

Abstract
Aims
Addition of isatuximab (Isa) to pomalidomide/dexamethasone (Pd) significantly improved progression‐free survival (PFS) in patients with relapsed/refractory multiple myeloma (RRMM). We aimed to characterize the relationship between serum M‐protein kinetics and PFS in the phase 3 ICARIA‐MM trial (NCT02990338), and to evaluate an alternative dosing regimen of Isa by simulation.
Methods
Data from the ICARIA‐MM trial comparing Isa 10 mg/kg weekly for 4 weeks then every 2 weeks (QW‐Q2W) in combination with Pd versus Pd in 256 evaluable RRMM patients were used. A joint model of serum M‐protein dynamics and PFS was developed. Trial simulations were then performed to evaluate whether efficacy is maintained after switching to a monthly dosing regimen.
Results
The model identified instantaneous changes (slope) in serum M‐protein as the best on‐treatment predictor for PFS and baseline patient characteristics impacting serum M‐protein kinetics (albumin and β2‐microglobulin on baseline levels, non‐IgG type on growth rate) and PFS (presence of plasmacytomas). Trial simulations demonstrated that switching to a monthly Isa regimen at 6 months would shorten median PFS by 2.3 weeks and induce 42.3% patients to progress earlier.
Conclusions
Trial simulations supported selection of the approved Isa 10 mg/kg QW‐Q2W regimen and showed that switching to a monthly regimen after 6 months may reduce clinical benefit in the overall population. However, patients with good prognostic characteristics and with a stable, very good partial response may switch to a monthly regimen after 6 months without compromising the risk of disease progression. This hypothesis will be tested in a prospective clinical trial.
Keywords: modelling and simulation, monoclonal antibodies, pharmacodynamics
What is already known about this subject
Addition of isatuximab (Isa) 10 mg/kg weekly for 4 weeks then every 2 weeks (QW‐Q2W) to pomalidomide/dexamethasone (Pd) significantly improved progression‐free survival (PFS) in patients with relapsed/refractory multiple myeloma (MM) in the phase 3 ICARIA‐MM trial.
Serum M‐protein is known as a surrogate marker of tumour burden in MM and can predict long‐term clinical benefit such as PFS.
What this study adds
Association of M‐protein kinetics, patient characteristics and PFS was well characterized by joint modelling.
The simulation demonstrated that switching to an Isa monthly regimen after 6 months may reduce clinical benefit, supporting FDA‐approved Isa QW‐Q2W dosing. However, patients with good prognosis may switch to monthly dosing after 6 months without compromising risk of disease progression.
1. INTRODUCTION
Despite significant therapeutic advances and prolongation in overall survival (OS), multiple myeloma (MM) remains incurable, with many patients relapsing and requiring additional treatment. 1 Isatuximab (Isa) is an immunoglobin G1 (IgG1) monoclonal antibody that targets the CD38 transmembrane glycoprotein. Isa kills MM cells via multiple mechanisms, including antibody‐dependent cell‐mediated cytotoxicity, complement‐dependent cytotoxicity, direct induction of apoptosis without cross‐linking and inhibition of CD38 enzymatic activity. In a phase 1b study of patients with relapsed/refractory MM (RRMM), a 10 mg/kg weekly for 4 weeks then every 2 weeks (QW‐Q2W) dose of Isa in combination with pomalidomide (P) and low‐dose dexamethasone (d) (Isa‐Pd) achieved an overall response rate (ORR) of 64.5% and median progression‐free survival (PFS) of 17.6 months. These results, combined with exposure‐response and disease modelling of tumour burden (serum M‐protein), provided the justification for the 10 mg/kg QW‐Q2W of Isa‐Pd. 2 , 3 This combination was then assessed in the phase 3 ICARIA‐MM study (NCT02990338), which showed that addition of Isa to Pd significantly improved PFS in RRMM patients. 4 Based on this pivotal study, Isa in combination with Pd is approved in multiple countries for RRMM patients with ≥2 prior lines. Furthermore, to date, Isa in combination with carfilzomib/dexamethasone is approved in the United States for relapsed MM patients with 1‐3 prior lines, and in the European Union for MM patients with ≥1 prior therapy, based on the phase 3 IKEMA study. 5 , 6 , 7
Efforts have been made to develop tumour growth inhibition (TGI) models and predict clinical responses, OS or PFS rates in cancer patients from various clinical settings. 8 , 9 TGI models are used to find early changes in tumour size that would predict OS or PFS. Joint models have emerged as a promising framework for concurrently investigating the relationship between continuous disease progression through longitudinal outcomes such as biomarkers, tumour size and incidence of clinical events such as progression and death. These models provide precise and unbiased estimation of the parameters in an informative censoring context. 10 Mechanistic joint models adequately predicted OS in clinical trials for atezolizumab in urothelial carcinoma, cabazitaxel in metastatic prostate cancer and aflibercept in metastatic colorectal cancer. 11 , 12 , 13
In most patients, MM is characterized by the secretion of a monoclonal Ig protein (M‐protein), which is produced by the abnormal plasma cells. Similar to tumour burden for solid tumours, serum M‐protein levels are an important part of the response criteria for MM patients 14 and thus their dynamic change can predict long‐term clinical benefit (PFS, OS). Several examples in MM showed that TGI modelling based on longitudinal M‐protein can be used to predict OS or PFS. 15 , 16 , 17 , 18
For Isa, the joint modelling framework was used to integrate early drug development results with later‐stage clinical data from phase 1‐2 monotherapy and combination studies. 2 , 3 , 19 Disease progression was initially captured together with serum M‐protein dynamics using a joint model and accounting for dropout. Longitudinal serum M‐protein modelling provided more insights in patient response over time and supported phase 2 and phase 3 dosing‐regimen selection in MM patients. 20 This framework and modelling approach can be extended to account for PFS since PFS is a composite endpoint, which depends not only on serum M‐protein but also radiological progression or death. This joint modelling therefore improves the predictive and simulation value of the models in exploring the benefits of a different dosing strategy, eg, switching to a monthly regimen after 6 months to reduce dosing intensification in a context of long treatment duration.
The objectives of this work were therefore to (i) quantitatively evaluate the association between serum M‐protein kinetics, baseline covariates and PFS in RRMM patients in both Isa‐Pd and Pd arms of the ICARIA‐MM study, and (ii) simulate longitudinal serum M‐protein and PFS when switching to a hypothetical monthly Isa dosing regimen after 6 months.
2. METHODS
2.1. Study design and data
Data were obtained from the ICARIA‐MM study. Isa was administered intravenously at 10 mg/kg QW for 4 weeks followed by Q2W for 28‐day cycles in combination with standard pomalidomide (4 mg orally on days 1‐21/each cycle) and dexamethasone (40 or 20 mg for patients aged ≥75 years orally or intravenously on days 1, 8, 15 and 22 in each cycle). The study was conducted following the principles of the Declaration of Helsinki and ICH Good Clinical Pratice (GCP) Guidelines. The protocol was approved by institutional review boards and independent ethics committees at the participating institutions. All patients provided written informed consent. The primary study endpoint was PFS, defined as the time from randomization to first documentation of progressive disease or death from any cause, whichever came first. Response and disease progression were determined by an independent response committee using the International Myeloma Working Group (IMWG) criteria, based on central, M‐protein laboratory assessments and radiology review. 14 Patients with ≥2 serum M‐protein values, including one baseline value and for whom response could be evaluated by serum M‐protein, were included in the analysis. Serum M‐protein was assessed by serum protein electrophoresis and immunofixation electrophoresis. 20 Per protocol, serum M‐protein was measured at baseline, end of each cycle, and study end. No lower limit of quantification (LOQ) could be provided for serum M‐protein measurement because M‐protein and its structure are patient‐specific. For this analysis, LOQ was considered as half of the actual lowest value for serum M‐protein in the ICARIA‐MM study (ie, 0.5 g/L).
2.2. Model development
Serum M‐protein longitudinal data from both study arms and PFS data were first modelled separately. To account for dose effect, treatment exposure over time was introduced in the longitudinal model using concentrations predicted by the individual PK parameters for Isa and a kinetic‐pharmacodynamic (K‐PD) model for pomalidomide and dexamethasone. Several joint models were then used to find the best link between serum M‐protein kinetics and PFS.
2.2.1. Population PK model for Isa
A two‐compartment PK model with parallel linear and nonlinear (Michaelis‐Menten) elimination from the central compartment and time‐varying linear clearance function was used to describe the Isa plasma concentrations versus time data collected from 476 MM patients who received 1‐20 mg/kg Isa alone or in combination with Pd in four phase 1‐3 clinical trials, including ICARIA‐MM. 21 The equations of this structural PK model are presented in the Supporting Information. Individual PK parameters for ICARIA‐MM patients were obtained as post hoc estimates and typical PK parameters were attributed for patients without PK data.
2.2.2. K‐PD model for pomalidomide and dexamethasone
Since concentrations of combined Pd were not measured in this study, the kinetics of these drugs were simplified using a K‐PD modelling approach. 22 Their PK was therefore described by a simple, virtual one compartment with bolus input and fixed elimination‐rate constant derived from their central distribution volume and clearance value estimates in the literature. 23 , 24
2.2.3. TGI model for M‐protein data and covariate selection
Structural model
A TGI model accounting for tumour growth dynamics, antitumour drug effect and resistance to drug effect was developed by Claret et al. 25 This model was also successfully applied to describe serum M‐protein data as a surrogate of tumour growth in MM patients. 15 , 16 , 18 , 26 , 27 In this analysis, a mechanism‐based model drawn from the Claret's TGI model was proposed to describe the underlying disease progression and exposure‐driven drug effect of Isa and Pd on the serum M‐protein time‐course. The structural model for this TGI model, shown in Figure 1, is described by the following differential equation:
where is serum M‐protein at time , M 0 is the baseline serum M‐protein, KL is the tumour growth rate, and are the shrinkage rate due to Isa and combined Pd exposure respectively, and are the rate constant of resistance appearance to Isa and combined Pd, respectively, and , and are the molar concentrations of Isa, pomalidomide and dexamethasone at time , respectively. The contribution of and in increasing M‐protein shrinkage rate was assumed to be equal based on the response rates of a randomized phase 2 study comparing pomalidomide alone or combined with dexamethasone. 28
FIGURE 1.
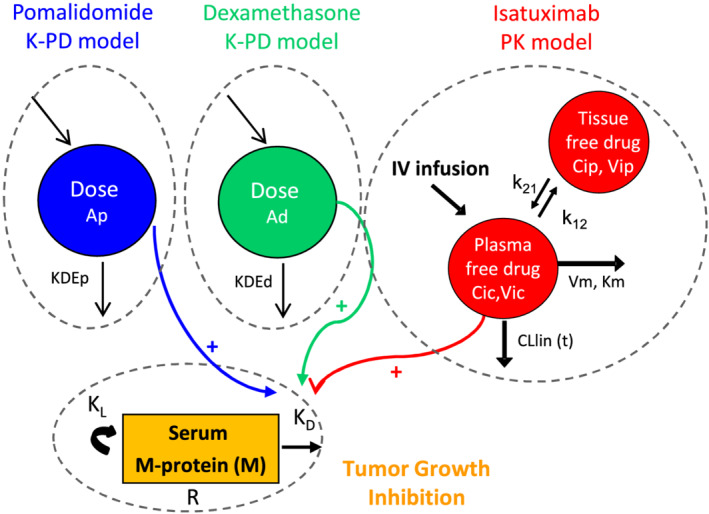
Schematic representation of the integrated drug disease model. It integrates kinetic‐pharmacodynamic models (K‐PD) for pomalidomide and dexamethasone, the pharmacokinetic (PK) model for isatuximab, and the tumour growth inhibition (TGI) model for serum M‐protein
Statistical model
An exponential interindividual model implying a log‐normal distribution was included on all parameters. The variance‐covariance matrix was modelled using a diagonal matrix. The residual variability was modelled using a combined additive and proportional model.
Covariate analysis
Covariate analysis was performed after obtaining the base model. Twenty‐six baseline covariates were tested: demographics, baseline laboratory measurements and disease‐related patient characteristics (Supporting Information Table S1). In the case of missing data, the median value was input for continuous covariates; missing was considered as an additional category for categorical covariates. The parameter‐covariate relationship was first explored graphically using individual parameter estimates. The Conditional Sampling for Stepwise Approach based on Correlation tests (COSSAC) covariate selection algorithm was then used for automatic building of the covariate model. 29 , 30 The best covariate model was selected using the corrected version of Bayesian Information Criteria (BICc). 31 Additionally, only significant covariates with Wald test P value <.05 were kept in the final model.
2.2.4. PFS model and covariate selection
PFS was modelled using a parametric proportional‐hazard model with log‐logistic distribution for baseline hazard: where Te is the scale parameter (characteristic time) and s the shape parameter. The exponential and Weibull distribution were also tested. The baseline covariates were tested as potential prognostic factors using the classical stepwise covariate modelling method. The same criteria for covariate selection in the longitudinal M‐protein model development were used.
2.3. Joint modelling of serum M‐protein and PFS
Longitudinal and PFS models were built separately; thereafter several joint models were used to find the best link between serum M‐protein kinetics and PFS, relying on the following link functions:
no link
current serum M‐protein: M(t)
current M‐protein slope (instantaneous rate of change in serum M‐protein): dM/dt
area under the M‐protein.
Significant covariates found in the longitudinal and PFS submodels were evaluated, and only covariates significant with the Wald test (P value <.05) were kept in the joint model.
2.4. Parameter estimation
Parameter estimation of all models was performed using the Stochastic Approximation Expectation Maximization (SAEM) algorithm implemented in the software Monolix v.2019R1. Data below LOQ for serum M‐protein were accounted for using the extended SAEM algorithm implemented in Monolix.
2.5. Model selection and evaluation
Model selection was based on BIC for non‐nested models and the model giving the lowest BICc was retained. The likelihood ratio test was used to discriminate between nested models through the difference in log likehood (−2LL), computed using important sampling. A P value of .05 was considered statistically significant. Model evaluation was performed by investigating both residual‐ and simulation‐based diagnostics, including the individual weighted residuals (IWRES), visual predictive checks (VPC) for the longitudinal part, Cox‐Snell and deviance residuals, 32 de‐trended prediction discrepancies, 33 and Kaplan‐Meier VPC for PFS, respectively. Additional goodness‐of‐fit plots were assessed by visual inspection of individual fits or by comparing observations versus individual predictions. Longitudinal VPC accounted for risk of progression using the methods described by Friberg et al. 34 Briefly, this involved reproducing the event mechanisms in simulation and omitting simulations occurring after a simulated progression time. PFS VPC considered the design of each patient, ie, dose regimens and follow‐up duration. Indeed, simulated time to progression (TTP) was censored by the maximum time between duration of follow‐up, end of treatment and observed TTP.
2.6. Simulation of monthly dosing regimen
To evaluate longitudinal serum M‐protein and PFS after switching to a hypothetical monthly (Q4W) Isa dosing regimen after 6 months, 1000 trials (using the same design and patient characteristics as in the data) were simulated with both the Isa‐Pd and Pd arms for 80 weeks. All the drug‐related parameters, in particular the appearance rate of resistance to Isa, were assumed to be conserved when switching the dosing regimen to Q4W. Patients received Isa 10 mg/kg QW for 4 weeks then Q2W for 20 weeks, then monthly in the Isa‐Pd arm. The combination dosing regimen with standard Pd was the same as in ICARIA‐MM. Patients at risk at 6 months were evaluated for impact on TTP (increase >25% with absolute change of ≥5 g/L for serum M‐protein compared to nadir) and PFS. The original ICARIA‐MM Isa‐Pd arm was also simulated with patients receiving Isa 10 mg/kg QW‐Q2W to present results as median (5th‐95th percentiles) difference from the original arm. Hazard ratios (HR) for the two regimens vs the control arm were also compared.
3. RESULTS
3.1. Data used for model building
Among the 307 randomized patients in the ICARIA‐MM trial, 256 serum M‐protein evaluable patients (128 patients/arm) were considered in this analysis. In this serum M‐protein population (N = 256), median PFS was significantly longer with Isa‐Pd vs Pd (11.4 months [95% confidence interval {CI} 8.5‐13.8] vs 6.96 months [95% CI 4.4‐8.5], HR 0.618, 95% CI 0.44‐0.87, P = .0048). Similar observations on PFS and HR were obtained for the overall population (N = 307), although 16.6% of the ICARIA‐MM patients could not be included in this analysis because their MM disease was characterized by urine M‐protein instead of serum M‐protein or on‐treatment serum M‐protein values were not available.
Baseline patient characteristics were balanced across arms (Table 1). Median age was 67 years (50% female). Baseline, median serum β2‐microglobulin (B2MG) and serum albumin (ALB) were 3.5 mg/L and 37 g/L, respectively. High‐risk cytogenetics were present in 53 (21%) patients and median estimated glomerular filtration (eGFR) rate was 70 mL/min. Most patients were IgG MM type (190 [74%]), had no plasmacytomas (232 [91%]) and 64 (25%) and 164 (64%) patients had Revised International Staging System (R‐ISS) I or II stage at diagnosis, respectively. Baseline median serum M‐protein was 23 g/L with a wide value range (5‐95 g/L), together with various profiles during treatment. A total of 2637 serum M‐protein measurements in the 256 evaluable patients were considered, with a median of 14 (range 2‐22) assessments/patient. The data below LOQ accounted for 14% (22% in Isa‐Pd, 6% in Pd).
TABLE 1.
Baseline demographics and patient characteristics in serum M‐protein population
| Isatuximab plus pomalidomide and dexamethasone (Isa‐Pd) (n = 128) | Pomalidomide and dexamethasone (Pd) (n = 128) | |
|---|---|---|
| Age, years (range) | 68 (36‐83) | 66 (41‐86) |
| Sex, n (%) | ||
| Female | 56 (44) | 72 (56) |
| Male | 72 (56) | 56 (44) |
| Weight (kg), median (range) | 74 (34‐110) | 73 (39‐140) |
| eGFR (mL/min), median (range) | 69 (30‐177) | 71 (31‐135) |
| R‐ISS, n (%) | ||
| I | 36 (28) | 28 (22) |
| II a | 81 (63) | 83 (65) |
| III | 11 (9) | 17 (13) |
| Serum β2‐microglobulin (mg/L), median (range) | 3.5 (1.1‐27) | 3.5 (0.7‐55) |
| Serum albumin (g/L), median (range) | 36.9 (16‐48.7) | 37 (16.5‐46.4) |
| Serum M‐protein at baseline (g/L), median (range) | 22 (5‐95) | 23 (5‐83) |
| Type of myeloma, n (%) | ||
| IgG | 97 (76) | 93 (73) |
| Non‐IgG | 31 (24) | 35 (27) |
| Plasmacytomas, n (%) | ||
| Yes | 13 (10) | 11 (9) |
| No | 115 (90) | 117 (91) |
| Cytogenetic risk at study entry, n (%) | ||
| Standard | 86 (67) | 63 (49) |
| High | 20 (16) | 33 (26) |
| Missing | 22 (17) | 32 (25) |
Abbreviations: e‐GFR, estimated glomerular filtration rate; R‐ISS, Revised Multiple Myeloma International Staging System (derived based on the combination of serum β2‐microglobulin, albumin, cytogenetic risk and lactate dehydrogenase).
As pre‐specified in the study statistical analysis plan, patients with unknown cytogenetics at baseline were classified as R‐ISS stage II.
3.2. Modelling serum M‐protein kinetics and PFS
The proposed TGI model provided an adequate fit for the longitudinal serum M‐protein data of both study arms. It performed better than the Wang model. 35 In addition, the fit was improved when adding the PK of Isa compared to a K‐PD model only. Twenty‐six potential covariates were evaluated by testing their relationship with all the longitudinal model parameters. The final longitudinal model includes three covariates: the effects of baseline ALB and B2MG on baseline serum M‐protein levels, and the non‐IgG MM type on KL, the serum M‐protein growth rate.
Regarding PFS, a log‐logistic model best characterized the underlying baseline hazard distribution. Baseline covariates such as presence of plasmacytoma, ALB and serum M‐protein were significant (P < .005). Further information on the modelling results of longitudinal data and PFS is included in Supporting Information Tables S2 and S3.
3.3. Joint modelling of serum M‐protein and PFS
The joint model using the serum M‐protein slope outperformed all models relying on serum M‐protein in terms of BIC, with a 196‐point decrease compared with the no‐link model, that is, the parametric log‐logistic model with no association between serum M‐protein and PFS. The alternative models, based on current serum M‐protein value or cumulative serum M‐protein (area‐under‐serum M‐protein), led to a BIC improvement <103. Comparison of joint models with different link functions is provided in Supporting Information Table S4. In the best, final joint model, the longitudinal model still includes the same three covariates, but only the presence of plasmacytomas remains on the PFS part. Parameter estimates obtained with the serum M‐protein slope joint model are summarized in Table 2. They were reasonably well estimated with low relative standard error for both fixed effects and variance components. Although the shrinkage estimates for KDi and Ri are large (69% and 61.5%), they did not impact the covariate analysis because the sampling of conditional distribution was used instead of conditional mode estimates, allowing avoid the associated bias of large shrinkage to be avoided. 29
TABLE 2.
Parameter estimates values (relative standard error %) of the best joint final model
| Parameter estimates | Relative standard error (%) | Shrinkage (%) | P value Wald test | |
|---|---|---|---|---|
| Fixed effects | ||||
| Longitudinal submodel | ||||
| M0 (g/L) | 17 | 3.33 | 2.9 | |
| β1 ~ ALBN | −1.41 | 15.8 | 2.34E‐10 | |
| β2 ~ B2MG | 0.331 | 17 | 3.88E‐09 | |
| KL (d−1) | 0.00627 | 8.4 | 23.5 | |
| β3 ~ non_IgG | 0.55 | 27.5 | 0.000277 | |
| KDi (L mol−1 d−1) | 0.0138 | 8.73 | 69 | |
| Ri (day−1) | 0.00579 | 14.6 | 61.5 | |
| KDpd (L mol−1 d−1) | 0.188 | 7.36 | 24.1 | |
| Rpd (d−1) | 0.00952 | 10.6 | 34 | |
| Survival submodel | ||||
| Te (d−1) | 459 | 11.9 | ||
| S | 2.33 | 9.75 | ||
| β4 ~ PCYTOMA = Y | 0.858 | 36.3 | 0.00591 | |
| β5 ~ SlopeM | 11.9 | 7.66 | 4.46E‐39 | |
| Interindividual variability standard deviation | ||||
| ω_M0 | 50.5 | 4.73 | ||
| ω _KL | 88.7 | 6.76 | ||
| ω _KDi | 61.9 | 9.7 | ||
| ω _Ri | 105 | 9.76 | ||
| ω _KDpd | 92 | 6.81 | ||
| ω _Rpd | 110 | 9.33 | ||
| Residual variability | ||||
| σ additive (g/L) | 0.411 | 7.23 | ||
| σ proportional (%) | 15 | 3.65 | ||
Abbreviations: ALBN, baseline serum albumin normalized to the upper limit value; B2MG, baseline β2‐microglobulin; M0, serum M‐protein at baseline, PCYTOMA, Y, presence of plasmacytomas.
The estimated link between serum M‐protein slope and PFS is high at 11.9, consistent with IMWG criteria, in which the decrease in serum M‐protein in response to treatment is the main component directly impacting PFS. Thus, in case of initial response, the serum M‐protein decrease is associated with a current slope <0 and hence a reduced risk of progression. The relationship among serum M‐protein kinetics, slope and PFS is illustrated in Figure 2 for six representative patients who either had a PFS event or not. The PFS probability decreased during tumour growth, ie, when the serum M‐protein slope increased. Furthermore, the baseline covariates were found to modify parameters of serum M‐protein kinetics and PFS.
FIGURE 2.
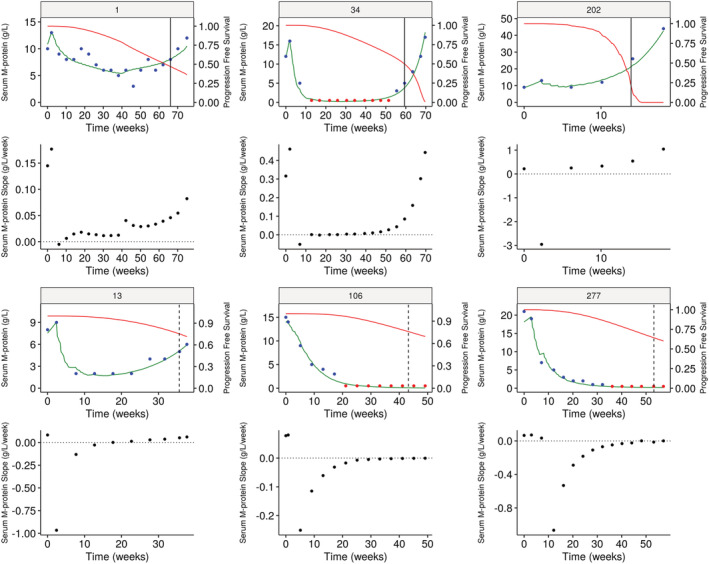
Individual fits of serum M‐protein time course and PFS probability in six illustrative patients, three with the observed event and three with the censored event. Patients in the Isa‐Pd arm are in the middle and on the left; patients in the Pd arm are on the right. Blue dots denote the serum M‐protein observations and red dots the BLQ observations. The green curves denote the longitudinal predictions using the joint model. The vertical lines show the status of the patients (solid, progression event occurred; dashed, censored). The red solid curves denote the PFS probability predicted by the joint model. The black curves represent the predicted value of the current slope of serum M‐protein kinetics. BLQ, below the limit of quantification; Isa, isatuximab; MP, M‐protein; Pd, pomalidomide and dexamethasone; PFS, progression‐free survival
3.4. Model evaluation
The various serum M‐protein kinetic patterns could be well captured by the model and predicted PFS probability is consistent with occurrent time of progression or censored event. Figure 3 shows VPC plots generated for both longitudinal and PFS models by simulation of 1000 clinical trials under the final joint model, using the same design and patient characteristics as in the data. The model described reasonably well the observed serum M‐protein and PFS data with observed median generally included in the 90% prediction interval. However, an unusual early event was observed, since the model did not capture a small group of patients who switched therapy without achieving PFS criteria. The final joint model also predicted well the HR observed between arms (Figure 4), with observed HR close to the predicted median HR. Additional goodness‐of‐fit plots are presented in Supporting Information Figure S1.
FIGURE 3.
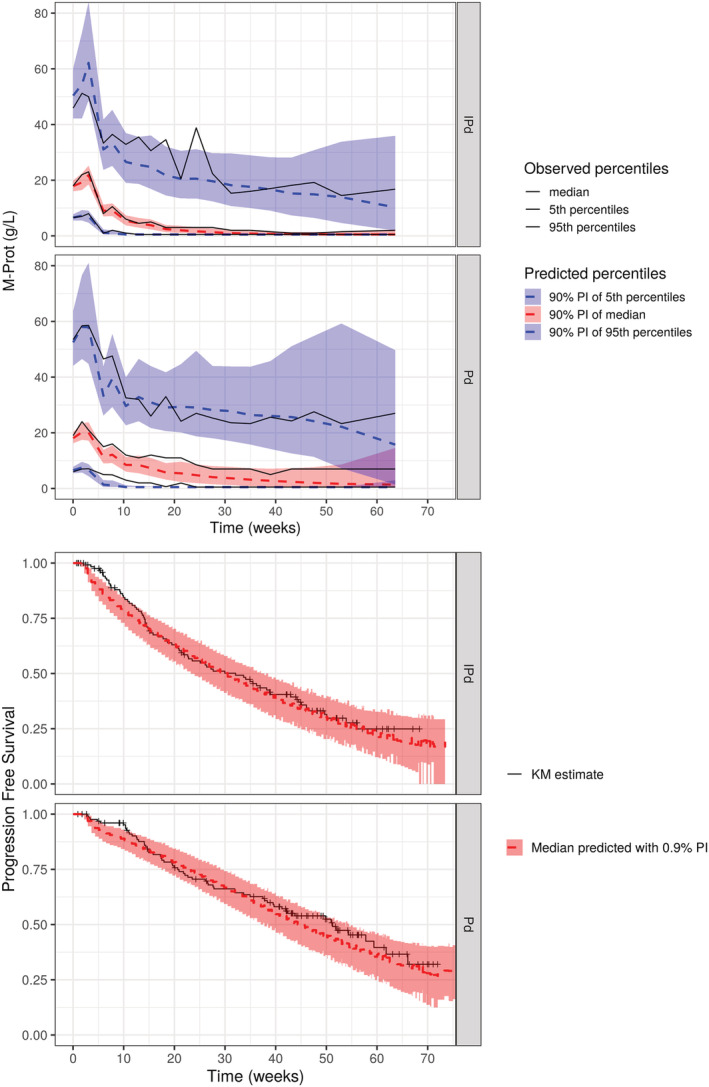
Visual predictive checks for the longitudinal part and PFS of the final joint model. Shaded area and the dotted lines represent the 90% prediction interval and the predicted median of the 5th, 50th and 95th percentiles of simulated M‐protein or PFS data (n = 1000). The solid lines represent the 5th, 50th and 95th percentiles of observed longitudinal data or the observed Kaplan‐Meier estimate. CI, confidence interval; I, isatuximab; KM, Kaplan‐Meier; M‐P, M‐protein; Pd, pomalidomide and dexamethasone; PFS, progression‐free survival; PI, prediction interval
FIGURE 4.
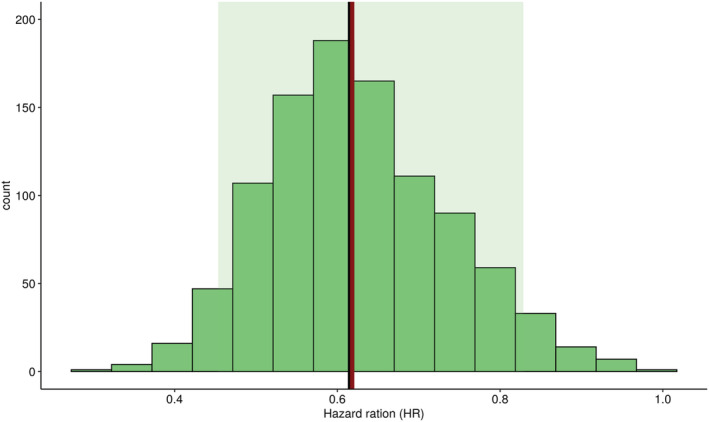
Posterior predictive check of PFS HR using the joint model. Green zone, 95% prediction interval; black bar, predicted median HR; red bar, observed HR; PFS, progression‐free survival; HR, hazard ratio
3.5. Assessment of covariate effects
Simulations were performed to quantify the impact of each covariate using the population parameters and visualized in a typical patient (Figure 5). The effect of covariates was assessed individually by setting others to median value for continuous covariates and for the most frequent class for categorical covariates (ie, IgG type). The effect of continuous covariates, baseline ALB and B2MG, were examined for variations within the 5th‐95th database percentiles.
FIGURE 5.
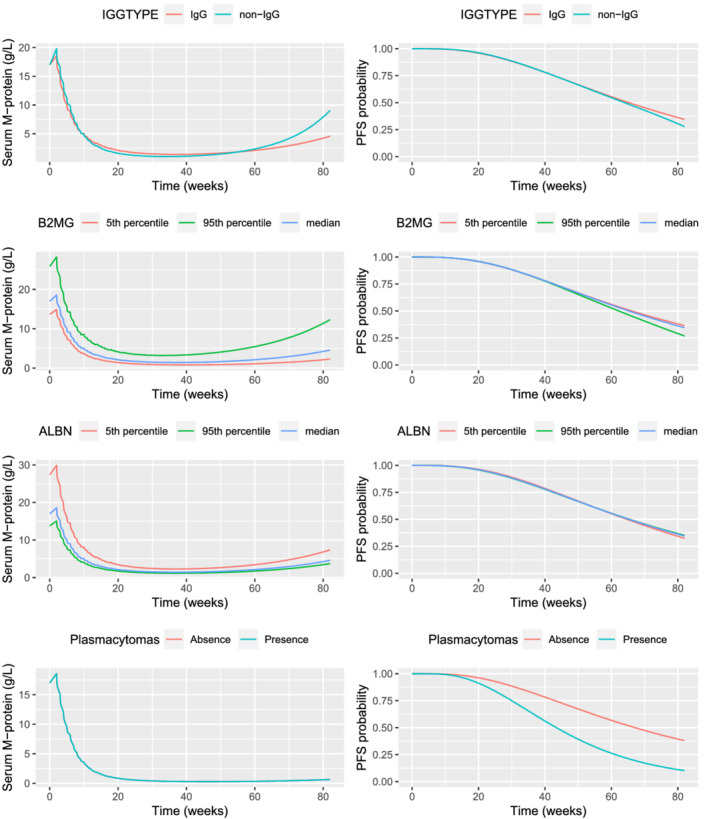
Impact of covariate effects on serum M‐protein kinetics and PFS probability of not progressed; total serum M‐protein population (N = 256). ALBN, albumin; B2MG, β2‐microglobulin; Ig, immunoglobulin; PFS, progression‐free survival
Non‐IgG MM patients had similar behaviour on serum M‐protein kinetics for the first 60 weeks even with higher Isa exposure and tended to have more rapid tumour regrowth (ie, re‐increase in serum M‐protein) afterwards compared with IgG MM patients (Supporting Information Figure S2). Similar PFS probability is predicted for non‐IgG compared to IgG MM patients. The VPC plots stratified by Ig MM type and treatment arm show agreement between model prediction and observed data (Supporting Information Figure S3).
Patients with low baseline ALB (<27 g/L) or high B2MG (>12.8 mg/L) levels are more likely to have 57% or 52% higher baseline serum M‐protein, respectively, compared with patients with normal ALB or B2MG levels. The impact on the shape of M‐protein profiles was a slightly faster tumour regrowth and slightly lower PFS probability at the end of treatment compared with other patients. Patients with plasmacytomas shared a similar PFS profile for 20 weeks, but tended to have lower PFS probability, up to 25% after 80 weeks.
3.6. Simulation of monthly dosing regimen
In the 1000 simulated trials, the median (min‐max) number of patients at risk after 6 months (ie, patients who had not progressed by 6 months) was 97 (86‐107) in the Isa‐Pd arm. In patients at risk at 6 months who switched to the Isa monthly dosing regimen after 6 months of QW‐Q2W, median (5th‐95th percentiles) progression was predicted to occur 2.29 (0.57‐4.73) weeks earlier and HR predicted to be greater (0.7 vs 0.66) compared with the original Isa‐Pd arm. Additionally, when considering the TTP criteria (increase in serum M‐protein >25% with absolute increase >5 g/L), 44/104 (42.3%) patients who had not progressed at 6 months in the original Isa‐Pd arm had their serum M‐protein regrow faster and lower PFS probability with significant difference in median PFS (Wilcoxon test, P value = .026) (Figure 6). Evaluation of baseline patient characteristics showed that these impacted patients have more baseline disease burden, ie, higher serum M‐protein, higher bone marrow plasma cells and worse prognostic characteristics, including lower eGFR, lower ALB and higher B2MG, with more frequent R‐ISS II/III stage disease (80% vs 53.2%), compared with patients who did not progress earlier (Supporting Information Figure S4). Conversely, patients with no risk of earlier progression tend to have lower tumour burden and better prognostic characteristics at baseline. Additionally, at 6 months they have significantly lower M‐protein (median, 0.31 vs 3.04 g/L) and more patients have reached their maximum response, with a serum M‐protein slope close to 0 (ie, M‐protein level reached a plateau, median −0.01 vs −0.06 g L−1 d−1) and 85% of them have predicted response status of at least very good partial response.
FIGURE 6.
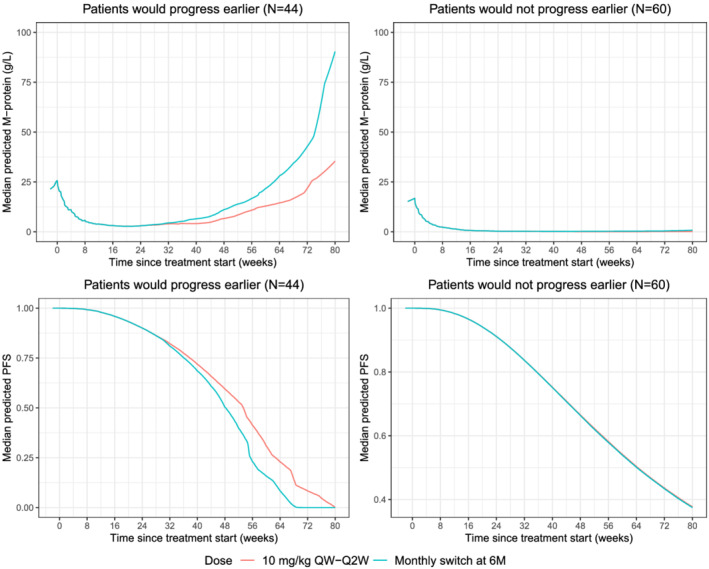
Median predicted M‐protein and PFS for patients who would progress earlier (N = 44) or would not progress earlier (N = 60). M, months; QW, weekly; Q2W every 2 weeks
4. DISCUSSION AND CONCLUSION
A nonlinear joint model was developed in a MM setting, as joint models can provide efficient estimates and reduced bias of treatment effects on both time‐to‐event and longitudinal markers. We developed the TGI model using serum M‐protein longitudinal data first and then the time‐to‐event model for PFS. Joint modelling was then performed to explore the best link between longitudinal serum M‐protein and PFS. The model building was based on 256 serum M‐protein evaluable patients (128 patients/arm) from the ICARIA‐MM trial.
The TGI model from Claret et al was developed to describe longitudinal serum M‐protein data in both arms of ICARIA‐MM and compared with the Wang model, which was used for elotuzumab plus lenalidomide/dexamethasone (ELOQUENT‐2) data. 25 , 35 We selected the Claret model, which provided a better fit and included the combined pomalidomide/dexamethasone dose and Isa PK exposure as predictors, allowing simulation of the serum M‐protein response under other dosing regimens. This model accounts for three important clinical features of tumour progression in anticancer drug treatment, including the dynamics of tumour growth/production of serum M‐protein, antitumour drug effect and resistance to drug effect. A more mechanism‐based model incorporating distinct subpopulations of MM malignant cells would be interesting for further evaluation but requires more information on cell data.
Furthermore, we studied the impact of numerous baseline covariates on serum M‐protein kinetics and risk of progression. The significant baseline covariates in the joint model were Ig MM type, ALB, B2MG and presence of plasmacytomas. Patients with low baseline ALB or high B2MG levels were more likely to have ~50% higher baseline serum M‐protein and slightly faster tumour regrowth rate at the end of treatment, resulting in similar PFS profiles compared to other patients. Of note, these laboratory tests are part of the ISS/R‐ISS staging systems, which are relevant for prognosis assessment, because patients with more advanced stage (ISS stage III) are less likely to respond to treatment. The presence of plasmacytomas likely induced lower PFS probability, consistent with results of exposure‐response analyses. 3 Furthermore, the serum M‐protein instantaneous slope was associated with PFS consistent with IMWG criteria, in which the serum M‐protein decrease in response to treatment is the main component directly impacting PFS.
The Ig MM type was found to correlate with M‐protein growth rate in the joint model, with faster growth rate for non‐IgG patients. Simulation of typical patients indicated that non‐IgG MM patients have similar behaviour on serum M‐protein kinetics for the first 60 weeks, even with two‐fold higher steady‐state Isa exposure, but tend to have more rapid tumour regrowth afterwards and similar PFS compared to IgG MM patients. The Ig MM type was also identified as the main contributor explaining Isa PK interindividual variability, with faster clearance in IgG MM patients resulting from competition for the neonatal Fc receptor, which protects IgG from degradation. 36 , 37 , 38 However, the impact of Ig MM type (IgG vs non‐IgG) on Isa exposure does not translate into difference in treatment effect, and therefore does not appear to be clinically meaningful. This finding is consistent with the results of exposure‐response and subgroup analyses which showed that there was no significant difference in treatment benefit with Isa‐Pd versus Pd on PFS or ORR for IgG versus non‐IgG patients. 39 Similar results were observed with daratumumab. Response rates between IgG and non‐IgG MM patients were similar despite the difference in linear clearance levels between these populations (~110% higher in IgG, leading to 70% higher predicted trough serum concentrations on day 1/cycle 3 in non‐IgG patients). 40
Cytogenetics was not a significant covariate in the joint model, on either serum M‐protein dynamics or PFS, in agreement with ICARIA‐MM subgroup analyses, which showed that Isa‐Pd provided consistent benefit versus Pd in RRMM patients, regardless of cytogenetic risk. 41
The drug‐disease modelling platform established based on ICARIA‐MM data was further applied to predict the impact of using a hypothetical monthly dosing regimen after 6 months of Isa QW‐Q2W in RRMM patients. In patients still on treatment, simulations of a hypothetical switch to monthly dosing after 6 months predicted progression to occur 2.3 weeks earlier compared with the original Isa‐Pd arm, with 42.3% of patients having their serum M‐protein regrow faster. Patients with no risk of earlier progression tended to have lower tumour burden and better prognostic characteristics at baseline, with a stable, at least very good partial response at 6 months. This modelling framework would be a useful tool to perform individual dynamic predictions in a Bayesian approach, to improve patient follow‐up and early identification of most‐at‐risk patients, similarly to what was done for PSA kinetics and OS in prostate cancer. 42 Nevertheless, the landmark times allowing a good individual prediction will need to be further evaluated.
A major assumption in the M‐protein model is that resistance develops over time and the resistance magnitude will not change when simulating alternative dosing regimens. However, resistance could be induced by drug exposure, resulting in a different resistance magnitude when switching to monthly dosing. Further investigation with more follow‐up data on the relationship between drug exposure and resistance will be necessary to confirm the simulation outcome based on the current assumption.
The results of this joint modelling and simulation confirm the approved Isa QW‐Q2W dosing regimen, which was selected for ICARIA‐MM. Further evaluation of on‐treatment switch to an Isa monthly dosing regimen should occur through a prospective clinical trial, perhaps exploring other response observations (such as minimal residual disease) as a trigger point for switching to a less frequent dosing regimen.
CONTRIBUTORS
H,‐T.T., G.A., B.S., H.vdV., D.S. and C.V,‐F. wrote the manuscript, H,‐T.T. and C.V,‐F. designed the research, H,‐T.T., B.S., H.vdV., D.S. and C.V,‐F. performed the research, H,‐T.T., N.G,‐D., and C.V,‐F. analysed the data. H.‐T.T., N.G.‐D., M.C., G.A. and J.‐B.F. contributed analytical tools.
COMPETING INTERESTS
H.‐T.T., N.G.‐D., M.C., J.‐B.F., B.S., H.vdV., D.S. and C.V.‐F. are employees of Sanofi. G.A. is an employee of Lixoft, a Simulations Plus company.
CLINICAL TRIAL REGISTRATION
Supporting information
Supporting Information Table S1 List of baseline patient characteristics tested as covariates
Supporting Information Table S2 Parameter estimates of the longitudinal model without covariates and with significant covariates. P values of covariate effects were computed by the Wald test
Supporting Information Table S3 Parameter estimates of the PFS log‐logistic model without covariates and with significant covariates. P values of covariate effects were computed by Wald test
Supporting Information Table S4 Parameter estimates of joint model with different links
ACKNOWLEDGEMENTS
The ICARIA‐MM study was sponsored by Sanofi. The authors thank the participating patients and their families, and the study centres and investigators, for their contributions to the study. The authors also thank Pascale Boittet and Solenn Le‐Guennec for their contributions. Medical writing support was provided by Stephanie Brillhart and Sara Mariani of Elevate Medical Affairs, contracted by Sanofi Genzyme for publication support services. This study was funded by Sanofi.
Thai H‐T, Gaudel N, Cerou M, et al. Joint modelling and simulation of M‐protein dynamics and progression‐free survival for alternative isatuximab dosing with pomalidomide/dexamethasone. Br J Clin Pharmacol. 2022;88(5):2052-2064. doi: 10.1111/bcp.15123
FUNDING INFORMATION
The ICARIA‐MM study was sponsored by Sanofi. This study was funded by Sanofi.
The authors confirm that the principal investigator for this paper is Dr van de Velde and he has direct clinical responsibility for patients.
Funding information Sanofi
DATA AVAILABILITY STATEMENT
Qualified researchers can request access to patient‐level data and related study documents, including the clinical study report, study protocol with any amendments, blank case report forms, statistical analysis plan, and dataset specifications. Patient‐level data will be anonymized and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi's data‐sharing criteria, eligible studies and process for requesting access are at https://www.clinicalstudydatarequest.com.
REFERENCES
- 1. Kumar SK, Rajkumar V, Kyle RA, et al. Multiple myeloma. Nat Rev Dis Primers. 2017;3(1):17046. [DOI] [PubMed] [Google Scholar]
- 2. Thai H‐T, Liu L, Koiwai K, et al. Exposure‐response analysis and disease modeling for selection of optimal dosing regimen of isatuximab as single agent in patients with multiple myeloma. European Hematology Association Annual Meeting 2019: Abstract PF645.
- 3. Rachedi F, Koiwai K, Gaudel‐Dedieu N, et al. Exposure‐response analyses and disease modeling for selection and confirmation of optimal dosing regimen of isatuximab in combination treatment in patients with multiple myeloma. Blood. 2019;134(Supplement_1):1897‐1897. [Google Scholar]
- 4. Attal M, Richardson PG, Rajkumar SV, et al. Isatuximab plus pomalidomide and low‐dose dexamethasone versus pomalidomide and low‐dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA‐MM): a randomised, multicentre, open‐label, phase 3 study. Lancet. 2019;394(10214):2096‐2107. [DOI] [PubMed] [Google Scholar]
- 5. Moreau P, Dimopoulos M‐A, Mikhael J, et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): a multicentre, open‐label, randomised phase 3 trial. Lancet. 2021;397(10292):2361‐2371. [DOI] [PubMed] [Google Scholar]
- 6. SARCLISA® (isatuximab‐irfc) injection, for intravenous use. Prescribing Information. March 2021. https://products.sanofi.us/Sarclisa/sarclisa.pdf. Accessed September 1, 2021.
- 7. European Medicines Agency . Sarclisa, INN‐Isatuximab. Summary of product characteristics. 2021. https://www.ema.europa.eu/en/documents/product-information/sarclisa-epar-product-information_en.pdf. Accessed September 1, 2021.
- 8. Ribba B, Holford N, Magni P, et al. A review of mixed‐effects models of tumor growth and effects of anticancer drug treatment used in population analysis. CPT Pharmacometrics Syst Pharmacol. 2014;3(5):e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bruno R, Bottino D, de Alwis DP, et al. Progress and opportunities to advance clinical cancer therapeutics using tumor dynamic models. Clin Cancer Res. 2020;26(8):1787‐1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Desmée S, Mentré F, Veyrat‐Follet C, Guedj J. Nonlinear mixed‐effect models for prostate‐specific antigen kinetics and link with survival in the context of metastatic prostate cancer: a comparison by simulation of two‐stage and joint approaches. AAPS J. 2015;17(3):691‐699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tardivon C, Desmée S, Kerioui M, et al. Association between tumor size kinetics and survival in patients with urothelial carcinoma treated with atezolizumab: implication for patient follow‐up. Clin Pharmacol Ther. 2019;106(4):810‐820. [DOI] [PubMed] [Google Scholar]
- 12. Desmée S, Guedj J, Veyrat‐Follet C, Comets E. Mechanistic joint modelling for longitudinal PSA and survival data in advanced metastatic prostate cancer. Annual Meeting of the Population Approach Group in Europe. 2017: Abstract 7154. https://www.page-meeting.org/?abstract=7154. Accessed September 1, 2021.
- 13. Thai H‐T, Fau J, France M, Comets E, Veyrat‐Follet C. Joint modeling of longitudinal tumor burden and time‐to‐event data to predict survival: application to aflibercept in second line metastatic colorectal cancer. Annual Meeting of the Population Approach Group in Europe. 2015: Abstract 3527. https://www.page-meeting.org/?abstract=3527. Accessed September 1, 2021.
- 14. Durie BGM, Harousseau J‐L, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20(9):1467‐1473. [DOI] [PubMed] [Google Scholar]
- 15. Chanu P, Claret L, Bruno R, et al. PK/PD relationship of the monoclonal anti‐BAFF antibody tabalumab in combination with bortezomib in patients with previously treated multiple myeloma: comparison of serum M‐protein and serum free light chains as predictors of progression free survival. Annual Meeting of the Population Approach Group in Europe. 2013: Abstract 2732. https://www.page-meeting.org/?abstract=2732. Accessed September 1, 2021.
- 16. Marchand M, Claret L, Losic N, Puchalski TA, Bruno R. Population pharmacokinetics and exposure‐response analyses to support dose selection of daratumumab in multiple myeloma patients. Annual Meeting of the Population Approach Group in Europe. 2013: Abstract 2668. https://www.page-meeting.org/?abstract=2668. Accessed September 1, 2021.
- 17. Bruno R, Jonsson F, Zaki M, et al. Simulation of clinical outcome for pomalidomide plus low‐dose dexamethasone in patients with refractory multiple myeloma based on week 8 M‐protein response. Blood. 2011;118(21):1881‐1881. [Google Scholar]
- 18. Jonsson F, Ou Y, Claret L, et al. A tumor growth inhibition model based on M‐protein levels in subjects with relapsed/refractory multiple myeloma following single‐agent carfilzomib use. CPT Pharmacometrics Syst Pharmacol. 2015;4(12):711‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thai H‐T, Liu L, Semiond D, et al. Model‐based drug development to support isatuximab dosing regimen selection in Phase II multiple myeloma patients. Annual Meeting of the Population Approach Group in Europe. 2016: Abstract 6018. https://www.page-meeting.org/?abstract=6018. Accessed September 1, 2021.
- 20. Koiwai K, El‐Cheikh R, Thai HT, et al. PK/PD modeling analysis for dosing regimen selection of isatuximab as single agent and in combination therapy in patients with multiple myeloma. CPT Pharmacometrics Syst Pharmacol. 2021;10(8):928‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fau J, El‐Cheikh R, Brillac C, et al. Drug‐disease interaction and time‐dependent population pharmacokinetics of isatuximab in relapsed/refractory multiple myeloma patients. CPT Pharmacometrics Syst Pharmacol. 2020;9(11):649‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jacqmin P, Snoeck E, van Schaick EA, et al. Modelling response time profiles in the absence of drug concentrations: definition and performance evaluation of the K‐PD model. J Pharmacokinet Pharmacodyn. 2007;34(1):57‐85. [DOI] [PubMed] [Google Scholar]
- 23. Li Y, Xu Y, Liu L, Wang X, Palmisano M, Zhou S. Population pharmacokinetics of pomalidomide. J Clin Pharmacol. 2015;55(5):563‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Spoorenberg SMC, Deneer VHM, Grutters JC, et al. Pharmacokinetics of oral vs. intravenous dexamethasone in patients hospitalized with community‐acquired pneumonia. Br J Clin Pharmacol. 2014;78(1):78‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Claret L, Girard P, Hoff PM, et al. Model‐based prediction of phase III overall survival in colorectal cancer on the basis of phase II tumor dynamics. J Clin Oncol. 2009;27(25):4103‐4108. [DOI] [PubMed] [Google Scholar]
- 26. Chanu P, Claret L, Marchand M, Losic N, Puchalski TA, Bruno R. Population pharmacokinetic/pharmacodynamic models to support dose selection of daratumumab in multiple myeloma patients. Annual Meeting of the Population Approach Group in Europe. 2014: Abstract 3281. https://www.page-meeting.org/?abstract=3281. Accessed September 1, 2021.
- 27. Jonsson F, Claret L, Knight R, et al. A longitudinal tumor growth inhibition model based on serum M‐protein levels in patients with multiple myeloma treated by dexamethasone. Annual Meeting of the Population Approach Group in Europe. 2010: Abstract 1705. https://www.page-meeting.org/default.asp?abstract=1705. Accessed September 1, 2021.
- 28. Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with low‐dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood. 2014;123(12):1826‐1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ayral G, Si Abdallah J, Magnard C, Chauvin J. A novel method based on unbiased correlations tests for covariate selection in nonlinear mixed effects models: The COSSAC approach. CPT Pharmacometrics Syst Pharmacol. 2021;10(4):318‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lavielle M, Ribba B. Enhanced method for diagnosing pharmacometric models: random sampling from conditional distributions. Pharm Res. 2016;33(12):2979‐2988. [DOI] [PubMed] [Google Scholar]
- 31. Traynard P, Ayral G, Twarogowska M, Chauvin J. Efficient pharmacokinetic modeling workflow with the MonolixSuite: A case study of remifentanil. CPT Pharmacometrics Syst Pharmacol. 2020;9(4):198‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Therneau TM, Grambsch PM, Fleming TR. Martingale‐based residuals for survival models. Biometrika. 1990;77(1):147‐160. [Google Scholar]
- 33. Cerou M, Lavielle M, Brendel K, Chenel M, Comets E. Development and performance of npde for the evaluation of time‐to‐event models. Pharm Res. 2018;35(2):30. [DOI] [PubMed] [Google Scholar]
- 34. Friberg L, de Greef R, Kerbusch T, Karlsson M. Modeling and simulation of the time course of asenapine exposure response and dropout patterns in acute schizophrenia. Clin Pharmacol Ther. 2009;86(1):84‐91. [DOI] [PubMed] [Google Scholar]
- 35. Wang Y, Sung C, Dartois C, et al. Elucidation of relationship between tumor size and survival in non‐small‐cell lung cancer patients can aid early decision making in clinical drug development. Clin Pharmacol Ther. 2009;86(2):167‐174. [DOI] [PubMed] [Google Scholar]
- 36. Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6(9):576‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu XS, Schecter JM, Jansson R, Yan X. Response to “The Role of FcRn in the Pharmacokinetics of Biologics in Patients with Multiple Myeloma”. Clin Pharmacol Ther. 2017;102(6):905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jacobs J, Mould D. The role of FcRn in the pharmacokinetics of biologics in patients with multiple myeloma. Clin Pharmacol Ther. 2017;102(6):903‐904. [DOI] [PubMed] [Google Scholar]
- 39. Food and Drug Administration Center For Drug Evaluation And Research . BLA Multi‐disciplinary Review and Evaluation BLA 761113 Sarclisa (isatuximab). 2016. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761113Orig1s000MultidisciplineR.pdf. Accessed September 1, 2021.
- 40. Yan X, Clemens PL, Puchalski T, et al. Influence of disease and patient characteristics on daratumumab exposure and clinical outcomes in relapsed or refractory multiple myeloma. Clin Pharmacokinet. 2018;57(4):529‐538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harrison SJ, Perrot A, Alegre A, et al. Subgroup analysis of ICARIA‐MM study in relapsed/refractory multiple myeloma patients with high‐risk cytogenetics. Br J Haematol. 2021;194(1):120‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Desmée S, Mentré F, Veyrat‐Follet C, Sébastien B, Guedj J. Nonlinear joint models for individual dynamic prediction of risk of death using Hamiltonian Monte Carlo: application to metastatic prostate cancer. BMC Med Res Methodol. 2017;17(1):105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Table S1 List of baseline patient characteristics tested as covariates
Supporting Information Table S2 Parameter estimates of the longitudinal model without covariates and with significant covariates. P values of covariate effects were computed by the Wald test
Supporting Information Table S3 Parameter estimates of the PFS log‐logistic model without covariates and with significant covariates. P values of covariate effects were computed by Wald test
Supporting Information Table S4 Parameter estimates of joint model with different links
Data Availability Statement
Qualified researchers can request access to patient‐level data and related study documents, including the clinical study report, study protocol with any amendments, blank case report forms, statistical analysis plan, and dataset specifications. Patient‐level data will be anonymized and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi's data‐sharing criteria, eligible studies and process for requesting access are at https://www.clinicalstudydatarequest.com.