

Abstract
Cobaloximes are promising, earth‐abundant catalysts for the light‐driven hydrogen evolution reaction (HER). Typically, these cobalt(III) complexes are prepared in situ or employed in their neutral form, for example, [Co(dmgH)2(py)Cl], even though related complex salts have been reported previously and could, in principle, offer improved catalytic activity as well as more efficient immobilization on solid support. Herein, we report an interdisciplinary investigation into complex salts [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]−, and [Co(dmgH)2(py)2]+BArF−. We describe their strategic syntheses from the commercially available complex [Co(dmgH)2(py)Cl] and demonstrate that these double and single complex salts are potent catalysts for the light‐driven HER. We also show that scanning electrochemical cell microscopy can be used to deposit arrays of catalysts [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]−, and [Co(dmgH)2(py)Cl] on supported and free‐standing amino‐terminated ∼1‐nm‐thick carbon nanomembranes (CNMs). Photocatalytic H2 evolution at such arrays was quantified with Pd microsensors by scanning electrochemical microscopy, thus providing a new approach for catalytic evaluation and opening up novel routes for the creation and analysis of “designer catalyst arrays”, nanoprinted in a desired pattern on a solid support.
Keywords: catalyst nanoarrays, cobaloximes, in situ amperometric H2 measurements, photocatalysis, SECCM
We report the synthesis of a new cobaloxime‐based double complex salt and its use as a catalyst for the homogeneous and heterogeneous, light‐driven hydrogen evolution reaction (HER). We successfully deposited these catalysts by scanning electrochemical cell microscopy (SECCM) on supported and free‐standing 2D amino‐terminated carbon nanomembranes (CNMs) and quantified hydrogen by using Pd microsensors.
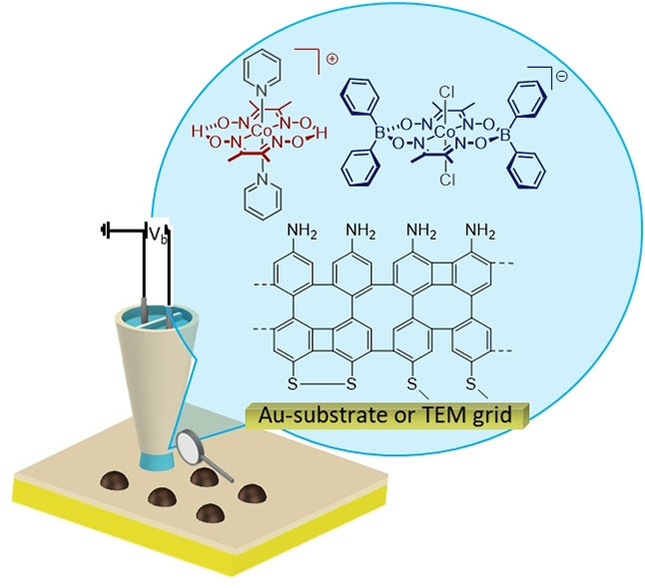
Introduction
Cobaloximes [1] have been recognized as catalysts for the hydrogen evolution reaction (HER) almost half a century ago. [2] Over the past two decades, an increased interest in photocatalytic water splitting [3] has led to a revival of syntheses of various earth‐abundant glyoxime‐based cobalt complexes. Notable structural variations include BF n ‐bridged derivatives, [4] modification of the axial pyridine ligand,[ 3a , 5 ] and changes to the equatorial ligand framework, [6] typically for the purpose of catalyst immobilization [7] or the generation of dinuclear[ 3k , 8 ] complexes. To the best of our knowledge, there is only one report of a BPh2‐bridged, organometallic cobaloxime complex (axial ligand: CH3) [9] and the immobilization of cobalt complexes on two‐dimensional (2D) material is an underexplored challenge. [10]
Herein we report the unexpected discovery of asymmetric double complex salt [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]− (Figure 1a) that we consistently obtained in good yields during attempts to synthesize BPh2‐bridged cobaloxime complexes. While double complex salts [11] based on the dmgH framework have been described as (unwanted) reaction by‐products, [12] we wondered whether the new salt, which was found to be stable in solution, could offer advantages for photocatalysis in solution or after immobilization. [13] Such a modification of 2D materials with HER catalysts and its characterization in respect to activity, possible degradation during illumination and associated activity losses are of importance for future technologically relevant heterogeneous water splitting devices. CNMs are highly attractive, mechanically stable molecular 2D materials with a thickness of ∼1 nm, obtained by low‐energy electron irradiation induced crosslinking of aromatic self‐assembled monolayers (SAMs) [14] such as 4’‐nitro‐1,1′‐biphenyl‐4‐thiol (NBPT). [15]
Figure 1.

a) Synthesis of [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]−, [Co(dmgH)2(py)2]+ BArF− and TBA+ [Co(dmgBPh2)2Cl2]−. b) 1H NMR (CD3CN, 400 MHz, RT). Top: [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]−; middle: [Co(dmgH)2(py)2]+ BArF−; bottom: TBA+ [Co(dmgBPh2)2Cl2]−. c) Solid state structure of [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]−. Space group: P‐1, crystal system: triclinic. The average equatorial Co−Noxime distance is 1.90 Å in the cationic complex [Co(dmgH)2(py)2]+, 1.86 Å in the anionic complex [Co(dmgBPh2)2Cl2]− and 1.89 Å in neutral cobaloximes. [29] Solvent and H atoms are omitted for clarity.
Depositing arrays by, for example, microspotting of catalysts for photoelectrochemical water splitting allows fast preparation and in‐situ screening experiments, as a pioneering study by Bard and co‐workers has shown for water oxidation catalysts. [16] Improved lateral resolution for local surface patterning can be achieved using scanning electrochemical cell microscopy (SECCM) with dual barrel nanopipettes, [17] as the positioning and timing of droplet formation can be precisely controlled. In SECCM, single barrel or double‐barrel nanopipettes with a quasi‐reference/counterelectrode (QRCE) in each barrel with orifices of the barrels as small as 30 nm are used to form a local electrochemical cell, when the nanopipette is approached to a surface to make meniscus contact. [18] As the distance can be precisely controlled and the status of surface contact of the meniscus monitored, sample damage is omitted, [19] which is important for surface modification of fragile samples such as free‐standing CNM on TEM grids. Compared to other surface modification techniques, like microspotting [20] and microcontact printing, [21] SECCM can generate nanoscale deposits which has been demonstrated for potential‐controlled electrodeposition of, for example, metals[ 17 , 22 ] or polymers[ 19 , 23 ] on conductive substrates. Surface modification using micro‐ and nanopipettes can also be obtained without electrochemically driving the deposition. When the droplet is in contact with the surface, molecules are delivered from the pipette to the surface. After retracting the pipette, the solvent evaporates quickly and the molecules are deposited and kept by van der Waals forces at the surface. [24] For example, deposition of Ni x B catalytic nanoparticles on a liquid‐cell transmission electron microscopy chip was recently shown by this approach. [24b]
Determining the photocatalytic activity at deposited catalyst micro‐ and nanoarrays by standard H2 head‐space gas chromatography is not suitable due to the low amount of H2 evolution at such arrays compared to the measurements in homogenous bulk photocatalysis. Scanning electrochemical microscopy (SECM), [25] as a well‐established method for studying solid/electrolyte interfaces in situ has been employed to electrochemically map reaction intermediates and final products like H2 and O2 at electrocatalysts for photoelectrochemical water oxidation and photocatalytically active materials,[ 16 , 26 ] mainly using generator/collector mode.[ 16 , 26a , 26e , 26f ] In this work, we therefore investigated the prospect of electrochemically immobilizing HER‐active cobaloxime complexes on two‐dimensional solid support using SECCM with the ultimate aim of conducting parameter screenings with SECM. As a proof‐of‐principle, we investigated the H2 evolution at microarrays of three catalysts in situ using Pd‐microsensors.
Results and Discussion
Complex salts: Synthesis and characterization
At the outset of this study, we wondered whether equatorial boron‐based bridges other than “BF2“ could improve catalytic performance of cobaloximes and allow the fine‐tuning of key properties as well as late‐stage introduction of functional groups. For these reasons, we treated commercially available [Co(dmgH)2(py)Cl] [27] with 2.0 equivalents of triphenylborane (BPh3) and were pleased when we were able to isolate a beige precipitate in a yield of 77 % (Figure 1a, left). The presence of two distinct methyl signals in the 1H NMR spectrum (Figure 1b, top) and the observation of both [Co(dmgH)2(py)2]+ (positive mode) and [Co(dmgBPh2)2Cl2]− (negative mode) fragments by high‐resolution mass spectrometry (HRMS) suggested that a double complex salt[ 11c , 28 ] had formed. Fortunately, we were able to obtain single crystals of the unknown compound by the slow vapor diffusion method. The X‐ray crystallographic data (Figure 1c) confirmed that the product is a double complex salt of the formula [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]−, which explains the presence of two signals (with 1 : 1 integral ratio) for the methyl groups “A −“ and “A +“ in the 1H NMR spectrum (Figure 1b, purple trace), as well as the observation of only one set of signals for phenyl substituents and pyridine ligands, respectively. A detailed overview of structural features in comparison with neutral complex [Co(dmgH)2(py)Cl] [29] is provided in Table S3 in the Supporting Information.
Before discussing the interesting question of why we were able to obtain this specific onefold BPh2‐bridged double complex salt in such high yield, it is important to note that equilibrium (1) between neutral [Co(dmgH)2(py)Cl] and the corresponding complex salt has been described before. [12]
| (1) |
In the same report, it has been proposed that this dynamic equilibrium requires the presence of CoII species and that the equilibrium position is solvent and concentration dependent. In light of a recent report by Verani and co‐workers, [30] we propose that BPh3 could act not only as an electrophile but also bind reversibly to pyridine and in this way assist in the necessary ligand exchange. Additionally, there seems to be a pronounced thermodynamic preference for the formation of the BPh2‐bridged anion [Co(dmgBPh2)2Cl2]− over the BPh2‐bridged cation [Co(dmgBPh2)2(py)2]+, which we have never observed even in traces (e. g., mass spectrometry). Once formed, the preferred boron‐capped anion [Co(dmgBPh2)2Cl2]− therefore precipitates from the reaction mixture as a salt with counter‐cation [Co(dmgH)2(py)2]+ until most starting material is consumed.
As shown in Figure 1a, right, we were able to transform [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]−, into “single complex salts”, in which either a cationic or anionic cobalt complex is paired with weakly coordinating counterions tetrakis[3,5‐bis(trifluoromethyl)phenyl]borate (BArF−) and tetrabutylammonium (TBA+). The 1H NMR spectra of the single complex salts [Co(dmgH)2(py)2]+ BArF− and TBA+ [Co(dmgBPh2)2Cl2]− (Figure 1b, middle and bottom) show, that these compounds can be obtained in high purity and provide further evidence for our structural assignment, because the spectrum of the double salt is essentially the sum of the spectra of single salts with only minor changes of the chemical shifts. A similar picture emerges from the electrochemical data (in acetonitrile), which is summarized in Table 1. The double complex salt displays four reduction potentials, which correspond to the two reduction potentials observed for the stepwise reduction of CoIII to CoI in the single salts.
Table 1.
Electrochemical data from cyclic voltammetry.[a]
|
|
Reduction potentials [V] |
|||
|---|---|---|---|---|
|
|
CoIII/CoII |
CoIII/CoII |
CoII/CoI |
CoII/CoI |
|
[Co(dmgH)2(py)2]+ [Co(dmgBPh2)2Cl2]− |
−0.63 |
−0.97 |
−1.17 |
−1.38 |
|
TBA+[Co(dmgBPh2)2Cl2]− |
|
−1.04 |
|
−1.46 (irr) |
|
[Co(dmgH)2(py)2]+ BArF− |
−0.62 |
|
−1.24 |
|
|
[Co(dmgH)2(py)Cl] |
−0.69 |
|
|
−1.47 |
[a] CV conditions: acetonitrile at RT, supporting electrolyte: TBAPF6 (0.1 M), potentials vs. Fc/Fc+. Scan rate 50 mV/s. [cobalt salt]=1.0×10−3 M. See also Figures S2–S4.
Light‐driven HER in solution
We proceeded with a preliminary study of the new double and single complex salts as catalysts for the light‐driven HER. Experiments were carried out in triplicates, using acetone as solvent, irradiation with blue LED light (460 nm), ruthenium trisbipyridine hexafluorophosphate [Ru(bpy)3](PF6)2 as commercially available photosensitizer, and a non‐aqueous sacrificial electron donor system ([HNEt3]+[BF4]−/NEt3) previously used by Artero and co‐workers. [3a] To allow a meaningful comparison between double and single complex salts, turnover numbers (TON) were calculated as moles of generated dihydrogen per mole cobalt, and not per mole catalyst. As Figure 2 shows, all three complex salts reached similar TONs of about 60, whereas the neutral benchmark complex only reaches a TON of 35. We therefore conclude that, at least under the conditions investigated herein, all cobaloxime‐based complex salts have a higher longevity than the neutral benchmark complex, suggesting that there may be extra stability derived from an electrostatic charge and/or from the BPh2 bridge.
Figure 2.
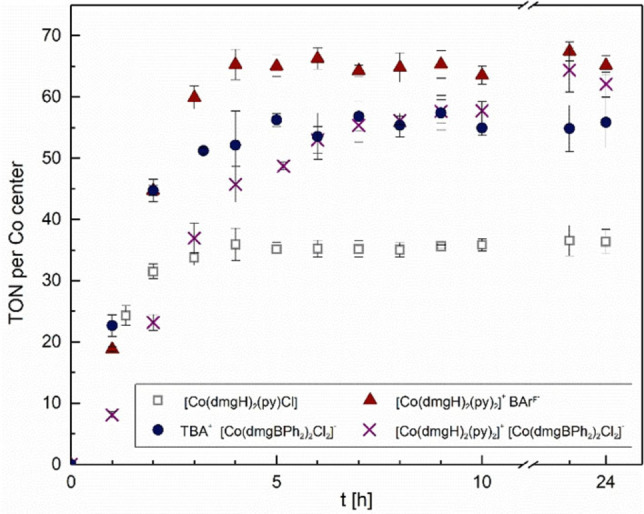
Monitoring of photocatalytic production of dihydrogen in acetone. Proton source/sacrificial electron donor [HNEt3][BF4]/NEt3 (1000 equiv.), [catalyst]=0.043 mM, [Ru(bpy)3](PF6)2=0.043 mM, light source: blue LED (460 nm, 50 mW/cm2). Error bars correspond to standard uncertainty of triplicate experiments.
When inspecting the kinetic profiles for hydrogen generation as well as the turnover frequency (TOF) observed for the first hour of the reaction (Figure 2 and Table 2), the double complex salt [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]− represents an obvious outlier. While the two single salts and the neutral complex furnish approximately 20 turnovers at the beginning of the reaction, the double complex salt significantly lags behind and only approaches its plateau for hydrogen production after about 12 h. We suggest that the slower initial rates observed for the double complex salt could perhaps be due to relatively strong ion pairing between the cation and the anion [Co(dmgBPh2)2Cl2]−, which could affect electron transfer with the charged photosensitizer and/or the turnover‐limiting step of HER. Future studies will be directed towards a deeper understanding of these effects.
Table 2.
Preliminary photocatalytic hydrogen evolution studies.[a]
|
|
Catalyst |
TON |
TOF [h−1] |
|---|---|---|---|
|
1 |
[Co(dmgH)2(py)2]+ [Co(dmgBPh2)2Cl2]− |
61±3 |
8±1 |
|
2 |
TBA+[Co(dmgBPh2)2Cl2]− |
56±3 |
23±2 |
|
3 |
[Co(dmgH)2(py)2]+ BArF− |
65±2 |
19±1 |
|
4 |
[Co(dmgH)2(py)Cl] |
35±2 |
18±2 |
[a] Turnover numbers (TON) and turnover frequencies (TOF) derived from the data shown in Figure 2.
Deposition on carbon nanomembranes
We further investigated the local deposition of two of the catalysts, ([Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]− and the commercially available [Co(dmgH)2(py)Cl]) already characterized in this study in respect to bulk photocatalysis (Table 2). We used SECCM to deposit arrays of micro‐ (Figure S16) or nanospots on NH2‐CNMs either directly formed on gold‐coated substrates [14c] or on NH2‐CNMs transferred to TEM grids [31] as schematically depicted in Figure 3a. Deposition of catalysts on bare gold substrates was unsuccessful, it seems that the amino‐terminated NH2‐CNM is crucial to obtain spots, a fact that has to be further investigated in a future study. The shape and size of such nanopipette induced electroless deposition is influenced by various factors such as meniscus size, solute concentration, evaporation of the solvent etc., as recently reviewed. [32] For depositing spots as shown in Figure 3b, a dual barrel nanopipette with an overall orifice of 100 nm (single barrel orifice: 50 nm) was filled with a solution of dissolved in acetonitrile. After forming the droplet at the surface, the SECCM pipette was kept in contact with the sample surface for 10 s followed by withdrawal and moving to a new area. Catalyst‐spot in the size of ∼1.5 times of the overall nanopipette orifice remained on the surface for the deposited [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]− catalyst, as shown in Figure 3b for a pattern with 4 spots, and 2 spots on a NH2‐CNM modified TEM grid (Figure 3b, d). The deposits appear round shaped (inset Figure 3b). TEM/EDX analysis of a [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]− spot shows uniform elemental distribution of cobalt, chloride, nitrogen and oxygen (Figure 3f). Note that this spot is only supported by a ∼1 nm thin 2D material, as it is located in an area with freestanding NH2‐CNM on a TEM grid hole. The neutral complex [Co(dmgH)2(py)Cl] formed structures with larger irregular shapes as shown Figure 3c (4 spots) and 3e (2 spots). TEM diffraction patterns of both deposited catalysts indicate amorphous nature (data not shown). The four [Co(dmgH)2(py)Cl] spots obtained by the same overall tip orifice (100 nm) gave spots with diameter in the range of 500 nm.
Figure 3.
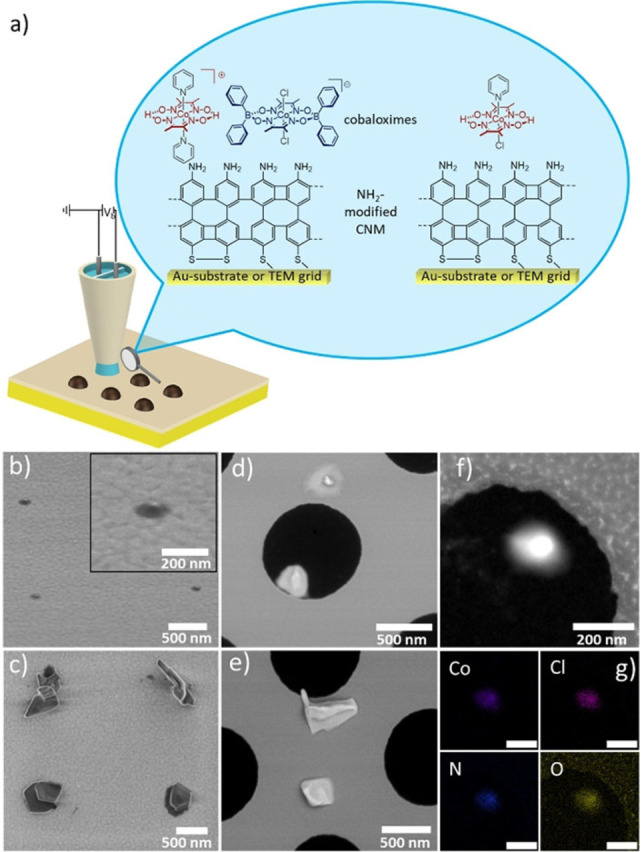
a) Scheme of the SECCM for the deposition of cobaloxime catalysts on NH2‐CNM‐modified substrates. A bias voltage (25 mV) was applied between the quasi reference/counter electrodes (QRCEs) located in the barrels of the nanopipette, and the resulting ion conductance current (i DC) across the meniscus was used as input for the positional feedback. Deposits on b) and c) NH2‐CNM/Au substrate and d) and e) immobilized on a NH2‐CNM modified TEM grid. b) four 100–150 nm diameter spots; inset: zoomed view of a single spot. d) two spots on a modified TEM grid. One of them is on the free‐standing NH2‐CNM area. c) Four 500 nm diameter [Co(dmgH)2(py)Cl] nanospots, and e) two spots on a modified TEM grid. f) A HAADF STEM image of a nanospot (ø 180 nm) and g) the corresponding color‐coded EDX elemental maps for the Co, Cl, N, and O distributions, as noted in the images; scale bars: 200 nm.
Stability studies of the immobilized catalyst spots
We investigated the stability of the immobilized spots on NH2‐CNM/Au substrates under photocatalytic conditions. The samples were immersed in an aqueous solution containing [Ru(tbbpy)2(mmip)]Cl3 (tbbpy=4,4’‐di‐tert‐butyl‐2,2’‐bipyridine; mmip=1,3‐dimethyl‐1H‐imidazol[4,5‐f][1,10]phenanthrolinium, abbreviated “Ru(mmip)”) as photosensitizer and ascorbic acid as sacrificial electron donor at pH 4. Ru(mmip) was chosen, as it was reported to give high catalytic activity in light‐driven homogeneous photocatalysis with [Co(dmgH)2(py)Cl] as catalyst. [33] High‐resolution AFM images of the spots were recorded before and after illumination with a blue LED (470 nm) two times for a period of 15 h (total 30 h). AFM images of the same spots were recorded prior, after 15 and 30 h.
Figure 4 shows exemplarily the AFM topography images of a [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]− spot (Figure 4a) and a [Co(dmgH)2(py)Cl] spot (Figure 4b), which were deposited with a dual barrel nanopipette (overall opening of ca. 400 nm) prior to the illumination experiments. As already visible in the SEM images, [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]− has a more circular shape which approximates the pipette opening, in contrast to [Co(dmgH)2(py)Cl] that forms irregular structures with larger sizes. An overlay of line scans derived from the single spot for both catalysts (before and after the two illumination steps) (shown in Figure 4c and d, respectively) reveal no significant height change for the [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]− spot after 30 h illumination. Whereas the height of the [Co(dmgH)2(py)Cl] spot decreased by approximately 40 nm after 30 h of illumination. Figure S17 shows the topography and deflection images of the [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]− and [Co(dmgH)2(py)Cl] prior to illumination (a, d), after 15 h of illumination (b, e) and 30 h of illumination (c, f). In total, we investigated eight spots for each catalyst; whereby 4 spots from two different samples were investigated. Figure 4e and f show the bar diagrams reflecting height changes. A mean change in height of only 3±2 % (n=7) after 30 h illumination was determined for [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]− with a maximum observed height change of 12 nm. Given experimental uncertainties in the measurements, we conclude that the catalyst spots maintained their original height and shape, as no trend in height change could be observed in respect to the illumination time. Small variations visible for some of the spots are within the experimental error. For the [Co(dmgH)2(py)Cl] spots, a decrease in height after 15 and 30 h was recorded for all investigated spots with an average height decrease of 21±5 % (n=8) after 30 h illumination and a maximum observed height change of 98 nm. We hypothesize that either higher solubility of the deposited neutral Co complex [Co(dmgH)2(py)Cl] in solution (pH 4) or structural changes of the catalyst under photocatalytic conditions lead to the observed changes.
Figure 4.

AFM topography images recorded in air; a) and b) [Co(dmgH)2(py)Cl] spots. c) and d) Height profiles before illumination (grey), after 15 h of illumination (red) and after 30 h of illumination (blue) in 0.5 mM Ru(mmip) and 0.1 M ascorbic acid solution (pH 4). e) and f) The change in height of 8 spots for each catalyst. (Note: in (e) only data before and after 15 h were recorded for spot 1).
We also observed morphological changes after 30 h illumination in SEM images (data not shown). To exclude that higher solubility of the neutral Co complex [Co(dmgH)2(py)Cl] was responsible for the observed height changes of the catalyst, the sample was immersed for the same period of time in 0.5 mM Ru(mmip) and 0.1 M ascorbic acid solution (pH 4) under dark conditions. Figure S19a shows the height profiles of eight [Co(dmgH)2(py)Cl] spots and in Figure S19b, a bar diagram is depicted comparing the average height of the spots after illumination (H2 measurements) and after immersing the sample for 30 h under dark conditions. A mean change in height of only 2±3 % (n=8) after 30 h in the reaction solution was determined with a maximum observed height change of 12 nm. We conclude that the [Co(dmgH)2(py)Cl] spots are stable in the reaction solution and the immobilized catalyst is more stable than the immobilized [Co(dmgH)2(py)Cl] under photocatalytic conditions.
In‐situ hydrogen measurements on NH2‐CNM immobilized Co catalyst spots
For the in situ determination of the photocatalytic activity of such catalyst arrays, we used amperometric H2 microsensors which were positioned approx. 30 μm above the catalyst array as schematically shown in Figure 5a. For this proof of principle studies, larger arrays (9×9 spots) were deposited, which consisted of 81 spots (diameter of the spots and spacing between the spots: 1 μm) covering an area of 17×17 μm as shown in Figure S16. Larger arrays in the size of the active electrode area of the microsensor were initially chosen to ensure that the produced H2 amount was not below the detection limit of the sensors. For in‐situ H2 measurements at the catalyst arrays, we used Pd‐modified microelectrodes (diameter of 25 μm), as Pd forms a Pd hydride adsorbing H2 into its lattice, leading to a change in its electronic properties. [34] The Pd layers were deposited onto roughened gold microelectrodes (active surface area 5.0×10 4 μm2) and calibrated towards their H2 response as shown in Figure 5b). The limit of detection of the H2 microsensors is in the range of 10–30 μM (determined for the three microsensors used in the presented experiments). After positioning the H2 microsensor above the catalyst array, measurements were performed in solution with Ru(mmip) as sensitizer and ascorbic acid as electron donor. H2 measurements were recorded for a period of 1 h, which reflects the first measurement point in the bulk photocatalytic measurements (Table 2). Based on the calibration data, a hydrogen concentration of 22.33±4.71 mmol L−1 (n=3) for the catalyst arrays was obtained, where the triplicate was performed at two different samples (one sample was measured twice). The same experiments were performed at [Co(dmgH)2(py)Cl] arrays (Figure S16b), again on two different samples, where one array was illuminated twice, as well as at an array of TBA+[Co(dmgBPh2)2Cl2]− (SEM image not shown). The measurements at the [Co(dmgH)2(py)Cl] arrays resulted in a hydrogen concentration of 1.27±0.46 mmol L−1 (n=3) (Figure 5c). Hydrogen measurements at the TBA+[Co(dmgBPh2)2Cl2]− catalyst array resulted in a concentration of 15.36±4.40 mmol L−1 (n=2). The and the TBA+[Co(dmgBPh2)2Cl2]− array revealed a significant higher HER activity with 18 and 12 times higher H2 concentration, respectively compared to [Co(dmgH)2(py)Cl]. Interestingly, measurements at the same [Co(dmgH)2(py)Cl] array for a second time resulted in significantly reduced H2 evolution, which may be associated with the degradation of the [Co(dmgH)2(py)Cl] spots. It is reported in the literature that the [Co(dmgH)2(py)Cl] catalyst is less stable at pH values below pH 5. [35] Therefore, we repeated the H2 measurements at the [Co(dmgH)2(py)Cl] array in a solution of 0.5 mM Ru(mmip) and 0.1 M ascorbic acid solution adjusted to pH 5 (n=3). The obtained H2 concentration obtained in pH 5 is only slightly higher (1.69±0.49 mmol L−1, n=3) compared to pH 4, as shown in Figure S21. There is apparently no statistical difference between the measurements carried out at pH 4 and at pH 5. Based on this finding as well as the dark control experiment described above (Figure S19), we believe that ligand degradation is highly unlikely to occur during the short time periods (60 min) of the in‐situ HER experiments.
Figure 5.

a) Schematic of the SECM illumination experiment. b) Example calibrations curve for a Pd microsensor. c) Bar diagram of the H2 concentration yield after 1 h of illumination of three catalysts. Abbreviations of the x‐coordinate labelling is as follows: [Co(dmgH)2(py)Cl]=Co(dmgH), [Co(dmgH)2(py)2]+[Co(dmgBPh2)2Cl2]−=[Co]+[Co]−, and TBA+[Co(dmgBPh2)2Cl2]−=[Co]−. All experiments were performed under argon. Error bars reflect three measurements for [Co]+[Co]− and Co(dmgH), and two measurements for [Co]−.
We also compared the obtained results from the in‐situ H2 measurements with the results obtained by GC headspace from the homogeneous catalysis experiments, where a lower maximal H2 concentration of 5.23 mmol L−1 for the catalyst and 2.40 mmol L−1 for the TBA+[Co(dmgBPh2)2Cl2]− catalyst was derived from the TON values reported in Table 2. It should be noted that this comparison is only a rough estimate as the experimental parameters (e. g., used sacrificial electron donor, sensitizer, pH value, solvent, etc.) were different between the homogeneous and heterogeneous photocatalytic experiments. Comparing the results of the [Co(dmgH)2(py)Cl] catalyst, the bulk measurements yielded a maximal H2 concentration of 1.50 mmol L−1, which is slightly higher than the obtained values from the in‐situ measurements.
We performed the following control experiments (Figure S20a) for the same time period (60 min) to ensure that the observed cathodic response at the Pd microsensor was related to the HER. Control experiments included measurements under dark conditions (dotted) and illumination (solid), where only ascorbic acid (black solid and dotted line) or only Ru(mmip) (red solid and dotted line) was added to solution. In these cases, no HER was detected with or without illumination (Figure S20a). A further control experiment was done at a bare gold substrate with donor and sensitizer in solution under dark condition and under illumination, leading under illumination to a shift in the stable current response due to the re‐oxidation of the sacrificial donor but no observable H2 detection, as expected. Only if the solution contains both components and is illuminated at the immobilized Co‐catalyst arrays, an increase in cathodic current over the period of illumination is observed, indicating HER (see example i–t curves in Figure S20b, c).
Conclusion
In conclusion, we have developed straightforward syntheses of cobalt‐based double and single complex salts with a BPh2 bridge. Photocatalytic HER experiments in solution showed that all three complex salts described herein outperform the neutral benchmark complex [Co(dmgH)2(py)Cl] in terms of TON. Characteristic differences in TOF suggest that the reported salts might open the door to further uses of electrostatic effects [36] in order to modulate catalytic activity. We further demonstrated that complementary scanning probe microscopy techniques – SECCM, AFM, and SECM – are attractive methods for producing tailored photoelectrode catalysts for heterogenous light‐driven water splitting and assessing their performance. By using SECCM, cobaloxime catalyst arrays were successfully immobilized on supported and freestanding 1 nm thick NH2‐CNM (on Au substrates or TEM grids). Catalyst deposition on bare Au substrates was unsuccessful. We could show in situ measurements of H2 due to the HER activity of immobilized , TBA+[Co(dmgBPh2)2Cl2]− and [Co(dmgH)2(py)Cl] catalyst arrays for the first time. We correlate the difference in HER activity among other effects to the stability of the deposited catalysts, under illumination, which was investigated by AFM. The catalyst spots remained stable under the photocatalytic conditions, while the [Co(dmgH)2(py)Cl] nanospots showed a clear decrease in size (amount) after 30 h of illumination.
Experimental Section
Catalyst deposition via SECCM: SECCM experiments were performed on a home build set‐up. Samples were placed on a x–y piezo table (P‐541.2CD, Physik Instrumente, Karlsruhe, Germany) while the nanopipette was attached to a z‐piezo positioner (P‐753.2CD, PI) mounted on a stepper motor (MTS25‐Z8, Thorlabs GmbH, Bergkirchen, Germany) and connected to a digital piezo controller (E‐727, Physik Instrumente, Karlsruhe, Germany). Current measurements were performed with a low‐noise current preamplifier (SR570, Stanford Research Systems, USA). The instrumentation was controlled, and data was collected by a FPGA card (PCIe‐7852R, National Instruments, Austin, USA). The experiments were controlled using the Warwick Electrochemical Scanning Probe Microscopy (WEC‐SPM) software. The set‐up is built on a vibration isolation table (Benchmate 2210, Kinetic Systems, Boston, USA) The instrument was set up in a faraday cage. Further a digital camera (PL‐B776 U, PixeLink, Ottawa, Canada) and a cold light source (MI‐150, Edmund Optics, Mainz, Germany) were used to aid the initial positioning of the nanopipette tip around 10–20 μm above the substrate. Nanopipettes with orifices in the range of 100 nm to 1.0 μm (measured accurately by SEM) were made from quartz theta capillaries (1.2 mm OD, 0.9 mm ID, Sutter Instruments, Novato, USA) using a laser pipette puller (P‐2000, Sutter Instruments). The nanopipettes were back‐filled with either TBA+[Co(dmgBPh2)2Cl2]− or [Co(dmgH)2(py)Cl] catalyst (0.4 mM solved in acetonitrile). Ag wires were inserted in the back opening of each barrel to serve as QRCEs. The nanopipette was then approached via a piezo positioner to the surface (50 nm s−1) while a bias of 25 mV was applied between the two QRCEs. The DC ion current, measured between the QRCEs in the two barrels, was used as feedback for detecting meniscus contact. [37] The nanopipette was kept for 10 s at the surface before withdrawing the nanopipette and moving to the next position where it was again approached towards the surface in an approach, hold, withdraw, move protocol.
In‐situ H2 measurements via SECM: For HER measurements, Pd‐modified Au/Ni (etched) microelectrodes were used as H2 microsensors. The SECM studies were performed in a three‐electrode setup with a Pd‐modified microelectrode as working electrode, an Ag/AgCl QRE and a Pt wire as counter electrode. For the electrochemical experiments, a Palmsens4 potentiostat (Palmsens, Houten, Netherlands) was used. The Pd microsensor was positioned at a distance of 30 μm above the catalyst arrays, by recording approach curves in 5 mM 1,1’‐ferrocendimethanol/0.1 M KCl solution. To measure the photocatalytic activity, the solution was removed, and the sample was washed 3 times with high purity water. Then the electrochemical cell was filled with 0.5 mM [Ru(tbbpy)2(mmip)]Cl3 in 0.1 M ascorbic acid solution (pH 4 or pH 5) and purged with argon for at least 15 min to remove dissolved oxygen. A constant tip potential of −600 mV vs. Ag/AgCl was applied to the Pd‐modified microelectrode. All measurements were performed under argon atmosphere. A 400 μm optical fibre (MT‐28L01, Thorlabs GmbH, Bergkirchen, Germany) connected to a 21.8 mW blue LED (M470F3, Thorlabs GmbH) was used for illumination.
Additional Information
Deposition Number 1958254 (for contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The project is funded by the Deutsche Forschungsgemeinschaft (DFG – German Research Foundation) – project number 364549901 – TRR 234, subprojects A4, B2, B7 and C4. We thank Dr. Simon Clausing for helpful discussions. Simon Schauer is acknowledged for AFM measurements. Elisabeth Hofmeister, Jisoo Woo and Jie Zhao are acknowledged for contributions to synthesis and characterization. We like to thank Kevin Hanus for his support during the electrochemical measurements. Gregor Neusser is acknowledged for support with SEM measurements. P. R. U. thanks the Royal Society for a Wolfson Research Merit Award and the funding from grant EPSRC EP/R018820/1. Open Access funding enabled and organized by Projekt DEAL.
E. Oswald, A.-L. Gaus, J. Kund, M. Küllmer, J. Romer, S. Weizenegger, T. Ullrich, A. K. Mengele, L. Petermann, R. Leiter, P. R. Unwin, U. Kaiser, S. Rau, A. Kahnt, A. Turchanin, M. von Delius, C. Kranz, Chem. Eur. J. 2021, 27, 16896.
Contributor Information
Prof. Dr. Max von Delius, Email: max.vondelius@uni-ulm.de.
Prof. Dr. Christine Kranz, Email: christine.kranz@uni-ulm.de.
References
- 1.
- 1a. Dreos R., Geremia S., Randaccio L., Siega P., Properties, Structure and Reactivity of Cobaloximes, Wiley, Hoboken, 2010; [Google Scholar]
- 1b. Bresciani-Pahor N., Forcolin M., Marzilli L. G., Randaccio L., Summers M. F., Toscano P. J., Coord. Chem. Rev. 1985, 63, 1–125. [Google Scholar]
- 2.
- 2a. Hawecker J., Lehn J.-M., Ziessel R., Nouv. J. Chim. 1983, 7, 271–277; [Google Scholar]
- 2b. Connolly P., Espenson J. H., Inorg. Chem. 1986, 25, 2684–2688. [Google Scholar]
- 3.
- 3a. Fihri A., Artero V., Razavet M., Baffert C., Leibl W., Fontecave M., Angew. Chem. Int. Ed. 2008, 47, 564–567; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 574–577; [Google Scholar]
- 3b. Dempsey J. L., Brunschwig B. S., Winkler J. R., Gray H. B., Acc. Chem. Res. 2009, 42, 1995–2004; [DOI] [PubMed] [Google Scholar]
- 3c. Cartwright K. C., Davies A. M., Tunge J. A., Eur. J. Org. Chem. 2020, 2020, 1245–1258; [Google Scholar]
- 3d. Dolui D., Khandelwal S., Majumder P., Dutta A., Chem. Commun. 2020, 56, 8166–8181; [DOI] [PubMed] [Google Scholar]
- 3e. Lazarides T., McCormick T., Du P., Luo G., Lindley B., Eisenberg R., J. Am. Chem. Soc. 2009, 131, 9192–9194; [DOI] [PubMed] [Google Scholar]
- 3f. Losse S., Vos J. G., Rau S., Coord. Chem. Rev. 2010, 254, 2492–2504; [Google Scholar]
- 3g. Artero V., Chavarot-Kerlidou M., Fontecave M., Angew. Chem. Int. Ed. 2011, 32, 7238–7266; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 132, 7376–74055; [Google Scholar]
- 3h. Solis B. H., Hammes-Schiffer S., J. Am. Chem. Soc. 2011, 133, 19036–19039; [DOI] [PubMed] [Google Scholar]
- 3i. Marinescu S. C., Winkler J. R., Gray H. B., Proc. Natl. Acad. Sci. USA 2012, 109, 15127–15131; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3j. Du P., Eisenberg R., Energy Environ. Sci. 2012, 5, 6012–6021; [Google Scholar]
- 3k. Hogue R. W., Schott O., Hanan G. S., Brooker S., Chem. Eur. J. 2018, 24, 9820–9832; [DOI] [PubMed] [Google Scholar]
- 3l. Willkomm J., Reisner E., Bull. Japan Soc. Coord. Chem. 2018, 71, 18–29. [Google Scholar]
- 4.
- 4a. Hu X., Cossairt B. M., Brunschwig B. S., Lewis N. S., Peters J. C., Chem. Commun. 2005, 1, 4723–4725; [DOI] [PubMed] [Google Scholar]
- 4b. Zhang P., Wang M., Dong J., Li X., Wang F., Wu L., Sun L., J. Phys. Chem. C 2010, 114, 15868–15874; [Google Scholar]
- 4c. Anxolabéhère-Mallart E., Costentin C., Fournier M., Nowak S., Robert M., Savéant J.-M., J. Am. Chem. Soc. 2012, 134, 6104–6107. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Du P., Schneider J., Luo G., Brennessel W. W., Eisenberg R., Inorg. Chem. 2009, 48, 4952–4962; [DOI] [PubMed] [Google Scholar]
- 5b. Peuntinger K., Lazarides T., Dafnomili D., Charalambidis G., Landrou G., Kahnt A., Sabatini R. P., McCamant D. W., Gryko D. T., Coutsolelos A. G., Guldi D. M., J. Phys. Chem. C 2013, 117, 1647–1655; [Google Scholar]
- 5c. Bartelmess J., Francis A. J., El Roz K. A., Castellano F. N., Weare W. W., Sommer R. D., Inorg. Chem. 2014, 53, 4527–4534; [DOI] [PubMed] [Google Scholar]
- 5d. Lentz C., Schott O., Auvray T., Hanan G., Elias B., Inorg. Chem. 2017, 56, 10875–10881. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Jacques P.-A., Artero V., Pécaut J., Fontecave M., Proc. Natl. Acad. Sci. USA 2009, 106, 20627–20632; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Pati P. B., Zhang L., Philippe B., Fernández-Téran R., Ahmadi S., Tian L., Rensmo H., Hammarström L., Tian H., ChemSusChem 2017, 10, 2480–2495; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Bold S., Zedler L., Zhang Y., Massin J., Artero V., Chavarot-Kerlidou M., Dietzek B., Chem. Commun. 2018, 54, 10594–10597. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Muresan N. M., Willkomm J., Mersch D., Vaynzof Y., Reisner E., Angew. Chem. Int. Ed. 2012, 51, 12749–12753; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12921–12925; [Google Scholar]
- 7b. Kaeffer N., Massin J., Lebrun C., Renault O., Chavarot-Kerlidou M., Artero V., J. Am. Chem. Soc. 2016, 138, 12308–12311; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Gottschling K., Savasci G., Vignolo-González H., Schmidt S., Mauker P., Banerjee T., Rovó P., Ochsenfeld C., Lotsch B. V., J. Am. Chem. Soc. 2020, 142, 12146–12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Szymczak N. K., Berben L. A., Peters J. C., Chem. Commun. 2009, 44, 6729–6731; [DOI] [PubMed] [Google Scholar]
- 8b. Valdez C. N., Dempsey J. L., Brunschwig B. S., Winkler J. R., Gray H. B., Proc. Natl. Acad. Sci. USA 2012, 109, 15589–15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dreos R., Tauzher G., Vuano S., Asaro F., Pellizer G., Nardin G., Randaccio L., Geremia S., J. Organomet. Chem. 1995, 505, 135–138. [Google Scholar]
- 10.
- 10a. Song X.-W., Wen H.-M., Ma C.-B., Cui H.-H., Chen H., Chen C.-N., RSC Adv. 2014, 4, 18853–18861; [Google Scholar]
- 10b. Cai M., Zhang F., Zhang C., Lu C., He Y., Qu Y., Tian H., Feng X., Zhuang X., J. Mater. Chem. A 2017, 6, 138–144. [Google Scholar]
- 11.
- 11a. Fujita I., Kobayashi H., J. Chem. Phys. 1973, 2902, 2902–2908; [Google Scholar]
- 11b. Tsuchiya R., Joba S., Uehara A., Kyuno E., Bull. Chem. Soc. Jpn. 1973, 46, 169–175; [Google Scholar]
- 11c. Doerrer L. H., Dalton Trans. 2010, 39, 3543–3553. [DOI] [PubMed] [Google Scholar]
- 12. Trogler W. C., Stewart R. C., Epps L. A., Marzilli L. G., Inorg. Chem. 1974, 13, 1564–1570. [Google Scholar]
- 13.
- 13a. Krawicz A., Yang J., Anzenberg E., Yano J., Sharp I. D., Moore G. F., J. Am. Chem. Soc. 2013, 135, 11861–11868; [DOI] [PubMed] [Google Scholar]
- 13b. Andreiadis E. S., Jacques P.-A., Tran P. D., Leyris A., Chavarot-Kerlidou M., Jousselme B., Matheron M., Pécaut J., Palacin S., Fontecave M., Artero V., Nat. Chem. 2013, 5, 48–53; [DOI] [PubMed] [Google Scholar]
- 13c. Donck S., Fize J., Gravel E., Doris E., Artero V., Chem. Commun. 2016, 4, 21–36; [DOI] [PubMed] [Google Scholar]
- 13d. Reuillard B., Warnan J., Leung J. J., Wakerley D. W., Reisner E., Angew. Chem. Int. Ed. 2016, 55, 3952–3957; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4020–4025; [Google Scholar]
- 13e. Ma B., Chen G., Fave C., Chen L., Kuriki R., Maeda K., Ishitani O., Lau T.-C., Bonin J., Robert M., J. Am. Chem. Soc. 2020, 142, 6188–6195; [DOI] [PubMed] [Google Scholar]
- 13f. Sévery L., Szczerbiński J., Taskin M., Tuncay I., Nunes F. B., Cignarella C., Tocci G., Blacque O., Osterwalder J., Zenobi R., Iannuzzi M., Tilley S. D., Nat. Chem. 2021, 13, 523–529. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Turchanin A., Gölzhäuser A., Prog. Surf. Sci. 2012, 87, 108–162; [Google Scholar]
- 14b. Angelova P., Vieker H., Weber N. E., Matei D., Reimer O., Meier I., Kurasch S., Biskupek J., Lorbach D., Wunderlich K., Chen L., Terfort A., Klapper M., Müllen K., Kaiser U., Gölzhäuser A., Turchanin A., ACS Nano 2013, 7, 6489–6497; [DOI] [PubMed] [Google Scholar]
- 14c. Turchanin A., Gölzhäuser A., Adv. Mater. 2016, 28, 6075–6103. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Eck W., Stadler V., Geyer W., Zharnikov M., Gölzhäuser A., Grunze M., Adv. Mater. 2000, 12, 805–808; [Google Scholar]
- 15b. Neumann C., Wilhelm R. A., Küllmer M., Turchanin A., Faraday Discuss. 2021, 227, 61–79. [DOI] [PubMed] [Google Scholar]
- 16. Lee J., Ye H., Pan S., Bard A. J., Anal. Chem. 2008, 80, 7445–7450. [DOI] [PubMed] [Google Scholar]
- 17. Momotenko D., Page A., Adobes-Vidal M., Unwin P. R., ACS Nano 2016, 10, 8871–8878. [DOI] [PubMed] [Google Scholar]
- 18. Ebejer N., Güell A. G., Lai S. C. S., McKelvey K., Snowden M. E., Unwin P. R., Annu. Rev. Anal. Chem. 2013, 6, 329–351. [DOI] [PubMed] [Google Scholar]
- 19. McKelvey K., O'Connell M. A., Unwin P. R., Chem. Commun. 2013, 49, 2986–2988. [DOI] [PubMed] [Google Scholar]
- 20. Shumaker-Parry J. S., Zareie M. H., Aebersold R., Campbell C. T., Anal. Chem. 2004, 76, 918–929. [DOI] [PubMed] [Google Scholar]
- 21. Gunawan C. A., Ge M., Zhao C., Nat. Commun. 2014, 5, 3744. [DOI] [PubMed] [Google Scholar]
- 22. Lai S. C. S., Lazenby R. A., Kirkman P. M., Unwin P. R., Chem. Sci. 2015, 6, 1126–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Aaronson B. D. B., Garoz-Ruiz J., Byers J. C., Colina A., Unwin P. R., Langmuir 2015, 31, 12814–12822; [DOI] [PubMed] [Google Scholar]
- 23b. Oseland E. E., Ayres Z. J., Basile A., Haddleton D. M., Wilson P., Unwin P. R., Chem. Commun. 2016, 52, 9929–9932. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Kim J. H., Chang W. S., Kim D., Yang J. R., Han J. T., Lee G.-W., Kim J. T., Seol S. K., Adv. Mater. 2015, 27, 157–161; [DOI] [PubMed] [Google Scholar]
- 24b. Tarnev T., Cychy S., Andronescu C., Muhler M., Schuhmann W., Chen Y., Angew. Chem. Int. Ed. 2020, 59, 5586–5590; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 5634–5638. [Google Scholar]
- 25. Amemiya S., Bard A. J., Fan F.-R. F., Mirkin M. V., Unwin P. R., Annu. Rev. Anal. Chem. 2008, 1, 95–131. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Bae J. H., Nepomnyashchii A. B., Wang X., Potapenko D. V., Mirkin M. V., Anal. Chem. 2019, 91, 12601–12605; [DOI] [PubMed] [Google Scholar]
- 26b. Barwe S., Andronescu C., Engels R., Conzuelo F., Seisel S., Wilde P., Chen Y.-T., Masa J., Schuhmann W., Electrochim. Acta 2019, 297, 1042–1051; [Google Scholar]
- 26c. Bhattacharya C., Lee H. C., Bard A. J., J. Phys. Chem. C 2013, 117, 9633–9640; [Google Scholar]
- 26d. Conzuelo F., Sliozberg K., Gutkowski R., Grützke S., Nebel M., Schuhmann W., Anal. Chem. 2017, 89, 1222–1228; [DOI] [PubMed] [Google Scholar]
- 26e. Ye H., Lee J., Jang J. S., Bard A. J., J. Phys. Chem. C 2010, 114, 13322–13328; [Google Scholar]
- 26f. Ye H., Park H. S., Bard A. J., J. Phys. Chem. C 2011, 115, 12464–12470. [Google Scholar]
- 27.
- 27a. Giese B., Hartung J., Pérez B. M., Hartung J., Chlorobis(Dimethylglyoximato)(Pyridine)Cobalt(III), Wiley, Hoboken, 2008; [Google Scholar]
- 27b. Kahnt A., Peuntinger K., Dammann C., Drewello T., Hermann R., Naumov S., Abel B., Guldi D. M., J. Phys. Chem. A 2014, 118, 4382–4391. [DOI] [PubMed] [Google Scholar]
- 28. Mandai T., Masu H., Johansson P., Dalton Trans. 2015, 44, 11259–11263. [DOI] [PubMed] [Google Scholar]
- 29. Geremia S., Dreos R., Randaccio L., Tauzher G., Antolini L., Inorg. Chim. Acta 1994, 216, 125–129. [Google Scholar]
- 30. Basu D., Mazumder S., Niklas J., Baydoun H., Wanniarachchi D., Shi X., Staples R. J., Poluektov O., Schlegel H. B., Verani C. N., Chem. Sci. 2016, 7, 3264–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tang Z., Neumann C., Winter A., Turchanin A., Nanoscale 2020, 12, 8656–8663. [DOI] [PubMed] [Google Scholar]
- 32. Je J. H., Kim J.-M., Jaworski J., Small 2017, 13, 1600137. [DOI] [PubMed] [Google Scholar]
- 33. Petermann L., Staehle R., Pfeifer M., Reichardt C., Sorsche D., Wächtler M., Popp J., Dietzek B., Rau S., Chem. Eur. J. 2016, 22, 8240–8253. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Flanagan T. B., Oates W. A., Annu. Rev. Mater. Sci. 1991, 21, 269–304; [Google Scholar]
- 34b. Hubkowska K., Łukaszewski M., Czerwiński A., Electrochim. Acta 2011, 56, 2344–2350; [Google Scholar]
- 34c. Hubkowska K., Soszko M., Symonowicz M., Łukaszewski M., Czerwiński A., Electrocatalysis 2017, 8, 295–300; [Google Scholar]
- 34d. Koster D., Gutkowski R., Masa J., Schuhmann W., J. Electroanal. Chem. 2018, 812, 207–212. [Google Scholar]
- 35.
- 35a. Coutard N., Kaeffer N., Artero V., Chem. Commun. 2016, 52, 13728–13748; [DOI] [PubMed] [Google Scholar]
- 35b. Dolui D., Khandelwal S., Shaik A., Gaat D., Thiruvenkatam V., Dutta A., ACS Catal. 2019, 9, 10115–10125; [Google Scholar]
- 35c. Natali M., ACS Catal. 2017, 7, 1330–1339. [Google Scholar]
- 36.
- 36a. Dong J., Wang M., Zhang P., Yang S., Liu J., Li X., Sun L., J. Phys. Chem. C 2011, 115, 15089–15096; [Google Scholar]
- 36b. Xu Y., Chen R., Li Z., Li A., Han H., Li C., ACS Appl. Mater. Interfaces 2017, 9, 23230–23237. [DOI] [PubMed] [Google Scholar]
- 37. Daviddi E., Chen Z., Beam Massani B., Lee J., Bentley C. L., Unwin P. R., Ratcliff E. L., ACS Nano 2019, 13, 13271–13284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information