

Abstract
A wide phenotypic spectrum of neurological diseases is associated with KCNA1 (Kv1.1) variants. To investigate the molecular basis of such a heterogeneous clinical presentation and identify the possible correlation with in vitro phenotypes, we compared the functional consequences of three heterozygous de novo variants (p.P403S, p.P405L, and p.P405S) in Kv1.1 pore region found in four patients with severe developmental and epileptic encephalopathy (DEE), with those of a de novo variant in the voltage sensor (p.A261T) identified in two patients with mild, carbamazepine‐responsive, focal epilepsy. Patch‐clamp electrophysiology was used to investigate the functional properties of mutant Kv1.1 subunits, both expressed as homomers and heteromers with wild‐type Kv1.1 subunits. KCNA1 pore mutations markedly decreased (p. P405S) or fully suppressed (p. P403S, p. P405L) Kv1.1‐mediated currents, exerting loss‐of‐function (LoF) effects. By contrast, channels carrying the p.A261T variant exhibited a hyperpolarizing shift of the activation process, consistent with a gain‐of‐function (GoF) effect. The present results unveil a novel correlation between in vitro phenotype (GoF vs LoF) and clinical course (mild vs severe) in KCNA1‐related phenotypes. The excellent clinical response to carbamazepine observed in the patients carrying the A261T variant suggests an exquisite sensitivity of KCNA1 GoF to sodium channel inhibition that should be further explored.
Keywords: developmental encephalopathies, epilepsy, gain‐of‐function variants, KCNA1, loss‐of‐function variants, potassium channels
1. INTRODUCTION
A wide phenotypic spectrum of neurological diseases is associated with variants in the KCNA1 gene encoding for neuronal Kv1.1 voltage‐gated potassium channels. Although about 50% of all variants have been associated with isolated episodic ataxia type 1 (EA1), 1 the remaining 50% are associated with additional phenotypes, with limited overlapping, including EA1 with epilepsy, severe developmental and epileptic encephalopathy (DEE), myokymia, hyperthermia, and hypomagnesemia. 2 , 3
Here, we describe the functional consequences of four pathogenic KCNA1 variants, of which three, located within the highly conserved Pro‐Val‐Pro (PVP) pore motif and identified in patients with severe epilepsy or early onset DEE 2 , resulted in a loss‐of‐function (LoF) effect, and one, located in the voltage sensor and identified in a patient with mild, drug‐responsive focal epilepsy, caused gain‐of‐function (GoF) effects in vitro.
2. MATERIALS AND METHODS
2.1. Patients
Patients 1–4 were reported previously in a clinical and genetic study describing severe, early onset epilepsy phenotypes associated with KCNA1 genetic variants. 2 Patient 5 was referred for diagnostic workup and treatment of childhood‐onset focal epilepsy and is described here for the first time. Clinical findings of the five patients are summarized in Table 1 and a detailed description of Patient 5 is presented in the “Results” section. This study was approved by the Paediatric Ethics Committees of the Tuscany Region, Italy.
TABLE 1.
Clinical description of patients with KCNA1 variants functionally characterized
| Patient | Genetic findings | Clinical findings | Described in: | Additional references | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change (NM_000217) | Amino acid substitution | gnomAD frequency | Inheritance | Functional domain | Phenotype | Age at seizure onset | Age at last \follow‐up | Seizure types/treatments used | EEG findings | Brain MRI | Additional clinical features | |||
| 1/M | c.1213C > T | p. Pro405Ser | — | de novo | S6 – PVP motif | Epileptic encephalopathy with severe cognitive impairment | 3 m | 5 y | 3 m: back arching spells; 5 m: brief staring spell that was followed by flaccidity of the arms and legs; 18 m: prolonged episodes (>30 min) with unresponsiveness and leftward eye deviation, with or without clonic movements; 5 y: 3 to 4 prolonged seizures per year, often precipitated by fever; /PB, Clob, CBZ, ZSM and VPA, all ineffective | 3 m: generalized slowing;18 m: subclinical seizure with left parietal onset, high‐amplitude, multifocal epileptiform abnormalities and generalised slowing | 3 m: symmetric subdural fluid collections and prominent subarachnoid spaces; 5 y: normal | Developmental delay, mild to moderate axial and appendicular hypotonia, increased insertional activity consistent with irritable motor nerves at electromyography but without classic myokymic discharges. | Rogers et al., 2018 | — |
| 2/F | c.1214C > T | p. Pro405Leu | — | de novo | S6 – PVP motif | Epileptic encephalopathy with severe cognitive impairment | 18 d | 7 y 6 m | 18 d: generalized clonic; 2 m: generalized tonic‐clonic seizures; 4 y: focal onset impaired awareness seizures; often precipitated by fever; PB, CBZ, LEV, and PHT all ineffective or very short benefit | 18 d: severe epileptiform discharges on both hemispheres; 4 y: multifocal abnormalities, with left temporal predominance; 7 y: severe bilateral independent epileptiform discharges, with status epilepticus during sleep (ESES) responsive to ACTH | normal | Developmental delay, autism spectrum disorder, moderate‐severe intellectual disability, distal tremor and impaired coordination of cerebellar origin | Parrini et al., 2017 | Russo et al., 2020 |
| 3/M (identical twin brother of Patient 4) | c.1207C > T | p. Pro403Ser | — | de novo | S6 – PVP motif | Generalized epilepsy, moderate cognitive impairment & myokymia | 6 m | 20 y | 6 m: generalized tonic‐clonic seizures; LTG partially effective, multiple additional medications not specified, all ineffective | NA | 6 m: normal | Developmental delay, ataxia, intellectual disability severe recurrent headaches | Rogers et al., 2018 | — |
| 4/M (identical twin brother of Patient 3) | c.1207C > T | p. Pro403Ser | — | de novo | S6 – PVP motif | Epilepsy, severe cognitive impairment & myokymia | 4 m | 20 y | Generalized tonic‐clonic and absence seizures; LTG, GVG, OXC, VPA, PHT, and Clob, all ineffective | NA | 2 y: high signal intensities in the globus pallidus, slightly delayed myelination | Developmental delay, ataxia, myokymia, moderate intellectual disability. Between 8–12 y, language loss and walk only with support | Rogers et al., 2018 | — |
| 5/F | c.781G > A | p. Ala261Thr | — | de novo | S3 | Focal epilepsy responsive to carbamazepine | 7 y | 18 y |
Focal occipital seizures with blurred vision, complex hallucinations and, at times, fading hearing; secondarily generalized tonic‐clonic seizures Seizure precipitation during fever; VPA transiently effective, CBZ effective |
Bilateral, right predominant, occipital spikes | 8 y: normal | None | This paper | Yuan et al., 2020 |
Abbreviations: CBZ, carbamazepine; Clob, clobazam; d, days; F, female; GVG, vigabatrin; LTG, lamotrigine; M, male; m, months; NA, not available; OXC, oxcarbazepine; PB, phenobarbital; PHT, phenytoin; VPA valproic acid; y, years; ZSM, zonisamide.
2.2. Genetic analysis
The methods for genetic testing in Patients 1–4 have been reported previously. 2 For Patient 5, genetic trio testing was performed using exome sequencing (ES) after informed consent. In brief, the Nextera Rapid Capture kit and a NextSeq 500 were used for the library preparation and the 2 × 100 bp paired‐end sequencing.
2.3. Mutagenesis and heterologous expression
Mutations were engineered in KCNA1 human comlementary DNA (cDNA) cloned into pCMV6 (#RC211000; OriGene) by QuikChange site‐directed mutagenesis (Agilent Technologies). 4 , 5 Channel subunits were expressed in Chinese Hamster Ovary (CHO) cells by transient transfection performed one day after seeding on glass coverslips using Lipofectamine 2000 (Invitrogen); a plasmid encoding for enhanced green fluorescent protein was used as a transfection marker; total cDNA in the transfection mixture was kept constant at 4 μg.
2.4. Whole‐cell electrophysiology
Currents were recorded at room temperature (20–22°C) 1–2 days after transfection, using an Axopatch 200B amplifier (Molecular Devices) and the whole‐cell configuration of the patch‐clamp technique, with glass micropipettes of 3–5 MΩ resistance. 4 , 5 The extracellular solution contained (in millimolar): 138 NaCl, 2 CaCl2, 5.4 KCl, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.4 with NaOH. The pipette (intracellular) solution contained (in millimolar): 140 KCl, 2 MgCl2, 10 EGTA, 10 HEPES, and 5 Mg‐ATP, pH 7.3–7.4 with KOH. The pCLAMP software (version 10.0.2) was used for data acquisition and analysis. Conductance‐voltage curves were obtained and analyzed as described previously. 6
2.5. Statistics
Data were analyzed using SigmaPlot (Systat Software Inc.). Values are expressed as the mean ± standard error of the mean (SEM) of cells recorded in at least three independent experimental sessions. Statistically significant differences between the data were evaluated with the Student's t test (p < .05).
3. RESULTS
3.1. Clinical and genetic findings
Updated clinical and genetic findings in Patients 1–4 are summarized in Table 1. 3 The findings for Patient 5, in addition to Table 1, are also presented in detail below.
Patient 5. This young lady, now age 18, manifested since age 7 focal occipital seizures with blurred vision, followed by secondary generalization, appearing in clusters precipitated by fever, and accompanied by focal interictal and ictal electroencephalography (EEG) abnormalities in the right occipital lobe. Cognitive level was normal and brain magnetic resonance imaging (MRI) was unrevealing. Seizures responded to initial valproate treatment, which was started at age 8 but she relapsed at age 12, with similar characteristics, again, during a febrile episode soon after treatment was suspended. Valproate was reintroduced but seizures persisted and were video‐EEG recorded as episodes of visual hallucinations of circles and blurred vision, lasting for about 30 s, accompanied by a sensation of fading hearing, at times followed by a secondary tonic‐clonic phase.
At age 15, valproate was replaced by carbamazepine monotherapy, with immediate and complete seizure control during the following 3 years, with drug plasma levels at around 8 μg/mL during the following 3 years, apart from a single seizure cluster after reduced compliance. Whole exome sequencing identified a c.781G > A heterozygous variant in the KCNA1 gene (NM_000217.3), confirmed to be de novo by Sanger sequencing and resulting in the missense substitution p.A261T. The variant was absent from the gnomAD population data set (gnomad.broadinstitute.org), not reported in the HGMD mutation database (portal.biobase‐international.com/hgmd/pro/start.php) and predicted as pathogenic by several computational tools. 7 We classified the variant as likely pathogenic (PS2, PM1, PM2, PP2, PP3) according to the American College of Medical Genetics classification. 8 No other relevant variant was detected in the patient.
3.2. Functional analysis
Kv1.1 subunits contain six transmembrane segments (S1–S6), with the S1–S4 region corresponding to the voltage sensing domain (VSD), and the S5–S6 segments and the intervening linker forming the ion selective pore. 9 The A261 residue is localized in the S3 segment of the VSD, whereas the P403 and P405 residues fall within the highly conserved PXP motif localized at the bottom of the pore‐forming S6 segment (Figure 1A). CHO cells expressing homomeric Kv1.1 channels generate robust voltage‐dependent K+‐selective currents that activate with fast kinetics with a threshold around −50 mV; when compared to Kv1.1, cells transfected with Kv1.1 A261T mutant cDNA displayed a 20 mV hyperpolarizing shift in steady‐state activation gating (Figure 1B). By contrast, although Kv1.1 P403S‐ or Kv1.1 P405L‐expressing cells failed to generate measurable currents, a marked (~70 mV) depolarizing shift in voltage‐dependent activation gating and a significant reduction in current density was observed in CHO cells expressing Kv1.1 P405S (Figure 1B–D). To reproduce in vitro the genetic balance of the patients who carry one wild‐type and one mutant allele, we transfected CHO cells with wild‐type and mutant cDNAs in a 1:1 ratio. When compared to Kv1.1, cells expressing Kv1.1+Kv1.1 A261T subunits showed a hyperpolarizing shift in voltage‐dependent activation, the extent of which was similar to that recorded in homomeric Kv1.1 A261T mutant channels (about ‐17 mV) (Figure 1D). By contrast, no currents could be recorded from Kv1.1 + Kv1.1 P403S‐ or Kv1.1 + Kv1.1 P405L‐transfected cells, whereas activation of currents from Kv1.1+Kv1.1 P405S‐transfected cells was positively shifted by 45 mV, displaying therefore a V1/2 intermediate between that of homomeric Kv1.1 and Kv1.1 P405S channels (Figure 1C, D). Table S1 summarizes the detailed biophysical properties of the currents carried by the described experimental groups.
FIGURE 1.
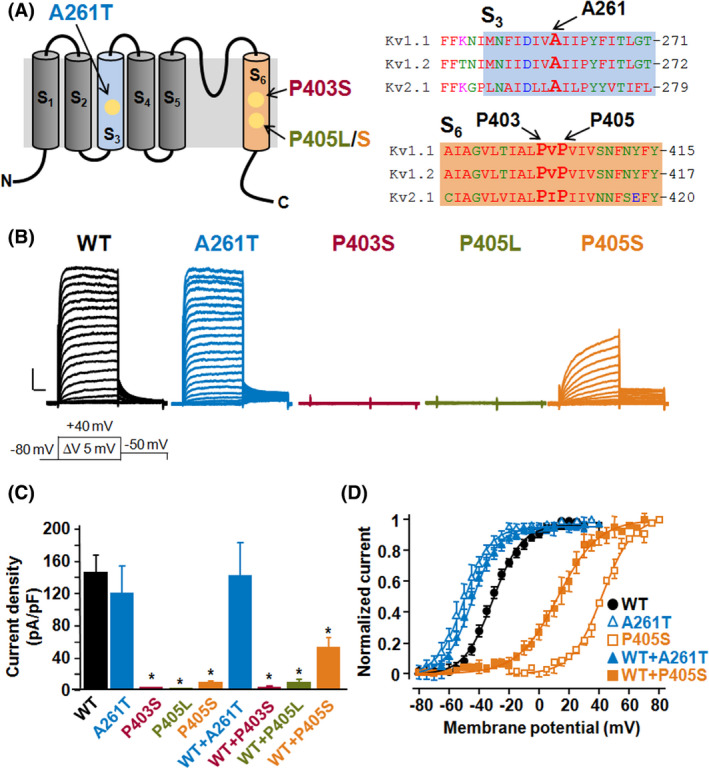
Location and functional characterization of Kv1.1 mutants. (A) Cartoon depicting the six transmembrane segments arrangement of a single Kv1.1 subunit (left panel) and sequence alignments of the S3 (top right panel) and S6 (lower right panel) region of Kv1.1, Kv1.2, and Kv2.1 subunits. (B) Macroscopic currents recorded from CHO cells transfected with plasmids encoding for wild‐type (WT) or the indicated mutant Kv1.1 subunits, in response to the voltage protocol shown below the Kv1.1 traces. Current scale, 500 pA; time scale, 40 ms. (C) Current density (expressed as pA/pF) at +20 mV from CHO cells expressing each of the indicated experimental group. Asterisks (*) indicate values significantly different from each respective control (p < .05). (D) Conductance/voltage curves for each of the indicated groups. Continuous lines are Boltzmann fits to the experimental data. Each data point is the mean ± SEM. of 13–21 cells recorded in at least three separate experimental sessions.
4. DISCUSSION
In vitro assessment of the functional consequences of rare variants in ion channel genes, can reveal pathogenic mechanisms and genotype‐phenotype correlations, and is critical for diagnostic evaluation, prognostic predictions, and, possibly, a more tailored therapeutic management of patients with a wide spectrum of neurological disorders. Mutations in the KCNA1 gene are among the most frequent cause of EA1, which is associated with epilepsy in some patients. 3 Additional, rare phenotypes associated with KCNA1 mutations include paroxysmal dyskinesias, severe developmental and epileptic encephalopathies, and hypomagnesemia. 2 , 3
Although the pathogenetic mechanisms for such a wide variety of symptoms for KCNA1‐associated phenotypes is only partly understood, in vitro functional analysis has consistently revealed that, regardless of the associated phenotype, incorporation of mutant subunits decreases channel function, suggesting a LoF pathogenetic mechanism. However, genotype‐phenotype correlations have been quite elusive due to both the rarity of variant occurrence (only about 50 KCNA1 variants have been described until now; HGMD 2021.2) and the lack of correlations between in vitro functional consequences and disease severity.
In this study, the functional consequence of a de novo KCNA1 variant (p. A261T) found in a patient with mild, carbamazepine‐sensitive, childhood‐onset focal epilepsy without ataxia was investigated. The same variant also occurred in a young girl with carbamazepine‐sensitive focal epilepsy, accompanied by rare ataxic and myokymic episodes. 10 Our findings indicate that channels incorporating mutant subunits, both in homomeric and heteromeric configuration with wild‐type subunits, were fully functional and displayed a marked increase in their voltage‐sensitivity, consistent with a GoF pathogenetic mechanism. Notably, since the first functional description of a disease‐causing variant over 25 years ago, 1 this is the first variant in KCNA1 in which GoF effects are demonstrated. By contrast, the three variants affecting two critical residues in the PVP pore motif (p. P403S, p. P405L, and p. P405S) occurring in patients with severe DEE, 2 , 11 , 12 all caused strong LoF in vitro changes. Notably, within the same PVP region, another KCNA1 variant (p. V404I) causing a dominantly transmitted, familial EA1 phenotype and responsible for milder in vitro LoF effects has been described. 13 Altogether these data are suggestive of novel genotype‐phenotype correlations for the KCNA1 gene, with GoF effects associated with drug‐sensitive epilepsy with no neurodevelopmental impairment, and strong LoF effects in the PVP region responsible for a severe phenotype of pharmacoresistant DEE with intellectual disability.
It is intriguing that a paralogous variant affecting the first proline of the PVP domain in KCNA2‐encoded Kv1.2 subunits (p. P405L), recurrently found in patients with DEE and mild‐moderate intellectual disability, also led to strong dominant‐negative LoF effects. 14 As shown herein for KCNA1, both LoF and GoF variants are associated with distinct clinical phenotypes within the KCNA2‐related diseases spectrum, 15 raising the possibility that the in vitro electrophysiological phenotype might help in predicting pathogenetic mechanisms and treatment possibilities. 4‐Aminopyridine has recently been shown to be a promising treatment for patients with GoF KCNA2‐encephalopathy 16 ; it remains to be investigated whether this drug might represent a potential therapeutic option also for patients with epilepsy due to GoF variants in KCNA1.
Structural data from highly homologous Kv1.2 subunits may allow speculation about the molecular basis for the LoF and GoF effects herein described. The conserved PVP sequence represents the pore activation gate. 9 In the closed channel, the four subunits come in close contact with each other at the PVP region to impede ion passage; during activation, the outward displacement of the VSD, via the S4–S5 linker, disrupts PVP intersubunit interactions and leads to pore opening. 17 Therefore, the PVP prolines provide the S6 region with the degree of mobility needed for channel gating 18 ; substitution of these prolines with nonpolar (L) or polar (S) residues might reduce S6 flexibility, thereby impeding the structural transition linking VSD movement to pore opening. On the other hand, the Kv1.2 residue corresponding to A261 (A262) in S3 is in a narrow (10 Å) hydrophobic region in the VSD where the transmembrane electric field is focused 19 ; introduction of a polar T residue at this position might affect such “focused field,” facilitating charge transfer during VDS displacement and causing GoF consequences.
In vivo, Kv1.1 subunits are abundantly expressed in neurons, where they regulate neuronal excitability; axonal expression of these channels repolarizes the action potential and regulates firing frequency, whereas presynaptic Kv1.1‐containing channels blunt neurotransmitter release. Given their inhibitory role on neuronal excitability, the herein found GoF effects of an epilepsy‐causing KCNA1 variant may seem paradoxical. However, GoF variants causing neuronal hyperexcitability have been described in several K+ channel–encoding genes, 20 and it has been suggested that faster action potential repolarization may lead to sodium channels repriming, 21 thereby increasing firing frequency and synchronization, hallmarks of neuronal hyperexcitability in the epileptic tissue. The marked anticonvulsant efficacy of the sodium‐channel blocker carbamazepine observed in our patient and in Patient 1 of Yuan et al., 10 who also carried the same p. A261T KCNA1 variant, but not in those with loss‐of‐function mutations, appears to support such a hypothesis.
CONFLICT OF INTEREST
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Table S1
ACKNOWLEDGMENTS
We thank all patients and family members for their participation in this study. RG and DM are members of the European Reference Network (ERN) for rare and complex epilepsies (EpiCARE). Open Access Funding provided by Universita degli Studi di Napoli Federico II. [Correction added on 7 June 2022, after first online publication CRUI funding statement has been added.]
Miceli F, Guerrini R, Nappi M, Soldovieri MV, Cellini E, Gurnett CA, et al. Distinct epilepsy phenotypes and response to drugs in KCNA1 gain‐ and loss‐of function variants. Epilepsia. 2022;63:e7–e14. 10.1111/epi.17118
Francesco Miceli and Renzo Guerrini contributed equally to this work.
Davide Mei and Maurizio Taglialatela contributed equally to this work.
Funding information
The study was funded by the EU 7th Framework Programme (FP7) under the project DESIRE grant N602531 (to RG); the Tuscany Region Call for Health 2018 (grant DECODE‐EE) (to RG) and Federazione Italiana Epilessie (to RG and DM); the Italian Ministry for University and Research (MIUR) (PRIN 2017ALCR7C to MT and MVS, and PRIN 2017YH3SXK to FM); the Italian Ministry of Health (Project RF‐2019‐12370491) (to MT); the European Commission H2020 (UNICOM – 875299) (to MT); and the European Joint Programme on Rare Disease JTC 2020 (TreatKCNQ) (to MT).
REFERENCES
- 1. Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–40. [DOI] [PubMed] [Google Scholar]
- 2. Rogers A, Golumbek P, Cellini E, Doccini V, Guerrini R, Wallgren‐Pettersson C, et al. De novo KCNA1 variants in the PVP motif cause infantile epileptic encephalopathy and cognitive impairment similar to recurrent KCNA2 variants. Am J Med Genet A. 2018;176:1748–52. [DOI] [PubMed] [Google Scholar]
- 3. Paulhus K, Ammerman L, Glasscock E. Clinical spectrum of KCNA1 mutations: new insights into episodic ataxia and epilepsy comorbidity. Int J Mol Sci. 2020;21:2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Millichap JJ, Miceli F, De Maria M, Keator C, Joshi N, Tran B, et al. Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain‐of‐function variant. Epilepsia. 2017;58:e10–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miceli F, Soldovieri MV, Ambrosino P, De Maria M, Migliore M, Migliore R, Taglialatela M. Early‐onset epileptic encephalopathy caused by gain‐of‐function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J Neurosci. 2015;35:3782–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Imbrici P, Altamura C, Gualandi F, Mangiatordi GF, Neri M, De Maria G, et al. A novel KCNA1 mutation in a patient with paroxysmal ataxia, myokymia, painful contractures and metabolic dysfunctions. Mol Cell Neurosci. 2017;83:6–12. [DOI] [PubMed] [Google Scholar]
- 7. Liu X, Li C, Mou C, Dong Y, Tu Y. dbNSFP v4: a comprehensive database of transcript‐specific functional predictions and annotations for human nonsynonymous and splice‐site SNVs. Genome Med. 2020;12:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bezanilla F. How membrane proteins sense voltage. Nat Rev Mol Cell Biol. 2008;9:323–32. [DOI] [PubMed] [Google Scholar]
- 10. Yuan H, Yuan H, Wang Q, Ye W, Yao R, Xu W, et al. Two novel KCNA1 variants identified in two unrelated Chinese families affected by episodic ataxia type 1 and neurodevelopmental disorders. Mol Genet Genomic Med. 2020;8:e1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parrini E, Marini C, Mei D, Galuppi A, Cellini E, Pucatti D, et al. Diagnostic targeted resequencing in 349 patients with drug‐resistant pediatric epilepsies identifies causative mutations in 30 different genes. Hum Mutat. 2017;38:216–25. [DOI] [PubMed] [Google Scholar]
- 12. Russo A, Gobbi G, Pini A, Møller RS, Rubboli G. Encephalopathy related to status epilepticus during sleep due to a de novo KCNA1 variant in the Kv‐specific Pro‐Val‐Pro motif: phenotypic description and remarkable electroclinical response to ACTH. Epileptic Disord. 2020;22:802–6. [DOI] [PubMed] [Google Scholar]
- 13. Eunson LH, Rea R, Zuberi SM, Youroukos S, Panayiotopoulos CP, Liguori R, et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol. 2000;48:647–56. [PubMed] [Google Scholar]
- 14. Syrbe S, Hedrich UBS, Riesch E, Djémié T, Müller S, Møller RS, et al. De novo loss‐ or gain‐of‐function mutations in KCNA2 cause epileptic encephalopathy. Nat Genet. 2015;47:393–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Masnada S, Hedrich UBS, Gardella E, Schubert J, Kaiwar C, Klee EW, et al. Clinical spectrum and genotype‐phenotype associations of KCNA2‐related encephalopathies. Brain. 2017;140:2337–54. [DOI] [PubMed] [Google Scholar]
- 16. Hedrich UBS, Lauxmann S, Wolff M, Synofzik M, Bast T, Binelli A, et al. 4‐Aminopyridine is a promising treatment option for patients with gain‐of‐function KCNA2‐encephalopathy. Sci Transl Med. 2021;13:eaaz4957. [DOI] [PubMed] [Google Scholar]
- 17. Peters CJ, Werry D, Gill HS, Accili EA, Fedida D. Mechanism of accelerated current decay caused by an episodic ataxia type‐1‐associated mutant in a potassium channel pore. J Neurosci. 2011;31:17449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–8. [DOI] [PubMed] [Google Scholar]
- 19. Chen X, Wang Q, Ni F, Ma J. Structure of the full‐length Shaker potassium channel Kv1.2 by normal‐mode‐based X‐ray crystallographic refinement. Proc Natl Acad Sci USA. 2010;107:11352–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Niday Z, Tzingounis AV. Potassium channel gain of function in epilepsy: an unresolved paradox. Neuroscientist. 2018;24:368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Du W, Bautista JF, Yang H, Diez‐Sampedro A, You S‐A, Wang L, et al. Calcium‐sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37:733–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1