

Abstract
Arginine vasopressin (AVP) serves as a neuromodulator in the brain. The hippocampus is one of the major targets for AVP, as it has been demonstrated that the hippocampus receives vasopressinergic innervation and expresses AVP receptors. The dentate gyrus (DG) granule cells (GCs) serve as a gate governing the inflow of information to the hippocampus. High densities of AVP receptors are expressed in the DG GCs. However, the roles and the underlying cellular and molecular mechanisms of AVP in the DG GCs have not been determined. We addressed this question by recording from the DG GCs in rat hippocampal slices. Our results showed that application of AVP concentration-dependently evoked an inward holding current recorded from the DG GCs. AVP depolarized the DG GCs and increased their action potential firing frequency. The excitatory effects of AVP were mediated by activation of V1a receptors and required the function of phospholipase Cβ (PLCβ). Whereas intracellular Ca2+ release and protein kinase C activity were unnecessary, PLCβ-induced depletion of phosphatidylinositol 4,5-bisphosphate was involved in AVP-evoked excitation of the DG GCs. AVP excited the DG GCs by depression of the ATP-sensitive K+ channels, which were required for AVP-elicited facilitation of long-term potentiation at the perforant path–GC synapses. Our results may provide a cellular and molecular mechanism to explain the physiological functions of AVP, such as learning and memory, and pathologic disorders like anxiety.
Keywords: action potential, depolarization, hippocampus, peptide, receptors, signal transduction
Significance Statement
Dentate gyrus is the first station of the hippocampus and serves as the gate governing the inflow of information to the hippocampus. Modification of the excitability of the dentate gyrus granule cells likely plays a significant role in the expression of hippocampal functions. We showed that activation of V1a receptors excites dentate gyrus granule cells by phospholipase Cβ-mediated depression of ATP-sensitive K+ channels and that this cellular mechanism is responsible for arginine vasopressin (AVP)-elicited facilitation of long-term potentiation. Our results may provide a cellular and molecular mechanism to explain the physiological functions of AVP, such as learning and memory, and pathologic disorders like anxiety.
Introduction
Arginine vasopressin (AVP) is a nonapeptide synthesized in the paraventricular and supraoptic nuclei of the hypothalamus. AVP is then transported along the axons of these neurosecretory cells to the posterior pituitary where it is released into the bloodstream to exert its hormonal functions in the periphery on blood vessels, kidney, and uterus (Stoop, 2012). Additionally, vasopressinergic fibers from the parvocellular neurons of the hypothalamus project to discrete extrahypothalamic limbic brain regions including the hippocampus, subiculum, amygdala, and nucleus accumbens (Buijs et al., 1978; Buijs and Swaab, 1979; Lang et al., 1983; DeVries et al., 1985; Hawthorn et al., 1985). While the hypothalamus and pituitary form the major source of AVP in the brain, AVP immunoreactivity has also been detected in neurons in the extrahypothalamic structures, including the bed nucleus of stria terminalis, septal region, medial amygdala, and locus coeruleus (Caffé and van Leeuwen, 1983; van Leeuwen and Caffé, 1983; Sofroniew, 1985), although the targets of these vasopressinergic projections have not been clearly defined.
The biological functions of AVP are mediated by interacting with three types of vasopressin receptors: V1a, V1b, and V2 receptors. V1a and V1b receptors are coupled to the Gαq/11 proteins activating phospholipase Cβ (PLCβ), which further breaks down phosphatidylinositol 4,5-bisphosphate (PIP2) to generate 1,4,5-trisphosphate (IP3) to elevate intracellular Ca2+ release and diacylglycerol (DAG) to activate protein kinase C (PKC). V2 receptors are coupled to Gs-proteins, increasing the activity of adenylyl cyclase to elevate cyclic AMP levels. In the brain, AVP serves as a neuromodulator that regulates a variety of physiological functions including social behaviors (Cilz et al., 2019; Kompier et al., 2019), learning and memory (de Wied et al., 1993; Caldwell et al., 2008), nociception (Koshimizu and Tsujimoto, 2009), circadian rhythms (Gizowski et al., 2017), and neurologic diseases such as anxiety (Caldwell et al., 2008; Neumann and Landgraf, 2012). However, the cellular and molecular mechanisms whereby AVP modulates these physiological functions and pathologic disorders have not been completely determined.
The hippocampus is one of the major biological targets for AVP because high densities of vasopressin receptors have been detected in the hippocampus (Biegon et al., 1984; Brinton et al., 1984; De Kloet et al., 1985; Lawrence et al., 1988) and the hippocampus also receives vasopressinergic innervation (Buijs, 1980; Caffé et al., 1987; Metzger et al., 1993). In line with the distributions of both AVP-containing fibers and AVP receptors in the hippocampus, activation of V1a receptors excites both pyramidal neurons (Hu et al., 2022) and interneurons (Ramanathan et al., 2012) in the CA1 region. However, the highest densities of AVP receptors (Brinton et al., 1984; De Kloet et al., 1985; van Leeuwen et al., 1987; Campbell et al., 2009), especially the V1a receptors (Ostrowski et al., 1994; Szot et al., 1994), have been detected in the dentate gyrus (DG), which serves as the gate governing the inflow of information to the hippocampus. Consistent with the anatomic expression of AVP receptors in the DG, bath application of AVP modulates the slope of the field potentials in the DG recorded from in vitro slices, depending on the extracellular Ca2+ concentration (Chen et al., 1993). Furthermore, intracerebroventricular injection of AVP augments long-term potentiation (LTP) in the DG in intact anesthetized rats (Dubrovsky et al., 2003) and induces Fos protein expression in the DG (Paban et al., 1999), suggesting that AVP increases the neuronal excitability in the DG. However, the cellular and molecular mechanisms whereby AVP modulates neuronal excitability and synaptic transmission and plasticity in the DG have not been determined. In this study, we studied the effects of AVP on the excitability of the granule cells (GCs) in the DG. Our results indicate that the activation of V1a receptors increases the excitability of the DG GCs via PLCβ-mediated depression of the ATP-sensitive K+ (KATP) channels. AVP did not modulate glutamatergic transmission but augmented the LTP at the perforant path (PP)–GC synapses. Our results may provide a cellular and molecular mechanism to explain the functions of AVP in vivo.
Materials and Methods
Slice preparation
Horizontal brain slices (350 μm) were prepared from both male and female Sprague Dawley rats (25–40 d old) purchased from Envigo RMS. Animals were housed in the institutional animal center with food and water available ad libitum until use. The animal rooms were maintained on a 14/10 h light/dark cycle (lights on at 7:00 A.M.), with a room temperature of 22°C. All procedures and experiments presented in this study were approved by the Institutional Animal Care and Use Committee and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The number of males and females for each experiment was kept as equal as possible. After being deeply anesthetized with isoflurane, an animal was decapitated and the brain was dissected out. Slices were cut in ice-cold saline solution that contained the following (in mm): 250 glycerol, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 26 NaHCO3, and 11 glucose, at ∼330 mOsm, as described previously (Ye et al., 2006). After incubation at 35°C for 60 min in the extracellular solution containing (in mm) 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 2.5 CaCl2, 1.5 MgCl2, and 10 glucose, saturated with 95% O2 and 5% CO2, slices were kept at room temperature until use. All animal procedures conformed to the guidelines approved by the Institutional Animal Care and Use Committee.
Recordings of action potentials, resting membrane potentials, and holding currents from the DG GCs
Whole-cell recordings using a Multiclamp 700B amplifier (Molecular Devices) in voltage-clamp or current-clamp mode were made from the DG GCs visually identified with infrared video microscopy (model BX51WI microscope, Olympus) and differential interference contrast optics. During recordings, the bath temperature was maintained at 33–34°C by an inline heater and an automatic temperature controller (model TC-324C, Warner Instruments). The bath solution was the above-mentioned incubation extracellular solution. The recording electrodes were filled with the following (in mm): 120 K+-gluconate, 10 KCl, 2 MgCl2, 10 HEPES, 0.6 EGTA, 2 ATPNa2, 0.4 GTPNa, and 5 phosphocreatine, at pH 7.3, unless stated otherwise. Holding currents at −60 mV and resting membrane potentials (RMPs) were recorded in the extracellular solution supplemented with tetrodotoxin (TTX; 0.5 μm), kynurenic acid (1 mm), and picrotoxin (100 μm) to block action potential (AP) firing, glutamatergic transmission, and GABAergic transmission, respectively. APs evoked by injections of a series of positive currents from 25 to 400 pA at an interval of 25 pA were recorded in the above solution without TTX. AVP was dissolved in the extracellular solution and bath applied. To avoid potential desensitization induced by repeated applications of the agonist, one slice was limited to only one application of AVP. Pharmacological inhibitors were applied to the cells either extracellularly or intracellularly via the recording pipettes. For extracellular application, slices were pretreated for at least 1 h to ensure permeation of reagents into the cells in the slices and the extracellular solution continuously contained the same concentration of the reagents, unless stated otherwise. For intracellular application, we waited for >15 min after the formation of whole-cell configuration to ensure the diffusion of the inhibitors into the cells. Data were filtered at 2 kHz, digitized at 10 kHz, acquired, and analyzed subsequently using pCLAMP 10.7 software (Molecular Devices).
Recordings of AMPA EPSCs and LTP
Whole-cell recordings were used to record AMPA EPSCs at −65 mV from the DG GCs by placing a concentric bipolar stimulation electrode [model MX21XES(DB9), FHC] in the middle to the inner one-third of the molecular layer of the DG to stimulate the medial PP. The intracellular solution was the above K+-gluconate internal solution supplemented with QX-314 (1 mm) to block AP firing. For a subset of experiments, a Cs+-containing intracellular solution was prepared by replacing the K+ in the above solution with the same concentration of Cs+ that was used. The extracellular solution was supplemented with 10 μm bicuculline to block GABAergic transmission. The stimulation intensity was set to the level that produced 30–40% of the maximal amplitude of EPSCs. After recording basal AMPA EPSCs at −65 mV in 0.1 Hz, we applied a protocol by paring presynaptic stimulation (1 Hz, for 40 pulses) with postsynaptic depolarization to −30 mV to induce LTP, as described previously (Colino and Malenka, 1993). Recordings of AMPA EPSCs (at −65 mV in 0.1 Hz) were resumed after the protocol to monitor LTP. The amplitudes of AMPA EPSCs were normalized to the average of those recorded in control condition for 5 min. Series resistance was rigorously monitored by the delivery of 5 mV voltage steps after each evoked current. Experiments were discontinued if the series resistance changed by >15%.
Data analysis
Data were presented as the mean ± SEM. The concentration–response curve of AVP was fit by the Hill equation: I = Imax × {1/[1 + (EC50/[ligand])n]}, where Imax is the maximum response, EC50 is the concentration of ligand producing a half-maximal response, and n is the Hill coefficient. N numbers in the text were the numbers of cells used for each experiment. To minimize potential influences of variation from individual animals, at least four animals were used for each experiment. Because the maximal response occurred within 5 min during the application of AVP, we measured the peak response of AVP for statistical analysis. A Wilcoxon matched-pairs signed-rank test (abbreviated as “Wilcoxon test” in the text), Mann–Whitney test, one-way ANOVA followed by Dunnett’s multiple-comparisons test, or two-way repeated-measures ANOVA followed by a Sidak multiple-comparisons test was used as appropriate for statistical analysis. The control data for AVP-induced inward currents were pooled results from the control experiments performed for each individual pharmacological experiment. A one-way ANOVA followed by Dunnett’s multiple-comparisons test was used for statistical analysis when the pooled control data were used for comparison. The p values were reported throughout the text, and significance was set as p < 0.05.
Chemicals
The following chemicals were products of R&D Systems: AVP, TTX, kynurenic acid, picrotoxin, SR49059, U73122, heparin, thapsigargin, bisindolylmaleimide II (Bis II), RHC 80267, and ML 133. Glibenclamide was purchased from MedChemExpress. diC8-PIP2 was purchased from Echelon Biosciences. Drugs were initially prepared in stock solution, aliquoted, and stored at −20°C. For those chemicals requiring dimethylsulfoxide (DMSO) as a solvent, the concentration of DMSO was <0.1%.
Results
AVP elicits an inward current in the DG GCs via activation of V1a receptors
We probed the effects of AVP on the DG GCs by recording the holding currents at −60 mV in voltage clamp. Bath application of AVP (0.3 μm) evoked an inward current (−19.7 ± 2.1 pA, n = 25; p < 0.0001 vs baseline, Wilcoxon test; Fig. 1A,C). The effect of AVP was mediated by activation of V1a receptors because pretreatment of slices with and continuous bath application of the selective V1a receptor antagonist SR49059 (1 μm) significantly reduced AVP-evoked inward currents (−4.6 ± 1.1 pA, n = 18; p = 0.002 vs baseline, Wilcoxon test; p < 0.0001 vs AVP alone, Mann–Whitney test; Fig. 1B,C), suggesting the involvement of V1a receptors. The EC50 of AVP was calculated to be 0.015 μm (Fig. 1D). We used AVP at 0.3 μm for the remaining experiments because this is a near-saturating concentration.
Figure 1.
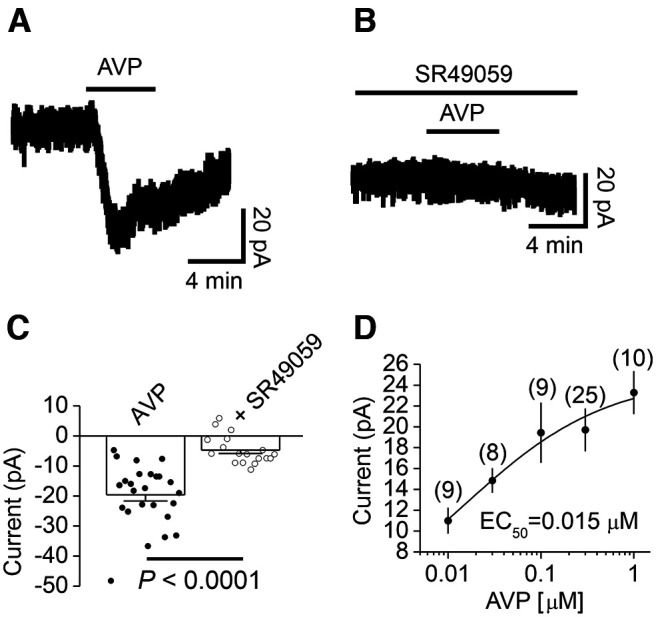
Bath application of AVP elicits an inward current in DG GCs. A, Current trace recorded from a DG GC in response to bath application of AVP (0.3 μm). B, Current trace recorded from a DG GC in a slice pretreated with the selective V1a receptor antagonist SR49095 (1 μm). The extracellular solution continuously contained the same concentration of SR49095. C, Summary graph showing AVP-induced inward currents in control condition or in the presence of SR49095 (1 μm). The circles represent the values from individual cells, and the bars are their averages. D, Concentration–response curve of AVP constructed by measuring AVP-induced inward currents. The numbers within the parentheses were the numbers of cells recorded at each concentration.
AVP-elicited inward currents in the DG GCs are mediated by depressing KATP channels
We further determined the ionic mechanisms underlying AVP-elicited inward currents in the DG GCs. We measured the input resistance (Rin) of the DG GCs before and during the application of AVP by injecting negative currents from 0 to −75 pA with 25 pA steps for a duration of 600 ms. We fit the current–voltage relationship (I–V) with a linear function for each cell to obtain Rin, which equals the slope of the linear fitting. Bath application of AVP depolarizes DG GCs (see below) and increased Rin (control, 155 ± 15 MΩ; AVP, 188 ± 18 MΩ; n = 9; p = 0.004, Wilcoxon test; Fig. 2A,B), suggesting that AVP decreases a membrane conductance. We further measured the I–V of the currents generated by AVP. The extracellular solution was supplemented with TTX (0.5 μm) to block voltage‐gated Na+ channels. Cells were held at −60 mV and stepped from −140 to −40 mV for 400 ms at a voltage interval of 10 mV every 10 s. Steady‐state currents were measured within 5 ms before the end of the step voltage protocol. Under these circumstances, the I–V curve of the AVP‐elicited currents recorded from the DG GCs showed inward rectification with a reversal potential of −90.4 ± 3.4 mV (n = 12; Fig. 2C–E), resembling that of the inwardly rectifying K+ (Kir) channels. This result suggests that AVP-induced inward currents are mediated by inhibiting a Kir channel.
Figure 2.
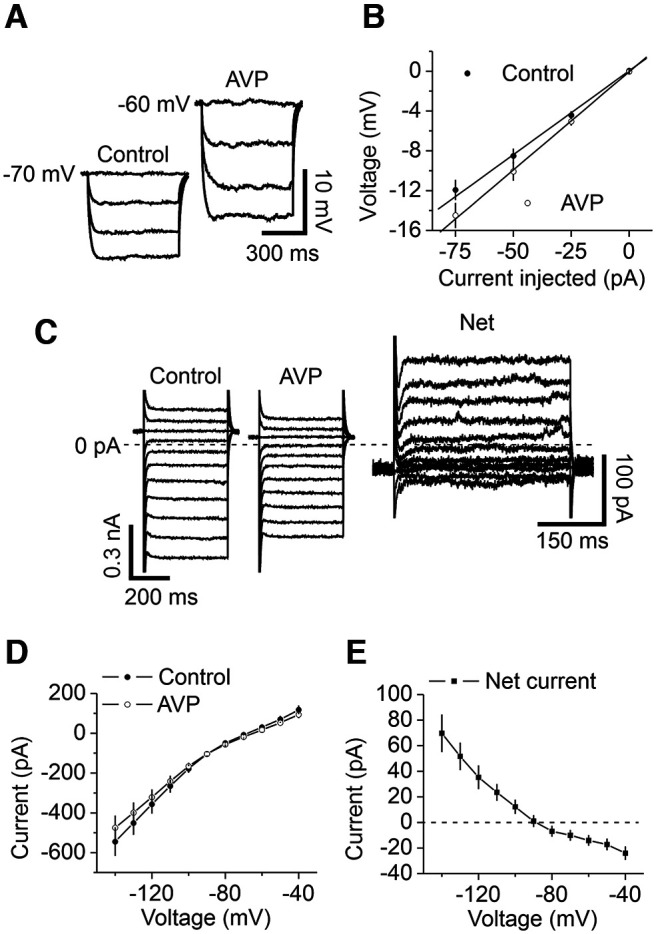
AVP-elicited inward currents are mediated by depression of Kir channels. A, B, AVP increased the input resistance of DG GCs. A, Voltage responses evoked by the injection of negative currents from 0 to −75 pA at an interval of 25 pA before (left) and during (right) the application of AVP from a DG GC. B, The current–voltage relationship averaged from nine cells. Input resistance was obtained by linear fitting of the current–voltage relationship. C, Currents elicited by a voltage step protocol before (left) and during (middle) bath application of AVP and the net current obtained by subtraction (right) from a GC. Cells were held at −60 mV and stepped from −140 to −40 mV for 400 ms at a voltage interval of 10 mV every 10 s. Steady-state currents were measured within 5 ms before the end of the step voltage protocols. Note the differences in the scale bars. The dashed line was the zero current level. D, I–V curve averaged from 12 GCs before and during the application of AVP. E, I–V curve of the net current obtained by subtracting the currents in control condition from those during the application of AVP. Note that the net currents showed inward rectification, suggesting the involvement of Kir channels.
We further confirmed the involvement of Kir channels in AVP-induced inward currents in the DG GCs with Kir channel blockers. Bath application of Ba2+ (500 μm) induced an inward current by itself (−55.0 ± 10.7 pA, n = 11; p = 0.001 vs baseline, Wilcoxon test; Fig. 3A), suggesting that the DG GCs express functional Kir channels. Following application of AVP in the presence of Ba2+ evoked a significantly smaller inward current (−8.5 ± 2.3 pA, n = 11; p = 0.002 vs Ba2+ alone, Wilcoxon test; p = 0.018 vs AVP alone, one-way ANOVA followed by Dunnett’s test; Fig. 3A,E), further confirming the involvement of Kir channels. Kir channels are classified into four functional groups including Kir2, Kir3 [G-protein-gated Kir (GIRK) channels], Kir6 (KATP channels) and K+ transport channels (Hibino et al., 2010). We used ML 133, a specific antagonist for Kir2 subfamily channels (Wang et al., 2011; Kim et al., 2015; Ford and Baccei, 2016; Sonkusare et al., 2016; Huang et al., 2018), to test the roles of the Kir2 subfamily channels in AVP-elicited inward currents. Bath application of ML 133 (30 μm) by itself evoked a small inward current (−6.7 ± 1.3 pA, n = 12; p = 0.001 vs baseline, Wilcoxon test; Fig. 3B). Subsequent application of AVP still elicited a comparable inward current (−24.3 ± 3.0 pA, n = 12; p = 0.0005 vs ML 133 alone, Wilcoxon test; p = 0.808 vs AVP alone, one-way ANOVA followed by Dunnett’s test; Fig. 3B,E), suggesting that the Kir2 subfamily is not involved in AVP-induced inward currents.
Figure 3.

Effects of Kir channel blockers on AVP-elicited inward currents recorded from DG GCs. A, Current trace recorded from a DG GC in response to bath application of Ba2+ (500 μm) alone and concomitant application of AVP. B, Current trace recorded from a DG GC in response to bath application of ML 133 (30 μm) alone and coapplication of AVP. C, Current trace recorded from a DG GC in response to bath application of SCH23390 (40 μm) alone and coapplication of AVP. D, Current trace recorded from a DG GC in response to bath application of glibenclamide (100 μm) alone and coapplication of AVP. E, Summary graph showing the effects of Kir channel blockers on AVP-induced inward currents. The shaded bar was the averaged inward currents evoked by AVP in control condition pooled from the control experiment conducted for each individual pharmacological experiment. *p = 0.018, ****p < 0.0001 versus AVP alone, one-way ANOVA followed by Dunnett’s test.
We further tested the roles of the Kir3 subfamily in AVP-elicited inward currents in the DG GCs. Bath application of the Kir3 channel blocker SCH23390 (40 μm; Kuzhikandathil and Oxford, 2002) evoked an inward current by itself (−29.7 ± 5.1 pA, n = 13; p = 0.0002 vs baseline, Wilcoxon test; Fig. 3C), suggesting the expression of Kir3 channels in the DG GCs. However, application of AVP in the presence of SCH23390 still elicited a comparable inward current (−20.5 ± 2.5 pA, n = 13; p = 0.0002 vs SCH23390 alone, Wilcoxon test; p = 0.999 vs AVP alone, one-way ANOVA followed by Dunnett’s test; Fig. 3C,E), suggesting that the Kir3 subfamily is not involved in AVP-induced inward currents.
Whereas our intracellular solution in the recording pipettes contained 2 mm ATP, which should exert inhibition on KATP channels, the effects of KATP channels on neuronal excitability are not fully blocked by an intracellular solution containing 4 mm ATP (Lemak et al., 2014), possibly because the open probability of KATP channels reflects activity-dependent fluctuations of ATP/ADP concentrations within local submembrane domains that are not entirely controlled by the solution in the patch pipette (Haller et al., 2001; Mollajew et al., 2013). Furthermore, as will be shown below, PLCβ-mediated depletion of PIP2 was involved in AVP-mediated inward currents, and PIP2 alters the sensitivity of KATP channels to ATP (Hilgemann and Ball, 1996; Fan and Makielski, 1997; Baukrowitz et al., 1998; Shyng and Nichols, 1998). Another rationale to test the roles of KATP channels is that high densities of KATP channels are expressed in the DG GCs (Mourre et al., 1990; Zawar et al., 1999; Pelletier et al., 2000; Tanner et al., 2011). We thus probed the roles of KATP channels in AVP-induced inward currents by testing the hypothesis that activation of V1a receptors generates an inward current by depressing KATP channels in the DG GCs. The premise of this hypothesis is that KATP channels should be open in the resting condition. We therefore used the selective KATP channel blocker glibenclamide. Bath application of glibenclamide (100 μm) by itself induced an inward current (−22.8 ± 3.8 pA, n = 18; p < 0.0001 vs baseline, Wilcoxon test; Fig. 3D), suggesting a tonic activation of KATP channels in the DG GCs. Following application of AVP in the continuous presence of glibenclamide generated a significantly smaller inward current (−5.6 ± 1.1 pA, n = 18; p = 0.0002 vs glibenclamide alone, Wilcoxon test; p < 0.0001 vs AVP alone, one-way ANOVA followed by Dunnett’s test; Fig. 3D,E), suggesting that AVP-generated inward currents were mediated by depressing KATP channels in the DG GCs.
PLCβ, but not PKC or intracellular Ca2+ release is necessary for AVP-elicited inward currents in the DG GCs
V1a receptors are coupled to Gαq/11 proteins elevating the activity of PLCβ, which hydrolyzes PIP2 to generate IP3 to increase intracellular Ca2+ release and DAG to activate PKC. We tested the roles of these signaling molecules in AVP-mediated inward currents in the DG GCs. Pretreatment of slices with and continuous bath application of the PLC inhibitor U73122 (5 μm) significantly reduced AVP-induced inward currents (−7.6 ± 2.0 pA, n = 15; p = 0.003 vs baseline, Wilcoxon test; p = 0.002 vs AVP alone, one-way ANOVA followed by Dunnett’s test; Fig. 4B,H), suggesting the involvement of PLCβ. We then tested the roles of intracellular Ca2+ released from the IP3 store and PKC in AVP-elicited inward currents. Dialysis of the IP3 receptor blocker heparin at an effective concentration (0.5 mg/ml; Saleem et al., 2014), via the recording pipettes, failed to alter significantly AVP-induced inward currents (−25.6 ± 3.2 pA, n = 12; p = 0.0005 vs baseline, Wilcoxon test; p = 0.523 vs AVP alone, one-way ANOVA followed by Dunnett’s test; Fig. 4C,H), suggesting that Ca2+ released from the IP3 store is not required for AVP-induced inward currents. Likewise, intracellular application of the endoplasmic reticulum Ca2+-ATPase inhibitor thapsigargin (10 μm) via the recording pipettes did not significantly change AVP-induced inward currents (−26.1 ± 2.7 pA, n = 13; p = 0.0002 vs baseline, Wilcoxon test; p = 0.378 vs AVP alone, one-way ANOVA followed by Dunnett’s test; Fig. 4D,H), suggesting that intracellular Ca2+ release is unnecessary for AVP-mediated inward currents. We further examined the roles of PKC in AVP-induced inward currents. Pretreatment of slices with and continuous bath application of the selective PKC inhibitor Bis II (1 μm) failed to block AVP‐elicited inward currents (−26.9 ± 5.5 pA, n = 12; p = 0.0005 vs baseline, Wilcoxon test; p = 0.282 vs AVP alone, one-way ANOVA followed by Dunnett’s test; Fig. 4E,H), suggesting that the function of PKC is not involved in AVP-induced inward currents.
Figure 4.

AVP-elicited inward currents depend on PLCβ and depletion of PIP2, but do not require the functions of intracellular Ca2+ release and PKC activity. A, Current trace recorded from a DG GC in response to bath application of AVP alone. B, Current trace recorded from a DG GC in response to bath application of AVP in a slice pretreated with the PLC inhibitor U73122 (5 μm). C, Current trace recorded from a DG GC before, during, and after bath application of AVP in the intracellular solution containing heparin (0.5 mg/ml). D, Current trace recorded from a DG GC in response to bath application of AVP in the intracellular solution supplemented with thapsigargin (10 μm). E, AVP-induced inward current recorded from a DG GC in a slice pretreated with Bis II (2 μm). The extracellular solution continuously contained the same concentration of Bis II. F, AVP-elicited inward current trace recorded from a DG GC in a slice pretreated with RHC 80267 (25 μm), a DAG lipase inhibitor. The extracellular solution contained the same concentration of RHC 80267. G, Current trace recorded from a DG GC dialyzed with the intracellular solution containing diC8-PIP2 (50 μm). H, Summary graph. The shaded bar was the averaged inward currents evoked by AVP in control condition pooled from the control experiment conducted for each individual pharmacological experiment. **p = 0.002, ***p = 0.0002 versus AVP alone, one-way ANOVA followed by Dunnett’s test.
DAG generated in response to the activation of Gq-coupled receptors can be metabolized by DAG lipase to produce 2-arachidonoylglycerol (2-AG), which has been reported to inhibit A-type K+ channels to excite midbrain dopamine neurons (Gantz and Bean, 2017). We next tested the role of DAG lipase in AVP-elicited excitation of DG GCs. To exclude contributions from differing isoforms of DAG lipase, we used RHC 80267 to inhibit both α and β DAG lipase. Pretreatment of slices with and continuous bath application of RHC 80267 (25 μm) had no significant effect on AVP-induced inward currents (−17.9 ± 2.2 pA, n = 16; p < 0.0001 vs baseline, Wilcoxon test; p = 0.999 vs AVP alone, one-way ANOVA followed by Dunnett’s test; Fig. 4F,H), suggesting that AVP-elicited excitation of DG GCs is not mediated by 2-AG.
Depletion of PIP2 is required for AVP-evoked inward currents in the DG GCs
PIP2 has been shown to modulate numerous ion channels (Suh and Hille, 2008; Rodríguez-Menchaca et al., 2012), including the KATP channels (Hilgemann and Ball, 1996; Fan and Makielski, 1997; Baukrowitz et al., 1998; Shyng and Nichols, 1998). We therefore studied the roles of PIP2 depletion elicited by activation of PLCβ in response to V1a receptor activation. Inclusion of the short-chain, water-soluble analog diC8-PIP2 (50 μm) in the recording pipettes significantly reduced AVP-induced inward currents (−5.2 ± 1.4 pA, n = 14; p = 0.004 vs baseline, Wilcoxon test; p = 0.0002 vs AVP alone, one-way ANOVA followed by Dunnett’s test; Fig. 4G,H), suggesting that depletion of PIP2 is required for AVP-mediated inward currents in the DG GCs.
Activation of V1a receptors augments the excitability of the DG GCs
We tested the effects of AVP on the RMPs and AP firing numbers recorded from the DG GCs. Bath application of AVP induced significant depolarization of the DG GCs (control, −70.8 ± 4.1 mV; AVP, −65.6 ± 6.0 mV; net depolarization, 5.2 ± 3.2 mV; n = 24; p < 0.0001, Wilcoxon test; Fig. 5A,B). We further probed the effects of AVP on the excitability of GCs by measuring the number of APs evoked by injecting a series of positive currents from 25 to 400 pA at an interval of 25 pA. With this protocol, the application of AVP significantly enhanced the AP firing numbers (F(1,12) = 24.05, p < 0.001, two-way repeated-measures ANOVA followed by Sidak’s multiple-comparisons test; Fig. 5C,D).
Figure 5.
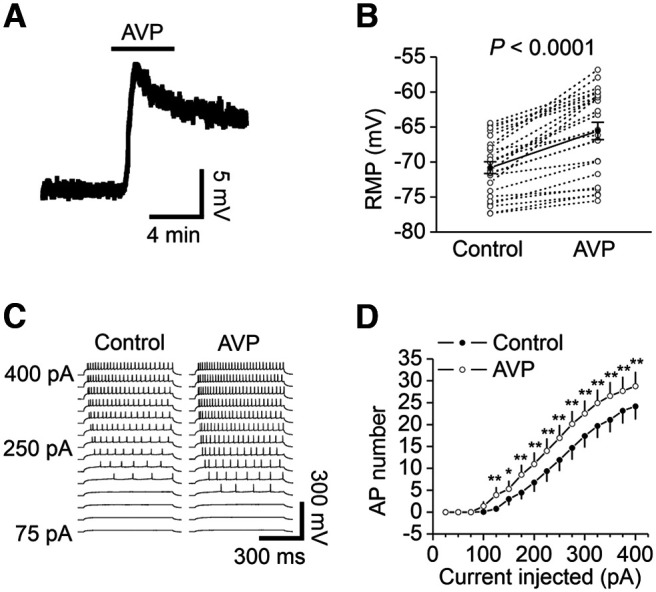
AVP depolarizes GCs and increases the number of APs elicited by injection of a series of positive currents. A, Resting membrane potential recorded from a GC before, during, and after the application of AVP. B, Summary data for AVP-induced depolarization. The empty circles represented the values from individual cells, and the solid symbols were their averages. C, APs elicited by injections of a series of positive currents from 25 to 400 pA in a GC before (left) and during (right) the application of AVP. D, Relationship between the injected currents and the elicited AP numbers from 13 GCs. *p < 0.05, **p < 0.001, two-way repeated-measures ANOVA followed by Sidak’s multiple-comparisons test.
AVP does not modulate glutamatergic transmission at the PP–GC synapses
The Ca2+ concentration in the extracellular solution was 2.5 mm. At this extracellular Ca2+ concentration, bath application of AVP has been shown to depress the slope of field EPSPs recorded in the DG (Chen et al., 1993). We therefore recorded AMPA EPSCs from the GCs by placing a stimulation electrode in the molecular layer to stimulate the PP. Bath application of AVP did not significantly modify AMPA EPSCs at the PP–GC synapses (104 ± 7% of control, n = 11; p = 0.577, Wilcoxon test; Fig. 6A), suggesting that AVP exerts no significant effect on basal glutamatergic transmission at the PP–GC synapses.
Figure 6.
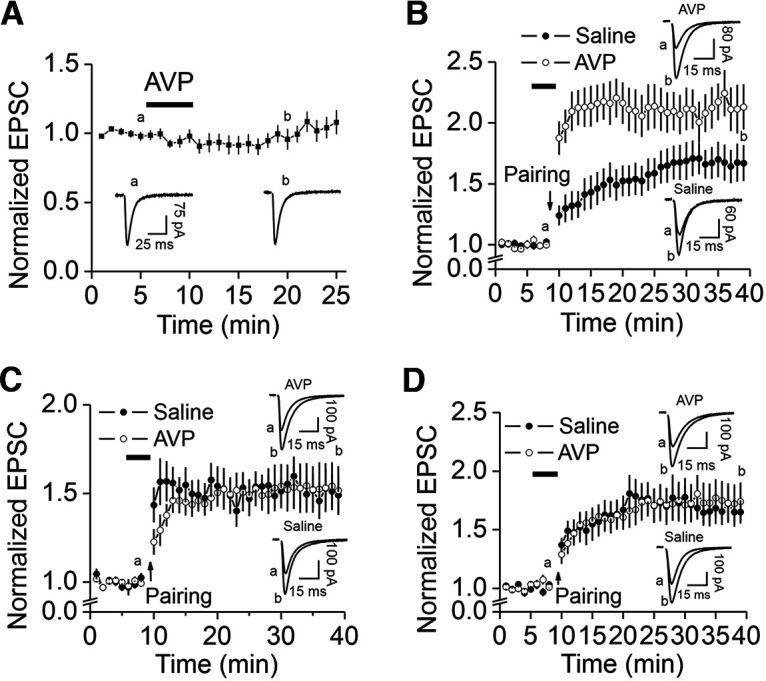
AVP does not modulate basal glutamatergic transmission but enhances LTP at the PP–GC synapses. A, Bath application of AVP (0.3 μm) did not alter significantly AMPA EPSCs recorded at the PP–GC synapses at −65 mV. The stimulation frequency was 0.1 Hz. The extracellular solution contained 10 μm bicuculline, and the intracellular solution was the K+-gluconate solution supplemented with 1 mm QX-314. The current traces were the averages of 1 min indicated at the time points shown in the figure. The stimulation artifacts were blanked. B, Bath application of AVP (0.3 μm) significantly enhanced LTP induced by pairing presynaptic stimulation (1 Hz, 40 pulses) with postsynaptic depolarization (−30 mV) recorded with K+-gluconate-containing intracellular solution. After recording basal AMPA EPSCs at −65 mV with the stimulation frequency of 0.1 Hz for 5 min, the bath was perfused with the extracellular solution containing AVP (0.3 μm) or saline (0.9% NaCl used to dissolve AVP) for 3 min, and the pairing protocol (1 Hz, 40 pulses, postsynaptic depolarization to −30 mV) was applied in the presence of AVP or saline. Recordings of AMPA EPSCs (−65 mV, 0.1 Hz) were resumed in the extracellular solution to observe the expression of LTP. Current traces were the averages in 1 min at the time points indicated in the figure. C, Application of AVP failed to enhance LTP when Cs+-gluconate-intracellular solution was used. D, Application of AVP did not augment LTP in the extracellular solution containing glibenclamide (100 μm) when K+-gluconate-intracellular solution was used.
AVP increases LTP at the PP–GC synapses
In pancreatic β-cells, hyperglycemia results in the closure of KATP channels, leading to membrane depolarization. Membrane depolarization opens Ca2+ channels to increase insulin release to decrease blood glucose concentration. Our results indicate that AVP-mediated activation of V1a receptors elicited subthreshold depolarization of the DG GCs via depression of KATP channels. At the PP–GC synapses, administration of a pairing protocol has been shown to induce LTP (Colino and Malenka, 1993). We therefore tested the effect of AVP on LTP at the PP–GC synapses by using a protocol of pairing presynaptic stimulation (1 Hz, 40 pulses) with postsynaptic depolarization to −30 mV. With K+-gluconate-containing intracellular solution, application of the protocol induced LTP (30 min after the protocol, 167 ± 15% of control, n = 13; p = 0.0002 vs baseline, Wilcoxon test; Fig. 6B). We further explored the effects of AVP on LTP at the PP–GC synapses. After recording basal AMPA EPSCs for 5 min, AVP (0.3 μm) dissolved in the extracellular solution was applied for 3 min because our results showed that the maximal effect of AVP on DG GCs could be observed in this time period. We then applied the protocol in the continuous presence of AVP. Under these circumstances, the level of LTP was significantly increased (213 ± 18% of control, n = 14; p = 0.0001 vs baseline, Wilcoxon test; F(1,950) = 194.8; p < 0.0001 vs control LTP, two-way ANOVA; Fig. 6B), suggesting that AVP augments LTP.
Because our results indicate that the activation of V1a receptors depolarizes the DG GCs via depression of KATP channels, we then tested the roles of K+ channels by using Cs+-gluconate-containing intracellular solution to annul the contribution of K+ channels. In this condition, the application of AVP did not significantly increase LTP (152 ± 14% of control, n = 11; p = 0.001 vs baseline, Wilcoxon test; Fig. 6C), compared with saline (151 ± 15% of control, n = 10; p = 0.002 vs baseline, Wilcoxon test; F(1,722) = 0.106; p = 0.745 vs LTP in response to AVP, two-way ANOVA; Fig. 6C), suggesting that AVP-mediated depression of K+ channels is responsible for AVP-elicited augmentation of LTP. We further probed the roles of KATP channels in AVP-induced enhancement of LTP with the K+-containing intracellular solution. In the presence of the KATP channel blocker glibenclamide (100 μm), the application of AVP did not significantly alter LTP induced by the administration of the pairing protocol (174 ± 16% of control, n = 11; p = 0.001 vs baseline, Wilcoxon test; Fig. 6D), compared with the LTP in response to bath application of saline (165 ± 10%, n = 11; p = 0.001 vs baseline, Wilcoxon test; F(1,760) = 0.039; p = 0.843 vs the LTP in response to AVP, two-way ANOVA; Fig. 6D). These results together suggest that AVP-induced depression of KATP channels contributes to its facilitatory effect on LTP.
Discussion
Our results indicate that application of AVP induces an inward current recorded from the DG GCs in voltage clamp. In current-clamp mode, AVP depolarizes the DG GCs and increases the action potential firing numbers. The effects of AVP are mediated by activation of V1a receptors and require the function of PLCβ. Whereas intracellular Ca2+ release and PKC activity are unnecessary, PLCβ-elicited depletion of PIP2 is responsible for AVP-elicited excitation of the DG GCs. AVP-induced excitation of the DG GCs is mediated by the depression of KATP channels. Activation of V1a receptors augments LTP at the PP–GC synapses, which is also mediated by the depression of KATP channels. Our results provide a cellular and molecular mechanism to explain the roles of V1a receptor activation in learning and memory and anxiety.
Our results show that AVP-elicited excitation of the DG GCs is mediated by the depression of KATP channels. Consistent with our electrophysiological results, high densities of KATP channels are expressed in the DG GCs (Mourre et al., 1990; Zawar et al., 1999; Pelletier et al., 2000; Tanner et al., 2011). KATP channels play a key role in the coupling between cellular metabolism and electrical activity in a wide range of tissues. KATP channels are formed from an ATP-binding cassette protein, the sulfonylurea receptor (SUR1, SUR2), and a Kir channel (Kir6.1, Kir6.2). Both subunits assemble in a 1:1 stoichiometry, with four SUR and four Kir subunits required to form functional KATP channels (Inagaki et al., 1995, 1997; Clement et al., 1997; Shyng and Nichols, 1997). While Kir6 acts as the pore-forming part in the channel complex that determines its single-channel conductance, its blockade by polyamines, and its inhibition by ATP, SUR has been identified as the regulatory subunit of KATP channels that confers sensitivity to sulfonylureas, channel openers, and Mg-ADP (Baukrowitz and Fakler, 2000). The AVP-sensitive currents in the DG GCs show inward rectification, and application of the KATP channel blocker glibenclamide induces an inward current by itself and blocks the effects of AVP, suggesting that the activation of V1a receptors excites the DG GCs via inhibition of KATP channels.
Our results further demonstrate that AVP-elicited excitation of the DG GCs is mediated by the activation of V1a receptors, consistent with the expression of high densities of AVP receptors in the GC (Brinton et al., 1984; De Kloet et al., 1985; van Leeuwen et al., 1987; Campbell et al., 2009). Our results further demonstrate that PLCβ is required, whereas intracellular Ca2+ release and PKC are dispensable for AVP-elicited inward currents in the DG GCs. In line with our results, exogenous application of IP3 had no effect on KATP channel activity (Fan and Makielski, 1997; Shyng and Nichols, 1998). However, it is controversial as to whether PKC is involved in modulating KATP channels. Whereas PKC has been shown to inhibit recombinant (Thorneloe et al., 2002) and native (Bonev and Nelson, 1993; Hatakeyama et al., 1995; Nuttle and Farley, 1997; Jun et al., 2001) KATP channels, PKC is not required for muscarinic suppression of KATP channels mediated by the M3/Gq/11/PLC pathway in mouse ileal smooth muscle cells (Wang et al., 2018).
PIP2 has been shown to modulate numerous ion channels (Suh and Hille, 2008; Rodríguez-Menchaca et al., 2012), including the KATP channels (Baukrowitz and Fakler, 2000). PIP2 is known to increase the open probability and decrease the ATP sensitivity of the KATP channels (Hilgemann and Ball, 1996; Fan and Makielski, 1997; Baukrowitz et al., 1998; Shyng and Nichols, 1998). Low-micromolar ATP is sufficient to inhibit KATP channels following patch excision, whereas millimolar concentrations of the nucleotide are required for channel inhibition after PIP2 is applied to inside-out patches for a few seconds and prolonged exposure to PIP2 renders the channels completely insensitive to 1 mm ATP (Baukrowitz et al., 1998; Shyng and Nichols, 1998). Membrane PIP2 content increases when PI and PI 4-monophosphate (PIP) are consecutively phosphorylated by PI 4-kinase and PIP 5-kinase (Anderson et al., 1999), whereas dephosphorylation of PIP2 mediated by inositolpolyphosphate phosphatase decreases PIP2 content in the membrane (Majerus et al., 1999). In addition, PIP2 is hydrolyzed by PLC to generate IP3 and DAG in response to G-protein-coupled receptors or tyrosine kinase receptors, resulting in the reduction of membrane PIP2 content by ∼85% (Willars et al., 1998). Because PIP2 has been shown to augment the open probability and decrease the ATP sensitivity of the KATP channels (Hilgemann and Ball, 1996; Fan and Makielski, 1997; Baukrowitz et al., 1998; Shyng and Nichols, 1998), PLCβ-elicited depletion of PIP2 in response to V1a receptor activation likely decreases open probability and increases the ATP sensitivity of the KATP channels. The outcome would be the depression of KATP channels and excitation of the DG GCs.
In CA1 pyramidal neurons of the hippocampus, the activation of V1a receptors increases neuronal excitability by the inhibition of GIRK channels (Hu et al., 2022), whereas the results in this study indicate that the activation of V1a receptors excites the DG GCs by depressing KATP channels. The discrepancy may be because of the distinct expression of KATP channels between CA1 pyramidal neurons and the DG GCs. KATP channels are expressed in 89% of the GCs, whereas only 26% of CA1 pyramidal neurons express KATP channels (Zawar et al., 1999).
In the DG, bath application of AVP increased the slope of field potentials when the extracellular Ca2+ concentration was 1.5 mm, but decreased it when the extracellular concentration was 2.5 mm (Chen et al., 1993). With whole-cell recordings, we failed to observe significant alteration of AMPA EPSCs in response to bath application of AVP in our extracellular solution containing 2.5 mm Ca2+. One explanation for the discrepancy of the results is that field potentials may represent the combined effects of AVP from many synapses, whereas AMPA EPSCs recorded by whole-cell recordings reflect the action of AVP at the synapses onto a single GC. If the effects of AVP on synaptic transmission are subtle, they may have been missed with whole-cell recordings from single cells. However, we have indeed observed that the bath application of AVP significantly increases the level of LTP at the PP–GC synapses by depressing KATP channels. Because the induction of LTP at the PP–GC synapses is dependent on NMDA receptors (Colino and Malenka, 1993) and NMDA receptors are voltage-dependently blocked by Mg2+, AVP-induced depolarization could facilitate NMDA receptor opening and thus augments LTP. An alternative mechanism is that V1a receptor-mediated depression of KATP channels could depolarize the DG GCs to open voltage-gated Ca2+ channels, resulting in the augmentation of Ca2+ influx to facilitate LTP. Further studies are required to determine the cellular and molecular mechanisms underpinning AVP-mediated augmentation of LTP. Consistent with our results, the depression of KATP channels enhances hippocampal LTP (Schröder et al., 2004; Moriguchi et al., 2018, 2021).
The physiological functions underlying V1a receptor-elicited excitation of the DG GCs and facilitation of LTP may be related to the effects of AVP on learning and memory (Alescio-Lautier and Soumireu-Mourat, 1998). For example, microinjection of AVP into the DG facilitates (Kovács et al., 1979), whereas microinjection of AVP antiserum into the dorsal DG attenuates (Kovács et al., 1982), passive avoidance behavior in rats. Intracerebroventricular injection of vasopressin-(4–9), a major metabolite C-terminal fragment of AVP, ameliorates scopolamine-induced impairments of rat spatial memory (Mishima et al., 2001). Subcutaneous injection of NC-1900, an active fragment analog of AVP, improves learning and memory deficits induced by β-amyloid protein in rats (Tanaka et al., 1998). However, the cellular and molecular mechanisms underlying AVP-mediated augmentation of learning and memory have not been determined. Our results that activation of V1a receptors excites the DG GCs and augments LTP at the PP–GC synapses could serve as a cellular mechanism to explain the effects of AVP on memory. Furthermore, activation of V1a receptors exerts anxiogenic effects (Landgraf et al., 1995; Bielsky et al., 2004, 2005; Egashira et al., 2007), and the ventral hippocampus is closely involved in anxiety-like behaviors (Charney and Deutch, 1996; Kjelstrup et al., 2002; Bannerman et al., 2003; Engin and Treit, 2007; Fanselow and Dong, 2010; Adhikari, 2014; Strange et al., 2014; Calhoon and Tye, 2015; Jimenez et al., 2018). Because the DG GCs are glutamatergic neurons and elevation of glutamatergic functions underlies the generation of anxiety and reduction of glutamatergic functions represents a novel treatment for anxiety (Kent et al., 2002; Gorman, 2003; Bergink et al., 2004; Simon and Gorman, 2006; Sanacora et al., 2008; Riaza Bermudo-Soriano et al., 2012; Sanacora et al., 2012), our results may represent a cellular and molecular mechanism whereby the activation of V1a receptors facilitates anxiety responses.
Synthesis
Reviewing Editor: Lindsay De Biase, University of California Los Angeles
Decisions are customarily a result of the Reviewing Editor and the peer reviewers coming together and discussing their recommendations until a consensus is reached. When revisions are invited, a fact-based synthesis statement explaining their decision and outlining what is needed to prepare a revision will be listed below. The following reviewer(s) agreed to reveal their identity: Hau-Jie Yau.
In the article “PLCβ-mediated depletion of PIP2 and ATP-sensitive K+ channels are involved in arginine vasopressin-induced facilitation of neuronal excitability and LTP in the dentate gyrus” the authors investigate the ability of arginine vasopressin (AVP) to modulate intrinsic activity and synaptic transmission of dentate gyrus (DG) granule cells, the mechanisms of which remain elusive. They combine pharmacological tools and bath-application of AVP to acute hippocampal brain slices, and find that this manipulation increases excitability of DG granule cells. Their finding is supported by AVP-induced inward currents measured with whole-cell voltage-clamp recordings from granule cells, and by AVP-induced depolarization and facilitation of action potential firing, determined in current-clamp recordings from granule cells. They found AVP-induced no changes to glutamatergic synaptic signaling onto DG granule cells. These results advance knowledge based on previous studies of AVP effects on hippocampal DG electrical signaling using extracellular field recordings. The authors investigate the underlying mechanism by determining the pharmacological profile of the AVP-induced inward current. They found that PLC activation is required but not IP3-dependent ER Ca2+ release or PKC activation. Instead, they can inhibit AVP effects by inhibiting KATP channels or by providing an excess of the PIP2 analog diC8-PIP2. From these experiments, and the reported high expression of V1A in DG granule cells, they conclude that the AVP effect on granule cell excitability may be explained by V1A-mediated depletion of PIP2, which might render tonically active KATP channels more sensitive to inhibition by ATP and thereby reduce the resting potassium conductance. Finally, they demonstrate that AVP facilitated the expression of PP-GC LTP in a KATP-dependent manner.
The manuscript is well written and organized. With the limitation that the authors did not employ any genetic manipulation of V1a in DG GCs, the experiments appear technically well-designed and the proposed model appears plausible based on prior literature and the reported experiments. The novelty is limited and more incremental due to similar AVP-induced effects reported for CA1 pyramidal neurons, albeit by targeting a different potassium channel (GIRK). Nevertheless, by linking KATP channel function to AVP effects on the gatekeeper of hippocampal function this study advances our insight into the underlying molecular and cellular mechanisms. These findings could also help shed light on molecular mechanisms for AVP modulation of DG-relevant behavioral functions. However, reviewers agree that several concerns in the data need to be addressed.
Major concerns:
1) For Figs. 1, 3, 4 and 5 the authors used the same AVP control effect as reference for statistical comparison (10 direct comparisons). Ideally, each experiment should have its own control group. This reveals critical information about how repeatable and consistent the data are in the control condition if experiments are executed at different times as the project evolves and progresses. Can the authors repeat at least one key experiment (e.g. for Fig. 5) and include a new control condition? At a minimum, it is recommended that in all figures following Fig. 1, the AVP control data be presented as grayed out and the authors explicitly comment on the recycling of the control data in the respective population data description in the figure legends. It should also be clarified whether the recycling has been accounted for in multiple comparison compensations of the statistical analyses.
2) The authors should be more rigorous in choosing their representative trace examples for panels Fig. 1B, Fig. 3A, Fig. 4A and Fig. 5E. The representative examples should be close to the mean value for that entire group.
3) The control experiments in Fig. 5 convincingly demonstrate that ER Ca2+ release and/or PKC activation are not required for the AVP effect studied here. It has been found that 2-arachidonoylglycerol (2-AG) and arachidonic acid, two diacylglycerol (DAG) lipase-dependent downstream products of DAG, can right-shift the voltage-dependence of voltage-gated potassium channels underlying the A-type current (PMID:28262417). It is recommended to include inhibition of DAG lipase as control experiment in Fig. 5.
4) The authors have provided the evidence that there was tonic activation of KATP channels in DG GCs in experimental condition. Therefore, it is unexpected that the manuscript did not provide the key direct evidence that AVP would increase membrane resistance by closing active KATP channels.
Minor concerns:
1) In the presentation and discussion of the LTP results it appears that the authors did not take sufficient efforts to distinguish between induction and expression. Fig. 7 legend emphasizes LTP expression. Specifically, on page 9 lower half they state: “We further explored the effects of AVP on the induction of LTP at the PP-GC synapses. [...], suggesting that AVP augments the expression of LTP.” If the investigators claim that they tested the effect of AVP on induction of LTP they would clearly need to explain how this was possible with the GC membrane potential clamped to -30 mV during induction. Since the LTP experiment was conducted in voltage clamp mode, how can AVP-elicited depolarization ‘facilitate the opening of Ca2+ channels or relieve the Mg2+ blockade of NMDA receptors’? They need to come up with some other reasonable explanations or they could test the AVP modulation of NMDAR-mediated EPSC.
2) page 12, middle: “reflex” probably means “reflects”
3) It is suggested to combine Figure 3 and 4.
References
- Adhikari A (2014) Distributed circuits underlying anxiety. Front Behav Neurosci 8:112. 10.3389/fnbeh.2014.00112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alescio-Lautier B, Soumireu-Mourat B (1998) Role of vasopressin in learning and memory in the hippocampus. Prog Brain Res 119:501–521. 10.1016/s0079-6123(08)61590-3 [DOI] [PubMed] [Google Scholar]
- Anderson RA, Boronenkov IV, Doughman SD, Kunz J, Loijens JC (1999) Phosphatidylinositol phosphate kinases, a multifaceted family of signaling enzymes. J Biol Chem 274:9907–9910. 10.1074/jbc.274.15.9907 [DOI] [PubMed] [Google Scholar]
- Bannerman DM, Grubb M, Deacon RMJ, Yee BK, Feldon J, Rawlins JNP (2003) Ventral hippocampal lesions affect anxiety but not spatial learning. Behav Brain Res 139:197–213. 10.1016/S0166-4328(02)00268-1 [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Fakler B (2000) K(ATP) channels: linker between phospholipid metabolism and excitability. Biochem Pharmacol 60:735–740. 10.1016/s0006-2952(00)00267-7 [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, Ruppersberg JP, Fakler B (1998) PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 282:1141–1144. 10.1126/science.282.5391.1141 [DOI] [PubMed] [Google Scholar]
- Bergink V, van Megen HJ, Westenberg HG (2004) Glutamate and anxiety. Eur Neuropsychopharmacol 14:175–183. 10.1016/S0924-977X(03)00100-7 [DOI] [PubMed] [Google Scholar]
- Biegon A, Terlou M, Voorhuis TD, de Kloet ER (1984) Arginine-vasopressin binding sites in rat brain: a quantitative autoradiographic study. Neurosci Lett 44:229–234. 10.1016/0304-3940(84)90027-2 [DOI] [PubMed] [Google Scholar]
- Bielsky IF, Hu SB, Szegda KL, Westphal H, Young LJ (2004) Profound impairment in social recognition and reduction in anxiety-like behavior in vasopressin V1a receptor knockout mice. Neuropsychopharmacology 29:483–493. 10.1038/sj.npp.1300360 [DOI] [PubMed] [Google Scholar]
- Bielsky IF, Hu SB, Ren X, Terwilliger EF, Young LJ (2005) The V1a vasopressin receptor is necessary and sufficient for normal social recognition: a gene replacement study. Neuron 47:503–513. 10.1016/j.neuron.2005.06.031 [DOI] [PubMed] [Google Scholar]
- Bonev AD, Nelson MT (1993) Muscarinic inhibition of ATP-sensitive K+ channels by protein kinase C in urinary bladder smooth muscle. Am J Physiol 265:C1723–C1728. 10.1152/ajpcell.1993.265.6.C1723 [DOI] [PubMed] [Google Scholar]
- Brinton RE, Gee KW, Wamsley JK, Davis TP, Yamamura HI (1984) Regional distribution of putative vasopressin receptors in rat brain and pituitary by quantitative autoradiography. Proc Natl Acad Sci U|S|A 81:7248–7252. 10.1073/pnas.81.22.7248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buijs RM (1980) Immunocytochemical demonstration of vasopressin and oxytocin in the rat brain by light and electron microscopy. J Histochem Cytochem 28:357–360. 10.1177/28.4.6989899 [DOI] [PubMed] [Google Scholar]
- Buijs RM, Swaab DF (1979) Immuno-electron microscopical demonstration of vasopressin and oxytocin synapses in the limbic system of the rat. Cell Tissue Res 204:355–365. 10.1007/BF00233648 [DOI] [PubMed] [Google Scholar]
- Buijs RM, Swaab DF, Dogterom J, van Leeuwen FW (1978) Intra- and extrahypothalamic vasopressin and oxytocin pathways in the rat. Cell Tissue Res 186:423–435. 10.1007/BF00224932 [DOI] [PubMed] [Google Scholar]
- Caffé AR, van Leeuwen FW (1983) Vasopressin-immunoreactive cells in the dorsomedial hypothalamic region, medial amygdaloid nucleus and locus coeruleus of the rat. Cell Tissue Res 233:23–33. 10.1007/BF00222229 [DOI] [PubMed] [Google Scholar]
- Caffé AR, van Leeuwen FW, Luiten PG (1987) Vasopressin cells in the medial amygdala of the rat project to the lateral septum and ventral hippocampus. J Comp Neurol 261:237–252. 10.1002/cne.902610206 [DOI] [PubMed] [Google Scholar]
- Caldwell HK, Lee HJ, Macbeth AH, Young WS 3rd (2008) Vasopressin: behavioral roles of an “original” neuropeptide. Prog Neurobiol 84:1–24. 10.1016/j.pneurobio.2007.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoon GG, Tye KM (2015) Resolving the neural circuits of anxiety. Nat Neurosci 18:1394–1404. 10.1038/nn.4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell P, Ophir AG, Phelps SM (2009) Central vasopressin and oxytocin receptor distributions in two species of singing mice. J Comp Neurol 516:321–333. 10.1002/cne.22116 [DOI] [PubMed] [Google Scholar]
- Charney DS, Deutch A (1996) A functional neuroanatomy of anxiety and fear: implications for the pathophysiology and treatment of anxiety disorders. Crit Rev Neurobiol 10:419–446. 10.1615/critrevneurobiol.v10.i3-4.70 [DOI] [PubMed] [Google Scholar]
- Chen C, Díaz Brinton RD, Shors TJ, Thompson RF (1993) Vasopressin induction of long-lasting potentiation of synaptic transmission in the dentate gyrus. Hippocampus 3:193–203. 10.1002/hipo.450030211 [DOI] [PubMed] [Google Scholar]
- Cilz NI, Cymerblit-Sabba A, Young WS (2019) Oxytocin and vasopressin in the rodent hippocampus. Genes Brain Behav 18:e12535. 10.1111/gbb.12535 [DOI] [PubMed] [Google Scholar]
- Clement JPt, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, Bryan J (1997) Association and stoichiometry of K(ATP) channel subunits. Neuron 18:827–838. 10.1016/S0896-6273(00)80321-9 [DOI] [PubMed] [Google Scholar]
- Colino A, Malenka RC (1993) Mechanisms underlying induction of long-term potentiation in rat medial and lateral perforant paths in vitro. J Neurophysiol 69:1150–1159. 10.1152/jn.1993.69.4.1150 [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Rotteveel F, Voorhuis TA, Terlou M (1985) Topography of binding sites for neurohypophyseal hormones in rat brain. Eur J Pharmacol 110:113–119. 10.1016/0014-2999(85)90036-6 [DOI] [PubMed] [Google Scholar]
- DeVries GJ, Buijs RM, Van Leeuwen FW, Caffé AR, Swaab DF (1985) The vasopressinergic innervation of the brain in normal and castrated rats. J Comp Neurol 233:236–254. 10.1002/cne.902330206 [DOI] [PubMed] [Google Scholar]
- de Wied D, Diamant M, Fodor M (1993) Central nervous system effects of the neurohypophyseal hormones and related peptides. Front Neuroendocrinol 14:251–302. 10.1006/frne.1993.1009 [DOI] [PubMed] [Google Scholar]
- Dubrovsky B, Tatarinov A, Gijsbers K, Harris J, Tsiodras A (2003) Effects of arginine-vasopressin (AVP) on long-term potentiation in intact anesthetized rats. Brain Res Bull 59:467–472. 10.1016/s0361-9230(02)00961-9 [DOI] [PubMed] [Google Scholar]
- Egashira N, Tanoue A, Matsuda T, Koushi E, Harada S, Takano Y, Tsujimoto G, Mishima K, Iwasaki K, Fujiwara M (2007) Impaired social interaction and reduced anxiety-related behavior in vasopressin V1a receptor knockout mice. Behav Brain Res 178:123–127. 10.1016/j.bbr.2006.12.009 [DOI] [PubMed] [Google Scholar]
- Engin E, Treit D (2007) The role of hippocampus in anxiety: intracerebral infusion studies. Behav Pharmacol 18:365–374. 10.1097/FBP.0b013e3282de7929 [DOI] [PubMed] [Google Scholar]
- Fan Z, Makielski JC (1997) Anionic phospholipids activate ATP-sensitive potassium channels. J Biol Chem 272:5388–5395. 10.1074/jbc.272.9.5388 [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Dong HW (2010) Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65:7–19. 10.1016/j.neuron.2009.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford NC, Baccei ML (2016) Inward-rectifying K(+) (Kir2) leak conductance dampens the excitability of lamina I projection neurons in the neonatal rat. Neuroscience 339:502–510. 10.1016/j.neuroscience.2016.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz SC, Bean BP (2017) Cell-autonomous excitation of midbrain dopamine neurons by endocannabinoid-dependent lipid signaling. Neuron 93:1375–1387.e2. 10.1016/j.neuron.2017.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizowski C, Trudel E, Bourque CW (2017) Central and peripheral roles of vasopressin in the circadian defense of body hydration. Best Pract Res Clin Endocrinol Metab 31:535–546. 10.1016/j.beem.2017.11.001 [DOI] [PubMed] [Google Scholar]
- Gorman JM (2003) New molecular targets for antianxiety interventions. J Clin Psychiatry 64 [Suppl 3]:28–35. [PubMed] [Google Scholar]
- Haller M, Mironov SL, Karschin A, Richter DW (2001) Dynamic activation of K(ATP) channels in rhythmically active neurons. J Physiol 537:69–81. 10.1111/j.1469-7793.2001.0069k.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama N, Wang Q, Goyal RK, Akbarali HI (1995) Muscarinic suppression of ATP-sensitive K+ channel in rabbit esophageal smooth muscle. Am J Physiol 268:C877–C885. 10.1152/ajpcell.1995.268.4.C877 [DOI] [PubMed] [Google Scholar]
- Hawthorn J, Ang VT, Jenkins JS (1985) Effects of lesions in the hypothalamic paraventricular, supraoptic and suprachiasmatic nuclei on vasopressin and oxytocin in rat brain and spinal cord. Brain Res 346:51–57. 10.1016/0006-8993(85)91093-5 [DOI] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90:291–366. 10.1152/physrev.00021.2009 [DOI] [PubMed] [Google Scholar]
- Hilgemann DW, Ball R (1996) Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science 273:956–959. 10.1126/science.273.5277.956 [DOI] [PubMed] [Google Scholar]
- Hu B, Boyle CA, Lei S (2022) Roles of PLCβ, PIP2, and GIRK channels in arginine vasopressin-elicited excitation of CA1 pyramidal neurons. J Cell Physiol 237:660–674. 10.1002/jcp.30535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Lee SH, Lu H, Sanders KM, Koh SD (2018) Molecular and functional characterization of inwardly rectifying K(+) currents in murine proximal colon. J Physiol 596:379–391. 10.1113/JP275234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP 4th, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J (1995) Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270:1166–1170. 10.1126/science.270.5239.1166 [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Seino S (1997) Subunit stoichiometry of the pancreatic beta-cell ATP-sensitive K+ channel. FEBS Lett 409:232–236. 10.1016/s0014-5793(97)00488-2 [DOI] [PubMed] [Google Scholar]
- Jimenez JC, Su K, Goldberg AR, Luna VM, Biane JS, Ordek G, Zhou P, Ong SK, Wright MA, Zweifel L, Paninski L, Hen R, Kheirbek MA (2018) Anxiety cells in a hippocampal-hypothalamic circuit. Neuron 97:670–683.e6. 10.1016/j.neuron.2018.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JY, Kong ID, Koh SD, Wang XY, Perrino BA, Ward SM, Sanders KM (2001) Regulation of ATP-sensitive K(+) channels by protein kinase C in murine colonic myocytes. Am J Physiol Cell Physiol 281:C857–C864. 10.1152/ajpcell.2001.281.3.C857 [DOI] [PubMed] [Google Scholar]
- Kent JM, Mathew SJ, Gorman JM (2002) Molecular targets in the treatment of anxiety. Biol Psychiatry 52:1008–1030. 10.1016/s0006-3223(02)01672-4 [DOI] [PubMed] [Google Scholar]
- Kim KS, Jang JH, Lin H, Choi SW, Kim HR, Shin DH, Nam JH, Zhang YH, Kim SJ (2015) Rise and fall of Kir2.2 current by TLR4 signaling in human monocytes: PKC-dependent trafficking and PI3K-mediated PIP2 decrease. J Immunol 195:3345–3354. 10.4049/jimmunol.1500056 [DOI] [PubMed] [Google Scholar]
- Kjelstrup KG, Tuvnes FA, Steffenach HA, Murison R, Moser EI, Moser MB (2002) Reduced fear expression after lesions of the ventral hippocampus. Proc Natl Acad Sci U|S|A 99:10825–10830. 10.1073/pnas.152112399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kompier NF, Keysers C, Gazzola V, Lucassen PJ, Krugers HJ (2019) Early life adversity and adult social behavior: focus on arginine vasopressin and oxytocin as potential mediators. Front Behav Neurosci 13:143. 10.3389/fnbeh.2019.00143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshimizu TA, Tsujimoto G (2009) New topics in vasopressin receptors and approach to novel drugs: vasopressin and pain perception. J Pharmacol Sci 109:33–37. 10.1254/jphs.08r18fm [DOI] [PubMed] [Google Scholar]
- Kovács GL, Bohus B, Versteeg DH, de Kloet ER, de Wied D (1979) Effect of oxytocin and vasopressin on memory consolidation: sites of action and catecholaminergic correlates after local microinjection into limbic-midbrain structures. Brain Res 175:303–314. 10.1016/0006-8993(79)91009-6 [DOI] [PubMed] [Google Scholar]
- Kovács GL, Buijs RM, Bohus B, van Wimersma Greidanus TB (1982) Microinjection of arginine8-vasopressin antiserum into the dorsal hippocampus attenuates passive avoidance behavior in rats. Physiol Behav 28:45–48. 10.1016/0031-9384(82)90099-3 [DOI] [PubMed] [Google Scholar]
- Kuzhikandathil EV, Oxford GS (2002) Classic D1 dopamine receptor antagonist R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23390) directly inhibits G protein-coupled inwardly rectifying potassium channels. Mol Pharmacol 62:119–126. 10.1124/mol.62.1.119 [DOI] [PubMed] [Google Scholar]
- Landgraf R, Gerstberger R, Montkowski A, Probst JC, Wotjak CT, Holsboer F, Engelmann M (1995) V1 vasopressin receptor antisense oligodeoxynucleotide into septum reduces vasopressin binding, social discrimination abilities, and anxiety-related behavior in rats. J Neurosci 15:4250–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang RE, Heil J, Ganten D, Hermann K, Rascher W, Unger T (1983) Effects of lesions in the paraventricular nucleus of the hypothalamus on vasopressin and oxytocin contents in brainstem and spinal cord of rat. Brain Res 260:326–329. 10.1016/0006-8993(83)90690-x [DOI] [PubMed] [Google Scholar]
- Lawrence JA, Poulin P, Lawrence D, Lederis K (1988) [3H]arginine vasopressin binding to rat brain: a homogenate and autoradiographic study. Brain Res 446:212–218. 10.1016/0006-8993(88)90879-7 [DOI] [PubMed] [Google Scholar]
- Lemak MS, Voloshanenko O, Draguhn A, Egorov AV (2014) KATP channels modulate intrinsic firing activity of immature entorhinal cortex layer III neurons. Front Cell Neurosci 8:255. 10.3389/fncel.2014.00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majerus PW, Kisseleva MV, Norris FA (1999) The role of phosphatases in inositol signaling reactions. J Biol Chem 274:10669–10672. 10.1074/jbc.274.16.10669 [DOI] [PubMed] [Google Scholar]
- Metzger D, Alescio-Lautier B, Bosler O, Devigne C, Soumireu-Mourat B (1993) Effect of changes in the intrahippocampal vasopressin on memory retrieval and relearning. Behav Neural Biol 59:29–48. 10.1016/0163-1047(93)91131-6 [DOI] [PubMed] [Google Scholar]
- Mishima K, Tsukikawa H, Inada K, Fujii M, Iwasaki K, Matsumoto Y, Abe K, Egawa T, Fujiwara M (2001) Ameliorative effect of vasopressin-(4-9) through vasopressin V(1A) receptor on scopolamine-induced impairments of rat spatial memory in the eight-arm radial maze. Eur J Pharmacol 427:43–52. 10.1016/s0014-2999(01)01200-6 [DOI] [PubMed] [Google Scholar]
- Mollajew R, Toloe J, Mironov SL (2013) Single KATP channel opening in response to stimulation of AMPA/kainate receptors is mediated by Na+ accumulation and submembrane ATP and ADP changes. J Physiol 591:2593–2609. 10.1113/jphysiol.2012.248369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi S, Ishizuka T, Yabuki Y, Shioda N, Sasaki Y, Tagashira H, Yawo H, Yeh JZ, Sakagami H, Narahashi T, Fukunaga K (2018) Blockade of the KATP channel Kir6.2 by memantine represents a novel mechanism relevant to Alzheimer's disease therapy. Mol Psychiatry 23:211–221. 10.1038/mp.2016.187 [DOI] [PubMed] [Google Scholar]
- Moriguchi S, Inagaki R, Fukunaga K (2021) Memantine improves cognitive deficits via KATP channel inhibition in olfactory bulbectomized mice. Mol Cell Neurosci 117:103680. 10.1016/j.mcn.2021.103680 [DOI] [PubMed] [Google Scholar]
- Mourre C, Widmann C, Lazdunski M (1990) Sulfonylurea binding sites associated with ATP-regulated K+ channels in the central nervous system: autoradiographic analysis of their distribution and ontogenesis, and of their localization in mutant mice cerebellum. Brain Res 519:29–43. 10.1016/0006-8993(90)90057-i [DOI] [PubMed] [Google Scholar]
- Neumann ID, Landgraf R (2012) Balance of brain oxytocin and vasopressin: implications for anxiety, depression, and social behaviors. Trends Neurosci 35:649–659. 10.1016/j.tins.2012.08.004 [DOI] [PubMed] [Google Scholar]
- Nuttle LC, Farley JM (1997) Muscarinic receptors inhibit ATP-sensitive K+ channels in swine tracheal smooth muscle. Am J Physiol 273:L478–L484. 10.1152/ajplung.1997.273.2.L478 [DOI] [PubMed] [Google Scholar]
- Ostrowski NL, Lolait SJ, Young WS 3rd (1994) Cellular localization of vasopressin V1a receptor messenger ribonucleic acid in adult male rat brain, pineal, and brain vasculature. Endocrinology 135:1511–1528. 10.1210/endo.135.4.7925112 [DOI] [PubMed] [Google Scholar]
- Paban V, Alescio-Lautier B, Devigne C, Soumireu-Mourat B (1999) Fos protein expression induced by intracerebroventricular injection of vasopressin in unconditioned and conditioned mice. Brain Res 825:115–131. 10.1016/s0006-8993(99)01232-9 [DOI] [PubMed] [Google Scholar]
- Pelletier MR, Pahapill PA, Pennefather PS, Carlen PL (2000) Analysis of single K(ATP) channels in mammalian dentate gyrus granule cells. J Neurophysiol 84:2291–2301. 10.1152/jn.2000.84.5.2291 [DOI] [PubMed] [Google Scholar]
- Ramanathan G, Cilz NI, Kurada L, Hu B, Wang X, Lei S (2012) Vasopressin facilitates GABAergic transmission in rat hippocampus via activation of V(1A) receptors. Neuropharmacology 63:1218–1226. 10.1016/j.neuropharm.2012.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaza Bermudo-Soriano C, Perez-Rodriguez MM, Vaquero-Lorenzo C, Baca-Garcia E (2012) New perspectives in glutamate and anxiety. Pharmacol Biochem Behav 100:752–774. 10.1016/j.pbb.2011.04.010 [DOI] [PubMed] [Google Scholar]
- Rodríguez-Menchaca AA, Adney SK, Zhou L, Logothetis DE (2012) Dual regulation of voltage-sensitive ion channels by PIP(2). Front Pharmacol 3:170. 10.3389/fphar.2012.00170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem H, Tovey SC, Molinski TF, Taylor CW (2014) Interactions of antagonists with subtypes of inositol 1,4,5-trisphosphate (IP3) receptor. Br J Pharmacol 171:3298–3312. 10.1111/bph.12685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Zarate CA, Krystal JH, Manji HK (2008) Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov 7:426–437. 10.1038/nrd2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Treccani G, Popoli M (2012) Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 62:63–77. 10.1016/j.neuropharm.2011.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder UH, Hock FJ, Wirth K, Englert HC, Reymann KG (2004) The ATP-regulated K+-channel inhibitor HMR-1372 affects synaptic plasticity in hippocampal slices. Eur J Pharmacol 502:99–104. 10.1016/j.ejphar.2004.08.046 [DOI] [PubMed] [Google Scholar]
- Shyng S, Nichols CG (1997) Octameric stoichiometry of the KATP channel complex. J Gen Physiol 110:655–664. 10.1085/jgp.110.6.655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Nichols CG (1998) Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 282:1138–1141. 10.1126/science.282.5391.1138 [DOI] [PubMed] [Google Scholar]
- Simon AB, Gorman JM (2006) Advances in the treatment of anxiety: targeting glutamate. NeuroRx 3:57–68. 10.1016/j.nurx.2005.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV (1985) Vasopressin- and neurophysin-immunoreactive neurons in the septal region, medial amygdala and locus coeruleus in colchicine-treated rats. Neuroscience 15:347–358. 10.1016/0306-4522(85)90217-9 [DOI] [PubMed] [Google Scholar]
- Sonkusare SK, Dalsgaard T, Bonev AD, Nelson MT (2016) Inward rectifier potassium (Kir2.1) channels as end-stage boosters of endothelium-dependent vasodilators. J Physiol 594:3271–3285. 10.1113/JP271652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoop R (2012) Neuromodulation by oxytocin and vasopressin. Neuron 76:142–159. 10.1016/j.neuron.2012.09.025 [DOI] [PubMed] [Google Scholar]
- Strange BA, Witter MP, Lein ES, Moser EI (2014) Functional organization of the hippocampal longitudinal axis. Nat Rev Neurosci 15:655–669. 10.1038/nrn3785 [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B (2008) PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys 37:175–195. 10.1146/annurev.biophys.37.032807.125859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szot P, Bale TL, Dorsa DM (1994) Distribution of messenger RNA for the vasopressin V1a receptor in the CNS of male and female rats. Brain Res Mol Brain Res 24:1–10. 10.1016/0169-328x(94)90111-2 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Yamada K, Senzaki K, Narimatsu H, Nishimura K, Kameyama T, Nabeshima T (1998) NC-1900, an active fragment analog of arginine vasopressin, improves learning and memory deficits induced by beta-amyloid protein in rats. Eur J Pharmacol 352:135–142. 10.1016/s0014-2999(98)00344-6 [DOI] [PubMed] [Google Scholar]
- Tanner GR, Lutas A, Martínez-François JR, Yellen G (2011) Single K ATP channel opening in response to action potential firing in mouse dentate granule neurons. J Neurosci 31:8689–8696. 10.1523/JNEUROSCI.5951-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorneloe KS, Maruyama Y, Malcolm AT, Light PE, Walsh MP, Cole WC (2002) Protein kinase C modulation of recombinant ATP-sensitive K(+) channels composed of Kir6.1 and/or Kir6.2 expressed with SUR2B. J Physiol 541:65–80. 10.1113/jphysiol.2002.018101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen F, Caffé R (1983) Vasopressin-immunoreactive cell bodies in the bed nucleus of the stria terminalis of the rat. Cell Tissue Res 228:525–534. 10.1007/BF00211473 [DOI] [PubMed] [Google Scholar]
- van Leeuwen FW, van der Beek EM, van Heerikhuize JJ, Wolters P, van der Meulen G, Wan YP (1987) Quantitative light microscopic autoradiographic localization of binding sites labelled with [3H]vasopressin antagonist d(CH2)5Tyr(Me)VP in the rat brain, pituitary and kidney. Neurosci Lett 80:121–126. 10.1016/0304-3940(87)90640-9 [DOI] [PubMed] [Google Scholar]
- Wang B, Murakami Y, Ono M, Fujikawa S, Matsuyama H, Unno T, Naitou K, Tanahashi Y (2018) Muscarinic suppression of ATP-sensitive K(+) channels mediated by the M3/Gq/11/phospholipase C pathway contributes to mouse ileal smooth muscle contractions. Am J Physiol Gastrointest Liver Physiol 315:G618–G630. 10.1152/ajpgi.00069.2018 [DOI] [PubMed] [Google Scholar]
- Wang HR, Wu M, Yu H, Long S, Stevens A, Engers DW, Sackin H, Daniels JS, Dawson ES, Hopkins CR, Lindsley CW, Li M, McManus OB (2011) Selective inhibition of the K(ir)2 family of inward rectifier potassium channels by a small molecule probe: the discovery, SAR, and pharmacological characterization of ML133. ACS Chem Biol 6:845–856. 10.1021/cb200146a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willars GB, Nahorski SR, Challiss RA (1998) Differential regulation of muscarinic acetylcholine receptor-sensitive polyphosphoinositide pools and consequences for signaling in human neuroblastoma cells. J Biol Chem 273:5037–5046. 10.1074/jbc.273.9.5037 [DOI] [PubMed] [Google Scholar]
- Ye JH, Zhang J, Xiao C, Kong JQ (2006) Patch-clamp studies in the CNS illustrate a simple new method for obtaining viable neurons in rat brain slices: glycerol replacement of NaCl protects CNS neurons. J Neurosci Methods 158:251–259. 10.1016/j.jneumeth.2006.06.006 [DOI] [PubMed] [Google Scholar]
- Zawar C, Plant TD, Schirra C, Konnerth A, Neumcke B (1999) Cell-type specific expression of ATP-sensitive potassium channels in the rat hippocampus. J Physiol 514:327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]