

Abstract
TNFR2+ regulatory T cells preferentially accumulate in the tumor microenvironment, express high levels of immunosuppressive molecules and possess strong suppressive activity. Our study aimed to explore the characteristics and role of TNFR2+ Tregs in the microenvironment and progression of gastric cancer via polychromatic immunofluorescence, single‐cell RNA sequencing and flow cytometry assays. The TNFR2+ Treg infiltration level in the tumor microenvironment increased significantly as gastric cancer progressed and was demonstrated to be a prognostic marker. Single‐cell RNA sequencing revealed high levels of TNFR2 in tumor‐infiltrating Tregs. The TNF‐α/TNFR2 signaling pathway was activated, accompanied by the upregulation of costimulatory molecules. Unlike blood Tregs, tumor‐infiltrating Tregs existed in activated and effector states. In addition to expressing costimulatory molecules such as TNFR2, 4‐1BB, OX40 and GITR, tumor‐infiltrating Tregs were also characterized by high expression levels of immune checkpoints such as CTLA‐4 and TIGIT and chemokines such as CCR6. In vitro studies showed that the TNF‐α/TNFR2 pathway increased the Foxp3 expression in CD4+CD25+ T cells and the latent TGF‐β production in Tregs as well as enhanced the immunosuppressive function of Tregs. In summary, our study revealed high infiltration levels of TNFR2+ Tregs that were in activated and effector states in the tumor microenvironment. The infiltration level of TNFR2+ Tregs is a prognostic marker and an independent risk factor for gastric cancer. Activation of the TNF‐α/TNFR2 pathway promotes the immunosuppressive phenotype and function of Tregs. Our study provides a new theoretical basis for TNFR2+ Tregs as a therapeutic target in gastric cancer.
Keywords: gastric cancer, single‐cell RNA sequencing, TNFR2+ , Tregs, tumor microenvironment
What's new?
TNFR2+ regulatory T cells play a key role in mediating the immunosuppressive tumour microenvironment. However, little is known about the role of TNFR2+ Tregs in gastric cancer. The authors found that the TNFR2+ Treg infiltration level in the tumour microenvironment increased significantly as gastric cancer progressed and acted as a prognostic marker. In vitro studies showed that the TNF‐α/TNFR2 pathway can increase Foxp3 levels in CD4+CD25+ T cells and latent TGF‐β production in Tregs and enhance the immunosuppressive function of Tregs. Overall, this study provides a new theoretical basis for using TNFR2+ Tregs as a therapeutic target in gastric cancer.
Abbreviations
- 4‐1BB
TNFRSF9, TNF receptor superfamily member 9
- CCR6
C‐C motif chemokine receptor 6
- CL
cell label
- CTLA‐4
cytotoxic T‐lymphocyte associated protein 4
- DFS
disease‐free survival
- FACS
fluorescence‐activated cell sorter
- Foxp3
forkhead box P3
- GC
gastric cancer
- GITR
TNFRSF18, TNF receptor superfamily member 18
- GO
Gene Ontology
- Helios
IKZF2, IKAROS family zinc finger 2
- ICOS
inducible T cell costimulator
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LAG3
lymphocyte activating 3
- LAP
latency‐associated peptide
- MACS
magnetic cell sorting
- MALAT1
metastasis‐associated lung adenocarcinoma transcript 1
- MDSCs
myeloid‐derived suppressor cells
- MKI67
marker of proliferation Ki‐67
- OS
overall survival
- PBMC
peripheral blood mononuclear cell
- scRNA‐seq
single‐cell RNA sequencing
- TAMs
tumor‐associated macrophages
- TCR
T cell receptor
- TGF‐β
transforming growth factor beta 1
- TIGIT
T cell immunoreceptor with Ig and ITIM domains
- TNFR1
TNFRSF1A, TNF receptor superfamily member 1A
- TNFR2
TNFRSF1B, TNF receptor superfamily member 1B
- TNF‐α
tumor necrosis factor
- TOP2A
DNA topoisomerase II alpha
- Tregs
regulatory T cells
- UMAP
uniform manifold approximation and projection
- UMI
unique molecular index
1. INTRODUCTION
Although the worldwide incidence of gastric cancer (GC) has decreased over the past 50 years, GC remains the third leading cause of cancer‐related death. 1 , 2 , 3 Tumor cells can escape immune surveillance via the downregulation of antigens and major histocompatibility (MHC) molecules on the cell surface. 4 In addition, immune suppressors such as regulatory T cells (Tregs), myeloid‐derived suppressor cells (MDSCs) and tumor‐associated macrophages (TAMs) in the tumor microenvironment can strongly suppress antitumor responses, which play an important role in tumor progression. 5
Tregs, characterized by the expression of the transcription factor Foxp3, are powerful immune suppressors that are critical for the regulation of immune responses and immune homeostasis. 4 , 5 , 6 In tumor immunity, Tregs mediate tumor development and progression by inhibiting antitumor immunity. 7 , 8 Researchers have developed therapies based on the combination of Treg modulation or depletion with cancer vaccines and adoptive cell technologies to induce robust antitumor immune responses that can effectively suppress tumor progression. 9 , 10
Xin Chen et al reported that the tumor necrosis factor (TNF)‐α/TNF receptor superfamily member 1B (TNFR2) signaling pathway promoted the activation and expansion of Tregs in mice and that the expression of TNFR2 identified the subset of mouse CD4+CD25+Foxp3+ Tregs with maximal suppressive activity. 11 , 12 In human peripheral blood, the combination of CD4, CD25 and TNFR2 can be used to identify a large population of functional Tregs 13 with immunosuppressive functions that can significantly inhibit the proliferation and cytokine production of effector T cells. 13 In 2013, Xin Chen et al reported that TNFR2 was critical for maintaining Foxp3 expression and sustaining the phenotypic and functional stability of Tregs in an inflammatory environment. 14 Recently, an increasing number of studies have demonstrated increased numbers of TNFR2+ Tregs in patients with tumors, especially in the tumor microenvironment, and that this subset is responsible for the immunosuppressive tumor microenvironment. 15 , 16 , 17 , 18 , 19 , 20 In our previous study, approximately 90% of Foxp3+ Tregs in the peripheral blood of lung cancer patients were to found to express TNFR2. 21 TNFR2+ Tregs expressed high levels of CTLA‐4 and low levels of CD127, indicating their immunosuppressive phenotype. 21 In addition, compared to TNFR2− Tregs, TNFR2+ Tregs significantly inhibited the proliferation of CD8+ T cells and IFN‐γ production. 21 Moreover, the expression level of TNFR2 in Tregs has been found to be correlated with the clinicopathological progression and metastasis of lung cancer. 21 Compelling evidence supports the use of targeting TNFR2 to abolish the immunosuppressive activity of tumor‐infiltrating Tregs and enhance antitumor immune responses. 15 , 16 , 20 , 22 However, little is currently known about the role of TNFR2+ Tregs in the GC tumor microenvironment. In our study, we confirmed the accumulation of TNFR2+ Tregs in the tumor tissues of GC patients and analyzed the correlations of this cell population with clinical indicators. In addition, we elucidated the transcriptomic characteristics of Tregs by single‐cell RNA sequencing (scRNA‐seq), focused on the role of the TNF‐α/TNFR2 signaling pathway in Tregs and explored the phenotypic characteristics of TNFR2+ Tregs. These findings can improve our understanding of the role of TNFR2+ Tregs in the microenvironment and progression of GC.
2. MATERIALS AND METHODS
2.1. Clinical sample collection
A total of 145 patients with gastric adenocarcinoma were enrolled in our study according to the following inclusion criteria: (1) the age of the patient was >18 years; (2) the diagnosis of gastric adenocarcinoma had been histologically or cytologically confirmed; and (3) the patient had primary gastric cancer. The exclusion criteria were as follows: (1) the patient had gastric nonadenocarcinoma, and (2) no follow‐up information for the patient was available after the operation had been performed. All human GC tissues were obtained from surgically resected samples harvested from patients who had not received prior chemotherapy or radiotherapy at Tianjin Cancer Hospital from January 2010 to December 2012.
For scRNA‐seq, tumor tissues and peripheral blood were obtained from three GC patients who had been pathologically diagnosed with gastric adenocarcinoma. None of the patients received neoadjuvant radiotherapy or chemotherapy before surgery. All patients were diagnosed with Stage III cancer. The available clinical information is summarized in Table S1.
2.2. Polychromatic immunofluorescence
We collected paraffin sections of GC tumor tissues and assessed Treg and TNFR2+ Treg infiltration in the tumor microenvironment using an Opal 7‐Color Manual IHC Kit (PerkinElmer, NEL811001KT). The following primary antibodies were used: rabbit anti‐human CD4 (Abcam, ab133616, 1:1200), rabbit anti‐human Foxp3 (CST, #98377, 1:250) and mouse anti‐human TNFR2 (Santa Cruz, sc‐8041, 1:250). Polychromatic immunofluorescence staining was performed according to the kit protocol. In addition, single‐color immunofluorescence staining was performed to effectively distinguish the overlapping signals during the analysis.
2.3. Collection of single cells and Treg sorting
Tumor tissues were digested with an enzyme solution containing 0.05 mg/mL DNase I (Sigma), 1 mg/mL collagenase IV (Sigma), 0.1 mg/mL hyaluronidase (Sigma) and 0.05 mL/mL trypsin‐EDTA solution (0.25%, Shanghai Yuanpei) for 1 hour in a shaking incubator at 37°C. Then, the dissociated tissues were passed through a 100 μm cell strainer. We purified lymphocytes from the cell suspension with Ficoll solution (DAKEWE). Peripheral blood was diluted 1:1 with PBS, and lymphocytes were then obtained with Ficoll solution as described above. The suspensions of lymphocytes from tumor tissues and peripheral blood were stained with antibodies against CD4 (FITC), CD25 (APC) and CD127 (BV421) (all from BD Biosciences). CD4+CD25highCD127− Tregs were sorted by FACS (BD FACS Aria III).
2.4. Single‐cell capture, cDNA library construction and sequencing
Single‐cell suspensions were loaded into the BD Rhapsody Cartridge, which could effectively capture and separate single cells. Then, we loaded magnetic beads carrying a cell‐specific sequence and unique molecular index (UMI) into the cartridge. In each well, a single cell was lysed, and the released mRNA was captured by a barcoded bead. We collected all beads carrying mRNA in an Eppendorf tube and then conducted cDNA synthesis, transcriptome amplification and library amplification via PCR with a cDNA kit. The libraries were analyzed on the NovaSeq PE150 platform. The sequencing coverage and quality statistics for each sample are summarized in Table S2.
2.5. Quality control and processing of the scRNA‐seq data
Quality control of the scRNA‐seq data included removing low‐quality reads, removing junctions, calculating the sequencing error rate, eliminating batch differences, filtering out cells with a proportion of mitochondrial genes exceeding 0.25, and filtering out cells with fewer than 200 genes.
Transcript comparison: Reads that were aligned to the exon region were compared to transcripts with known annotations. Only reads that were compared to transcripts were counted as having a UMI. Cell counting: The reads of each cell were distinguished by cell label (CL) input data. The number of cells in the sample, number of reads and number of genes were counted after filtering and screening. Samples containing a mixture of cells with an extremely abnormal RNA content were excluded.
2.6. Analysis of scRNA‐seq data
Dimensionality reduction and cluster analysis: The dimensionality reduction method was used to project high‐dimensional data into low‐dimensional space by optimizing the preservation of key features in the original data so that the data could be presented in two or three dimensions. In the two‐dimensional space projected by the uniform manifold approximation and projection (UMAP) algorithm, cells closer to each other have more similar gene expression profiles. Each cell was analyzed by a clustering algorithm, and after clustering, the cells with similar expression profiles were grouped together.
Pseudotime analysis: We used Monocle 2 to determine the pseudotime trajectories of these cells. The R toolkit algorithm was used to elucidate the trajectory of gene expression changes in each cell during state transition. Once the overall trajectory of gene expression changes was understood, each cell was placed at the appropriate location in the trajectory.
Differential expression analysis: This experiment aimed to identify genes with higher expression in each cluster relative to the other clusters and was performed by Seurat. After differential gene expression analysis, gene functional enrichment analysis was carried out.
2.7. Cell cultures
Tregs were purified by magnetic cell sorting (MACS, CD4+CD25+ Regulatory T Cell Isolation Kit, Miltenyi) from peripheral blood mononuclear cells (PBMCs) and cultivated in 96‐well flat plates (1 × 105 cells/well) in complete medium composed of RPMI 1640 and 10% fetal bovine serum, which was supplemented with IL‐2 (500 U/mL, PeproTech), TGF‐β (5 ng/mL, Thermo Fisher) and β‐mercaptoethanol (1:1000, Sigma) in all cultures. Tregs were activated with anti‐CD3/CD28 beads (1:1, Thermo Fisher) for 72 hours. Tregs were stimulated either with TNF‐α (20 ng/mL, Novoprotein) or with TNF‐α and the TNFR2 neutralizing antibody Mab726 (1.5 μg/mL, R&D Systems) for 72 hours. The phenotypes of Tregs were detected by flow cytometry using the following antibodies: anti‐CD4 (FITC, BD), anti‐CD25 (APC, BD), anti‐Foxp3 (PE, BD), anti‐TNFR2 (PE‐CF594, BD), anti‐CTLA‐4 (PerCP‐Cy5.5, Biolegend), anti‐TIGIT (PerCP‐Cy7, Biolegend), anti‐CCR6 (BV510, Biolegend), anti‐ICOS (BV421, Biolegend), anti‐CD44 (PerCP‐Cy5.5, Biolegend), anti‐OX40 (BV510, Biolegend), anti‐41‐BB (BV421, Biolegend), anti‐GITR (PerCP‐Cy7, Biolegend) and anti‐LAP (BV421, Biolegend).
2.8. Suppression assay
CD8+ T cells labeled with carboxyfluorescein succinimidyl ester (CFSE, FITC, Thermo Fisher) were coincubated with TNF‐α, Tregs, Tregs and TNF‐α, or Tregs, TNF‐α, and Mab726 for 72 hours. The proliferation percentage of CD8+ T cells was detected by flow cytometry, and the secretion of IFN‐γ by CD8+ T cells was detected by ELISA (DAYOU).
3. RESULTS
3.1. A high level of TNFR2 + Treg infiltration can indicate poor prognosis in GC patients
Polychromatic immunofluorescence staining and spectral imaging techniques were used to assess the infiltration levels of Tregs (CD4+Foxp3+ Tregs/CD4+ T cells) and TNFR2+ Tregs (CD4+Foxp3+TNFR2+ Tregs/CD4+Foxp3+ Tregs) in the GC tumor microenvironment (Figure 1A). The level of TNFR2+ Tregs in GC patients was significantly correlated with both the TNM stage (P < .001) and N stage (P < .001) (Table 1). In different TNM stages, Treg infiltration exhibited an increasing trend as the tumor progressed, but the differences among the different stages were not significant (Figure 1B). However, the TNFR2+ Treg infiltration level was significantly higher in patients with Stage III disease than in patients with Stage I and II disease (P < .05) (Figure 1B). Regarding progression at the N stage, the infiltration levels of Tregs and TNFR2+ Tregs were increased in stages N2 and N3 compared to Stage N0 (P < .001; P < .0001) (Figure 1C).
FIGURE 1.
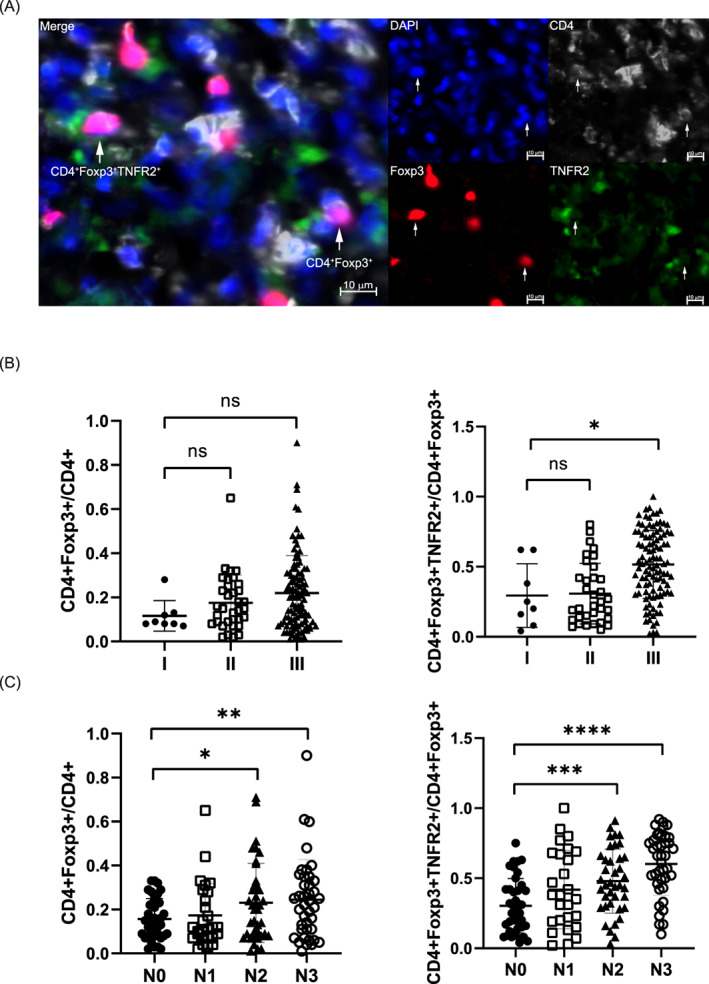
Distributions of CD4+ Foxp3+ Tregs and CD4+ Foxp3+TNFR2+ Tregs in the tissues of GC patients in different TNM and N stages. (A) Polychromatic immunofluorescence staining with anti‐CD4, anti‐Foxp3 and anti‐TNFR2 antibodies to validate the distributions of CD4+Foxp3+ Tregs and CD4+Foxp3+TNFR2+ Tregs in tumor tissue (n = 145). Scale bar, 10 μM. Polychromatic immunofluorescence was analyzed with ZEISS ZEN 3.0. (B) Infiltration levels of Tregs and TNFR2+ Tregs at different TNM stages and analysis of their statistical correlations with the TNM stage (Wilcoxon test, *P < .05). All tests were performed with GraphPad Prism 8.0.1. (C) Infiltration levels of Tregs and TNFR2+ Tregs at different N stages and analysis of their statistical correlations with the TNM stage (Wilcoxon test, *P < .05; **P < .01; ***P < .001; ****P < .0001). All tests were performed with GraphPad Prism 8.0.1 [Color figure can be viewed at wileyonlinelibrary.com]
TABLE 1.
Infiltration levels of Tregs and TNFR2+ Tregs and their correlation with clinical indicators in GC patients
| Cases | CD4+Foxp3+Tregs in CD4+T cells % | P | CD4+Foxp3+TNFR2+Tregs in CD4+Foxp3+Tregs % | P | |
|---|---|---|---|---|---|
| Variables | Median (IQR) | Median (IQR) | |||
| Age | .248 | .653 | |||
| ≤60 | 77 | 0.160 (0.070‐0.270) | 0.430 (0.275‐0.710) | ||
| >60 | 68 | 0.185 (0.090‐0.298) | 0.455 (0.203‐0.638) | ||
| Gender | .968 | .06 | |||
| Male | 99 | 0.150 (0.080‐0.310) | 0.420 (0.200‐0.640) | ||
| Female | 46 | 0.185 (0.090‐0.260) | 0.550 (0.308‐0.718) | ||
| TNM stage | .407 | <.001 | |||
| I‐II | 42 | 0.130 (0.080‐0.235) | 0.235 (0.128‐0.440) | ||
| IIIa | 19 | 0.120 (0.070‐0.250) | 0.390 (0.200‐0.690) | ||
| IIIb | 39 | 0.200 (0.080‐0.300) | 0.430 (0.300‐0.630) | ||
| IIIc | 45 | 0.205 (0.073‐0.325) | 0.625 (0.443‐0.750) | ||
| N stage | .104 | <.001 | |||
| N0 | 39 | 0.140 (0.080‐0.230) | 0.270 (0.140‐0.420) | ||
| N1 | 27 | 0.120 (0.070‐0.250) | 0.380 (0.190‐0.680) | ||
| N2 | 39 | 0.200 (0.080‐0.310) | 0.450 (0.300‐0.650) | ||
| N3 | 40 | 0.230 (0.110‐0.345) | 0.660 (0.468‐0.785) |
Abbreviation: IQR, interquartile range.
To evaluate the abilities of the Treg and TNFR2+ Treg infiltration levels to serve as prognostic markers, we divided the patients into a high‐infiltration group and a low‐infiltration group according to the median infiltration ratio of these two cell populations and analyzed the correlations between the infiltration levels in the tumor microenvironment and the prognosis of the GC patients. The overall survival (OS) and disease‐free survival (DFS) rates of GC patients with high infiltration levels of Tregs or TNFR2+ Tregs were significantly lower than those of patients with low infiltration levels, which indicated poor prognosis (P < .05) (Figures 2A,D and S1A,D). In the subgroup analysis, high infiltration of Tregs indicated poor prognosis only in patients with Stage I‐II (OS: P < .05) or Stage N1 disease (OS and DFS: P < .05) (Figures 2B,C and S1C). In the subgroup analysis, the OS and DFS curves showed that high infiltration levels of TNFR2+ Tregs were predictive of poor prognosis at Stage III or various N stages (P < .05) (Figures 2E,F and S1E,F). These results suggest that the expression of TNFR2 in Tregs increases throughout tumor progression.
FIGURE 2.
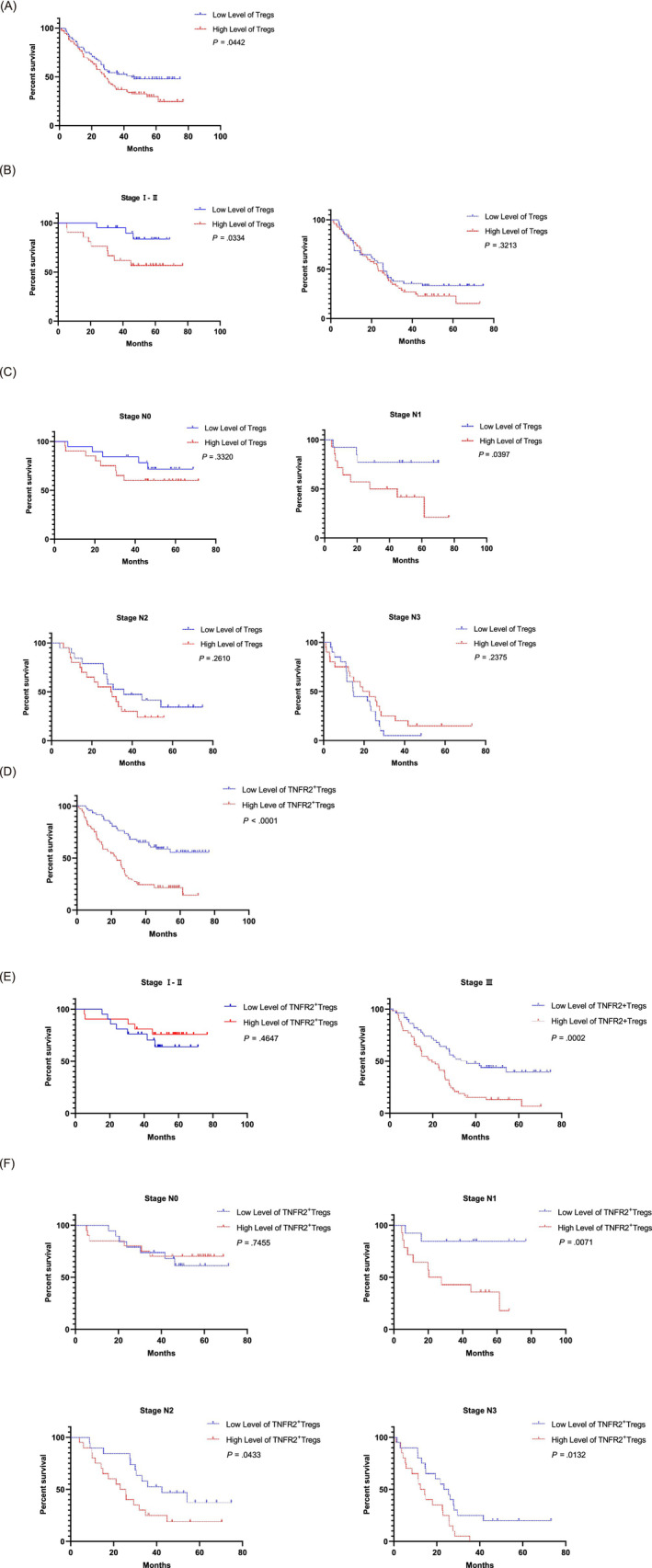
The correlations between Treg and TNFR2+ Treg infiltration levels and OS. (A) Patients were divided into high‐ and low‐infiltration groups according to the median Treg infiltration ratio. Kaplan‐Meier survival curves showed a difference in the OS between patients with high and low levels of tumor‐infiltrating Tregs (log‐rank test, P < .05). (B) Subgroup analysis. The OS curves for patients stratified by TNM stage were compared between patients with high and low levels of tumor‐infiltrating Tregs (log‐rank test, P < .05). (C) Subgroup analysis. The OS curves for patients stratified by N stage were compared between patients with high and low levels of tumor‐infiltrating Tregs (log‐rank test, P < .05). (D) Patients were divided into high‐ and low‐infiltration groups according to the median TNFR2+ Treg infiltration ratio. The Kaplan‐Meier survival curves showed a difference in the OS between patients with high and low levels of tumor‐infiltrating TNFR2+ Tregs (log‐rank test, P < .05). (E) Subgroup analysis. The OS curves for patients stratified by TNM stage were compared between patients with high and low levels of tumor‐infiltrating TNFR2+ Tregs (log‐rank test, P < .05). (F) Subgroup analysis. The OS curves for patients stratified by N stage were compared between patients with high and low levels of tumor‐infiltrating TNFR2+ Tregs (log‐rank test, P < .05). All tests were performed with GraphPad Prism 8.0.1 [Color figure can be viewed at wileyonlinelibrary.com]
Cox regression analyses of the age, sex, N stage, TNM stage and TNFR2+ Treg infiltration level parameters indicated that the infiltration level of TNFR2+ Tregs was an independent risk factor that affected the prognosis of GC patients (Table S3). Cox regression analysis of the overall Treg infiltration level showed no statistical significance for this parameter (P > .05) (data not shown).
The above data show that high infiltration levels of TNFR2+ Tregs in the GC tumor microenvironment can be to indicate poor prognosis, especially for patients with Stage III or Stage N1‐N3 disease.
3.2. TNFR2 is ubiquitously expressed and the TNF‐α/TNFR2 signaling pathway is activated in tumor‐infiltrating Tregs as determined by ScRNA‐seq
The above results showed that the infiltration level of TNFR2+ Tregs was related to the prognosis of GC patients. We next used scRNA‐seq to analyze the transcriptomic characteristics and possible mechanism of action of TNFR2+ Tregs in the GC tumor microenvironment. We isolated CD4+CD25+CD127− Tregs from tumor tissues and matched peripheral blood samples from three GC patients by flow cytometry (Figure 3A). After quality control, normalization and batch correction, we finally captured 7252 tumor‐infiltrating Tregs and 2340 peripheral blood Tregs.
FIGURE 3.
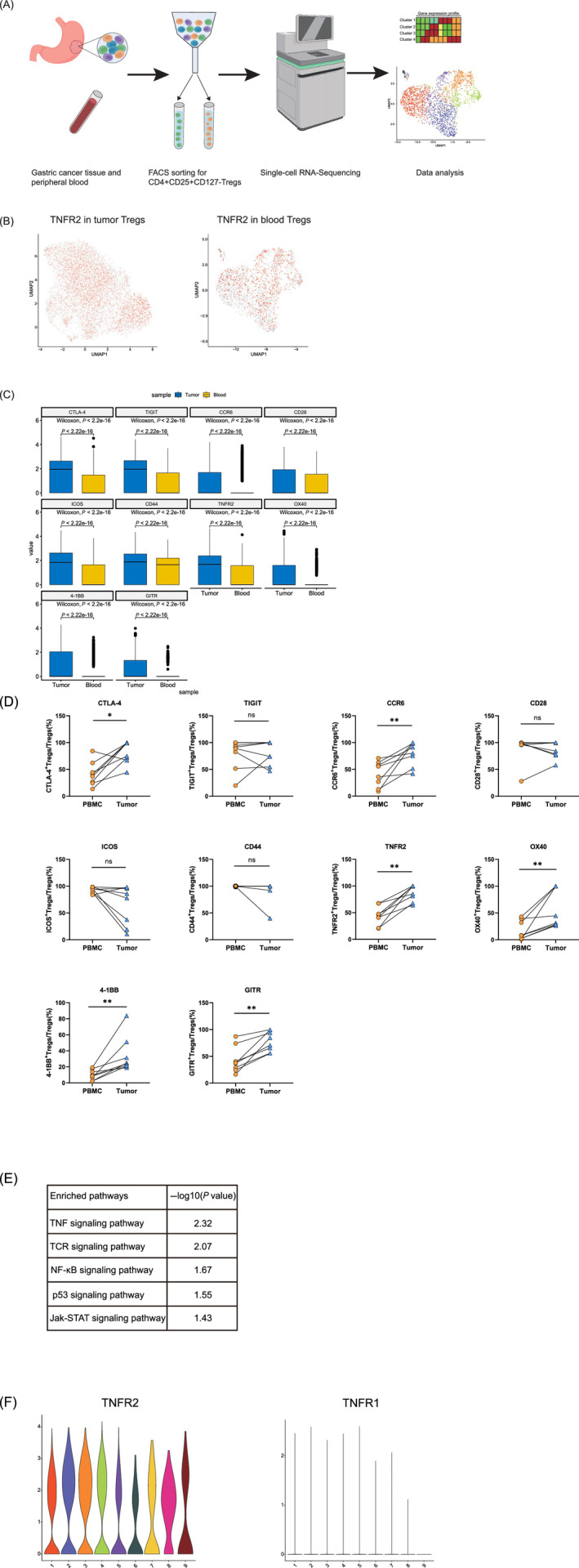
TNFR2 is ubiquitously highly expressed and the TNF‐α/TNFR2 signaling pathway is activated in tumor‐infiltrating Tregs as determined by scRNA‐seq analysis. (A) Flowchart of scRNA‐seq. scRNA‐seq (BD Rhapsody) was performed on CD4+CD25+CD127− Tregs derived from the peripheral blood and tumors of GC patients. The flowchart was created with BioRender.com. (B) UMAP plots showing the expression levels of TNFR2 in tumor‐infiltrating Tregs (left) and blood Tregs (right). UMAP plots colored red indicate high expression of TNFR2, and UMAP plots colored gray indicate low expression of TNFR2. All UMAP plot graphics were generated with Seurat. (C) Boxplot showing the expression levels of immune‐related genes in tumor‐infiltrating Tregs and blood Tregs (Wilcoxon test, P < .05). The boxplot was generated with Python. (D) The expression levels of the immune‐related genes in the tumor‐infiltrating Tregs and blood Tregs of GC patients were compared by flow cytometry. Independent experiments included eight biological duplicates. Statistical analyses were performed using GraphPad Prism 8.0.1. The statistical tests used are indicated in the figure legends: *P < .05, **P < .01. (E) Pathways enriched with upregulated genes in tumor‐infiltrating Tregs. (F) Violin plots showing high expression of TNFR2 and low expression of TNFR1 in tumor‐infiltrating Tregs. The violin plots were generated with R package [Color figure can be viewed at wileyonlinelibrary.com]
We compared the expression levels of TNFR2 in tumor‐infiltrating Tregs and blood Tregs and found that 62.8% of the tumor‐infiltrating Tregs and only 34.6% of the blood Tregs expressed TNFR2 (Figure 3B). TNFR2 was ubiquitously expressed in tumor‐infiltrating Tregs rather than concentrated in a cluster (Figure 3B). Moreover, tumor‐infiltrating Tregs expressed significantly higher levels of the immune checkpoint genes CTLA‐4 (P < .0001) and TIGIT (P < .0001), the chemokine receptor CCR6 (P < .0001), and some costimulatory signaling molecules (P < .0001), including CD28, ICOS, CD44, TNFR2, 4‐1BB, OX40 and GITR (Figure 3C), than blood Tregs. The expression levels of the immune‐related genes in the tumor‐infiltrating Tregs and blood Tregs of 8 GC patients were compared by flow cytometry. Results showed that tumor‐infiltrating Tregs expressed higher levels of CTLA‐4 (P < .05), CCR6 (P < .01), TNFR2 (P < .01), OX40 (P < .01), 41‐BB (P < .01) and GITR (P < .01) than blood Tregs (Figure 3D). These gene expression characteristics suggested that the tumor‐infiltrating Tregs were activated and had migratory and immunosuppressive activities. Gene functional enrichment analysis was performed for the genes highly expressed in tumor‐infiltrating Tregs. These genes were enriched in the response to the TNF pathway, indicating that the TNF‐α signaling pathway was activated (Figure 3E). In addition, tumor‐infiltrating Tregs expressed high levels of TNFR2 and low levels of TNF receptor superfamily member 1A (TNFR1, another receptor for TNF‐α (Figure 3F). Therefore, the TNF‐α/TNFR2 signaling pathway rather than the TNF‐α/TNFR1 pathway was activated. Researchers have reported that TNF‐α can promote the activation and expansion of CD4+Foxp3+ Tregs through TNFR2, preferentially upregulate the expression levels of TNFRSF molecules and facilitate the optimal activation of Tregs. 23 , 24 , 25 Therefore, activation of the TNF‐α/TNFR2 signaling pathway likely plays an important role in promoting the activated state and immunosuppressive function of Tregs in tumor tissues.
3.3. Unlike blood Tregs, tumor‐infiltrating Tregs are in an active and effector state
We observed that the tumor‐infiltrating Tregs were activated and exhibited an effector status. Thus, their functional status was further assessed by performing gene functional enrichment analysis. The genes upregulated in blood Tregs were enriched mainly in ribosome‐related and protein transport pathways (Figure 4A). However, almost no other signaling pathways were activated, possibly because these cells did not respond to the external environment. In contrast, the genes upregulated in tumor‐infiltrating Tregs were enriched mainly in the TNF‐α signaling pathway, TCR signaling pathway, NF‐κB signaling pathway, and biological processes such as response to cytokines and cytokine production (Figure 4A). These results suggest that tumor‐infiltrating Tregs respond actively to cytokines in the tumor microenvironment and are in an effector state.
FIGURE 4.
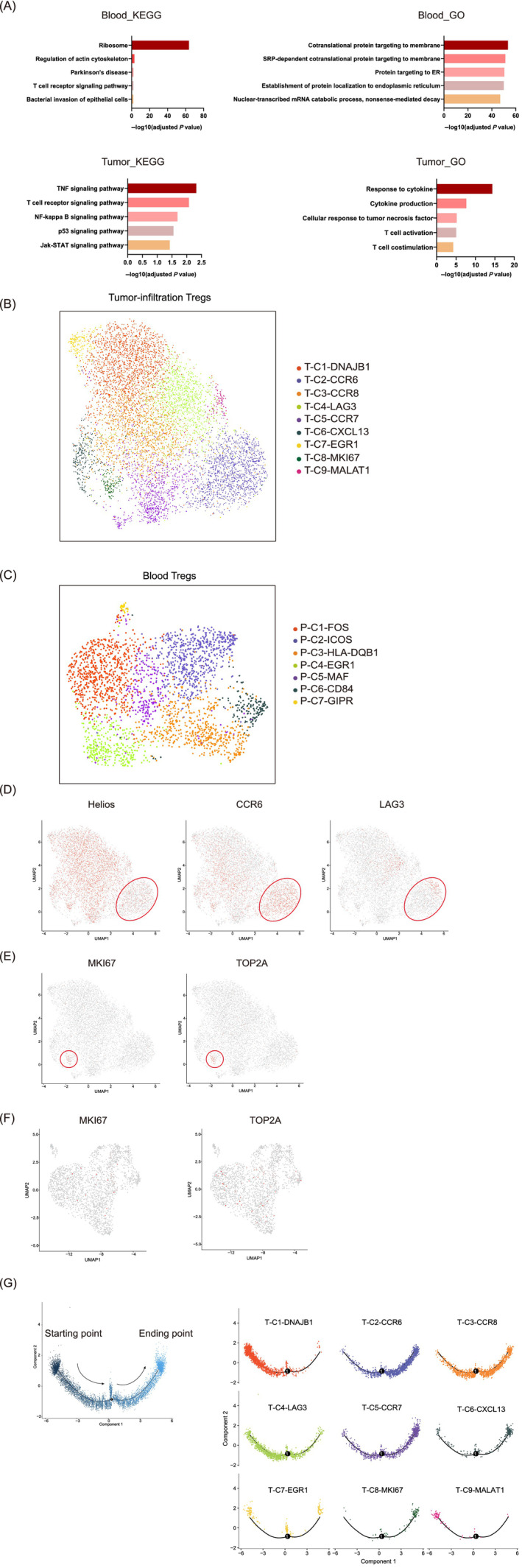
Differences in the transcriptomic characteristics of blood Tregs and tumor‐infiltrating Tregs. (A) Pathways most highly enriched with upregulated genes in blood Tregs and tumor‐infiltrating Tregs. The graphic was created with GraphPad Prism 8.0.1. (B) UMAP plots showing nine clusters of tumor‐infiltrating Tregs identified by integrated analysis (colored by cluster). (C) UMAP plots showing seven clusters of blood Tregs identified by integrated analysis (colored by cluster). (D) Feature plots of the expression distributions of Helios, CCR6 and LAG3 in tumor‐infiltrating Tregs. (E) Feature plots of the expression distributions of MKI67 and TOP2A in tumor‐infiltrating Tregs. (F) Feature plots of the expression distributions of MKI67 and TOP2A in blood Tregs. (G) Pseudotime analysis was performed to elucidate the trajectories of state transitions among tumor‐infiltrating Tregs. The pattern diagram (left) shows the direction of cell state transformation. Each cluster is located at the corresponding position on the pseudotime axis according to the cell state (right). Pseudotime analysis was performed with Monocle 2 [Color figure can be viewed at wileyonlinelibrary.com]
Next, we analyzed the distributions of different Treg subgroups in the tumor microenvironment and peripheral blood on the basis of the previous analysis. Our data revealed a set of nine clusters of tumor‐infiltrating Tregs and seven clusters of blood Tregs (Figure 4B,C). We defined these subgroups according to the differentially expressed genes in each cluster. Tregs in T‐C2‐CCR6 expressed higher levels of the effector/memory‐like Treg marker CCR6 26 , 27 and low levels of the natural Treg (nTreg) marker Helios, thus exhibiting the characteristics of effector/memory‐like Tregs (Figure 4D). In addition, a small fraction of Tregs in T‐C2‐CCR6 cells expressed high levels of LAG3 (Figure 4D). Since LAG3 is highly expressed in activated inducible Tregs (iTregs), 6 these Helios−LAG3+ Tregs may be in the transitional state between iTregs and effector/memory‐like Tregs. Tregs in T‐C8‐MKI67 expressed high levels of MKI67 and TOP2A, indicating that the Tregs in this cluster were in an active state of proliferation (Figure 4E). Unlike tumor‐infiltrating Tregs, almost no blood Tregs expressed MKI67 and TOP2A, suggesting that few blood Tregs were in a proliferative state (Figure 4F).
Pseudotime analysis was used to elucidate the trajectory of transformation and succession among tumor‐infiltrating Tregs. 28 , 29 On the pseudotime axis, the T‐C9‐MALAT1 cluster was located at the origin (Figure 4G). Among the differentially expressed genes, the expression level of ribosomal protein in T‐C9‐MALAT1 was very low compared to that in the other clusters, indicating that this cluster was transcriptionally and translationally inactive and that the cells were in the initial state. Cluster T‐C2‐CCR6, representing the abovementioned effector/memory‐like Tregs, was largely located further along the axis, consistent with the active state of cells in this cluster. Cells in T‐C8‐MKI67 expressed high mRNA levels of MKI67 and TOP2A and were in a state of robust proliferation; thus, most cells in this cluster were located at the end of the pseudotime axis. Other clusters were distributed along the axis at different positions according to their active state. These results indicated that the tumor‐infiltrating Treg population contained cells at various stages from the resting state to the activated state to the effector/memory state.
3.4. Tumor‐derived clusters have higher functional diversity than blood Tregs
To investigate the existence of a specific Treg subset in the GC microenvironment, we integrated and reclustered all Tregs derived from tumor tissue and peripheral blood. The UMAP plot shows that the total Treg population was divided into 18 clusters (Figure 5A). Most Tregs derived from tumor tissue and peripheral blood overlapped on the UMAP plot, but a small number of subgroups existed specifically in tumor tissue or peripheral blood (Figure 5A). Although we captured more tumor‐infiltrating Tregs than peripheral blood Tregs, the Tregs derived from peripheral blood accounted for the vast majority of Tregs in Clusters 6 and 8, with proportions of 96% and 79%, respectively (Figure 5B). Therefore, we identified these two clusters as blood‐specific Tregs. In contrast, most Tregs in Cluster 2 (96%) were derived from tumor tissue, and Cluster 2 was thus identified as a tumor tissue‐specific subgroup in GC (Figure 5B).
FIGURE 5.
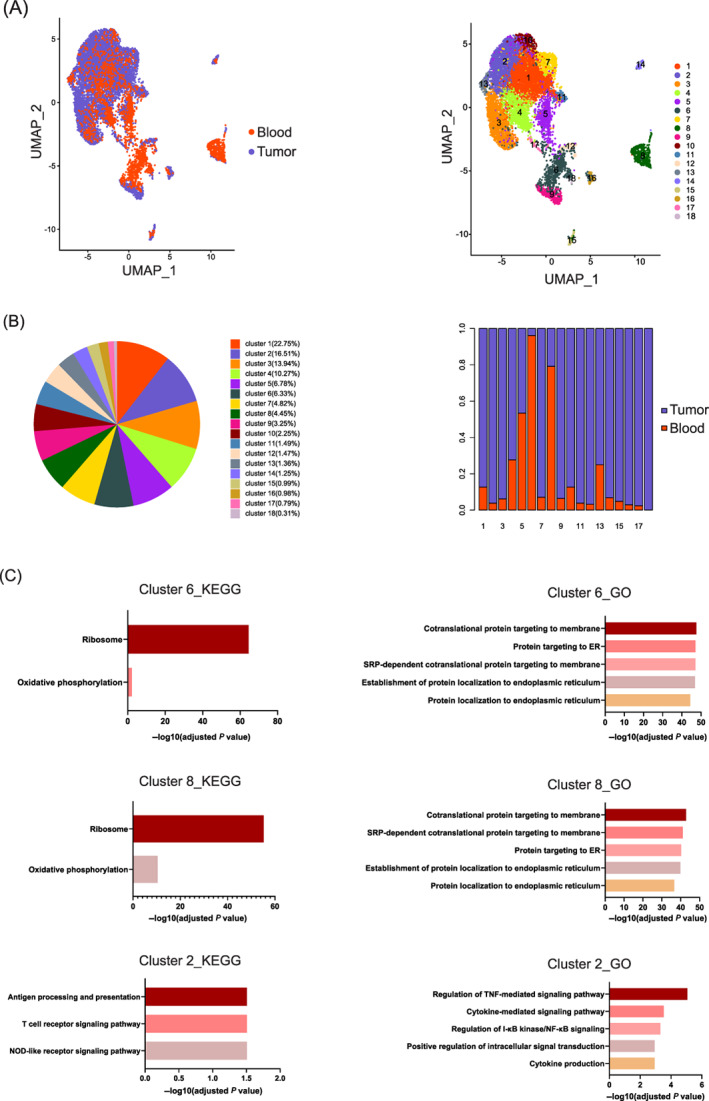
Transcriptome characteristics of tissue‐specific subgroups among blood‐ and tumor‐infiltrating Tregs. (A) Tumor‐infiltrating Tregs and blood Tregs were merged and reclustered. The UMAP plots on the left show 18 clusters of Tregs identified by integrated analysis (colored by cluster). The UMAP plots on the right display the distributions of tumor‐infiltrating Tregs and blood Tregs. (B) The proportion of each cluster within the total Treg population (left) and the proportions of Tregs from tumor tissue and peripheral blood within each cluster. (C) KEGG and GO enrichment analyses of genes upregulated in blood‐specific Clusters 6 and 8 and in tumor‐specific Cluster 2 [Color figure can be viewed at wileyonlinelibrary.com]
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis showed that the upregulated genes expressed in Clusters 6 and 8 were significantly enriched in the ribosome pathway, and Gene Ontology (GO) enrichment analysis showed that these upregulated genes were enriched mainly in protein transportation and cellular components related to ribosomal subunits, indicating robust protein synthesis in these blood‐specific Tregs (Figure 5C). However, no other signaling pathways were activated, and we believe that these blood‐specific Tregs were nonresponsive to the external environment. Similarly, KEGG and GO enrichment analyses of tumor tissue‐specific Cluster 2 showed that the upregulated genes were enriched mainly in antigen processing and presentation, the T cell receptor signaling pathway and the NOD‐like receptor signaling pathway. These genes were also enriched in the TNF‐mediated signaling pathway, cytokine‐mediated signaling pathway, and I‐κB kinase/NF‐κB signaling pathway (Figure 5C). Unlike blood‐specific Tregs, the tumor tissue‐specific Tregs were shown to participate in various biological processes and to be in activated and effector states.
Tumor‐infiltrating Tregs accounted for more than 95% of the total Treg population in several clusters in addition to the above clusters, including Clusters 11, 12, 15, 16 and 17. The Tregs in this cluster were involved in lymphocyte activation, cell proliferation and differentiation (Figure S2A‐E).
The above results indicate that compared to blood Tregs, the Tregs in the tumor microenvironment exhibited an increased diversity and played roles in different functions, including TNF signaling pathway processes, cell activation, proliferation, and immune responses.
3.5. The TNF‐α/TNFR2 signaling pathway enhances the immunosuppressive phenotype and function of Tregs
The scRNA‐seq results showed that tumor‐infiltrating Tregs were activated and that the differentially expressed genes were enriched in the TNF‐α/TNFR2 signaling pathway. We isolated CD4+CD25+ Tregs in vitro to validate that the TNF‐α/TNFR2 signaling pathway plays a role in the phenotype and function of Tregs. Flow cytometry showed a significant increase in the proportion of Foxp3+ Tregs in CD4+ T cells after in vitro (P < .0001) (Figure 6A). Interestingly, the proportion of Foxp3+ Tregs in CD4+CD25+ T cells was increased by TNF‐α stimulation in the absence of anti‐CD3/CD28 beads (P < .001) (Figure 6B), and this effect was neutralized by the TNFR2 neutralizing antibody Mab726 (P < .001) (Figure 6B). This result suggests that the TNF‐α/TNFR2 signaling pathway can promote the expression of Foxp3 in CD4+CD25+ T cells. Compared to unactivated Tregs, activated Tregs expressed higher levels of TNFR2 (P < .0001), CTLA‐4 (P < .001), and ICOS (P < .0001) (Figure 6A), which was consistent with the scRNA‐seq results. During the culture of Tregs, we added either TNF‐α or TNF‐α plus Mab726, and the phenotype of Tregs was detected by flow cytometry. We found that the latency‐associated peptide (LAP) expression in Tregs was significantly increased by TNF‐α stimulation (P < .01) and that this effect was eliminated by Mab726 (P < .01) (Figure 6C), which indicated that the TNF‐α/TNFR2 signaling pathway could enhance the immunosuppressive function of Tregs by increasing the level of latent TGF‐β. To verify the effect of the TNF‐α/TNFR2 signaling pathway on the immunosuppressive function of Tregs, the effects of activated Tregs on the proliferation and IFN‐γ production of CD8+ T cells were evaluated. Compared to Tregs not pretreated with TNF‐α, those stimulated by TNF‐α further inhibited the proliferation and production of IFN‐γ by CD8+ T cells (P < .05), while Mab726 eliminated this effect (P < .05) (Figure 6D,E). These results suggest that the TNF‐α/TNFR2 signaling pathway can further enhance the immunosuppressive function of activated Tregs.
FIGURE 6.
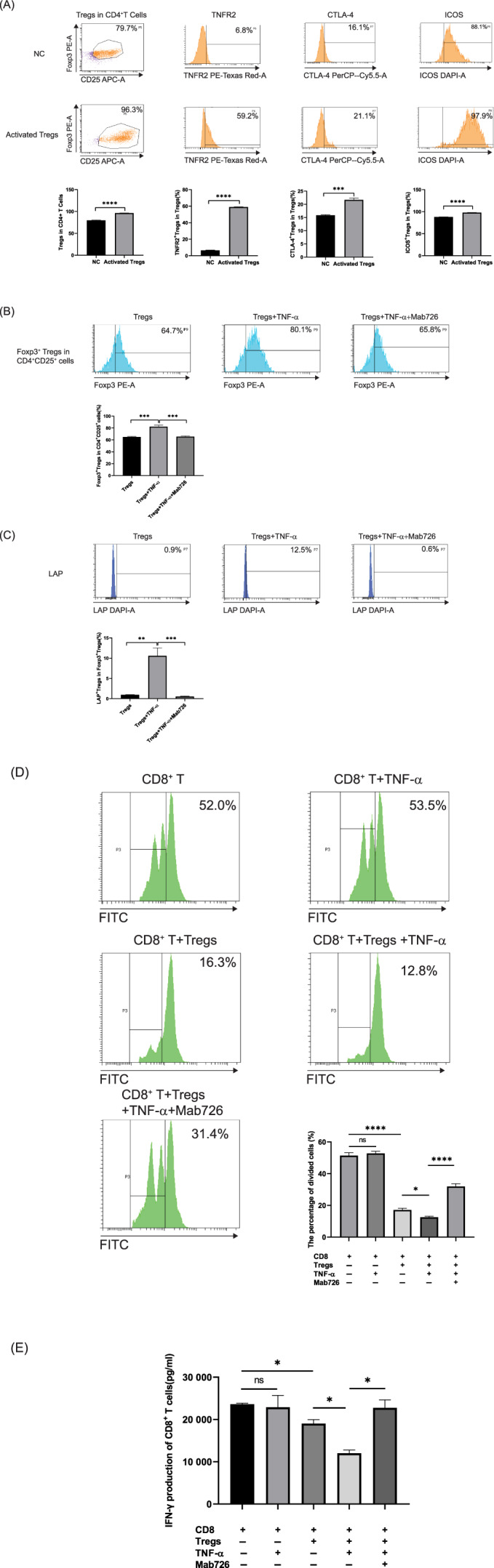
The TNF‐α/TNFR2 signaling pathway promotes the proliferation and enhances the immunosuppressive phenotype and function of Tregs. (A) The proportions of Foxp3+ Tregs in CD4+ T cells and the expression levels of TNFR2, CTLA‐4 and ICOS in control Tregs and activated Tregs were compared by flow cytometry analysis. Three biological duplicates of independent experiments were carried out. (B) Tregs were stimulated by either TNF‐α or TNF‐α plus Mab726 for 72 hours in the absence of anti‐CD3/CD28 beads. The proportion of Foxp3+ Tregs in CD4+CD25+ T cells was determined by flow cytometry. Representatives of three independent experiments with biological duplicates are shown. (C) Tregs were stimulated with either TNF‐α or TNF‐α plus Mab726 for 72 hours in the absence of anti‐CD3/CD28 beads. The level of LAP expression in activated Tregs was determined by flow cytometry. (D) Proliferation suppression assay. CFSE‐labeled CD8+ T cells were cultured alone or coincubated with activated Tregs and stimulated with either TNF‐α or TNF‐α plus Mab726 for 72 hours to quantify the proliferation percentage by flow cytometry. (E) Purified CD8+ T cells were cultured alone or coincubated with activated Tregs and stimulated by either TNF‐α or TNF‐α plus Mab726 for 72 hours. The IFN‐γ production in CD8+ T cells was detected by ELISA. Statistical analyses were performed using GraphPad Prism 8.0.1. The statistical tests used are indicated in the figure legends: *P < .05; **P < .01; ***P < .001; ****P < .0001 [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
Our study demonstrated that TNFR2+ Tregs accumulate in the tumor microenvironment as GC progresses. The infiltration level of TNFR2+ Tregs can be used as a prognostic marker for patients with GC and serves as an independent risk factor for their prognosis. ScRNA‐seq analysis revealed that TNFR2 was ubiquitously highly expressed in tumor‐infiltrating Tregs and that the TNF‐α/TNFR2 pathway was activated in these cells. The tumor‐infiltrating Treg population contained cells that were more active and effective and participated in more biological processes than the blood Treg population. In addition, the TNF‐α/TNFR2 pathway was shown to promote the expression of Foxp3 in CD4+ T cells and the production of latent TGF‐β in activated Tregs. The TNF‐α/TNFR2 pathway can further enhance the immunosuppressive function of activated Tregs.
According to research reports, the utility of Tregs as a prognostic marker remains controversial. The correlations between the density of tumor‐infiltrating Tregs and the prognosis of patients are not consistent across human cancers. 30 Treg assessments should be subdivided based on their localization in the tumor tissue, and the prognostic impact of each group should be individually examined. 31 , 32 In GC, we found that although the infiltration level of Tregs indicated the prognosis of patients with Stage I‐II disease, the prognostic value of Tregs gradually decreased as the tumor progressed. The proportion of TNFR2+ Tregs among Tregs increased significantly as the tumor progressed and could thus serve as a prognostic marker for GC patients. Since TNFR2+ Tregs have been reported to be a Treg subset with maximal immunosuppressive activity, the increased expression of TNFR2 in Tregs indicates an increasingly favorable environment for tumor progression. We speculate that in GC patients, changes in the proportions of TNFR2+ subgroups among Tregs are more important for tumor progression than the expression level of TNFR2.
We conducted scRNA‐seq to further explore the transcriptomic characteristics of Tregs and TNFR2+ Tregs isolated from the tumor tissues and peripheral blood of GC patients. A previous report indicated that human Tregs are phenotypically and functionally heterogeneous, composed of distinct, although developmentally related, subpopulations. 33 The results showed that in GC patients with Stage III disease, the TNF‐α signaling pathway was activated in tumor‐infiltrating Tregs. TNF‐α mediates its biological functions through two structurally distinct receptors: TNFR1 and TNFR2. TNFR1 is ubiquitously expressed on nearly all cells, while TNFR2 is restricted to T lymphocytes and other cells. 34 , 35 , 36 Our results showed that the TNF‐α/TNFR2 signaling pathway rather than the TNF‐α/TNFR1 signaling pathway was activated in tumor‐infiltrating Tregs. In addition, tumor‐infiltrating Tregs expressed significantly higher levels of the immune checkpoint genes CTLA‐4, the chemokine CCR6, and costimulatory molecules, including TNFR2, OX40, 4‐1BB, and GITR, than blood Tregs. The suppressive function of Tregs is largely derived from their ability to control and restrict the availability of cosignaling molecules to other T cells. However, Tregs themselves also depend on many of the same cosignaling molecules for their own homeostasis, constituting a complex feedback system. 37 The TNFRSF members TNFR2, 4‐1BB, OX40, and GITR can activate canonical NF‐κB signaling and promote the proliferation and activation of Tregs. 37 According to a research report, TNF‐α, in concert with IL‐2, preferentially upregulates the expression of costimulatory TNFRSF family members, such as TNFR2, 4‐1BB and OX40, on Tregs, resulting in positive feedback amplification of the stimulatory effect of TNF‐α on Tregs. 23 Upregulation of 4‐1BB and OX40 by TNF‐α/IL‐2 on Tregs can further promote their proliferation while preserving or even enhancing their potent suppressive activity. 23 Considering the above analysis results collectively, we can infer that the TNF‐α/TNFR2 signaling pathway is activated in tumor‐infiltrating Tregs, accompanied by the upregulated expression of TNFRSF members. Moreover, Tregs express high levels of immune checkpoints, chemokines, and other costimulatory molecules, which indicates their highly immunosuppressive and activated phenotypic characteristics. The biological effects of the TNF‐α/TNFR2 signaling pathway on Tregs include activation, proliferation and enhancement of inhibitory functions and survival. 38 In summary, tumor‐infiltrating Tregs ubiquitously express high levels of TNFR2 in GC patients, and activation of the TNF‐α/TNFR2 signaling pathway plays an important role in promoting the activation and immunosuppressive function of tumor‐infiltrating Tregs.
In addition to the TNF‐α signaling pathway, numerous activation‐related signaling pathways are activated in tumor‐infiltrating Tregs. Genes highly expressed in tumor‐infiltrating Tregs mediate a variety of biological processes, and tumor‐infiltrating Tregs actively respond to cytokines in the tumor microenvironment. Tumor‐infiltrating Tregs, including clusters in different states, are more heterogeneous than blood Tregs. The clusters of tumor‐infiltrating Tregs are distributed on the pseudotime axis from the origin (initial state) to the endpoint (activated state). Most cells are distributed along the time axis and are in an intermediate state of transition to the activated state. Tumor tissue‐specific Tregs are involved in more biological processes, including those related to lysosome and phagosome pathways, exocytosis and endocytosis, cell movement and migration, T cell activation and proliferation, and immune response‐related pathways. We can infer that Tregs infiltrating the GC tumor microenvironment are in an activated state and can respond to stimuli. These cells perform immunosuppressive functions in the tumor microenvironment to facilitate the progression of GC.
Our in vitro studies showed that the TNF‐α/TNFR2 pathway enhanced the proportion of Foxp3+ Tregs in CD4+ T cells in the absence of other inductive conditions. TNFR2 is reported to be critical for maintaining robust Foxp3 expression by preventing the aberrant methylation of CpG motifs at the Foxp3 promoter and subsequent Foxp3 gene silencing. 39 TNFR2 agonists demonstrated a robust ability to increase Treg proliferation and survival and maintain the functional stability of human Treg cells, which involves canonical NF‐κB activation. 40 , 41 , 42 In addition, activation of the TNF‐α/TNFR2 signaling pathway significantly increased the production of latent TGF‐β in activated Tregs. TGF‐β released from Tregs can inhibit the proliferation and differentiation of effector T cells (Teffs) to suppress the immune response. 43 TGF‐β can also provide a shield to protect and maintain Tregs from apoptosis and destabilization. 44 However, the mechanism by which the TNF‐α/TNFR2 signaling pathway increases the production of latent TGF‐β in Tregs still needs to be further explored. Our study confirmed that the TNF‐α/TNFR2 signaling pathway enhanced the immunosuppressive function of activated Tregs, as determined by a suppression assay. After blocking TNFR2 with a neutralizing antibody, the immunosuppressive function of Tregs was significantly inhibited. These results indicate that the TNF‐α/TNFR2 signaling pathway plays an important role in the activation and proliferation, the production of inhibitory molecules and the immunosuppressive function of Tregs. Researchers have proposed the use of TNFR2 as a new target for cancer immunotherapies. 45 , 46 TNFR2 inhibitors might be safer and more targeted alternatives to immune checkpoint inhibitor cancer treatment because the expression of TNFR2 is limited to Tregs or various cancer cells. 45 Blocking TNFR2 might target abundant TNFR2+ tumor‐infiltrating Tregs and directly kill TNFR2‐expressing tumor cells. 45 Thus far, there is no evidence that TNFR2 blockade interferes with antitumor immunity. 46 Our study confirmed that in patients with advanced GC, TNFR2+ Tregs can be used as not only a prognostic marker but also a selective immunotherapeutic target.
In conclusion, our study demonstrated the effects of TNF‐a/TNFR2 in Tregs on the microenvironment and progression of GC and revealed that the TNF‐a/TNFR2 pathway can promote the proliferation and survival, immunosuppressive phenotype and function of Tregs. This finding provides not only new insight into the role of TNF‐α/TNFR2 in the regulation of Tregs in the tumor immune microenvironment but also a new theoretical basis for targeting TNFR2+ Tregs as an immunotherapeutic strategy for GC.
CONFLICT OF INTEREST
The authors declare no potential conflict of interests.
AUTHORS CONTRIBUTION
Yang Qu: Experiments, data processing, writing–original draft preparation. Xianhao Wang: Experiments, data processing. Shuai Bai: Experiments, data processing. Liling Niu: Data processing, writing–reviewing and editing. Gang Zhao: Polychromatic immunofluorescence. Yuan Yao: Flow cytometry sample sorting and detection. Bin Li: Clinical sample collection. Hui Li: Supervision, writing–reviewing and editing.
ETHICS STATEMENT
The present study was approved by the local Research Ethics Committee of the Academic Medical Center of Tianjin Cancer Hospital (approval number: EK2017002). Informed consent was obtained in all cases.
Supporting information
Appendix S1: Supporting Information
Qu Y, Wang X, Bai S, et al. The effects of TNF‐α/TNFR2 in regulatory T cells on the microenvironment and progression of gastric cancer. Int. J. Cancer. 2022;150(8):1373‐1391. doi: 10.1002/ijc.33873
Funding informationThis work was supported by the National Natural Science Foundation of China #1 under grant number 81772620 and the Tianjin Science and Technology Major Project of Chronic Diseases Prevention and Control #2 under grant number 17ZXMFSY00130.
Yang Qu, Xianhao Wang and Shuai Bai contributed equally to this paper.
DATA AVAILABILITY STATEMENT
The raw single‐cell RNA‐seq data generated in our study are available in the Gene Expression Omnibus (GEO) Genome Sequence Archive (GSA) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE172131) under accession number GSE172131. Other data that support the findings of our study are available from the corresponding author upon request.
REFERENCES
- 1. Danaei G, Vander Hoorn S, Lopez AD, Murray CJ, Ezzati M, Comparative Risk Assessment Collaborating Group . Causes of cancer in the world: comparative risk assessment of nine behavioural and environmental risk factors. Lancet. 2005;366:1784‐1793. [DOI] [PubMed] [Google Scholar]
- 2. Niccolai E, Taddei A, Prisco D, Amedei A. Gastric cancer and the epoch of immunotherapy approaches. World J Gastroenterol. 2015;21:5778‐5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359‐E386. [DOI] [PubMed] [Google Scholar]
- 4. Savage PA, Malchow S, Leventhal DS. Basic principles of tumor‐associated regulatory T cell biology. Trends Immunol. 2013;34:33‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hall BM. T cells: soldiers and spies—the surveillance and control of effector T cells by regulatory T cells. Clin J Am Soc Nephrol. 2015;10:2050‐2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frydrychowicz M, Boruczkowski M, Kolecka‐Bednarczyk A, Dworacki G. The dual role of Treg in cancer. Scand J Immunol. 2017;86:436‐443. [DOI] [PubMed] [Google Scholar]
- 7. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci. 2019;110:2080‐2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kumar P, Saini S, Prabhakar BS. Cancer immunotherapy with check point inhibitor can cause autoimmune adverse events due to loss of Treg homeostasis. Semin Cancer Biol. 2020;64:29‐35. [DOI] [PubMed] [Google Scholar]
- 9. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yan S, Zhang Y, Sun B. The function and potential drug targets of tumour‐associated Tregs for cancer immunotherapy. Sci China Life Sci. 2019;62:179‐186. [DOI] [PubMed] [Google Scholar]
- 11. Chen X, Baumel M, Mannel DN, Howard OM, Oppenheim JJ. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol. 2007;179:154‐161. [DOI] [PubMed] [Google Scholar]
- 12. Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, Oppenheim JJ. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: applicability to tumor‐infiltrating T regulatory cells. J Immunol. 2008;180:6467‐6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen X, Subleski JJ, Hamano R, Howard OM, Wiltrout RH, Oppenheim JJ. Co‐expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+ regulatory T cells in human peripheral blood. Eur J Immunol. 2010;40:1099‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J Immunol. 2013;190:1076‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He J, Li R, Chen Y, Hu Y, Chen X. TNFR2‐expressing CD4(+)Foxp3(+) regulatory T cells in cancer immunology and immunotherapy. Prog Mol Biol Transl Sci. 2019;164:101‐117. [DOI] [PubMed] [Google Scholar]
- 16. Nie Y, He J, Shirota H, et al. Blockade of TNFR2 signaling enhances the immunotherapeutic effect of CpG ODN in a mouse model of colon cancer. Sci Signal. 2018;11(511):eaan0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kampan NC, Madondo MT, McNally OM, Stephens AN, Quinn MA, Plebanski M. Interleukin 6 present in inflammatory ascites from advanced epithelial ovarian cancer patients promotes tumor necrosis factor receptor 2‐expressing regulatory T cells. Front Immunol. 2017;8:1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang M, Zhang C, Tian T, et al. Increased regulatory T cells in peripheral blood of acute myeloid leukemia patients rely on tumor necrosis factor (TNF)‐alpha‐TNF receptor‐2 pathway. Front Immunol. 2018;9:1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang T, Jiao J, Jiao X, et al. Aberrant frequency of TNFR2 Treg and related cytokines in patients with CIN and cervical cancer. Oncotarget. 2018;9:5073‐5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chang LY, Lin YC, Chiang JM, et al. Blockade of TNF‐alpha signaling benefits cancer therapy by suppressing effector regulatory T cell expansion. Onco Targets Ther. 2015;4:e1040215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yan F, Du R, Wei F, et al. Expression of TNFR2 by regulatory T cells in peripheral blood is correlated with clinical pathology of lung cancer patients. Cancer Immunol Immunother. 2015;64:1475‐1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Williams G, Mistry B, Guillard S, et al. Phenotypic screening reveals TNFR2 as a promising target for cancer immunotherapy. Oncotarget. 2016;7:68278‐68291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hamano R, Huang J, Yoshimura T, Oppenheim JJ, Chen X. TNF optimally activatives regulatory T cells by inducing TNF receptor superfamily members TNFR2, 4‐1BB and OX40. Eur J Immunol. 2011;41:2010‐2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elpek KG, Yolcu ES, Franke DD, Lacelle C, Schabowsky RH, Shirwan H. Ex vivo expansion of CD4+CD25+FoxP3+ T regulatory cells based on synergy between IL‐2 and 4‐1BB signaling. J Immunol. 2007;179:7295‐7304. [DOI] [PubMed] [Google Scholar]
- 25. Ruby CE, Yates MA, Hirschhorn‐Cymerman D, et al. Cutting edge: OX40 agonists can drive regulatory T cell expansion if the cytokine milieu is right. J Immunol. 2009;183:4853‐4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kleinewietfeld M, Puentes F, Borsellino G, Battistini L, Rötzschke O, Falk K. CCR6 expression defines regulatory effector/memory‐like cells within the CD25(+)CD4+ T‐cell subset. Blood. 2005;105:2877‐2886. [DOI] [PubMed] [Google Scholar]
- 27. Lee JJ, Kao KC, Chiu YL, et al. Enrichment of human CCR6(+) regulatory T cells with superior suppressive activity in Oral cancer. J Immunol. 2017;199:467‐476. [DOI] [PubMed] [Google Scholar]
- 28. Wei J, Zhou T, Zhang X, Tian T. SCOUT: a new algorithm for the inference of pseudo‐time trajectory using single‐cell data. Comput Biol Chem. 2019;80:111‐120. [DOI] [PubMed] [Google Scholar]
- 29. Stubbington M, Rozenblatt‐Rosen O, Regev A, Teichmann S. Single‐cell transcriptomics to explore the immune system in health and disease. Science (New York, NY). 2017;358:58‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. de Leeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3+ tumor‐infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res. 2012;18:3022‐3029. [DOI] [PubMed] [Google Scholar]
- 31. Saleh R, Elkord E. FoxP3(+) T regulatory cells in cancer: prognostic biomarkers and therapeutic targets. Cancer Lett. 2020;490:174‐185. [DOI] [PubMed] [Google Scholar]
- 32. Vlad C, Kubelac P, Fetica B, Vlad D, Irimie A, Achimas‐Cadariu P. The prognostic value of FOXP3+ T regulatory cells in colorectal cancer. J BUON. 2015;20:114‐119. [PubMed] [Google Scholar]
- 33. Miyara M, Sakaguchi S. Human FoxP3(+)CD4(+) regulatory T cells: their knowns and unknowns. Immunol Cell Biol. 2011;89:346‐351. [DOI] [PubMed] [Google Scholar]
- 34. Chen X, Oppenheim JJ. TNF‐alpha: an activator of CD4+FoxP3+TNFR2+ regulatory T cells. Curr Dir Autoimmun. 2010;11:119‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang S, Xie C, Chen Y, et al. Differential roles of TNFalpha‐TNFR1 and TNFalpha‐TNFR2 in the differentiation and function of CD4(+)Foxp3(+) induced Treg cells in vitro and in vivo periphery in autoimmune diseases. Cell Death Dis. 2019;10:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mehta AK, Gracias DT, Croft M. TNF activity and T cells. Cytokine. 2018;101:14‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wing JB, Tay C, Sakaguchi S. Control of regulatory T cells by co‐signal molecules. Adv Exp Med Biol. 2019;1189:179‐210. [DOI] [PubMed] [Google Scholar]
- 38. Chen X, Oppenheim JJ. The phenotypic and functional consequences of tumour necrosis factor receptor type 2 expression on CD4(+) FoxP3(+) regulatory T cells. Immunology. 2011;133:426‐433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tseng WY, Huang YS, Clanchy F, et al. TNF receptor 2 signaling prevents DNA methylation at the Foxp3 promoter and prevents pathogenic conversion of regulatory T cells. Proc Natl Acad Sci U S A. 2019;116:21666‐21672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lubrano di Ricco M, Ronin E, Collares D, et al. Tumor necrosis factor receptor family costimulation increases regulatory T‐cell activation and function via NF‐kappaB. Eur J Immunol. 2020;50:972‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He X, Landman S, Bauland SC, van den Dolder J, Koenen HJ, Joosten I. A TNFR2‐agonist facilitates high purity expansion of human low purity Treg cells. PLoS One. 2016;11:e0156311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Urbano PCM, Koenen H, Joosten I, He X. An autocrine TNFalpha‐tumor necrosis factor receptor 2 loop promotes epigenetic effects inducing human Treg stability in vitro. Front Immunol. 2018;9:573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miyazono K, Katsuno Y, Koinuma D, Ehata S, Morikawa M. Intracellular and extracellular TGF‐beta signaling in cancer: some recent topics. Front Med. 2018;12:387‐411. [DOI] [PubMed] [Google Scholar]
- 44. Tran DQ. TGF‐beta: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol. 2012;4:29‐37. [DOI] [PubMed] [Google Scholar]
- 45. Vanamee ES, Faustman DL. TNFR2: a novel target for cancer immunotherapy. Trends Mol Med. 2017;23:1037‐1046. [DOI] [PubMed] [Google Scholar]
- 46. Medler J, Wajant H. Tumor necrosis factor receptor‐2 (TNFR2): an overview of an emerging drug target. Expert Opin Ther Targets. 2019;23:295‐307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
The raw single‐cell RNA‐seq data generated in our study are available in the Gene Expression Omnibus (GEO) Genome Sequence Archive (GSA) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE172131) under accession number GSE172131. Other data that support the findings of our study are available from the corresponding author upon request.