pore.[ 29 , 35 ] SC‐AIDA‐I alone can meet these requirements and can therefore be successfully presented on the cell surface, as shown in this study. However, covalent binding of SC‐AIDA‐I to BM3‐ST probably blocks translocation through the autotransporter pore, as the co‐expressed BM3‐ST is presumably fully folded already inside the cell. The AIDA‐I anchor might therefore not be suitable for use with the S3D system. In contrast, the PgsA membrane anchor enabled successful display of the BM3 passenger via the S3D system as revealed by immunostaining. Importantly, the S3D system provided significant advantages over the conventional direct genetic fusion to PgsA, which resulted in no detectable surface display of BM3 due to absent expression of the PgsA‐BM3 fusion protein, similar to previously reported results for the Lpp‐OmpA membrane anchor. [14] Although display capacity and whole‐cell activity were lower than when Lpp‐OmpA‐SC was used, expression of PgsA‐SC appeared to have a slightly reduced impact on cell growth and membrane permeability. Depending on the intended applications, the use of one or the other membrane anchor could therefore be beneficial. The results indicate that our novel S3D system could also be transferred to other membrane anchors in order to extend the toolbox. However, care should be taken to select anchors which use a mechanism that allows translocation of already folded SC/ST conjugates. On the side of the passenger only a simple modification with a ST is sufficient to enable fast and convenient screening of different membrane anchors. This will significantly contribute to minimizing the workload required to identify the most suitable membrane anchor for different applications. Furthermore, our study is the first to compare different approaches for surface display of the challenging BM3 passenger enzyme and may provide guidance to researchers considering the use of whole‐cell biocatalysis with P450 BM3 variants.
Experimental Section
Plasmids
All fusion proteins containing membrane anchors were encoded on pTF16 based plasmids employing a p15A ori, a chloramphenicol resistance gene and the tight L‐arabinose‐inducible araB promoter. BM3‐ST fusion proteins expressed in the cytosol were encoded on fully orthogonal pET based plasmids employing a pBR322 ori, an ampicillin resistance gene and an IPTG inducible T7 promoter. Details about plasmid construction and amino acid sequences are given in the Supporting Information.
Bacterial growth conditions
E. coli BL21(DE3) cells were transformed with the respective plasmids using electroporation. The cells were selected overnight on LB/agar plates containing appropriate antibiotics (30 μg/mL chloramphenicol and/ or 100 μg/mL ampicillin) at 37 °C. Liquid cultures of 5 mL LB+antibiotics were generated from clones of the LB/agar plates and cultured overnight for 14–18 h at 37 °C, 180 rpm in culture tubes. Flasks with 20 mL LB+antibiotics where inoculated with 0.5 mL overnight culture. The cultures were incubated at 37 °C, 180 rpm until an OD600 of 0.6 was reached. Subsequently protein expression was induced with 1 mM L‐arabinose (pTF16 based plasmids) and/or 1 mM IPTG (pET based plasmids) and the cultures were incubated at 25 °C, 180 rpm for 20 h.
Expression of different membrane anchors fused to SC
Cells were transformed with plasmid pET‐EXPn1_his6‐SC to generate a control strain with intracellular SC or with plasmids pTF16_lpp‐ompA‐SC/pTF16_pgsA‐SC/pTF16_inpNC‐SC/pTF16_ctxB‐SC‐AIDA to generate strains with surface displayed SC. Cultures were grown and induced as described above. 20 h after induction the OD600 was measured to determine the cell growth. Subsequently cells were harvested by centrifugation and resuspended in PBS (11.5 mM NaH2PO4, 50 mM NaCl, pH 7.3) as described below in order to analyze the expression of fusion proteins and to investigate on the membrane permeability as well as on the surface exposure and functionality of the SC domain.
Protein expression and western blot analysis
Cells were resuspended in PBS and the OD600 was adjusted to 10. Laemmli sample buffer (4×) was added and samples were sonicated for 15 s and incubated at 95 °C for 10 min. Proteins were separated on SDS‐PAGE (8 % or 15 % (w/v) acrylamide) and visualized by staining with Coomassie Brilliant Blue G‐250. For western blot analysis an identical gel was transferred onto a PVDF membrane (VWR) via electroblotting. The membrane was incubated with a mouse anti‐Myc antibody (Myc Tag Monoclonal Antibody (Myc.A7) MA1‐21316, Thermo Fisher Scientific) at a 1 : 1000 dilution and a secondary anti‐mouse antibody conjugated to alkaline phosphatase (Goat Anti‐Mouse IgG Alkaline Phosphatase AP‐112, Columbia Biosciences) at a 1 : 5000 dilution. Subsequently, the blot was developed with a colorimetric reagent (AP Conjugate substrate kit, Biorad).
Membrane permeability
Cells were resuspended in PBS and the OD600 was adjusted to 1. 100 μL of the cell suspensions were transferred into a microtiter plate (F96 Polysorb Nunc Plate, Thermo Fisher Scientific) and incubated with 1 μL propidium iodide solution (0.1 mg/mL in ddH2O) for 10 min at room temperature. Subsequently the fluorescence of DNA bound propidium iodide was measured with the aid of a monochromator‐based multi‐mode microplate reader (Synergy H1, BioTek Instruments GmbH) using an excitation wavelength of 535 nm and an emission wavelength of 617 nm. A schematic representation of the assay read‐out as well as exemplary data for intact and permeabilized cells can be found in Figure S1.
Surface exposure and functionality of SC domains
Cells were resuspended in PBS and the OD600 was adjusted to 5. 100 μL of the cell suspensions were mixed with 100 pmol mRFP‐ST‐his6 (see below for expression and purification of this protein probe) and incubated for 60 min at room temperature under continuous shaking. After two cycles of pelleting and washing the cells in 0.5 mL PBS buffer, the cells were resuspended in 100 μL PBS and incubated with a fluorescently labeled antibody against His6 (6x‐His Tag Monoclonal Antibody (4E3D10H2/E3) Alexa Fluor488 MA1‐135‐A488, Invitrogen) at a 1 : 100 dilution for 1 h at room temperature under continuous shaking. After three cycles of pelleting and washing the cells in 0.5 mL PBS buffer, the cells were finally resuspended in 100 μL PBS and transferred into a microtiter plate (F96 Polysorb Nunc Plate, Thermo Fisher Scientific). Subsequently the Alexa Fluor488 fluorescence was measured with the aid of a monochromator‐based multi‐mode microplate reader (Synergy H1, BioTek Instruments GmbH) using an excitation wavelength of 490 nm and an emission wavelength of 510 nm. Finally 10 μL of the cell suspensions were analyzed by fluorescence microscopy using a Rhodamine filter set for mRFP fluorescence (red channel) and a FITC filter set for Alexa Fluor488 fluorescence (green channel).
Surface display of BM3
Cells were transformed with different plasmids depending on the surface display system. For the control cells with intracellular BM3: pET_BM3‐ST‐myc. For the S3D system: pET_BM3‐ST‐myc along with either pTF16_lpp‐ompA‐SC, pTF16_pgsA‐SC, pTF16_inpNC‐SC or pTF16_ctxB‐SC‐AIDA‐I. For the conventional system: pTF16_lpp‐ompA‐BM3‐myc, pTF16_pgsA‐BM3‐myc, pTF16_inpNC‐BM3‐myc or pTF16_ctxB‐myc‐BM3‐AIDA‐I. Cultures were grown and induced as described above. 20 h after induction the OD600 was measured to determine the cell growth. Subsequently cells were harvested by centrifugation and resuspended in Tris buffer (100 mM Tris, pH 8.1) as described below for subsequent investigation on the BM3 surface display capacity and the BM3 whole‐cell biocatalytic activity. Alternatively cells were resuspended in PBS as described above for western blot analysis.
BM3 surface display capacity
The OD600 of the cell suspensions was adjusted to 5 and 100 μL were incubated with a fluorescently labeled antibody against Myc (Myc Tag Monoclonal Antibody (Myc.A7) DyLight488 MA1‐21316‐D488, Invitrogen) at a 1 : 100 dilution for 1 h at room temperature under continuous shaking. After three cycles of pelleting and washing the cells in 0.5 mL Tris buffer, the cells were finally resuspended in 100 μL Tris and transferred into a microtiter plate (F96 Polysorb Nunc Plate, Thermo Fisher Scientific). Subsequently the DyLight488 fluorescence was measured with the aid of a monochromator‐based multi‐mode microplate reader (Synergy MX, BioTek Instruments GmbH) using an excitation wavelength of 490 nm and an emission wavelength of 510 nm. Finally, 10 μL of the cell suspensions were analyzed by fluorescence microscopy using a FITC filter set (green channel).
BM3 whole‐cell biocatalytic activity
Activities were determined using a surrogate substrate described in a report of Neufeld et al. [15] The cell suspensions were diluted in microtiter plates (F96 Polysorb Nunc Plate, Thermo Fisher Scientific) to an OD600 of 0.25 in a total volume of 200 μL Tris buffer with 100 μM 12‐(4‐trifluoromethylcoumarin‐7‐yloxy)dodecanoic acid (1) and the reaction was started by addition of 1 mM NADPH. Stock solutions of substrate (1) were prepared in dimethyl sulfoxide (DMSO) and accounted for 1 % of the final assay samples. Samples were incubated at 30 °C and formation of the fluorescent product 7‐hydroxy‐4‐trifluoromethylcoumarin (3) was followed in time intervals of 60 s with the aid of a monochromator‐based multi‐mode microplate reader (Synergy H1, BioTek Instruments GmbH) using an excitation wavelength of 420 nm and an emission wavelength of 500 nm. Samples were shaken between measurements to avoid cell sedimentation. Fluorescence intensities were converted to product amounts using calibration samples of the fluorescent product (3) (Sigma‐Aldrich) in total amounts ranging from 0 to 400 pmol.
Expression and purification of mRFP‐ST‐his6
E. coli BL21(DE3) cells were transformed with the plasmid pET_mRFP‐ST‐his6 using heat shock. The cells were selected overnight on LB/agar plates containing 100 μg/mL ampicillin at 37 °C. Liquid cultures of 100 mL LB+Amp were generated from clones of the LB/agar plates and cultured overnight at 37 °C, 180 rpm. Two flasks with 2 l LB+Amp where inoculated with 40 mL overnight culture. The cultures were incubated at 37 °C, 180 rpm until an OD600 of 0.6 was reached. Subsequently the culture was cooled down to 25 °C for at least 15 min and mRFP‐ST‐his6 expression was induced with 0.5 mM IPTG. After incubation at 25 °C overnight the cells were harvested by centrifugation (10,000×g, 10 min, 4 °C), resuspended in NPi10 buffer (50 mM NaH2PO4, 500 mM NaCl, 10 mM imidazole, pH 8) and frozen at −80 °C until further processing. The cell suspension was thawed at 25 °C in a water bath and incubated with DNaseI (AppliChem) and lysozyme (AppliChem) for 30 min at room temperature. After further cell disruption using ultrasonication, debris were pelleted by centrifugation (45,000×g, 1 h, 4 °C) and the protein rich supernatant filtered through a 0.45 μm Durapore PVDF membrane (Steriflip, Millipore) and loaded on a 5 mL His60 Ni Superflow Cartridge (Clontech) mounted on an Äkta Pure liquid chromatography system (GE Healthcare). The cartridge was washed with a mixture of 2 % NPi500 (50 mM NaH2PO4, 500 mM NaCl, 500 mM imidazole, pH 8) and 98 % NPi10. Subsequently the protein was eluted using a linear gradient (2 % to 100 % NPi500). The column outflow was collected in 900 μL fractions and protein containing fractions (detected at 280 nm) were pooled. Finally, the buffer was exchanged to PBS using Vivaspin Turbo 15, 10000 MWCO concentrators (Sartorius).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported through the Helmholtz program “Materials Systems Engineering” under the topic “Adaptive and Bioinstructive Materials Systems”. S.G. is grateful for support by a Kekulé fellowship by Fonds der Chemischen Industrie (FCI). We thank Ishtiaq Ahmed for synthesis of the fluorogenic BM3 substrate and Martin Peng and Sebastian Häcker for construction of the plasmid pET_SC‐his6. Open Access funding enabled and organized by Projekt DEAL.
S. Gallus, E. Mittmann, K. S. Rabe, ChemBioChem 2022, 23, e202100472.
References
- 1. Becker S., Schmoldt H.-U., Adams T. M., Wilhelm S., Kolmar H., Curr. Opin. Biotechnol. 2004, 15, 323. [DOI] [PubMed] [Google Scholar]
- 2. van Bloois E., Winter R. T., Kolmar H., Fraaije M. W., Trends Biotechnol. 2011, 29, 79. [DOI] [PubMed] [Google Scholar]
- 3. Georgiou G., Stathopoulos C., Daugherty P. S., Nayak A. R., Iverson B. L., Curtiss R., Nat. Biotechnol. 1997, 15, 29. [DOI] [PubMed] [Google Scholar]
- 4. Han L., Zhao Y., Cui S., Liang B., Appl. Biochem. Biotechnol. 2018, 185, 396. [DOI] [PubMed] [Google Scholar]
- 5. Schüürmann J., Quehl P., Festel G., Jose J., Appl. Microbiol. Biotechnol. 2014, 98, 8031. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Samuelson P., Gunneriusson E., Nygren P.-Å., Ståhl S., J. Biotechnol. 2002, 96, 129; [DOI] [PubMed] [Google Scholar]
- 6b. Lee S. Y., Choi J. H., Xu Z., Trends Biotechnol. 2003, 21, 45. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Csibra E., Renders M., Pinheiro V. B., ChemBioChem 2020, 21, 2844–2853; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Chung M.-E., Goroncy K., Kolesnikova A., Schönauer D., Schwaneberg U., Biotechnol. Bioeng. 2020, 117, 3699–3711; [DOI] [PubMed] [Google Scholar]
- 7c. Heyde S. A. H., Arnling Bååth J., Westh P., Nørholm M. H. H., Jensen K., Microb. Cell Fact. 2021, 20, 93; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7d. Nicchi S., Giuliani M., Giusti F., Pancotto L., Maione D., Delany I., Galeotti C. L., Brettoni C., Microb. Cell Fact. 2021, 20, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zakeri B., Fierer J. O., Celik E., Chittock E. C., Schwarz-Linek U., Moy V. T., Howarth M., Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Sutherland A. R., Alam M. K., Geyer C. R., ChemBioChem 2019, 20, 319; [DOI] [PubMed] [Google Scholar]
- 9b. Keeble A. H., Howarth M., Chem. Sci. 2020, 11, 7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Jia L., Minamihata K., Ichinose H., Tsumoto K., Kamiya N., Biotechnol. J. 2017, 12; [DOI] [PubMed] [Google Scholar]
- 10b. Bao J., Liu N., Zhu L., Xu Q., Huang H., Jiang L., J. Agric. Food Chem. 2018, 66, 8061; [DOI] [PubMed] [Google Scholar]
- 10c. Peschke T., Bitterwolf P., Gallus S., Hu Y., Oelschlaeger C., Willenbacher N., Rabe K. S., Niemeyer C. M., Angew. Chem. Int. Ed. 2018, 57, 17028; [DOI] [PubMed] [Google Scholar]
- 10d. Zhang G., Quin M. B., Schmidt-Dannert C., ACS Catal. 2018, 8, 5611; [Google Scholar]
- 10e. Mittmann E., Gallus S., Bitterwolf P., Oelschlaeger C., Willenbacher N., Niemeyer C. M., Rabe K. S., Micromachines 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Peschke T., Rabe K. S., Niemeyer C. M., Angew. Chem. Int. Ed. 2017, 56, 2183; [DOI] [PubMed] [Google Scholar]
- 11b. Ma W., Saccardo A., Roccatano D., Aboagye-Mensah D., Alkaseem M., Jewkes M., Di Nezza F., Baron M., Soloviev M., Ferrari E., Nat. Commun. 2018, 9, 1489; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Peng M., Siebert D. L., Engqvist M. K. M., Niemeyer C. M., Rabe K. S., ChemBioChem 2021, 10.1002/cbic.202100468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Brune K. D., Leneghan D. B., Brian I. J., Ishizuka A. S., Bachmann M. F., Draper S. J., Biswas S., Howarth M., Sci. Rep. 2016, 6, 19234; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Thrane S., Janitzek C. M., Matondo S., Resende M., Gustavsson T., de Jongh W. A., Clemmensen S., Roeffen W., van de Vegte-Bolmer M., van Gemert G. J., Sauerwein X., Schiller J. T., Nielsen M. A., Theander T. G., Salanti A., Sander A. F., J. Nanobiotechnol. 2016, 14, 30; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. van den Berg van Saparoea H. B., Houben D., de Jonge M. I., Jong W. S. P., Luirink J., Appl. Environ. Microbiol. 2018, 84, e02567–e02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Alam M. K., Gonzalez C., Hill W., El-Sayed A., Fonge H., Barreto K., Geyer C. R., ChemBioChem 2017, 18, 2217; [DOI] [PubMed] [Google Scholar]
- 13b. Alam M. K., El-Sayed A., Barreto K., Bernhard W., Fonge H., Geyer C. R., Mol. Imaging Biol. 2019, 21, 54. [DOI] [PubMed] [Google Scholar]
- 14. Gallus S., Peschke T., Paulsen M., Burgahn T., Niemeyer C. M., Rabe K. S., ChemBioChem 2020, 21, 2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Neufeld K., zu Berstenhorst S. M., Pietruszka J., Anal. Biochem. 2014, 456, 70. [DOI] [PubMed] [Google Scholar]
- 16. Wan H.-M., Chang B.-Y., Lin S.-C., Biotechnol. Bioeng. 2002, 79, 457. [DOI] [PubMed] [Google Scholar]
- 17. Kajiwara K., Aoki W., Koike N., Ueda M., Sci. Rep. 2021, 11, 11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Narita J., Okano K., Tateno T., Tanino T., Sewaki T., Sung M.-H., Fukuda H., Kondo A., Appl. Microbiol. Biotechnol. 2006, 70, 564. [DOI] [PubMed] [Google Scholar]
- 19. Jung H.-C., Park J.-H., Park S.-H., Lebeault J.-M., Pan J.-G., Enzyme Microb. Technol. 1998, 22, 348. [DOI] [PubMed] [Google Scholar]
- 20. Maurer J., Jose J., Meyer T. F., J. Bacteriol. 1997, 179, 794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Francisco J. A., Earhart C. F., Georgiou G., Proc. Natl. Acad. Sci. USA 1992, 89, 2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Richins R. D., Kaneva I., Mulchandani A., Chen W., Nat. Biotechnol. 1997, 15, 984; [DOI] [PubMed] [Google Scholar]
- 22b. Yang C., Zhao Q., Liu Z., Li Q., Qiao C., Mulchandani A., Chen W., Environ. Sci. Technol. 2008, 42, 6105; [DOI] [PubMed] [Google Scholar]
- 22c. Jo J.-H., Han C.-W., Kim S.-H., Kwon H.-J., Lee H.-H., J. Microbiol. 2014, 52, 856. [DOI] [PubMed] [Google Scholar]
- 23. Georgiou G., Stephens D. L., Stathopoulos C., Poetschke H. L., Mendenhall J., Earhart C. F., Protein Eng. Des. Sel. 1996, 9, 239. [DOI] [PubMed] [Google Scholar]
- 24. Ashiuchi M., Nawa C., Kamei T., Song J.-J., Hong S.-P., Sung M.-H., Soda K., Yagi T., Misono H., Eur. J. Biochem. 2001, 268, 5321. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Ryu S., Karim M. N., Appl. Microbiol. Biotechnol. 2011, 91, 529; [DOI] [PubMed] [Google Scholar]
- 25b. Chen Y.-P., Hwang I.-E., Lin C.-J., Wang H.-J., Tseng C.-P., J. Appl. Microbiol. 2012, 112, 455; [DOI] [PubMed] [Google Scholar]
- 25c. Fujii T., Tochio T., Hirano K., Tamura K., Tonozuka T., Biosci. Biotechnol. Biochem. 2018, 82, 1599. [DOI] [PubMed] [Google Scholar]
- 26. Jung H. C., Lebeault J. M., Pan J. G., Nat. Biotechnol. 1998, 16, 576. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. van Bloois E., Winter R. T., Janssen D. B., Fraaije M. W., Appl. Microbiol. Biotechnol. 2009, 83, 679; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Liang B., Li L., Tang X., Lang Q., Wang H., Li F., Shi J., Shen W., Palchetti I., Mascini M., Liu A., Biosens. Bioelectron. 2013, 45, 19. [DOI] [PubMed] [Google Scholar]
- 28. Yim S.-K., Kim D. H., Jung H. C., Pan J. G., Kang H. S., Ahn T., Yun C. H., J. Microbiol. Biotechnol. 2010, 20, 712. [DOI] [PubMed] [Google Scholar]
- 29. Jose J., Meyer T. F., Microbiol. Mol. Biol. Rev. 2007, 71, 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.
- 30a. Li C., Zhu Y., Benz I., Schmidt M. A., Chen W., Mulchandani A., Qiao C., Biotechnol. Bioeng. 2008, 99, 485; [DOI] [PubMed] [Google Scholar]
- 30b. Kranen E., Detzel C., Weber T., Jose J., Microb. Cell Fact. 2014, 13, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Strohle F. W., Kranen E., Schrader J., Maas R., Holtmann D., Biotechnol. Bioeng. 2016, 113, 1225. [DOI] [PubMed] [Google Scholar]
- 32. van Gerven N., Sleutel M., Deboeck F., de Greve H., Hernalsteens J.-P., Microbiol. 2009, 155, 468. [DOI] [PubMed] [Google Scholar]
- 33. Wagner S., Bader M. L., Drew D., de Gier J.-W., Trends Biotechnol. 2006, 24, 364. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Wagner S., Baars L., Ytterberg A. J., Klussmeier A., Wagner C. S., Nord O., Nygren P.-A., van Wijk K. J., de Gier J.-W., Mol. Cell. Proteomics 2007, 6, 1527; [DOI] [PubMed] [Google Scholar]
- 34b. Narayanan N., Chou C. P., Biotechnol. Prog. 2008, 24, 293. [DOI] [PubMed] [Google Scholar]
- 35. van Ulsen P., Zinner K. M., Jong W. S. P., Luirink J., FEMS Microbiol. Lett. 2018, 365. [DOI] [PubMed] [Google Scholar]
- 36.
- 36a. Wu P.-H., Giridhar R., Wu W.-T., Biotechnol. Bioeng. 2006, 95, 1138; [DOI] [PubMed] [Google Scholar]
- 36b. Yu F., Liu X., Tao Y., Zhu K., FEMS Microbiol. Lett. 2013, 345, 141; [DOI] [PubMed] [Google Scholar]
- 36c. Wee M. Y. J., A. Murad A. M., Abu Bakar F. D., Low K. O., Illias R. M., Process Biochem. 2019, 78, 25. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information