

Abstract
Background
Hidradenitis suppurativa (HS) is a chronic, inflammatory disease of the apocrine gland‐rich (AGR) skin region. The initial steps of disease development are not fully understood, despite intense investigations into immune alterations in lesional HS skin.
Objectives
We aimed to systematically investigate the inflammatory molecules involved in three stages of HS pathogenesis, including healthy AGR, non‐lesional HS and lesional HS skin, with the parallel application of multiple mRNA and protein‐based methods.
Methods
Immune cell counts (T cells, dendritic cells, macrophages), Th1/Th17‐related molecules (IL‐12B, TBX21, IFNG, TNFA, IL‐17, IL10, IL‐23A, TGFB1, RORC, CCL20), keratinocyte‐related sensors (TLR2,4), mediators (S100A7, S100A8, S100A9, DEFB4B, LCN2, CAMP, CCL2) and pro‐inflammatory molecules (IL1B, IL6, TNFA, IL‐23A) were investigated in the three groups by RNASeq, RT‐qPCR, immunohistochemistry and immunofluorescence.
Results
Epidermal changes were already detectable in non‐lesional HS skin; the epidermal occurrence of antimicrobial peptides (AMPs), IL‐1β, TNF‐α and IL‐23 was highly upregulated compared with healthy AGR skin. In lesional HS epidermis, TNF‐α and IL‐1β expression remained at high levels while AMPs and IL‐23 increased even more compared with non‐lesional skin. In the dermis of non‐lesional HS skin, signs of inflammation were barely detectable (vs. AGR), while in the lesional dermis, the number of inflammatory cells and Th1/Th17‐related mediators were significantly elevated.
Conclusions
Our findings that non‐lesional HS epidermal keratinocytes produce not only AMPs and IL‐1β but also high levels of TNF‐α and IL‐23 confirm the driver role of keratinocytes in HS pathogenesis and highlight the possible role of keratinocytes in the transformation of non‐inflammatory Th17 cells (of healthy AGR skin) into inflammatory cells (of HS) via the production of these mediators. The fact that epidermal TNF‐α and IL‐23 appear also in non‐lesional HS seems to prove these cytokines as excellent therapeutic targets.
Introduction
Hidradenitis suppurativa (HS) is a chronic inflammatory, debilitating skin disease characteristically localized to the apocrine gland‐rich (AGR) skin regions. Although the immune phenotype of lesional HS skin is relatively well‐characterized, little is known about the initial drivers of inflammation during the development of the disease. 1 , 2 , 3 , 4 , 5 , 6 Growing evidence suggests that healthy‐looking, non‐lesional skin represents an intermediate stage between healthy skin and lesional skin of patients with immune‐mediated skin diseases (e.g. psoriasis). 7 Non‐lesional skin displays a prediseased phenotype and provides a tissue environment to support disease manifestation. 8 , 9 The same intermediate stage may occur in HS, as a subclinical inflammatory state was observed before the formation of active lesions. 10 To demonstrate alterations that drive disease pathogenesis, we compared the immune characteristics of 3 sample groups: healthy AGR skin, asymptomatic non‐lesional HS skin (representing subclinical inflammation during HS development) and lesional HS skin.
In our previous study, differences between healthy AGR and lesional HS samples have been determined and 34 immune‐related cellular components and molecules were significantly upregulated in lesional HS samples. The majority of differentially expressed molecules were linked to T helper (Th)1/Th17 signalling pathways. 1 In another study, we detected 16 HS markers, including proteins associated with interleukin (IL)‐17 signalling, antimicrobial peptides (AMPs), S100 calcium binding proteins S100A8 and S100A9, and serpin 3 and 4, with follicular upregulation in HS‐involved skin. 11
In the current study, occurrences of the immune alterations were investigated in healthy AGR, non‐lesional HS and lesional HS samples (Table 1). We focused on those cellular components, keratinocyte (KC)‐related mediators and sensors, and Th1/Th17‐related mediators and transcription factors that were found significantly different earlier between healthy AGR and lesional HS samples (Table 2). 1 The number of CD4+ T cells, CD11c+ myeloid dendritic cells (DCs) and CD163+ macrophages and the expression of dermal Th1/Th17‐related molecules, KC‐related factors, pro‐inflammatory molecules and chemokines were investigated by RNASeq, quantitative real‐time PCR (RT‐qPCR), as well as immunohistochemistry (IHC) and immunofluorescent staining (IF) to detect precise localization too.
Table 1.
Characteristics of skin samples from apocrine gland‐rich (AGR) skin regions of healthy individuals and hidradenitis suppurativa (HS) patients
| Healthy individuals (n = 8) | ||||
|---|---|---|---|---|
| Healthy individuals | Sex | Age | Localization | |
| AGR1 | F | 48 | Axilla | |
| AGR2 | F | 60 | Axilla | |
| AGR3 | F | 38 | Axilla | |
| AGR4 | F | 45 | Axilla | |
| AGR5 | F | 55 | Axilla | |
| AGR6 | F | 57 | Axilla | |
| AGR7 | F | 41 | Axilla | |
| AGR8 | F | 60 | Axilla | |
| Median age (IQR) | 51.5 (42–59.25) | |||
| HS individuals | Sex | Age | Localization | Modified sartorius |
|---|---|---|---|---|
| HS1 | F | 29 | Axilla | 28 |
| HS2 | F | 25 | Axilla | 35 |
| HS3 | M | 30 | Axilla | 30 |
| HS4 | M | 26 | Axilla | 17 |
| HS5 | M | 16 | Axilla | 23 |
| HS6 | F | 20 | Axilla | 16 |
| HS7 | M | 31 | Axilla | 28 |
| HS8 | M | 43 | Axilla | 45 |
| HS9 | F | 22 | Axilla | 18 |
| HS10 | F | 33 | Axilla | 18 |
| Median age (IQR) | 27.5 (21.5–31.5) |
Table 2.
Comparison of immune component expression in healthy AGR skin and non‐lesional and lesional HS by RNASeq, RT‐qPCR and IHC
| Non‐lesional HS vs. healthy AGR skin | Lesional HS vs. Non‐lesional HS skin | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variable | RNAseq | qRT‐PCR | IHC Epidermis | IHC dermis | Variable | RNAseq | qRT‐PCR | IHC Epidermis | IHC dermis | ||||||||
| P‐value | FC | P‐value | FC | P‐value | FC | P‐value | FC | P‐value | FC | P‐value | FC | P‐value | FC | P‐value | FC | ||
| KC‐related mediators and sensors | KC‐related mediators and sensors | ||||||||||||||||
| S100A7 | 0.0020 | 10.18↑ | 0.0168 | 22.01 ↑ | nd | ‐ | S100A7 | 0.0367 | 4.62↑ | 0.0008 | 2.43↑ | nd | ‐ | ||||
| S100A8 | 0.0056 | 4.54↑ | 0.0389 | 7.62 ↑ | 0.0704 | 3.99 | ‐ | S100A8 | 0.0002 | 10.68↑ | <0.0001 | 5.54↑ | 0.0318 | 3.78 ↑ | ‐ | ||
| S100A9 | 0.0034 | 4.87↑ | 0.0076 | 5.32 ↑ | nd | ‐ | S100A9 | 0.0002 | 10.23↑ | <0.0001 | 4.16↑ | nd | ‐ | ||||
| DEFB4B | NS | 0.9996 | 11.99 | 0.2539 | 13.03 | ‐ | DEFB4B | 0.0002 | 109.26↑ | <0.0001 | 294.59↑ | 0.0159 | 2.81↑ | ‐ | |||
| LCN2 | NS | 0.215 | 5.7 | 0.0969 | 1.74 | ‐ | LCN2 | NS | 0.0007 | 3.33↑ | 0.0402 | 1.52↑ | ‐ | ||||
| CAMP | NS | 0.0271 | 2.55 ↑ | nd | ‐ | CAMP | NS | 0.7267 | 3.97 | nd | ‐ | ||||||
| TLR2 | NS | 0.8701 | 1.49 | nd | ‐ | TLR2 | 0.0002 | 9.15↑ | 0.0335 | 8.3 ↑ | nd | ‐ | |||||
| TLR4 | 0.0320 | −2.08 | 0.2673 | −1.76 | nd | ‐ | TLR4 | 0.0002 | 11.15↑ | 0.0007 | 5.82↑ | nd | ‐ | ||||
| IL‐1B | NS | 0.8744 | 1.7 | 0.0042 | 2.62↑ | ‐ | IL‐1B | 0.0002 | 81.66↑ | 0.0067 | 29.05↑ | >0.9999 | 1.16 | ‐ | |||
| IL‐6 | NS | 0.3816 | −13.63 | 0.2071 | 1.40 | ‐ | IL‐6 | 0.0002 | 136.13↑ | 0.0053 | 36.69↑ | >0.9999 | 2.31 | ‐ | |||
| TNFA | NS | >0.9999 | −1.24 | 0.037 | 1.79↑ | ‐ | TNFA | 0.0082 | 3.1↑ | 0.2058 | 2.36 | 0.6294 | 1.79 | ‐ | |||
| IL‐23A | NS | >0.9999 | 1.31 | 0.0454 | 1.65↑ | ‐ | IL‐23A | 0.0004 | 5.16↑ | 0.0213 | 6.24 ↑ | 0.2277 | 2.2 | ‐ | |||
| CCL2 | NS | >0.9999 | −1.55 | 0.1632 | 5.04 | ‐ | CCL2 | 0.0004 | 12.75↑ | 0.0062 | 5.17 ↑ | 0.1452 | 2.44 | ‐ | |||
| Infiltrating cells | Infiltrating cells | ||||||||||||||||
| CD4 | NS | nd | ‐ | 0.5401 | 1.44 | CD4 | 0.0026 | 5.15↑ | nd | ‐ | <0.0001 | 3.04↑ | |||||
| CD11c | NS | nd | ‐ | 0.5686 | 1.62 | CD11c | NS | nd | ‐ | <0.0001 | 4.81↑ | ||||||
| CD83 | NS | 0.3407 | −1.65 | ‐ | nd | CD83 | 0.0061 | 5.63↑ | 0.001 | 4.24 ↑ | ‐ | nd | |||||
| CD163 | NS | nd | ‐ | 0.311 | 3.52 | CD163 | 0.0002 | 5.47↑ | nd | ‐ | 0.0153 | 3.59↑ | |||||
| Th1‐related mediators and transcription factors | Th1‐related mediators and transcription factors | ||||||||||||||||
| IL‐12B | NS | 0.6175 | 2.4 | ‐ | 0.1312 | 15.6 | IL‐12B | 0.0179 | 3.17↑ | >0.9999 | 1.89 | ‐ | 0.0372 | 5.29↑ | |||
| TBX21 | NS | 0.9319 | −1.03 | ‐ | nd | TBX21 | 0.0002 | 8.77↑ | 0.1045 | 3.06 | ‐ | nd | |||||
| IFNG | NS | >0.9999 | −1.42 | ‐ | 0.0962 | 2.05 | IFNG | 0.0002 | 24.36↑ | 0.0015 | 28.70↑ | ‐ | 0.0484 | 7.12↑ | |||
| TNFA | NS | >0.9999 | −1.24 | ‐ | 0.0702 | 1.33 | TNFA | 0.0082 | 3.1↑ | 0.2058 | 2.36 | ‐ | <0.0001 | 2.39 ↑ | |||
| Th17‐related mediators and transcription factors | Th17‐related mediators and transcription factors | ||||||||||||||||
| IL‐17A | NS | >0.9999 | −1.82 | ‐ | 0.8469 | 1.06 | IL‐17A | 0.0002 | 27.03↑ | 0.0022 | 40.10↑ | ‐ | <0.0001 | 2.27↑ | |||
| IL‐10 | NS | 0.9464 | −1.58 | ‐ | 0.43 | 1.21 | IL‐10 | 0.0002 | 12.03↑ | 0.0026 | 7.66↑ | ‐ | 0.0163 | 2.23↑ | |||
| IL‐23A | NS | >0.9999 | 1.31 | ‐ | 0.53 | 1.46 | IL‐23A | 0.0004 | 5.16↑ | 0.0213 | 6.24↑ | ‐ | 0.0002 | 2.54↑ | |||
| TGFB1 | 0.0143 | −1.5↓ | 0.9452 | 1.21 | ‐ | 0.9577 | 1.31 | TGFB1 | 0.0002 | 2.55↑ | 0.0418 | 2.3↑ | ‐ | 0.0047 | 3.58↑ | ||
| RORC | NS | 0.0607 | −1.76 | ‐ | nd | RORC | 0.0002 | −6.26↓ | 0.1614 | −2.10 | ‐ | nd | |||||
| CCL20 | NS | 0.6095 | 1.45 | ‐ | 0.2676 | 1.43 | CCL20 | 0.0084 | 7.33↑ | 0.371 | 3.81 | ‐ | <0.0001 | 2.19↑ | |||
Epidermal and dermal TNF‐α and IL‐23 protein levels were quantified independently. Statistical analyses between protein and mRNA levels were determined by one‐way analysis of variance followed by Sidak’s post hoc test in case of normal data distribution or Kruskal–Wallis test followed by Dunn’s post hoc test when data distribution was not normal. Bold type indicates data with significant differences. Arrows indicate the direction of significant changes.
Abbreviations: AGR, apocrine gland‐rich; CCL, chemokine (C‐C motif) ligand; DC, dendritic cell; FC, fold change; HS, Hidradenitis suppurativa; IHC, immunohistochemistry; KC, keratinocyte; nd, not determined; NS, not significant; qRT‐PCR, quantitative real‐time PCR; RNASeq, RNA sequencing; Th, T helper.
To date, a comprehensive study investigating the above‐mentioned series of molecules in the three stages of HS pathogenesis (healthy AGR, non‐lesional HS and lesional HS skin) with the parallel application of multiple mRNA and in situ protein‐based methods has not been reported.
Materials and methods
Skin biopsies
Skin biopsies were collected from normal skin of 8 healthy individuals (samples from axillary region representing AGR) and from lesional and perilesional skin of 10 patients with HS, after obtaining written, informed consent, according to the Declaration of Helsinki principles (Table 1). The study was approved by the local ethics committee of the University of Debrecen, Hungary. Lesional HS skin was harvested from a nodule localized in a Hurley stage II area with no visible sinus/fistulae formation, as epithelial tunnels can be a source of inflammatory mediators causing tissue heterogeneity. 12 , 13 Clinically unaffected, normal‐appearing perilesional skin was obtained at least 5 cm away from lesions. HS patients were included in the study according to the following criteria: individuals (over 18 years) with clinically diagnosed moderate‐to‐severe HS with at least 6 months of disease duration. All patients were biological therapy naïve before skin biopsy, and any previous conventional systemic therapies were discontinued for 4 weeks while topical treatments were discontinued for 4 days before skin biopsy. One part of each biopsy was stored in RNAlater (Qiagen, Hilden, Germany) at −70°C until RNA isolation for RT‐PCR, another part of the biopsies was formalin‐fixed, paraffin‐embedded and used for IHC and IF.
RNA isolation, reverse transcription
All samples were homogenized in TriReagent solution (Sigma‐Aldrich, Dorset, UK) with Tissue Lyser (QIAGEN) using previously autoclaved metal beads (QIAGEN). Total RNA was isolated from the biopsies. RNA concentrations and purities were measured using a NanoDrop spectrophotometer (Thermo Scientific, Bioscience, Budapest, Hungary). RNA quality was checked using an Agilent 2100 Bioanalyser. Samples were treated with DNase I (Applied Biosystems, Foster City, CA, USA). RNA was reverse transcribed into complementary DNA (cDNA) using the High Capacity cDNA Archive Kit (Invitrogen, Life Technologies, San Francisco, CA, USA), according to the manufacturer’s instructions.
RNASeq
Normalization and export of whole transcriptomic data were performed as described previously. 14 The matrix data containing the expression levels of the study target molecules were collected manually. A heatmap was generated using Morpheus (Morpheus, https://software.broadinstitute.org/morpheus).
Real‐time quantitative PCR
qRT‐PCR measurements were carried out in triplicate using predesigned FAM‐MGB assays and TaqMan® Gene Expression Master Mix ordered from Applied Biosystems (Life Technologies). The following oligo sets were used: PPIA (Hs99999904_m1), IL17A (Hs00174383_m1), IL10 (Hs00174086_m1), IL1B (Hs00174097_m1), CD83 (Hs00188486_m1), IL6 (Hs00985639_m1), TGFB1 (Hs00171257_m1), RORC (Hs01076112_m1), IL‐12B (Hs01011518_m1), TBX21 (Hs00203436_m1), TNFA (Hs00174128_m1), IL‐23A (Hs00900829_g1), CCL2 (Hs00234140_m1), CCL20 (Hs00355476_m1), S100A7 (Hs00161488_m1), S100A8 (Hs00374264_g1), S100A9 (Hs00610058_m1), DEFB4B (hBD‐2) (Hs00175474_m1), LCN2 (Hs01008571_m1), CAMP(LL‐37) (Hs00189038_m1), TLR2 (Hs01872448_s1), TLR4 (Hs00152939_m1) and IFNγ (Hs00174143_m1). All reactions were performed with a LightCycler® 480 System (Roche, Basel, Switzerland).
Relative mRNA levels were calculated using either the comparative Ct method or based on a standard curve and normalized to the expression of PPIA mRNA.
Immunohistochemistry
For IHC analyses, freshly prepared formalin‐fixed paraffin‐embedded (FFPE) sections from HS patients and healthy control skins were used. After deparaffinizing and rehydrating the samples, endogenous peroxidase activity was eliminated with 3% H2O2 for 15 min. Subsequently, heat‐induced antigen retrieval was performed. After blocking in 1% bovine serum albumin (BSA) solution, sections were incubated with primary antibodies overnight at 4°C. Primary antibodies included human IL‐17 (rabbit polyclonal IgG [bs‐2140R]: Bioss Antibodies, Woburn, MA, USA), human IFN‐γ (rabbit polyclonal [NBP1‐19761]: Novus Biologicals, Centennial, CO, USA), human CCL2/MCP1 (mouse monoclonal IgG1 [NBP2‐22115]: Novus Biologicals), human CCL20/MIP‐3‐α (rabbit polyclonal IgG [ab9829] Abcam, Cambridge, UK), human S100A8 (rabbit polyclonal IgG [HPA024372]: Sigma‐Aldrich, Budapest, Hungary), human lipocalin/NGAL (rabbit polyclonal IgG [PA5‐32476]: Invitrogen, Carlsbad, CA, USA), human TNF‐α (mouse monoclonal IgG [SAB1404480‐100UG]: Sigma‐Aldrich), human CD4 (rabbit monoclonal IgG [ab133616]: Abcam), human CD11c (rabbit monoclonal IgG [ab52632]: Abcam), human CD163 (mouse monoclonal IgG [BM4041B]: Origene, Rockville, MD, USA), human IL‐10 (mouse monoclonal IgG [mab30207]: Covalab, Bron, France), IL‐1β (rabbit polyclonal IgG [ab9722]: Abcam), IL‐6 (mouse polyclonal IgG [SAB1400139‐50UG]: Sigma‐Aldrich), IL‐23 (rabbit polyclonal IgG [PA5‐20239]: Thermo Fisher Scientific, Waltham, MA, USA), TGFβ‐1 (mouse monoclonal IgG [MA1‐34093]: Invitrogen), IL‐12 (mouse monoclonal IgG [CF808081]: Origene) and beta 2 defensin (rabbit, polyclonal IgG [ab63982]: Abcam). Subsequently, anti‐mouse/rabbit HRP‐conjugated secondary antibodies (Dako, Santa Clara, CA, USA) were employed. Before and after incubating with antibodies, samples were washed 3 times with TBST for 5 min. Signals were detected with the Vector® ImmPACT™ NovaRED™ Kit (VECTOR Laboratories, Burlingame, CA, USA). The background was stained with methylene green. The detection of each protein was carried out on all sections in parallel to evaluate comparable protein levels. Positive, Ig and isotype controls were also used to normalize staining against all proteins. Protein levels were quantified by Pannoramic Viewer as previously described. 14
Immunofluorescent staining
Immunofluorescent staining was performed similarly on FFPE sections as described in the immunohistochemistry section till the time point of the application of secondary antibodies. After incubating with primary antibodies against TNF‐α and IL‐23, Alexa FluorTM 555 goat anti‐mouse IgG (H + L) and Alexa FluorTM 488 goat anti‐rabbit IgG (H + L) secondary antibodies were applied (Thermo Fisher Scientific).
Statistical analysis
To determine the statistical significance between the groups, one‐way analysis of variance followed by Sidak’s post hoc test (in case of normal data distribution) or Kruskal–Wallis test followed by Dunn’s post hoc test (when data distribution was not normal) was used (*P < 0.05; **P < 0.01; ***P < 0.001), focusing on comparisons of healthy AGR vs. non‐lesional HS and non‐lesional HS vs. lesional HS samples. Graphs demonstrate the mean and the corresponding 95% confidence intervals (boxes). Statistical analyses were performed using GraphPad Prism software version 8 (GraphPad Software Inc., San Diego, CA, USA).
Results
RNASeq investigation of the immune characteristics in healthy AGR, non‐lesional and lesional HS skin samples
First, we collected the normalized gene expression levels of the previously mentioned target molecules from our newly assessed RNASeq data set derived from the 3 sample groups (Fig. 1, Table 2). The details of the whole transcriptomic analysis are not the subject of our current publication (manuscript under preparation). According to the heatmap, mRNA expression of most target molecules was similar at first glance between non‐lesional HS and healthy AGR skin (Fig. 1). Among KC‐related mediators and sensors, the mRNA levels of S100A7, S100A8 and S100A9 AMPs were highly and significantly upregulated, while TLR4 was significantly lower in non‐lesional HS skin compared with healthy AGR skin (Fig. 1, Table 2). No significant differences were found in the immune cell surface markers (CD4, CD11c, CD83 and CD163) and Th1/Th17‐related mediators and transcription factors between the two groups, except for the slightly but significantly downregulated TGFB1 in non‐lesional HS compared with healthy AGR skin (Fig. 1, Table 2).
Figure 1.
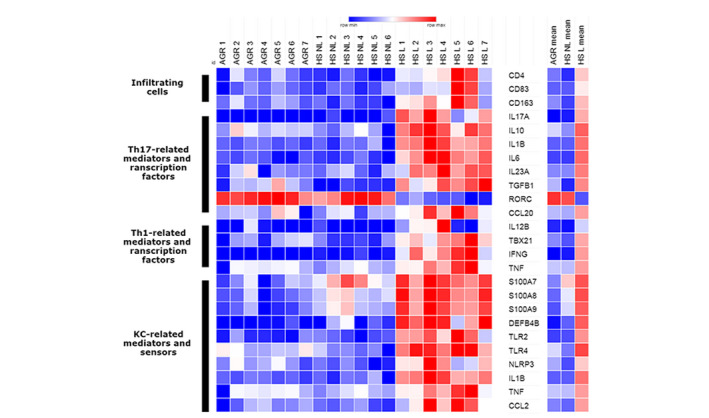
Heatmap indicating keratinocytes as a possible disease driver of HS. The heatmap was generated from the normalized gene expression levels of significantly differentially expressed target molecules derived from our RNASeq data set of healthy and non‐lesional and lesional HS samples. The highly increased expression levels in lesional HS were obvious while non‐lesional HS and healthy AGR skin seemed to be similar. KC‐related AMPs were highly upregulated even in non‐lesional HS skin, indicating keratinocytes as a possible disease driver cell. Abbreviations: KC, keratinocyte; Th, T helper.
The differential expression of target molecules was obvious when comparing lesional HS and non‐lesional HS samples (Fig. 1). The KC‐related mediators and sensors were highly and significantly upregulated in lesional HS, except for LCN2 and CAMP where the difference was not significant (Fig. 1, Table 2). Similarly, mRNA levels for cell surface markers and Th1/Th17‐related mediators and transcription factors were significantly elevated in lesional HS compared with non‐lesional HS samples, including an 8.77‐fold increase in TBX21, the master transcription factor of inflammatory Th17 and Th1 cells 15 (Fig. 1, Table 2). Notably, the expression of the master transcription factor of non‐inflammatory Th17 cells 15 RORC changed in the opposite direction and decreased 6.26‐fold in HS lesions compared with non‐lesional HS skin (Fig. 1, Table 2).
Comparison of non‐lesional HS and healthy AGR skin by RT‐qPCR and IHC
To further investigate the initial drivers of inflammation during HS development, healthy AGR and non‐lesional HS skin samples were compared using RT‐qPCR and IHC. No significant differences were detected in the number of dermal cellular components, including T cells, DCs and macrophages. In addition, the expression levels of mediators related to Th1/Th17 signalling were similar at the mRNA and protein levels (Table 2, Figs S1, S3 and S4, Supporting Information). In contrast, all investigated KC‐related AMPs were highly upregulated at the mRNA level with four of them reaching a significant level (Table 2, Fig. S2, Supporting Information). In addition, the presence of S100A8, LCN2 and human β‐defensin‐2 (hBD‐2) was prominent in the interfollicular epidermis of non‐lesional HS; however, the differences were not significant (Table 2, Fig. 2). Regarding pro‐inflammatory cytokines, beside the significantly upregulated epidermal occurrence of IL‐1β, the levels of TNF‐α and IL‐23 were significantly increased in the non‐lesional HS interfollicular epidermis (Table 2, Fig. 2) but not in the dermis.
Figure 2.
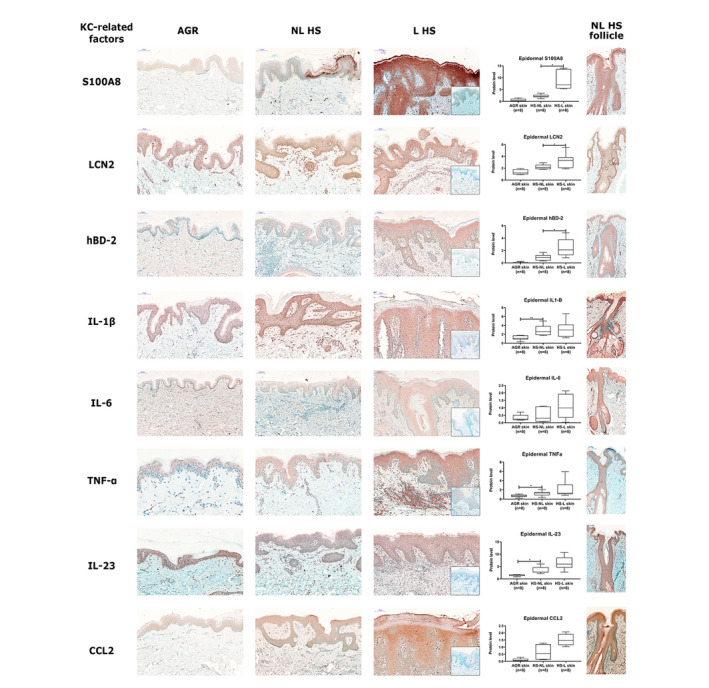
Representative images for immunostaining and epidermal quantification of KC‐related mediators in healthy AGR, non‐lesional HS and lesional HS skin samples. Protein levels were blindly analysed by Pannoramic Viewer software. Negative control staining is presented in the bottom right corner of lesional HS images. In the last column, representative images for the follicular epidermal pattern of KC‐related mediators in non‐lesional HS skin samples were demonstrated. Abbreviations: hBD‐2, human beta‐defensin‐2; IL, interleukin; LCN2, lipocalin 2; TNF, tumour necrosis factor. Size bars = 100 μm. The graphs show the median ± 95% confidence interval of measured protein levels (*P < 0.05; **P < 0.01; ***P < 0.001, as determined by one‐way analysis of variance followed by Sidak’s post hoc test in case of normal distribution or Kruskal–Wallis test followed by Dunn’s post hoc test when data distribution was not normal). Abbreviations: AGR, apocrine gland‐rich; HS, Hidradenitis suppurativa; HS‐L, HS lesional skin; HS‐NL, HS non‐lesional skin; IL, interleukin; KC, keratinocyte; TNF, tumour necrosis factor.
Regarding the staining patterns in the follicular epidermis, the above‐mentioned molecules showed strong positivity in non‐lesional HS. In addition, although protein levels in the follicular epithelia could not be quantified due to the uneven distribution of hair follicles in the skin specimens, their levels tended to increase compared with interfollicular epidermis (see also the last column in Fig. 2).
Comparison of lesional and non‐lesional HS skin by RT‐qPCR and IHC
When comparing non‐lesional and lesional HS skin samples by RT‐qPCR and IHC methods, the majority of KC‐related factors, which were already highly upregulated in non‐lesional HS (AMP mRNA levels, IL1B, TNFA and IL23) further increased in lesional HS at the mRNA level; however, the changes were not significant in the cases of TNFA and CAMP (Table 2, Fig. S2, Supporting Information). Regarding IL‐1β, IL‐23 and TNF‐α protein levels in the interfollicular epidermis, IL‐23 tended to increase while TNF‐α and IL‐1β expression remained at high levels without further increases compared with non‐lesional skin, probably indicating a decline in the speed of their expression (Table 2, Fig. 2). Parallel with these changes, in the dermis, the number of T cells, DCs and macrophages and the expression of Th1/Th17‐related mediators (CD83, IFNG, IL‐17A, IL‐10, IL23A and TGFB1) and IL‐12+, IFN‐γ+, TNF‐α+, IL‐17A+, IL‐10+, IL‐23+, TGFβ+ and CCL20+ cell counts were significantly elevated in lesional HS skin compared with non‐lesional HS skin (Table 2, Figs S1, S3 and S4, Supporting Information). Since, as a consequence of robust immune activation, the disruption of hair follicles characterizes lesional HS skin, we were unable to assess follicular staining patterns.
Immunofluorescent detection of IL‐23 and TNF‐α in HS epidermis
The unexpected early epidermal presence of IL‐23 and TNF‐α in non‐lesional HS samples led us to confirm these results with IF to demonstrate the staining pattern of these proteins more precisely in a Horseradish peroxidase‐independent system (Fig. 3). IF studies confirmed the increased IL‐23 and TNF‐α epidermal expression in non‐lesional HS samples, while their dermal occurrence was less prominent. According to our morphological findings, TNF‐α was localized mostly in the upper, apical part of the epidermis with decreasing levels towards the basal epidermal layer. In contrast, the proliferating basal KC layers showed strong cytoplasmic staining for IL‐23, while slight positivity could be detected through the whole epidermis (Fig. 3). In lesional HS, remarkable numbers of IL‐23+ and TNF‐α+ dermal infiltrate were present with similar epidermal staining.
Figure 3.
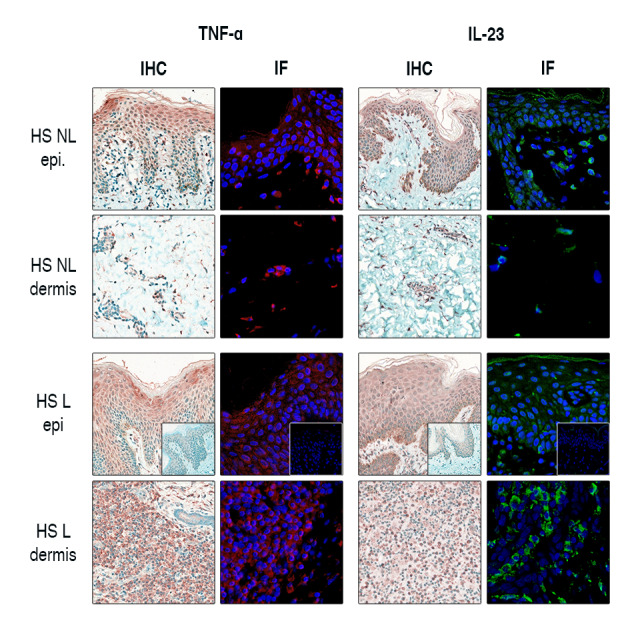
Immunofluorescent staining further confirms the epidermal presence of IL‐23 and TNF‐α in HS. The presence of IL‐23 and TNF‐α in non‐lesional HS samples was validated by the parallel application of IHC and IF staining in HS samples. IL‐23 and TNF‐α staining was prominent in the epidermis of non‐lesional HS, while their dermal occurrence was weak. In lesional HS, IL‐23+ and TNF‐α+ dermal cell counts were remarkable, while epidermal presence became even more pronounced. Negative control staining is presented in the bottom right corner of images in the third row.
Discussion
In good concordance with our previous report, 11 our present findings confirm that the activation of epidermal KCs drives immunological events in the development of HS, since all investigated AMPs and IL‐1β, IL‐23 and TNF‐α were expressed in the epidermis of non‐lesional HS skin (Fig. 4). On the other hand, the dermal production of IL‐23 and TNF‐α was significantly enhanced only in the lesional HS skin, together with significantly increased T cell, dendritic cell and macrophage influx and elevated IL‐12, IFN‐γ, IL‐17A, IL‐10, TGF‐β and CCL20 expression levels. In lesional skin, epidermal IL‐1β, IL‐23, TNF‐α and CCL2 protein levels also remained high (without further significant increase), while S100A8 and LCN2 levels significantly increased even further compared with non‐lesional HS. Altogether, these results confirm KCs as drivers of HS pathophysiology (Fig. 4).
Figure 4.
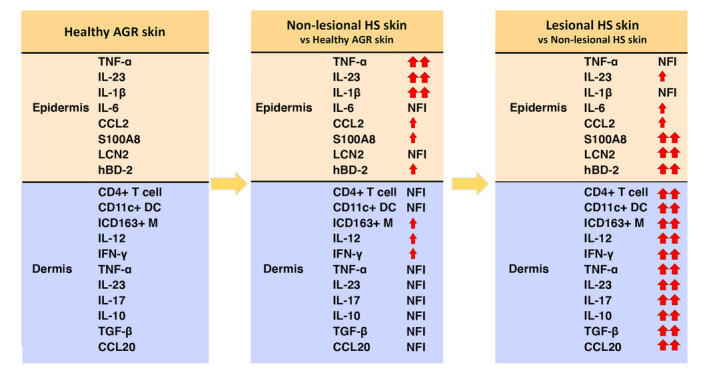
Keratinocytes can be considered as the key driver cells in HS pathophysiology According to our findings, we propose that epidermal, KC‐mediated immune activity is the first step in HS development since all investigated AMPs and the pro‐inflammatory cytokines, IL‐1β, IL‐23 and TNF‐α, are already highly expressed in non‐lesional HS skin by KCs. During the disease progression, when HS lesions develop, the dermal production of IL‐23 and TNF‐α is also significantly enhanced supplemented with an increased influx of inflammatory cells and elevated protein levels of Th1/Th17‐related cytokines and chemokines in the dermis. At the same time, the epidermal presence of IL‐1β, IL‐23 and TNF‐α proteins remains high without further significant increase, compared with non‐lesional HS. Altogether, these results confirm KCs as the key driver cells of HS pathogenesis. The data presented are based on our findings at the protein level. Small red arrows mean nonsignificant upregulation with FC ≥ 2, while duplicated bigger red arrows indicate significant upregulation. NFI means ‘no further increase’ and represents nonsignificant changes with fold change (FC) lower than 2. Abbreviations: AGR, apocrine gland‐rich; CD163+ M, CD163+ macrophage; DC, dendritic cell; HS, Hidradenitis suppurativa; HS‐L, lesional HS; HS‐NL, non‐lesional HS; IF, immunofluorescence; IHC, immunohistochemistry; KC, keratinocyte).
The prominent role of KCs in the early phases of HS lesions has already been raised by other research groups. Hotz et al. detected increased gene expression of inflammatory cytokines, chemokines, and AMPs by RT‐qPCR and Luminex assay, although their findings were mainly based on investigations of isolated outer root sheath KCs collected from HS patients. 16 Similarly, Coates et al. identified elevated AMP levels in HS lesional skin and suggested that the pathogenesis of HS may be driven by changes in AMP expression. 17 AMPs in non‐lesional and lesional HS were also investigated by other authors either at the mRNA or protein levels; however, these studies did not consequently include healthy skin samples as controls. 18 , 19 Regarding the presence of IL‐1β, TNF‐α, and IL‐23, several studies focused only on their dermal presence without highlighting their epidermal expression, while other studies applied methods that were not suitable for the in situ detection of the target molecules. 10 , 20 , 21
In our current investigation, the activity of KCs in non‐lesional HS skin was very pronounced not only in the interfollicular epidermis, but also in the follicular epidermis. The prominent role of follicular KCs in the pathogenesis of HS has been suggested in our previous study. This previous study provided the first evidence that dysregulation of the immune response of follicular KCs could be a key driver of HS, focusing on follicular AMP production in lesional and non‐lesional HS 11 (see also Fig. 2).
Our current findings are in line with the above‐mentioned studies, even more, our approach may provide evidence that molecular alterations in KCs precede dermal activation. By comparing healthy, non‐lesional HS and lesional HS skin samples and applying morphological staining methods parallel with RNASeq and RT‐qPCR, we could observe the localization of immunological changes that characterize the three stages of disease pathogenesis. In the case of psoriasis, healthy‐looking, non‐lesional skin represents an intermediate stage between healthy and lesional skin and displays a prediseased phenotype. By applying this same concept in the present study, we not only differentiated the epidermal and dermal localization of mediators in skin tissue, but also detected the possible order of immune‐related alterations during HS evolution. 7 , 8 , 9 Our findings also emphasize the importance of the combined application of RNA and in situ protein‐based methods, instead of their separate use; these results supplement each other and offer valuable comprehensive data.
In our previous study, high numbers of non‐inflammatory Th17 cells were detected in healthy AGR skin, while in lesional HS skin, the dominant presence of inflammatory type Th17 cells was demonstrated. 1 In good concordance with the previous study, our current findings suggest that the enhanced epidermal production of IL‐1β and IL‐23 may initiate the phenotypic switch of resident T cells from IL‐17/IL‐10‐producing non‐inflammatory Th17 [Th17(β)] to IL‐17/IFN‐γ‐producing inflammatory Th17 [Th17(23)] cells. This switch was also confirmed by the RNASeq data (heatmap) showing highly downregulated mRNA levels of RORC, the master transcription factor of Th17(β) cells, while the gene expression level of the master transcription factor of Th17(23) cells, TBX21, was prominently upregulated (Fig. 1). Several authors published that parallel with the high occurrence of inflammatory Th17 cells, the effector Th17/Treg ratio becomes imbalanced and increased in HS and can be considered as an important step of disease pathogenesis. 22 , 23 On the other hand, the epidermal production of TNF‐α can increase the inflammatory potential of IL‐17 since recent immunological data proved that the combined presence of TNF‐α and IL‐17 is a strong inducer of inflammatory mediators in a synergistic manner while IL‐17 alone cannot initiate inflammation effectively. 24
The importance of TNF‐α and IL‐23 cytokines is strengthened by the approval of biologicals against TNF‐α for HS therapy, while anti‐IL‐23 monoclonal antibodies are under clinical investigation. Although these therapeutics possess significant efficacy in HS patients, their effectiveness is not as strong as their superb efficacy in psoriasis patients. The cause of this phenomenon is unknown. However, in addition to the extremely high dermal TNF‐α and IL‐23 production in HS, another explanation can be their high epidermal production. A detailed investigation of the retrograde penetration of these high‐molecular‐weight (anti‐TNF: 148 kDa; anti‐IL‐23: 143 kDa) monoclonal antibodies into the epidermis in HS would be beneficial.
In summary, we performed a comprehensive analysis of healthy AGR, non‐lesional HS and lesional HS skin by multiple mRNA and protein‐based methods to better understand the pathogenesis of the disease. The results highlighted the importance of KCs as drivers of HS pathology, due to their prominent production of mediators playing key role in the switch from non‐inflammatory Th17 to inflammatory type Th17 cells and amplify inflammation.
Supporting information
Fig S1. Gene expression levels of Th1 and Th17‐related mediators in healthy AGR, non‐lesional HS, and lesional HS skin examined by qRT‐PCR
Fig S2. Gene expression levels of KC‐related mediators in healthy AGR, non‐lesional HS, and lesional HS skin examined by qRT‐PCR.
Fig S3. Representative images for immunostaining and dermal quantification of cellular components and Th1‐related mediators in healthy AGR, non‐lesional HS, and lesional HS skin samples.
Fig S4. Representative images for immunostaining and dermal quantification of Th17‐related mediators in healthy AGR, non‐lesional HS, and lesional HS skin samples.
Acknowledgement
The publication is supported by Hungarian Research Grants (NKFIH K‐128250 and NKFIH PD‐131689) and EFOP‐3.6.1‐16‐2016‐00022 projects. The project is cofinanced by the European Union and the European Social Fund. This project was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (AK and DZ) and is also supported by the ÚNKP‐20‐5 New National Excellence Program of the Ministry for Innovation and Technology (DZ and TD). The Department of Dermatology, Faculty of Medicine, University of Debrecen, Debrecen, Hungary and the Departments of Dermatology, Venereology, Allergology and Immunology, Dessau Medical Center, Dessau, Germany, are healthcare providers of the European Reference Network for Rare and Complex Skin Diseases (ERN Skin – ALLOCATE Skin group).
Z. Dajnoki and O. Somogyi contributed equally to this work.
A. Kapitány and A. Szegedi contributed equally to this work.
Conflict of interest
Regarding the submitted work the authors state no conflict of interest. CCZ reports personal fees from Idorsia, Incyte, Inflarx, Janssen, Novartis, Regeneron and UCB outside the submitted work. His departments have received grants from AbbVie, Inflarx, Novartis and UCB for his contribution as a clinical investigator.
Funding sources
The study received funding from Hungarian Research Grants (NKFIH K‐128250 and NKFIH PD‐131689), EFOP‐3.6.1‐16‐2016‐00022 project, János Bolyai Research Scholarship of the Hungarian Academy of Sciences and ÚNKP‐20‐5 New National Excellence Program of the Ministry for Innovation and Technology.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Jenei A, Dajnoki Z, Medgyesi B et al. Apocrine gland‐rich skin has a non‐inflammatory IL‐17‐related immune milieu, that turns to inflammatory IL‐17‐mediated disease in Hidradenitis Suppurativa. J Invest Dermatol 2019; 139: 964–968. [DOI] [PubMed] [Google Scholar]
- 2. Gudjonsson JE, Tsoi LC, Ma F et al. Contribution of plasma cells and B cells to hidradenitis suppurativa pathogenesis. JCI Insight 2020; 5: e139930. 10.1172/jci.insight.139930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lowe MM, Naik HB, Clancy S et al. Immunopathogenesis of hidradenitis suppurativa and response to anti‐TNF‐alpha therapy. JCI Insight 2020; 5: e139932. 10.1172/jci.insight.139932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zouboulis CC, Benhadou F, Byrd AS et al. What causes hidradenitis suppurativa ?‐15 years after. Exp Dermatol 2020; 29: 1154–1170. [DOI] [PubMed] [Google Scholar]
- 5. Ghias MH, Hyde MJ, Tomalin LE, et al. Role of the complement pathway in inflammatory skin diseases: a focus on Hidradenitis Suppurativa. J Invest Dermatol 2020; 140: 531–536 e531. [DOI] [PubMed] [Google Scholar]
- 6. Prens E, Deckers I. Pathophysiology of hidradenitis suppurativa: an update. J Am Acad Dermatol 2015; 73: S8–11. [DOI] [PubMed] [Google Scholar]
- 7. Szel E, Bozó R, Hunyadi‐Gulyás É et al. Comprehensive proteomic analysis reveals intermediate stage of non‐lesional psoriatic skin and points out the importance of proteins outside this trend. Sci Rep 2019; 9: 11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bozo R, Flink LB, Belső N et al. Could basement membrane alterations, resembling micro‐wounds at the dermo‐epidermal junction in psoriatic non‐lesional skin, make the skin susceptible to lesion formation? Exp Dermatol 2021; 30: 765–772. [DOI] [PubMed] [Google Scholar]
- 9. Nosbaum A, Dahel K, Goujon C, Nicolas JF, Mengeaud V, Vocanson M. Psoriasis is a disease of the entire skin: non‐lesional skin displays a prepsoriasis phenotype. Eur J Dermatol 2021; 31: 143–154. [DOI] [PubMed] [Google Scholar]
- 10. Kelly G, Hughes R, McGarry T et al. Dysregulated cytokine expression in lesional and nonlesional skin in hidradenitis suppurativa. Br J Dermatol 2015; 173: 1431–1439. [DOI] [PubMed] [Google Scholar]
- 11. Zouboulis CC, Nogueira da Costa A, Makrantonaki E et al. Alterations in innate immunity and epithelial cell differentiation are the molecular pillars of hidradenitis suppurativa. J Eur Acad Dermatol Venereol 2020; 34: 846–861. [DOI] [PubMed] [Google Scholar]
- 12. Navrazhina K, Garcet S, Gonzalez J, Grand D, Frew JW, Krueger JG. In‐depth analysis of the Hidradenitis Suppurativa serum proteome identifies distinct inflammatory subtypes. J Invest Dermatol 2021; 141: 2197–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Navrazhina K, Frew JW, Gilleaudeau P, Sullivan‐Whalen M, Garcet S, Krueger JG. Epithelialized tunnels are a source of inflammation in hidradenitis suppurativa. J Allergy Clin Immunol 2021; 147: 2213–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Medgyesi B, Dajnoki Z, Béke G et al. Rosacea is characterized by a profoundly diminished skin barrier. J Invest Dermatol 2020; 140: 1938–1950 e1935. [DOI] [PubMed] [Google Scholar]
- 15. Nomura T, Kabashima K, Miyachi Y. The panoply of alphabetaT cells in the skin. J Dermatol Sci 2014; 76: 3–9. [DOI] [PubMed] [Google Scholar]
- 16. Hotz C, Boniotto M, Guguin A et al. Intrinsic defect in keratinocyte function leads to inflammation in Hidradenitis Suppurativa. J Invest Dermatol 2016; 136: 1768–1780. [DOI] [PubMed] [Google Scholar]
- 17. Coates M, Mariottoni P, Corcoran DL et al. The skin transcriptome in hidradenitis suppurativa uncovers an antimicrobial and sweat gland gene signature which has distinct overlap with wounded skin. PLoS One 2019; 14: e0216249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lima AL, Karl I, Giner T et al. Keratinocytes and neutrophils are important sources of proinflammatory molecules in hidradenitis suppurativa. Br J Dermatol 2016; 174: 514–521. [DOI] [PubMed] [Google Scholar]
- 19. Bechara FG, Sand M, Skrygan M, Kreuter A, Altmeyer P, Gambichler T. Acne inversa: evaluating antimicrobial peptides and proteins. Ann Dermatol 2012; 24: 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schlapbach C, Hanni T, Yawalkar N, Hunger RE. Expression of the IL‐23/Th17 pathway in lesions of hidradenitis suppurativa. J Am Acad Dermatol 2011; 65: 790–798. [DOI] [PubMed] [Google Scholar]
- 21. van der Zee HH, de Ruiter L, Van Den Broecke DG, Dik WA, Laman JD, Prens EP. Elevated levels of tumour necrosis factor (TNF)‐alpha, interleukin (IL)‐1beta and IL‐10 in hidradenitis suppurativa skin: a rationale for targeting TNF‐alpha and IL‐1beta. Br J Dermatol 2011; 164: 1292–1298. [DOI] [PubMed] [Google Scholar]
- 22. Moran B, Sweeney CM, Hughes R et al. Hidradenitis Suppurativa is characterized by dysregulation of the Th17: treg cell axis, which is corrected by anti‐TNF therapy. J Invest Dermatol 2017; 137: 2389–2395. [DOI] [PubMed] [Google Scholar]
- 23. Melnik BC, John SM, Chen W, Plewig G. T helper 17 cell/regulatory T‐cell imbalance in hidradenitis suppurativa/acne inversa: the link to hair follicle dissection, obesity, smoking and autoimmune comorbidities. Br J Dermatol 2018; 179: 260–272. [DOI] [PubMed] [Google Scholar]
- 24. Veldhoen M. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol 2017; 18: 612–621. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Gene expression levels of Th1 and Th17‐related mediators in healthy AGR, non‐lesional HS, and lesional HS skin examined by qRT‐PCR
Fig S2. Gene expression levels of KC‐related mediators in healthy AGR, non‐lesional HS, and lesional HS skin examined by qRT‐PCR.
Fig S3. Representative images for immunostaining and dermal quantification of cellular components and Th1‐related mediators in healthy AGR, non‐lesional HS, and lesional HS skin samples.
Fig S4. Representative images for immunostaining and dermal quantification of Th17‐related mediators in healthy AGR, non‐lesional HS, and lesional HS skin samples.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.