

Abstract
Introduction/Aims
Currently, there are no straightforward guidelines for the clinical and diagnostic management of hyperCKemia, a frequent and nonspecific presentation in muscle diseases. Therefore, we aimed to describe our diagnostic workflow for evaluating patients with this condition.
Methods
We selected 83 asymptomatic or minimally symptomatic patients with persistent hyperCKemia for participation in this Italian multicenter study. Patients with facial involvement and distal or congenital myopathies were excluded, as were patients with suspected inflammatory myopathies or predominant respiratory or cardiac involvement. All patients underwent a neurological examination and nerve conduction and electromyography studies. The first step of the investigation included a screening for Pompe disease. We then evaluated the patients for myotonic dystrophy type II–related CCTG expansion and excluded patients with copy number variations in the DMD gene. Subsequently, the undiagnosed patients were investigated using a target gene panel that included 20 genes associated with isolated hyperCKemia.
Results
Using this approach, we established a definitive diagnosis in one third of the patients. The detection rate was higher in patients with severe hyperCKemia and abnormal electromyographic findings.
Discussion
We have described our diagnostic workflow for isolated hyperCKemia, which is based on electrodiagnostic data, biochemical screening, and first‐line genetic investigations, followed by successive targeted sequencing panels. Both clinical signs and electromyographic abnormalities are associated with increased diagnostic yields.
Keywords: creatine kinase, diagnostic workflow, hyperCKemia, muscle disease, next‐generation sequencing
Abbreviations
- AR
autosomal recessive
- CK
creatine kinase
- CPT2
carnitine palmitoyltransferase 2
- DBS
dried blood spot
- DM2
myotonic dystrophy type II
- DR
detection rate
- EFNS
European Federation of the Neurological Societies
- EMG
electromyography
- GAA
α‐1,4‐glucosidase
- HMGCR
anti‐3‐hydroxy‐3‐methylglutaryl coenzyme A reductase
- LGMD
limb‐girdle muscular dystrophy
- MLPA
multiplex ligation probe amplification
- MRC
Medical Research Council
- NADH
reduced nicotinamide adenine dinucleotide
- NGS
next‐generation sequencing
- PCR
polymerase chain reaction
- ULN
upper limit of normal
- VLCAD
very long‐chain acyl‐coenzyme A dehydrogenase
- VUS
variant of uncertain significance
1. INTRODUCTION
Muscle diseases encompass a broad range of disorders and, in most cases, they lead to motor impairment, often due to a genetic defect. 1 In some patients, the clinical features are straightforward, making it possible to establish an accurate diagnosis immediately. In most cases, however, the clinical clues are limited due to overlapping and nonspecific presentations, making it challenging to establish a precise diagnosis. 2 In cases of milder signs or symptoms, such as isolated hyperCKemia, the clinical presentation may be even less specific, complicating the diagnostic process.
With the advent of next‐generation sequencing (NGS) technology, the limitations of the previous gene‐by‐gene approach have largely been overcome. The simultaneous analysis of numerous genes is time‐saving and allows for the analysis of rare genes associated with heterogeneous phenotypes. 3 Therefore, compared with Sanger sequencing, targeted gene panels are more cost‐effective and result in a larger number of diagnoses. 4 Overall, massively parallel sequencing has improved the diagnostic approach in genetic disorders, as confirmed by several reports describing its diagnostic efficacy worldwide. 3 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20
Accordingly, the reported diagnostic rate of a massively parallel sequencing‐based approach in muscle diseases is higher than that obtained using traditional strategies, such as Sanger sequencing, 21 and an increasing number of studies using targeted sequencing panels for the molecular characterization of muscular disorders have reported detection rates ranging from 15% to 65%. 16 , 22 Target gene panels are frequently used in clinical practice, but a consensus has not yet been reached on when they should be used during the diagnostic process. For instance, it has been proposed that massively parallel sequencing should be performed before other investigations, 16 thereby endorsing target gene panels or genome sequencing as the universal first‐tier test for these heterogeneous genetic conditions. 11 , 23 However, the massively parallel sequencing approach presents some limitations because the large volume of generated data requires time for data analysis and management, and the process involves some risk of errors, including false‐negative results owing to missed repeat expansions or false‐positive results. 24 In addition, gene panels may lead to incidental findings in unrelated genes. 25
Therefore, we aimed to describe our diagnostic approach, which includes a combination of different steps for overcoming the limitations of NGS, in patients with asymptomatic or minimally symptomatic hyperCKemia.
2. METHODS
Patients were recruited from neuromuscular clinics at the IRCCS Polyclinic San Martino in Genoa, IRCCS Giannina Gaslini Institute in Genoa, AOU Hospital Federico II in Naples, and Molinette Hospital in Turin, during a period of approximately 3 years from March 2017 to January 2020. All the patients were older than 16 years of age. The inclusion criteria were presence of hyperCKemia (confirmed in two independent examinations) alone or in association with mild signs or symptoms of muscle disease. HyperCKemia was defined as a creatine kinase (CK) level greater than 1.5 times the upper limit of normal (ULN), as defined in the European Federation of the Neurological Societies (EFNS) guidelines. 26 , 27
All patients were evaluated by taking a clinical history and performing a neurological examination that included the Medical Research Council (MRC) scale for assessing muscle strength.
We included minimally symptomatic patients who presented with mild signs of muscle disease, such as mild muscle weakness (MRC ≥4) present in fewer than four muscles, rippling phenomena, and episodes of rhabdomyolysis. We also considered minimally symptomatic patients who presented with only vague symptoms, such as myalgia, undue fatigue, exercise intolerance, cramps, and stiffness. 26 We excluded patients with a family history of diagnosed myopathy or with a clear phenotype suggestive of myotonic dystrophy type 1 or facioscapulohumeral muscular dystrophy, which could easily lead to a diagnosis, as well as patients with predominant distal weakness or congenital onset myopathy. Patients exhibiting the acute or subacute onset of muscle weakness or a rapid disease course were suspected of having inflammatory myopathy and were required to undergo a specific work‐up. 28 The medical causes of hyperCKemia, such as statins or other drug‐based treatment associated with CK elevation, abnormalities in thyroid function, and other endocrine or metabolic causes, were ruled out. 29 Furthermore, we excluded patients with primary involvement of the respiratory or cardiac system.
Figure 1 summarizes the diagnostic workflow. All patients underwent a confirmatory laboratory test to evaluate the levels of CK, aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, and lactic acid, as well as a urine analysis. We classified hyperCKemia as follows: mild, less than fivefold the ULN; moderate, five‐ to tenfold the ULN; and severe, greater than tenfold the ULN. Electrodiagnostic studies, consisting of both nerve conduction studies and electromyography (EMG), were part of the routine evaluation. Electrodiagnostic data were used to stratify patients into different groups: patients with normal EMG findings; patients with EMG findings compatible with a muscular disorder; and patients with neurogenic signs. In some patients, a muscle biopsy was performed previously and did not aid in establishing a diagnosis. Based on clinical data and electrodiagnostic results, the patients were examined using our diagnostic workflow, and the previous muscle biopsy was not an exclusion criterion. In these cases, the electrodiagnostic studies were retrospective.
FIGURE 1.
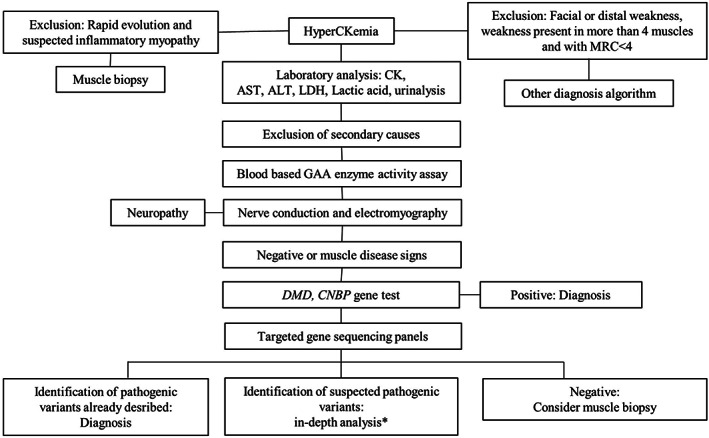
Diagnostic algorithm for hyperCKemia. Asterisk indicates evaluation of segregation study, MLPA and cDNA analysis, and muscle biopsy results to establish the pathogenicity of a VUS. Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; cDNA, complementary deoxyribose nucleic acid; CK, creatine kinase; GAA, α‐1,4‐glucosidase; LDH, lactate dehydrogenase; MLPA, multiplex ligation probe amplification; MRC, Medical Research Council
After obtaining written consent for genetic testing, we performed a dried blood spot (DBS) test for evaluating α‐glucosidase (GAA) activity. We then tested our patients for myotonic dystrophy type II (DM2) and copy number variations in the DMD gene, which is responsible for most of the Duchenne and Becker forms of muscular dystrophy. An analysis through a target gene panel was performed in patients without a diagnosis after these initial steps. The target gene panel was performed in the Laboratory of Neurogenetics and Neuroscience of Giannina Gaslini Institute (Genova) and included 20 different genes associated with metabolic myopathies or with limb‐girdle muscular dystrophy (LGMD) (AGL, ENO3, GAA, LAMP2, LDHA, PFKM, PGAM2, PGK1, PGM1, PYGM, ACADVL, CPT2, LPIN1, ANO5, LMNA, CAPN3, FKRP, FKTN, CAV3, and RYR1).
The AmpliSeq/Ion Torrent PGM technology was used to perform the NGS study, with the minimum fraction of targeted regions set at 95% to be covered by at least 20×. The median coverage was 300×. After the removal of duplicates, the paired‐end reads were mapped to the reference human genome sequence (GRch37/hg19) using IOn Reporter and CLC Bio Genomics Workbench 7.5.1 software (CLC Bio, Aarhus, Denmark). Single‐nucleotide polymorphisms (SNPs) and short deletion or insertion (indels) variants were called with CLC Bio Workbench software using a specific variant calling plugin and dbSNP147 databases. The validation of variants was performed using Sanger sequencing. In selected cases, to exclude gene microdeletions/duplications, multiplex ligation probe amplification (MLPA) was performed for specific genes (CAPN3 and GAA). Standards and guidelines for the interpretation of sequence variants were used. 30
Based on the results, we re‐evaluated the muscle biopsy slides of patients who had previously undergone muscle biopsy. The specimens were examined with routine staining (hematoxylin and eosin; modified Gomori trichrome; cytochrome c oxidase; succinate dehydrogenase; reduced nicotinamide adenine dinucleotide [NADH] dehydrogenase; adenosine triphosphatase at pH 10.4, 4.6, and 4.3; periodic acid‐Schiff; and Sudan) and reactions to myoadenylate deaminase, myophosphorylase, and phosphofructokinase. An immunofluorescence analysis was performed using antibodies against dystrophin (Dys 1, 2, and 3), sarcoglycans, α‐dystroglycan, collagen VI, caveolin, dysferlin, and merosin.
All patients or their parents or guardians provided written informed consent, and the study was approved by the local ethics committee.
3. RESULTS
This study included 83 patients, 28 (34%) of whom were females. All patients were Italian (except for 1 man of African origin) and between 16 and 71 years of age (median age, 38.5 ± 16.8 years).
Forty‐five patients (54%) had previously undergone a muscle biopsy, which did not lead to a definitive diagnosis. Thirty‐eight patients (46%) were new patients who underwent examinations based on the established protocol after being referred.
We found that 36% of the patients were affected by isolated hyperCKemia (n = 30), whereas the others had hyperCKemia associated with mild signs of muscle involvement, including mild muscle weakness (n = 9, 11%), rhabdomyolysis (n = 7, 8%), rippling (n = 1, 1%), or other symptoms, such as myalgia, cramps, exercise intolerance, or “second‐wind” phenomenon (n = 36, 43%). Further, the hyperCKemia was mild in 35%, moderate in 39% and severe in 26%. On average, our patients had hyperCKemia for at least 10 years. Patients’ demographics, clinical phenotypes, muscle biopsy results, and genetic findings are presented in Table S1.
Through the electrodiagnostic studies, one patient who was affected by axonal neuropathy was identified and excluded from further investigations, leaving our cohort with 82 patients. Needle EMG revealed alterations suggestive of myopathy in 31 patients (38%), whereas findings were normal in the remaining patients. A blood‐based GAA enzyme assay showed reduced activity in two patients, which was later confirmed by GAA gene sequencing, leading to a diagnosis of late‐onset Pompe disease in both patients. Through the first set of genetic tests, we identified a female patient carrying the Duchenne/Becker muscular dystrophy gene, three male patients carrying pathogenic variants in the DMD gene, and a patient with DM2. Thus, our first step in the investigation allowed for the diagnosis of seven patients with well‐known muscle diseases.
In the remaining 75 patients, we performed a massively parallel sequencing analysis with our target panel. A conclusive genetic diagnosis was reached in 18 patients (24%): ANO5 pathogenic variants (n = 8) including isolated hyperCKemia (n = 2), myalgia (n = 2), rhabdomyolysis (n = 2), exercise intolerance (n = 1), and mild muscle weakness causing anoctamin 5–related LGMD (LGMDR12) (n = 1); rippling muscle disease due to a CAV3 pathogenic variant (n = 1); McArdle disease (n = 3); carnitine palmitoyltransferase 2 (CPT2) deficiency (n = 1); very long‐chain acyl‐CoA dehydrogenase (VLCAD) deficiency (n = 2); and RYR1 pathogenic variants (n = 3). One of the patients with a VLCAD deficiency was diagnosed because he was found to be a carrier of two variants of the ACADVL gene: one pathogenic variant and one variant of uncertain significance (VUS) predicted to be likely pathogenic. As the patient's clinical history was consistent with a VLCAD deficiency, the patient was considered to have been diagnosed.
No variants were found in 24 patients, and at least one VUS was found in 33 patients. Among these patients, 12 were found to be carriers of a heterozygous pathogenic variant in a recessive gene (6 CAPN3, 1 ACADVL, 1 ANO5, 1 GAA, and 1 FKTN), and two were carriers of two pathogenic variants in two different recessive genes (CPT2 and ANO5 and ENO3 and ANO5). The variants are listed in Table S1. Thus, our protocol enabled us to reach a diagnosis in 25 patients, with a global detection rate (DR) of 30%. The distribution of positive diagnostic results is graphically summarized in Figure 2.
FIGURE 2.
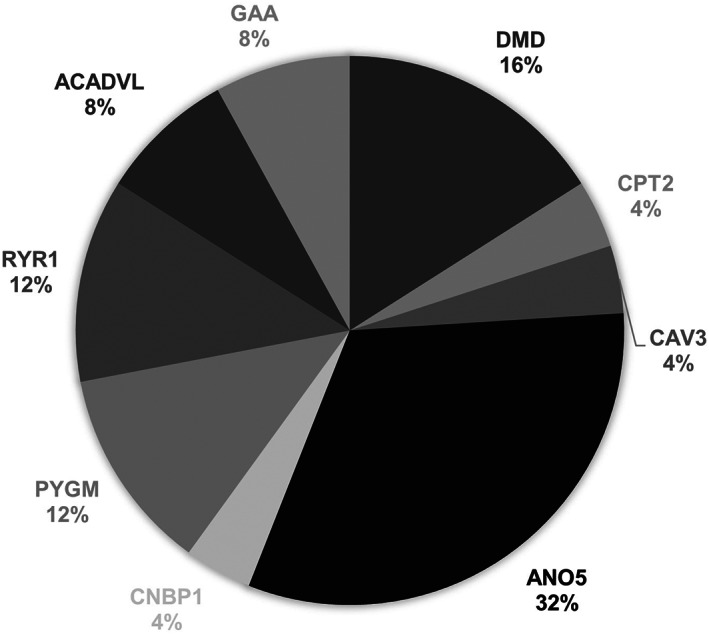
Distribution of diagnostic results. A diagnosis was achieved in 25 patients (30%): Pompe disease (n = 2); pathogenic variants of DMD (n = 4); a female patient who was a carrier and three patients with Becker muscular dystrophy), DM2 (n = 1), ANO5 pathogenic variants (n = 8), rippling muscle disease (n = 1), McArdle disease (n = 3), CPT2 deficiency (n = 1), VLCAD deficiency (n = 2), and RYR1 pathogenic variants (n = 3). Abbreviations: CPT2, carnitine palmitoyltransferase 2; DM2, myotonic dystrophy type II, VLCAD, very long‐chain acyl‐CoA dehydrogenase
We found no differences in DR (29% vs 31%) between patients who developed hyperCKemia during childhood to adolescence (<18 years of age, n = 31) and those who developed hyperCKemia during adulthood (≥18 years of age, n = 51).
When we evaluated the DR in relation to the severity of hyperCKemia, we observed a low diagnostic yield in patients with mild hyperCKemia (CK of <5× ULN, n = 29, DR = 14%). No differences in the DR were observed in patients with either severely (n = 21, DR = 48%) or moderately (n = 32, DR = 34%) elevated CK levels. The DR was higher in patients with altered EMG (n = 31, DR = 42%) than in patients with normal EMG (n = 51, DR = 24%). Among these patients, the DR was higher in those with severe hyperCKemia (Figure 3). No difference in the DR was evident in patients who presented with mild muscular signs or symptoms compared with those who presented with isolated hyperCKemia (33% vs 27%). However, in the subgroup of patients with mild muscle weakness, DR increased to 44%.
FIGURE 3.
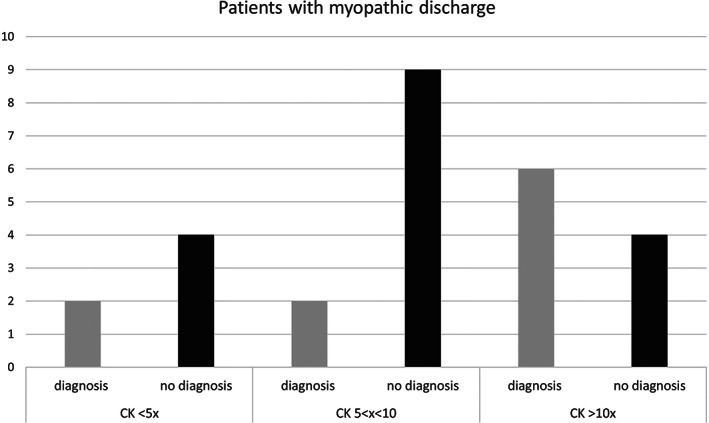
Diagnosis of patients with myopathic discharges and evaluation of the different severity levels of hyperCKemia. The abscissa represents patients with myopathic discharges subdivided based on the hyperCKemia severity. Ordinates represent the number of patients. The black columns represent patients without a diagnosis, and the gray columns represent the diagnosed patients
Previous muscle biopsies were performed in 45 patients and revealed nonspecific signs of myopathy (fiber size variability, degenerating fibers, intrafibral vacuoles, and increased connective tissue) in 32 patients and normal findings in the remaining 13 patients. Among the patients who previously underwent a muscle biopsy, a final diagnosis was established in 11 patients through genetic analysis: ANO5 pathogenic variants (n = 3) and VLCAD deficiency (n = 1) among patients with normal muscle biopsy findings, and ANO5‐related myopathies (n = 2), Pompe disease (n = 1), RYR1 pathogenic variants (n = 2), VLCAD deficiency (n = 1), and DMD pathogenic variants (n = 1) among patients whose biopsy revealed nonspecific signs of muscle damage. In the last patient, the muscle biopsy was performed in 2012 and showed only nonspecific signs of myopathy. After analyzing the genetic results, the specimens were re‐evaluated using more specific antibodies, and a Western blot revealed a reduction in the dystrophin‐related protein complex levels. The diagnosis of a patient affected by late‐onset Pompe disease was missed despite having undergone a previous muscle biopsy. The re‐evaluation of the biopsy specimens in the two patients carrying pathogenic variants in RYR1 revealed the presence of corelike areas with NADH staining. Cores are typical of RYR1 mutations but are not a specific and exclusive finding; thus, the muscle biopsy in these cases was not considered diagnostic.
Among the 38 patients who did not undergo a muscle biopsy, we established a diagnosis in 14 patients (36%): pathogenic variants in dystrophin (n = 2, male patients), DMD mutation (n = 1, female patient), DM2 (n = 1), CPT2 deficiency (n = 1), ANO5 pathogenic variants (n = 3), ring muscle disease due to CAV3 mutations (n = 1), McArdle disease (n = 3), Pompe disease (n = 1), and RYR1 pathogenic variant (n = 1). In these patients, we could easily establish a diagnosis with the help of a muscle biopsy (eg, McArdle disease or DMD mutations). At the time of this writing, none of the patients who were undiagnosed underwent a subsequent muscle biopsy.
4. DISCUSSION
Our study has introduced an algorithm for diagnosing hyperCKemia with the aim of improving the diagnostic yield in patients with this condition.
The initial steps of our protocol were focused on excluding Pompe disease, pathogenic deletions in DMD, and DM2. These conditions are relatively common, easy to diagnose with DBS in the case of Pompe disease, and, most importantly, potentially missed during the massively parallel sequencing analysis, which does not reliably detect large deletions/duplications or nucleotide repeat expansions. 7 In addition, the most common GAA pathogenic variant is the splice site mutation, c.‐32‐13T>G, 31 which can also be missed in a routine NGS analysis. Vacuoles and glycogen accumulation in both juveniles and adults with Pompe disease may not be prominent and could easily be missed 32 in a muscle biopsy, as happened in one of our patients. Indeed, with our screening process, we identified two patients with an α‐glucosidase defect successively confirmed by polymerase chain reaction (PCR) Sanger sequencing of the GAA gene, three patients with a DMD pathogenic variant, one female patient who was a DMD carrier, and one patient with DM2. Interestingly, this last patient had normal EMG findings, thus highlighting that this disease is probably underdiagnosed 33 , 34 and should be evaluated even when myotonia is not evident.
Overall, the combination of targeted PCR sequencing, MLPA, and massively parallel sequencing in the entire cohort enabled us to reach a molecular diagnosis in one third of our patients within the first year of follow‐up (25 diagnoses in 82 patients). It is important to highlight that the target panel approach alone would have been less effective, only enabling a diagnosis in 18 patients (24%).
Two studies evaluated the role of a target panel in the diagnosis of hyperCKemia. Wu 16 evaluated the diagnostic value of a target panel in a population of patients with muscle weakness (n = 135), rhabdomyolysis (n = 18), or asymptomatic hyperCKemia (n = 16). Both pathogenic and likely pathogenic variants were considered in the calculation of a DR of 36.09%, which is similar to our results. ANO5‐related myopathies were the most common diagnoses. More recently, Rubegni et al 17 described the role of NGS in patients with undiagnosed asymptomatic hyperCKemia (n = 34) or mildly symptomatic hyperCKemia (mild limb‐girdle muscle weakness [n = 19] and occasional exercise intolerance and myalgia [n = 13]). A diagnosis was reached in 33 patients (50%), among whom 11 harbored a pathogenic variant in the RYR1 gene. Both of the studies just noted highlighted the efficacy of a target panel in establishing a diagnosis, whereas our report showed its role in combination with other genetic techniques as part of a diagnostic algorithm.
The size of our target panel, comprising 20 genes, is a limitation that could be overcome by a larger target gene panel, but cost‐effectiveness and time‐efficiency make targeted sequencing panels an effective approach as the first‐line screening process for studying heterogeneous disorders, such as muscle diseases. 19 Interestingly, Thuriot et al22 recently compared the use of target gene panel testing and exome sequencing in patients with suspected muscle disorders from outpatient clinics. They noted that, in comparison with gene panel testing, exome sequencing resulted in a lower risk of missed diagnoses, while potentially increasing the diagnostic yield. At the same time, they found that almost half of the diagnoses were based on a few genes (DMD, RYR1, CAPN3, PYGM, DYSF, and FKRP), suggesting a role for target gene panels as a first‐line approach. Alternatively, whole exome sequencing, followed by filtering for defined genes, could be a valid strategy. 35 , 36 An important limitation is that the DMD gene was not included in our target gene panel, potentially missing rare DMD point mutations causing isolated hyperCKemia. 37 , 38 Moreover, it must be noted that gene panels must be periodically updated due to the frequent identification of novel causative genes. 39
In our analysis, the most frequent pathogenic variants were in the ANO5 gene, which were identified in eight patients. This finding is in line with those in previous reports, according to which ANO5‐related muscle diseases frequently manifest in patients with a long‐standing history of hyperCKemia without muscle weakness and commonly present in association with exercise intolerance, myalgia, and, more rarely, episodes of rhabdomyolysis, 40 , 41 in addition to the muscle biopsy not showing specific pathological signs. 42
Three patients presented with a pathogenic RYR1 variant, further confirming the frequency of this disorder in hyperCKemia patients. 17 All the RYR1 pathogenic variants identified in our study have been associated with malignant hyperthermia. 43 , 44 , 45 None of our patients developed malignant hyperthermia, but they are still at risk.
We found single causative variants in recessive genes in 16% of patients (12 of 75 patients who underwent panel analysis), which is also a common finding. 11 In such patients, further genetic investigations are warranted to exclude the presence of a hidden variant in the second allele. In this group, CAPN3 was the most frequently mutated gene (42%), as seen in six patients. MLPA was performed for the CAPN3 gene in all the patients with a single variant and yielded negative results. Autosomal dominant transmission has been described for CAPN3‐related myopathy. 46 , 47 , 48 , 49 Interestingly, one of the pathogenic variants identified (c.1706 T>C) was recently described by González‐Mera et al 49 as a cause for dominant calpainopathy. However, our patient (patient number 59) did not have a family history compatible with autosomal dominant transmission and had not yet undergone a muscle biopsy analysis to investigate calpain‐3 expression; therefore, at present, he remains undiagnosed. Even if several studies suggested that carriers of single heterozygous pathogenic variants in genes associated with autosomal recessive (AR) disorders can present with milder forms of the disease, further epidemiological and genetic studies on larger populations are required. 50
One of the pitfalls of the massively parallel sequencing approach is the high prevalence of VUSs. Most VUSs are variants that have not been previously reported or have been reported less frequently but without established pathogenicity studies. VUSs were found in 28% (n = 21) of the patients evaluated with parallel sequencing tests (n = 75) and were mostly in the RYR1 gene (40%). This gene is highly polymorphic and the list of associated variants continues to grow 51 ; these variants could be common in hyperCKemia cohorts. 17 However, given that the role of these variants is still unclear, all cases involving VUSs were considered unsolved.
The high prevalence of VUSs and single heterozygous pathogenic variants in AR genes highlights the importance of further investigations. Of note is that two of the three patients with pathogenic variants of RYR1 were found to have core areas in the muscle biopsy specimens after careful reanalysis of the muscle sections. In this context, muscle biopsy could be used to establish the pathogenicity of new variants. 13
Historically, muscle biopsies have played a fundamental role in the diagnostic algorithm for elevated CK levels. According to Morandi et al, 52 patients with asymptomatic hyperCKemia should be evaluated by first excluding systemic disorders and performing an electrodiagnostic study. Muscle biopsy is recommended after these steps. In our protocol, we performed a target genetic analysis before muscle biopsy. However, our protocol could lead to missed mitochondrial myopathies, which can present with isolated hyperCKemia, 53 , 54 or atypical inflammatory myopathies, such as anti–3‐hydroxy‐3‐methylglutaryl coenzyme A reductase (HMGCR) myopathy, which can be present with an asymptomatic elevation of CK for several years before the appearance of weakness. 55 Therefore, we believe that a muscle biopsy should always be performed in unsolved cases or to confirm new pathogenic variants or validate candidate genes. 56
In our patient cohort, there were no differences in the DR with regard to the age of onset of hyperCKemia (childhood or adolescence vs adulthood); this finding differs from the expected finding according to the EFNS guidelines, which suggest a higher probability of diagnosis in younger patients. 26 However, the fact that the two groups were not equally represented, as the number of adults was larger, could have influenced our results.
According to Prelle et al, 57 the probability of establishing a diagnosis in patients with hyperCKemia is positively associated with needle EMG results (P < .05; odds ratio = 2.9). Similarly, in our study, the DR in patients with EMG signs of myopathy was higher than that in patients with normal EMG results. Moreover, among these patients, the DR was higher in those with severe hyperCKemia. This observation supports the application of extensive genetic analyses, particularly in patients with a severe increase in CK levels and EMG abnormalities. The nerve conduction study was as important as a screening investigation, because it enabled us to identify a patient with neuropathy who needed to undergo a different diagnostic process.
In this study, we have described our diagnostic algorithm for asymptomatic or mildly symptomatic hyperCKemia, which enabled us to establish a diagnosis in approximately one third of our patients. The steps presented in the flowchart improved the efficacy of focused massively parallel sequencing in the diagnostic process for muscle diseases.
CONFLICT OF INTEREST
The authors declare no potential conflicts of interest.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on all of the issues involved in ethical publication, and we affirm that this report is consistent with those guidelines.
Supporting information
TABLE S1 Individual patient data
ACKNOWLEDGMENTS
The authors thank the staff of the Laboratory of Medical Genetics of the IRCCS G. Gaslini Institute and IRCCS Policlinico San Martino for performing the genetic analyses. Open Access Funding provided by Universita degli Studi di Genova within the CRUI‐CARE Agreement.
Gemelli C, Traverso M, Trevisan L, et al. An integrated approach to the evaluation of patients with asymptomatic or minimally symptomatic hyperCKemia . Muscle & Nerve. 2022;65(1):96‐104. doi: 10.1002/mus.27448
Chiara Gemelli, Monica Traverso, Chiara Fiorello, and Marina Grandis contributed equally to this work.
The preliminary data for this study, “Diagnostic Approach in Muscle Diseases in the Era of NGS Medicine,” were presented orally at the XLIX Congresso SIN, Rome, Italy, in 2018, and at the XIX Congresso Nazionale AIM, Bergamo, Italy, in 2019.
Funding information Genzyme
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.
REFERENCES
- 1. Laing NG. Genetics of neuromuscular disorders. Crit Rev Clin Lab Sci. 2012;49:33‐48. [DOI] [PubMed] [Google Scholar]
- 2. Liewluck T, Milone M. Untangling the complexity of limb‐girdle muscular dystrophies. Muscle Nerve. 2018;58:167‐177. [DOI] [PubMed] [Google Scholar]
- 3. Evilä A, Arumilli M, Udd B, Hackman P. Targeted next‐generation sequencing assay for detection of mutations in primary myopathies. Neuromuscul Disord. 2016. Published online: 2016;26:7‐15. doi: 10.1016/j.nmd.2015.10.003 [DOI] [PubMed] [Google Scholar]
- 4. Schofield D, Alam K, Douglas L, et al. Cost‐effectiveness of massively parallel sequencing for diagnosis of paediatric muscle diseases. NPJ Genom Med. 2017;2:4. doi: 10.1038/s41525-017-0006-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dai Y, Wei X, Zhao Y, et al. A comprehensive genetic diagnosis of Chinese muscular dystrophy and congenital myopathy patients by targeted next‐generation sequencing. Neuromuscul Disord. 2015. Published online: 2015;25:617‐624. doi: 10.1016/j.nmd.2015.03.002 [DOI] [PubMed] [Google Scholar]
- 6. Chae JH, Vasta V, Cho A, et al. Utility of next generation sequencing in genetic diagnosis of early onset neuromuscular disorders. J Med Genet. 2015. Published online: 2015;52:208‐216. doi: 10.1136/jmedgenet-2014-102819 [DOI] [PubMed] [Google Scholar]
- 7. Lévesque S, Auray‐Blais C, Gravel E, et al. Diagnosis of late‐onset Pompe disease and other muscle disorders by next‐generation sequencing. Orphanet J Rare Dis. 2016. Published online: 2016;11:8. doi: 10.1186/s13023-016-0390-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sevy A, Cerino M, Gorokhova S, Dionnet E, et al. Improving molecular diagnosis of distal myopathies by targeted next‐generation sequencing. J Neurol Neurosurg Psychiatry. 2016;87:340‐342. [DOI] [PubMed] [Google Scholar]
- 9. Seong MW, Cho A, Park HW, et al. Clinical applications of next‐generation sequencing‐based gene panel in patients with muscular dystrophy: Korean experience. Clin Genet. 2016. Published online: 2016;89:484‐488. doi: 10.1111/cge.12621 [DOI] [PubMed] [Google Scholar]
- 10. O'Grady GL, Lek M, Lamande SR, et al. Diagnosis and etiology of congenital muscular dystrophy: we are halfway there. Ann Neurol. 2016. Published online: 2016;80:101‐111. doi: 10.1002/ana.24687 [DOI] [PubMed] [Google Scholar]
- 11. Savarese M, di Fruscio G, Torella A, et al. The genetic basis of undiagnosed muscular dystrophies and myopathies. Neurology. 2016. Published online: 2016;87:71‐76. doi: 10.1212/WNL.0000000000002800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kitamura Y, Kondo E, Urano M, Aoki R, Saito K. Target resequencing of neuromuscular disease‐related genes using next‐generation sequencing for patients with undiagnosed early‐onset neuromuscular disorders. J Hum Genet. 2016. Published online: 2016;61:931‐942. doi: 10.1038/jhg.2016.79 [DOI] [PubMed] [Google Scholar]
- 13. Kuhn M, Gläser D, Joshi PR, et al. Utility of a next‐generation sequencing‐based gene panel investigation in German patients with genetically unclassified limb‐girdle muscular dystrophy. J Neurol. 2016. Published online: 2016;263:743‐750. doi: 10.1007/s00415-016-8036-0 [DOI] [PubMed] [Google Scholar]
- 14. Yu M, Zheng Y, Jin S, et al. Mutational spectrum of Chinese LGMD patients by targeted next‐generation sequencing. PLoS One. 2017. Published online: 2017;12:e0175343. doi: 10.1371/journal.pone.0175343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nallamilli BRR, Chakravorty S, Kesari A, et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol. 2018. Published online: 2018;5:1574‐1587. doi: 10.1002/acn3.649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu L, Brady L, Shoffner J, Tarnopolsky MA. Next‐generation sequencing to diagnose muscular dystrophy, rhabdomyolysis, and hyperCKemia. Can J Neurol Sci. 2018. Published online: 2018;45:262‐268. doi: 10.1017/cjn.2017.286 [DOI] [PubMed] [Google Scholar]
- 17. Rubegni A, Malandrini A, Dosi C, et al. Next‐generation sequencing approach to hyperCKemia. Neurol Genet. 2019. Published online: 2019;5:e352. doi: 10.1212/nxg.0000000000000352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beecroft SJ, Yau KS, Allcock RJN, et al. Targeted gene panel use in 2249 neuromuscular patients: the Australasian referral center experience. Ann Clin Transl Neurol. 2020. Published online: 2020;7:353‐362. doi: 10.1002/acn3.51002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gonzalez‐Quereda L, Rodriguez MJ, Diaz‐Manera J, et al. Targeted next‐generation sequencing in a large cohort of genetically undiagnosed patients with neuromuscular disorders in Spain. Genes. 2020. Published online: 2020;11. doi: 10.3390/genes11050539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Winder TL, Tan CA, Klemm S, et al. Clinical utility of multigene analysis in over 25,000 patients with neuromuscular disorders. Neurol Genet. 2020. Published online: 2020;6:e412. doi: 10.1212/NXG.0000000000000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ankala A, Da Silva C, Gualandi F, et al. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann Neurol. 2015. Published online: 2015;77:206‐214. doi: 10.1002/ana.24303 [DOI] [PubMed] [Google Scholar]
- 22. Thuriot F, Gravel E, Buote C, et al. Molecular diagnosis of muscular diseases in outpatient clinics: a Canadian perspective. Neurol Genet. 2020. Published online: 2020;6:e408. doi: 10.1212/NXG.0000000000000408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haskell GT, Adams MC, Fan Z, et al. Diagnostic utility of exome sequencing in the evaluation of neuromuscular disorders. Neurol Genet. 2018. Published online: 2018;4:e212. doi: 10.1212/NXG.0000000000000212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vasli N, Laporte J. Impacts of massively parallel sequencing for genetic diagnosis of neuromuscular disorders. Acta Neuropathol. 2013;125:173‐185. [DOI] [PubMed] [Google Scholar]
- 25. Fernandez‐Marmiesse A, Gouveia S, Couce ML. NGS technologies as a turning point in rare disease research, diagnosis and treatment. Curr Med Chem. 2017;25. Published online: 2017:404‐432. doi: 10.2174/0929867324666170718101946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kyriakides T, Angelini C, Schaefer J, et al. EFNS guidelines on the diagnostic approach to pauci‐ or asymptomatic hyperCKemia. Eur J Neurol. 2010;17:767‐773. [DOI] [PubMed] [Google Scholar]
- 27. Kyriakides T, Angelini C, Vilchez J, Hilton‐Jones D. European Federation of the Neurological Societies guidelines on the diagnostic approach to paucisymptomatic or asymptomatic hyperCKemia. Muscle Nerve. 2020;61:E14‐E15. [DOI] [PubMed] [Google Scholar]
- 28. Schmidt J. Current classification and management of inflammatory myopathies. J Neuromuscul Dis. 2018;5:109‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kley RA, Schmidt‐Wilcke T, Vorgerd M. Differential diagnosis of hyperCKemia. Aktuel Neurol. 2018;02:E72‐E83. [Google Scholar]
- 30. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015. Published online: 2015;17:405‐423. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peruzzo P, Pavan E, Dardis A. Molecular genetics of Pompe disease: a comprehensive overview. Ann Transl Med. 2019. Published online: 2019. doi: 10.21037/atm.2019.04.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vissing J, Lukacs Z, Straub V. Diagnosis of Pompe disease muscle biopsy vs blood‐based assays. JAMA Neurol. 2013;70:923. [DOI] [PubMed] [Google Scholar]
- 33. Young NP, Daube JR, Sorenson EJ, Milone M. Absent, unrecognized, and minimal myotonic discharges in myotonic dystrophy type 2. Muscle Nerve. 2010. Published online: 2010;41:758‐762. doi: 10.1002/mus.21615 [DOI] [PubMed] [Google Scholar]
- 34. Hilbert JE, Ashizawa T, Day JW, et al. Diagnostic odyssey of patients with myotonic dystrophy. J Neurol. 2013. Published online: 2013;260:2497‐2504. doi: 10.1007/s00415-013-6993-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Westra D, Schouten MI, Stunnenberg BC, et al. Panel‐based exome sequencing for neuromuscular disorders as a diagnostic service. J Neuromuscul Dis. 2019. Published online: 2019;6:241‐258. doi: 10.3233/JND-180376 [DOI] [PubMed] [Google Scholar]
- 36. Töpf A, Johnson K, Bates A, et al. Sequential targeted exome sequencing of 1001 patients affected by unexplained limb‐girdle weakness. Genet Med. 2020. Published online: 2020;22:1478‐1488. doi: 10.1038/s41436-020-0840-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Magri F, del Bo R, D'Angelo MG, et al. Clinical and molecular characterization of a cohort of patients with novel nucleotide alterations of the dystrophin gene detected by direct sequencing. BMC Med Genet. 2011;12. Published online: 2011. doi: 10.1186/1471-2350-12-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Neri M, Rossi R, Trabanelli C, et al. The genetic landscape of dystrophin mutations in Italy: a Nationwide study. Front Genet. 2020;11. Published online: 2020. doi: 10.3389/fgene.2020.00131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barp A, Mosca L, Sansone VA. Facilitations and hurdles of genetic testing in neuromuscular disorders. Diagnostics. 2021;11. Published online: 2021. doi: 10.3390/diagnostics11040701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vázquez J, Lefeuvre C, Escobar RE, et al. Phenotypic spectrum of myopathies with recessive anoctamin‐5 mutations. J Neuromuscul Dis. 2020 Published online. 2020;7:443‐451. doi: 10.3233/JND-200515 [DOI] [PubMed] [Google Scholar]
- 41. Papadopoulos C, LaforÊt P, Nectoux J, et al. Hyperckemia and myalgia are common presentations of anoctamin‐5‐related myopathy in French patients. Muscle Nerve. 2017. Published online: 2017;56:1096‐1100. doi: 10.1002/mus.25608 [DOI] [PubMed] [Google Scholar]
- 42. Panadés‐de Oliveira L, Bermejo‐Guerrero L, de Fuenmayor‐Fernández de la Hoz CP, et al. Persistent asymptomatic or mild symptomatic hyperCKemia due to mutations in ANO5: the mildest end of the anoctaminopathies spectrum. J Neurol. 2020. Published online: 2020;267:2546‐2555. doi: 10.1007/s00415-020-09872-7 [DOI] [PubMed] [Google Scholar]
- 43. Vukcevic M, Broman M, Islander G, et al. Functional properties of RYR1 mutations identified in Swedish patients with malignant hyperthermia and central core disease. Anesth Analg. 2010. Published online: 2010;111:185‐190. doi: 10.1213/ANE.0b013e3181cbd815 [DOI] [PubMed] [Google Scholar]
- 44. Sambuughin N, McWilliams S, de Bantel A, Sivakumar K, Nelson TE. Single‐amino‐acid deletion in the RYR1 gene, associated with malignant hyperthermia susceptibility and unusual contraction phenotype. Am J Hum Genet. 2001. Published online: 2001;69:204‐208. doi: 10.1086/321270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Galli L, Orrico A, Lorenzini S, et al. Frequency and localization of mutations in the 106 exons of the RYR1 gene in 50 individuals with malignant hyperthermia. Hum Mutat. 2006. Published online: 2006;27:830. doi: 10.1002/humu.9442 [DOI] [PubMed] [Google Scholar]
- 46. Vissing J, Barresi R, Witting N, et al. A heterozygous 21‐bp deletion in CAPN3 causes dominantly inherited limb girdle muscular dystrophy. Brain. 2016;139. Published online: 2016. doi: 10.1093/brain/aww133 [DOI] [PubMed] [Google Scholar]
- 47. Cerino M, Campana‐Salort E, Salvi A, et al. Novel CAPN3 variant associated with an autosomal dominant calpainopathy. Neuropathol Appl Neurobiol. 2020. Published online: 2020;46:564‐578. doi: 10.1111/nan.12624 [DOI] [PubMed] [Google Scholar]
- 48. Martinez‐Thompson JM, Niu Z, Tracy JA, et al. Autosomal dominant calpainopathy due to heterozygous CAPN3 C.643_663del21. Muscle Nerve. 2018. Published online: 2018;57:679‐683. doi: 10.1002/mus.25970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. González‐Mera L, Ravenscroft G, Cabrera‐Serrano M, et al. Heterozygous CAPN3 missense variants causing autosomal‐dominant calpainopathy in seven unrelated families. Neuropathol Appl Neurobiol. 2021;47. Published online: 2021:283‐296. doi: 10.1111/nan.12663 [DOI] [PubMed] [Google Scholar]
- 50. Savarese M, Di Fruscio G, Tasca G, et al. Next generation sequencing on patients with LGMD and nonspecific myopathies: findings associated with ANO5 mutations. Neuromuscul Disord. 2015. Published online: 2015;25:533‐541. doi: 10.1016/j.nmd.2015.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gonsalves SG, Ng D, Johnston JJ, et al. Using exome data to identify malignant hyperthermia susceptibility mutations. Anesthesiology. 2013. Published online: 2013;119:1043‐1053. doi: 10.1097/ALN.0b013e3182a8a8e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Morandi L, Angelini C, Prelle A, et al. High plasma creatine kinase: review of the literature and proposal for a diagnostic algorithm. Neurol Sci. 2006;27:303‐311. [DOI] [PubMed] [Google Scholar]
- 53. Rubegni A, Cardaioli E, Chini E, et al. A case of 3243A>G mutation in mtDNA presenting as apparently idiopathic hyperCKemia. J Neurol Sci. 2014. Published online: 2014;338:232‐234. doi: 10.1016/j.jns.2014.01.010 [DOI] [PubMed] [Google Scholar]
- 54. Finsterer J, Kovacs GG, Rauschka H, Ahting U. Adult, isolated respiratory chain complex IV deficiency with minimal manifestations. Folia Neuropathol. 2015. Published online: 2015;2:153‐157. doi: 10.5114/fn.2015.52412 [DOI] [PubMed] [Google Scholar]
- 55. Mohassel P, Landon‐Cardinal O, Foley AR, et al. Anti‐HMGCR myopathy may resemble limb‐girdle muscular dystrophy. Neurol Neuroimmunol Neuroinflamm. 2019. Published online: 2019;6:e523. doi: 10.1212/NXI.0000000000000523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schoser B. Diagnostic muscle biopsy: is it still needed on the way to a liquid muscle pathology? Curr Opin Neurol. 2016;29:602‐605. [DOI] [PubMed] [Google Scholar]
- 57. Prelle A, Tancredi L, Sciacco M, et al. Retrospective study of a large population of patients with asymptomatic or minimally symptomatic raised serum creatine kinase levels. J Neurol. 2002. Published online: 2002;249:305‐311. doi: 10.1007/s004150200010 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Individual patient data
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.