

Abstract
Background and purpose
Using the treatment goal of “no evidence of disease activity” (NEDA) incorporating magnetic resonance imaging (MRI) re‐baselining, we aimed to assess the efficacy of ocrelizumab in patients with relapsing‐remitting multiple sclerosis with a prior suboptimal response, defined by MRI or relapse criteria, to one or two disease‐modifying therapies (DMTs).
Methods
CASTING was a prospective, international, multicenter, single‐arm, open‐label phase 3 trial (NCT02861014). Patients (Expanded Disability Status Scale [EDSS] score ≤ 4.0, with discontinued prior DMT of ≥6 months duration due to suboptimal disease control) received intravenous ocrelizumab 600 mg every 24 weeks for 96 weeks. The primary endpoint was NEDA (defined as absence of relapses, disability progression, and inflammatory MRI measures, with prespecified MRI re‐baselining at Week 8) over 96 weeks.
Results
A total of 680 patients were enrolled, 167 (24.6%) based on MRI activity only. At Week 96, 74.8% (95% confidence interval [CI] 71.3–78.0, n/N = 492/658) of patients had NEDA. NEDA was highest among patients enrolled due to MRI activity alone (80.6% [95% CI 68.6–89.6], n/N = 50/62) versus those enrolled for relapse (75.1% [95% CI 69.0–80.6], n/N = 172/229) or for relapse with MRI (70.5% [95% CI 60.0–79.0], n/N = 74/105). NEDA across subgroups was highest in patients with a baseline EDSS score <2.5 (77.2% [95% CI 72.8–81.2], n/N = 315/408). NEDA was higher in patients receiving one prior DMT (77.6% [95% CI 73.2–81.6], n/N = 312/402) versus two prior DMTs (70.3% [95% CI 64.3–75.8], n/N = 180/256).
Conclusions
In patients switching therapy due to suboptimal disease control, treatment with ocrelizumab led to an overall high NEDA rate across a wide range of disease‐related and demographic subgroups, regardless of prior treatment background, with no new safety signals detected.
Keywords: disease activity, disease‐modifying therapies, multiple sclerosis, ocrelizumab, relapsing‐remitting multiple sclerosis
Data from the phase IIIb CASTING trial in patients with relapsing‐remitting multiple sclerosis with a suboptimal response to one or two prior DMTs shows that switching to ocrelizumab led to a high proportion of patients with no evidence of disease activity (NEDA; defined as absence of relapses, disability progression, and inflammatory MRI measures, with prespecified magnetic resonance imaging re‐baselining at Week 8) at the end of the 96‐week treatment period.
![]()
INTRODUCTION
Patients with relapsing‐remitting multiple sclerosis (RRMS) often experience disease activity despite receiving a disease‐modifying therapy (DMT) [1, 2, 3]. Ocrelizumab, a recombinant humanized monoclonal antibody that selectively depletes CD20‐expressing B cells while preserving the capacity for B‐cell reconstitution and pre‐existing humoral immunity [4, 5], showed significant benefit on clinical and magnetic resonance imaging (MRI) measures of disease activity and disability progression in pivotal phase 3 studies in patients with relapsing multiple sclerosis (RMS) [6] or primary progressive multiple sclerosis (PPMS) [7]. The efficacy of ocrelizumab was sustained in the respective open‐label extension periods [8, 9].
In the double‐blind period of the pivotal studies, the majority of ocrelizumab‐treated patients (73.3% in OPERA I and OPERA II in RMS [6] [n = 605/825]; 88.7% in ORATORIO in PPMS [7] [n = 433/488]) were not receiving DMT at enrollment, and direct data on the efficacy and safety of ocrelizumab in patients with a suboptimal response to prior DMTs are limited. In those patients who had received prior DMTs in OPERA I and OPERA II, these were mainly limited to contemporaneous therapies, that is, interferon (IFN)‐based regimens (n = 161/220 [73%]) or glatiramer acetate (n = 77/220 [35%]) [6].
CASTING (NCT02861014) was a European‐based phase 3b trial to evaluate the efficacy and safety of ocrelizumab, designed specifically for patients with RRMS with a suboptimal response to one or two prior DMTs (a parallel study, CHORDS, was conducted in North America; NCT02637856). The CASTING study aims to assess the proportion of patients who have no evidence of disease activity (NEDA), which has become an important treatment goal for patients with multiple sclerosis (MS) [10]. Importantly, CASTING incorporates MRI re‐baselining at Week 8 to exclude MRI activity that occurs during the first 8 weeks of treatment before the potential treatment benefit of ocrelizumab is realized, based on results from the phase II study [11].
METHODS
Study design and procedures
CASTING (NCT02861014) was a prospective, international, multicenter, interventional, open‐label phase 3b study investigating the efficacy and safety of ocrelizumab in patients with RRMS who had a suboptimal response to an adequate course of DMT (Figure 1). Patients entered a screening period (up to 4 weeks), after which eligible patients received an intravenous infusion of ocrelizumab 600 mg every 24 weeks throughout the 96‐week open‐label treatment period (last dose on Week 72) for a maximum of four doses (first dose administered as two 300‐mg infusions, 14 days apart). Expanded Disability Status Scale (EDSS) score, relapse, and MRI assessments were conducted at least at baseline and at Weeks 24, 48, and 96 (NB, an additional MRI assessment was made at Week 8 for MRI re‐baselining purposes).
FIGURE 1.
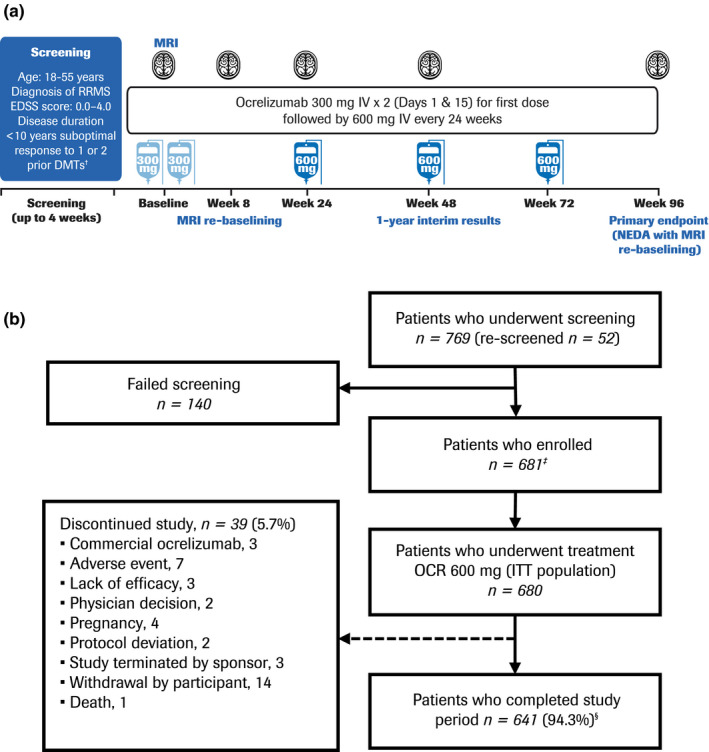
Study design (a) and patient disposition (b). †Patients were treated with a disease‐modifying therapy (DMT) for at least 6 months and with suboptimal efficacy within the last 12 months whilst on the DMT. ‡One patient discontinued before receiving treatment. §Of patients who completed the study treatment period, 154 discontinued for “other reasons” before safety follow‐up (of whom 152 took commercial ocrelizumab); 438 rolled over into the long‐term extension study (one additional patient also rolled over); 40 withdrew consent and did not enter safety follow‐up (of whom 25 took commercial ocrelizumab); eight patients entered safety follow‐up; and one patient discontinued before safety follow‐up by “physician decision” (this patient also took commercial ocrelizumab). Note: The per‐protocol (PP) population consisted of 560 patients. Reasons for exclusion from the PP population were: full course of ocrelizumab treatment not completed (n = 37); major protocol deviation deemed to potentially affect the efficacy endpoints (n = 56); and magnetic resonance imaging (MRI) performed after ocrelizumab infusion (n = 80). EDSS, Expanded Disability Status Scale; ITT, intention‐to‐treat; IV, intravenous; NEDA, no evidence of disease activity; OCR, ocrelizumab; RRMS, relapsing‐remitting multiple sclerosis
Patients were encouraged to enrol in a separate long‐term extension study at the end of the treatment period to further evaluate the efficacy and safety of ocrelizumab. Patients not enrolling in the long‐term extension study and those who discontinued treatment early entered a safety follow‐up for at least 96 weeks. Patients who discontinued and switched to commercially marketed ocrelizumab or other B‐cell‐depleting therapies, either after completion or early discontinuation of the 96‐week treatment period, were not allowed to enter the safety follow‐up period. The trial protocol (NCT02861014) was approved by the relevant institutional review boards/ethics committees. All patients provided written informed consent.
Patients
Key eligibility criteria included: age 18–55 years; diagnosis of RRMS according to the McDonald 2010 criteria [12]; EDSS score of 0.0–4.0 inclusive at screening; disease duration < 10 years from first symptom (if date of first symptom was unknown, then the diagnosis of RRMS was ≤ 5 years prior to enrollment); and prior suboptimal response to one or two DMTs. A suboptimal response was defined as: one or more clinically reported relapse(s); or one or more T1‐weighted contrast‐enhancing lesion(s) (CELs); or two or more new and/or enlarging T2‐weighted hyperintense lesions on MRI while the patient was receiving a stable dose of that DMT for at least 6 months; hence, patients could be enrolled where detection of a suboptimal response to DMT was based on MRI criteria only. In patients receiving stable doses of the same approved DMT for more than 1 year, at least one of the above events must have occurred within the last 12 months of treatment with this DMT. Patients were not withdrawn from DMTs for the sole purpose of participation in the study; discontinuation and washout of DMTs was guided by local prescribing labels. Exclusion criteria included previous treatment with natalizumab (to avoid risk of progressive multifocal leukoencephalopathy) unless natalizumab was discontinued because of persistent anti‐natalizumab antibodies.
Outcomes
The primary efficacy endpoint was the proportion of patients with NEDA (with MRI re‐baselining at Week 8) during the 96‐week treatment period. NEDA was defined as an absence of protocol‐defined relapse (PDR; defined as occurrence of new or worsening neurological symptoms that were attributable to MS), 24‐week confirmed disability progression (CDP; defined as an increase in EDSS score from baseline of at least 1.0 point), CELs, and new and/or enlarging T2‐weighted hyperintense lesions. An analysis of the proportion of patients with NEDA during the initial 24‐week and 48‐week periods was conducted as a secondary endpoint.
Additional secondary study endpoints included: time to the first event of disease activity (defined as the occurrence of the first event on any NEDA component); time to onset of the first PDR, 24‐week CDP, and on brain MRI, first new and/or enlarging T2‐weighted hyperintense lesion; mean change in EDSS score (from baseline to Week 96); proportion of patients with CDP, stable disability, or confirmed disability improvement (CDI; defined as a reduction in EDSS score ≥ 1.0 point compared with baseline, in patients with a baseline EDSS score ≥ 2); annualized rate of PDRs at Week 96 (ARR; calculated as the total number of PDRs for all patients divided by the total patient‐years of exposure to that treatment); total number of CELs (Weeks 24, 48, and 96); change in total T2‐weighted hyperintense lesion volume (Weeks 8–96); volume and number of new and/or enlarging T2‐weighted hyperintense lesions (Week 8 to Weeks 24, 48, and 96); and percentage changes in cerebral whole‐brain volume (WBV), white matter volume (WMV) and cortical gray matter volume (CGMV; Weeks 24, 48, and 96). Brain volume change was assessed using the percentage change from Week 8 in WBV using SIENA/X [13] and the percentage change in WMV and CGMV using paired Jacobian integration [14].
The primary and secondary endpoints were analyzed by subgroup (when n > 50) relating to patient demographics (age [≤ 40/> 40 years]), disease characteristics (EDSS score at baseline [< 2.5/≥ 2.5], number of relapses over the year prior to inclusion [≤ 1/> 1], time of the event leading to enrollment in relation to screening [<6 months/≥ 6 months]), reason for enrollment (MRI only, relapse only, MRI with relapse), and prior DMT before enrollment (IFNs, glatiramer acetate, teriflunomide, fingolimod, dimethyl fumarate, natalizumab).
Exploratory endpoints included the proportion of patients with NEDA during the treatment period (MRI from screening); predictors of NEDA and association with disability or other efficacy variables; and an epoch analysis of NEDA (Year 1 [Weeks 0–48] and Year 2 [Weeks 48–96]). Analyses of the proportion of patients with NEDA (with MRI re‐baselining at Week 8) by number of prior DMTs (1 vs. 2) were assessed post hoc.
Safety
Safety outcome measures included the incidence and nature of adverse events (AEs), serious AEs, discontinuations for AEs, vital sign measurements, physical and neurological examinations, clinical laboratory tests, locally reviewed MRI for safety (non‐MS central nervous system pathology), and concomitant medications. The severity of AEs was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events.
Statistical analyses
The analysis of this single‐arm noncomparative study was exploratory, and primarily based on descriptive statistical methods. No formal hypothesis was tested. The efficacy analyses were performed on the intention‐to‐treat (ITT) population, which included all enrolled patients who received any dose of ocrelizumab, including those who prematurely withdrew and did not undergo any assessments. A modified ITT population was defined as all patients from the ITT population excluding patients with both screening and baseline EDSS scores missing. The safety population consisted of all patients who received at least one dose of ocrelizumab.
For the primary outcome, the proportion of patients with NEDA during the 96‐week treatment period in the modified ITT population, descriptive statistics with the corresponding two‐sided Clopper‐Pearson 95% confidence interval (CI) were used; patients receiving any dose and who discontinued early without a protocol‐defined event were imputed as having an event if the treatment discontinuation reason was lack of efficacy, or death; others were excluded. The same approach as for primary endpoint analysis was used for secondary NEDA endpoints. The time to first protocol‐defined event of disease activity used Kaplan–Meier analysis, with Greenwood 95% CI using log–log transformation (for NEDA); patients who discontinued the study due to lack of efficacy, or death without confirmed progression of disease were imputed as having an event at the time of discontinuation. For the primary outcome, an additional analysis to identify baseline factors prognostic for NEDA at Week 96 used a logistic regression model, adjusted by duration since MS symptom onset (≤ 3 years, > 3 and ≤ 5 years, > 5 years and ≤ 10 years, > 10 years), T2‐weighted hyperintense lesion volume at Week 8, presence of T1‐weighted CELs at screening, presence of relapses prior to screening as per the case report form (Yes/No), number of previous DMTs (1, > 1), baseline EDSS, and gender.
Expanded Disability Status Scale score and mean change from baseline in EDSS score were analyzed using the longitudinal mixed‐effect model of repeated measures (MMRM). The proportion of patients who had CDP or CDI was analyzed using the ITT population. The ARR analysis (adjusted for duration since MS symptom onset, presence of T1 gadolinium‐enhancing lesions, presence of relapses prior to screening as per the case report form, number of previous DMTs, and log‐transformed years of drug exposure time) used a Poisson model and the ITT population. The total number of CELs and number of new or enlarging T2‐weighted hyperintense lesions were analyzed using a Poisson model and the ITT population. Descriptive analyses, change, and percentage change summary were used for volume of new or enlarging T2‐weighted hyperintense lesions in the ITT population. The percentage changes in WBV, CGMV, and WMV were analyzed using the MMRM and the ITT population.
RESULTS
The CASTING study (9 September 2016 to 25 October 2019) enrolled 681 of 769 screened patients, across 157 centers in 16 participating countries across Europe and Turkey. One patient who did not receive treatment was discontinued from the study and excluded from the ITT and modified ITT populations (n = 680 for both). A total of 641/680 patients (94.3%) completed treatment and 39/680 patients (5.7%) withdrew from study treatment prematurely (Figure 1). Baseline demographic data and disease characteristics are provided in Table 1. Patients were most frequently enrolled for reason of relapse (35.0%, n = 238/680), although 24.6% (n = 167/680) were enrolled due to MRI activity only. More patients had received one prior DMT (60.4%, n = 411/680) than two prior DMTs (39.6%, n = 269/680), with IFN‐based regimens being the most commonly used DMT in patients pretreated with one DMT (29.1%, n = 198/680) as well as the most commonly used first DMT (70.6%, n = 190/269) in patients who had received two prior DMTs. The overall mean (standard deviation [SD]) time from the last DMT treatment to the first ocrelizumab infusion was 1.88 (2.07) months, the washout period was based on the local label. Disease characteristics in patients who had received two DMTs prior to enrollment reflected a greater degree of disease progression than in those who had previously received only one prior DMT. Patients who received fingolimod typically had disease characteristics reflecting more advanced disease relative to other last prior DMTs.
TABLE 1.
Baseline demographics, disease characteristics, and prior disease‐modifying therapies
| Parameter | ITT population (N = 680) |
|---|---|
| Age, mean (SD), years | 34.2 (8.6) |
| Age ≤40/>40 years, n (%) | 511 (75.1)/169 (24.9) |
| Women, n (%) | 436 (64.1) |
| White, n (%) a | 625 (91.9) |
| BMI, mean (SD), kg/m2 | 25.0 (5.4) |
| Reason for enrollment, n (%) | |
| MRI activity only | 167 (24.6) |
| Relapse activity only | 238 (35.0) |
| MRI + relapse activity | 275 (40.4) |
| Duration since MS symptom onset, mean (SD), years | 5.0 (2.7) b |
| Duration since RMS diagnosis, mean (SD), years | 3.7 (2.5) |
| Number of relapses in last year, mean (SD) | 1.2 (0.9) c |
| Patients with ≤1 /> 1 relapse in the last year, n (%) | 488 (71.9)/191 (28.1) c |
| Baseline EDSS score, mean (SD) d | 2.1 (1.1) |
| Patients with EDSS score <2.5/≥2.5, n (%) | 422 (62.1)/258 (37.9) |
| Number of T1‐weighted CELs, mean (SD) e | 0.26 (0.88) |
| Patients with 0 lesion, n (%) | 566 (85.9) |
| Patients with 1 lesion, n (%) | 62 (9.4) |
| Patients with 2 lesions, n (%) | 12 (1.8) |
| Patients with 3 lesions, n (%) | 6 (0.9) |
| Patients with ≥ 4 lesions, n (%) | 13 (2.0) |
| Number of T2‐weighted hyperintense lesions, mean (SD) e | 43.88 (4120) f |
| Volume of T2‐weighted hyperintense lesions, mean (SD), µl e | 6133.48 (7840.24) g |
| Normalized brain volume, mean (SD), cm3 | 1436.88 (83.58) h |
| Duration of last DMT, mean (SD), months | 26.5 (20.6) |
| Duration between last DMT and OCR initiation, mean (SD), months i | 1.9 (2.1) |
| Patients pretreated with one/two unique DMTs, n (%) | 411 (60.4)/269 (39.6) |
| Last prior DMT, n (%) | |
| Interferon‐based regimens j | 198 (29.1) |
| Dimethyl fumarate | 168 (24.7) |
| Fingolimod | 129 (19.0) |
| Glatiramer acetate | 116 (17.1) |
| Teriflunomide | 65 (9.6) |
| Natalizumab | 4 (0.6) |
Baseline EDSS score is defined as the average of the EDSS scores at the screening and baseline visits.
Abbreviations: BMI, body mass index; CEL, contrast‐enhancing lesion; DMT, disease‐modifying therapy; EDSS, Expanded Disability Status Scale; IFN, interferon; ITT, intention‐to‐treat; MS, multiple sclerosis; OCR, ocrelizumab; RMS, relapsing multiple sclerosis; SD, standard deviation.
More than one response was possible.
N = 670.
N = 679.
Screening EDSS score if baseline EDSS score was not available.
At the Week‐8 baseline reset, N = 659 for lesion groups.
N = 668.
N = 669.
N = 635.
N = 647.
Includes IFN β‐1a (n = 134), IFN β‐1b (n = 43), and pegylated IFN β‐1a (n = 21).
Most patients (74.8% [95% CI 71.3–78.0], n/N = 492/658) had NEDA at the end of the treatment period (Week 96) when MRI outputs were re‐baselined at Week 8 (primary endpoint; Figure 2); 80.4% (95% CI 77.2–83.4, n/N = 529/658) of patients had no evidence of clinical activity (absence of PDR and 24‐week CDP) and 91.5% (95% CI 89.1–93.5, n/N = 602/658) of patients were free from MRI activity (absence of CELs and new/enlarging T2‐weighted hyperintense lesions). Comparable NEDA rates in the per‐protocol population confirmed the robustness of the primary analysis (76.6% [95% CI 79.2–80.1], n/N = 429/560). NEDA rates during the initial 24‐week (87.1% [95% CI 84.3–89.5], n/N = 586/673) and 48‐week periods (82.6% [95% CI 79.5–85.4], n/N = 549/665; secondary endpoints) were also high, and a higher proportion of patients had NEDA in Year 2 (87.0% [95% CI 84.2–89.5], n = 571/656) than in Year 1 (82.6% [95% CI 79.5–85.4], n/N = 549/665).
FIGURE 2.
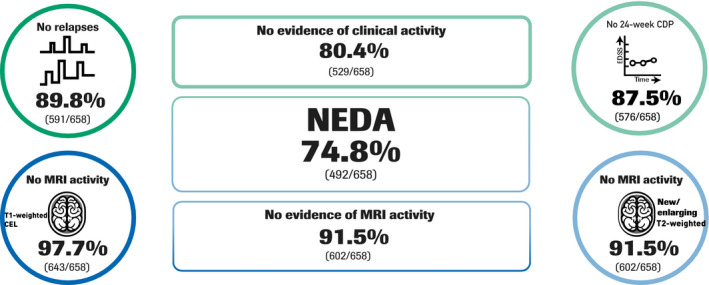
No evidence of disease activity (NEDA) at Week 96, with magnetic resonance imaging (MRI) re‐baselining at Week 8. Data are per prespecified primary analysis. CDP, confirmed disability progression; T1w‐CEL, T1‐weighted contrast‐enhancing lesion; T2w, T2‐weighted hyperintense lesions
The rate of NEDA (with re‐baselined MRI) was highest in patients enrolled due to MRI activity only (80.6% [95% CI 68.6–89.6], n/N = 50/62) versus patients enrolled for relapse (75.1% [95% CI 69.0–80.6], n/N = 172/229) or relapse with MRI (70.5% [95% CI 60.0–79.0], n/N = 74/105), and those with a baseline EDSS score < 2.5 (77.2% [95% CI 72.8–81.2], n/N = 315/408), ≤ 1 relapse prior to enrollment (78.2% [95% CI 74.2–81.9], n/N = 370/473) and the event leading to enrollment occurring ≥ 6 months prior to study entry (75.8% [95% CI 68.9–81.9], n/N = 135/178) versus their counterpart subgroups (Figure 3). The NEDA rate (with re‐baselined MRI) did not vary by age (≤ 40 years: 74.7% [95% CI 70.6–78.5],n/N = 366/490; > 40 years: 75.0% [95% CI 67.7–81.3], n/N = 126/168) or by T1‐weighted CEL status at baseline (absent: 75.8% [95% CI 72.0–79.3], n/N = 416/549; present: 73.0% [95% CI 62.6–81.9], n/N = 65/89 [Figure 3]). NEDA rates (with re‐baselined MRI) were higher in patients receiving one prior DMT (77.6% [95% CI 73.2–81.6], n/N = 312/402) versus two prior DMTs (70.3% [95% CI 64.3–75.8], n/N = 180/256) and remained generally high when stratified by the last prior DMT received before enrollment. NEDA rates were highest in patients receiving IFN‐based regimens as the last DMT (81.1% [95% CI 74.7–86.4], n/N = 154/190) and lowest in those receiving fingolimod (68.9% [95% CI 59.8–76.9], n/N = 84/122 [Figure 3]). A smaller proportion of patients had NEDA with MRI from screening (52.0% [95% CI 48.2–55.9], n/N = 346/665) versus Week 8 (74.8% [95% CI 71.3–78.0], n/N = 492/658), which was attributable to there being a lower proportion of patients with no new/enlarging T2‐weighted hyperintense lesions after re‐baselining at Week 8 (Week 8: n = 424/665 [63.8%]; Week 96: n = 602/658 [91.5%], respectively). Only gender was found to be a potential prognostic factor of NEDA and disability or other efficacy parameters (women vs. men: odds ratio 0.65 [95% CI 0.43–0.97]; p = 0.037 [Table S1]).
FIGURE 3.
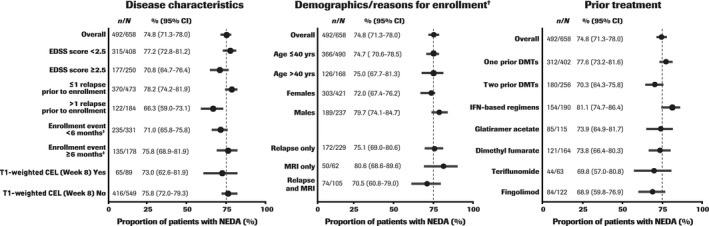
No evidence of disease activity (NEDA) subgroup analyses at Week 96 (magnetic resonance imaging [MRI] re‐baselined at Week 8). †Enrollment based on McDonald 2010 criteria. ‡Event timing in relation to study entry. Dashed line represents NEDA rate in the overall intention‐to‐treat population. CEL, contrast‐enhancing lesion; CI, confidence interval; DMT, disease‐modifying therapy; EDSS, Expanded Disability Status Scale; IFN, interferon
For secondary clinical outcomes, the Kaplan–Meier estimates of event‐free rates at intervals over the study duration are shown in Table 2. At Week 96, the chance of not having had an event of disease activity (event‐free rate) was 76.3% (95% CI 72.81–79.31, patients at risk n = 394) for first protocol‐defined event of disease activity, 88.4% (95% CI 85.7–90.6, patients at risk n = 439) for 24‐week CDP and 90.0% (95% CI 87.5–92.0, patients at risk n = 457) for first PDR, whereas in the population of patients with a baseline EDSS score ≥ 2, the chance of having had an event of 24‐week CDI was 16.5% (95% CI 13.1–20.5, n = 241). Mean (SD) EDSS scores over the treatment period were stable (baseline: 2.09 [1.06] vs. Week 96: 2.09 [1.30]; additional intervals shown in Table 2); this was reflected in the proportion of evaluable patients (n = 640; only patients with non‐missing values) with no change from baseline in EDSS score (72.2%, n = 462/640; change ≤ 0.5 and ≥ −0.5) at Week 96, while 13.4% (n = 86/640) had worse (> 0.5) and 14.4% (n = 92/640) had improved (< −0.5) EDSS scores. Over the study duration, a total of 79 PDRs were recorded; at Week 96 the adjusted ARR was 0.030 (95% CI 0.02–0.04).
TABLE 2.
Changes in clinical and magnetic resonance imaging parameters and event‐free rates over the duration of the CASTING study
| Parameter | Ocrelizumab (N = 680) | ||||
|---|---|---|---|---|---|
| Baseline | Week 24 | Week 48 | Week 72 | Week 96 | |
| EDSS score a (mITT), mean (SD), n | 2.09 (1.06) 680 | 2.08 (1.15) 679 | 2.11 (1.28) 665 | 2.07 (1.29) 650 | 2.09 (1.30) 640 |
| Change in EDSS b score from baseline (mITT), adjusted mean (95% CI), n | ‐ | −0.02 (−0.11, 0.07) 679 | 0.00 (−0.10, 0.10) 665 | −0.02 (−0.12, 0.08) 650 | 0.00 (−0.10, 0.10) 640 |
| Time to onset of clinical measure, KM event‐free rate estimate %, (95% CI) patients at risk, n | |||||
| First protocol‐defined event of disease activity (ITT) | ‐ | 91.45 (89.08, 93.32) 618 | 85.50 (82.61, 87.94) 575 | ‐ | 76.25 (72.81, 79.31) 394 |
| 24‐week CDP (ITT) | ‐ | 98.08 (96.71, 98.88) 661 | 95.25 (93.35, 96.62) 636 | ‐ | 88.39 (85.68, 90.62) 439 |
| 24‐week CDI (mITT), baseline EDSS score ≥ 2 | ‐ | 95.99 (93.54, 97.52) 385 | 89.96 (86.56, 92.53) 358 | ‐ | 83.53 (79.49, 86.85) 241 |
| First PDR (ITT) | ‐ | 94.70 (92.73, 96.15) 639 | 93.51 (91.38, 95.13) 625 | ‐ | 90.00 (87.47, 92.04) 457 |
| MRI measures, adjusted rate (95% CI), n | |||||
| Number of T1‐weighted CELs (ITT) c | ‐ | 0.004 (0.001, 0.014) 665 | 0.004 (0.001, 0.011) 658 | ‐ | NR d |
| Number of new and/or enlarging T2‐weighted hyperintense lesions (ITT) c | ‐ | 0.053 (0.038, 0.075) 671 | 0.009 (0.004, 0.017) 666 | ‐ | 0.011 (0.006, 0.020) 636 |
Abbreviations: CEL, contrast‐enhancing lesion; CDI, confirmed disability improvement; CI, confidence interval; CDP, confirmed disability progression; CRF, case report form; DMT, disease‐modifying therapy; EDSS, Expanded Disability Status Scale; ITT, intention‐to‐treat; KM, Kaplan–Meier; mITT, modified intention‐to‐treat; MMRM, mixed‐effect model of repeated measures; MRI, magnetic resonance imaging; MS, multiple sclerosis; NR, not recorded; PDR, protocol‐defined relapse; SD, standard deviation.
Baseline EDSS score defined as the average of the EDSS scores at the screening and baseline visits. If one of the EDSS scores from screening or baseline visits was missing, the other was used for baseline EDSS.
Estimates are from analysis based on MMRM using unstructured covariance matrix: change = visit + baseline EDSS score + duration since MS symptom onset (≤3 years, >3 and ≤5 years, >5 and ≤10 years, >10 years) + presence of T1 gadolinium‐enhanced lesions at screening (Yes/No) + number of previous DMTs (1, >1) + enrollment reasons (relapse only, MRI only, or both relapse and MRI).
The total number of T1 gadolinium‐enhanced or new and/or enlarging T2‐weighted hyperintense lesions for all patients divided by the total number of brain MRI scans, adjusted by duration since MS symptom onset (continuous variable), presence of T1 gadolinium‐enhanced lesions at screening (Yes/No), presence of relapses prior to screening as per CRF (Yes/No) and number of previous DMTs (= 1 vs. > 1). The total number of new and/or enlarging T2‐weighted hyperintense lesions adjusted rate for Weeks 24, 48, and 96 combined was 0.032 (95% CI 0.025, 0.042). The total number of T1 gadolinium‐enhanced lesions adjusted rate for Weeks 24, 48, and 96 combined was 0.004 (95% CI 0.002, 0.007).
Due to the very low number of T1‐weighted CELs at Week 96, the Poisson model did not converge.
A near‐complete suppression of MRI lesional activity was observed, with most patients having no CELs and no new and/or enlarging T2‐weighted hyperintense lesions from Weeks 8 to 96 (Figure S1). A total of 14 CELs were detected by brain MRI at Week 24 in all patients/brain MRIs assessed (n = 665), six at Week 48 (n = 658), and one at Week 96 (n = 629); adjusted CEL rates were low and stable over time (Table 2). There were 39 new and/or enlarging T2‐weighted hyperintense lesions at Week 24 (n = 671), 13 at Week 48 (n = 666), and 14 at Week 96 (n = 636); adjusted rates were low and decreased over time (Table 2). The mean (SD) T2‐weighted hyperintense lesion volume was 6133.5 (7840.2) µl at Week 8 (n = 669) and 5518.2 (7178.6) µl at Week 96 (n = 632); the total T2‐weighted hyperintense lesion volume decreased over time with a mean (SD) change from baseline to Week 96 (n = 624) of −558.6 (1194.6) μl and a mean (SD) percentage change of −8.5 (18.2)%. Normalized brain volume decreased over time, with mean percentage change from Week 8 baseline of –0.15% (95% CI −0.26, −0.05) at Week 24, –0.45% (95% CI −0.56, −0.33) at Week 48, and –0.81% (95% CI −0.94, −0.67) at Week 96. The mean percentage change from Week 8 baseline (95% CI) in WMV was –0.38% (–0.61, –0.15) at Week 48 and –0.75% (–0.98, –0.51) at Week 96, and in CGMV was –0.31% (–0.61, –0.02) at Week 48 and –0.62% (–0.93, –0.31) at Week 96.
Table 3 summarizes the incidence of AEs among all patients receiving ocrelizumab in CASTING. No new safety signals were identified. A total of 606 of 680 patients (89.1%) reported AEs. The most common AEs (occurring in > 10% of patients) were infusion‐related reactions (IRRs), nasopharyngitis, headache, influenza, and urinary tract infection. Most patients (77.2%, n = 525/680) had a maximum‐grade AE of mild to moderate (Grade 1/2). Seventy‐one patients (10.4%) and nine patients (1.3%) had a maximum of Grade 3 or 4 AEs, respectively; there was one Grade 5 AE (suicide; 0.1%). The only treatment‐related AEs occurring in ≥ 5% of patients were IRRs (43.2%, n = 294/680) and nasopharyngitis (7.6%, n = 52/680).
TABLE 3.
Adverse events, serious adverse events, and adverse events leading to discontinuation for patients receiving ocrelizumab 600 mg in the CASTING study
| Variable | Ocrelizumab (N = 680) |
|---|---|
| Number of patients (%) | |
| Any AE | 606 (89.1) |
| AEs leading to study treatment discontinuation | 7 (1.0) |
| AEs occurring in > 10% of patients | |
| Infusion‐related reaction | 294 (43.2) |
| Nasopharyngitis | 210 (30.9) |
| Headache | 154 (22.6) |
| Influenza | 92 (13.5) |
| Urinary tract infection | 70 (10.3) |
| Serious AE | 49 (7.2) |
| Serious infection or infestation | 11 (1.6) |
| Malignancies a | 3 (0.4) |
Adverse events were encoded using MedDRA v21.0.
Abbreviations: AE, adverse event; MedDRA, Medical Dictionary for Regulatory Activities.
Basal cell carcinoma (n = 1), squamous cell carcinoma of the cervix (n = 1), and benign neoplasm of the thymus (n = 1).
Overall, 49 of 680 patients (7.2%) reported a total of 69 serious AEs (SAEs) during the treatment period, of whom 32 patients (4.7%) had Grade 3 events, seven patients (1.0%) had Grade 4 events, and one patient had a Grade 5 event (suicide, 0.1%). Of the reported SAEs, 17 events in 13 patients were deemed related to treatment; the only treatment‐related SAE to occur in >1 patient was sinusitis (n = 3 [0.4%]). All events were reported as recovered/resolved, except for an event of guttate psoriasis, which was reported as recovered/resolved with sequelae.
Adverse events leading to interruptions during the infusions or delays in dosing occurred in 102 patients (15.0%), the majority of which (n = 97 [14.3%]) were IRRs. AEs leading to withdrawal of treatment were infrequent and occurred in seven patients (1.0%), of whom five patients (0.7%) had SAEs (anal abscess, guttate psoriasis, hepatitis [with negative viral hepatitis test] of unknown origin [the patient had experienced hepatitis before inclusion in the study], enterocolitis, and hallucination; each occurring in one patient); no event occurred in more than one patient.
Of safety events of additional interest, IRRs were the most common (570 IRRs occurring in 294/680 patients [43.2%], and most frequently during the first [n = 217; 31.9%]) versus subsequent infusions (between n = 58/644 [9.0%] at Week 72 and n = 104/672 [15.5%] at Week 24). Most IRRs were Grade 1 (n = 192/680 [28.2%]) or Grade 2 events (n = 97/680 [14.3%]); five patients (0.7%) had Grade 3 IRRs. There were no Grade 4/5 IRRs. A total of 1489 infections were experienced by 455 patients (66.9%), of which serious infections were experienced by 11 patients (1.6%); only sinusitis (n = 3 [0.4%]) and pneumonia (n = 2 [0.2%]) occurred in > 1 patient. Five patients had serious infections that were considered related to treatment, of whom three patients had sinusitis, one patient had bacterial esophagitis, oral bacterial infection, fungal superinfection, and sepsis, and one patient had a viral infection (varicella zoster). All serious infections, excluding a case of anal abscess unrelated to treatment (not recovered/not resolved), were reported as ‘recovered/resolved’. For neoplasms, three patients (0.4%) experienced SAEs of neoplasms, namely, one basal cell carcinoma (reported as related to ocrelizumab and resolved), one squamous cell carcinoma of the cervix (reported as related to ocrelizumab), and a benign neoplasm of the thymus (reported as unrelated to ocrelizumab and resolved).
There were five (0.7%) pregnancies, and no treatment‐related complications or abnormalities were reported. There were no notable findings in laboratory assessments, physical examinations, neurological examinations, vital signs, and non‐MS MRI pathology.
DISCUSSION
The CASTING study, unique for enrolling a population of patients with RRMS selected specifically for prior DMT failure, a quarter of which were based on MRI activity only, and utilizing MRI‐re‐baselining for the assessment of NEDA, showed that patients treated with ocrelizumab had generally low disease activity (87.1% within 6 months), with high rates maintained after 1 (82.6%) and 2 years (74.8%; primary endpoint).
Despite the available therapeutic interventions across the chronic continuum of MS [15, 16], breakthrough disease activity, defined using clinical and/or MRI measures according to clinical, guidelines, or DMT label specifications [17, 18, 19, 20, 21, 22], is often experienced during DMT, requiring a change in therapy [1, 2, 3, 23], and is associated with poor long‐term outcomes [20, 24, 25, 26]. The reappraisal of treatment pathways in MS notwithstanding [27, 28], there remains a need for highly effective therapies that can be used at any time during the disease course. In a post hoc analysis of the pooled population of the OPERA I and II studies comprising both treatment‐naive and previously treated patients receiving ocrelizumab, NEDA without re‐baselining was 47.7% in the overall population, and 49.5% and 42.8% in subgroups of patients with and without prior DMT, respectively [29]. Findings from the CHORDS study, which included patients treated with up to three previous DMTs, will further our understanding of ocrelizumab use in patients who respond poorly to other treatments, with preliminary results showing that 48.1% of patients had NEDA (without re‐baselining) after 2 years [30]. In CASTING, where the different baseline demographics and disease characteristics caveat direct comparisons with the CHORDS study, 52.0% of patients had NEDA without re‐baselining. However, in order to represent the full efficacy of a DMT unconfounded by disease activity carried over during the first 4–8 weeks from treatment initiation, particularly MRI‐related, a re‐baselining approach at first available MRI has been suggested [19, 20, 31], generating a NEDA rate of 72.2% in the pooled OPERA I and II population with re‐baselining at Week 24 (time of first post‐baseline MRI) [31]. Based on the rapid suppression of acute MRI and clinical disease activity within 8 weeks of treatment initiation with ocrelizumab in patients with RRMS and RMS [11, 32], MRI re‐baselining at Week 8 was incorporated into the CASTING study design. While it is of interest to consider re‐baselining of other NEDA components, very few patients in the present study exhibited relapse in the 8‐week re‐baselining window (2‐year NEDA with 8‐week MRI and relapse re‐baselining: 76.2% [95% CI 72.8–79.4], n/N = 500/656).
The NEDA rate remained high across a wide range of disease‐related and demographic subgroups, regardless of prior treatment background or reason for study inclusion, and, importantly, the rate did not vary by age, supporting the benefit of ocrelizumab across subgroups previously reported from the pooled OPERA I and II studies [29]. The findings that the proportion of patients with NEDA was higher in patients enrolled based on MRI activity only, in those with lower baseline disability scores and markers of less active disease, and in patients receiving ocrelizumab as the first switch versus second switch, demonstrate the importance of identifying and reacting to early suboptimal clinical or subclinical response to a DMT [27, 28], and that switching to ocrelizumab is never as effective as earlier treatment, and does not reverse lost function [8, 9].
Of the baseline factors analyzed, only gender was a prognostic factor for NEDA at Week 96, with women more likely to have evidence of disease progression. This may be related to the fact that women had slightly higher baseline inflammatory MRI activity. Other studies have shown that the risk of not achieving NEDA was associated with clinical and MRI measures of disease activity and progression (e.g., higher baseline EDSS score, number of relapses in the previous 12 months, number of CELs) [33, 34, 35]. Conversely, studies of the prognostic value of NEDA status over 2 years in predicting future disability progression up to 10 years have provided conflicting data, but this was related to the DMT used, with few patients maintaining NEDA over the long term in studies in which self‐injectable DMTs were used, while NEDA was enhanced in patients over the long term in studies utilizing more effective DMTs [31]. Indeed, data from the pooled OPERA I and II population suggest NEDA status over the short term may predict longer‐term benefits [31].
In patients treated with ocrelizumab, the high proportion with NEDA was reflected in related measures of clinical and MRI disease activity. After 2 years in the CASTING study, stable or improved EDSS score compared with baseline in most patients (86.6%) and an adjusted ARR (0.03) equivalent to one relapse approximately every 33 years were seen. This, combined with the near‐complete suppression of new MRI lesions and a normalized brain volume loss within the range of healthy controls [36], suggests ocrelizumab is effective in a population with disease poorly controlled by other DMTs, and supports the notion of an early intensive versus escalation approach to therapy in patients with MS [37].
The high proportion of patients with NEDA, largely stable disability scores, and decreases in MRI measures in patients treated with ocrelizumab over the 2‐year CASTING study were associated with a consistent safety profile and accompanied by a low rate of attrition, with a 94.3% patient‐completion rate. IRRs and infections were the most common AEs observed, and there were low rates of serious events and AE‐related discontinuations. A slightly higher rate of IRRs compared with that observed in the pivotal phase 3 trials [6, 7] was likely attributable to differences in reporting, as was also observed in the related CHORDS study, in patients with RRMS with suboptimal responses to prior DMTs [30].
The CASTING study addresses an important patient population not included in the pivotal phase 3 OPERA I and II studies, since, apart from IFNs and glatiramer acetate, no other DMTs were available at the start of the OPERA I and II studies. Furthermore, patients were selected specifically for prior DMT failure with only one or two prior DMTs (not > 2 DMTs), that is, they were experiencing breakthrough disease and hence were switched to ocrelizumab early in the disease course. Escalation therapy in MS is an area that still raises many questions [27, 28], and the results from the CASTING study in this specific patient population support the notion of an early intensive treatment approach rather than a gradual escalation to therapy in MS.
As CASTING is a single‐arm trial, the lack of a parallel group for comparison poses some limitations; it is not possible to assess whether NEDA rates were higher than might have been expected if patients continued on their previous DMT, and we cannot distinguish whether the high NEDA rates observed were attributable to the effect of ocrelizumab or to the natural history of the population. However, the proportion of patients with NEDA increased significantly relative to baseline, and these patients were previously experiencing disease activity with their prior DMT, thus supporting the benefit of ocrelizumab.
Overall, in patients treated with ocrelizumab we observed a high proportion with NEDA, reflecting preserved clinical function and tissue preservation, across a wide range of disease‐related and demographic subgroups, regardless of prior treatment background, with no new safety signals detected. This study highlights the importance of recognizing breakthrough disease and the value of reacting with a treatment switch to ocrelizumab in order to maintain disease control and optimize short‐ and long‐term patient outcomes. The highly positive benefit–risk profile observed in this and other clinical studies of ocrelizumab continues to be evaluated in long‐term extension studies.
CONFLICT OF INTERESTS
Patrick Vermersch has received honoraria or consulting fees from AB Science, Biogen, Celgene‐BMS, Imcyse, Merck, Novartis, Roche, Sanofi Genzyme, and Teva, and research support from Biogen, Merck, Novartis, F. Hoffmann‐La Roche, and Sanofi Genzyme. Celia Oreja‐Guevara has received payment or honoraria for lectures, presentations, speakers' bureaus, manuscript writing or educational events from Alexion, F. Hoffmann‐La Roche Ltd, Merck, and Novartis, and support for attending meetings and/or travel from Sanofi Genzyme. Aksel Siva has received honoraria or consultancy fees and/or travel and registration coverage for attending several national or international congresses or symposia from Biogen Idec/Gen Pharma of Turkey, F. Hoffmann‐La Roche Ltd, Sanofi Genzyme, Merck Serono, Novartis, and Teva, and grants/ contracts for research support from the Turkish MS Society and Istanbul University. Bart Van Wijmeersch has received consulting fees and/or speaker fees from BMS, Biogen, F. Hoffmann‐La Roche Ltd, Janssen Pharma, Merck, Novartis, and Sanofi Genzyme; and support for attending meetings and/or travel from Biogen, Merck, and Sanofi Genzyme. Heinz Wiendl has received grant/research support from Biogen Idec, Deutsche Forschungsgesellschaft, Else Kröner‐Fresenius Foundation, the German Federal Ministry of Education and Research, GlaxoSmithKline GmbH, Hertie Foundation, the Interdisciplinary Center for Clinical Studies in Münster, Germany, the European Union, NRW Ministry of Education and Research, Roche Pharma AG and Sanofi Genzyme; and has received consulting fees from Actelion, Biogen, Evgen, Genzyme IGES, Johnson & Johnson, Merck, Novartis, Hoffmann‐La Roche, Sanofi‐Aventis, the Swiss Multiple Sclerosis Society, and Serono. Jens Wuerfel is an employee of MIAC AG and has participated in Data Safety Monitoring Boards or Advisory Boards for Idorsia, INmuneBio, Johnson & Johnson, F. Hoffmann‐La Roche Ltd and Sanofi Genzyme. Regine Buffels and Karen Kadner are employees of F. Hoffmann‐La Roche Ltd. Thomas Kuenzel is an employee of/has stock or stock options from F. Hoffmann‐La Roche Ltd. Giancarlo Comi has received consulting fees or honoraria from Almirall, BMS, F. Hoffmann‐La Roche Ltd, Janssen Pharma, Merck, Novartis, and Sanofi Genzyme; and has participated in a data safety monitoring or advisory board for BMS, Janssen Pharma, and Sanofi Genzyme.
AUTHOR CONTRIBUTIONS
Patrick Vermersch: Formal analysis (equal); Methodology (equal); Supervision (equal); Validation (equal); Visualization (equal); Writing – original draft (equal); Writing – review and editing (equal). Celia Oreja‐Guevara: Conceptualization (equal); Formal analysis (equal); Writing – original draft (equal); Writing – review and editing (equal). Aksel Siva: Conceptualization (equal); Formal analysis (equal); Writing – original draft (equal); Writing – review and editing (equal). Bart Van Wijmeersch: Conceptualization (equal); Formal analysis (equal); Writing – original draft (equal); Writing – review and editing (equal). Heinz Wiendl: Conceptualization (equal); Formal analysis (equal); Writing – original draft (equal); Writing – review and editing (equal). Jens Wuerfel: Conceptualization (equal); Formal analysis (equal); Writing – original draft (equal); Writing – review and editing (equal). Regine Buffels: Conceptualization (equal); Formal analysis (equal); Supervision (equal); Writing – original draft (equal); Writing – review and editing (equal). Karen Kadner: Formal analysis (equal); Writing – original draft (equal); Writing – review and editing (equal). Thomas Kuenzel: Formal analysis (equal); Writing – original draft (equal); Writing – review and editing (equal). Giancarlo Comi: Conceptualization (equal); Formal analysis (equal); Writing – original draft (equal); Writing – review and editing (equal).
Supporting information
ACKNOWLEDGEMENTS
We thank all patients, their families, and the investigators who participated in this trial (including the CASTING study Steering Committee, which provided study oversight). We also thank Wei Wei of F. Hoffmann‐La Roche Ltd (Basel, Switzerland) for her support with data analysis.
Vermersch P, Oreja‐Guevara C, Siva A, et al; the CASTING Investigators . Efficacy and safety of ocrelizumab in patients with relapsing‐remitting multiple sclerosis with suboptimal response to prior disease‐modifying therapies: A primary analysis from the phase 3b CASTING single‐arm, open‐label trial. Eur J Neurol.2022;29:790–801. doi: 10.1111/ene.15171
Funding information
This research was funded by F. Hoffmann‐La Roche Ltd (Basel, Switzerland). Andrew Stead and Terence Smith of Articulate Science, UK provided editorial assistance, funded by F. Hoffmann‐La Roche Ltd. The authors had full editorial control of the manuscript and provided final approval
DATA AVAILABILITY STATEMENT
Qualified researchers may request access to individual patient‐level data through the clinical study data request platform (https://vivli.org/). Further details on Roche's criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
REFERENCES
- 1. Freedman MS. Treatment options for patients with multiple sclerosis who have a suboptimal response to interferon‐β therapy. Eur J Neurol. 2014;21:377‐387. [DOI] [PubMed] [Google Scholar]
- 2. Tanasescu R, Ionete C, Chou I‐J, Constantinescu CS. Advances in the treatment of relapsing‐remitting multiple sclerosis. Biomed J. 2014;37:41‐49. [DOI] [PubMed] [Google Scholar]
- 3. Ziemssen T, Stefano ND, Sormani MP, et al. Optimizing therapy early in multiple sclerosis: an evidence‐based view. Mult Scler Relat Disord. 2015;4:460‐469. [DOI] [PubMed] [Google Scholar]
- 4. DiLillo DJ, Hamaguchi Y, Ueda Y, et al. Maintenance of long‐lived plasma cells and serological memory despite mature and memory B cell depletion during CD20 immunotherapy in mice. J Immunol. 2008;180:361‐371. [DOI] [PubMed] [Google Scholar]
- 5. Klein C, Lammens A, Schäfer W, et al. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. mAbs. 2013;5:22‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hauser SL, Bar‐Or A, Comi G, et al. Ocrelizumab versus interferon beta‐1a in relapsing multiple sclerosis. N Engl J Med. 2017;376:221‐234. [DOI] [PubMed] [Google Scholar]
- 7. Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376:209‐220. [DOI] [PubMed] [Google Scholar]
- 8. Hauser SL, Kappos L, Arnold DL, et al. Five years of ocrelizumab in relapsing multiple sclerosis: OPERA studies open‐label extension. Neurology. 2020;95:e1854‐e1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wolinsky JS, Arnold DL, Brochet B, et al. Long‐term follow‐up from the ORATORIO trial of ocrelizumab in primary progressive multiple sclerosis: post‐hoc analysis from the ongoing open‐label extension of the randomised, placebo‐controlled, phase 3 trial. Lancet Neurol. 2020;19:998‐1009. [DOI] [PubMed] [Google Scholar]
- 10. Rotstein DL, Healy BC, Malik MT, et al. Evaluation of no evidence of disease activity in a 7‐year longitudinal multiple sclerosis cohort. JAMA Neurol. 2015;72:152‐158. [DOI] [PubMed] [Google Scholar]
- 11. Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing‐remitting multiple sclerosis: a phase 2, randomised, placebo‐controlled, multicentre trial. Lancet. 2011;378:1779‐1787. [DOI] [PubMed] [Google Scholar]
- 12. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith SM, Zhang Y, Jenkinson M, et al. Accurate, robust, and automated longitudinal and cross‐sectional brain change analysis. NeuroImage. 2002;17:479‐489. [DOI] [PubMed] [Google Scholar]
- 14. Nakamura K, Guizard N, Fonov VS, et al. Jacobian integration method increases the statistical power to measure gray matter atrophy in multiple sclerosis. Neuroimage Clin. 2013;4:10‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cree BAC, Mares J, Hartung HP. Current therapeutic landscape in multiple sclerosis: an evolving treatment paradigm. Curr Opin Neurol. 2019;32:365‐377. [DOI] [PubMed] [Google Scholar]
- 16. Derfuss T, Mehling M, Papadopoulou A, et al. Advances in oral immunomodulating therapies in relapsing multiple sclerosis. Lancet Neurol. 2020;19:336‐347. [DOI] [PubMed] [Google Scholar]
- 17. Simpson A, Mowry EM, Newsome SD. Early aggressive treatment approaches for multiple sclerosis. Curr Treat Options Neurol. 2021;23:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Río J, Ruiz‐Peña JL. Short‐term suboptimal response criteria for predicting long‐term non‐response to first‐line disease modifying therapies in multiple sclerosis: a systematic review and meta‐analysis. J Neurol Sci. 2016;361:158‐167. [DOI] [PubMed] [Google Scholar]
- 19. Freedman MS, Devonshire V, Duquette P, et al. Treatment optimization in multiple sclerosis: Canadian MS Working Group Recommendations. Can J Neurol Sci. 2020;47:437‐455. [DOI] [PubMed] [Google Scholar]
- 20. Montalban X, Gold R, Thompson AJ, et al. ECTRIMS/EAN guideline on the pharmacological treatment of people with multiple sclerosis. Eur J Neurol. 2018;25:215‐237. [DOI] [PubMed] [Google Scholar]
- 21. European Medicines Agency . Gilenya EU summary of product characteristics, 2011. Accessed October 12, 2020. Available at: http://www.ema.europa.eu/en/documents/product‐information/gilenya‐epar‐product‐information_en.pdf
- 22. European Medicines Agency . Tysabri EU summary of product characteristics, 2012. Accessed October 12, 2020. Available at: http://www.ema.europa.eu/en/documents/product‐information/tysabri‐epar‐product‐information_en.pdf
- 23. Gross RH, Corboy JR. Monitoring, switching, and stopping multiple sclerosis disease‐modifying therapies. Continuum (Minneap Minn). 2019;25:715‐735. [DOI] [PubMed] [Google Scholar]
- 24. Río J, Castilló J, Rovira A, et al. Measures in the first year of therapy predict the response to interferon beta in MS. Mult Scler. 2009;15:848‐853. [DOI] [PubMed] [Google Scholar]
- 25. Scalfari A, Neuhaus A, Degenhardt A, et al. The natural history of multiple sclerosis: a geographically based study 10: relapses and long‐term disability. Brain. 2010;133:1914‐1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jokubaitis VG, Spelman T, Kalincik T, et al. Predictors of long‐term disability accrual in relapse‐onset multiple sclerosis. Ann Neurol. 2016;80:89‐100. [DOI] [PubMed] [Google Scholar]
- 27. Ontaneda D, Tallantyre E, Kalincik T, et al. Early highly effective versus escalation treatment approaches in relapsing multiple sclerosis. Lancet Neurol. 2019;18:973‐980. [DOI] [PubMed] [Google Scholar]
- 28. Giovannoni G. Disease‐modifying treatments for early and advanced multiple sclerosis: a new treatment paradigm. Curr Opin Neurol. 2018;31:233‐243. [DOI] [PubMed] [Google Scholar]
- 29. Turner B, Cree BAC, Kappos L, et al. Ocrelizumab efficacy in subgroups of patients with relapsing multiple sclerosis. J Neurol. 2019;266:1182‐1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weinstock‐Guttman B, Bermel R & Cutter G et al. Ocrelizumab treatment for relapsing‐remitting multiple sclerosis after a suboptimal response to previous disease‐modifying therapy: A nonrandomized controlled trial. Mult Scler. 2021. 10.1177/13524585211035740. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Havrdová E, Arnold DL, Bar‐Or A, et al. No evidence of disease activity (NEDA) analysis by epochs in patients with relapsing multiple sclerosis treated with ocrelizumab vs interferon beta‐1a. Mult Scler J Exp Transl Clin. 2018;4:2055217318760642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barkhof F, Kappos L, Wolinsky JS, et al. Onset of clinical and MRI efficacy of ocrelizumab in relapsing multiple sclerosis. Neurology. 2019;93:e1778‐e1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Giuliani M, Logoteta A, Prosperini L, et al. Baseline characteristics associated with NEDA‐3 status in fingolimod‐treated patients with relapsing‐remitting multiple sclerosis. Mult Scler Dem Disord. 2017;2:10. [Google Scholar]
- 34. Arnold DL, Calabresi PA, Kieseier BC, et al. Peginterferon beta‐1a improves MRI measures and increases the proportion of patients with no evidence of disease activity in relapsing‐remitting multiple sclerosis: 2‐year results from the ADVANCE randomized controlled trial. BMC Neurol. 2017;17:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dominguez‐Mozo MI, Perez‐Perez S, Villar LM, et al. Predictive factors and early biomarkers of response in multiple sclerosis patients treated with natalizumab. Sci Rep. 2020;10:14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. De Stefano N, Stromillo ML, Giorgio A, et al. Establishing pathological cut‐offs of brain atrophy rates in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2016;87:93‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harding K, Williams O, Willis M, et al. Clinical outcomes of escalation vs early intensive disease‐modifying therapy in patients with multiple sclerosis. JAMA Neurol. 2019;76:536‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Qualified researchers may request access to individual patient‐level data through the clinical study data request platform (https://vivli.org/). Further details on Roche's criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).