

Abstract
We describe the operation and improvement of AlphaFold, the system that was entered by the team AlphaFold2 to the “human” category in the 14th Critical Assessment of Protein Structure Prediction (CASP14). The AlphaFold system entered in CASP14 is entirely different to the one entered in CASP13. It used a novel end‐to‐end deep neural network trained to produce protein structures from amino acid sequence, multiple sequence alignments, and homologous proteins. In the assessors' ranking by summed z scores (>2.0), AlphaFold scored 244.0 compared to 90.8 by the next best group. The predictions made by AlphaFold had a median domain GDT_TS of 92.4; this is the first time that this level of average accuracy has been achieved during CASP, especially on the more difficult Free Modeling targets, and represents a significant improvement in the state of the art in protein structure prediction. We reported how AlphaFold was run as a human team during CASP14 and improved such that it now achieves an equivalent level of performance without intervention, opening the door to highly accurate large‐scale structure prediction.
Keywords: AlphaFold, CASP, deep learning, machine learning, protein structure prediction
1. INTRODUCTION
In this paper, we describe the entry from team AlphaFold2 1 to the “human” category in the 14th Critical Assessment of Protein Structure Prediction (CASP14) and, in particular, how the system was applied and improved during the CASP assessment. The AlphaFold system used was an updated version of the one deployed in CASP‐Covid 1 and differs substantially to the version of AlphaFold deployed in CASP13. 2 , 3 The new system is based around a new neural network architecture, Evoformer, that we developed to process biological and physical information as well as a number of advances with the “structure module” that enable us to design and train a network that accurately produces a 3‐D structure as output. A full description of the method used is provided by Jumper et al. 4 Here, we will focus on the operation of the system, a very small number of manual interventions, and improvements made to the system during CASP14.
In the CASP14 assessors' ranking by summed z scores (>2.0), AlphaFold2 scored 244.0 compared to 90.8 by the next best group. 5 The system predicted high‐accuracy structures (GDT_TS 6 > 70, best of five) for 87 out of 92 domains, structures on par with experimental accuracy (GDT_TS > 90, best of five) for 58 domains, and a median GDT_TS of 92.4. This is the first time that this level of accuracy has been routinely achieved during CASP, especially on the more difficult Free Modeling targets, and represents a significant improvement in the state of the art in protein structure prediction.
2. METHODS
AlphaFold2, the model deployed in CASP14, is a deep neural network that directly processes the MSA and intermediate pairwise representations (including template information) using a new Evoformer architecture in an interleaved manner, rather than simple convolutions as in the previous AlphaFold, allowing long‐range interactions between residues. A novel rotationally and translationally equivariant neural network module was developed to directly generate the full atomic structure. The network is iterated multiple times in a “recycling” procedure to further refine the structure predictions. A full description of the model architecture and details of the training process are given by Jumper et al. 4
As well as the atomic coordinates, AlphaFold2 produces the distogram and “predicted lDDT‐Cα” (pLDDT) confidence measure as auxiliary outputs. The latter regresses the true per‐residue lDDT‐Cα 7 for the predicted structures during training.
To remove small stereochemical violations, we relaxed our predicted structures using gradient descent on Amber99sb restrained to the original prediction with harmonic restraints. This minimization produces an extremely small structure difference in most cases but removes distracting bond and steric violations.
2.1. Model selection and ranking
The CASP assessment allows competitors to submit up to five different predictions for each target. We found that generating five predictions using five sets of model parameters and then ranking by pLDDT gave the highest accuracy, as judged by the accuracy of the top ranked prediction.
2.2. Testing and monitoring
The system used during CASP14 was developed with the goal of minimizing the possibility of human mistakes leading to poor predictions, while making it possible to manually intervene, and to deploy new versions of AlphaFold during the CASP14 assessment if necessary.
Components of the model were covered by tests of individual components (unit tests), checking for issues, such as coding errors, stability, and numerical correctness. Furthermore, a daily test of the whole system (integration test) monitored for, otherwise, difficult‐to‐detect regressions in end‐to‐end model accuracy.
The system was run and analysis carried out by a single researcher, determined by a weekly rota for the duration of the CASP14 assessment. When a prediction was made, a standardized Colaboratory 8 notebook was used to visualize (using matplotlib 9 and NGL 10 ) and check multiple aspects of the prediction (Figure 1). First, the confidence expressed by the model as pLDDT for the overall structure as well as per residue was checked. High uncertainty across the entire chain was a cause for concern, though areas of uncertainty at the C‐ and N‐termini and domain‐linking loop regions could be expected, as they are expected to often be disordered and not resolved in experimental structures. Other significant areas of uncertainty could also be indicative of intrinsic disorder (rather than a poor prediction), which would not be resolved experimentally and so not scored in CASP14, though information about resolved regions is not known at the time of the prediction.
FIGURE 1.
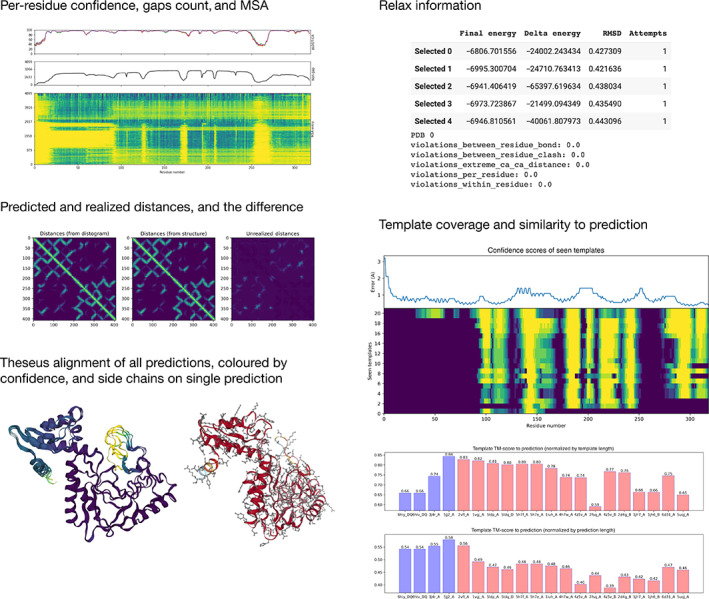
Examples of visualizations used in the prediction checking Colaboratory notebook, shown with CASP target T1101
Next, the expected distance matrix was computed from the predicted distogram. This is different from the distance matrix of the predicted structure as it is able to capture distance uncertainty, and also is not required to represent a single, concrete set of 3D coordinates. The difference between the expected distance matrix and the distance matrix of the predicted structure was examined to reveal predicted distances that were not realized in the predicted structure. This was considered an indicator of multiple conformations, and that the prediction might need manual intervention. All five predictions were aligned with TM‐align 11 against one another to check for diversity. Very high TM scores across the five models were considered problematic if we had reasons to believe either that the model should be less certain (e.g., if the pLDDT was low) or if there was a biological reason to believe that important conformations were missed as in T1024.
The result of the relaxation process was examined to check that the total energy and change in energy were negative, and that there was not a large RMSD change from the starting structure. Extreme Cɑ–Cɑ distances and inter/intra‐residue atomic clashes were also checked to ensure that there were not any stereochemical violations in the structure.
When a protein was predicted with low certainty or another issue was encountered, it was treated as both an immediate problem to find a workaround and submit an acceptable prediction, and as a research question to improve the automated system to avoid the need for manual intervention in the future. The speed of prediction with AlphaFold meant that predictions were created within a day, and usually much faster. This allowed time for a number of manual evaluations and re‐predictions, if necessary, before submitting to CASP ahead of the target deadline.
2.3. Manual interventions
Almost all of our submitted predictions in CASP14 were made using our automated system without any sort of manual intervention. In the case of a few predictions, our analysis of model quality suggested interventions (described below) that we applied to obtain the final predictions as well as improvements to AlphaFold to make the interventions unnecessary. While these sorts of manual interventions are not scalable to large sets of proteins, it is instructive to see how AlphaFold predictions could be interpreted, and see how insights led to improvements in the automated system, which does scale to large numbers of sequences without a significant difference in accuracy.
2.3.1. T1024
The target T1024 is active transporter LmrP, a member of the major facilitator superfamily, and so was expected to have both inward‐facing and outward‐facing conformations. Whether the experimental structure provided in CASP14 would be inward or outward facing was not deducible a priori, as the conformational state is influenced by biophysical context, and this information was not provided.
Our search of databases provided a large MSA (5702 alignments, good coverage) and good templates: 88 were found with sum of probabilities (sum_probs 12 ) across matching residues >100 and coverage of both domains. Low pLDDT in the linker region (Figure 2A) around residue 200 suggested flexibility between the domains. We observed a lack of diversity in our initial five predictions (all >0.99 TM score to one another), meaning that our submission would only capture one of the multiple possible conformations. We also noted that the expected distance matrix contained inter‐domain contacts unrealized in the structure (Figure 2B), indicating that the distogram possibly contained alternate conformations.
FIGURE 2.
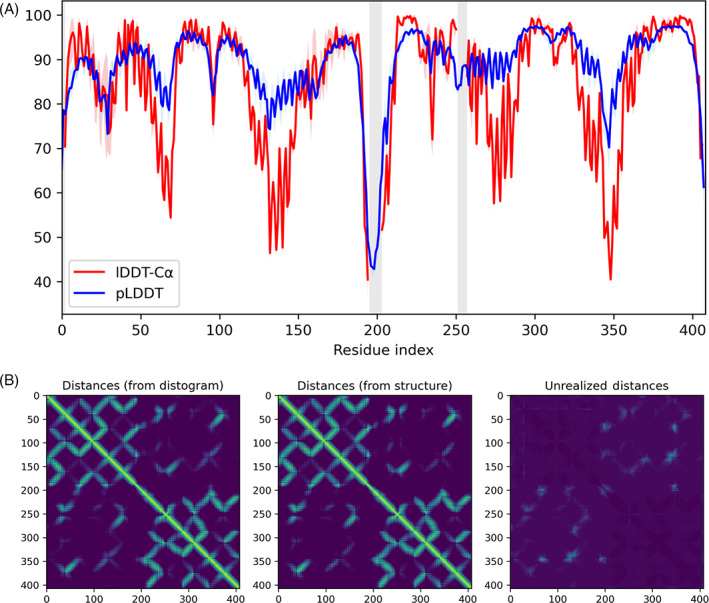
T1024: (A) Per‐residue lDDT‐Cα and pLDDT of T1024. Vertical gray shading indicates residues missing in the experimental structure, and colored shading indicates minimum and maximum values over five predictions. The pLDDT shows low confidence in the linker region indicating possible flexibility and qualitatively agrees with the true per‐residue lDDT‐Cα. (B) Unrealized distances in the expected distances of T1024 indicating possible alternate relative conformations of the two domains
We confirmed that within the set of high confidence templates found there was a diversity of inward‐facing and outward‐facing conformations. The initial predictions had high TM scores to some of these but not others, forming distinct “clusters” of templates. We attempted to force the model to produce the alternate conformation by passing in only the templates from the alternate cluster, but we found that the model largely ignored changes in the template inputs. We hypothesized this was due to the sufficiently large MSA for this target, and after removing the MSA entirely, we could force the model to follow the templates. Using pLDDT as a guide, we reintroduced the top 30 sequences (now known to be near the threshold of high confidence AlphaFold predictions) in the MSA to provide an adequate balance between following the templates and the MSA, and this successfully predicted a structure in the alternate conformation.
To automate creation of the template clusters, we clustered templates that had a value of sum_probs divided by the sequence length greater than 0.5. Clustering was performed by measuring similarity with TM score and then analyzed using scipy's cluster hierarchical linkage clustering algorithm 13 , 14 to give a maximum of three clusters. Three extra structure predictions using the three template clusters were created, in addition to the original five predictions. Predictions from the original five predictions that had high similarity (>97 LDDT) to another prediction and were lower ranked were removed and replaced with a maximally dissimilar template‐clustered prediction.
Our final submission for T1024 used two original predictions (in positions 1 and 2) and three template‐clustered predictions (in positions 3–5).
2.3.2. T1044
Targets T1031, T1033, T1035, T1037, T1039, T1040, T1041, T1042, and T1043 are all subsequences of S0A2C3, an RNA polymerase. The full sequence was included separately as a target, first as H1044 and then as T1044. We initially predicted the structures of these domains as separate sequences. In addition, to see if the extra context helped, we predicted the structure of the full 2180 amino acid sequence.
Several domain‐level targets (T1033, T1039, T1040, and T1043) were found to have very shallow MSAs, and the predictions had high uncertainty and model diversity, indicating poor model outputs. However, these domains had many more hits in their corresponding regions when using the full chain as the sequence for genetics search as opposed to per‐domain searches (Figure 3). It was also noted that the prediction for T1033 when cropped out of the full‐sequence prediction appeared to be superior to the domain‐level prediction of T1033 with higher confidence and more consistent structure.
FIGURE 3.
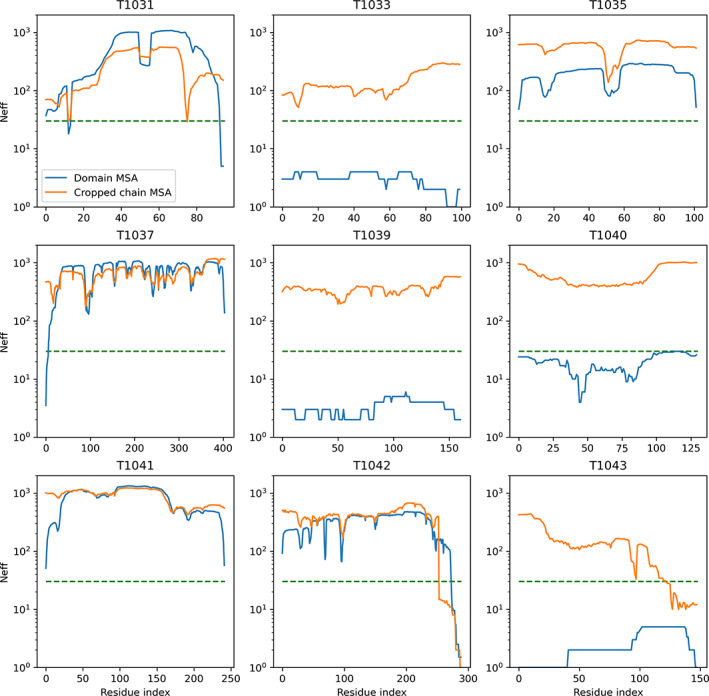
T1044: Comparison of the number of effective alignments (Neff) per residue for each MSA, derived both from domain sequences and from cropping the full sequence MSA. Four domains (T1033, T1039, T1040, and T1043) substantially benefit from using the full sequence MSA. The dashed green line shows the approximate 30 alignment threshold considered sufficient for a good prediction with AlphaFold
These results indicated that using the full sequence rather than individual domains as given by the CASP targets could give a superior prediction. However, it was noticed for T1040 that the prediction, when cropped out of the full‐sequence prediction, was lower confidence (and less compact) than when the same domain was predicted using just the domain‐level MSA. The final domain (T1043) was also nearly completely unfolded in the full‐sequence prediction. It was hypothesized that these apparent failures were “long‐chain collapse”—that the model failed to generalize to long sequence lengths due to making predictions far out of the distribution of sequence lengths that the model was trained on. We describe below both how we handled this for T1044 domain submissions and how this led to further research that ameliorated the problem in the final AlphaFold system.
To address this during the prediction window, new domain MSAs were created by cropping out the appropriate columns from the full‐sequence MSA, which captured the extra alignments that it provided. Structures were then predicted for just these domain‐sized sequences in order to avoid long‐chain collapse—“crop‐then‐fold.” The domain structure predictions using cropped full‐sequence MSAs appeared to be higher quality and more confident than the domain predictions using MSAs generated just from the domain sequence.
In order to create a full‐chain prediction that did not suffer from collapse and captured additional inter‐domain context, we passed the crop‐then‐fold domain predictions for T1031, T1033, T1041, and T1042 + T1043 (joined) as artificial templates for a full‐sequence prediction. The resulting prediction was a high quality model of the full sequence that did not suffer from collapse and gave the domains their full structural context. Domains cropped out of this model were then submitted as our predictions. Table 1A summarizes the pLDDT confidences that were used to guide the process of building these predictions.
TABLE 1.
T1044: (A) Confidence scores (pLDDT) for different prediction systems that were considered and (B) accuracy (GDT_TS) for predictions of domains in T1044
| (A) Confidence (pLDDT) a | |||||
|---|---|---|---|---|---|
| Original domain | Original full sequence | Crop‐then‐fold domain | Submitted full sequence | Final system full sequence | |
| T1031 | 80.6 | 64.4 | 84.1 | 71.9 | 69.4 |
| T1033 | 54.7 | 69.0 | 85.0 | 73.9 | 77.2 |
| T1035 | 81.7 | 78.8 | 83.2 | 77.9 | 82.6 |
| T1037 | 83.9 | 78.7 | 89.7 | 77.7 | 82.8 |
| T1039 | 75.0 | 68.2 | 89.3 | 67.9 | 71.6 |
| T1040 | 73.1 | 51.6 | 87.2 | 82.0 | 74.7 |
| T1041 | 83.7 | 78.4 | 86.6 | 77.8 | 80.0 |
| T1042 | 79.7 | 68.1 | 81.5 | 80.6 | 73.5 |
| T1043 | 37.5 | 47.4 | 79.7 | 82.8 | 64.1 |
| Average | 72.2 | 67.2 | 85.2 | 76.9 | 75.1 |
| Full sequence | N/A | 71.3 | N/A | 77.1 | 77.4 |
| (B) Accuracy (GDT_TS) b | |||||
|---|---|---|---|---|---|
| Original domain | Original full sequence | Crop‐then‐fold domain | Submitted full sequence | Final system full sequence | |
| T1031 | 87.6 | 86.6 | 86.8 | 87.1 | 88.4 |
| T1033 | 44.0 | 85.7 | 89.0 | 87.7 | 87.0 |
| T1035 | 92.6 | 94.1 | 93.1 | 94.9 | 95.8 |
| T1037 | 82.9 | 88.4 | 85.0 | 87.3 | 92.6 |
| T1039 | 79.2 | 81.4 | 78.9 | 82.3 | 82.9 |
| T1040 | 55.0 | 30.8 | 70.0 | 71.7 | 69.2 |
| T1041 | 86.6 | 89.7 | 85.8 | 90.5 | 89.6 |
| T1042 | 62.1 | 80.2 | 69.9 | 83.8 | 90.4 |
| T1043 | 16.6 | 53.2 | 76.2 | 83.3 | 82.3 |
| Average | 67.4 | 76.7 | 81.6 | 85.4 | 86.5 |
| Full sequence (TM score) | N/A | 0.807 | N/A | 0.878 | 0.960 |
T1044: Confidence scores (pLDDT) for different prediction systems that were considered. The mean full‐sequence pLDDT over a given domain cannot be directly compared to the mean pLDDT found just by folding that domain, as pLDDT will consider the effect of mispredicting inter‐domain distances as well as intra‐domain distances, which penalizes longer predictions. However, it can be seen that using “crop‐then‐fold” led to an improvement, often substantial, in confidence across all domains. The full sequence confidences of predictions made with the submitted (template‐patched) system were also superior to the original system. The final improved system gives an equivalent level of confidence to the submitted prediction.
Accuracy (GDT_TS) for predictions of domains in T1044. It can be seen that, using the original system, T1033 was predicted more accurately as part of the full chain, but T1040 was predicted more accurately when folded as an independent domain. Both crop‐then‐fold domains and submitted (template‐patched) full‐sequence predictions get the best of both worlds and give better mean domain accuracy. The final system gets equivalent performance with no complex interventions, and better chain‐level TM score.
This manual process is cumbersome so we also explored ways to improve our model to not suffer from long‐chain collapse. We found that fine‐tuning the model with 384 residue crops from the training set (256 originally) addressed the problem, producing a non‐collapsed full sequence prediction without manual intervention with quality equivalent to the template‐patched prediction. We have tested proteins up to 2700 residues in length with this fix, but expect even longer proteins (e.g., titin) to still suffer from the long‐chain collapse problem.
This improved model, the “Final system,” was used for targets T1080, T1091, and T1095 and later targets.
2.3.3. T1064
Target T1064 is the SARS‐CoV‐2 protein ORF8. The initial MSA contained very few alignments (19) and gave a low confidence prediction. ORF8 is known to be hypervariable compared to other SARS‐CoV‐2 proteins—it has only 30% identity to SARS‐1 ORF8. 15 , 16
Guided by pLDDT, we tried various MSAs using additional databases and chose the one that maximized pLDDT. We found that an extra five sequences from BLAST‐NR 17 —searched with Jackhmmer and deduplicated to have an edit distance of at least 3, to give a total of 24 aligned sequences—gave the best improvement. We also ran a model using a more recent version of UniRef90, 18 reasoning that it was likely that more Coronaviridae had been sequenced in the months since the 2020_03 release.
As with some other targets, given the high uncertainty of the structure, we were concerned that the target might have multiple viable structural modes. To address this, we developed an experimental “multi‐head” model based on the M‐heads method of Kohl et al. 19 This “multi‐head” model comprises four parallel copies of AlphaFold (including the Evoformer and Structure modules), each with identical tied parameters. However, each copy has a unique learnt embedding, which is added to the MSA embedding and pair representation on each iteration of recycling, allowing each copy to make a different prediction despite being, otherwise, identical. The loss was modified, such that the copy with the lowest total loss has weight 0.95, and the other copies have weight 0.016, thus backpropagating most of the gradients into the best model. This has the effect of allowing the model to produce a diverse set of predictions over several modes without being penalized as long as at least one head produced a good prediction.
Folding with the multi‐head model gave higher pLDDTs than the original model. However, it should be cautioned that the multi‐head training setup could lead to a bias toward high confidence. To check for this bias, we assembled a small validation set of five viral proteins with shallow MSAs. After confirming that pLDDT was rank correlated with prediction accuracy, even across different types of model, we decided to submit models ranked by pLDDT. In addition, literature evidence indicated that ORF8 has nine beta strands (Figure 2B by Tan et al. 16 ). On the validation set, we also found that higher non‐loop secondary structure percentage was indicative of higher lDDT‐Cα. We also found that higher pLDDT was correlated with higher beta strand content in ORF8, lending confidence to our ranking method.
Seeing a large range of pLDDTs produced by the multi‐head model, the multi‐head model was run five more times, creating the additional 20 structure predictions with a wide range of pLDDTs, some with substantially higher confidence. Re‐running AlphaFold multiple times can produce different predictions, especially when it is uncertain, due to the random processing (masking and clustering) of the input MSA.
Our final submission was a combination of methods, mostly ranked by pLDDT, to ensure a diversity of predictions. The top two predictions were the top‐ranked (by pLDDT) predictions from the multi‐head reruns. The next prediction was made by the original system with an updated Uniref90 database, and the fourth prediction was made by the original system without any interventions. For diversity, the final model was the third best multi‐head model prediction from our first run of it.
2.4. Other interventions
In two targets (T1074 and T1080), we found that our relaxation procedure did not remove all stereochemical violations for all five predictions. For these, we re‐ran the relaxation stage using weaker restraints until a violation‐free prediction was generated.
The multi‐head model was also used to predict the third ranked model for target T1100. The template clustering algorithm was also used for target T1057, which resulted in the substitution of the fifth ranked prediction. For T1055, models cropped out of a prediction of the full sequence (UniProt accession A0A2I6J1H4) were used for the fourth and fifth predictions.
An experimental model that used the provided homomeric stoichiometry generated the fourth and fifth predictions for T1070 and the fifth prediction for T1060s2.
3. RESULTS
Considering domain predictions by AlphaFold in CASP14 (N = 92), the mean domain GDT_TS over all five submitted models was 87.32, and over the top‐1 models was 88.01, an average increase of +0.69. This indicates that the ranking procedure was effective at selecting models that were better than the average prediction. The maximum improvement achievable by ranking would be +0.80 GDT_TS, as the mean domain GDT_TS of the best prediction from each chain is 88.81. This means that our selection procedure achieved 0.69/0.80 = 86% of the maximum improvement from model selection. We note, however, that perfect selection is likely impossible in practice since missing context may mean that the correct model is not predictable from sequence alone, such as for T1024.
In only two cases was the top ranked prediction more than five GDT_TS worse than the mean GDT_TS of the five predictions: T1030‐D1 (−6.29) and T1070‐D1 (−6.90); in three cases, the first ranked structure was more than five GDT_TS better: T1030‐D2 (+29.12), T1064‐D1 (+11.80), and T1086‐D1 (+7.95). In the case of T1030, it appears that that large improvement in placement of a helix in T1030‐D2 (present in models 1 and 2) was associated with a small deleterious change in helix placement in T1030‐D1, giving a more accurate chain overall.
Overall, our manual interventions on three predictions provided +1.14 GDT_TS top‐1 (+1.20 GDT_TS top‐5) improvement across all CASP14 domains versus the “unadjusted” automated system as it was at the time of the target release. Re‐running all targets using our automated “final” system as it was by the end of CASP14, manual interventions only gives +0.01 GDT_TS top‐1 (+0.35 GDT_TS top‐5) improvement, largely reflecting the impact of fine‐tuning AlphaFold on larger crop sizes.
The most significant gains for domain accuracy were for T1044 and T1064 (Figure 4). The individual domains of T1024 were already well predicted, but the best‐of‐five full‐sequence accuracy, which takes into account the relative placement of the domains, improved significantly from 0.682 TM score (60.8 GDT_TS) for the prediction by the original system to 0.929 (79.3 GDT_TS) for the submitted structure. The TM score was 0.965 (86.7 GDT_TS) for the final improved system.
FIGURE 4.
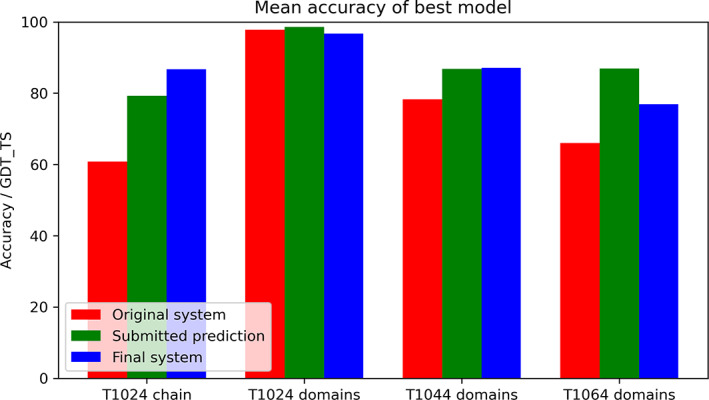
Comparison of three different prediction methods for the targets with significant interventions: “Original system” is the automated prediction system as it existed at target release. “Submitted prediction” is the submitted structure prediction. “Final system” is the automated system as it existed at the end of the CASP14 assessment, improved by experience
For T1024, in CASP14, it was revealed that, out of all submissions, our third model was the best match for the experimental structure. This was a model generated using template clustering. The experimental structure had a large ligand in the pore, forcing the outward‐facing conformation. This would be hard to predict in advance without the ligand context being provided with the protein sequence.
A recent paper 20 suggests, with the aid of distance restraints from double electron–electron resonance spectroscopy, that our top ranked structure correctly predicted an inward‐facing conformation of LmrP, for which a structure has so far not been determined experimentally.
For T1044, a full comparison of the accuracy of each prediction method on each domain is shown in Table 1B. One can see that the average domain GDT_TS is better for the submitted (templated‐patched) predictions than the original domain predictions, and that the final automated system is able to make predictions at an equivalent level of accuracy. The overall full‐sequence accuracy, as measured by TM score, is superior for the submitted prediction as compared to the original full‐sequence prediction, and the final improved system is superior to both.
For T1064, comparing all our predictions against the experimental structure of ORF8 (Figure 5), pLDDT is strongly correlated with lDDT‐Cα against the ground‐truth structures, even across different types of models. This validates our decision to select predictions by ranking by pLDDT.
FIGURE 5.
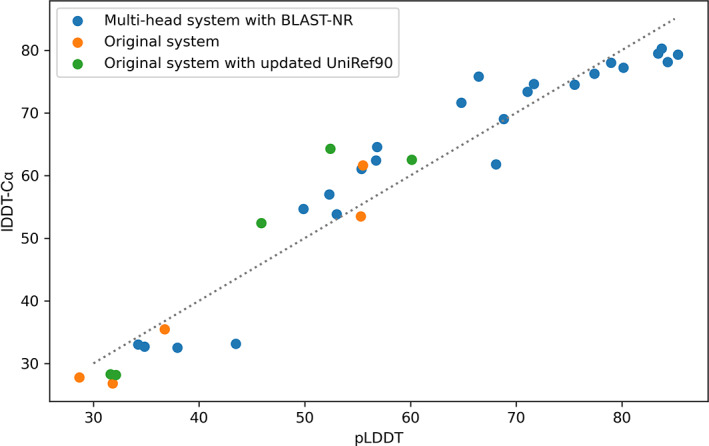
All models produced for T1064–pLDDT versus final lDDT‐Cα. The strong correlation indicates that ranking many predictions by pLDDT was a successful strategy for this target
Interventions for targets T1057, T1100, T1055, T1060s2, T1074, and T1080 had no effect on top‐1 or top‐5 accuracy metrics. The intervention for T1070 gave a small increase in top‐5 GDT_TS for T1070‐D1 (+4.28).
3.1. Lower accuracy targets
Despite overall exceptional performance, some predictions made by AlphaFold failed to reach a high level of accuracy. Analysis on a very large test set of recent PDB structures by Jumper et al. (Figure 5A) 4 and Tunyasuvunakool et al. (Figure 4D) 21 shows that when the model fails, it can often be explained by small MSA sizes or a high proportion of heterotypic contacts. The AlphaFold prediction for T1047s1‐D1 (best‐of‐5 GDT_TS = 50.47) showed a relatively unique failure mode where it captured the secondary structure of a very long beta sheet but put it at the wrong angle from the rest of the domain and improperly curled it. We speculate that the total lack of other intra‐domain structure in this region together with the very high oligomerization state, a homo‐78mer, contributed to this poor prediction.
We also note that AlphaFold can sometimes predict a single conformation with high certainty that was not the particular conformation solved by the experimentalist. This likely occurred for Model 1 of T1024, and the careful analysis of the original NMR data for T1027 by Huang et al. 22 suggests that it is possible that the AlphaFold produced a minor conformation present in the NMR data.
Finally, in some cases, discrepancies between AlphaFold and experiment will turn out to have been limitations or errors in the experimental model. In the same NMR analysis, the authors show that AlphaFold's prediction for T1055 and T1029 fit the NMR data better than the experimentalist's model. For T1029, the effect of reanalysis is quite dramatic, and the authors develop a novel procedure to reanalyze the NMR data in light of the AlphaFold model. After reanalysis, the new coordinates have a GDT_TS of 89 to the AlphaFold model submitted during CASP14.
4. DISCUSSION
In this work, we have described the process of entering AlphaFold in the CASP14 assessment. Almost all of our predictions were generated by the automated system without any intervention. As described, in a very small number of cases, we decided to adapt this system, with positive results as compared to our original prediction. We have described the investigations carried out for these cases and hope they prove instructive in how to run and use AlphaFold. In all cases, we found that the predicted lDDT‐Cα (“pLDDT”) proved to be a useful guide to understanding and improving the prediction, demonstrating the value of robust quality indicators for both manually improving models and finding opportunities to improve automated systems.
While we generally found that interventions that improve pLDDT resulted in higher accuracy models, caution should be taken to avoid a version of “Goodhart's law” by testing too many interventions and reducing the predictive power of pLDDT. We also note that low pLDDT can be an indicator of real disorder in the protein 21 rather than a modeling failure per se, especially when the protein is not, otherwise, known to fold to a compact structure.
The challenge of entering AlphaFold in CASP14 helped to make it better. We were able to use the insights gleaned from our manual interventions to improve the automated system to an equivalent level of performance. This makes it possible to create predictions at the same level of accuracy at larger scales without intervention, as demonstrated with proteome‐scale predictions by Tunyasuvunakool et al. 21
CONFLICT OF INTERESTS
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ronneberger, Russ Bates, Alex Bridgland, Simon A. A. Kohl, David Reiman, and Andrew W. Senior have filed provisional patent applications relating to machine learning for predicting protein structures. The remaining authors declare no competing interests.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/prot.26257.
ACKNOWLEDGMENTS
The authors would like to thank Alban Rrustemi, Albert Gu, Alexey Guseynov, Charlie Beattie, Craig Donner, Emilio Parisotto, Erich Elsen, Florentina Popovici, Hector Maclean, Jacob Menick, James Kirkpatrick, Jeff Stanway, Karen Simonyan, Laurent Sifre, Sam Blackwell, Shaobo Hou, Stephan Gouws, Steven Wheelwright, and Zachary Wu for their contributions; Milot Mirdita for his help with datasets; Mildred Piovesan‐Forster and Alexander Nelson for their help managing the project; and our colleagues at DeepMind for their support. Martin Steinegger acknowledges support from the National Research Foundation of Korea grant (2019R1A6A1A10073437 and 2020M3A9G7103933), and the New Faculty Startup Fund and the Creative‐Pioneering Researchers Program through Seoul National University. They would also like to thank the CASP14 organizers and the experimentalists whose structures enabled the assessment.
Jumper J, Evans R, Pritzel A, et al. Applying and improving AlphaFold at CASP14 . Proteins. 2021;89(12):1711-1721. doi: 10.1002/prot.26257
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ronneberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, Alex Bridgland, Clemens Meyer, Simon A. A. Kohl, Andrew J. Ballard, Andrew Cowie, Bernardino Romera‐Paredes, Stanislav Nikolov, Rishub Jain, and Demis Hassabis contributed equally.
Funding information National Research Foundation of Korea, Grant/Award Numbers: 2019R1A6A1A10073437, 2020M3A9G7103933; Seoul National University, Grant/Award Numbers: Creative‐Pioneering Researchers Program, New Faculty Startup Fund
Endnote
AlphaFold v2.0. For expediency, we refer to this model simply as AlphaFold throughout the rest of this article.
Contributor Information
John Jumper, Email: jumper@google.com.
Demis Hassabis, Email: demishassabis@google.com.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available from https://www.predictioncenter.org/casp14/. Unsubmitted predictions and auxiliary data are not shared.
REFERENCES
- 1. Kryshtafovych A, Moult J, Billings WM, et al. Modeling SARS‐CoV2 proteins in the CASP‐commons experiment. In review (2021). [DOI] [PMC free article] [PubMed]
- 2. Senior AW, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577:706‐710. [DOI] [PubMed] [Google Scholar]
- 3. Senior AW, Evans R, Jumper J, et al. Protein structure prediction using multiple deep neural networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins: Struct Funct Bioinf. 2019;87:1141‐1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical assessment of methods of protein structure prediction (CASP)—round XIV. In review (2021). [DOI] [PMC free article] [PubMed]
- 6. Zemla A. LGA – a method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31:3370‐3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mariani V, Biasini M, Barbato A, Schwede T. lDDT: a local superposition‐free score for comparing protein structures and models using distance difference tests. Bioinformatics. 2013;29:2722‐2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bisong E. Google Colaboratory. Building Machine Learning and Deep Learning Models on Google Cloud Platform: A Comprehensive Guide for Beginners. Apress; 2019:59‐64. [Google Scholar]
- 9. Hunter JD. Matplotlib: a 2D graphics environment. IEEE Ann Hist Comput. 2007;9:90‐95. [Google Scholar]
- 10. Rose AS, Bradley AR, Valasatava Y, Duarte JM, Prlić A, Rose PW. Web‐based molecular graphics for large complexes. Paper presented at: Proceedings of the 21st International Conference on Web3D Technology. Association for Computing Machinery; 2016:185‐186.
- 11. Zhang Y, Skolnick J. TM‐align: a protein structure alignment algorithm based on the TM‐score. Nucleic Acids Res. 2005;33:2302‐2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Steinegger M, Meier M, Mirdita M, Vöhringer H, Haunsberger SJ, Söding J. HH‐suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019;20:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Virtanen P, Gommers R, Oliphant TE, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. 2020;17:261‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Müllner D. Modern hierarchical, agglomerative clustering algorithms. arXiv [stat.ML] (2011).
- 15. Ceraolo C, Giorgi FM. Genomic variance of the 2019‐nCoV coronavirus. J Med Virol. 2020;92:522‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tan Y, Schneider T, Leong M, Aravind L, Zhang D. Novel immunoglobulin domain proteins provide insights into evolution and pathogenesis of SARS‐CoV‐2‐related viruses. MBio. 2020;11(3):e00760‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sayers EW, Barrett T, Benson DA, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2019;47:D23‐D28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, UniProt Consortium . UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015;31:926‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kohl SAA, Romera‐Paredes B, Meyer C, et al. A probabilistic U‐net for segmentation of ambiguous images. arXiv [cs.CV] (2018).
- 20. del Alamo D, Govaerts C, Mchaourab HS. AlphaFold2 predicts the inward‐facing conformation of the multidrug transporter LmrP. Proteins. 2021. ;89(9):1226–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tunyasuvunakool K, Adler J, Wu Z, et al. Highly accurate protein structure prediction for the human proteome. Nature. 2021;596:590‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang YJ, Zhang N, Bersch B, et al. Assessment of Prediction Methods for Protein Structures Determined by NMR in CASP14: Impact of AlphaFold2.2021;89(12):1959–1976. 10.1002/prot.26246 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are openly available from https://www.predictioncenter.org/casp14/. Unsubmitted predictions and auxiliary data are not shared.