

Abstract
Tuberculosis represents one of the ten most common courses of death worldwide and the emergence of multidrug‐resistant M. tuberculosis makes the discovery of novel anti‐tuberculosis active structures an urgent priority. Here, we show that (+)‐floyocidin B representing the first example of a novel dihydroisoquinoline class of fungus‐derived natural products, displays promising antitubercular hit properties. (+)‐Floyocidin B was identified by activity‐guided extract screening and its structure was unambiguously determined by total synthesis. The absolute configuration was deduced from a key synthesis intermediate by single crystal X‐ray diffraction analysis. A hit series was generated by the isolation of further natural congeners and the synthesis of analogs of (+)‐floyocidin B. Extensive biological and physicochemical profiling of this series revealed first structure‐activity relationships and set the basis for further optimization and development of this novel antitubercular scaffold.
Keywords: Natural products, Mycobacterium tuberculosis, total synthesis, structure-activity relationship, biological profiling
Anti‐TB hit series: Our search for new antitubercular natural products resulted in the discovery of the novel epoxyquinone (+)‐floyocidin B, which structure was unambiguously determined by total synthesis. Biological and physicochemical profiling of (+)‐floyocidin B and synthetic analogs revealed first structure‐activity relationships and set the basis for further optimization and development of this novel antitubercular scaffold.
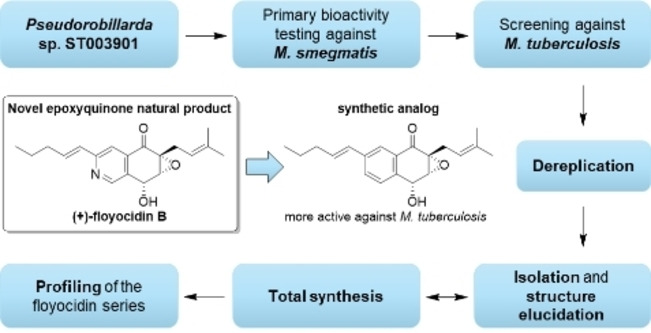
Introduction
Tuberculosis (TB) remains one of the top causes of mortality associated with infectious diseases. Despite some progress in decreasing the global incidence between 2015 and 2019 by 9 %, a key milestone of the End TB Strategy (20 % reduction between 2015 and 2020) was not met. To date, an enormous health burden with 1.4 million deaths caused by TB and 10 million new cases in 2019 persists. Compliance problems issuing from the long and complex standard treatment schedules for drug‐sensitive TB infections (a combination therapy with 4 drugs over 6 months or longer) have led to the emergence of rifampicin and multi‐drug resistant TB (RR‐TB/MDR‐TB). [1]
Therefore, the continuous search for new drugs to establish improved therapy options, either in combination with existing or newly designed schedules, is still urgently needed. Ideally, a new drug should lead to a simpler and shorter treatment and have the potential to overcome resistant TB by acting on a novel target.[ 2 , 3 ] While bacterial‐derived drugs, such as streptomycin and rifampicin, have been successfully introduced into the treatment of TB, natural products (NPs) of fungal origin have been less explored and therefore represent a promising starting point for the discovery of structurally novel lead structures acting by new antimycobacterial modes of action.
Thus, in the context of a public‐private partnership (PPP) between Sanofi and Fraunhofer we have set up a bioactivity‐guided screening campaign to identify new compounds with antibacterial activity in a collection of microbial extracts prepared from the Fraunhofer‐Sanofi strain collection. [4] Utilizing the established microfractionation screening process, fractions of the crude extract of the fungus Pseudorobillarda sp. ST003901 showed activities against the surrogate strain M. smegmatis (Msm) and the target pathogen M. tuberculosis (Mtb). Based on dereplication results, which indicated that the activity was due to the presence of potentially new compounds, a first fermentation campaign was initiated to obtain sufficient amounts of crude extract to isolate and identify the NPs responsible for the activities. Thereby, the two unknown epoxyquinones named (+)‐floyocidin A (3) and B (4) were isolated and their structures elucidated (Figure 1).
Figure 1.

Structures of isolated floyocidins A−D (3–6) and structurally related NPs (+)‐ambuic acid (1) and (−)‐avicennone C (2).
Results and Discussion
The relative stereochemistry of 3 was established by comparison with the structurally related NP (+)‐ambuic acid (1). [5] Proton and carbon chemical shifts as well as homonuclear coupling constants of the common epoxyquinone moiety are almost identical. In case of 4 the coupling constant between H1a and H2 (see Table 1) does not allow an unambiguous differentiation between the cis‐ and trans‐isomer. A direct comparison with the known NP (−)‐avicennone C (2) and its diastereomer [6] is hampered by the different aromatic system. The dihydroisoquinolinone scaffold of 4 displays an unprecedented structural motif within the epoxyquinone NP family.[ 7 , 8 ] Isolated 3 and 4 showed both weak activity against Msm with minimum inhibitory concentration (MIC) of 32–16 μg/mL and additionally (+)‐floyocidin B (4) exhibits to be active against Mtb (9.4 μg/mL). This Mtb activity qualified 4 as a starting point for an active to hit evaluation program, which includes investigations on structure‐activity relationships (SAR) and profiling with focus on parameters such as activity, pharmacokinetic properties, and toxicity.[ 1 , 2 ]
Table 1.
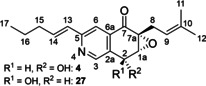
Comparison of NMR data and optical rotation of isolated and synthesized compounds 4 and 27.
|
| ||||||
|---|---|---|---|---|---|---|
|
|
Floyocidin‐B (isolated NP) |
4 (synthetic) |
27 (synthetic) |
|||
|
Position |
1H‐NMR |
13C‐NMR |
1H‐NMR |
13C‐NMR |
1H‐NMR |
13C‐NMR |
|
|
H (500 MHz)[a] |
C (125 MHz)[a] |
H (600 MHz)[a] |
C (150 MHz)[a] |
H (500 MHz)[a] |
C (125 MHz)[a] |
|
1a |
3.82, d (1.9) |
61.1 |
3.83, d (1.9) |
61.1 |
3.82, d (2.3) |
61.9 |
|
2 |
5.17, s, br |
65.3 |
5.18, s, br |
65.3 |
5.21, d (2.3) |
64.9 |
|
2a |
|
133.8 |
|
133.8 |
|
133.6 |
|
3 |
8.74, s |
150.0 |
8.75, s |
149.9 |
8.61, s |
152.5 |
|
5 |
|
157.5 |
|
157.5 |
|
158.5 |
|
6 |
7.67, s |
117.1 |
7.68, s |
117.1 |
7.72, s |
117.5 |
|
6a |
|
137.7 |
|
137.7 |
|
138.2 |
|
7 |
|
195.4 |
|
195.4 |
|
195.5 |
|
7a |
|
63.1 |
|
63.1 |
|
62.1 |
|
8 |
2.81, dd (15.2, 8.0) |
27.2 |
2.82, dd (15.2, 8.0) |
27.2 |
2.84, dd (15.2, 8.0) |
27.5 |
|
|
2.64, dd (15.2, 6.9) |
|
2.65, dd (15.2, 6.9) |
|
2.63, dd (15.2, 6.9) |
|
|
9 |
5.14, m |
117.8 |
5.14, m |
117.8 |
5.15, m |
117.8 |
|
10 |
|
137.0 |
|
137.0 |
|
137.0 |
|
11 |
1.69, s |
18.1 |
1.70, s |
18.1 |
1.69, s |
18.1 |
|
12 |
1.73, s |
26.0 |
1.74, s |
26.0 |
1.73, s |
26.0 |
|
13 |
6.55, dt (15.8, 1.5) |
130.1 |
6.56, dt (15.8, 1.5) |
130.0 |
6.56, dt (15.8, 1.5) |
130.1 |
|
14 |
6.78, dt (15.8, 7.1) |
138.5 |
6.79, dt (15.8, 7.1) |
138.5 |
6.82, dt (15.8, 7.1) |
139.0 |
|
15 |
2.27, m |
36.0 |
2.27, m |
36.0 |
2.27, m |
36.0 |
|
16 |
1.55, m |
23.2 |
1.56, m |
23.2 |
1.55, m |
23.2 |
|
17 |
0.98, t (7.4) |
14.1 |
0.99, t (7.4) |
14.1 |
0.98, t (7.4) |
14.1 |
|
7‐OH[b] |
– |
– |
|
– |
|
|
|
specific rotation |
= +160.0 (c 0.03; CHCl3) |
= +159.3 (c 0.27; CHCl3) |
= +179.3 (c 0.33; CHCl3) |
|||
|
specific rotation of ent‐series |
– |
=−206.0 (c 0.22; CHCl3) |
=−160.3 (c 1.20; CHCl3) |
|||
[a] MeOD (δ in ppm, multiplicity, J in Hz). [b] OH group not detectible in MeOD.
To gain access to additional quantities of 4 for initial profiling, a refermentation in liquid culture (8 L) was performed. Besides a significant amount of 3 (55 mg) only traces of 4 (0.8 mg) were obtained as an inseparable mixture of 3, 4, and the new compound floyocidin C (5), which structure was elucidated as a Z double bond isomer of 3. Additionally, a new derivative named floyocidin D (6) was isolated (Figure 1). [9] Due to the structural similarity of 3, 4, and 5, the same stereochemistry is assumed and fermentation kinetic studies indicated 3 as a biosynthetic precursor of 4. It can be hypothesized that these NPs are biosynthesized by a polyketide synthase (PKS).[ 7 , 10 ] In spite of efforts for fermentation optimization, e. g. by variation of media composition, pH value or concentration of oxygen, only low production titers of 4 were observed. Therefore, the focus was switched to total synthesis for structure elucidation and material supply.
In our retrosynthetic analysis we identified the attachment of the pentenyl side chain via a Suzuki reaction as a suitable late stage functionalization of chloropyridine intermediate 7. This reaction should be fully compatible with all functionalities and should allow for flexible decoration of the scaffold in a potential hit optimization program. The synthesis of 7 was developed in parallel to the synthesis of all four stereoisomers of (−)‐avicennone C (2) which served as a simplified model system [6] for route scouting. Thus, ring closure was envisaged via selective bromine‐lithium exchange of nitrile 8, which should be accessible by selective iodine‐magnesium exchange on trihalogenated pyridine 10 and reaction with epoxy aldehyde 9 followed by protecting group manipulations and oxidation to the nitrile (Scheme 1). With this synthetic strategy, all four stereoisomers of (+)‐floyocidin B (4) should be synthesized as references to elucidate the absolute stereochemistry and to support SAR investigations.
Scheme 1.

Retrosynthetic analysis of floyocidin B (4).
In spite of its structural simplicity, no references for the synthesis of 4‐bromo‐2‐chloro‐5‐iodopyridine (10) are described in literature. After investigation of different strategies to synthesize this trihalogenated pyridine, we finally identified a convenient access to 10 via a modification of the Sandmeyer reaction under anhydrous conditions, [11] which converted commercially available amine 11 into 10 in 5 g scale and excellent yields. As expected iodine‐magnesium exchange with i‐PrMgCl occurred with full chemoselectivity and trapping of the Grignard reagent with aldehyde 9 yielded the two diastereomeric alcohols 12 and 13 in good yields. 12 and 13 were acetate protected and the TBDPS group was cleaved by TBAF, which was accompanied by a spontaneous acetate migration to the primary position. Consecutive TIPS protection and saponification of the acetate resulted in primary alcohols 17 and 21, which were oxidized to nitriles 8 and 22 (Scheme 2). The reaction sequence was performed in close analogy to the total synthesis of (−)‐avicennone C [6] (2) with two significant differences: (1) a direct conversion of 12 and 13 to 17 and 21 via TIPS protection and selective TBDPS deprotection (not shown) was hampered by very low yields (<20 % over two steps) due to double deprotection of both silyl protecting groups and (2) the acetate migration in the conversion of 14 and 18 to 15 and 19 occurred without addition of a base.
Scheme 2.

Synthesis of cyclization precursors 8 and 22: (a) t‐BuONO, CuBr2, MeCN, 0 °C to rt, 97 %; (b) i‐PrMgCl, THF, −40 °C, 45 min; 9, −40 °C, 1.5 h; 49 % for 12, 42 % for 13; (c) Ac2O, pyridine, DMAP (0.1 eq.), CH2Cl2; 97 % for 14, 98 % for 18; (d) TBAF, HOAc, THF; 86 % for 15, 90 % for 19; (e) TIPSOTf, 2,6‐lutidine, CH2Cl2; (f) LiOH, THF/H2O 5 : 1; 55 % over two steps for 17, 71 % over two steps for 21; (g) TEMPO (0.2 eq.), NH4OAc, BAIB, MeCN/H2O 9 : 1; 70 % for 8, 60 % for 22.
The absolute stereochemistry of alcohol intermediate 17 was unambiguously assigned by single X‐ray crystal diffraction and the stereochemistry of the other stereoisomers was deduced thereof (Figure 2).
Figure 2.

Thermal ellipsoid plot of the molecular structure of 17. Thermal ellipsoid probability set to 50 %, only mostly occupied disorder part shown.
Selective bromine‐lithium exchange at low temperature and intramolecular reaction with the nitrile resulted in 7 and 25, respectively, after slightly acidic work‐up. While 25 was obtained in good yields and as a single product, conversion of 8 resulted in the formation of desired compound 7 and a second product, which was identified as cyclic imidate 23. This tendency was also observed for the synthesis of ent ‐7 and ent ‐25. Noteworthy, in the course of the synthesis of avicennone C this cyclization proceeded smoothly for both diastereomers. [6] Carefully monitored Suzuki reaction with trans‐1‐penten‐1‐ylboronic acid pinacol ester [12] and final TIPS deprotection yielded all four stereoisomers of floyocidin B. To compensate the loss of yield in the conversion of 8 to 7, the previously developed recycling strategy via Mitsunobu inversion and saponification of the acetate was successfully applied to the conversion of 27 to 4 (Scheme 3). [6]
Scheme 3.

Finalization of the total synthesis of floyocidin B (4) and 27: (a) n‐BuLi (1.0 eq.), THF, −100 °C, 20 min; 5 min without cooling; 26 % for 7, 81 % for 25; (b) APhos Pd G3 (0.1 eq.), Cs2CO3, trans‐1‐penten‐1‐ylboronic acid pinacol ester, 1,4‐dioxane/H2O 8 : 1, 100 °C; 51 % for 24, 84 % for 26; (c) TBAF, HOAc, THF; 60 % for 4, 93 % ee; 65 % for 27, 93 % ee; (d) PPh3, HOAc, DIAD, THF; 84 %; (e) LiOH, THF/H2O 5 : 1; 51 %.
Comparison of NMR spectra, specific rotations (Table 1), and chiral HPLC (see Supporting Information) elucidated the 1aR,2R,7aR‐isomer (4) as the correct structure of NP (+)‐floyocidin B.
To address the side product formation during Suzuki reaction of compounds 7 and 25, an alternative strategy based on an E‐selective formation of the pentenyl side chain at an early stage of the synthesis was developed. Since our first generation synthesis gave access to sufficient amounts of all four stereoisomers of (+)‐floyocidin B (4), formal avicennone C‐floyocidin B hybrids 35 and 36 were chosen as attractive targets for both route scouting and investigation of the SAR. These hybrids exhibit punctual mutations of 4 and 27 with increased lipophilicity and were expected to show higher activity on Mtb due to improved cell permeability. Central intermediate 32 was obtained starting from carboxylic acid 28 in four steps. 28 was converted to aldehyde 30 and served as starting material for a Julia‐Kocieński olefination with a high E‐selectivity. [13] Selective iodine‐magnesium exchange and reaction with aldehyde 9, protecting group manipulations, oxidation to the nitrile, cyclisation via bromine‐lithium exchange, and final TIPS deprotection were performed in close analogy to the synthesis of floyocidin B (Scheme 4, for details see Supporting Information).
Scheme 4.

Total synthesis of C‐analogs 35, ent ‐35, 36, and ent ‐36 of floyocidin‐B, exemplarily shown for one diastereomeric series: (a) (COCl)2, DMF, CH2Cl2; (b) LiBH4, THF; 93 % over two steps; (c) (COCl)2, DMSO, CH2Cl2, −78 °C, 15 min; 29, −78 °C, 90 min; DIPEA, −78 °C to 0 °C, 30 min; quant. used as a crude; (d) 31, KHMDS, THF, −55 °C, 1 h 10 min; 32, −55 °C, 1 h; 75 % (E/Z 94 : 6); (e) i‐PrMgCl, THF, −40 °C, 45 min; 9, −40 °C, 1.5 h; 42 % for 33, 42 % for 34; (f) Ac2O, pyridine, DMAP (0.1 eq.), CH2Cl2; 97 %/99 %; (g) TBAF, HOAc, THF; (h) DBU, CH2Cl2; 87 %/79 % over two steps; (i) TIPSOTf, 2,6‐lutidine, CH2Cl2; (j) LiOH, THF/H2O 5 : 1; 46 %/63 % over two steps; (k) TEMPO (0.2 eq.), NH4OAc, BAIB, MeCN/H2O 9 : 1; 48 %/41 %; (l) n‐BuLi, THF, −100 °C, 20 min; 5 min without cooling; (m) TBAF, HOAc, THF; 58 % over two steps for 35, 31 % over two steps for 36.
New NPs are often discovered as singletons or a set of compounds with a few congeners. In many cases there are only limited bioactivity data available, which are mainly related to disease‐relevant assays such as activity against a panel of cancer cell lines or pathogenic bacteria. However, from a medicinal chemistry perspective, assessment of a screening hit does not only require data with regard to potency at a target and off‐target effects, but also knowledge of key parameters concerning physicochemical and pharmacokinetic properties and potential liabilities thereof. [14] Ideally, SAR with these parameters that can be established on a small series of structurally related analogs, should allow for an efficient multi‐objective molecular optimization toward a valuable lead structure and clinical development candidate. [15]
To this end, the antibacterial activities of NP (+)‐floyocidin A (3) from isolation and in total 12 synthesized compounds (all four stereoisomers of (+)‐floyocidin B and (−)‐avicennone C [6] (2) as well as hybrids 35, ent ‐35, 36, and ent ‐36) were tested in vitro against a panel of seven indicator strains (E. coli, P. aeruginosa, M. catarrhalis, S. aureus, C. albicans, M. smegmatis, M. tuberculosis) (Table 2 and see Supporting Information). While none of the tested substrates inhibited the growth of E. coli, P. aeruginosa or C. albicans, only (+)‐floyocidin A (3) showed a weak activity against S. aureus (64 μg/mL). The stereoisomers of (+)‐floyocidin B (4), avicennone C as well as the hybrids inhibited the growth of the surrogate strain M. smegmatis with MICs down to 4 μg/mL. Both, isolated and synthetic (+)‐floyocidin B were active against M. tuberculosis H37Rv with a MIC of 9.4 μg/mL. The C‐analogs 35, ent ‐35, 36, and ent ‐36 displayed activity against M. tuberculosis H37Rv in the same range, pointing toward a positive correlation between lipophilicity and activity. Furthermore, the data indicate that the presence of a pentenyl side chain and the syn‐configuration of the epoxide and hydroxyl group seem to be important for the antibacterial activity. Unfortunately, cytotoxic effects (TC50) in both THP‐1 and HepG2 were observed in the same order of magnitude as the antitubercular activity, but the safety index may be improved with additional derivatives (see for example 36, which is three times less cytotoxic than 4 but still displays the same activity against M. tuberculosis). Undoubtedly, identification of the mode of action of both, the antibacterial and off‐target effects should be of high priority in further investigation.
Table 2.
Bioactivity data of NPs and SAR‐compounds against M. smegmatis ATCC607 (Msm) and M. tuberculosis H37Rv (Mtb).
|
| |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
MIC [μg/mL] |
||||||||||||
|
|
4 |
27 |
ent‐4 |
ent‐27 |
35 |
36 |
ent‐35 |
ent‐36 |
37 |
38 |
ent‐37 |
2 |
3 |
|
Msm |
64 |
16 |
32 |
16 |
32 |
4 |
32 |
8–4 |
128–64 |
NA |
128–64 |
NA |
32 |
|
Mtb |
9.4 |
NA |
NA |
NA |
9.4 |
9.4 |
9.4 |
9.4 |
NA |
NA |
NA |
NA |
NA |

[a] NA=not active.
The floyocidins and their analogs displayed a relatively high lipophilicity (log D from 2.8 to 4.7), which has turned out to be favorable in other compound optimization programs for a high compound exposure in tissues relevant to tuberculosis (lung, spleen).[ 14 , 16 ] The observed lipophilicity range, together with molecular weights around 300 Da and a low number of hydrogen bond donors and acceptors should translate into a good oral bioavailability (a requisite for TB therapy schemes), which is further underlined by the prediction of high human oral resorption through excellent permeability (Papp) values larger than 40 ⋅ 10−7 cm/s in the Caco‐2 intestinal epithelial barrier model [17] (Table 3). Since no increase of the apical to basolateral transport rate in the presence of elacridar was observed in the Caco‐2 assay, the permeability of the compounds is not affected by the PgP efflux transporter. [18] Metabolism was studied in liver microsomes. [14] While the compounds showed moderate to low clearance in human microsomes corresponding to an acceptable stability, rapid metabolization was observed in mice microsomes. This finding may hamper further investigations in mice models of tuberculosis infection and should be addressed early on in compound optimization programs after UPLC‐HRMS and/or NMR‐based identification of the metabolic soft spots.
Table 3.
Profiling data of NPs and SAR‐compounds.
|
|
Log D [d] |
Microsomal stability[d] |
Caco‐2 permeability[d] |
Cytotoxicity[d] |
|||
|---|---|---|---|---|---|---|---|
|
|
pH 7.4 |
CLint [μL/min/mg protein] (t1/2 [min]) |
Papp [10−7 cm s−1] (recovery [%]) |
TC50 [μM] |
|||
|
|
Human |
Mouse |
A2B |
+Elacridar |
THP1 |
HepG2 |
|
|
3 |
4.11 |
[c] |
[c] |
[b] |
[c] |
[c] |
[c] |
|
4 |
4.01 |
72.7 (19.1) |
349 (3.97) |
41.1 (10.1) |
56.2 (14.2) |
5.84 |
4.5 |
|
27 |
4.14 |
95.8 (14.5) |
1500 (0.924) |
117 (42.1) |
87.7 (38.5) |
10.6 |
10.7 |
|
ent‐4 |
3.97 |
26.9 (51.6) |
271 (5.12) |
73.5 (23.4) |
46.7 (28.7) |
8.78 |
5.97 |
|
ent‐27 |
4.16 |
44.3 (31.3) |
764 (1.81) |
40.5 (8.69) |
64.4 (15.3) |
11.5 |
10.4 |
|
35 |
4.7 |
45.5 (30.5) |
1040 (1.33) |
135 (76.0) |
181 (29.6) |
12.9 |
9.39 |
|
36 |
4.73 |
39.7 (34.9) |
259 (5.36) |
[b] (16.1) |
[b] (19.9) |
15.1 |
16.1 |
|
ent‐35 |
4.72 |
40.5 (34.3) |
561 (2.47) |
162 (37.2) |
[b] (46.1) |
12.9 |
14.7 |
|
ent‐36 |
4.74 |
29.9 (46.3) |
721 (1.92) |
8.55 (14.3) |
12.4 (12.0) |
18.2 |
11.3 |
|
37 |
2.82 |
54.6 (25.4) |
[a] |
326 (74.8) |
279 (80.9) |
27.2 |
30 |
|
38 |
2.84 |
76.0 (18.2) |
1070 (1.29) |
620 (127) |
331 (132) |
30 |
9.49 |
|
ent‐37 |
2.82 |
73.5 (18.9) |
370 (3.75) |
360 (81.3) |
300 (74.6) |
25.2 |
30 |
|
2 |
2.86 |
150 (9.26) |
[a] |
407 (86.7) |
393 (83.4) |
30 |
>30 |
[a] Test compound not detectable by 5 minutes. Unable to determine CLint. [b] No data obtained. [c] Not determined. [d] For measurement details and standard deviations of biological data see Supporting Information.
Conclusion
In conclusion, the floyocidins have been characterized as a novel NP scaffold for the potential lead discovery and optimization of new anti‐tuberculosis drugs. Besides isolation, a flexible and scalable synthetic entry into the floyocidin B stereoisomers in combination with single crystal X‐ray diffraction analysis was key for structure elucidation. The delivery of sufficient amounts of both, the NPs as well as a series of synthetic analogs that allowed for extensive biological profiling, provided insight into first SAR not only regarding antibacterial and cytotoxic activities but also related to pharmacokinetic properties. In this context, metabolic lability and off‐target effects were identified as key liabilities to be addressed in forthcoming multi‐objective compound optimization programs toward clinical development candidates. Further studies of SAR are required to increase the therapeutic window but in particular combination therapy as practiced in the current standard of care may give an opportunity for the introduction of a floyocidin‐based drug to achieve synergistic effects, overcome resistance and achieve a more favorable therapeutic window.
Experimental Section
General methods: All chemicals and solvents/anhydrous solvents were commercially supplied and used without further purification. For heating of reaction mixtures, aluminum flask carriers in different sizes from IKA were used. Reactions were monitored using thin layer chromatography (TLC) or using one of the following LC‐MS systems: 1100 HPLC (Agilent) with DAD and ELSD equipped with MSD (Agilent) ESI quadrupole MS, 1100 HPLC (Agilent) with DAD equipped with Amazon (Bruker) ESI trap MS, 1290 UPLC (Agilent) with DAD or ELSD equipped with micrOTOF (Bruker) ESI TOF MS or 1290 UPLC (Agilent) with DAD and ELSD equipped with maXis II (Bruker) ESI TOF MS. TLC was performed on pre‐coated silica gel glass plates (Merck TLC Silica gel 60 F254) and compounds were detected under UV light (254 nm) and/or by staining with an aqueous solution of KMnO4 with K2CO3 and NaOH or an aqueous solution of phosphomolybdic acid, cerium(IV) sulfate, and H2SO4 followed by heating with a heat gun. Products were purified by flash column chromatography using silica gel 60 M (Macherey‐Nagel) or by using an automated flash column chromatography system (Biotage® SP4 with ISOLUTE® Flash SI II) equipped with ISOLUTE® Flash SI II columns of different sizes from Biotage, PF‐15SIHC flash columns of different sizes from Interchim or Götec GX flash columns of different sizes from Götec‐Labortechnik (eluents are given in parentheses). The HPLC purifications were performed with a semi‐preparative 1100 HPLC system from Agilent or a semi‐preparative 1200 HPLC system from Agilent using RP columns (flow rate: 3 mL/min; columns and eluents are given in parentheses). The product containing fractions were identified using LC–MS, pooled and concentrated in vacuo or freeze‐dried. NMR spectra were recorded on a Bruker AVANCE II WB spectrometer (400 MHz), an AVANCE III HD spectrometer (400 MHz), an AVANCE III spectrometer (500 MHz) equipped with a TCI CryoProbe or an AVANCE III HD spectrometer (600 MHz) with CDCl3 or CD3OD as solvent with chemical shifts (δ) quoted in parts per million (ppm) and referenced to the solvent signal (δ1H/13C: CDCl3 7.26/77.16, CD3OD 3.31/49.00) or TMS (δ=0.00 ppm in CDCl3). Assignment was confirmed based on COSY, HSQC, HMBC, ROESY, and NOESY correlations. The absolute configuration of compound 17 was assigned by X‐ray diffraction analysis (for details regarding solvent and method for crystal growth see synthetic procedure). High resolution mass spectrometry was performed on the maXis II (Bruker) ESI TOF MS. Specific rotation was measured by a polarimeter (P 3000 series) from Krüss. Synthetic steps are described in detail for intermediates leading to the NP (+)‐floyocidin B (4) and the corresponding stereoisomers are listed afterward.
Initial fermentation and isolation for structure elucidation: Strain ST003901 from the Fraunhofer‐Sanofi strain collection [4] was cultivated on solid agar in 10×300 mL Erlenmeyer flasks filled with 75 mL medium comprising 20 g/L malt extract, 2 g/L yeast extract, 10 g/L glucose, 0.5 g/L (NH4)2HPO4, H2O (pH=6.0 before autoclaving). After incubation for 11 days as standing cultures at 25 °C, solid agar from fermentation was homogenized in water and extracted with 2x volume of ethyl acetate by stirring overnight. Evaporation of the ethyl acetate layer yielded 157 mg of extract. Pre‐purification was performed by semi‐preparative HPLC (column: Synergy Fusion‐RP 80 Å, 4 μm, 250×10 mm from Phenomenex; Eluent A: H2O+0.1 % formic acid, Eluent B: MeCN+0.1 % formic acid, 50 % B at 0 min, 50 % B at 5 min, 90 % B at 25 min, and 100 % B at 26 min). Elution of target compounds 3 and 4 was monitored using LC‐MS and fractionation was performed. Fractions containing the target compounds where pooled and further purified by HPLC separation (column: Jupiter® 4 μm Proteo 90 Å, 250×10 mm from Phenomenex; isocratic with 65 % MeCN in H2O+0.1 % formic acid). Two separate fractions yielded (+)‐floyocidin A (3, 1.3 mg) and (+)‐floyocidin B (4, 0.6 mg) as colorless solids.
8 L refermentation: Inoculation of 80 x 300 mL Erlenmeyer flasks filled with 100 mL medium comprising 20 g/L malt extract, 2 g/L yeast extract, 10 g/L glucose, 0.5 g/L (NH4)2HPO4, H2O (pH=6.0 before autoclaving) was performed with 5 % (v/v) pre‐culture of strain ST003901. After 13 days incubation at 25 °C with shaking at 180 rpm, the first 4 L of the fermentation were harvested, the remaining culture after 17 days. Cells and culture filtrate were separated by centrifugation and filtration. The filtrates were extracted with ethyl acetate (8 L) and evaporation of the ethyl acetate layer yielded 646 mg of extract. Cells were freeze‐dried and extracted with MeOH (3×250 mL per 4 L fermentation volume). The MeOH extract was concentrated under reduced pressure and the crude extract (6.56 g) was subsequently fractionated by stepwise elution (0 %, 20 %, 40 %, 60 %, 80 %, and 100 % MeOH in H2O) over a column (4 x 30 cm) with Amberlite® XAD‐7/XAD‐16 N (1 : 1, 125 g/125 g). Fractions containing target compounds 3 and 4 (80 % and 100 % MeOH in H2O) were merged, dried under reduced pressure, and pre‐purified together with dried ethyl acetate extract by semi‐preparative HPLC (column: Synergy Fusion‐RP 80 Å, 4 μm, 250 x 10 mm from Phenomenex; Eluent A: H2O+0.1 % formic acid, Eluent B: MeCN+0.1 % formic acid, 55 % B at 0 min, 55 % B at 5 min, 65 % B at 25 min, and 100 % B at 26 min). Fractions containing the target compounds where pooled and further purified by HPLC (column: Nucleodur C18 Gravity SB, 3 μm, 250×10 mm; isocratic with 57 % MeCN in H2O+0.1 % formic acid). Besides further (+)‐floyocidin A (3) (55 mg), a mixture of floyocidin C (5)/floyocidin B (4)/ floyocidin A (3) (0.8 mg, 2.4 : 1.2 : 1) was obtained. During the purification of nearly pure fractions with floyocidin A by semi‐preparative HPLC (column: Nucleodur C18 Gravity SB, 3 μm, 250×10 mm; isocratic with 57 % MeCN in H2O+0.1 % formic acid), formation of a further signal was observed. The structure was elucidated by NMR analysis and the compound was named floyocidin D (6). [19]
(+)‐Floyocidin A [20] / (1R,5R,6R)‐3‐((1E,3E)‐Hepta‐1,3‐dien‐1‐yl)‐5‐hydroxy‐4‐(hydroxymethyl)‐1‐(3‐methylbut‐2‐en‐1‐yl)‐7‐oxabicyclo[4.1.0]hept‐3‐en‐2‐one (3): 1 H‐NMR (MeOD, 500 MHz): δ=6.48 (dd, 1H, J=15.7, 10.5 Hz, Cq‐CH=CH), 6.24 (d, 1H, J=15.7 Hz, Cq‐CH=CH), 6.13 (dd, 1H, J=15.2, 10.4 Hz, Cq‐CH=CH‐CH), 5.80 (dt, 1H, J=15.2, 7.1 Hz, CH2‐CH2‐CH), 5.07 (m, 1H, Me2C=CH), 4.77 (s, br, 1H, CH‐OH), 4.48 (d, 1H, J=12.9 Hz, CH 2‐OH), 4.41 (d, 1H, J=12.9 Hz, CH 2‐OH), 3.67 (d, 1H, J=2.8 Hz, epoxy‐CH), 2.71 (dd, 1H, J=15.2, 7.9 Hz, Me2C=CH‐CH 2), 2.48 (dd, 1H, J=15.2, 6.9 Hz, Me2C=CH‐CH 2), 2.10 (m, 2H, CH3‐CH2‐CH 2), 1.70 (s, 3H, E‐CH 3), 1.65 (s, 3H, Z‐CH 3), 1.44 (m, 2H, CH3‐CH 2), 0.92 (t, 3H, J=7.4 Hz, CH 3‐CH2); 1 H‐NMR (CDCl3, 400 MHz): δ=6.48 (dd, 1H, J=15.7, 10.5 Hz, Cq‐CH=CH), 6.15 (d, 1H, J=15.7 Hz, Cq‐CH=CH), 6.09 (dd, 1H, J=15.2, 10.5 Hz, Cq‐CH=CH‐CH), 5.81 (dt, 1H, J=15.1, 7.0 Hz, CH2‐CH2‐CH), 5.02 (m, 1H, Me2C=CH), 4.84 (s, br, 1H, CH‐OH), 4.66–4.50 (m, 2H, J=12.9 Hz, CH 2‐OH), 3.72 (d, 1H, J=2.8 Hz, epoxy‐CH), 2.78 (dd, 1H, J=15.4, 7.9 Hz, Me2C=CH‐CH 2), 2.52 (dd, 1H, J=15.4, 6.7 Hz, Me2C=CH‐CH 2), 2.08 (m, 2H, CH3‐CH2‐CH 2), 1.70 (s, 3H, E‐CH 3), 1.63 (s, 3H, Z‐CH 3), 1.42 (m, 2H, CH3‐CH 2), 0.90 (t, 3H, J=7.4 Hz, CH 3‐CH2); 13 C‐NMR (MeOD, 125 MHz): δ=196.7 (C=O), 150.2 (OH‐CH2‐C q), 138.0 (Cq‐CH=CH), 138.0 (CH2‐CH2‐CH), 136.5 (C qMe2), 132.3 (Cq‐CH=CH‐CH), 131.8 (C q‐CH=CH), 122.9 (Cq‐CH=CH), 118.2 (CH=CMe2), 66.3 (CH‐OH), 61.9 (epoxy‐C q), 60.5 (epoxy‐CH), 60.0 (CH2‐OH), 36.0 (CH3‐CH2‐CH2), 27.6 (Me2C=CH‐CH2), 26.0 (E‐CH3), 23.5 (CH3‐CH2), 18.0 (Z‐CH3), 14.0 (CH3‐CH2); 13 C‐NMR (CDCl3, 100 MHz): δ=194.6 (C=O), 146.7 (OH‐CH2‐C q), 138.3 (Cq‐CH=CH), 137.8 (CH2‐CH2‐CH), 136.3 (C qMe2), 130.8 (Cq‐CH=CH‐CH), 130.4 (C q‐CH=CH), 120.7 (Cq‐CH=CH), 116.5 (CH=CMe2), 66.8 (CH‐OH), 61.5 (epoxy‐C q), 61.3 (epoxy‐CH), 58.7 (CH2‐OH), 35.0 (CH3‐CH2‐CH2), 26.6 (Me2C=CH‐CH2), 26.0 (E‐CH3), 22.4 (CH3‐CH2), 18.1 (Z‐CH3), 13.8 (CH3‐CH2); HRMS (ESI) m/z calcd. for C19H27O4: 319.1904 (M+H)+; found: 319.1905 (M+H)+; Rf (n‐heptane/EA 1 : 1): 0.34; Specific rotation =+174.5 (c=0.90; CHCl3).
(+)‐Floyocidin B [21] / (1aR,2R,7aR)‐2‐Hydroxy‐7 a‐(3‐methylbut‐2‐en‐1‐yl)‐5‐((E)‐pent‐1‐en‐1‐yl)‐1 a,7 a‐dihydrooxire‐no[2,3‐g]isoquinolin‐7(2H)‐one (4): 1 H‐NMR (MeOD, 500 MHz): δ=8.74 (s, 1H, N‐CH), 7.67 (s, 1H, pentenyl‐Cq‐CH), 6.78 (dt, 1H, J=15.8, 7.1 Hz, CH2‐CH=CH), 6.55 (dt, 1H, J=15.8, 1.5 Hz, CH2‐CH=CH), 5.17 (s, br, 1H, CH‐OH), 5.14 (m, 1H, Me2C=CH), 3.82 (d, 1H, J=1.9 Hz, epoxy‐CH), 2.81 (dd, 1H, J=15.2, 8.0 Hz, Me2C=CH‐CH 2), 2.64 (dd, 1H, J=15.2, 6.9 Hz, Me2C=CH‐CH 2), 2.27 (m, 2H, CH 2‐CH=CH), 1.73 (s, 3H, E‐CH 3), 1.69 (s, 3H, Z‐CH 3), 1.55 (m, 2H, CH3‐CH 2), 0.98 (t, 3H, J=7.4 Hz, CH 3‐CH2); 13 C‐NMR (MeOD, 125 MHz): δ=195.4 (C=O), 157.5 (N‐C q), 150.0 (N‐CH), 138.5 (CH2‐CH=CH), 137.7 (Ar‐C q‐CO), 137.0 (C qMe2), 133.8 (N‐CH‐C q), 130.1 (CH2‐CH=CH), 117.8 (CH=CMe2), 117.1 (pentenyl‐Cq ‐CH), 65.3 (CH‐OH), 63.1 (epoxy‐C q), 61.1 (epoxy‐CH), 36.0 (CH2‐CH=CH), 27.2 (Me2C=CH‐CH2), 26.0 (E‐CH3), 23.2 (CH3‐CH2), 18.1 (Z‐CH3), 14.1 (CH3‐CH2); HRMS (ESI) m/z calcd. for C19H24NO3: 314.1751 (M+H)+; found: 314.1751 (M+H)+; Rf (n‐heptane/ethyl acetate 1 : 1): 0.37; Specific rotation =+160.0 (c=0.03; CHCl3).
Floyocidin C [22] / (1R,5R,6R)‐3‐((1E,3Z)‐Hepta‐1,3‐dien‐1‐yl)‐5‐hydroxy‐4‐(hydroxymethyl)‐1‐(3‐methylbut‐2‐en‐1‐yl)‐7‐oxabicyclo[4.1.0]hept‐3‐en‐2‐one (5): 1 H‐NMR (MeOD, 500 MHz): δ=6.86 (dd, 1H, J=15.7, 11.2 Hz, Cq‐CH=CH), 6.32 (d, 1H, J=15.5 Hz, Cq‐CH=CH), 6.09 (dd, 1H, J=11.2, 10.8 Hz, Cq‐CH=CH‐CH), 5.55 (m, 1H, CH2‐CH2‐CH), 5.07 (m, 1H, Me2C=CH), 4.77 (s, br, 1H, CH‐OH), 4.48 (d, 1H, J=12.8 Hz, CH 2‐OH), 4.42 (d, 1H, J=12.8 Hz, CH 2‐OH), 3.68 (d, 1H, J=2.7 Hz, epoxy‐CH), 2.73 (dd, 1H, J=15.0, 7.6 Hz, Me2C=CH‐CH 2), 2.49 (dd, 1H, J=15.0, 6.9 Hz, Me2C=CH‐CH 2), 2.20 (m, 2H, CH3‐CH2‐CH 2), 1.70 (s, 3H, E‐CH 3), 1.66 (s, 3H, Z‐CH 3), 1.43 (m, 2H, CH3‐CH 2), 0.93 (t, 3H, J=7.4 Hz, CH 3‐CH2); 13 C‐NMR (MeOD, 125 MHz): δ=196.7 (C=O), 150.6 (OH‐CH2‐C q), 136.5 (br, C qMe2), 135.1 (CH2‐CH2‐CH), 133.0 (Cq‐CH=CH), 132.0 (C q‐CH=CH), 130.5 (Cq‐CH=CH‐CH), 124.7 (Cq‐CH=CH), 118.2 (CH=CMe2), 86.3 (CH‐OH), 62.0 (epoxy‐C q), 60.5 (epoxy‐CH), 60.0 (CH2‐OH), 31.0 (CH3‐CH2‐CH2), 27.6 (Me2C=CH‐CH2), 26.0 (E‐CH3), 23.9 (CH3‐CH2), 18.1 (Z‐CH3), 14.0 (CH3‐CH2); HRMS (ESI) m/z calcd. for C19H27O4: 319.1904 (M+H)+; found: 319.1903 (M+H)+; retardation factor and specific rotation was not determined due to low isolated quantity and the mixture of three compounds.
Floyocidin D [22] / (R)‐3‐((1E,3E)‐Hepta‐1,3‐dien‐1‐yl)‐5‐hydroxy‐2‐(hydroxymethyl)‐5‐(3‐methylbut‐2‐en‐1‐yl)cyclo‐hex‐2‐ene‐1,4‐dione (6): 1 H‐NMR (MeOD, 500 MHz): δ=7.08 (dd, 1H, J=15.5, 10.7 Hz, Cq‐CH=CH), 6.60 (d, 1H, J=15.5 Hz, Cq‐CH=CH), 6.29 (dd, 1H, J=15.1, 10.7 Hz, Cq‐CH=CH‐CH), 6.04 (dt, 1H, J=15.1, 7.1 Hz, CH2‐CH2‐CH), 5.14 (m, 1H, Me2C=CH), 4.49 (d, 1H, J=11.7 Hz, CH 2‐OH), 4.43 (d, 1H, J=11.7 Hz, CH 2‐OH), 2.99 (d, 1H, J=16.6 Hz, CH 2‐C=O), 2.81 (d, 1H, J=16.6 Hz, CH 2‐C=O), 2.55 (dd, 1H, J=14.5, 8.7 Hz, Me2C=CH‐CH 2), 2.38 (dd, 1H, J=14.5, 6.5 Hz, Me2C=CH‐CH 2), 2.17 (m, 2H, CH3‐CH2‐CH 2), 1.64 (s, 3H, E‐CH 3), 1.48 (s, 3H, Z‐CH 3), 1.47 (m, 2H, CH3‐CH 2), 0.94 (t, 3H, J=7.4 Hz, CH 3‐CH2); 13 C‐NMR (MeOD, 125 MHz): δ=203.2 (br, OH‐Cq‐C=O), 196.4 (CH2‐C=O), 146.5 (C q‐CH=CH), 144.4 (Cq‐CH=CH), 143.4 (CH2‐CH2‐CH), 142.1 (OH‐CH2‐C q), 136.9 (C qMe2), 133.0 (Cq‐CH=CH‐CH), 122.8 (Cq‐CH=CH), 118.8 (CH=CMe2), 80.2 (OH‐C q), 55.1 (CH2‐OH), 51.5 (CH2‐C=O), 38.7 (Me2C=CH‐CH2), 36.2 (CH3‐CH2‐CH2), 26.1 (E‐CH3), 23.2 (CH3‐CH2), 18.1 (Z‐CH3), 14.0 (CH3‐CH2); HRMS (ESI) m/z calcd. for C19H27O4: 319.1904 (M+H)+; found: 319.1901 (M+H)+; retardation factor and specific rotation was not determined due to low isolated quantity and impurity of the compound.
4‐Bromo‐2‐chloro‐5‐iodopyridine (10): To a suspension of copper(II) bromide (4.18 g, 18.7 mmol, 1.20 eq.) in MeCN (60 mL), t‐BuONO (90 %, 3.10 mL, 23.4 mmol, 1.50 eq.) was added slowly. The suspension was stirred for 15 min at room temperature and was then cooled to 0 °C. Amine 11 (3.96 g, 15.6 mmol, 1.00 eq.), dissolved in MeCN (40 mL) by gentle warming, was added at 0 °C. After 1 h stirring at 0 °C, the mixture was allowed to warm to room temperature and stirred overnight. After concentration in vacuo, [23] aqueous ammonia solution (15 %, 40 mL) was added. The mixture was extracted with DCM (3 x 30 mL) and the combined organic layers were washed with saturated aqueous NaCl (30 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. [23] The crude product was purified by flash column chromatography (100 % DCM) to give product 10 (4.84 g, 15.2 mmol, 97 %) as colorless solid. 1 H‐NMR (CDCl3, 400 MHz): δ=8.69 (s, 1H, N‐CH), 7.65 (s, 1H, ClC‐CH); 13 C‐NMR (CDCl3, 100 MHz): δ=157.3 (N‐CH), 151.6 (C–Cl), 141.5 (CBr), 128.4 (ClC‐CH), 99.1 (CI); HRMS (ESI) m/z calcd. for C5H3BrClIN: 317.8177 (M+H)+; found: 317.8172 (M+H)+; Rf (DCM): 0.62.
(R)‐(4‐Bromo‐6‐chloropyridin‐3‐yl)((2R,3S)‐3‐(((tert‐butyldiphenylsilyl)oxy)methyl)‐3‐(3‐methylbut‐2‐en‐1‐yl) oxiran‐2‐yl)methanol (12) and (S)‐(4‐Bromo‐6‐chloropyridin‐3‐yl)((2R,3S)‐3‐(((tert‐butyldiphenylsilyl)oxy)methyl)‐3‐(3‐methylbut‐2‐en‐1‐yl)oxiran‐2‐yl)methanol (13): The reaction was carried out in moisture‐free glassware under argon atmosphere. To a solution of iodopyridine 10 (1.91 g, 6.01 mmol, 1.40 eq.) dissolved in anhydrous THF (30 mL), i‐PrMgCl in THF (2.0 M, 2.79 mL, 5.58 mmol, 1.30 eq.) was added dropwise at −40 °C. After stirring for 45 min, epoxy aldehyde 9 (1.75 g, 4.29 mmol, 1.00 eq.) in anhydrous THF (10 mL) was added. The reaction mixture was stirred for 1 h 30 min at −40 °C and was then diluted with saturated aqueous NH4Cl (50 mL). The mixture was extracted with ethyl acetate (2×150 mL) and the combined organic layers were washed with saturated aqueous NaCl (75 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (0–20 % ethyl acetate in n‐heptane) to give addition products 12 (1.25 g, 2.09 mmol, 49 % over two steps) and 13 (1.09 g, 1.89 mmol, 42 % over two steps) as colorless oils. For 12: 1 H‐NMR (CDCl3, 400 MHz): δ=8.54 (s, 1H, N‐CH), 7.73–7.66 (m, 4H, phenyl‐H), 7.53 (s, 1H, ClC‐CH), 7.51–7.38 (m, 6H, phenyl‐H), 4.99 (m, 1H, Me2C=CH), 4.96 (d, 1H, J=8.2 Hz, CH‐OH), 4.03 (d, 1H, J=11.2 Hz, TBDPSO‐CH 2), 3.79 (d, 1H, J=11.0 Hz, TBDPSO‐CH 2), 3.29 (s, 1H, OH), 3.07 (d, 1H, J=7.9 Hz, epoxy‐CH), 2.44 (m, 2H, Me2C=CH‐CH 2), 1.66 (s, 3H, E‐CH 3), 1.57 (s, 3H, Z‐CH 3), 1.12 (s, 9H, t Bu‐CH 3); 13 C‐NMR (CDCl3, 100 MHz): δ=151.3 (Cl–C), 149.3 (N‐CH), 136.0 (C qMe2), 135.9/135.7 (phenyl‐CH), 135.5 (N‐CH‐C q), 134.6 (C‐Br), 132.4/132.2 (phenyl‐C q), 130.4 (phenyl‐CH), 128.2/128.1 (phenyl‐CH), 127.7 (ClC‐CH), 117.3 (CH=CMe2), 69.6 (CH‐OH), 65.5 (TBDPSO‐CH2), 64.3 (epoxy‐CH), 63.8 (epoxy‐C q), 32.0 (Me2C=CH‐CH2), 27.1 ( t Bu‐CH3), 25.9 (E‐CH3), 19.3 ( t Bu‐C q), 18.1 (Z‐CH3); HRMS (ESI) m/z calcd. for C30H36BrClNO3Si: 600.1331 (M+H)+; found: 600.1327 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.32; Specific rotation =+15.1 (c=1.53; CHCl3). For 13: 1 H‐NMR (CDCl3, 400 MHz): δ=8.49 (s, 1H, N‐CH), 7.72–7.61 (m, 4H, phenyl‐H), 7.51 (s, 1H, ClC‐CH), 7.49–7.36 (m, 6H, phenyl‐H), 4.99 (m, 1H, Me2C=CH), 4.86 (d, 1H, J=6.1 Hz, CH‐OH), 3.86 (d, 1H, J=11.4 Hz, TBDPSO‐CH 2), 3.80 (d, 1H, J=11.5 Hz, TBDPSO‐CH 2), 3.09 (d, 1H, J=6.0 Hz, epoxy‐CH), 2.81 (d, 1H, J=2.6 Hz, OH), 2.67 (dd, 1H, J=15.0, 8.1 Hz Me2C=CH‐CH 2), 2.30 (dd, 1H, J=15.0, 6.8 Hz Me2C=CH‐CH 2), 1.67 (s, 3H, E‐CH 3), 1.57 (s, 3H, Z‐CH 3), 1.07 (s, 9H, t Bu‐CH 3); 13 C‐NMR (CDCl3, 100 MHz): δ=151.4 (Cl−C), 149.2 (N‐CH), 136.0 (C qMe2), 135.9/135.8 (phenyl‐CH), 135.5 (N‐CH‐C q), 134.0 (C‐Br), 133.1/132.9 (phenyl‐C q), 130.1/130.1 (phenyl‐CH), 127.9/127.9 (phenyl‐CH), 127.7 (ClC‐CH), 117.3 (CH=CMe2), 67.8 (CH‐OH), 65.8 (epoxy‐C q), 64.0 (TBDPSO‐CH2), 63.8 (epoxy‐CH), 31.3 (Me2C=CH‐CH2), 26.9 ( t Bu‐CH3), 25.9 (E‐CH3), 19.4 ( t Bu‐C q), 18.1 (Z‐CH3); HRMS (ESI) m/z calcd. for C30H36BrClNO3Si: 600.1331 (M+H)+; found: 600.1329 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.25; Specific rotation =<‐M‐>2.2 (c=1.39; CHCl3).
Ent ‐12 (1.21 g, 2.01 mmol, 50 % over two steps) and ent ‐13 (1.01 g, 1.69 mmol, 42 % over two steps) were synthesized in analogous manner to 12 and 13 starting from epoxy aldehyde ent ‐9 (1.65 g, 4.05 mmol) and iodide 10 (1.80 g, 5.66 mmol). The NMR data are identical to the ones reported for 12 and 13. For ent ‐12: HRMS (ESI) m/z calcd. for C30H36BrClNO3Si: 600.1331 (M+H)+; found: 600.1326 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.32; Specific rotation =−25.5 (c=0.94; CHCl3). For ent ‐13: HRMS (ESI) m/z calcd. for C30H36BrClNO3Si: 600.1331 (M+H)+; found: 600.1332 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.25; Specific rotation =+4.2 (c=1.42; CHCl3).
(R)‐(4‐Bromo‐6‐chloropyridin‐3‐yl)((2R,3S)‐3‐(((tert‐butyldiphenylsilyl)oxy)methyl)‐3‐(3‐methylbut‐2‐en‐1‐yl)oxiran‐2‐yl)methyl acetate (14): To a solution of alcohol 12 (0.243 g, 0.404 mmol, 1.00 eq.) in anhydrous DCM (7 mL), anhydrous pyridine (0.098 mL, 1.2 mmol, 3.00 eq.), Ac2O (0.115 mL, 1.21 mmol, 3.00 eq.) and DMAP (0.005 g, 0.04 mmol, 0.10 eq.) were added. The reaction mixture was stirred for 2 h 30 min until the TLC showed complete conversion. Toluene (10 mL) was added and the reaction mixture was concentrated in vacuo. The crude product was purified by flash column chromatography (0–30 % ethyl acetate in n‐heptane) to give 14 (0.249 g, 0.387 mmol, 97 %) as a colorless oil. 1 H‐NMR (CDCl3, 400 MHz): δ=8.34 (s, 1H, N‐CH), 7.70–7.63 (m, 4H, Ar‐H), 7.49 (s, 1H, ClC‐CH), 7.47–7.34 (m, 6H, Ar‐H), 5.79 (d, 1H, J=7.9 Hz, CH‐OAc), 5.05 (m, 1H, Me2C=CH), 3.91 (d, 1H, J=11.2 Hz, TBDPSO‐CH 2), 3.84 (d, 1H, J=11.4 Hz, TBDPSO‐CH 2), 3.21 (d, 1H, J=7.9 Hz, epoxy‐CH), 2.67 (dd, 1H, J=15.1, 7.6 Hz, Me2C=CH‐CH 2), 2.41 (dd, 1H, J=15.1, 6.9 Hz, Me2C=CH‐CH 2), 2.02 (s, 3H, CO‐CH3), 1.71 (s, 3H, E‐CH 3), 1.62 (s, 3H, Z‐CH 3), 1.07 (s, 9H, t Bu‐CH 3); 13 C‐NMR (CDCl3, 100 MHz): δ=169.1 (C=O), 151.7 (Cl–C), 149.5 (N‐CH), 135.8 (C qMe2), 135.9/135.8 (Ar‐C), 135.5 (C‐Br), 133.2/133.1 (Ar‐C q), 132.9 (N‐CH‐C q), 130.0 (Ar‐C), 128.0 (ClC‐CH), 128.0/127.9 (Ar‐C), 117.5 (CH=CMe2), 69.4 (CH‐OAc), 65.6 (epoxy‐C q), 64.0 (TBDPSO‐CH2), 61.9 (epoxy‐CH), 31.1 (Me2C=CH‐CH2), 26.9 ( t Bu‐CH3), 26.0 (E‐CH3), 20.8 (CO‐CH3), 19.4 ( t Bu‐C q), 18.2 (Z‐CH3); HRMS (ESI) m/z calcd. for C32H38BrClNO4Si: 642.1437 (M+H)+; found: 642.1441 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.46; Specific rotation =+15.0 (c=1.07; CHCl3).
(S)‐(4‐Bromo‐6‐chloropyridin‐3‐yl)((2R,3S)‐3‐(((tert‐butyldiphenylsilyl)oxy)methyl)‐3‐(3‐methylbut‐2‐en‐1‐yl)oxiran‐2‐yl)methyl acetate (18): 18 (0.229 g, 0.356 mmol, 98 %) was synthesized in analogous manner to 14 starting from 13 (0.218 g, 0.363 mmol) and was obtained as colorless oil. 1 H‐NMR (CDCl3, 400 MHz): δ=8.32 (s, 1H, N‐CH), 7.65–7.56 (m, 4H, Ar‐H), 7.48 (s, 1H, ClC‐CH), 7.46–7.34 (m, 6H, Ar‐H), 5.81 (d, 1H, J=8.1 Hz, CH‐OAc), 4.92 (m, 1H, Me2C=CH), 3.76 (d, 1H, J=11.4 Hz, TBDPSO‐CH 2), 3.70 (d, 1H, J=11.5 Hz, TBDPSO‐CH 2), 3.37 (d, 1H, J=8.1 Hz, epoxy‐CH), 2.66 (dd, 1H, J=15.1, 8.0 Hz, Me2C=CH‐CH 2), 2.26 (dd, 1H, J=15.0, 6.7 Hz, Me2C=CH‐CH 2), 2.04 (s, 3H, CO‐CH3), 1.66 (s, 3H, E‐CH 3), 1.56 (s, 3H, Z‐CH 3), 1.03 (s, 9H, t Bu‐CH 3); 13 C‐NMR (CDCl3, 100 MHz): δ=169.4 (C=O), 152.0 (Cl–C), 149.6 (N‐CH), 136.0 (C qMe2), 135.9/135.8 (Ar‐C), 135.0 (C‐Br), 133.2/132.8 (Ar‐C q), 132.5 (N‐CH‐C q), 130.0/130.0 (Ar‐C), 128.4 (ClC‐CH), 127.9/127.9 (Ar‐C), 117.3 (CH=CMe2), 71.5 (CH‐OAc), 64.8 (TBDPSO‐CH2), 64.6 (epoxy‐C q), 61.5 (epoxy‐CH), 30.8 (Me2C=CH‐CH2), 26.9 ( t Bu‐CH3), 25.9 (E‐CH3), 20.9 (CO‐CH3), 19.4 ( t Bu‐C q), 18.2 (Z‐CH3); HRMS (ESI) m/z calcd. for C32H38BrClNO4Si: 642.1437 (M+H)+; found: 642.1437 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.34; Specific rotation =+2.6 (c=1.15; CHCl3).
Ent ‐14 (1.25 g, 1.94 mmol, 97 %) and ent ‐18 (1.06 g, 1.65 mmol, 98 %) were synthesized in analogous manner to 14 and 18 starting from ent ‐12 (1.20 g, 2.00 mmol) and ent ‐13 (1.01 g, 1.68 mmol). The NMR data are identical to the ones reported for 14 and 18. For ent ‐14: HRMS (ESI) m/z calcd. for C32H38BrClNO4Si: 642.1437 (M+H)+; found: 642.1440 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.46; Specific rotation =−16.0 (c=2.25; CHCl3). For ent ‐18: HRMS (ESI) m/z calcd. for C32H38BrClNO4Si: 642.1437 (M+H)+; found: 642.1449 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.34; Specific rotation =−5.5 (c=1.27; CHCl3).
((2S,3R)‐3‐((R)‐(4‐Bromo‐6‐chloropyridin‐3‐yl)(hydroxy)methyl)‐2‐(3‐methylbut‐2‐en‐1‐yl)oxiran‐2‐yl)methyl acetate (15): In a 50 mL falcon tube, acetic acid glacial (0.164 mL, 2.87 mmol, 1.40 eq.) followed by TBAF in THF (1 M in THF, 2.50 mL, 2.46 mmol, 1.20 eq.) were added to a solution of TBDPS‐protected alcohol 14 (1.32 g, 2.05 mmol, 1.00 eq.) in THF (25 mL). After 2 h, a second portion of TBAF (0.20 eq.) was added. Complete consumption of the starting material, which was monitored by TLC, was achieved after 1 h. The acetate migration from secondary to primary hydroxy was monitored by LC–MS. After stirring overnight at room temperature, LC–MS indicated an acyl migration to the primary hydroxy group of >90 %. [24] The reaction mixture was loaded on silica and purified via flash column chromatography (n‐heptane / ethyl acetate 3 : 1) to give 15 (0.715 g, 1.77 mmol, 86 %) as colorless oil. 1 H‐NMR (CDCl3, 400 MHz): δ=8.57 (s, 1H, N‐CH), 7.56 (s, 1H, ClC‐CH), 5.12 (d, 1H, J=8.2 Hz, CH‐OH), 5.09 (m, 1H, Me2C=CH), 4.37 (d, 1H, J=12.0 Hz, CH 2‐OAc), 4.28 (d, 1H, J=12.2 Hz, CH 2‐OAc), 3.70 (s, br, 1H, OH), 3.11 (d, 1H, J=8.1 Hz, epoxy‐CH), 2.41 (dd, 1H, J=15.0, 7.4 Hz, Me2C=CH‐CH 2), 2.30 (dd, 1H, J=15.0, 7.7 Hz, Me2C=CH‐CH 2), 2.15 (s, 3H, CO‐CH3), 1.72 (s, 3H, E‐CH 3), 1.62 (s, 3H, Z‐CH 3); 13 C‐NMR (CDCl3, 100 MHz): δ=171.9 (C=O), 151.4 (Cl–C), 149.5 (N‐CH), 136.4 (C qMe2), 135.4 (N‐CH‐C q), 134.7 (C‐Br), 127.7 (ClC‐CH), 116.8 (CH=CMe2), 68.4 (CH‐OH), 65.4 (epoxy‐CH), 63.1 (CH2‐OAc), 61.9 (epoxy‐C q), 32.0 (Me2C=CH‐CH2), 26.0 (E‐CH3), 21.0 (CO‐CH3), 18.1 (Z‐CH3); HRMS (ESI) m/z calcd. for C16H20BrClNO4: 404.0259 (M+H)+; found: 404.0255 (M+H)+; Rf (n‐heptane/ethyl acetate 2 : 1): 0.31; Specific rotation =−8.5 (c=0.82; CHCl3).
((2S,3R)‐3‐((S)‐(4‐Bromo‐6‐chloropyridin‐3‐yl)(hydroxy)methyl)‐2‐(3‐methylbut‐2‐en‐1‐yl)oxiran‐2‐yl)methyl acetate (19): 19 (0.694 g, 1.71 mmol, 90 %) [24] was synthesized in analogous manner to 15 starting from 18 (1.23 g, 1.91 mmol) and was obtained as colorless oil. 1 H‐NMR (CDCl3, 400 MHz): δ=8.56 (s, 1H, N‐CH), 7.59 (s, 1H, ClC‐CH), 5.07–4.96 (m, 2H, Me2C=CH, CH‐OH), 4.32 (d, 1H, J=12.2 Hz, CH 2‐OAc), 4.29 (d, 1H, J=12.1 Hz, CH 2‐OAc), 3.16 (d, 1H, J=6.2 Hz, epoxy‐CH), 2.86 (s, br, 1H, OH), 2.55 (dd, 1H, J=15.0, 8.1 Hz, Me2C=CH‐CH 2), 2.26 (dd, 1H, J=15.0, 7.0 Hz, Me2C=CH‐CH 2), 2.08 (s, 3H, CO‐CH3), 1.70 (s, 3H, E‐CH 3), 1.58 (s, 3H, Z‐CH 3); 13 C‐NMR (CDCl3, 100 MHz): δ=170.7 (C=O), 151.4 (Cl–C), 149.3 (N‐CH), 136.8 (C qMe2), 135.1 (N‐CH‐C q), 134.0 (C‐Br), 127.8 (ClC‐CH), 116.5 (CH=CMe2), 68.1 (CH‐OH), 64.2 (epoxy‐CH), 63.9 (CH2‐OAc), 63.5 (epoxy‐C q), 31.9 (Me2C=CH‐CH2), 26.0 (E‐CH3), 20.9 (CO‐CH3), 18.1 (Z‐CH3); HRMS (ESI) m/z calcd. for C16H20BrClNO4: 404.0259 (M+H)+; found: 404.0255 (M+H)+; Rf (n‐heptane/ethyl acetate 2 : 1): 0.29; Specific rotation =+12.3 (c=0.98; CHCl3).
Ent ‐15 (0.670 g, 1.66 mmol, 86 %) [24] and ent ‐19 (0.600 g, 1.48 mmol, 90 %) [24] were synthesized in analogous manner to 15 and 19 starting from ent ‐14 (1.24 g, 1.93 mmol) and ent ‐18 (1.06 g, 1.65 mmol). The NMR data are identical to the ones reported for 15 and 19. For ent ‐15: HRMS (ESI) m/z calcd. for C16H20BrClNO4: 404.0259 (M+H)+; found: 404.0258 (M+H)+; Rf (n‐heptane/ethyl acetate 2 : 1): 0.31; Specific rotation =+5.1 (c=1.17; CHCl3). For ent ‐19: HRMS (ESI) m/z calcd. for C16H20BrClNO4: 404.0259 (M+H)+; found: 404.0262 (M+H)+; Rf (n‐heptane/ethyl acetate 2 : 1): 0.29; Specific rotation =−11.8 (c=1.19; CHCl3).
((2S,3R)‐3‐((R)‐(4‐Bromo‐6‐chloropyridin‐3‐yl)((triisopropylsilyl)oxy)methyl)‐2‐(3‐methylbut‐2‐en‐1‐yl)oxiran‐2‐yl)methanol (17): 2,6‐Lutidine (1.20 mL, 10.4 mmol, 6.00 eq.) and then TIPSOTf (1.40 mL, 5.17 mmol, 3.00 eq.) were added to a solution of alcohol 15 (0.698 g, 1.73 mmol, 1.00 eq.) in anhydrous DCM (20 mL) at 0 °C. The reaction mixture was allowed to stir at room temperature. After 1 h 30 min and 4 h, additional portions of 2,6‐lutidine (4.00 eq.) and TIPSOTf (2.00 eq.) were added to drive the reaction to complete conversion of starting material 15. After stirring overnight at room temperature, the TLC showed complete conversion. Saturated aqueous NH4Cl (30 mL) was added. The mixture was extracted with ethyl acetate (2 x 100 mL) and the combined organic layers were washed with saturated aqueous NaCl (70 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product of 16 was pre‐purified by flash column chromatography (0–18 % ethyl acetate in n‐heptane).
Pre‐purified crude 16 was dissolved in THF/H2O (5 : 1, 60 mL/12 mL) and LiOH ⋅ H2O (0.145 g, 3.45 mmol, 2.00 eq.) was added. After stirring for 7 h at room temperature, the TLC showed complete conversion. Saturated aqueous NaHCO3 (50 mL) was added. The mixture was extracted with ethyl acetate (2 x 100 mL) and the combined organic layers were washed with saturated aqueous NaCl (70 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (0–25 % ethyl acetate in n‐heptane) to give 17 (0.491 g, 0.946 mmol, 55 % over two steps) as a colorless solid. In a 4 mL vial, a small portion of 17 was dissolved in DCM (1 mL) and n‐heptane (1.5 mL) was added. The system was left at room temperature for one night. A crystal was obtained for structure elucidation by X‐ray crystal diffraction (see chapter 4 of Supporting Information). 1 H‐NMR (CDCl3, 400 MHz): δ=8.57 (s, 1H, N‐CH), 7.51 (s, 1H, ClC‐CH), 5.29 (d, 1H, J=7.7 Hz, CH‐OTIPS), 5.08 (m, 1H, Me2C=CH), 3.94 (s, 2H, CH 2‐OH), 3.09 (d, 1H, J=7.9 Hz, epoxy‐CH), 2.64 (dd, 1H, J=14.7, 8.2 Hz, Me2C=CH‐CH 2), 2.12 (dd, 1H, J=14.7, 7.0 Hz, Me2C=CH‐CH 2), 1.81 (s, br, 1H, OH), 1.70 (s, 3H, E‐CH 3), 1.63 (s, 3H, Z‐CH 3), 1.09–0.96 (m, 21H, TIPS‐H); 13 C‐NMR (CDCl3, 100 MHz): δ=151.1 (Cl–C), 150.3 (N‐CH), 137.2 (C‐Br or N‐CH‐C q), 136.1 (C qMe2), 133.8 (N‐CH‐C q or C‐Br), 127.4 (ClC‐CH), 117.5 (CH=CMe2), 68.3 (CH‐OTIPS), 67.3 (epoxy‐CH), 65.9 (epoxy‐C q), 62.2 (CH2‐OH), 32.0 (Me2C=CH‐CH2), 25.9 (E‐CH3), 18.1 (Z‐CH3), 17.9/17.9 (CH(CH3)2), 12.3 (CH(CH3)2); HRMS (ESI) m/z calcd. for C23H38BrClNO3Si: 518.1487 (M+H)+; found: 518.1482 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.32; Specific rotation =−19.3 (c=1.61; CHCl3).
((2S,3R)‐3‐((S)‐(4‐Bromo‐6‐chloropyridin‐3‐yl)((triisopropylsilyl)oxy)methyl)‐2‐(3‐methylbut‐2‐en‐1‐yl)oxiran‐2‐yl)methanol (21): 21 (0.527 g, 1.02 mmol, 71 % over two steps) was synthesized in analogous manner to 17 starting from 19 (0.583 g, 1.44 mmol) and was obtained as colorless oil. 1 H‐NMR (CDCl3, 400 MHz): δ=8.50 (s, 1H, N‐CH), 7.54 (s, 1H, ClC‐CH), 5.20 (d, 1H, J=7.7 Hz, CH‐OTIPS), 4.93 (m, 1H, Me2C=CH), 3.72 (dd, 1H, J=12.1, 6.2 Hz, CH 2‐OH), 3.66 (dd, 1H, J=12.3, 5.7 Hz, CH 2‐OH), 3.31 (d, 1H, J=7.7 Hz, epoxy‐CH), 2.46 (dd, 1H, J=15.0, 7.7 Hz, Me2C=CH‐CH 2), 2.14 (dd, 1H, J=14.9, 7.2 Hz, Me2C=CH‐CH 2), 1.73 (t, 1H, J=6.3 Hz, OH), 1.64 (s, 3H, E‐CH 3), 1.53 (s, 3H, Z‐CH 3), 1.15–0.95 (m, 21H, TIPS‐H); 13 C‐NMR (CDCl3, 100 MHz): δ=151.2 (Cl–C), 150.6 (N‐CH), 136.4 (C‐Br), 136.0 (C qMe2), 133.3 (N‐CH‐C q), 127.8 (ClC‐CH), 117.3 (CH=CMe2), 71.0 (CH‐OTIPS), 66.9 (epoxy‐CH), 64.9 (epoxy‐C q), 62.7 (CH2‐OH), 32.0 (Me2C=CH‐CH2), 25.9 (E‐CH3), 18.1 (Z‐CH3), 17.9/17.9 (CH(CH3)2), 12.3 (CH(CH3)2); HRMS (ESI) m/z calcd. for C23H38BrClNO3Si: 518.1487 (M+H)+; found: 518.1490 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.32; Specific rotation =+6.0 (c=1.34; CHCl3).
Ent ‐17 (0.442 g, 0.852 mmol, 53 % over two steps) and ent ‐21 (0.434 g, 0.836 mmol, 57 % over two steps) were synthesized in analogous manner to 17 and 21 starting from ent ‐15 (0.655 g, 1.62 mmol) and ent ‐19 (0.593 g, 1.47 mmol). The NMR data are identical to the ones reported for 17 and 21. For ent ‐17: HRMS (ESI) m/z calcd. for C23H38BrClNO3Si: 518.1487 (M+H)+; found: 518.1491 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.32; Specific rotation =+35.9 (c=1.03; CHCl3). For ent ‐21: HRMS (ESI) m/z calcd. for C23H38BrClNO3Si: 518.1487 (M+H)+; found: 518.1479 (M+H)+; Rf (n‐heptane/ethyl acetate4 : 1): 0.32; Specific rotation =−8.5 (c=1.41; CHCl3).
(2S,3R)‐3‐((R)‐(4‐Bromo‐6‐chloropyridin‐3‐yl)((triisopropylsilyl)oxy)methyl)‐2‐(3‐methylbut‐2‐en‐1‐yl)oxirane‐2‐carbonitrile (8): Alcohol 17 (0.382 g, 0.735 mmol, 1.00 eq.) was dissolved in MeCN/H2O 9 : 1 (27 mL/3.0 mL) and NH4OAc (0.227 g, 2.94 mmol, 4.00 eq.), BAIB (0.711 g, 2.21 mmol, 3.00 eq.), as well as TEMPO (0.023 g, 0.15 mmol, 0.20 eq.) were added. The reaction solution was allowed to stir for 6 h 30 min at room temperature, until the TLC and the LC–MS showed complete conversion to the nitrile (n.b. the starting material is first converted to the intermediate aldehyde and then converted to the nitrile in a two‐step fashion). Saturated aqueous Na2SO3 (40 mL) was added. The mixture was extracted with ethyl acetate (80 mL) and the organic layer was washed with saturated aqueous NaCl (50 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified via flash column chromatography (0–15 % ethyl acetate in n‐heptane) to give nitrile 8 (0.265 g, 0.515 mmol, 70 %) as a colorless oil. 1 H‐NMR (CDCl3, 400 MHz): δ=8.62 (s, 1H, N‐CH), 7.56 (s, 1H, ClC‐CH), 5.34 (d, 1H, J=6.8 Hz, CH‐OTIPS), 5.11 (m, 1H, Me2C=CH), 3.18 (d, 1H, J=6.8 Hz, epoxy‐CH), 2.62 (dd, 1H, J=15.0, 7.9 Hz, Me2C=CH‐CH 2), 2.48 (dd, 1H, J=15.1, 7.3 Hz, Me2C=CH‐CH 2), 1.75 (s, 3H, E‐CH 3), 1.65 (s, 3H, Z‐CH 3), 1.21–0.99 (m, 21H, TIPS‐H); 13 C‐NMR (CDCl3, 100 MHz): δ=151.7 (Cl–C), 150.0 (N‐CH), 139.3 (C qMe2), 135.7 (N‐CH‐C q), 134.2 (C‐Br), 127.7 (ClC‐CH), 116.8 (CN), 114.2 (CH=CMe2), 69.3 (CH‐OTIPS), 65.4 (epoxy‐CH), 55.0 (epoxy‐C q), 32.6 (Me2C=CH‐CH2), 26.0 (E‐CH3), 18.2 (Z‐CH3), 18.0/17.9 (CH(CH3)2), 12.2 (CH(CH3)2); HRMS (ESI) m/z calcd. for C23H35BrClN2O2Si: 513.1334 (M+H)+; found: 513.1331 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.59; Specific rotation =+37.8 (c=1.14; CHCl3).
(2S,3R)‐3‐((S)‐(4‐Bromo‐6‐chloropyridin‐3‐yl)((triisopropylsilyl)oxy)methyl)‐2‐(3‐methylbut‐2‐en‐1‐yl)oxirane‐2‐carbonitrile (22): 22 (0.244 g, 0.475 mmol, 60 %) was synthesized in analogous manner to 8 starting from 21 (0.410 g, 0.789 mmol) and was obtained as colorless oil. 1 H‐NMR (CDCl3, 400 MHz): δ=8.55 (s, 1H, N‐CH), 7.59 (s, 1H, ClC‐CH), 5.14 (d, 1H, J=7.7 Hz, CH‐OTIPS), 5.03 (m, 1H, Me2C=CH), 3.40 (d, 1H, J=7.6 Hz, epoxy‐CH), 2.52 (dd, 1H, J=15.0, 7.6 Hz, Me2C=CH‐CH 2), 2.43 (dd, 1H, J=15.0, 7.3 Hz, Me2C=CH‐CH 2), 1.71 (s, 3H, E‐CH 3), 1.58 (s, 3H, Z‐CH 3), 1.19–0.96 (m, 21H, TIPS‐H); 13 C‐NMR (CDCl3, 100 MHz): δ=151.9 (Cl–C), 150.7 (N‐CH), 139.2 (C qMe2), 134.7 (N‐CH‐C q), 133.8 (C‐Br), 128.2 (ClC‐CH), 117.2 (CN), 114.2 (CH=CMe2), 72.1 (CH‐OTIPS), 65.1 (epoxy‐CH), 53.7 (epoxy‐C q), 32.9 (Me2C=CH‐CH2), 25.9 (E‐CH3), 18.3 (Z‐CH3), 17.9/17.9 (CH(CH3)2), 12.3(CH(CH3)2); HRMS (ESI) m/z calcd. for C23H35BrClN2O2Si: 513.1334 (M+H)+; found: 513.1329 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.59; Specific rotation =+10.3 (c=0.97; CHCl3).
Ent ‐8 (0.101 g, 0.197 mmol, 25 %) and ent ‐22 (0.129 g, 0.251 mmol, 31 %) were synthesized in analogous manner [25] to 8 and 22 starting from ent ‐17 (0.409 g, 0.788 mmol) and ent ‐21 (0.426 g, 0.821 mmol). The NMR data are identical to the ones reported for 8 and 22. For ent ‐8: HRMS (ESI) m/z calcd. for C23H35BrClN2O2Si: 513.1334 (M+H)+; found: 513.1340 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.59; Specific rotation =−40.7 (c=0.91; CHCl3). For ent ‐22: HRMS (ESI) m/z calcd. for C23H35BrClN2O2Si: 513.1334 (M+H)+; found: 513.1329 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.59; Specific rotation =−5.5 (c=1.09; CHCl3).
(1aR,2R,7aR)‐5‐Chloro‐7 a‐(3‐methylbut‐2‐en‐1‐yl)‐2‐((triisopropylsilyl)oxy)‐1 a,7 a‐dihydrooxireno[2,3‐g]isoquinolin‐7(2H)‐one (7): The halogen‐lithium exchange was carried out in a moisture‐free glassware under argon atmosphere. To a solution of nitrile 8 (0.154 g, 0.300 mmol, 1.00 eq.) in anhydrous THF (10 mL), prediluted n‐BuLi solution (0.30 mmol, 1.00 eq.) was added at −100 °C. [26] The obtained red solution was stirred for 20 min at –100 °C and for 5 min without cooling bath. For hydrolysis of the intermediate imine, aqueous citric acid (10 %, 15 mL) was added and the mixture was extracted with ethyl acetate (2 x 50 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (20 mL) as well as with saturated aqueous NaCl (20 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (0–15 % ethyl acetate in n‐heptane) to give desired product 7 (0.034 g, 0.077 mmol, 26 %) as colorless oil and the unexpected product 23 (0.046 g, 0.10 mmol, 35 %) as slightly yellow oil. 1 H‐NMR (CDCl3, 400 MHz): δ=8.76 (s, 1H, N‐CH), 7.65 (s, 1H, ClC‐CH), 5.35 (s, br, 1H, CH‐OTIPS), 5.07 (m, 1H, Me2C=CH), 3.79 (d, 1H, J=1.5 Hz, epoxy‐CH), 2.89 (dd, 1H, J=15.4, 8.1 Hz, Me2C=CH‐CH 2), 2.63 (dd, 1H, J=15.7, 7.3 Hz, Me2C=CH‐CH 2), 1.74 (s, 3H, E‐CH 3), 1.67 (s, 3H, Z‐CH 3), 1.34–1.14 (m, 21H, TIPS‐H); 13 C‐NMR (CDCl3, 100 MHz): δ=193.5 (C=O), 151.9 (Cl–C), 149.9 (N‐CH), 138.1 (Ar‐C q‐CO), 137.2 (C qMe2), 132.5 (N‐CH‐C q), 120.2 (ClC‐CH), 116.0 (CH=CMe2), 66.3 (CH‐OTIPS), 62.2 (epoxy‐C q), 59.0 (epoxy‐CH), 25.9 (Me2C=CH‐CH2), 25.9 (E‐CH3), 18.2 (Z‐CH3), 18.3/18.2, (CH(CH3)2), 12.8 (CH(CH3)2); HRMS (ESI) m/z calcd. for C23H35ClNO3Si: 436.2069 (M+H)+; found: 436.2066 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.60; Specific rotation =+9.1 (c=0.12; CHCl3).
(1R,4R,5R)‐4‐(6‐Chloro‐4‐(triisopropylsilyl)pyridin‐3‐yl)‐1‐(3‐methylbut‐2‐en‐1‐yl)‐3,6‐dioxabicyclo[3.1.0]hexan‐2‐ imine (23): 1 H‐NMR (CDCl3, 400 MHz): 8.13 (s, 1H, N‐CH), 7.45 (s, 1H, ClC‐CH), 5.35 (s, 1H, pyridine‐CHO), 5.16 (m, 1H, Me2C=CH), 3.78 (s, 1H, epoxy‐CH), 3.13–2.92 (m, 1H, Me2C=CH‐CH 2), 2.74 (dd, 1H, J=15.6, 7.2 Hz, Me2C=CH‐CH 2), 1.74 (s, 3H, E‐CH 3), 1.66 (s, 3H, Z‐CH 3), 1.46 (m, 3H, CH(CH3)2), 1.17–1.06 (m, 18H, CH(CH3)2); 13 C‐NMR (CDCl3, 100 MHz): δ=165.2 (NH), 152.3 (Cl–C), 150.4 (C q‐TIPS), 148.5 (N‐CH), 137.3 (C qMe2), 136.4 (N‐CH‐C q), 131.1 (ClC‐CH), 115.8 (CH=CMe2), 78.5 (pyridine‐CHO), 63.5 (epoxy‐C q), 63.4 (epoxy‐CH), 25.9 (E‐CH3), 24.9 (Me2C=CH‐CH2), 18.9/18.8 (CH(CH3)2), 18.2 (Z‐CH3), 12.5 (CH(CH3)2); HRMS (ESI) m/z calcd. for C23H36ClNO3Si: (M+H)+, 436.2069; found: 436.2072 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.28.
(1aR,2S,7aR)‐5‐Chloro‐7 a‐(3‐methylbut‐2‐en‐1‐yl)‐2‐((triisopropylsilyl)oxy)‐1 a,7 a‐dihydrooxireno[2,3‐g] isoquinolin‐7(2H)‐one (25): 25 (0.065 g, 0.15 mmol, 81 %) was synthesized in analogous manner to 7 starting from 22 (0.095 g, 0.18 mmol) and was obtained as colorless oil. [27] 1 H‐NMR (CDCl3, 400 MHz): δ=8.47 (s, 1H, N‐CH), 7.72 (s, 1H, ClC‐CH), 5.42 (d, 1H, J=2.7 Hz, CH‐OTIPS), 5.10 (m, 1H, Me2C=CH), 3.82 (d, 1H, J=2.7 Hz, epoxy‐CH), 2.97 (dd, 1H, J=15.3, 7.9 Hz, Me2C=CH‐CH 2), 2.55 (dd, 1H, J=15.5, 6.8 Hz, Me2C=CH‐CH 2), 1.72 (s, 3H, E‐CH 3), 1.68 (s, 3H, Z‐CH 3), 1.16–0.96 (m, 21H, TIPS‐H); 13 C‐NMR (CDCl3, 100 MHz): δ=193.6 (C=O), 152.7 (Cl–C), 151.2 (N‐CH), 139.4 (Ar‐C q‐CO), 136.6 (C qMe2), 132.7 (N‐CH‐C q), 121.3 (ClC‐CH), 116.2 (CH=CMe2), 65.5 (CH‐OTIPS), 60.9 (epoxy‐C q), 60.4 (epoxy‐CH), 26.4 (Me2C=CH‐CH2), 26.0 (E‐CH3), 18.2 (Z‐CH3), 18.1/18.0 (CH(CH3)2), 12.6 (CH(CH3)2); HRMS (ESI) m/z calcd. for C23H35ClNO3Si: 436.2069 (M+H)+; found: 436.2070 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.59; Specific rotation =+64.0 (c=0.47; CHCl3).
Ent ‐7 (0.020 g, 0.046 mmol, 23 %) and ent ‐25 (0.088 g, 0.20 mmol, 81 %) were synthesized in analogous manner to 7 and 25 starting from ent ‐8 (0.100 g, 0.195 mmol) and ent ‐22 (0.128 g, 0.249 mmol). The low yield of ent ‐7 was caused by the formation of ent ‐23 (0.023 g, 0.052 mmol, 27 %). The NMR data are identical to the ones reported for 7, 25, and 23. For ent ‐7: HRMS (ESI) m/z calcd. for C23H35ClNO3Si: 436.2069 (M+H)+; found: 436.2070 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.60; Specific rotation =−126.6 (c=0.96; CHCl3). For ent ‐25: HRMS (ESI) m/z calcd. for C23H35ClNO3Si: 436.2069 (M+H)+; found: 436.2073 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.59; Specific rotation =−121.9 (c=0.97; CHCl3). For ent ‐23: HRMS (ESI) m/z calcd. for C23H36ClNO3Si: (M+H)+, 436.2069; found: 436.2070 (M+H)+; Rf (n‐heptane/ethyl acetate 4 : 1): 0.28.
(1aR,2R,7aR)‐7 a‐(3‐Methylbut‐2‐en‐1‐yl)‐5‐((E)‐pent‐1‐en‐1‐yl)‐2‐((triisopropylsilyl)oxy)‐1 a,7 a‐dihydrooxireno[2,3‐g]isoquinolin‐7(2H)‐one (24): The reaction was carried out under argon atmosphere. 7 (0.017 g, 0.039 mmol, 1.00 eq.) was dissolved in 1,4‐dioxane/H2O 8 : 1 (5.00 mL/0.625 mL). trans‐1‐Penten‐1‐ylboronic acid pinacol ester (0.018 g, 0.16 mmol, 4.00 eq.) as well as Cs2CO3 (0.038 g, 0.12 mmol, 3.00 eq.) were added and the solution was degassed by bubbling a stream of argon through it for 15 min. Then APhos Pd G3 (0.001 g, 0.002 mmol, 0.10 eq.) was added. The resulting mixture was stirred for 2 h 30 min at 100 °C, [28] then the reaction mixture was allowed to get to room temperature, diluted with saturated aqueous NH4Cl (15 mL) and extracted with ethyl acetate (2×30 mL). The combined organic layers were washed with saturated aqueous NaCl (20 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (0–15 % ethyl acetate in n‐heptane) to give 24 (0.009 g, 0.02 mmol, 51 %) as colorless oil. 1 H‐NMR (CDCl3, 400 MHz): δ=8.89 (s, 1H, N‐CH), 7.56 (s, 1H, pentenyl‐Cq‐CH), 6.79 (dt, 1H, J=15.6, 7.1 Hz, CH2‐CH=CH), 6.52 (dt, 1H, J=15.5, 1.3 Hz, CH2‐CH=CH), 5.36 (s, br, 1H, CH‐OTIPS), 5.09 (m, 1H, Me2C=CH), 3.77 (d, 1H, J=1.6 Hz, epoxy‐CH), 2.89 (dd, 1H, J=15.3, 8.0 Hz, Me2C=CH‐CH 2), 2.63 (dd, 1H, J=15.3, 7.0 Hz, Me2C=CH‐CH 2), 2.25 (m, 2H, CH 2‐CH=CH), 1.73 (s, 3H, E‐CH 3), 1.67 (s, 3H, Z‐CH 3), 1.58–1.50 (m, 2H, CH3‐CH 2), 1.34–1.09 (m, 21H, TIPS‐H), 0.96 (t, 3H, J=7.4 Hz, CH 3‐CH2); 13 C‐NMR (CDCl3, 100 MHz): δ=195.1 (C=O), 156.8 (N‐C q), 149.2 (N‐CH), 137.3 (CH2‐CH=CH), 136.8 (C qMe2), 135.8 (Ar‐C q‐CO), 131.1 (N‐CH‐C q), 129.4 (CH2‐CH=CH), 116.4 (CH=CMe2), 116.3 (pentenyl‐Cq ‐CH), 66.5 (CH‐OTIPS), 62.1 (epoxy‐C q), 59.1 (epoxy‐CH), 35.1 (CH2‐CH=CH), 26.1 (Me2C=CH‐CH2), 25.9 (E‐CH3), 22.2 (CH3‐CH2), 18.2 (Z‐CH3), 18.3/18.2 (CH(CH3)2), 12.9 (CH(CH3)2), 13.9 (CH3‐CH2); HRMS (ESI) m/z calcd. for C28H44NO3Si: 470.3085 (M+H)+; found: 470.3079 (M+H)+; Rf (n‐heptane/ethyl acetate 8 : 1): 0.41; Specific rotation =+146.2 (c=0.47; CHCl3).
(1aR,2S,7aR)‐7 a‐(3‐Methylbut‐2‐en‐1‐yl)‐5‐((E)‐pent‐1‐en‐1‐yl)‐2‐((triisopropylsilyl)oxy)‐1 a,7 a‐dihydrooxireno[2,3‐g]isoquinolin‐7(2H)‐one (26): 26 (0.102 g, 0.217 mmol, 84 %) was synthesized in analogous manner to 24 starting from 25 (0.114 g, 0.261 mmol) and was obtained as colorless oil. 1 H‐NMR (CDCl3, 600 MHz): δ=8.60 (s, 1H, N‐CH), 7.63 (s, 1H, pentenyl‐Cq‐CH), 6.79 (dt, 1H, J=15.7, 7.0 Hz, CH2‐CH=CH), 6.53 (dt, 1H, J=15.7, 1.5 Hz, CH2‐CH=CH), 5.41 (d, 1H, J=2.6 Hz, CH‐OTIPS), 5.12 (m, 1H, Me2C=CH), 3.79 (d, 1H, J=2.6 Hz, epoxy‐CH), 2.98 (dd, 1H, J=15.2, 7.9 Hz, Me2C=CH‐CH 2), 2.55 (dd, 1H, J=15.4, 6.8 Hz, Me2C=CH‐CH 2), 2.26 (m, 2H, CH 2‐CH=CH), 1.72 (s, 3H, E‐CH 3), 1.68 (s, 3H, Z‐CH 3), 1.53 (m, 2H, CH3‐CH 2), 1.07–0.98 (m, 21H, TIPS‐H), 0.96 (t, 3H, J=7.4 Hz, CH 3‐CH2); 13 C‐NMR (CDCl3, 100 MHz): δ=195.2 (C=O), 157.9 (N‐C q), 150.9 (N‐CH), 137.9 (CH2‐CH=CH), 137.0 (Ar‐C q‐CO), 136.3 (C qMe2), 131.3 (N‐CH‐C q), 129.5 (CH2‐CH=CH), 116.9 (pentenyl‐Cq ‐CH), 116.7 (CH=CMe2), 65.8 (CH‐OTIPS), 60.9 (epoxy‐C q), 60.5 (epoxy‐CH), 35.1 (CH2‐CH=CH), 26.6 (Me2C=CH‐CH2), 26.0 (E‐CH3), 22.2 (CH3‐CH2), 18.2 (Z‐CH3), 18.2/18.1 (CH(CH3)2), 12.7 (CH(CH3)2), 13.9 (CH3‐CH2); HRMS (ESI) m/z calcd. for C28H44NO3Si: (M+H)+, 470.3085; found: 470.3127 (M+H)+; Rf (n‐heptane/ethyl acetate 10 : 1): 0.27; Specific rotation =+179.3 (c=0.33; CHCl3).
Ent ‐24 (0.012 g, 0.026 mmol, 54 %) and ent ‐26 (0.066 g, 0.141 mmol, 70 %) were synthesized in analogous manner to 24 and 26 starting from ent ‐7 (0.021 g, 0.047 mmol) and ent ‐25 (0.088 g, 0.20 mmol). The NMR data are identical to the ones reported for 24 and 26. For ent ‐24: HRMS (ESI) m/z calcd. for C28H44NO3Si: 470.3085 (M+H)+; found: 470.3086 (M+H)+; Rf (n‐heptane/ethyl acetate 8 : 1): 0.41; Specific rotation =−130.2 (c=0.57; CHCl3). For ent ‐26: HRMS (ESI) m/z calcd. for C28H44NO3Si: 470.3085 (M+H)+; found: 470.3097 (M+H)+; Rf (n‐heptane/ethyl acetate 10 : 1): 0.27; Specific rotation =−119.1 (c=0.98; CHCl3).
(1aR,2R,7aR)‐2‐Hydroxy‐7 a‐(3‐methylbut‐2‐en‐1‐yl)‐5‐((E)‐pent‐1‐en‐1‐yl)‐1 a,7 a‐dihydrooxireno[2,3‐g]isoquinolin‐7(2H)‐one (4): Condition 1: In a 15 mL falcon tube, TBAF (1 M in THF, 0.030 mL, 0.030 mmol, 1.20 eq.), followed by glacial acetic acid (0.001 mL, 0.03 mmol, 1.00 eq.) were added to a solution of 24 (0.012 g, 0.025 mmol, 1.00 eq.) in THF (1.5 mL). After 1 h 30 min reaction time, the TLC showed complete conversion of the starting material. The reaction mixture was loaded on silica and purified via flash column chromatography (n‐heptane/ ethyl acetate 3 : 1, 5 g silica). Product 4 (0.005 g, 0.02 mmol, 60 %) was obtained as colorless solid.
Condition 2 (For synthesis and structure of SI1 see Supporting Information): To a solution of SI1 (0.027 g, 0.077 mmol, in THF/H2O (5 : 1, 4.0 mL/0.8 mL) LiOH ⋅ H2O (0.007 g, 0.2 mmol, 2.00 eq.) was added. The TLC showed a complete conversion after 1 h 30 min reaction time. Saturated aqueous NaHCO3 (20 mL) was added. The mixture was extracted with ethyl acetate (2×30 mL) and the combined organic layers were washed with saturated aqueous NaCl (20 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (0–75 % ethyl acetate in n‐heptane) to give 4 (0.012 g, 0.040 mmol, 51 %) as a colorless solid. 1 H‐NMR (MeOD, 600 MHz): δ=8.75 (s, 1H, N‐CH), 7.68 (s, 1H, pentenyl‐Cq‐CH), 6.79 (dt, 1H, J=15.8, 7.1 Hz, CH2‐CH=CH), 6.56 (dt, 1H, J=15.8, 1.5 Hz, CH2‐CH=CH), 5.18 (s, br, 1H, CH‐OH), 5.14 (m, 1H, Me2C=CH), 3.83 (d, 1H, J=1.9 Hz, epoxy‐CH), 2.82 (dd, 1H, J=15.2, 8.0 Hz, Me2C=CH‐CH 2), 2.65 (dd, 1H, J=15.2, 6.9 Hz, Me2C=CH‐CH 2), 2.27 (m, 2H, CH 2‐CH=CH), 1.74 (s, 3H, E‐CH 3), 1.70 (s, 3H, Z‐CH 3), 1.56 (m, 2H, CH3‐CH 2), 0.99 (t, 3H, J=7.4 Hz, CH 3‐CH2); 13 C‐NMR (MeOD, 150 MHz): δ=195.4 (C=O), 157.5 (N‐C q), 149.9 (N‐CH), 138.5 (CH2‐CH=CH), 137.7 (Ar‐C q‐CO), 137.0 (C qMe2), 133.8 (N‐CH‐C q), 130.0 (CH2‐CH=CH), 117.8 (CH=CMe2), 117.1 (pentenyl‐Cq ‐CH), 65.3 (CH‐OH), 63.1 (epoxy‐C q), 61.1 (epoxy‐CH), 36.0 (CH2‐CH=CH), 27.2 (Me2C=CH‐CH2), 26.0 (E‐CH3), 23.2 (CH3‐CH2), 18.1 (Z‐CH3), 14.1 (CH3‐CH2); 1 H‐NMR (CDCl3, 400 MHz): δ=8.90 (s, 1H, N‐CH), 7.63 (s, 1H, pentenyl‐Cq‐CH), 6.83 (dt, 1H, J=15.8, 7.1 Hz, CH2‐CH=CH), 6.55 (dt, 1H, J=15.8, 1.5 Hz, CH2‐CH=CH), 5.18 (s, br, 1H, CH‐OH), 5.08 (m, 1H, Me2C=CH), 3.88 (d, 1H, J=2.1 Hz, epoxy‐CH), 2.91 (dd, 1H, J=15.3, 8.0 Hz, Me2C=CH‐CH 2), 2.70 (s, br, 1H, OH), 2.62 (dd, 1H, J=15.4, 6.9 Hz, Me2C=CH‐CH 2), 2.26 (m, 2H, CH 2‐CH=CH), 1.73 (s, 3H, E‐CH 3), 1.38 (s, 3H, Z‐CH 3), 1.54 (m, 2H, CH3‐CH 2), 0.96 (t, 3H, J=7.4 Hz, CH 3‐CH2); 13 C‐NMR (CDCl3, 100 MHz): δ=193.9 (C=O), 156.9 (N‐C q), 149.4 (N‐CH), 138.4 (CH2‐CH=CH), 137.0 (C qMe2), 135.8 (Ar‐C q‐CO), 131.0 (N‐CH‐C q), 128.8 (CH2‐CH=CH), 116.5 (pentenyl‐Cq ‐CH), 116.0 (CH=CMe2), 65.0 (CH‐OH), 62.8 (epoxy‐C q), 59.7 (epoxy‐CH), 35.1 (CH2‐CH=CH), 26.5 (Me2C=CH‐CH2), 26.0 (E‐CH3), 22.2 (CH3‐CH2), 18.2 (Z‐CH3), 13.9 (CH3‐CH2); HRMS (ESI) m/z calcd. for C19H24NO3: 314.1751 (M+H)+; found: 314.1751 (M+H)+; Rf (n‐heptane/ethyl acetate 1 : 1): 0.37; chiral HPLC (Chiralpak IC; hexane:isopropyl alcohol 85 : 15): 4 (t R=13.8 min) : ent ‐4 (t R=11.1 min) 96.6 : 3.4 (93 % ee); Specific rotation =+159.3 (c=0.27; CHCl3).
(1aR,2S,7aR)‐2‐Hydroxy‐7 a‐(3‐methylbut‐2‐en‐1‐yl)‐5‐((E)‐pent‐1‐en‐1‐yl)‐1 a,7 a‐dihydrooxireno[2,3‐g]isoquinolin‐7(2H)‐one (27): 27 (0.044 g, 0.14 mmol, 65 %) was synthesized in analogous manner to 4 utilizing condition 1 starting from 26 (0.102 g, 0.217 mmol) and was obtained as colorless oil. 1 H‐NMR (MeOD, 500 MHz): δ=8.61 (s, 1H, N‐CH), 7.72 (s, 1H, pentenyl‐Cq‐CH), 6.82 (dt, 1H, J=15.8, 7.1 Hz, CH2‐CH=CH), 6.56 (dt, 1H, J=15.8, 1.5 Hz, CH2‐CH=CH), 5.21 (d, 1H, J=2.3 Hz, CH‐OH), 5.15 (m, 1H, Me2C=CH), 3.82 (d, 1H, J=2.3 Hz, epoxy‐CH), 2.84 (dd, 1H, J=15.2, 8.0 Hz, Me2C=CH‐CH 2), 2.63 (dd, 1H, J=15.2, 6.9 Hz, Me2C=CH‐CH 2), 2.27 (m, 2H, CH 2‐CH=CH), 1.73 (s, 3H, E‐CH 3), 1.69 (s, 3H, Z‐CH 3), 1.55 (m, 2H, CH3‐CH 2), 0.98 (t, 3H, J=7.4 Hz, CH 3‐CH2); 13 C‐NMR (MeOD, 125 MHz): δ=195.5 (C=O), 158.5 (N‐C q), 152.5 (N‐CH), 139.0 (CH2‐CH=CH), 138.2 (Ar‐C q‐CO), 137.0 (C qMe2), 133.6 (N‐CH‐C q), 130.1 (CH2‐CH=CH), 117.8 (CH=CMe2), 117.5 (pentenyl‐Cq ‐CH), 64.9 (CH‐OH), 62.1 (epoxy‐C q), 61.9 (epoxy‐CH), 36.0 (CH2‐CH=CH), 27.5 (Me2C=CH‐CH2), 26.0 (E‐CH3), 23.2 (CH3‐CH2), 18.1 (Z‐CH3), 14.1 (CH3‐CH2); 1 H‐NMR (CDCl3, 400 MHz): δ=8.53 (s, 1H, N‐CH), 7.62 (s, 1H, pentenyl‐Cq‐CH), 6.76 (dt, 1H, J=15.7, 7.0 Hz, CH2‐CH=CH), 6.46 (dt, 1H, J=15.7, 1.6 Hz, CH2‐CH=CH), 5.27 (d, 1H, J=2.3 Hz, CH‐OH), 5.09 (m, 1H, Me2C=CH), 3.85 (d, 1H, J=2.3 Hz, epoxy‐CH), 3.18 (s, br, 1H, OH), 2.84 (dd, 1H, J=15.5, 7.9 Hz, Me2C=CH‐CH 2), 2.64 (dd, 1H, J=15.4, 6.7 Hz, Me2C=CH‐CH 2), 2.24 (m, 2H, CH 2‐CH=CH), 1.73 (s, 3H, E‐CH 3), 1.68 (s, 3H, Z‐CH 3), 1.53 (m, 2H, CH3‐CH 2), 0.96 (t, 3H, J=7.4 Hz, CH 3‐CH2); 13 C‐NMR (CDCl3, 100 MHz): δ=194.5 (C=O), 157.9 (N‐C q), 151.1 (N‐CH), 138.8 (CH2‐CH=CH), 136.8 (Ar‐C q‐CO), 136.6 (C qMe2), 131.1 (N‐CH‐C q), 128.9 (CH2‐CH=CH), 116.9 (pentenyl‐Cq ‐CH), 116.1 (CH=CMe2), 64.6 (CH‐OH), 61.1 (epoxy‐C q), 60.4 (epoxy‐CH), 35.1 (CH2‐CH=CH), 26.5 (Me2C=CH‐CH2), 26.0 (E‐CH3), 22.2 (CH3‐CH2), 18.2 (Z‐CH3), 13.9 (CH3‐CH2); HRMS (ESI) m/z calcd. for C19H24NO3: 314.1751 (M+H)+; found: 314.1755 (M+H)+; Rf (n‐heptane/ethyl acetate 1 : 1): 0.44; chiral HPLC (Chiralpak IC; hexane:isopropyl alcohol 85 : 15): 27 (t R=6.6 min) : ent ‐27 (t R=4.9 min) 96.5 : 3.5 (93 % ee); Specific rotation =+179.3 (c=0.33; CHCl3).
Ent ‐4 (0.005 g, 0.01 mmol, 59 %) and ent ‐27 (0.032 g, 0.10 mmol, 74 %) were synthesized in analogous manner to 4 and 27 utilizing condition 1 and starting from ent ‐24 (0.012 g, 0.026 mmol) and ent ‐26 (0.066 g, 0.14 mmol). The NMR data are identical to the ones reported for 4 and 27. For ent ‐4: HRMS (ESI) m/z calcd. for C19H24NO3: 314.1751 (M+H)+; found: 314.1750 (M+H)+; Rf (n‐heptane/ethyl acetate 1 : 1): 0.37; chiral HPLC (Chiralpak IC; hexane:isopropyl alcohol 85 : 15): ent ‐4 (t R=11.1 min): 4 (t R=13.8 min) 98.5 : 1.5 (97 % ee); Specific rotation =−206.0 (c=0.22; CHCl3). For ent ‐27: HRMS (ESI) m/z calcd. for C19H24NO3: 314.1751 (M+H)+; found: 314.1748 (M+H)+; Rf (n‐heptane/ethyl acetate 1 : 1): 0.44; chiral HPLC (Chiralpak IC; hexane:isopropyl alcohol 85 : 15): ent ‐27 (t R=4.9 min) : 27 (t R=6.6 min) 96.5 : 3.5 (93 % ee); Specific rotation =−160.3 (c=1.20; CHCl3).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This study was financially supported by the Hessen State Ministry of Higher Education, Research, and the Arts (HMWK) via generous grand for the LOEWE Research Center Insect Biotechnology and Bioresources. Sanofi and Evotec International contributed in the framework of Sanofi‐Fraunhofer Natural Product Center of Excellence/Fraunhofer Evotec Natural Products Excellence Center. This work was supported in part by the Bill & Melinda Gates Foundation [INV‐001913]. For generous support we thank the NMR department of the JLU Gießen, Stefan Bernhardt (chiral HPLC analysis), and Christoph Hartwig (technical assistance) as well as Jennifer Kuhn, Christine Wehr, and Dr. Luigi Toti (fermentation). For performing the assays and for providing the data of compound profiling we thank colleagues at Fraunhofer, Evotec, and Cyprotex and here especially Kirsten‐Susann Bommersheim, Stéphanie Sans, Kerry Frost, Corinne Lafon, Marion Martignac and their teams. We thank Dr. Cédric Couturier, Dr. Frédéric Jeannot, Dr. Eric Bacqué, Dr. Laurent Goullieux, Dr. Jean‐Michel Culouscou, and Prof. Dr. Peter Schreiner for valuable discussions.
Y. Kleiner, C. Pöverlein, J. Klädtke, M. Kurz, H. F. König, J. Becker, S. Mihajlovic, F. Zubeil, M. Marner, A. Vilcinskas, T. F. Schäberle, P. Hammann, S. M. M. Schuler, A. Bauer, ChemMedChem 2022, 17, e202100644.
Contributor Information
Dr. Sören M. M. Schuler, Email: soeren.schuler@evotec.com.
Dr. Armin Bauer, Email: armin.bauer@sanofi.com.
References
- 1.World Health Organization, 2020, Global tuberculosis report 2020, Geneva.
- 2. Seung K. J., Hewison C., Lancet Glob. Health 2019, 7, e706. [DOI] [PubMed] [Google Scholar]
- 3. Mdluli K., Kaneko T., Upton A., Cold Spring Harbor Perspect. Med. 2015, 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fox J. L., Nat. Biotechnol. 2014, 32, 305. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Li J. Y., Harper J. K., Grant D. M., Tombe B. O., Bashyal B., Hess W. M., Strobel G. A., Phytochemistry 2001, 56, 463; [DOI] [PubMed] [Google Scholar]
- 5b. Jung S. H., Hwang G.-S., Lee S. I., Ryu D. H., J. Org. Chem. 2012, 77, 2513. [DOI] [PubMed] [Google Scholar]
- 6. Kleiner Y., Hammann P., Becker J., Bauer A., Pöverlein C., Schuler S. M. M., J. Org. Chem. 2020, 85, 13108. [DOI] [PubMed] [Google Scholar]
- 7. Mehta G., Sengupta S., Tetrahedron 2017, 73, 6223. [Google Scholar]
- 8.
- 8a. Miyashita K., Imanishi T., Chem. Rev. 2005, 105, 4515; [DOI] [PubMed] [Google Scholar]
- 8b. Marco-Contelles J., Molina M. T., Anjum S., Chem. Rev. 2004, 104, 2857. [DOI] [PubMed] [Google Scholar]
- 9.The increasing amount of 6 during acidic HPLC repurification of 3 clearly indicates 6 as an artificial degradation product of 3.
- 10.
- 10a.For a proposal of a plausible biosynthetic pathway see Supporting Information;
- 10b. Yu X., Gao Y., Frank M., Mándi A., Kurtán T., Müller W. E., Kalscheuer R., Guo Z., Zou K., Liu Z., et al., Tetrahedron 2021, 79, 131876. [Google Scholar]
- 11. Honraedt A., Gallagher T., Synlett 2016, 27, 67. [Google Scholar]
- 12.Prolonged reaction times resulted in a significant reduction of isolated yields. By careful monitoring of the reaction progress by liquid chromatography mass spectrometry (LC–MS), an acceptable compromise between consumption of starting material and side product formation was achieved.
- 13.A base screening for high E-selectivity was performed, for details see Supporting Information.
- 14. Lakshminarayana S. B., Huat T. B., Ho P. C., Manjunatha U. H., Dartois V., Dick T., Rao S. P. S., J. Antimicrob. Chemother. 2015, 70, 857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Nicolaou C. A., Brown N., Pattichis C. S., Curr. Opin. Drug Discov. Devel. 2007, 10, 316; [PubMed] [Google Scholar]
- 15b. Ekins S., Honeycutt J. D., Metz J. T., Drug Discovery Today 2010, 15, 451. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Machado D., Girardini M., Viveiros M., Pieroni M., Front. Microbiol. 2018, 9, 1367; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Kling A., Lukat P., Almeida D. V., Bauer A., Fontaine E., Sordello S., Zaburannyi N., Herrmann J., Wenzel S. C., König C., et al., Science 2015, 348, 1106. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Artursson P., Karlsson J., Biochem. Biophys. Res. Commun. 1991, 175, 880; [DOI] [PubMed] [Google Scholar]
- 17b. Press B., Di Grandi D., Curr. Drug Metab. 2008, 9, 893. [DOI] [PubMed] [Google Scholar]
- 18. Peng Y., Yadava P., Heikkinen A. T., Parrott N., Railkar A., Eur. J. Pharm. Sci. 2014, 56, 120. [DOI] [PubMed] [Google Scholar]
- 19.SF008-D was isolated as main component in a 0.6 mg mixture.
- 20.The absolute stereochemistry of (+)-floyocidin A (3) was not unambiguously determined. The showed structure is our hypothesis deduced from the absolute stereochemistry of (+)-floyocidin B (4).
- 21.The shown absolute stereochemistry of (+)-floyocidin B (4) was unambiguously determined by a combination of total synthesis of synthetic references, single X-ray crystal diffraction of intermediate 17, and comparison with an authentic sample of NP (+)-floyocidin B (4). For details see chapter 1.2 of Supporting Information and main manuscript.
- 22.The absolute stereochemistry of floyocidin C (5) and floyocidin D (6) was not unambiguously determined. The showed structures are our hypothesis deduced from the absolute stereochemistry of (+)-floyocidin B (4).
- 23.Because of sublimation of the product, careful concentration in vacuo (≥200 mbar) is necessary.
- 24.A stagnation of the acyl migration was observed after stirring overnight and the reaction was therefore stopped. The two regioisomers could not be separated by chromatography. The minor isomer was finally removed on a later stage by flash chromatography after TIPS-protection and saponification.
- 25.After full conversion to the nitrile, the reaction mixtures were treated by a simple aqueous work-up (ethyl acetate vs. saturated aqueous NaCl). Thereby, oxidative side reactions occurred and led to significantly reduced yields of isolated products ent -8 and ent -22 after flash chromatography. This emphasize the importance of a reductive aqueous work-up as described for the synthesis of the enantiomers 8 and 22. Since sufficient amounts of ent -8 and ent -22 were obtained to progress the synthesis, the reactions where not repeated under optimized conditions.
- 26.A 2.5 M solution of n-BuLi in hexane from a commercially supplier was diluted by a factor of four at −78 °C with anhydrous THF for a better handling. For the −100 °C cooling bath a mixture of ethanol and liquid N2 was used.
- 27.No side product formation was observed for the trans-isomer.
- 28.The progress of the reaction was carefully monitored by LC-MS measurement every 30 minutes since prolonged reaction times led to degradation of the desired product and significant reduction of isolated yield. Usually, durations of 1 h 30 min to 4 h were identified as a good compromise of reaction progress and product degradation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information