

Abstract
Background
An appropriate clinical diagnosis of von Willebrand disease (VWD) can be challenging because of a variable bleeding pattern and laboratory phenotype. Genotyping is a powerful diagnostic tool and may have an essential role in the diagnostic field of VWD.
Objectives
To unravel the clinical and laboratory heterogeneity of genetically confirmed VWD type 2M patients and to investigate their relationship.
Methods
Patients with a confirmed VWD type 2M genetic variant in the A1 or A3 domain of von Willebrand factor (VWF) and normal or only slightly aberrant VWF multimers were selected from all subjects genotyped at the Radboud university medical center because of a high suspicion of VWD. Bleeding scores and laboratory results were analyzed.
Results
Fifty patients had a clinically relevant genetic variant in the A1 domain. Median bleeding score was 5. Compared with the nationwide Willebrand in the Netherlands study type 2 cohort, bleeding after surgery or delivery was reported more frequently and mucocutaneous bleedings less frequently. Median VWF activity/VWF antigen (VWF:Act/VWF:Ag) ratio was 0.32, whereas VWF collagen binding activity/VWF antigen (VWF:CB/VWF:Ag) ratio was 0.80. Variants in the A3 domain were only found in two patients with low to normal VWF:Act/VWF:Ag ratios (0.45, 1.03) and low VWF:CB/VWF:Ag ratios (0.45, 0.63).
Conclusion
Genetically confirmed VWD type 2M patients have a relatively mild clinical phenotype, except for bleeding after surgery and delivery. Laboratory phenotype is variable and depends on the underlying genetic variant. Addition of genotyping to the current phenotypic characterization may improve diagnosis and classification of VWD.
Keywords: genotype; hemorrhage; hemostasis; phenotype; von Willebrand disease, type 2
Essentials.
Genotype‐phenotype correlation was assessed in VWD type 2M patients with a variant in the A1 or A3 domain of VWF and normal or only slightly abnormal multimers.
Bleeding after surgery or delivery is common in VWD type 2M patients.
The most prevalent genetic variants in the A1 domain in our cohort each had a unique laboratory phenotype.
Combining functional VWF assays with genetic analysis may result in an improved VWD classification and thereby personalized treatment strategies.
1. INTRODUCTION
Von Willebrand disease (VWD) is the most common inherited bleeding disorder and is caused by quantitative and/or qualitative deficiencies of von Willebrand factor (VWF). 1 , 2 Because VWF is the main protein involved in the attachment of platelets to a damaged vessel wall, any change in level or function of this protein may lead to a disorder of primary hemostasis. 3 Moreover, VWF acts as a carrier protein for coagulation factor VIII (FVIII), protecting FVIII from premature degradation. 3 , 4 Therefore, a deficiency of VWF may also hamper secondary hemostasis.
Von Willebrand disease is characterized by a heterogeneous clinical presentation, ranging from only minor bleeding episodes to severe bleeding problems that require direct hemostatic control. Most common symptoms are mucocutaneous bleeding, menorrhagia, bleeding from minor wounds, and bleeding episodes after dental extractions, surgery, and trauma. 2 , 3 , 4 The largest group of VWD patients are categorized as type 1 (an average of 70%–80%), which comprises a group with mostly mild bleeding problems. In general, patients with type 2 VWD (20%) have more pronounced bleeding problems. Only a very small number of VWD patients (<5%) is diagnosed with type 3, which can cause severe bleeding episodes comparable to hemophilia A. 4 , 5 , 6
Type 2 VWD is generally subdivided into four distinct disorders: type 2A is characterized by a reduction in (the most biologically active) high molecular weight VWF multimers, type 2B by an increased binding of VWF to platelet glycoprotein Ib (GPIb), and type 2M by a reduced binding of VWF to platelet GPIb or collagen. All have an autosomal dominant inheritance. Type 2N is characterized by a reduced binding of FVIII to VWF and penetrates by an autosomal recessive trait. 7 , 8 In contrast to type 2A and 2B, multimer distribution in type 2M is (approximately) normal. 8
Von Willebrand disease type 2M is mainly caused by missense mutations in the A1 domain and less frequently in the A3 domain of VWF. 7 , 9 Because the A1 domain contains the main binding site for platelet GPIb, genetic variants in this domain lead to a reduced VWF‐dependent platelet adhesion resulting in a disproportionately low level of VWF activity (VWF:Act) compared with VWF antigen level (VWF:Ag) and thus a low VWF:Act/VWF:Ag ratio. 7 , 8 , 10 Collagen binding sites are incorporated in both the A1 and A3 domain. 11 , 12 , 13 , 14 , 15 The principal binding sites for collagen types I and III are located in the A3 domain, 11 , 14 , 15 whereas the binding sites for collagen types IV and VI are located in the A1 domain. 11 , 12 , 13 , 14 , 16 In A3 genetic variants, VWF collagen binding activity level (VWF:CB) can be markedly reduced with a (near) normal VWF:Act and VWF:Ag level. 15
The clinical diagnosis of VWD is based on the combination of bleeding tendency, family history, and abnormalities in levels of VWF:Act, VWF:Ag, VWF:CB, and FVIII, ratios between several of these levels, and the VWF multimer pattern. In clinical practice, diagnosing VWD can be challenging because of variability in laboratory parameters, heterogeneity of bleeding symptoms, and the age‐related gradual increase in VWF levels. Genotyping is a powerful diagnostic instrument and may play an important role in improving the diagnostic trajectory of VWD.
The aim of this study is to characterize patients with a suspicion of VWD in VWD type 2M based on genetic analysis and multimeric pattern, and to investigate the relationship between genotype and phenotype in this population.
2. METHODS
2.1. Patient inclusion
All subjects who underwent genotyping of the VWF gene in the Radboud university medical center from 2010 to 2020 were retrospectively screened for eligibility to participate in this study. This genotyped cohort consisted of: (1) patients with a high suspicion of VWD based on clinical bleeding tendency (Tosetto bleeding score ≥4 in males and ≥6 in females) and/or laboratory phenotype (VWF:Ag <30 IU dl−1 and/or VWF:Act <30 IU dl−1 and/or VWF:CB <30 IU dl−1 and/or VWF:Act/VWF:Ag ratio <0.7 and/or VWF:CB/VWF:Ag ratio <0.7), (2) family members of affected patients, and (3) patients in whom the treating physician had determined that genotyping was necessary in the diagnostic trajectory.
Genotyped subjects were included in this study when they met the following criteria: (1) at least one genetic variant located in the A1 or A3 domain of VWF classified as pathogenic (class 5), likely pathogenic (class 4) or as variant of unknown significance (class 3), according to the ACMG guidelines, 17 and (2) VWF multimers classified as normal or only slightly abnormal but objectively different from the aberrant multimer patterns observed in VWD type 2A and 2B. Informed consent was obtained from all participants.
2.2. Genotype
DNA was isolated from peripheral blood leucocytes following standard protocols. All 52 exons of the VWF gene, including exon‐intron boundaries (± 20 nucleotides intronic), were amplified and sequenced by polymerase chain reaction and Sanger sequencing, respectively. M13‐labeled VWF gene‐specific polymerase chain reaction primers (Biolegio BV) were used for sequencing in a fully automated system (Hamilton). 18 Primer sequences were manually inspected using the ensemble genome browser to detect known polymorphisms, variants, and pseudogene binding. 19 In cases lacking any pathogenic variant or when the phenotype could not be directly related to the detected genetic variant, multiplex ligation‐dependent probe amplification (MRC Holland) was carried out to detect exonic deletions and/or duplications. The clinical significance of unknown variants was assessed by in silico analysis with variant interpretation software Alamut (Interactive Biosoftware). The VWF protein variant nomenclature used in this manuscript is deduced from the VWF NCBI Reference Sequence (transcript number) NM_000552.3.
2.3. Clinical phenotype
To achieve a reliable evaluation of bleeding severity and bleeding pattern, the Tosetto bleeding score (BS) was calculated for each participant. 20 The BS was retrospectively assessed by extracting data from electronic patient reports. These data were entered in a standardized note template during a regular outpatient clinic visit. In case of missing data, a nurse practitioner or hematologist contacted patients by telephone to perform a new complete bleeding score.
To avoid bias from prophylactic administration of hemostatic products, bleeding episodes after tooth extraction, surgery, and delivery were only scored for patients who had not received antifibrinolytics, desmopressin, or FVIII/VWF concentrates before these procedures.
The clinical phenotype of our study population was compared to the phenotype in a nationwide cohort of 347 VWD type 1 and 216 VWD type 2 patients including 140 type 2A patients, 37 type 2B patients, 23 type 2M patients, and 16 type 2N patients (“Willebrand in the Netherlands” [WiN] study). 21 , 22 In the WiN study group, only patients ≥16 years with hemorrhagic symptoms or family history of VWD and VWF:Ag and/or VWF:Act levels ≤30 IU dl−1 and/or FVIII ≤40 IU dl−1 (only in case of VWD type 2N), were included. Their bleeding symptoms were analyzed using a condensed version of the Tosetto BS. 21
2.4. Laboratory phenotype
VWF:Ag levels were measured with an enzyme‐linked immunoabsorbent assay (ELISA) using Asserachrom ELISA kits of Stago 18 or the STA Liatest using the STA Evolution analyzer (Stago Gennevilliers). Validation did not show any variation between these two methods.
As part of our cohort was treated in other Dutch hemophilia treatment centers (HTCs), VWF:Act level was determined with different assays. VWF:RCo was used in 35 patients, VWF:Ab in 9 patients, VWF:GPIbM in 3 patients, and VWF:GPIbR in 5 patients. 23
Asserachrom ELISA kits of Stago containing collagen type III (Gennevilliers, France) were used to measure the binding potential of VWF to collagen. 18
FVIII activity was performed with a one‐stage clotting assay using STA Evolution analyzer (Stago Gennevilliers). The standard assay (range 3–150 IU dl−1) was performed with Cephascreen (Stago Gennevilliers). FVIII levels in the low range (0.5–5 IU dl−1) were determined with PTT‐LA (Stago, Gennevilliers).
An in‐house method using SaeKem HGT 1.5% and 3% agarose gels (Lonza) containing 0.1% sodium dodecyl sulphate was performed to evaluate VWF multimers. The fast system (Pharmacia Biotech) was used at variable voltage (100–250 V, 1.0–10 A) during 3 h for electrophoresis. The gels were blotted onto a polyvinylidene difluoride membrane with a blotter at 1.0 A for 90 min and usage of a 0.5 M phosphate blot buffer. After incubation with rabbit polyclonal anti‐human VWF‐antibody (No. A0082; Dako), polyvinylidene difluoride membranes were incubated with peroxidase goat anti‐rabbit HRP‐labelled IgG antibodies (No. 1706515; Bio‐Rad). Hereafter, the VWF bands were stained and all blots were visually inspected. 18 , 24
All VWF‐related laboratory results were collected from electronic patient files in the patients' HTCs. When multiple results were available, the lowest historical values were used.
2.5. Statistical analyses
Statistical analyses were performed with IBM SPSS Statistics, version 25. When laboratory values crossed reference values, highest or lowest reference values were used (e.g., VWF:Act 5 IU dl−1 in case of VWF:Act < 5 IU dl−1). Data were presented as median values and interquartile ranges (IQR, 25%–75%) because of a non‐normal distribution of data. Mann‐Whitney U tests were used to compare differences between two independent groups. Kruskal‐Wallis tests were performed to compare more than two independent groups, followed by Mann‐Whitney U tests for post hoc analyses. p values ≤.05 were considered statistically significant. A Bonferroni correction was applied to the post hoc Mann‐Whitney U tests. Results of the post hoc analyses were therefore considered statistically significant at p ≤ .017. No subgroup analyses according to the clinical reason for genotyping were performed because of the small number of patients due to the rarity of the disease.
3. RESULTS
3.1. General characteristics
A total of 79 patients had a genetic variant in the A1 or A3 domain of the VWF gene: 72 patients in the A1 domain and 7 patients in the A3 domain (Figure 1). Two patients were excluded because they were also diagnosed with moderate severe hemophilia A. One patient passed away before start of the study. Two patients did not give informed consent. Nineteen patients could not be contacted for informed consent or did not respond. Therefore, the multimer patterns of 53 patients with an A1 variant and two patients with an A3 variant were analyzed.
FIGURE 1.
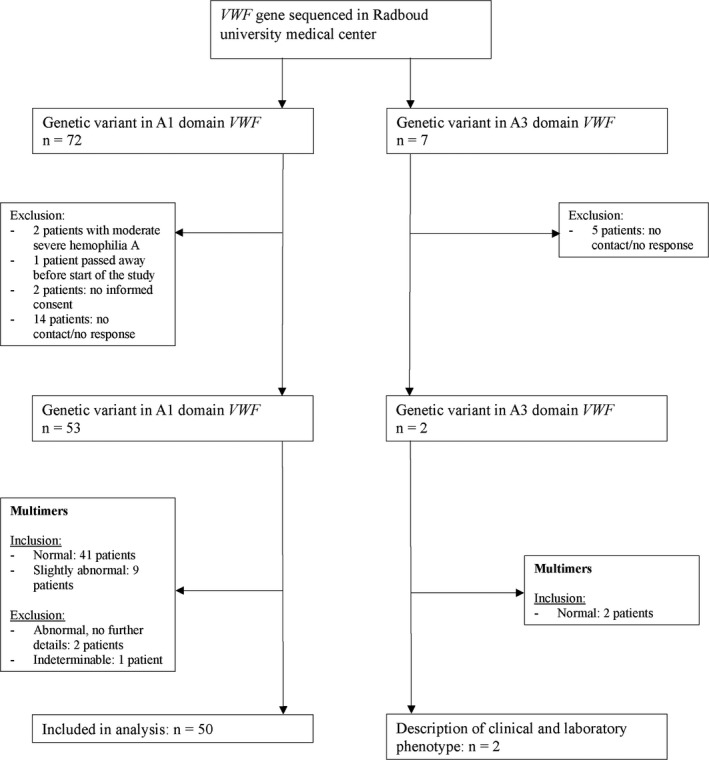
Flow chart of patients who met inclusion/exclusion criteria
The heterogeneous nature of the genetic variants is highlighted by the variance in multimeric pattern in some patients. In two patients with an A1 variant, multimer patterns were described as abnormal without providing any further information. In another patient, multimer pattern could not be definitely established because of low VWF levels. These three patients were excluded from our analysis. Eventually, 50 patients with a genetic variant in the A1 domain were analyzed, of whom 41 patients had normal multimers and nine had slightly abnormal multimers but clearly different from VWD type 2A or 2B (Table S1). These last patients were therefore also classified as VWD type 2M.
Of the patients with an A1 variant, 26 were women (52%) and 24 were men (48%). Median age was 39 years (range 7–81 years). Four patients were children (7–17 years; Table 1). We found no relevant differences in phenotype that can skew our analysis between included families (data not shown).
TABLE 1.
Characteristics of the study population
| Genetic variant in A1 domain | Genetic variant in A3 domain | |
|---|---|---|
| Number of patients, n (%) | 50 (96) | 2 (4) |
| Sex | ||
| Female, n (%) | 26 (52) | 2 (100) |
| Male, n (%) | 24 (48) | 0 (0) |
| Age, y | ||
| Total, median (range) | 39 (7–81) | 42–49 f |
| Adults, n (%) | 46 (92) | 2 (100) |
| Children, a n (%) | 4 (8) | 0 (0) |
| Bleeding score | ||
| Women, median (IQR) | 7 (3–13) b | 8–21 f |
| Men, median (IQR) | 5 (3–9) c | NA |
| Total, median (IQR) | 5 (3–11) d | 8–21 f |
| Laboratory phenotype | ||
| VWF:Ag, IU dl−1; median (IQR) | 29 (19–60) | 11–38 f |
| VWF:Act, IU dl−1; median (IQR) | 7 (5–30) | 5–39 f |
| VWF:CB, IU dl−1; median (IQR) | 20 (12–55) e | 5–24 f |
| VWF:Act/VWF:Ag ratio, median (IQR) | 0.32 (0.24–0.48) | 0.45–1.03 f |
| VWF:CB/VWF:Ag ratio, median (IQR) | 0.80 (0.65–1.01) e | 0.45–0.63 f |
| FVIII, IU dl−1,median (IQR) | 47 (30–76) | 55–57 f |
| Normal multimers, n (%) | 41 (82) | 2 (100) |
| Slightly abnormal multimers, n (%) | 9 (18) | 0 (0) |
| Blood group | ||
| O, n (%) | 28 (56) | 2 (100) |
| A, n (%) | 11 (22) | 0 (0) |
| B, n (%) | 6 (12) | 0 (0) |
| AB, n (%) | 0 (0) | 0 (0) |
| Unknown, n (%) | 5 (10) | 0 (0) |
All data are represented in numbers and in percentages between parentheses unless otherwise stated.
Abbreviations: FVIII, factor VIII; IQR, interquartile range; NA, not applicable; VWF, von Willebrand factor; VWF:Act, VWF activity; VWF:Ag, VWF antigen; VWF:CB, VWF collagen binding activity.
Aged 17 years or younger.
Data available for 25 patients.
Data available for 21 patients.
Data available for 46 patients.
Data available for 40 patients.
Absolute values were presented for each patient as only two patients with a variant in the A3 domain were included.
Both patients with an A3 variant had normal multimers (Figure 1). Patients with genetic variants in the A1 and A3 domain were analyzed separately (Table 1). A complete overview of patient characteristics is presented in Table S2. Fifteen patients with an A1 variant and one patient with an A3 variant from our cohort also participated in the WiN study. 21
3.2. Genotype
In this study, 14 different genetic variants were identified in the A1 domain of exon 28 of the VWF gene. In the A3 domain of the VWF gene, two different genetic variants were identified (Table S3). All subjects were heterozygous for the genetic variant.
Some patients also carried other variants outside the A1 or A3 domain. The polymorphism p. Asp1472His in exon 28 was detected in 10 patients. This polymorphism affects the ristocetin binding activity in the VWF:RCo assay mostly used in this study without altering the in vivo activity, leading to an underestimation of the VWF‐platelet binding capacity. 25 Previous research showed that this polymorphism was present in 63% of African American controls and 17% of Caucasian controls. 25
Two of the patients with the p. Asp1472His variant also had a variant in exon 37 of the VWF gene (p. Arg2185Gln). This variant was previously reported in 18.2% of healthy African Americans. 26 In another study, this variant was associated with lower levels of VWF (13 IU dl−1) and FVIII (7 IU dl−1). 27 One patient had a pathogenic gene conversion with VWF and its pseudogene (VWFP1) including the pathogenic variant c.3797C>T (p. Pro1266Leu) and coupled variants c.3789G>A and c.3835G>A (p. Val1279Ile). This patient also had the variant c.6553C>T (p. Arg2185Trp). It is not known whether this variant was in cis or in trans with the gene conversion variants.
Of nine patients with a small loss of only highest molecular weight multimers, six had the p. Arg1374Cys variant, two the p. Arg1374His variant, and one the p. Arg1315Cys variant (Table S1).
3.3. Clinical phenotype
Data about BS were available in 46 patients with an A1 variant and in both patients with an A3 variant (Table 1). These last two patients had a BS of 8 and 21.
In patients with an A1 variant, median BS was 5 (IQR 3–11). BS was not significantly different between women (7, IQR 3–13) and men (5, IQR 3–9; p = .48). An abnormal BS was found in 52% of women (BS ≥ 6), and 71% of men (BS ≥ 4). Most common symptoms were menorrhagia (92%), cutaneous bleedings (63%), bleeding from minor wounds (59%), epistaxis (41%), and gum bleeding (28%). Two patients (4%) had experienced a central nervous system bleeding (subarachnoid hemorrhage and traumatic cerebral bleeding).
Eight of 15 women with an A1 variant who gave birth (53%) did not receive hemostatic therapy during delivery because their diagnosis of VWD was not yet established. Six of them (75%) experienced a postpartum hemorrhage. Bleeding was reported in the majority of patients who underwent surgery without hemostatic treatment (n = 19, 83%) and in 12 patients (50%) who underwent dental procedures without hemostatic treatment.
The clinical phenotype of VWD type 2M patients with an A1 variant was compared with a nationwide cohort of 347 VWD type 1 and 216 VWD type 2 patients from the WiN study (Figure 2). 21 , 22 As mentioned earlier, our VWD type 2M patients suffered mainly from mucocutaneous bleedings as epistaxis, cutaneous bleeding, bleeding from minor wounds, gum bleeding, and menorrhagia. However, the prevalence of these bleeding symptoms in our VWD type 2M cohort was relatively low compared with the VWD type 1 and general VWD type 2 cohort, except for menorrhagia. Bleeding complications after surgery and delivery were more frequently reported in VWD type 2M patients, whereas bleeding after tooth extractions was more frequently mentioned in the VWD type 1 and general VWD type 2 cohort.
FIGURE 2.
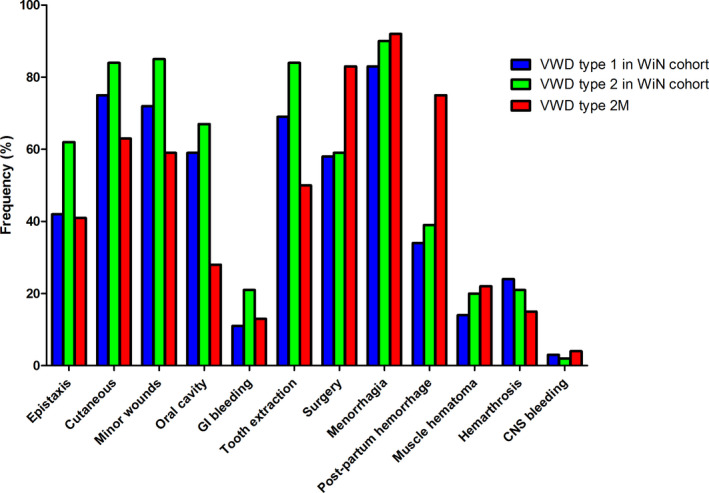
Frequency of bleeding symptoms in our cohort of VWD type 2M patients compared with a cohort of VWD type 1 and 2 patients from the WiN study. 22 A reported bleeding was defined as a subscore ≥1. In the VWD type 2M cohort, 24 patients underwent tooth extraction without hemostatic treatment, 23 patients underwent surgery without hemostatic treatment, 25 women have been menstruating, and 8 women gave birth without hemostatic treatment. VWD, von Willebrand disease; WiN, Willebrand in the Netherlands
3.4. Laboratory phenotype
First, we analyzed the laboratory phenotype of patients with an A1 variant. Median VWF:Ag level was 29 IU dl−1 (IQR 19–60), median VWF:Act 7 IU dl−1 (IQR 5–30), and median VWF:Act/VWF:Ag ratio 0.32 (IQR 0.24–0.48). Median levels of VWF:CB, VWF:CB/VWF:Ag ratio, and FVIII were 20 IU dl−1 (IQR 12–55), 0.80 (IQR 0.65–1.01), and 47 IU dL−1 (IQR 30–76), respectively (Table 1).
Most patients had blood group O (n = 28; 56%), 11 had blood group A (22%), and 6 had blood group B (12%). Blood group was unknown in 5 patients (10%; Table 1). No statistically significant differences in laboratory parameters were found between patients with blood group O, A, and B.
Furthermore, the laboratory phenotype of the two patients with an A3 variant was analyzed. The patient with the p. Ser1731Thr variant had a low VWF:CB/VWF:Ag ratio (0.63) but a normal VWF:Act/VWF:Ag ratio (1.03). VWF:CB/VWF:Ag and VWF:Act/VWF:Ag ratio were both decreased in the patient with the p. Lys1794Glu variant (0.45 each; Table S2).
None of the included patients had a multimer pattern characteristic of VWD type 2A or 2B. Multimers were normal in 43 patients. In nine patients, only minor abnormalities were found in the upper part of the multimer pattern, which were clearly different from that seen in VWD type 2A or 2B, as indicated by the red box in Figure 3.
FIGURE 3.

Multimeric analysis of plasma VWF by using 1.5% SDS‐agarose gel. N demonstrates multimers in a normal subject, I demonstrates multimers representative for the slightly aberrant multimer patterns observed in nine VWD type 2M patients in our cohort, and 2A demonstrates multimers in a patient with a well‐defined VWD type 2A. VWD, von Willebrand disease; VWF, von Willebrand factor
3.5. Most prevalent genetic variants
The clinical and laboratory phenotype of patients with the three most prevalent genetic variants in the A1 domain (p. Phe1293Leu, p. Val1360Ala, and p. Arg1374Cys) were compared. Figure 4A–E represents the VWF:Ag, VWF:Act, VWF:CB, VWF:Act/VWF:Ag ratio, and VWF:CB/VWF:Ag ratio, Figure 4F shows the FVIII activity level, and Figure 4G the BS for these patient groups and for the two patients with a genetic variant in the A3 domain.
FIGURE 4.
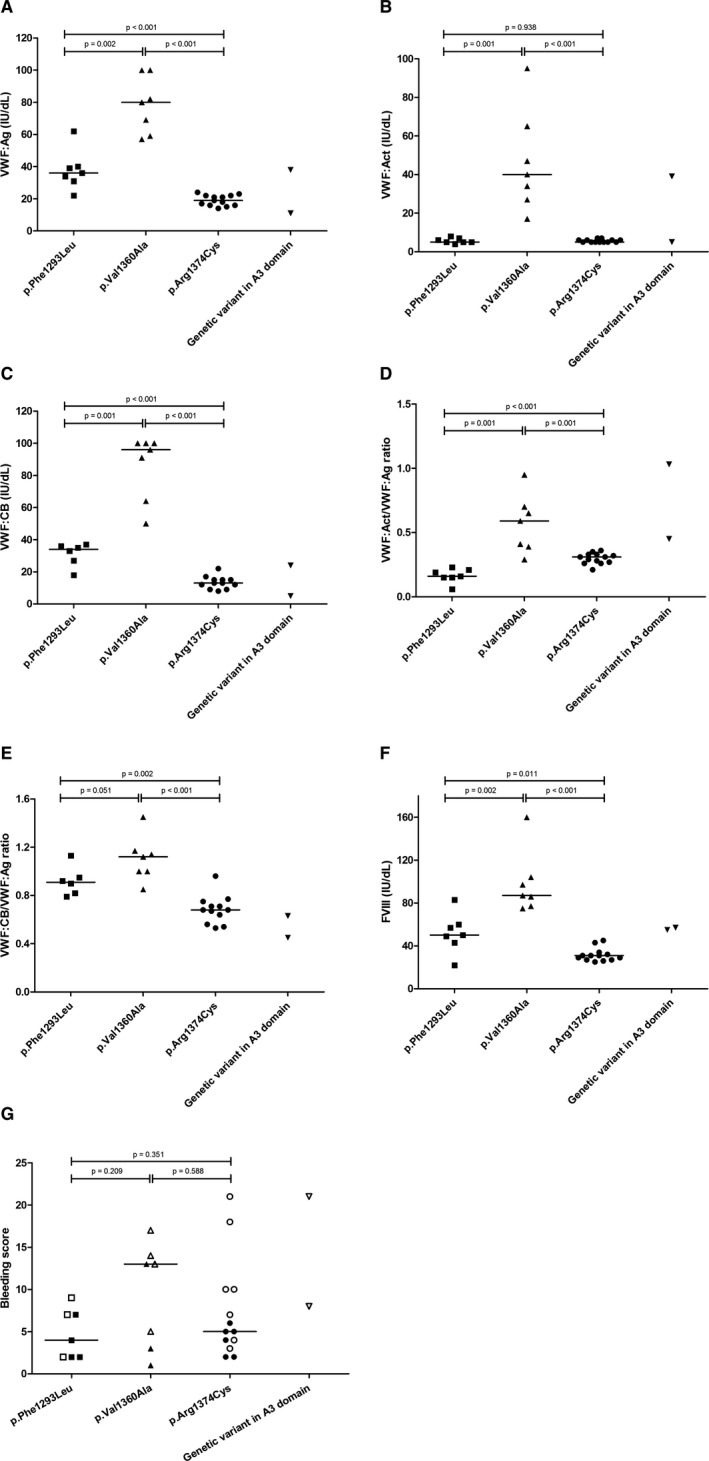
Laboratory and clinical phenotype in the three most prevalent VWD type 2M variants in the A1 domain and in the two included variants in the A3 domain. (A) VWF:Ag, (B) VWF:Act, (C) VWF:CB, (D) VWF:Act/VWF:Ag ratio, (E) VWF:CB/VWF:Ag ratio, (F) FVIII, and the (G) bleeding score. Horizontal lines represent median values. Each dot, box, and (reversed) triangle represent a single patient. In panel G, open figures represent females and closed figures represent males. VWD, von Willebrand disease; VWF, von Willebrand factor; VWF:Act, VWF activity; VWF:Ag, VWF antigen; VWF:CB, VWF collagen binding activity; FVIII, factor VIII
Significantly higher levels of VWF:Ag, VWF:Act, VWF:CB, and VWF:Act/VWF:Ag ratio were found in patients with the p. Val1360Ala variant compared with patients with the p. Phe1293Leu variant (p ≤ .002) and the p. Arg1374Cys variant (p ≤ .001; Figure 4A–D). Moreover, the VWF:CB/VWF:Ag ratio was significantly higher in patients with the p. Val1360Ala variant than in patients with the p. Arg1374Cys variant (p < .001, Figure 4E). The p. Phe1293Leu variant was characterized by significantly lower levels of the VWF:Act/VWF:Ag ratio and significantly higher levels of the VWF:CB/VWF:Ag ratio compared to the p. Arg1374Cys variant (p < .001 and p = .002, respectively; Figure 4D,E).
The clinical phenotype of patients with these A1 variants was heterogeneous, but did not differ significantly (Figure 4G). Higher BS was more frequently seen in female patients in all three genetic variants.
Three subjects with the p. Val1360Ala variant showed a (near) normal laboratory phenotype with a normal VWF:CB/VWF:Ag ratio (all ≥ 1.00) and a VWF:Act/VWF:Ag ratio of 0.95, 0.65, and 0.70 (Table S2: patients 16, 17, and 19). They reported a BS of 13, 17, and 5, respectively. These patients were all family members of a female patient with the p. Val1360Ala variant who was extensively analyzed for other coagulation disorders because of a discrepancy between near‐normal VWF parameters and clinical bleeding phenotype (BS 14, Table S2: patient 18). She was diagnosed recently with a thrombocytopathy (abnormal light transmission aggregometry results with epinephrine, adenosine diphosphate, and thrombin receptor activating protein). Patient 19 was tested negative for a thrombocytopathy; patients 16 and 17 were not tested.
4. DISCUSSION
In this study, we explored the clinical and laboratory heterogeneity in genetically confirmed VWD type 2M patients with variants in the A1 and A3 domain of the VWF gene, categorized as pathogenic, likely pathogenic or variant of unknown significance, without evident loss of multimers. In contrast to previous studies, we used the results of genotyping and VWF multimeric analysis to define our VWD type 2M subpopulation from a preselected cohort of patients with (a high suspicion of) VWD in whom genotyping was already performed in the regular diagnostic trajectory.
In most patients with A1 variants, the affinity of VWF to GPIb was decreased. Two patients had an A3 variant. The A3 domain of the VWF gene plays an important role in the binding of VWF to collagen because it contains the main binding sites for collagen types I and III. 28 , 29 Both patients with an A3 variant had a low VWF:CB/VWF:Ag ratio. However, one of these patients with the p. Lys1794Glu variant also had a low VWF:Act/VWF:Ag ratio (0.45). This genetic variant was previously only described in a patient with a VWD type 1 phenotype (VWF:Act/VWF:Ag ratio 0.76 and VWF:CB/VWF:Ag ratio 0.88). 30 Previous research related to the p. Ser1731Thr variant showed a significantly reduced binding to collagen type I, but a normal or only slightly decreased binding to collagen type III in static and flow‐based VWF:CB assays, respectively. 31 Another functional study on the p. Ser1731Thr variant demonstrated a reduced binding to both type I and type III collagen (45% and 50% of wild‐type, respectively). 32 In our patient with the p. Ser1731Thr variant, VWF:CB/VWF:Ag was 0.63, but we only measured the binding potential to collagen type III.
In the entire cohort of patients who underwent genotyping of the VWF gene in the Radboud university medical center, 72 patients had an A1 genetic variant, but only 7 had an A3 variant (Figure 1). Also in the literature, many variants in the A1 domain but only few in the A3 domain have been described. 12 , 16 This observation questions the importance of the function of the A3 domain in the binding of VWF to the vascular wall. 16 A possible theory is that the A1 domain might be able to compensate for a defective A3 domain by increasing collagen IV and VI binding via the A1 domain. Another previously hypothesized theory is the existence of a functional cross‐talk between the A1 and A3 domain in which the affinity of the A1 domain for collagen is negatively influenced by the A3 domain. In that situation, collagen binding via the A1 domain can be fully used in case of a defective A3 domain. 16
The mild averaged bleeding phenotype of our patient group (median BS 5) is comparable with previous studies in patients with VWD type 2M. 21 , 33 Type 2M patients in the WiN study (n = 23) had a slightly higher median BS of 9. 21 This discrepancy can be explained by the smaller 2M study population in the WiN study, that they used a slightly different self‐administered version of the condensed Tosetto BS, and their strict inclusion criteria concerning VWF:Ag and VWF:Act level. They only included patients with a VWF:Ag ≤ 30 IU dl−1 or VWF:Act ≤ 30 IU dl−1, and excluded children (<16 years). By applying these criteria, median BS in our cohort was 6. Furthermore, patients in the WiN study were classified based on their laboratory phenotype regardless of the specific genetic variants that were found. In contrast to this study, in which the genotype was used for the diagnostic subdivision of type 2M. Fifteen patients with an A1 variant from our cohort also participated in the WiN study. After exclusion of these WiN participants, BS in our cohort did not change (median BS 5, IQR 3–11). In another Italian cohort of 169 VWD type 2M patients, median BS was also 5. 33 However, only 46% of them had a severe laboratory phenotype (VWF:Act <10 IU dL−1) compared with 60% in our cohort.
Moreover, we compared the BS of our VWD type 2M cohort with the BS of patients with low VWF and VWD type 1 in two different cohorts because these patients are also known to have a milder bleeding phenotype. First, in an Irish cohort of 126 patients with a diagnosis of low VWF, median BS in women and men was 8 and 3, respectively, compared with 7 and 5 in our VWD type 2M cohort. 34 However, these data can be skewed because a significant personal bleeding history was a diagnostic criterium for low VWF. Furthermore, they used the ISTH BAT in contrast to the Tosetto BS used in our study. Second, in the WiN study, 347 VWD type 1 patients had a higher median BS of 9. 21 As described earlier, this discrepancy can be (partly) explained by the slightly different methods and strict inclusion criteria used in the WiN study.
In our study, mucocutaneous bleedings were most regularly reported but frequencies were low compared with other VWD type 2 patients. However, BS may be underestimated because family members of affected patients were also included in our study and patients were relatively young. The median age of women and men in our cohort was 39 (range 16–81) and 30 years (range 2–82) respectively.
A distinct difference in bleeding symptoms of patients with VWD type 2M and the general VWD type 2 population was the increased bleeding after surgery and postpartum, emphasizing the importance of a proper hemostatic treatment in VWD type 2M patients who will undergo surgery or give birth. An explanation is that the binding of VWF to platelet GPIb and the subsequent attachment to the injured endothelial wall is disturbed, which predisposes these VWD type 2M patients for ongoing bleeding. However, the number of patients in our cohort who underwent a surgical procedure or gave birth without hemostatic treatment was low (23 patients (46%) and 8 patients (16%), respectively).
Comparison of the phenotype of the three most prevalent A1 variants demonstrated that there were distinct differences in laboratory phenotype. The p. Val1360Ala variant was in our study accompanied with near normal VWF antigen and activity levels but with profound bleeding scores. Highest VWF antigen and activity levels were observed in elderly patients, but still with concomitantly high bleeding scores (Table S2). These patients would not have been diagnosed if only VWF parameters had been assessed in the diagnostic trajectory. Genetic analysis was essential for making a correct diagnosis of VWD type 2M in these patients. This observation raises the question whether VWF levels do increase with aging in patients with VWD type 2M, in contrast to VWD types 2A and 2B.
The previously mentioned patients with the p. Val1360Ala variant were family members of a patient who was diagnosed with both VWD type 2M and a thrombocytopathy. The elderly patients with the highest bleeding scores were not tested for a thrombocytopathy. These specific cases illustrate the importance of performing a thorough diagnostic trajectory if a bleeding phenotype does not match with a single hemostatic dysfunction. Thrombocytopathy and VWD type 2M are both disorders of primary hemostasis and thus can present with a severe bleeding diathesis once patients undergo an invasive procedure or event.
The characterization of some genetic variants, particularly p. Arg1374Cys and p. Arg1315Cys, as either VWD type 2A or 2M is debated in literature. 35 , 36 According to the updated classification, 8 VWF‐dependent platelet adhesion is reduced in both VWD types 2A and 2M. Multimeric distribution is approximately normal in VWD type 2M, whereas there is a clear deficiency of high molecular weight multimers in VWD type 2A. 8 We identified slightly abnormal multimer patterns in nine patients with VWD type 2M caused by an A1 genetic variant. The almost minor abnormalities in these patients comprised a small loss of only the highest molecular weight multimers, which clearly differed from the abnormalities seen in VWD type 2A or type 2B (Figure 3 and Table S1). These subtle aberrancies will probably only be detected by sophisticated multimeric analysis in specialized laboratories.
Despite the wide range of available laboratory assays, the phenotypic characterization of VWD is complex and currently imperfect, particularly the type 2 subclassification. Consequently, there are discrepancies in VWD diagnoses among clinical laboratories. 37 VWD type 2M is frequently underrecognized and misidentified as VWD type 1 or type 2A. 38 , 39 An accurate distinction between VWD type 1 and type 2 is relevant because of a generally more severe bleeding phenotype in VWD type 2 and a differential response to desmopressin. Patients with VWD type 1 usually respond well to desmopressin, whereas type 2 patients have a variable response. 40 , 41 Previous data suggest that the desmopressin response is generally better in patients with VWD type 2M than in patients with type 2A. 9 , 38 A trial to determine the response and effectiveness of desmopressin is nevertheless always recommended in these patients. 9 Besides the different response to desmopressin, current treatment of patients with VWD types 2A and 2M is almost identical for invasive procedures and major bleeding as they are all treated with VWF concentrates. 4 Maybe in the future, more sophisticated recombinant VWF concentrates will become available. A better differentiation of VWD type 2A and type 2M can than lead to more personalized tailor‐made treatment plans. A VWF concentrate containing mainly high molecular weight multimers may than be used in patients with VWD type 2A because they experience a loss of the high molecular weight multimers, whereas such a VWF concentrate is less relevant for patients with VWD type 2M. 38 Furthermore, VWD type 2M patients with a genetic variant in the A1 or A3 domain could be treated with different VWF concentrates aimed at improving either the VWF‐platelet or VWF‐collagen binding.
Additional to potential (future) treatment dissimilarities, an accurate diagnosis of either VWD type 2A or 2M can assist in an accurate prediction of the bleeding risk. It was demonstrated that patients with VWD type 2A have a more severe bleeding phenotype than patients with type 2M, and a higher incidence of gastrointestinal bleedings. 42
According to the current guidelines, analysis of VWF multimers and/or VWF:Act/VWF:Ag and VWF:CB/VWF:Ag ratio plays a crucial role in the classification of VWD types 2A and 2M. 43 However, multimeric analysis is a highly specialized assay with a high rate of diagnostic errors because of an inadequate performance or interlaboratory differences in interpretation. 38 In these cases, VWF:CB can be helpful as VWF:CB is a marker for the presence of high molecular weight VWF multimers, but also this assay is not broadly available. 43 Moreover, the type of collagen that is used varies between laboratories (mostly type I and/or type III). 43 Despite the presence of binding sites for collagen types IV and VI in the A1 domain of VWF and the fact that genetic variants influencing the interaction with these collagens have been described, binding to these types of collagen is not regularly tested. 13
These examples emphasize the need for further improvement in either the diagnostic strategies or classification of VWD. Despite occasional difficulties in the interpretation of genetic variants as being pathogenic or a polymorphism, genetic analysis is less prone to subjective interpretation compared to functional VWF assays. Because we observed that the three most prevalent genetic variants in our VWD type 2M cohort each have a unique laboratory phenotype, performing genetic analysis along with functional VWF assays can improve our understanding of the pathophysiology of VWD and may ultimately lead to more advanced classification schemes and individualized treatment plans. The distinct differences in laboratory phenotype between patients with the same genetic variant, and our observation that some patients with a genetic variant in the A1 domain did not have a typical VWD type 2M phenotype, also imply that the current VWD classification system is suboptimal.
A great strength of this study was the assessment of clinical and laboratory phenotype in a genetically confirmed VWD type 2M population. A potential prophylaxis‐bias in calculating the BS was eluded by not scoring bleeding symptoms when patients had received prophylaxis before invasive procedures or delivery. 21 , 44 Our study had some limitations. Because we included patients from an already genotyped cohort, a potential selection bias cannot be excluded. The use of a BS to determine clinical phenotype has inherent limitations. BS are cumulative and therefore they generally increase with advancing age. Novel (severe) bleeding events cannot be scored when a patient already has reached a maximum score in that specific bleeding category. Moreover, bleeding symptoms are self‐reported and consequently some recall bias may occur. BS remained unknown in four patients and blood group in five patients. Our results, however, did not change after subgroup analyses of patients with blood group O, indicating that blood group was not a confounding factor in our study. Not all VWF parameters were assessed in the same laboratory because some patients were treated in another center. However, all assays were performed in an HTC following standardized and validated protocols. The polymorphism p. Asp1472His was identified in 10 patients. Ristocetin independent assays were performed in six of these patients. In the remaining four patients, ristocetin dependent assays were used, potentially leading to an artificially reduced VWF:Act. Finally, because of the rarity of the disease, our study population was relatively small, particularly for A3 genetic variants. In the future, larger cohort studies are needed to investigate possible discrepancies in clinical phenotype between VWD type 2M patients with A1 and A3 genetic variants.
In conclusion, in this study, we have elucidated the mild clinical bleeding phenotype of patients with genetically confirmed VWD type 2M and observed that some genetic variants were associated with a more characteristic VWD type 2M laboratory phenotype (p. Phe1293Leu), whereas some other variants such as p. Arg1374Cys and p. Arg1374His, are not. The assessment of VWD, particularly in case of type 2, is challenging because of heterogeneity in bleeding phenotype, suboptimal functional laboratory tests with large interindividual and interlaboratory variability, and a currently insufficient classification system. Simultaneous performance of genetic analysis and functional VWF assays may lead to improvement of our current diagnostic capacities and subsequently can result in more advanced VWD classification and treatment schemes.
CONFLICT OF INTEREST
Dr. Atiq received the CSL Behring‐professor Heimburger Award 2018 and a travel grant from Sobi. Dr. Laros‐van Gorkom has received unrestricted educational grants from Baxter and CSL Behring. Dr. Leebeek received research support from CSL Behring and Shire/Takeda for performing the Willebrand in the Netherlands (WiN) study and Sobi and uniQure for studies not related to this manuscript, and is consultant for uniQure, Sobi, Biomarin, and Shire/Takeda, of which the fees go to the institution, and has received a travel grant from Sobi. He is also a DSMB member for a study by Roche. Dr. van Heerde reports speaker and consultant and travel fees from Takeda, Bayer, CSL Behring, and Sobi. He is also cofounder and CSO of Enzyre. Dr. Schols has received travel grants from Bayer and Takeda, and consultancy grants from Takeda and Novo Nordisk. None of the other authors has a conflict of interest to declare.
AUTHOR CONTRIBUTIONS
Dominique P. M. S. M. Maas designed the study, performed statistical analysis, interpreted data, and wrote the manuscript. Ferdows Atiq, Paul P.T. Brons, Britta A.P. Laros‐van Gorkom, Frank W.G. Leebeek, and Laurens Nieuwenhuizen recruited participants, collected data, and critically revised the manuscript. Nicole M.A. Blijlevens, Sandy Krouwel, Selene C.M. Schoormans, and Annet Simons interpreted data and critically revised the manuscript. Daniëlle Meijer, Waander L. van Heerde, and Saskia E.M. Schols designed the study, interpreted data, and critically revised the manuscript. All authors gave their consent to the final version of the manuscript.
Supporting information
Supplementary Material
Maas DPMSM, Atiq F, Blijlevens NMA, et al. Von Willebrand disease type 2M: Correlation between genotype and phenotype. J Thromb Haemost. 2022;20:316–327. doi: 10.1111/jth.15586
Manuscript handled by: David Lillicrap
REFERENCES
- 1. von Lillicrap D. Willebrand disease: advances in pathogenetic understanding, diagnosis, and therapy. Blood. 2013;122:3735‐3740. 10.1182/blood-2013-06-498303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fogarty H, Doherty D, O'Donnell JS. New developments in von Willebrand disease. Br J Haematol. 2020;191:329‐339. 10.1111/bjh.16681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baronciani L, Peyvandi F. How we make an accurate diagnosis of von Willebrand disease. Thromb Res. 2020;196:579‐589. 10.1016/j.thromres.2019.07.010 [DOI] [PubMed] [Google Scholar]
- 4. Leebeek FW, Eikenboom JC. Von Willebrand's disease. N Engl J Med. 2016;375:2067‐2080. 10.1056/NEJMra1601561 [DOI] [PubMed] [Google Scholar]
- 5. Leebeek FWG, Atiq F. How I manage severe von Willebrand disease. Br J Haematol. 2019;187:418‐430. 10.1111/bjh.16186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tosetto A, Badiee Z, Baghaipour MR, et al. Bleeding symptoms in patients diagnosed as type 3 von Willebrand disease: results from 3WINTERS‐IPS, an international and collaborative cross‐sectional study. J Thromb Haemost. 2020;18:2145‐2154. 10.1111/jth.14886 [DOI] [PubMed] [Google Scholar]
- 7. James PD, Lillicrap D. The molecular characterization of von Willebrand disease: good in parts. Br J Haematol. 2013;161:166‐176. 10.1111/bjh.12249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103‐2114. 10.1111/j.1538-7836.2006.02146.x [DOI] [PubMed] [Google Scholar]
- 9. Tosetto A, Castaman G. How I treat type 2 variant forms of von Willebrand disease. Blood. 2015;125:907‐914. 10.1182/blood-2014-08-551960 [DOI] [PubMed] [Google Scholar]
- 10. Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev. 2010;24:123‐134. 10.1016/j.blre.2010.03.003 [DOI] [PubMed] [Google Scholar]
- 11. Legendre P, Navarrete AM, Rayes J, et al. Mutations in the A3 domain of von Willebrand factor inducing combined qualitative and quantitative defects in the protein. Blood. 2013;121:2135‐2143. 10.1182/blood-2012-09-456038 [DOI] [PubMed] [Google Scholar]
- 12. Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood. 2014;124:1412‐1425. 10.1182/blood-2014-05-378638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Jong A, Eikenboom J. Von Willebrand disease mutation spectrum and associated mutation mechanisms. Thromb Res. 2017;159:65‐75. 10.1016/j.thromres.2017.09.025 [DOI] [PubMed] [Google Scholar]
- 14. Bryckaert M, Rosa JP, Denis CV, Lenting PJ. Of von Willebrand factor and platelets. Cell Mol Life Sci. 2015;72:307‐326. 10.1007/s00018-014-1743-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Riddell AF, Gomez K, Millar CM, et al. Characterization of W1745C and S1783A: 2 novel mutations causing defective collagen binding in the A3 domain of von Willebrand factor. Blood. 2009;114:3489‐3496. 10.1182/blood-2008-10-184317 [DOI] [PubMed] [Google Scholar]
- 16. Bonnefoy A, Romijn RA, Vandervoort PA, Van rompaey I, Vermylen J, Hoylaerts MF. von Willebrand factor A1 domain can adequately substitute for A3 domain in recruitment of flowing platelets to collagen. J Thromb Haemost. 2006;4:2151‐2161. 10.1111/j.1538-7836.2006.02111.x [DOI] [PubMed] [Google Scholar]
- 17. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Meegeren ME, Mancini TL, Schoormans SC, et al. Clinical phenotype in genetically confirmed von Willebrand disease type 2N patients reflects a haemophilia A phenotype. Haemophilia. 2015;21:e375‐e383. doi: 10.1111/hae.12733 [DOI] [PubMed] [Google Scholar]
- 19. http://www.ensembl.org.
- 20. Tosetto A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM‐1 VWD). J Thromb Haemost. 2006;4:766‐773. 10.1111/j.1538-7836.2006.01847.x [DOI] [PubMed] [Google Scholar]
- 21. de Wee EM, Sanders YV, Mauser‐Bunschoten EP, et al. Determinants of bleeding phenotype in adult patients with moderate or severe von Willebrand disease. Thromb Haemost. 2012;108:683‐692. 10.1160/th12-04-0244 [DOI] [PubMed] [Google Scholar]
- 22. Sanders YV, de Wee EM, Meijer K, et al. Von Willebrand disease in the Netherlands: the WiN study. Ned Tijdschr Geneeskd. 2014;158:A6518. [PubMed] [Google Scholar]
- 23. Higgins RA, Goodwin AJ. Automated assays for von Willebrand factor activity. Am J Hematol. 2019;94:496‐503. 10.1002/ajh.25393 [DOI] [PubMed] [Google Scholar]
- 24. Ledford‐Kraemer MR. Analysis of von Willebrand factor structure by multimer analysis. Am J Hematol. 2010;85:510‐514. 10.1002/ajh.21739 [DOI] [PubMed] [Google Scholar]
- 25. Flood VH, Gill JC, Morateck PA, et al. Common VWF exon 28 polymorphisms in African Americans affecting the VWF activity assay by ristocetin cofactor. Blood. 2010;116:280‐286. 10.1182/blood-2009-10-249102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bellissimo DB, Christopherson PA, Flood VH, et al. VWF mutations and new sequence variations identified in healthy controls are more frequent in the African‐American population. Blood. 2012;119:2135‐2140. 10.1182/blood-2011-10-384610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnsen JM, Auer PL, Morrison AC, et al. Common and rare von Willebrand factor (VWF) coding variants, VWF levels, and factor VIII levels in African Americans: the NHLBI Exome Sequencing Project. Blood. 2013;122:590‐597. 10.1182/blood-2013-02-485094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huizinga EG, Martijn van der Plas R, Kroon J, Sixma JJ, Gros P. Crystal structure of the A3 domain of human von Willebrand factor: implications for collagen binding. Structure. 1997;5:1147‐1156. 10.1016/s0969-2126(97)00266-9 [DOI] [PubMed] [Google Scholar]
- 29. Reininger AJ. Function of von Willebrand factor in haemostasis and thrombosis. Haemophilia. 2008;14(Suppl 5):11‐26. 10.1111/j.1365-2516.2008.01848.x [DOI] [PubMed] [Google Scholar]
- 30. Goodeve A, Eikenboom J, Castaman G, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease (MCMDM‐1VWD). Blood. 2007;109:112‐121. 10.1182/blood-2006-05-020784 [DOI] [PubMed] [Google Scholar]
- 31. Shida Y, Rydz N, Stegner D, et al. Analysis of the role of von Willebrand factor, platelet glycoprotein VI‐, and α2β1‐mediated collagen binding in thrombus formation. Blood. 2014;124:1799‐1807. 10.1182/blood-2013-09-521484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Flood VH, Lederman CA, Wren JS, et al. Absent collagen binding in a VWF A3 domain mutant: utility of the VWF:CB in diagnosis of VWD. J Thromb Haemost. 2010;8:1431‐1433. 10.1111/j.1538-7836.2010.03869.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Federici AB, Bucciarelli P, Castaman G, et al. The bleeding score predicts clinical outcomes and replacement therapy in adults with von Willebrand disease. Blood. 2014;123:4037‐4044. 10.1182/blood-2014-02-557264 [DOI] [PubMed] [Google Scholar]
- 34. Lavin M, Aguila S, Schneppenheim S, et al. Novel insights into the clinical phenotype and pathophysiology underlying low VWF levels. Blood. 2017;130:2344‐2353. 10.1182/blood-2017-05-786699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Penas N, Perez‐Rodriguez A, Torea JH, et al. von Willebrand disease R1374C: type 2A or 2M? A challenge to the revised classification. High frequency in the northwest of Spain (Galicia). Am J Hematol. 2005;80:188‐196. 10.1002/ajh.20470 [DOI] [PubMed] [Google Scholar]
- 36. Casana P, Martinez F, Espinos C, Haya S, Lorenzo JI, Aznar JA. Search for mutations in a segment of the exon 28 of the human von Willebrand factor gene: new mutations, R1315C and R1341W, associated with type 2M and 2B variants. Am J Hematol. 1998;59:57‐63. [DOI] [PubMed] [Google Scholar]
- 37. DiGiandomenico S, Christopherson PA, Haberichter SL, Abshire TC, Montgomery RR, Flood VH. Laboratory variability in the diagnosis of type 2 VWD variants. J Thromb Haemost. 2021;19:131‐138. 10.1111/jth.15129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Favaloro EJ, Pasalic L, Curnow J. Type 2M and Type 2A von Willebrand disease: similar but different. Semin Thromb Hemost. 2016;42:483‐497. 10.1055/s-0036-1579641 [DOI] [PubMed] [Google Scholar]
- 39. Favaloro EJ. Von Willebrand disease: local diagnosis and management of a globally distributed bleeding disorder. Semin Thromb Hemost. 2011;37:440‐455. 10.1055/s-0031-1281028 [DOI] [PubMed] [Google Scholar]
- 40. Federici AB. The use of desmopressin in von Willebrand disease: the experience of the first 30 years (1977–2007). Haemophilia. 2008;14(Suppl 1):5‐14. 10.1111/j.1365-2516.2007.01610.x [DOI] [PubMed] [Google Scholar]
- 41. Favaloro EJ. Towards personalised therapy for von Willebrand disease: a future role for recombinant products. Blood Transfus. 2016;14:262‐276. 10.2450/2016.0258-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Castaman G, Federici AB, Tosetto A, et al. Different bleeding risk in type 2A and 2M von Willebrand disease: a 2‐year prospective study in 107 patients. J Thromb Haemost. 2012;10:632‐638. 10.1111/j.1538-7836.2012.04661.x [DOI] [PubMed] [Google Scholar]
- 43. James PD, Connell NT, Ameer B, et al. Guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021;2021(5):280‐300. 10.1182/bloodadvances.2020003265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tosetto A, Castaman G, Rodeghiero F. Bleeding scores in inherited bleeding disorders: clinical or research tools? Haemophilia. 2008;14:415‐422. 10.1111/j.1365-2516.2007.01648.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material