

Abstract
The US Food and Drug Administration (FDA) is open to accepting real‐world evidence (RWE) to support its assessment of medical products. However, RWE stakeholders lack a shared understanding of FDA’s evidentiary expectations for the use of RWE in applications for new drugs and biologics. We conducted a systematic review of publicly available FDA approval documents from January 2019 to June 2021. We sought to quantify, by year, how many approvals incorporated RWE in any form, and the intended use of RWE in those applications. Among approvals with RWE intended to support safety and/or effectiveness, we classified whether and how those studies impacted FDA’s benefit‐risk considerations, whether those studies were incorporated into the product label, and the therapeutic area of the medical product. Finally, we qualified FDA’s documented feedback where available. We found that 116 approvals incorporated RWE in any form, with the proportion of approvals incorporating RWE increasing each year. Of these approvals, 88 included an RWE study intended to provide evidence of safety or effectiveness. Among these 88 approvals, 65 of the studies influenced FDA’s final decision and 38 were included in product labels. The 88 approvals spanned 18 therapeutic areas. FDA’s feedback on RWE study quality included methodological issues, sample size concerns, omission of patient level data, and other limitations. Based on these findings, we would anticipate that future guidance on FDA’s evidentiary expectations of RWE use will incorporate fit‐for‐purpose real‐world data selection and careful attention to study design and analysis.
Biopharmaceutical companies and their development partners increasingly use data from real‐world settings to generate evidence that can support regulatory decision making and approvals of their manufactured medical products. The use of such real‐world evidence (RWE) can complement 1 or, in some cases, serve as an alternative to 2 evidence traditionally yielded by randomized controlled trials (RCTs). For example, the use of external control arms 3 can have considerable benefits, including accelerating the development process or reducing burden on trial participants. RWE can also provide investigators the opportunity to ask more questions and to understand broader, more diverse populations, as compared to RCTs.
The US Food and Drug Administration (FDA; also referred to here as the Agency) has taken significant steps to advance the use of RWE in regulatory decision making. This momentum has grown after the 21st Century Cures Act passed in December 2016; the act required FDA to develop a program for evaluating the use of RWE to support new indications for already‐approved drugs and fulfill postapproval study requirements. In 2018, FDA published a framework for its RWE program and is currently drafting guidance on its regulatory expectations regarding the use of RWE in medical product approvals. 4 As part of its broad impact, FDA’s framework has helped to promote common definitions for real‐world data (RWD; defined as “data relating to patient health status and/or the delivery of health care routinely collected from a variety of sources” 4 ) and RWE (defined as “the clinical evidence about the usage and potential benefits or risks of a medical product derived from analysis of RWD” 4 ). This is part of a worldwide interest in the use of RWE, including by regulatory agencies, such as the European Medicines Agency (EMA), 1 , 5 the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan, 6 and the National Medical Products Administration (NMPA) in China. 7
Much of FDA’s regulatory use of RWE to date has been in the context of postmarket surveillance through programs like FDA’s Sentinel initiative, a system initially designed to aid the Agency in evaluating medical product safety, 8 and which has more recently expanded in scope. Current goals include enhancing Sentinel’s ability to evaluate effectiveness 8 and extending the evaluation of safety. 9 In parallel, FDA also created a pilot project in 2008 to provide for the evaluation of vaccine effectiveness 10 and, in 2017, launched the Biologics Effectiveness and Safety (BEST) system to enhance FDA’s use and analysis of data to assure the safety and effectiveness of biological products. 11
Beyond postmarket surveillance, FDA considers RWE studies as part of the evidence package for submissions seeking authorization to market new medical products, including new drug applications (NDAs) and biologics license applications (BLAs). 4 , 12 These submissions are reviewed and decisions are rendered primarily by two centers within the Agency: the Center for Drug Evaluation and Research (CDER), which focuses on drug products and therapeutic biological products, and the Center for Biologics Evaluation and Research (CBER), which focuses on biological products including vaccines. Studies submitted as part of a sponsor’s overall evidence package will be considered in decision making. Approvals will then be based on, among other things, studies that provide “substantial evidence” (in CDER decisions) or “primary evidence” (in CBER decisions), which make the primary case for product safety and effectiveness, and “supportive evidence,” which can serve to bolster the case. Submitted studies providing therapeutic context can help reviewers understand the landscape of the disease (such as disease prevalence and incidence) and any current standard of care, but may not directly affect decision making. Many submitted studies will be presented in the FDA‐approved product label of an approved or licensed drug or biological product. As such, RWE studies have the potential to be directly influential in prescribers’ decision making if FDA approves a product label that includes submitted RWE studies. 13 , 14
FDA and others have directly and indirectly provided insights into FDA’s current approaches by speaking to and publishing select examples of successful and unsuccessful uses of RWE studies in medical product approvals, 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 and FDA has provided publicly available guidance for industry and staff. 23 , 24 , 25 , 26 However, RWE methodology is evolving, and on many important topics, RWE stakeholders lack a shared understanding of FDA’s expectations around the use of RWE, particularly in the context of NDAs and BLAs.
To date, there are no published systematic assessments of the use of RWE studies in FDA‐approved NDAs and BLAs, nor on FDA’s feedback on and acceptance of such studies. In this paper, we address this gap by presenting a systematic review of FDA’s written approval documents involving RWE studies in NDAs and BLAs to determine whether and how FDA incorporated that evidence in its final decision for approval. We summarize trends in recent use of RWE in FDA‐approved NDAs and BLAs, as well as whether and how RWE supported FDA’s approval decision. By distilling these reviews, we also seek to identify best practices for avoiding common design and analysis pitfalls identified by the Agency.
METHODS
Data
We examined a variety of FDA’s public resources to identify approvals from January 2019 through June 2021, and extracted the public documents available about such approvals. Our sources included FDA CDER’s Drugs@FDA, an Agency database of approved drugs; 27 CDER’s list of novel drug approvals; 28 CDER’s manual on NDA classification codes; 29 CBER’s list of biological approvals by year; 30 and publicly accessible review and approval documents. We also referenced FDA’s Spectrum of Diseases by Therapeutic Area Found in Written Requests 31 to align to their taxonomy of therapeutic areas.
For products approved by CDER, the publicly accessible approval documents 27 we reviewed were FDA approval letters, product labels, advisory committee meeting materials, and published review documents. The FDA review documents we examined were the Multi‐Discipline Reviews, Integrated Reviews, Summary Reviews, Clinical Reviews, Other Reviews, Administrative and Correspondence Documents, and Statistical Reviews.
For products licensed by CBER, the publicly accessible review and approval documents 32 we reviewed were FDA approval letters, product labels, Summary Basis for Regulatory Action (product review document), and related items. Examples of related items included BLA Clinical Review Memoranda, Statistical Review Memoranda, Team Meeting Summaries, Summary of Mid‐Cycle Communication Teleconferences with CBER, and Records of Telephone Conversation.
Approval inclusion and exclusion criteria
We first included all NDAs approved by CDER for new molecular entities (NMEs) from January 1, 2019, through June 30, 2021. Based on FDA’s classification system, 29 this included NDA type 1 (“new molecular entity”) and type 9 (“new indication or claim, drug not to be marketed under type 9 NDA after approval”), as these are the core mechanisms for FDA to consider marketing authorization for new molecules. We excluded the remainder of FDA’s NDA classifications. Specifically, we excluded types 2 through 5, as these classifications pertain to new formulations, new combinations, or other administrative or “technical” modifications to existing molecular entities. We excluded type 6 as it is no longer actively used, and type 7 because it pertains to drugs marketed prior to 1962. We excluded type 8, as those pertain to drugs moving from prescription to over‐the‐counter. Finally, we excluded type 10, as it is used for duplicates of drug products that are included in pending or approved NDAs. We also excluded approval documents for non‐NME 505(b)(2) applications, which are non‐NMEs with a new indication, dosage form, or regimen supported by evidence packages not generated by the applicant. Additionally, we excluded medical gas approvals and unidentified NDAs.
We included all approval documents for original biologic products licensed by CBER or CDER over the same time period. To focus the analysis principally on novel drugs examined by the CDER, and vaccines and biologics examined by the CBER and CDER, we excluded assays, solutions, and blood products, including blood grouping reagents, coagulation factors, antihemophilic factors, pooled immunoglobulin, plasminogen, and source plasma.
We did not consider FDA‐approved efficacy supplements (e.g., additions or modifications of indications or claims) because FDA does not routinely release approval documents for these supplements to the general public; indeed, of the 443 FDA‐approved efficacy supplements, FDA only published approval documents for 29 medical products. Finally, for practical reasons, we excluded FDA‐approved NDAs and BLAs where the associated approval documents were not yet available on our study cutoff date of June 30, 2021.
Review of approvals and data abstraction
In analyzing approvals, we sought (i) to quantify what proportion of approvals incorporated RWE studies in any manner, and (ii) of those, how the applicant intended to make use of the RWE studies (i.e., to support therapeutic context, safety, and/or effectiveness). Among the approvals that included an RWE study to support medical product safety and/or effectiveness, we sought to classify (3a) how, if at all, the RWE study supported FDA’s benefit‐risk considerations, (3b) whether the RWE study and/or its findings were referenced in the product label, and (3c) in which therapeutic area the medical product with an RWE study fell. We further (4) qualified FDA’s documented feedback on these RWE studies to understand FDA’s evidence expectations. We additionally reported all findings stratified by year to observe any evolving time trends in FDA’s use of RWE.
Proportion of approvals incorporating RWE
To measure the proportion of included approvals that incorporated RWE studies (item 1), we evaluated whether the approval drew upon RWD in any form. Following FDA’s definition of RWD, 4 we considered an approval to include RWD if the approval’s included studies contained data from medical claims, electronic health records, disease and product registries, and/or used secondary analysis of patient‐ and physician‐reported outcomes. We examined all associated documents associated with each approval, reviewing each section and each reference for RWD. In some cases, the Agency clearly denoted a study as using RWD. In other cases, we assessed the underlying publication or study to determine whether it should be characterized as RWD. Only completed studies were considered.
Intended use of RWE
To classify how the applicant intended to make use of the RWE studies (item 2) in the context of the application, we categorized each RWE study by the applicant’s presumed intent. We developed three non‐mutually exclusive categories: RWE used to support the application’s therapeutic context (e.g., prevalence and incidence of a disease), RWE studies to support the demonstration of product safety, and RWE studies to support the demonstration of product effectiveness. To determine how to categorize the study, we used the applicant’s expressed intent of the RWE study where it was explicitly stated. Where the applicant did not state the intent, we categorized the study based on which section of the approval documents the RWE study was discussed (e.g., therapeutic context section or benefit‐risk section; Table 1 ).
Table 1.
Categorization applied to applicants’ intended use of RWE in NDAs and BLAs
| Category | Sources of information on RWE study | Examples |
|---|---|---|
| RWE supports therapeutic context |
RWE study appeared in either of:
|
|
| RWE supports the demonstration of product safety |
RWE study appeared in any of:
|
|
| RWE supports the demonstration product effectiveness |
RWE study appeared in any of:
|
|
BLA, biologics license application; CDER, Center for Drug Evaluation and Research; NDA, new drug application; RWD, real‐world data; RWE, real‐world evidence.
Use of RWE to support of benefit‐risk considerations
We further examined the subset of studies that we classified as intending to support product safety and/or effectiveness. For our item (3a), we quantified how, if at all, the submitted RWE study supported FDA’s benefit‐risk considerations (Table 2 ). 24 Benefit‐risk assessment is an FDA framework for regulatory decisions determined based on whether the benefit of a product outweighs known and potential risks, and relies on underlying assessments of safety and effectiveness. 24
Table 2.
Categorization applied to how submitted RWE supported FDA’s benefit‐risk considerations
| Category | Definition | Source documentation considered |
|---|---|---|
| Substantial evidence (CDER) 23 or primary evidence (CBER) | FDA’s documents explicitly used the phrase “substantial evidence” (CDER) or “primary evidence” (CBER) |
For CDER‐approved products, published reviews section on Conclusions on the Substantial Evidence of Effectiveness (Section 1.2) For CBER‐licensed products, Summary Basis for Regulatory Action sections on Introduction (Section 1) and Recommendations and Risk/ Benefit Assessment (Section 11) Product label |
| Supportive evidence | FDA’s documents noted how the RWE study influenced its decision and/or the RWE study was referenced in the label |
For CDER‐approved products, published reviews sections Benefit‐Risk Assessment (Section 1.3), Sources of Clinical Data and Review Strategy (Section 7), (8.2) Review of Safety (Section 8.2), and Conclusions and Recommendations (Section 8.4) For CBER‐licensed products, Summary of Basis for Regulatory Action sections Clinical/Statistical/Pharmacovigilance (Section 6) and Safety (Section 7) Product label |
| Not adequate for decision making | FDA’s documents affirmatively stated that the RWE study was not evidentiary; in some cases FDA spoke to the study limitations | Approval documents in full |
| RWE studies that FDA did not address | We did not identify any comments on or references to the RWE study in FDA documentation | Approval documents in full |
CBER, Center for Biologics Evaluation and Research; CDER, Center for Drug Evaluation and Research; FDA, US Food and Drug Administration; RWE, real‐world evidence.
To measure whether the study was supportive of the benefit‐risk assessment, we applied methods developed through a preliminary review of FDA documentation (see below) and classified studies as informing FDA’s benefit‐risk assessment if they provided substantial evidence (by CDER), primary evidence (by CBER), or supportive evidence. Studies that we classified as not informing benefit‐risk assessment were those that, from the FDA reviews, appeared inadequate to support FDA’s decision making, or were simply not addressed in the reviews.
From the learnings of our preliminary review, we identified studies that served as substantial or primary evidence by looking specifically for those phrases (Table 2 ). 24 As an example, in CDER’s multi‐discipline review of the tuberculosis medication pretomanid, we noted the phrase “the Applicant has provided substantial evidence of effectiveness… in a single phase 3 clinical trial… compared to [RWD] historical controls.” 33 (In this application, the controls were drawn as individual patients observed in an electronic medical record system over a 6 year period, and were considered both as a group and as individually matched to patients in the treated arm.) Similarly, in CBER’s Summary Basis for Regulatory Action on onasemnogene abeparvovec‐xioi, a gene therapy for spinal muscular atrophy (SMA), we noted the phrase “comparison of the results of the ongoing clinical trial to available natural history data of infants with SMA provides primary evidence of the effectiveness of [the product]” 34 (bolding added by the authors).
Categorization as supportive evidence was more qualitative (Table 2 ), 24 with phrases such as “sufficient evidence,” “no formal statistical comparisons were made,” and “the study did, nevertheless, demonstrate…” taken as indicators. For example, in a multi‐discipline review of selumetinib, a therapy for children with neurofibromatosis type 1 (NF1) and symptomatic, inoperable plexiform neurofibromas (PNs)—a rare disease— FDA wrote “a natural history [RWE] study of NF1… was submitted with the application to provide external control data; however, no formal statistical comparisons were made by the FDA. The study did, nevertheless, demonstrate that a key characteristic of NF1 PN is the uncommon occurrence of spontaneous regression such that the observed tumor responses in [the] SPRINT [clinical study] are concluded to be the effect of the drug.” 35 In another example, in CDER’s summary review of the coronavirus disease 2019 (COVID‐19) treatment remdesivir, the compiled safety database was comprised of a phase III clinical trial as well as data from administration of the medication under Emergency Use Authorization and in a compassionate use program. We noted FDA’s direct use of the word “support”: “The Agency deemed the safety database adequate to support the NDA.” 36
Categorization as not adequate to support decision making was also qualitative based on direct statements and/or documented issues with submitted RWE. For example, one review contained both, stating “Due to major methodological issues (including immortal time bias, selection bias, misclassification, confounding, and missing data), the FDA does not consider these [RWE] results adequate to support regulatory decision making.” 37
Finally, for our category of studies that went unaddressed, we noted cases where FDA neither directly addressed the submitted RWE positively or negatively, nor made reference to the RWE in the approval documents. More broadly, not using or addressing the submitted RWE did not necessarily imply that FDA did not approve the application, but rather that the RWE portion did not appear to affect the decision making.
Use of RWE in product labels
Among the approvals considered, we assessed the associated product labels to identify which included RWE submitted as part of the application (item 3b). If a product label included either the RWE study name or its findings, we categorized the approval as having RWE referenced in the product label.
Use of RWE in therapeutic areas
Among the approvals considered, we cross‐referenced the medication with FDA and external resources to identify the product’s primary therapeutic area (item 3c). Our main source was FDA’s Spectrum of Diseases by Therapeutic Area Found in Written Requests document, 31 which maps many diseases to one of 20 therapeutic areas. Based on each drug’s primary indication, we categorized therapeutic area according to this FDA taxonomy; if the disease treated by the medication was not included in FDA’s list, we categorized the medical product’s therapeutic area based on our understanding of the disease and how applicants titled their clinical development programs and marketing franchises.
FDA’s documented feedback on use of RWE
Finally, to categorize FDA’s publicly available feedback according to key themes (item 4), we reviewed FDA documents on the approvals that included an RWE study to inform medical product safety or effectiveness, either successfully or not. We sought to better understand whether FDA deemed the evidence adequate in its decision making, and if not, why not.
We developed four non‐mutually exclusive categories: feedback noting methodological issues (e.g., immortal time bias, confounding, and lack of comparability between trial data and RWD); sample size concerns (e.g., underpowered or unstable estimates); omission of patient‐level data (i.e., lack of transparency, lack of ability for FDA to re‐analyze the data and/or perform subsequent analysis or re‐analysis); and other limitations that did not fit the categories above. We applied the categorization after considering FDA’s documented feedback on the reported reasons for an RWE study's not being adequate for decision making.
The data abstraction for all of our classifications was conducted by two investigators (C.P. and N.H.) who independently assessed the approval documents for each medical product. As necessary, reviewers resolved any discordance via a discussion of relevant passages with input solicited from other investigators not otherwise involved.
RESULTS
Selection of approvals
Among the 378 FDA‐approved NDAs or BLAs identified from January 1, 2019, to June 30, 2021, we determined that 136 (36%) of applications met our inclusion criteria. Among the 242 that did not, 237 (98%) were excluded because they were outside our criteria for new molecular entities or original biologic products (Figure 1 , Table S1 ), and an additional 5 (2%) were excluded because documentation was not yet available for public review as of the study’s June 30, 2021 cutoff (Table S3 ).
Figure 1.
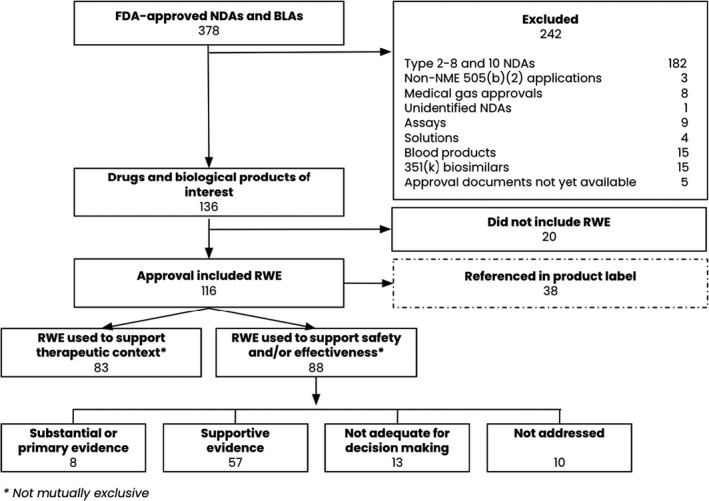
Inclusion of FDA‐approved NDAs and BLAs between January 2019 and June 2021. BLA, biologics license application; FDA, US Food and Drug Administration; NDA, new drug application; NME, new molecular entity; RWE, real‐world evidence.
We found that 116 approvals among the 136 (85%) included RWE in any form (item 1). The proportion of approvals including an RWE study increased from 2019 to 2021, with 38 of 51 (75%) approvals including an RWE study in 2019, 53 of 59 (90%) in 2020, and 25 of 26 (96%) in the first half of 2021 (Table 3 ).
Table 3.
Observed intended use of RWE in included NDAs/BLAs, January 2019 to June 2021
| Included NDAs and BLAs |
2019 n = 51 approvals |
2020 n = 59 approvals |
2021 through June 30 n = 26 approvals |
Total n = 136 approvals |
|---|---|---|---|---|
| Incorporated RWE for any purpose | 38 (75%) | 53 (90%) | 25 (96%) | 116 (85%) |
| Used RWE to provide therapeutic context | 25 (49%) | 36 (61%) | 22 (85%) | 83 (61%) |
| Used RWE to support safety and/or effectiveness | 27 (53%) | 46 (78%) | 15 (58%) | 88 (65%) |
| Safety only | 17 (33%) | 21 (36%) | 5 (19%) | 43 (32%) |
| Effectiveness only | 7 (14%) | 6 (10%) | 2 (8%) | 15 (11%) |
| Safety and effectiveness | 3 (6%) | 19 (32%) | 8 (31%) | 30 (22%) |
Categories are not mutually exclusive.
BLA, biologics license application; NDA, new drug application; RWE, real‐world evidence.
In categorizing the applicants’ intended use of RWE studies (item 2), we determined that a high proportion supplied RWE with the intent to provide evidence of product safety or effectiveness, with 88 of 136 (65%) of approvals using RWE studies to provide such evidence. 83 of 136 (61%) used RWE studies with the intent to provide therapeutic context (Table 3 ). The use of RWE studies to provide therapeutic context appeared to be trending upwards from 2019 to 2021 (from 49% in 2019 to 85% in the first half of 2021), whereas use of RWE studies to provide evidence of safety or effectiveness increased from 27 of 51 (53%) in 2019, to 46 of 59 (78%) in 2020, but decreased to 15 of 16 (58%) in the first half of 2021.
Table S4 notes the 20 FDA‐approved NDAs and BLAs without an RWE study, as well as the 28 FDA‐approved NDAs and BLAs where the applicant only intended to use RWE studies to support therapeutic context.
For item (3a), we further categorized the 88 approvals intended to support product safety or effectiveness by assessing whether and how the RWE studies supported FDA’s assessment of benefit‐risk. We found that among these approvals, FDA appeared to use 65 of the 88 RWE studies (74%) in its assessments, with 8 (9%) classified as substantial/primary evidence, and 57 (65%) as supportive evidence. The RWE studies appeared inadequate for FDA decision making in 13 (15%) cases, whereas in the remaining 10 (11%) cases, FDA did not directly address the provided RWE study in its published reviews (Table 4 ). Although the relatively small numbers make it difficult to identify trends, it appears that RWE is serving at least as supportive evidence in a growing proportion of approved applications over the time period studied. Separately, among the 88 approvals, 38 (43%) of the product labels referenced the RWE studies and/or findings (item 3b).
Table 4.
Observed FDA use of applicant‐submitted RWE in considered approvals, January 2019 to June 2021
| FDA’s use of RWE |
2019 n = 27 approvals |
2020 n = 46 approvals |
2021 through June 30 n = 15 approvals |
Total n = 88 approvals |
|---|---|---|---|---|
| Substantial or primary evidence | 3 (11%) | 4 (9%) | 1 (7%) | 8 (9%) |
| Supportive evidence | 16 (59%) | 30 (65%) | 11 (73%) | 57 (65%) |
| Not adequate for decision making | 6 (22%) | 4 (9%) | 3 (20%) | 13 (15%) |
| RWE studies that FDA did not address | 2 (7%) | 8 (17%) | 0 (0%) | 10 (11%) |
FDA, US Food and Drug Administration; RWE, real‐world evidence.
These 88 approvals spanned 16 therapeutic areas (Tables 5 , S5 ), with RWE submitted most commonly in oncology (30 of 43 approvals in that therapeutic area), infectious disease (16 of 17), neuroscience (13 of 25), and endocrinology and metabolism (8 of 14). FDA considered the evidence substantial/primary or supportive most commonly in the same therapeutic areas: oncology (16 of 43 approvals in that therapeutic area), infectious disease (12 of 17), neuroscience (11 of 25), and endocrinology and metabolism (6 of 14). However, when including RWE in the product label, the most common therapeutic areas differed somewhat: infectious disease (11 of 17 approvals in that therapeutic area), neuroscience (9 of 25), endocrinology and metabolism (4 of 14), oncology (3 of 43), and radiology (3 of 6).
Table 5.
Observed use of RWE in FDA approvals by therapeutic area, January 2019 to June 2021
| Therapeutic area | Total approvals per therapeutic area of n = 136 approvals | RWE intended to support safety and/or effectiveness in n = 88 approvals | RWE used as substantial/primary or supportive evidence in n = 65 approvals | RWE studies referenced in product label in n = 38 approvals |
|---|---|---|---|---|
| Oncology | 43 | 30 (70%) | 16 (37%) | 3 (7%) |
| Neuroscience | 25 | 13 (52%) | 11 (44%) | 9 (36%) |
| Infectious disease | 17 | 16 (94%) | 12 (71%) | 11 (65%) |
| Endocrinology and metabolism | 14 | 8 (57%) | 7 (50%) | 4 (29%) |
| Radiology | 6 | 6 (100%) | 6 (100%) | 3 (50%) |
| Hematology | 5 | 2 (40%) | 2 (40%) | 1 (20%) |
| Dermatology | 4 | 2 (2%) | 2 (3%) | 1 (3%) |
| Ophthalmology | 4 | 0 (0%) | 0 (0%) | 0 (0%) |
| Gastroenterology | 3 | 2 (66%) | 2 (66%) | 2 (66%) |
| Allergy | 2 | 1 (50%) | 0 (0%) | 0 (0%) |
| Anesthesiology | 2 | 1 (50%) | 1 (50%) | 1 (50%) |
| Cardiovascular | 2 | 2 (100%) | 2 (100%) | 1 (100%) |
| Gynecology | 2 | 1 (50%) | 1 (50%) | 1 (50%) |
| Inflammation and immunology | 2 | 0 (0%) | 0 (0%) | 0 (0%) |
| Urology | 2 | 1 (50%) | 1 (50%) | 1 (50%) |
| Autoimmune | 1 | 1 (1%) | 1 (2%) | 0 (0%) |
| Cosmetic | 1 | 1 (100%) | 0 (0%) | 0 (0%) |
| Respiratory | 1 | 1 (100%) | 1 (100%) | 0 (0%) |
FDA, US Food and Drug Administration; RWE, real‐world evidence.
FDA provided publicly documented feedback on the RWE studies in 37 of 88 approvals that included an RWE study to support medical product safety and/or effectiveness (item 4). Across all documented studies, the issues noted in FDA’s feedback were methodological issues (n = 23), sample size concern (n = 8), omission of patient‐level data (n = 3), and other limitations (n = 13). Some studies had multiple issues identified. Examples of each category are in Table 6 , with further detail in Tables S6 and S7 .
Table 6.
Examples of FDA’s feedback on use of RWE and/or the submitted RWE study
| Category of FDA’s feedback | Product’s primary basis of approval | FDA feedback on example studies |
|---|---|---|
| Methodological issues | Phase II, single‐arm study | Inadequate due to missing data, differences in follow‐up and response assessment, population heterogeneity, and bias in end point assessment 51 |
| Single‐arm study | Supportive with critiques: small sample size; lack of comparator arm; limitations of retrospective observational data; time‐to‐event end points not interpretable 52 | |
|
Phase II, single‐arm cohort Phase I/II study |
Inadequate due to determination and validation of endpoints, selection bias, confounding factors, and generalizability 53 , 54 | |
| Phase II study | Inadequate due to variability in natural history of disease, imprecision of population matching, and selection bias 55 , 56 | |
| General limitations | Phase I study | Inadequate without detail 57 |
| Phase III study | RWE did not overrule safety signal from clinical trials 58 | |
| Phase II study | Premise of RWE study deemed controversial 59 | |
| Sample size concern | Phase II trial and phase I/II study | Inadequate data collection 60 , 61 |
| Phase II, single‐arm study | Inadequate due to limited data 62 | |
| Three phase III studies | Limited sample size precluded it from FDA’s consideration for safety and effectiveness 63 , 64 | |
| Omission of patient‐level data | Phase II study | Supportive because it strengthened clinical relevance, although “… FDA cannot independently verify these results” 65 |
FDA, US Food and Drug Administration; RWE, real‐world evidence.
DISCUSSION
Regulatory agencies worldwide are considering how they may incorporate RWE into their decision‐making processes. In this study, we detailed how over the past two and a half years, both the quantity of RWE submitted and its impact on FDA’s decision making—particularly as supportive evidence—have significantly increased and, downstream, that RWE studies are appearing in US product labels. However, we also saw that not all RWE was accepted as the applicant intended; in a large number of cases, FDA either did not consider the RWE at all or found it inadequate to support an approval. In these cases, a consistent theme emerged: methodologically‐sound RWE rooted in principled study design and analysis supported FDA’s decisions, whereas studies that FDA found not to meet this bar bore far less impact.
Most approvals where RWE studies impacted decision making were in oncology, infectious disease, and neuroscience. Although we are not able to draw a direct line between these therapeutic areas and why they make up the largest portion of RWE use, we note that they have some common features: there is substantial drug development effort in each of these areas, the seriousness of many diseases and conditions in these areas commonly leads to eligibility for expedited programs 38 (which may in turn look to rapid evidence generation), and they may use expanded access programs 39 (which may generate data on real‐world experience). Further, in certain diseases within these areas, small patient populations, impracticality of running randomized trials, or ethical issues with using a placebo control arm may be favorable for the use of RWE.
Studies that support regulatory approvals are rooted in good study design, appropriate data collection, and thoughtful data analysis; this is true whether the study is a randomized or a nonrandomized, real‐world design. We saw instances where FDA directly addressed this in their comments. For example, in one case, an FDA document stated, “The Agency agreed to the use of the external contemporaneous control in the matched analysis because the trial investigators and Applicant took adequate measures to minimize potential sources of bias. The trial investigators undertook rigorous efforts to identify patients with [Hutchinson‐Gilford progeria syndrome] globally and offered all patients the same opportunity to participate in the natural history study, reducing potential selection bias. Known potential confounders (age at treatment initiation, variant status, sex, and continent of residence) were adequately addressed in the analyses.” 40
However, as noted above, FDA also cited instances where the submitted evidence did not meet the threshold to be included in their decision making, either directly stating that and/or identifying methodological issues in RWE generation. One review stated, “There are several methodological limitations of this comparative [RWE] analysis outlined below that impact the interpretability of the study results,” then went on to list four specific issues in detail: missing data, differences in the endpoint definitions or follow‐up period used to ascertain them, treatment heterogeneity among multiple standard of care therapies, and selection bias due to lack of comparability. 41
Reliable RWE is built on using fit‐for‐purpose RWD, and data‐related challenges were cited in FDA’s documented feedback. In one example, FDA found that patients represented in the selected RWD source were not comparable to those in an associated clinical trial, saying “differential selection of comparison groups, specifically, that trial patients were enrolled from academic medical centers primarily from Europe, while [RWD] controls were enrolled from community oncology centers in the US.” 42 To avoid issues like this, a focus on identifying RWD that meet expectations around relevance and reliability, 43 ensuring the data speak to the question at hand and are high‐quality, is critical. In assessing relevancy, applicants can ensure that the data represent the intended population, that key variables such as exposure, outcome, and confounders are captured and measurable, and that data are sufficiently longitudinal for the study. In assessing quality, applicants can assess accuracy, completeness, and consistency. Our research suggests that spending substantial effort on this phase can avoid some of the downstream issues that lead to RWE studies not being adequate for decision making.
With fit‐for‐purpose data in hand, identifying an appropriate study design is a key next step. Framing questions in a meaningful way for the ultimate decision‐making audience and enforcing designs and analyses that are transparent, auditable, and reproducible is crucial. 44 , 45 One reliable design strategy applicants can use is designing the RWE study to emulate either an actual or hypothetical target trial. 46 , 47 , 48 In emulating a target trial, researchers are advised to mimic all design aspects of a randomized trial, and use confounding identification and adjustment to mitigate bias in the absence of randomization. This ensures that key areas of design, including eligibility criteria, treatment strategies, start and end of follow‐up, outcomes, causal contrast, and the analysis plan are methodologically sound.
In some instances in the documented feedback, the importance of communication with FDA in the planning, design, and review phases of an RWE study was referenced. In the planning phase, early and frequent communication with FDA helps to ensure “good RWE” 49 by providing a channel for study design feedback and an opportunity to adjust any issues. Feedback provided in formal meetings provides an early signal on whether a particular RWE approach is likely to support FDA’s benefit‐risk assessment. In one case where the submitted RWD was deemed not adequate for decision making, FDA highlighted that lack of prior communication about the RWD: “As previously mentioned, without having reviewed and consented to a protocol and statistical analysis plan (SAP), FDA cannot be certain that the protocol and SAP were pre‐specified and unchanged during the data selection and analyses. This uncertainty and the knowledge that subsequent unmasked analyses have been performed could lead to overly optimistic conclusions.” 37
However, in another case where the RWE was ultimately deemed substantial, FDA took specific note of how the applicant heeded the Agency's early feedback. In this case, the applicant had originally published the mortality data of their medical product prior to creation of a SAP. As a result, the Agency stated in their meeting to discuss the initial clinical study, “Ideally, analysis plans for marketing applications should be submitted to the FDA for agreement before analyzing the data; this was a missed opportunity for the program and may be a review issue.” 40 Noting this, the applicant formally proposed to conduct the same analysis again as a part of the SAP. In the final approval, FDA stated: “The Applicant agreed with the Agency’s recommendation and revised the SAP with a cut‐off date of June 1, 2019 for follow‐up. The SAP was also revised based on Agency feedback pertaining to the matching approach and censoring. The revised SAP was deemed acceptable by the Agency prior to NDA submission, with the caveat that analyses were retrospectively planned and considered post‐hoc. During the course of the NDA review, issues with the analyses were identified and adequately resolved.” The RWE, in this case, was deemed substantial evidence. 40
In the review phase, applicants should allow FDA reviewers transparent access to the analyses (e.g., ability to trace data provenance, understand all data transformations, and see the order in which methods are applied), and provide them the option to reanalyze and/or conduct post hoc sensitivity analyses on the data. In our review, we found that FDA may place importance on being able to independently analyze data, in one case stating “While FDA considers data from Study X2102 and [RWD] Study X2401 to be supportive, [the sponsor] did not submit this data, and thus FDA cannot independently verify these results.” 50
We note three limitations. First, our systematic review covered all of 2019 and 2020, but only the first half of 2021. In past years, the majority of decisions have occurred in the second half of the year, so trends including 2021 data should be interpreted in this light. Second, our evidence base is limited to FDA‐approved NDAs and BLAs because complete response letters (which FDA sends when it cannot approve an application in its current form) are confidential between the applicant and the Agency. In addition, documents regarding efficacy supplements are not necessarily confidential but are also not routinely made public, although they may be available through a Freedom of Information Act request. Third, due to the low sample size, we were not able to draw strong conclusions about the role of RWE (e.g., “RWE was most likely to be deemed substantial evidence if…”), and we did not feel it appropriate to identify trends by design or analytic choice (e.g., “X type of RWE most often provided substantial evidence”), largely because of the heterogeneity in uses of RWE.
CONCLUSIONS
As the use of RWE in regulatory applications increases, there is opportunity to improve both the understanding of the expectations of worldwide regulatory agencies around the utility, substance and quality of RWE studies, as well as applicants’ adherence to such expectations. Over the next years, we expect that further clarification of these expectations will emerge, but feel confident that they will be built upon the foundations of fit‐for‐purpose RWD selection and careful attention to study design and analysis. As questions remain as to what specific guidances will be provided or how well they are being followed, further studies such as these, augmented with specific demonstration projects, can continue to build empirical evidence on how RWE can meet regulatory agencies’ expectations and improve public health.
FUNDING
This study was funded internally by Aetion.
CONFLICT OF INTEREST
All authors are employees of and hold stock options or equity in Aetion, Inc. No other interests are declared.
Supporting information
Tables S1–S9
ACKNOWLEDGMENTS
The authors would like to thank Amanda Patrick, Anthony Louder, Elizabeth Dabrowski, Ilker Oztop, Jennifer Polinski, Michelle Skornicki, Ray Harvey, Shannon Reynolds, and William Murk (Aetion, Inc.) for their scientific review and discussion of some of the extracted data used in this study; Liz DeMatteis and Nicolle Gatto (Aetion, Inc.), and Sebastian Schneeweiss (Harvard Medical School and Brigham and Women’s Hospital) for their input and feedback; and Jennifer Thorburn, Lexie Rubens, and Olivia Atlas (Aetion, Inc.) for their contributions to the writing and citations.
- 1. Eichler, H.‐G. et al. Randomized controlled trials versus real world evidence: neither magic nor myth. Clin. Pharmacol. Ther. 109, 1212–1218 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Franklin, J.M. et al. Emulating randomized clinical trials with nonrandomized real‐world evidence studies: first results from the RCT DUPLICATE initiative. Circulation. 143, 1002–1013 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burcu, M. et al. Real‐world evidence to support regulatory decision‐making for medicines: Considerations for external control arms. Pharmacoepidemiol. Drug Saf. 29, 1228–1235 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. FDA . Framework for FDA’s Real‐World Evidence Program. Published December 2018 <https://www.fda.gov/media/120060/download>. Accessed July 26, 2021.
- 5. EMA regulatory science to 2025. Published online 2020 <https://www.ema.europa.eu/en/documents/regulatory‐procedural‐guideline/ema‐regulatory‐science‐2025‐strategic‐reflection_en.pdf>. Accessed August 9, 2021.
- 6. Ando, T. Recent trend on utilization of real world data ‐ challenges in Japan <https://www.pmda.go.jp/files/000226214.pdf>. Accessed August 9, 2021.
- 7. NMPA: attachment: “guiding principles of real world data used to generate real world evidence (trial).” Published online April 2021 <https://redica.com/wp‐content/uploads/NMPA_‐Attachment_‐_Guiding‐Principles‐of‐Real‐World‐Data‐Used‐to‐Generate‐Real‐World‐Evidence‐Trial_.pdf>. Accessed August 9, 2021.
- 8. FDA sentinel system five‐year strategy 2019‐2023; 2019. <https://www.fda.gov/media/120333/download>. Accessed August 1, 2021.
- 9. FDA . FDA In Brief: FDA announces a new Sentinel System contract, affirming its commitment to harnessing Real‐World Data to improve the safety and effectiveness of drugs. Published September 27, 2019 <https://www.fda.gov/news‐events/fda‐brief/fda‐brief‐fda‐announces‐new‐sentinel‐system‐contract‐affirming‐its‐commitment‐harnessing‐real‐world>. Accessed August 6, 2021.
- 10. Ball, R. , Robb, M. , Anderson, S. & Dal Pan, G. The FDA’s sentinel initiative ‐ a comprehensive approach to medical product surveillance. Clin. Pharmacol. Ther. 99, 265–268 (2016). [DOI] [PubMed] [Google Scholar]
- 11. CBER Biologics Effectiveness and Safety (BEST) System . Published online December 4, 2020 <https://www.fda.gov/vaccines‐blood‐biologics/safety‐availability‐biologics/cber‐biologics‐effectiveness‐and‐safety‐best‐system>. Accessed August 10, 2021.
- 12. FDA . Examples of Real‐World Evidence (RWE) used in medical device regulatory decisions <https://www.FDA.gov/media/146258/download>. Accessed August 4, 2021.
- 13. Takada, M. , Demizu, M. & Shibakawa, M. Physicians’ prescribing attitudes to combined therapy with statins and fibrates: Physicians’ prescribing attitudes to combined therapy with statins and fibrates. J. Clin. Pharm. Ther. 28, 445–450 (2003). [DOI] [PubMed] [Google Scholar]
- 14. Rosenberg, M. et al. fda postmarketing safety labeling changes: What have we learned since 2010 about impacts on prescribing rates, drug utilization, and treatment outcomes. Pharmacoepidemiol. Drug Saf. 29, 1022–1029 (2020). [DOI] [PubMed] [Google Scholar]
- 15. Brennan, Z. FDA discusses RWD, RWE with industry, academia. Endpoint News. Published July 22, 2019 <https://endpts.com/fda‐discusses‐rwd‐rwe‐with‐industry‐academia/>. Accessed July 23, 2021.
- 16. Opportunities and challenges of using real‐world evidence to support regulatory decisions for drugs & drug & medical products educational conference; April 13, 2021 <https://www.afdo.org/wp‐content/uploads/2021/04/final‐Concato_Morales‐AFDO_13_April_2021_cleared.pdf>. Accessed July 26, 2021.
- 17. Baumfeld Andre, E. , Reynolds, R. , Caubel, P. , Azoulay, L. & Dreyer, N.A. Trial designs using real‐world data: the changing landscape of the regulatory approval process. Pharmacoepidemiol. Drug Saf. 29, 1201–1212 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goring, S. et al. Characteristics of non‐randomised studies using comparisons with external controls submitted for regulatory approval in the USA and Europe: a systematic review. BMJ Open 9, e024895 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Silverman, B. A Baker’s dozen of US FDA efficacy approvals using real world evidence. Pink Sheet. Published August 7, 2018 <https://pink.pharmaintelligence.informa.com/PS123648/A‐Bakers‐Dozen‐Of‐US‐FDA‐Efficacy‐Approvals‐Using‐Real‐World‐Evidence>. Accessed July 26, 2021.
- 20. Bolislis, W.R. , Fay, M. & Kühler, T.C. Use of real‐world data for new drug applications and line extensions. Clin. Ther. 42, 926–938 (2020). [DOI] [PubMed] [Google Scholar]
- 21. Mahendraratnam, N. , Mercon, K. , Gill, M. , Benzing, L. & McClellan, M.B. Understanding use of real‐world data and real‐world evidence to support regulatory decisions on medical product effectiveness. Clin. Pharmacol. Ther. 111, 150–154 (2022). [DOI] [PubMed] [Google Scholar]
- 22. Jahanshahi, M. et al. The use of external controls in FDA regulatory decision making. Ther. Innov. Regul. Sci. 55, 1019–1035 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. FDA . Submitting documents using real‐world data and real‐world evidence to the Food and Drug Administration for drugs and biologics; Draft Guidance for Industry; Availability. National Archives Federal Register. Published May 9, 2019 <https://www.federalregister.gov/documents/2019/05/09/2019‐09529/submitting‐documents‐using‐real‐world‐data‐and‐real‐world‐evidence‐to‐the‐food‐and‐drug>. Accessed July 27, 2021.
- 24. FDA . Demonstrating substantial evidence of effectiveness for human drug and biological products; Draft Guidance for Industry; Availability. National Archives Federal Register. Published December 20, 2019 <https://www.federalregister.gov/documents/2019/12/20/2019‐27524/demonstrating‐substantial‐evidence‐of‐effectiveness‐for‐human‐drug‐and‐biological‐products‐draft>. Accessed July 27, 2021.
- 25. Guidance for industry: E10 choice of control group and related issues in clinical trials. Published online May 2021 <https://www.fda.gov/media/71349/download>. Accessed August 10, 2021.
- 26. Best practices for conducting and reporting pharmacoepidemiologic safety studies using electronic healthcare data. Published online May 2013 <https://www.fda.gov/media/79922/download>. Accessed August 9, 2021.
- 27. FDA . Drugs@FDA: FDA‐approved drugs <https://www.accessdata.fda.gov/scripts/cder/daf/>. Accessed July 26, 2021.
- 28. FDA . Novel drug approvals for 2021. Published July 20, 2021 <https://www.fda.gov/drugs/new‐drugs‐fda‐cders‐new‐molecular‐entities‐and‐new‐therapeutic‐biological‐products/novel‐drug‐approvals‐2021>. Accessed July 27, 2021.
- 29. Center For Drug Evaluation and Research . NDA classification codes. FDA. Published November 4, 2015 <https://www.fda.gov/media/94381/download>. Accessed July 26, 2021.
- 30. FDA . Biologics approval by year. Published February 22, 2021 <https://www.fda.gov/vaccines‐blood‐biologics/development‐approval‐process‐cber/biological‐approvals‐year>. Accessed July 26, 2021.
- 31. FDA . Spectrum of diseases, conditions. Published December 31, 2019 <https://www.fda.gov/drugs/development‐resources/spectrum‐diseasesconditions>. Accessed August 4, 2021.
- 32. FDA . Licensed biological products with supporting documents. Published July 27, 2021 <https://www.fda.gov/vaccines‐blood‐biologics/licensed‐biological‐products‐supporting‐documents>. Accessed August 1, 2021.
- 33. Pretomanid NDA/BLA multi‐disciplinary review and evaluation. Published online December 14, 2018 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212862Orig1s000MultidisciplineR.pdf>. Accessed August 2, 2021.
- 34. Zolgensma (onasemnogene abeparvovec‐xioi) summary basis for regulatory action. Published online May 24, 2019 <https://www.fda.gov/media/127961/download>. Accessed August 4, 2021.
- 35. Koselugo (selumetinib) NDA/BLA multi‐disciplinary review and evaluation. Published online December 14, 2018 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213756Orig1s000MultidisciplineR.pdf>. Accessed August 2, 2021.
- 36. Veklury (remdesivir) summary review. Published online June 9, 2015 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/214787Orig1s000Sumr.pdf>. Accessed August 10, 2021.
- 37. Xpovio (Selinexor) NDA/BLA multi‐disciplinary review and evaluation. Published online July 1, 2019 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212306Orig1s000MultidisciplineR.pdf>. Accessed July 28, 2021.
- 38. Guidance for industry: expedited programs for serious conditions ‐ drugs and biologics. Published online May 2014 <https://www.fda.gov/media/86377/download>. Accessed August 10, 2021.
- 39. FDA . Expanded access. Published March 23, 2021 <https://www.fda.gov/news‐events/public‐health‐focus/expanded‐access>. Accessed August 10, 2021.
- 40. Zokinvy (lonafarnib) integrated review. Published online November 20, 2020 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213969Orig1s000IntegratedR.pdf>. Accessed August 10, 2021.
- 41. Abecma (idecabtagene vicleucel) clinical review memo. Published online March 26, 2021 <https://www.fda.gov/media/147740/download>. Accessed August 5, 2021.
- 42. Balversa (erdafitinib) other review(s). Published online April 11, 2019 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212018Orig1s000OtherR.pdf>. Accessed August 5, 2021.
- 43. Daniel, G. , Silcox, C. , Bryan, J. , McClellan, M. , Romine, M. & Frank, K. Characterizing RWD quality and relevancy for regulatory purposes.pdf. Published online October 1, 2018 <https://healthpolicy.duke.edu/sites/default/files/2020‐03/characterizing_rwd.pdf>. Accessed August 10, 2021.
- 44. Gatto, N.M. , Reynolds, R.F. & Campbell, U.B. A structured preapproval and postapproval comparative study design framework to generate valid and transparent real‐world evidence for regulatory decisions. Clin. Pharmacol. Ther. 106, 103–115 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang, S.V. et al. Reporting to improve reproducibility and facilitate validity assessment for healthcare database studies V1.0. Pharmacoepidemiol. Drug Saf. 26, 1018–1032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hernán, M.A. & Robins, J.M. Using big data to emulate a target trial when a randomized trial is not available: table 1. Am. J. Epidemiol. 183, 758–764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rubin, D.B. Estimating causal effects of treatments in randomized and nonrandomized studies. J. Educ. Psychol. 66(5), 688–701 (1974). [Google Scholar]
- 48. Cochran, W.G. & Chambers, S.P. The planning of observational studies of human populations. J. R. Stat. Soc. Ser. Gen. 128, 234–266 (1965). [Google Scholar]
- 49. Schneeweiss, S. & Avorn, J. A review of uses of health care utilization databases for epidemiologic research on therapeutics. J. Clin. Epidemiol. 58, 323–337 (2005). [DOI] [PubMed] [Google Scholar]
- 50. Tabrecta (Capmatinib) NDA/BLA multi‐disciplinary review and evaluation. Published online May 6, 2020 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213591Orig1s000MultidisciplineR.pdf>. Accessed July 28, 2021.
- 51. Abecma (idecabtagene vicleucel) clinical review memo. Published online March 26, 2021 <https://www.fda.gov/media/147740/download>. Accessed August 5, 2021.
- 52. Monjuvi (tafasitamab‐cxix) multi‐disciplinary review and evaluation. Published online July 31, 2020 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761163Orig1s000MultidisciplineR.pdf>. Accessed August 5, 2021.
- 53. Tazverik (tazemetostat) multi‐disciplinary review and evaluation. Published online January 23, 2020 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/211723Orig1s000MultidisciplineR.pdf>. Accessed August 5, 2021.
- 54. Tazverik (tazemetostat) multi‐disciplinary review and evaluation. Published online June 18, 2021 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213400Orig1s000MultidisciplineR.pdf>. Accessed August 5, 2021.
- 55. Viltepso (viltolarsen) summary review. Published online August 12, 2020 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/212154Orig1s000SumR.pdf>. Accessed August 5, 2021.
- 56. Viltepso (viltolarsen) clinical review. Published online August 12, 2020 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/212154Orig1s000MedR.pdf>. Accessed August 5, 2021.
- 57. Rybrevant (amivantamab‐vmjw) multi‐disciplinary review and evaluation. Published online May 21, 2021 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761210Orig1s000MultidisciplineR.pdf>. Accessed August 5, 2021.
- 58. Sarclisa (isatuximab) multi‐disciplinary review and evaluation. Published online March 2, 2020 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761113Orig1s000MultidisciplineR.pdf>. Accessed August 5, 2021.
- 59. Truseltiq (infigratinib) multi‐disciplinary review and evaluation. Published online May 28, 2021 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/214622Orig1s000MultidisciplineR.pdf>. Accessed August 5, 2021.
- 60. Danyelza (naxitamab‐gqgk) multi‐disciplinary review and evaluation. Published online November 22, 2020 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761171Orig1s000MultidisciplineR.pdf>. Accessed August 5, 2021.
- 61. Danyelza (naxitamab‐gqgk) administrative and correspondence documents. Published online November 22, 2020 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761171Orig1s000AdminCorres.pdf>. Accessed August 5, 2021.
- 62. Tepmetko (tepotinib) multi‐disciplinary review and evaluation. Published online February 3, 2021 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/214096Orig1s000MultidisciplineR.pdf>. Accessed August 5, 2021.
- 63. Veklury (remdesivir) clinical review. Published online October 22, 2020 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/214787Orig1s000MedR.pdf>. Accessed August 5, 2021.
- 64. Veklury (remdesivir) other review(s). Published online October 22, 2020 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/214787Orig1s000OtherR.pdf>. Accessed August 5, 2021.
- 65. Tabrecta (capmatinib) multi‐disciplinary review and evaluation. Published online May 6, 2020 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213591Orig1s000MultidisciplineR.pdf>. Accessed August 5, 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S9