

Summary
Plants evolved in association with a diverse community of microorganisms. The effect of plant phylogeny and domestication on host–microbiome co‐evolutionary dynamics are poorly understood.
Here we examined the effect of domestication and plant lineage on the composition of the endophytic microbiome of 11 Malus species, representing three major groups: domesticated apple (M. domestica), wild apple progenitors, and wild Malus species.
The endophytic community of M. domestica and its wild progenitors showed higher microbial diversity and abundance than wild Malus species. Heirloom and modern cultivars harbored a distinct community composition, though the difference was not significant. A community‐wide Bayesian model revealed that the endophytic microbiome of domesticated apple is an admixture of its wild progenitors, with clear evidence for microbiome introgression, especially for the bacterial community. We observed a significant correlation between the evolutionary distance of Malus species and their microbiome.
This study supports co‐evolution between Malus species and their microbiome during domestication. This finding has major implications for future breeding programs and our understanding of the evolution of plants and their microbiomes.
Keywords: bacterial community, endophytes, fungal community, microbial introgression, microbiota, phylosymbiosis
Introduction
The evolution of plants has occurred with diverse microbial communities inhabiting their tissues (Yeoh et al., 2017; Delaux & Schornack, 2021). Collectively called the plant microbiome, these microorganisms fulfill important functions for their host’s health by manipulating its gene expression, hormonal pathways and increasing its tolerance to biotic and/or abiotic stresses (Berg et al., 2020). Empirical evidence shows that the phenotypic expression of host traits is due to the combined genetic expression of the host and the host‐associated microbiome (Matsumoto et al., 2021; Ravanbakhsh et al., 2021).
The long and intimate history of plant–microbiome associations, the importance of host genetics in shaping the microbiome, and the profound effect that microbiomes have on the traits of their hosts indicate that plants and their associated microbiomes are co‐evolving (Krings et al., 2007; Delaux & Schornack, 2021). Such co‐evolutionary dynamics may be reflected in a tendency of closely related plant species to host similar microbial communities, also known as phylosymbiosis (Brucker & Bordenstein, 2013; Theis et al., 2016; Mazel et al., 2018). Phylosymbiosis has been demonstrated in several plant groups, with stronger phylosymbiotic patterns for endophytic than epiphytic or rhizosphere‐associated microbiomes (Bouffaud et al., 2014; Schlaeppi et al., 2014; Vincent et al., 2016; Mazel et al., 2018; Mendes et al., 2018; Abdullaeva et al., 2020; Kim et al., 2020). These observations were suggested to be governed by two mutually nonexclusive mechanisms, namely heritability and inheritance (Peiffer et al., 2013; Beilsmith et al., 2019). Heritability refers to how the host genotype affects the assembly of the plant microbiome from the environment (Beilsmith et al., 2019; Wagner, 2021), and inheritance refers to the microbial community that is vertically transmitted to subsequenct generations via seeds (Beilsmith et al., 2019; Abdelfattah et al., 2021b; Wagner, 2021). In this context, it is important to understand the impact of domestication on the co‐evolutionary dynamics between plants and their microbiomes, and test whether phylosymbiotic patterns extend from the phylogeny of wild species to their domesticated progenies.
Domestication and breeding history can have a major impact on the diversity, abundance, and composition of the plant microbiome. Some domesticated plants were found to have a distinct microbial community composition and a lower capacity to interact with microbial symbionts, as compared to their wild relatives (Mutch & Young, 2004; Kiers et al., 2007; Leff et al., 2017; Pérez‐Jaramillo et al., 2018; Porter & Sachs, 2020; Favela et al., 2021). The majority of studies have shown a decrease in microbial species diversity with domestication (Bulgarelli et al., 2015; Coleman‐Derr et al., 2016). However, others have reported increased diversity, e.g. in lettuce, cereal seeds (wheat and barley) and common bean (Cardinale et al., 2015; Abdullaeva et al., 2020), or no effect as in the case of wheat and sunflower (Leff et al., 2017; Spor et al., 2020). Among domesticated plants, phylosymbiosis has been observed, for example in Poaceae roots, seeds, and rhizosphere (Bouffaud et al., 2014; Abdullaeva et al., 2020; Favela et al., 2021). Moreover, recent studies on microbiomes of breeding lines showed that hybrid plants share a large fraction of their bacterial community with their parents as for example in the case of Cucurbita seeds and apple shoot endophytes (Adam et al., 2018; Liu et al., 2018; Kusstatscher et al., 2021a). Yet, it is unclear if the propotional contrubution of the microbiome from parents to offpring correspond to amount of genetic material contributed by each parent during breeding and/or domestication.
One particularly suitable system to explore the impact of domestication on the diversity and composition of the microbiome, as well as phylosymbiosis, is apple. Apple (Malus × domestica), was primarily domesticated about 4000–10 000 yr ago from Malus sieversii (Ldb.) Roem, whose center of origin is the Tian Shan Mountains of Central Asia. Apple germplasm thereafter moved westwards by people traveling along the Silk Route (Cornille et al., 2012; Duan et al., 2017), during which time other Malus species, such as M. prunifolia (Willd.) Borkh in Asia, M. orientalis Uglitz. in the Caucasus, and M. sylvestris Mill. in Europe contributed to the genome of M. domestica through introgressive hybridization (Cornille et al., 2014; Volk et al., 2015; Volk, 2019). The domestication of apple has resulted in thousands of cultivars with large fruits, high yield, firm flesh, and high sugar content (Duan et al., 2017). Heirloom cultivars are older varieties for which genetic pedigree information is lacking, are known to have existed for long periods of time but have not necessarily served as founding genotypes for the more modern cultivars that have been developed. The microbiome of domesticated apple is highly diverse, and has been shown to be affected by plant genotype, management practices, soil composition, postharvest treatments, geographical location, and health status (Shade et al., 2013; Abdelfattah et al., 2016, 2020, 2021a; Shen et al., 2018; Wassermann et al., 2019a, 2019b,2019a, 2019b; Cui et al., 2021; Whitehead et al., 2021). Little is known, however, about the composition of the endophytic microbiome of wild Malus species or the impact of domestication on the microbiome of M. domestica and its many cultivars.
The current study examined the endophytic microbial communities of Malus species collected from the United States Department of Agriculture – Agricultural Research Service (USDA‐ARS) Apple Germplasm Repository in Geneva, NY, USA. The common garden setup was used to overcome geographical variation in environmental factors, such as soil properties, environmental microbiome, and climatic conditions. The study focused on endophytes, since they are less likely to be environmental contaminants than epiphytes and are expected to have the most intimate relationship with their host (Hardoim et al., 2015). The microbial communities were identified using amplicon sequencing and their abundance was quantified by quantitative real‐time polymerase chain reaction (qPCR). The aims of the study were to:
investigate the effect of apple domestication on the diversity, abundance and composition of the endophytic microbial community by comparing domesticated apple to wild progenitor and wild Malus species, and by comparing heirloom and modern cultivars
determine whether phylosymbiotic patterns can be observed among Malus species
determine the origin of the M. domestica microbiome by estimating the proportional contribution of wild Malus species to domesticated apple
Regarding species diversity, richness, and evenness, we assumed three scenarios to be equally likely during apple domestication (Scenarios 1–3 in Fig. 1). A reduction in species interactions has been associated with domestication, which would result in a less diverse microbiome in M. domestica than in wild Malus species (Mutch & Young, 2004; Kiers et al., 2007; Leff et al., 2017; Pérez‐Jaramillo et al., 2018; Porter & Sachs, 2020; Favela et al., 2021) (Scenario 1 in Fig. 1). Secondly, introgressive hybridization during the domestication of apple may have been accompanied by the introgression of the microbiome, resulting in higher diversity in M. domestica (Cardinale et al., 2015; Abdullaeva et al., 2020) (Scenario 2 in Fig. 1). Lastly, the microbiome may have been reshuffled during domestication, without an increase or decrease in the number of species (Scenario 3 in Fig. 1). Importantly, these scenarios are not mutually exclusive, and processes within each scenario could counteract or complement each other. Similar scenarios are expected to play out in the case of heirloom and modern cultivars. We also expected to find a strong correlation between the phylogenetic distance among Malus species and the community composition of their associated microbiomes (Bouffaud et al., 2014; Schlaeppi et al., 2014; Vincent et al., 2016) (Fig. 1). Specifically, we anticipated (1) the M. domestica microbiome to be more similar to its wild progenitors, especially to its main ancestor M. sieversii, than to wild, nonprogenitor species; (2) a correlation between the genetic distance of wild species of North American and Asian origin and their microbial community composition; and (3) that the introgression events consisted of both genetic and microbial contributions, which together shaped the microbiome of the domesticated apple (Fig. 1).
Fig. 1.
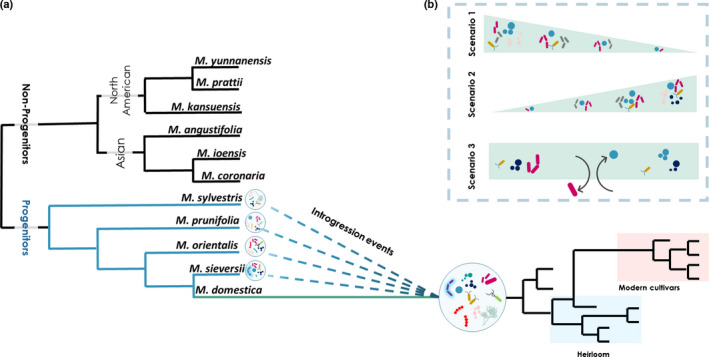
A conceptual figure on the impact of domestication on the plant endophytic microbiome. (a) A phylogenetic distance among Malus species which contains wild species (black branches) and progenitor wild species (blue branches). The extended green branch represents Malus domestica with its close affiliation its main ancestor (M. sieversii). Dashed lines indicate introgression events between Malus progenitors which contributed to the formation of M. domestica. (b) The predicted three scenarios: Scenario 1, reduction in species diversity due to loss in microbial species; Scenario 2, increase in microbial diversity due to introgressive hybridization during the apple domestication; Scenario 3, diversity was not affected by domestication.
Materials and Methods
Plant material and experimental design
Samples were collected from an orchard of the Apple Germplasm Repository of the USDA‐ARS, located in Geneva, NY, USA, to limit the effects of geographical location or climate on the microbiome. The orchard is characterized by high lime developed soil on glacial till with an average annual temperature of 9.4°C and rainfall of 89 cm. The selection of accessions was based on chloroplast haplotypes classification reported by Volk et al. (2015), in which the accessions were grouped into three clades: North American, Asian, and M. domestica admixture (Supporting Information Table S1). The present experiment was set up to include 61 apple accessions from 11 Malus species, representing three major groups: (1) domesticated apple cultivars (M. domestica), (2) wild progenitors of domesticated apple, and (3) nonprogenitors wild Malus species that did not contribute to apple domestication. While both the wild progenitors and the nonprogenitors represent wild apple species, for the purpose of simplicity and readability, we will use the term ‘wild’ only for the nonprogenitor species. The domesticated apple was represented by 18 accessions, representing seven heirloom cultivars (Fenouillet Gris, Miron Sacharanij, Coat Jersey, Landsberger Reinette, Borowitsky, Taylors, Fillbarrel) and six modern cultivars (Delicious, Golden Delicious, Honeycrisp, Northern Spy, Splendour, Frostbite). Apple wild progenitors included M. sieversii (nine accessions), M. sylvestris (three accessions), M. orientalis (seven accessions) and M. prunifolia (six accessions). Lastly, the wild Malus species consisted of M. kansuensis (three accessions), M. prattii (three accessions), M. yunnanensis (three accessions), M. angustifolia (three accessions), M. ioensis (three accessions), and M. coronaria (three accessions). The majority of the collected accessions were originally planted as seeds, self‐rooted cuttings or grafted onto EMLA7 (Table S1).
Collection and processing of samples
Shoots, c. 7–10 mm in diameter and 15 cm in length, were collected on November 2018 from current‐year growth. Four shoots of every accession were surface sterilized with 5% sodium hypochlorite (v/v) and rinsed three times in sterile water. The bark was removed with a sterile razor, as described in Liu et al. (2018). After peeling the bark, shoots were cut into 0.5–1.0 cm segments, and transferred to 50‐ml Falcon tubes with 40 ml sterile double distilled water (ddH2O) and shaken horizontally at 200 rpm for 30 min. The suspension was then transferred to centrifuge tubes and centrifuged at 12 000 g for 30 min. Pelleted solutions were used for DNA extraction with DNAeasy PowerSoil Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instruction. DNA quality and integrity were assessed using a NanoDrop UV‐spectrophotometer (ThermoFisher Inc., Grand Island, NY, USA). The same DNA extracts were used for amplicon library preparation and qPCR measurements.
Library preparation and sequencing
Library preparation and sequencing was conducted as previously described (Abdelfattah et al., 2020). Briefly, bacterial 16S rDNA region was amplified using the universal primers 515F and 806R in conjunction with peptide nucleic acids (PNAs) added to reduce the amplification of plant chloroplast and mitochondrial sequences. For fungal amplification, internal transcribed spacer (ITS) amplicons were produced using ITS3/KYO2 and ITS4 primers along with a custom‐designed blocking oligo designed to inhibit amplification of host apple sequences. For prokaryote 16S and fungal ITS amplicon generation, PCR reactions were conducted in a total volume of 25 μl containing 12.5 μl of KAPA HiFi HotStart ReadyMix (Kapa Biosystems, Wilmington, MA, USA), 1.0 μl of each primer (10 μM), and 2.5 μl of DNA template. For the 16S amplification, 2.5 μl of mitochondrial PNA (5 μM), 2.5 μl of plastid PNA (5 μM), and 3 μl nuclease‐free water, and in ITS amplification 1.0 μl of blocking oligo (10 μM) and 7 μl nuclease‐free water were added.
Reactions were incubated in a T100 thermal cycler (Bio‐Rad, Hercules, CA, USA) at 95°C for 5 min followed by 30 cycles of 95°C for 30 s, 78°C for 5 s, 55°C for 30 s, 72°C for 30 s, and a final extension step at 72°C for 5 min. For fungal (ITS) amplicon generation, at 95°C for 5 min followed by 30 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s and concluding with a final extension at 72°C for 5 min. Library preparation following amplicon PCR was performed as specified in the Illumina 16S Metagenomic Sequencing Library Preparation guide as outlined in conjunction with the use of a Nextera Index Kit (Illumina, San Diego, CA, USA) containing 96 indexes. Subsequent library size, quality, and confirmation of the absence of adapter dimers was checked on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Paired‐end sequencing of amplicons was done on an Illumina MiSeq (Illumina) sequencer with a V3 600‐cycle Reagent Kit (Illumina).
Bioinformatic and statistical analyses
Demultiplexing, quality trimming of low‐quality reads and creation of amplicon sequence variants (ASV) were done using the default parameters in Dada2 algorithm as integrated in Callahan et al. (2016) and Bolyen et al. (2019). Taxonomic assignment of the ASVs was done using Blast algorithm against the Silva 138 and UNITE databases for 16S and ITS reads, respectively (Abarenkov et al., 2010; Quast et al., 2012). MetagenomeSeq’s cumulative sum scaling (CSS) (Paulson et al., 2013) was used to account for uneven sequencing depth and then used for downstream analyses including Bray–Curtis dissimilarity metrics (Bray & Curtis, 1957), hierarchical clustering analysis and permutational multivariate analysis of variance (PERMANOVA). The rarefy_even_depth function implemented in the R package phyloseq v.1.32.0 was used to resample the ITS and 16S ASV tables to an even library size of 5000 and 1000, respectively. The rarefied tables were then used to calculate fungal and bacterial diversity using Shannon index (McMurdie & Holmes, 2013).
To evaluate the effect of domestication on the richness, evenness, diversity, and composition of the fungal and bacterial community, we modeled each response variable as a function of the fixed effect ‘domestication group’ (domesticated, wild progenitor and wild species) and species identity. Models with the univariate response variables were implemented using the function aov in R Package stats v.4.0.1 in R v.3.6.2 (R Core Team, 2020). Group means were compared using Tukey multiple comparisons of means.
Community composition was modeled using the function adonis2 (PERMANOVA) in the package vegan (Oksanen et al., 2007). Pairwise comparisons were made using Pairwise Multilevel Comparison R package pairwiseadonis (Arbizu, 2019). To understand whether the change in microbial community composition is due to species turnover or species loss or gain, we calculated community dissimilarity using binary Jaccard dissimilarity index for each domestication group and then partitioned the calculated indexes into species turnover (βJTU) and species gain or loss (βJNE) (Baselga & Orme, 2012). To understand the effect of domestication on the plant core microbiome, we first calculated the core microbiome after transforming the ASV table into compositional data, keeping ASVs present in at least 70% of all samples within each domestication category, i.e. M. domestica, wild progenitors, and wild species, using microbiome R package v.1.10.0 (Leo Lahti & Shetty, 2017). We then compare the number of fungal and bacterial ASVs among the three domestication groups to determine if they all share the same core species, increase, or decrease along the chronosequence of Malus germplasm.
To quantify the relationship between Malus phylogeny and its microbiome (Brooks et al., 2016), we first used hierarchical clustering based on Bray–Curtis dissimilarity distances with ‘average’ as the clustering method. This was performed using hclust in R package stats v.4.0.1 and the results were visualized using fviz_dend function in the R package factoextra v.1.0.7 (Bray & Curtis, 1957; Kassambara & Mundt, 2020). Second, to calculate phylogenetic distances among Malus species, the ITS regions of the investigated species in addition to Pyrus communis as outgroup, were retrieved from National Center for Biotechnology Information (NCBI), aligned using the MUSCLE algorithm (list of accessions with alignment is available in Table S2), and the phylogenetic distance was inferred by using the neighbor‐joining tree estimation in R package phangorn (Saitou & Nei, 1987; Schliep, 2010). To quantify the congruence between Malus phylogeny and the microbiome, the topologies of the constructed dendrograms of the fungal and bacterial community were compared to the phylogenetic tree of Malus species, using Procrustes test in vegan (Peres‐Neto & Jackson, 2001).
To estimate the potential contribution of Malus species to the microbiome of domesticated apple, we used SourceTracker2, a Bayesian approach originally developed to estimate the environmental sources of the microbial community (Knights et al., 2011). In this analysis, each Malus species was assigned as a potential source and M. domestica was assigned as the only sink. Both sources and sink were rarefied to 1500 reads per sample.
Quantitative real‐time polymerase chain reaction
Bacterial and fungal abundance was measured by qPCR using the primer pairs ITS1–ITS2 for fungi (White et al., 1990) and 515f–927r for bacteria (Köberl et al., 2011). Reaction mixtures contained 5 μl KAPA SYBR Green, 0.5 μl (10 μM each) of each primer, 1 μl template DNA, adjusted with PCR‐grade water to a final volume of 10 µl. A Rotor‐Gene 6000 real‐time rotary analyzer (Corbett Research, Sydney, NSW, Australia) was used to detect fluorescence intensity using the following cycling conditions: bacteria: 95°C for 3 min, 30 cycles of 95°C for 5 s, 54°C for 20 s, 72°C for 5 s, and a final melt curve of 72–96°C; fungi: 95°C for 3 min, 40 cycles of 95°C for 5 s, 58°C for 35 s, 72°C for 5 s with a final melt at 72°C for 10 min and a final melt curve of 72–96°C. For each sample replicate, three individual qPCR runs were conducted, and standard curves were constructed to determine the efficiency and linear range of the assay. Intermittently occurring gene copy numbers that were detected in negative control samples were subtracted from the respective run. Nonparametric Kruskal–Wallis test was applied to calculate statistical differences in bacterial and fungal abundance between groups and the P‐values were corrected using Bonferroni multiple test correction (Kruskal & Wallis, 1952).
Results
Sequencing results and the microbiome composition of Malus species
MiSeq sequencing yielded a total of 5813 667 ITS and 609 147 16S high‐quality reads after the removal of chimeras, plant chloroplast and mitochondrial sequences, and low‐quality reads. The clean reads were assigned to 2693 ITS fungal ASVs and 2688 bacterial ASVs. The number of sequences varied among samples, ranging from 81 147 to 1902 518 ITS reads and 10 578 to 187 894 16S reads. The fungal community was dominated by Ascomycota (71.2%) and Basidiomycota (27.0%), in which the genera Aureobasidium (45.4%), an unidentified genus of Pleosporaceae (13%), and Filobasidium (9.2%), prevailed. The bacterial community was dominated by Proteobacteria (82.9%), Actinobacteria (10.5%), and Bacteroidetes (2.8%). The predominant genera were Sphingomonas (23.5%), Pseudomonas (15.5%), and Methylobacterium‐Methylorubrum (15%) (Fig. S1).
The effect of domestication on the microbial community
Fungal species richness and diversity differed significantly among M. domestica, wild progenitors, and wild species (P = 0.0048 and P = 0.0012, respectively). Both richness and diversity were significantly higher in domesticated apple (M. domestica) and its wild progenitors than in wild species (Fig. 2a,b; Table S3). These estimates correspond also to absolute abundance of fungal gene copies numbers, measured via qPCR: endophytes of domesticated apple (P < 0.001) and its wild progenitors (P = 0.04) were significantly more abundant than those of wild apples, while no difference was found between progenitor and domesticated species (Fig. S2a; Table S4). The fungal community composition differed significantly among domesticated apple, progenitor species, and wild species (R 2 = 0.052, P = 0.001) (Fig. 2c), which was confirmed by pairwise comparisons (Table S4). Partitioning of beta diversity indicated that species turnover was the dominant factor in the differences observed in the fungal communities of wild, progenitor, and domesticated species (Fig. S2).
Fig. 2.
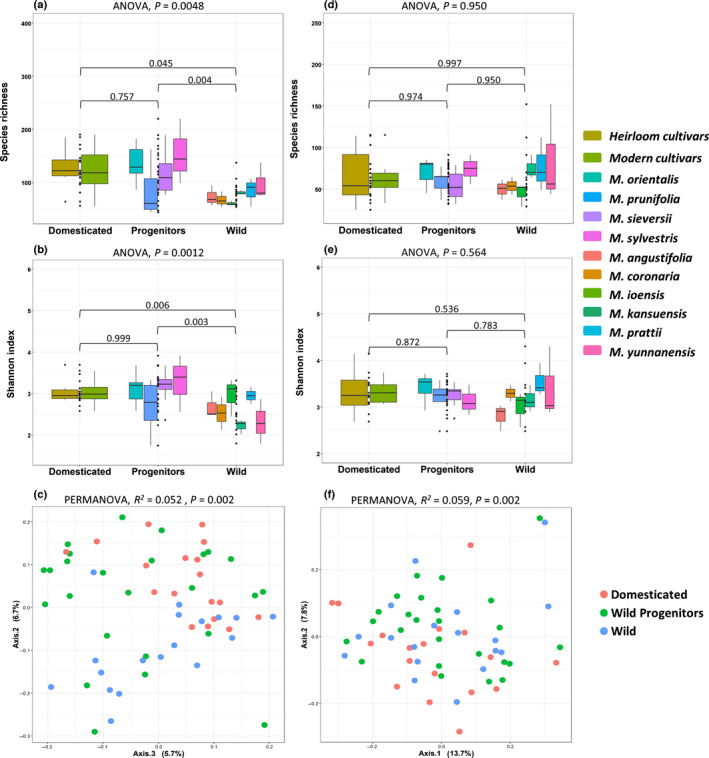
Box plots showing fungal (a–c) and bacterial (d–f) richness, Shannon diversity and community composition. The presented species were grouped into three groups from left to right: heirloom and modern cultivars of domesticated apple (Malus × domestica), wild progenitors (M. orientalis, M. prunifolia, M. sieversii, and M. sylvestris), and nonprogenitor Malus species (M. angustifolia, M. coronaria, M. ioensis, M. kansuensis, M. prattii, and M. yunnanensis). Superimposed on the box plots are the horizontally jittered raw data points combined for each domestication group. Box plots show the median (horizontal line), the lower and upper bounds of each box plot denote the first and third quartiles, and whiskers above and below the box plot show 1.5 times the interquartile range. The points located outside of the whiskers of the box plot represent the outliers. Ordination plots of fungal (c) and bacterial (f) community composition of Malus × domestica, wild progenitors and wild Malus species, based Bray–Curtis dissimilarity index. Results of the global statistical analyses are reported at the top of each panel and pairwise comparisons for alpha diversity are added onto the box plots.
In contrast to fungi, bacterial species richness, evenness, and diversity did not differ among the domesticated apple, progenitor species, and wild species (Fig. 2d; Table S3). However, domestication had a significant effect on the bacterial community composition (R 2 = 0.059, P = 0.002) (Fig. 2e). Pairwise comparison showed that nonprogenitor wild species differed significantly from both wild progenitors and domesticated apples (R 2 = 0.047, P = 0.003, R 2 = 0.062, P = 0.003), whereas the comparison between wild progenitors and domesticated apple showed no significant differences (R 2 = 0.029, P = 0.123) (Table S4). Measurements of qPCR, however, revealed bacterial endophytes to be significantly less abundant in wild species compared to domesticated apples (P < 0.001); no difference in bacterial abundance was observed between the other domestication groups (Fig. S2b).
The apple core microbiome (defined as ASVs present in 70% of the replicates of each domestication group), comprised 18 fungal and 12 bacterial ASVs (Fig. 3c). These core taxa represented a high fraction of the total microbial community, measured via qPCR, with ASVs annotated to Sphingomonas, Pseudomonas, Methylobacterium and Aureobasidium being most abundant among all apple groups. Core fungi accounted for 75%, 62%, and 63% of total ITS copy numbers detected in wild, progenitor, and domesticated apple, respectively. Abundance of core bacteria was slightly less for the three domestication groups, representing 48%, 45%, and 49% of total 16S rRNA gene copy numbers, respectively. In addition to those shared ASVs, six bacterial ASVs were unique to the wild species core microbiome, three bacterial and one fungal ASVs were unique to progenitor species, and three bacterial and 18 fungal ASVs were only detected in the domesticated apple core microbiome. The fraction of the unique core taxa in wild species was < 0.01% for fungi and 4.2% for bacteria, in progenitor species it was 0.2% for fungi and 1.2% for bacteria, and in domesticated apple it was 21% for fungi and 2.6% for bacteria.
Fig. 3.
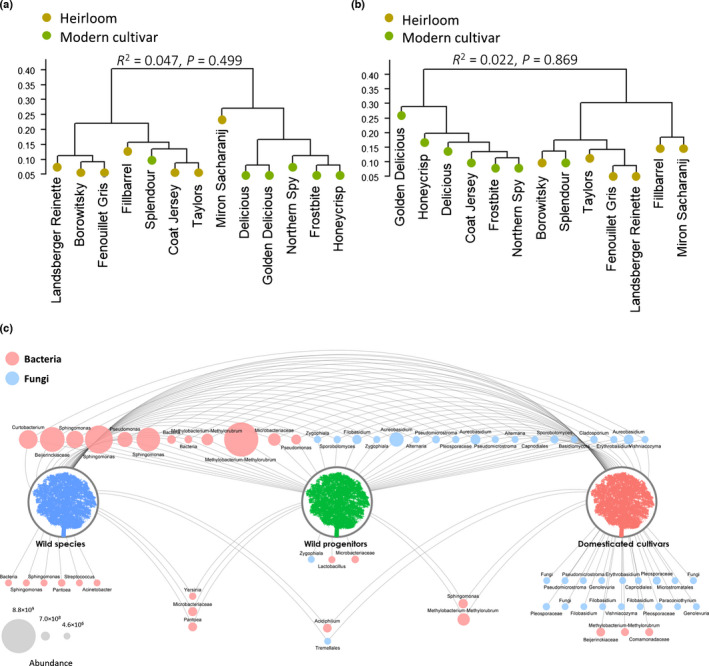
Dendrogram based on the similarity of the core fungal (a) and bacterial (b) community composition, according to Bray–Curtis index among Malus domestica cultivars, highlighting the difference between heirloom and modern cultivars. The dendrograms were visualized using the fviz_dend function in the R package factoextra v.1.0.7. Results of the global statistical analyses are reported at the top of each panel. (c) Network analysis showing the core microbiome distribution from wild Malus species, to progenitors, to domesticated apple. Blue and red circles (nodes) represent fungal and bacterial taxa, respectively. The core microbiome was calculated for each Malus group separately as amplicon sequence variants present in at least 70% of the samples. Node size corresponds to bacterial and fungal abundance, i.e. gene copy numbers measured by qPCR, as indicated in the legend on the lower left.
Within M. domestica, heirloom and modern cultivars did not differ significantly in their species richness (fungi: P = 0.999 and bacteria: P = 0.681) nor Shannon diversity (fungi: P = 0.832 and bacteria: P = 0.474) (Fig. 2). This was also true regarding community composition of their core microbiomes (fungi: R 2 = 0.047, P = 0.499 and bacteria: R 2 = 0.022, P = 0.869). Despite the lack of statistical differences, a hierarchical analysis revealed a clear distinction between the modern and heirloom cultivars, with two exceptions (Splendour and Miron Sacharanij), for both the fungal and bacterial core communities (Fig. 3a,b).
The effect of phylogeny on the community composition of Malus species
Hierarchical clustering, based on Bray–Curtis dissimilarity metrics of the fungal and bacterial community composition, revealed that M. domestica and its progenitor species (M. sieversii, M. prunifolia, and M. orientalis) clustered separately from wild Malus species. The sole exception to this clustering was M. sylvestris which clustered with M. yunnanensis and M. kansuensis in the fungal and bacterial trees, respectively (Fig. 4a,b). This pattern of clustering largely corresponded with the phylogeny of Malus species (Fig. 4c). The correlation between the evolutionary distance of Malus species and their associated microbial communities was found to be significant for both fungal (R 2 = 0.471, P = 0.024) and bacterial (R 2 = 0.465, P = 0.035) communities, based on Procrustes analysis. The correlation between the phylogenetic distance and the core microbiome of each Malus species, however, was not significant for either fungi (R 2 = 0.5624, P = 0.119) or bacteria (R 2 = 0.297, P = 0.158).
Fig. 4.
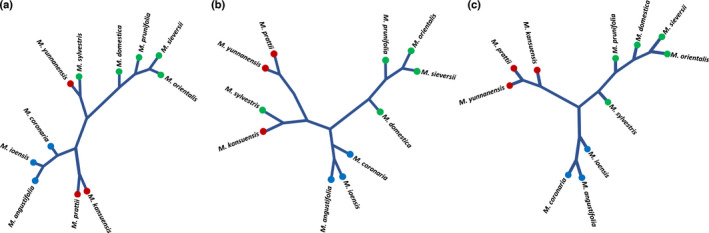
Results of hierarchical clustering based on Bray–Curtis dissimilarity distances of the fungal (a) and bacterial (b) community composition using clustering method ‘average’. (c) Shows the phylogenetic tree based on Malus ITS gene. Malus phylogenetic distance was inferred by using the neighbor‐joining tree estimation in R package phangorn. The leaf color indicates Malus groups: green = Malus × domestica and its wild progenitors (M. sieversii, M. orientalis, M. prunifolia, and M. sylvestris), blue = North American species (M. angustifolia, M. coronaria, and M. ioensis), and red = Asian species (M. kansuensis, M. yunnanensis, and M. prattii). The phylogenic plots were visualized using the fviz_dend function in the R package factoextra v.1.0.7.
Estimating the origin of the Malus microbiome
The community‐wide Bayesian model estimated that the majority of the fungal community of M. domestica originated from its progenitor species M. sieversii (51%), with an equal contribution from M. sylvestris (7%), M. yunnanensis (7%), and wild M. angustifolia (7%), followed by M. prattii (5%), M. ioensis (5%), and M. orientalis (5%) (Fig. 5a). The three progenitor species M. sieversii (23%), M. orientalis (21%), and M. sylvestris (16%) were the main source of the M. domestica bacterial community (Fig. 5b). A detailed description of ASVs estimated to have contributed to M. domestica from other Malus species is presented in Fig. S3.
Fig. 5.
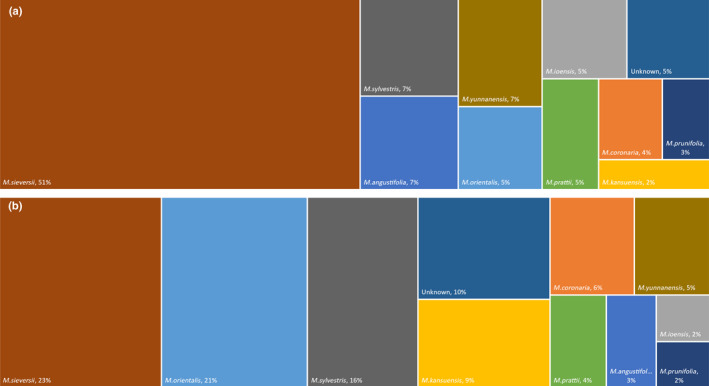
Treemap charts showing the estimated sources of the fungal (a) and bacterial (b) communities in Malus domestica. The estimates were calculated using Bayesian approach as implemented in SourceTracker2 by setting M. domestica as the sole sink and all the other Malus species (M. sieversii, M. orientalis, M. prunifolia, M. sylvestris, M. kansuensis, M. yunnanensis, M. angustifolia, M. coronaria, M. ioensis, and M. prattii) as potential sources. An unknown source was added automatically by the algorithm to allocate taxa in M. domestica with low probability to have originated from any of the assigned sources. The fungal and bacterial communities were rarefied to 1500 reads per sample in both the sink and sources.
Discussion
We examined the effects of domestication and species identity on the endophytic microbiome of Malus, including wild apple species, apple wild progenitors, and heirloom and modern cultivars of domesticated apple. We demonstrated (1) that the fungal community associated with domesticated apples and their wild progenitors had a higher species richness, diversity, abundance and distinct fungal and bacterial community compared to wild species, (2) strong evidence of phylosymbiosis for both fungal and bacterial communities, and (3) the microbiome of the domesticated apple to be an admixture of its wild progenitors with clear evidence of introgression for the bacterial community.
Impact of domestication on microbial diversity and composition
We observed significantly higher fungal richness, diversity, and abundance in domesticated apple compared to its wild ancestors. While this contrasts with the common hypothesis that domestication reduces microbial diversity (Mutch & Young, 2004; Kiers et al., 2007; Leff et al., 2017; Pérez‐Jaramillo et al., 2018; Porter & Sachs, 2020; Favela et al., 2021), it is in agreement with some previous reports (Cardinale et al., 2015; Abdullaeva et al., 2020). The increase in fungal diversity indicates that domestication resulted in a greater rate of species gain than loss. However, when considering that beta diversity was mainly explained by species turnover patterns, it is apparent that species gain was accompanied by considerable species turnover. The higher species diversity might be due to an increased niche size or amount of resources in domesticated apple. The increase in the resource availability is supported by the qPCR assays, which showed that domesticated Malus, both heirloom and modern cultivars, had a significantly higher quantity of microbial cells than their wild relatives. It might be that plant domestication and breeding for desirable traits have indirectly facilitated an increase in microbial population size. This hypothesis fits the evidence that a focus during domestication and breeding on increased yield, fruit size, water and sugar content results in lower levels of defense chemicals and stress resistance (Cornille et al., 2014; Whitehead & Poveda, 2019; Porter & Sachs, 2020). Indeed, wild apples have been shown to contain higher total phenolic concentrations and a higher diversity of metabolites than domesticated apples and are known to be more resistant to major apple diseases (Ballester et al., 2017; Sun et al., 2017; Whitehead & Poveda, 2019; Singh et al., 2021).
The bacterial core microbiome appeared to be relatively stable along the chronosequence of Malus germplasm, i.e. from wild to wild progenitor, and from wild progenitor to domesticated apples. However, the number of fungal core species was significantly higher in M. domestica compared to wild species. Collectively, the core microbiome represented a large fraction of the microbial community of wild, progenitor, and domesticated apple, accounting for two‐thirds and almost half of the abundance of the fungal and bacterial communities, respectively. The existence of such core microbiome that spans the Malus phylogeny suggests an evolutionary conservation of the core microbiome and is in agreement with an ecological role across evolutionary boundaries (Yeoh et al., 2017). The mechanism(s) by which such a community is maintained across the Malus phylogeny is difficult to pinpoint without further studies. Nevertheless, we speculate that continuous transmission of the core microorganisms across generations through vertical transmission, along with positive selection on retention of the core microbiome, represents a plausible explanation (Gundel et al., 2011; Hodgson et al., 2014; Shade et al., 2017; Bergna et al., 2018; Shahzad et al., 2018; Kim et al., 2020; Abdelfattah et al., 2021b). Our hypothesis is supported by recent findings including: (1) similar core species were reported in a global survey of the apple fruit microbiome, and (2) the role of microbial inheritance (vertical transmission) is increasingly being recognized to play an essential role in the continuity of the plant microbiome (Saikkonen et al., 2002; Gundel et al., 2011; Hodgson et al., 2014; Shade et al., 2017; Bergna et al., 2018; Shahzad et al., 2018; Kim et al., 2020; Abdelfattah et al., 2021a, 2021b,2021a, 2021b).
Evidence of phylosymbiosis
We found a significant correlation between the evolutionary distance of Malus species and their associated fungal and bacterial endophytic communities, which explained 47% of the observed differences. Based solely on the microbial community composition, we were able to distinguish between wild species from North America and Asia. However, the most intriguing result was the clustering of domesticated cultivars with their wild progenitors. These results demonstrate that phylosymbiosis exists in the genus Malus. Previous studies have reported phylosymbiosis between root‐associated bacterial communities and diverse groups of plants including lycopods, ferns, gymnosperms, and angiosperms (Bouffaud et al., 2014; Schlaeppi et al., 2014; Vincent et al., 2016), but, as far as we know, no such relationship has been reported for the fungal and bacterial endophytic community of Malus. These findings match the expectation that the genetic makeup of plants, driven by evolution and domestication, shapes the structure of the plant microbiome (Leff et al., 2017; Kim et al., 2020; Spor et al., 2020; Deng et al., 2021; Wagner, 2021). Conversely, the correlation between Malus phylogeny and the microbial community composition was not significant when considering the core microbiome. This is in agreement with the notion that rare taxa are important for structuring communities and for distinguishing between closely related plant species (Li et al., 2018; Ramirez et al., 2018; Berg et al., 2020). Rare taxa are also hypothesized to offer a pool of genetic resources that may be activated under the appropriate conditions (Jousset et al., 2017).
The origin of the M. domestica microbiome
The results of the Bayesian approach showed that M. sieversii, the main ancestor of M. domestica, accounted for 51% and 23% of the fungal and bacterial communities of domesticated apple, respectively. These results are in strong agreement with the genetic origin of domesticated apple. Although the microbiome of M. sylvestris clustered separately from the other wild progenitors, the Bayesian model showed it had contributed 16% of the bacterial community of M. domestica. This could be explained by the fact that Sphingomonas and Methylobacterium‐Methylorubrum, two highly abundant ASVs in both M. sylvestris and M. domestica were identified by the algorithm to originate from M. sylvestris. Such findings indicate that introgressive hybridization that occurred during domestication between apple progenitors comprised both genetic and microbial features, including some of the most important genera in domesticated apple. Although genetic hybridization is known as the incorporation of alleles from one species into the gene pool of another, the mechanisms by which microbial hybridization occurs has not, to the best of our knowledge, been studied. However, early studies on breeding show that similar mechanisms could apply for the transmission and hybridization of the microbiome (Adam et al., 2018; Kusstatscher et al., 2021a; Sahu & Mishra, 2021).
Evidence of co‐evolution
Whether plants and their microbiomes are co‐evolving or evolving together is a question that is still under debate (Theis et al., 2016; Limborg & Heeb, 2018). This is mainly because co‐evolution, in sensu stricto, is expected to result in reciprocal changes in the involved parties as, for example, in the case of pea‐aphid and its endosymbiotic bacteria Buchnera sp., whereby amino acid synthesis occurs through cooperation between host and symbiont (Brundrett, 2002; Russell et al., 2013). Another example, is the evolution of complementary traits between plants and mycorrhizal fungi, where fungi depend on the host plant carbon for energy consumption and hosts providing a more hospitable environment for fungi (Brundrett, 2002; Hoeksema, 2010). However, there is a growing body of literature that considers the microbiome as a superorganism and single unit of selection, in particular within the context of the holobiont framework (Theis et al., 2016; Ravanbakhsh et al., 2021; Tan et al., 2021). When taking this perspective, we think that the patterns of increased diversity with domestication, phylosymbiosis, and a strongly conserved core microbiome across the Malus phylogeny, together with evidence for concurrent plant and microbiome admixture and introgression, provide support for co‐evolution between Malus species and their microbiome during domestication. Yet, we caution that these patterns could also be explained by other, nonmutually exclusive mechanisms. For instance, phylosymbiosis can readily emerge from a simple ecological filtering process, where a host trait that varies with host phylogeny could act as a filter for preadapted microbes (Theis et al., 2016; Mazel et al., 2018; Beilsmith et al., 2019; Wagner, 2021). In which case, the metacommunity is also expected to play an important role since it represents the species pool of which plants and microorgamisns can establish their associations. To determine the underlying (co‐)evolutionary processes shaping the current patterns, we suggest that future studies use shotgun metagenomics and/or genome‐wide association studies to identify changes in the genetic composition of microbial species during domestication, combined with experiments that identify the functional role of any identified genetic variation.
Conclusion
Several recent initiatives have called for intentional manipulation of the plant microbiome to enhance crop performance and sustainability, especially in the framework of sustainable agroecosystems (Berg, 2009; D’Hondt et al., 2021; Favela et al., 2021; French et al., 2021). The pattern of phylosymbiosis, and the congruent pattern of plant and microbial admixture, indicates that changes in the plant microbiome can be predicted, and further supports the idea that microbiome‐based breeding strategies are feasible (Adam et al., 2018; Chen et al., 2020; Favela et al., 2021; Kusstatscher et al., 2021b). Overall, the increase in diversity, phylogenetic correlation, and the estimated origin of the apple microbiome indicate strongly towards microbial introgression events that occurred during Malus domestication and supports co‐evolution between plants and their microbiomes.
Author contributions
AA, JN, SD and MW conceptualized and designed the experiment. JL performed sample preparation and DNA extractions. BW performed and interpreted qPCR assays. AA analyzed and interpreted the amplicon data. AA wrote the first draft, and AJMT made a major contribution to the final version. GB, JN, SD, and MW contributed to the interpretation of the results and writing of the manuscript.
Supporting information
Fig. S1 Stacked bar charts showing the fungal (a) and bacterial (b) community composition of the most abundant taxa with a relative abundance > 0.1% across all the investigated Malus species.
Fig. S2 Microbial gene copy numbers in apple shoots determined by qPCR and the estimated beta diversity partitioning.
Fig. S3 Bar chart showing the estimated sources of the fungal (a) and bacterial (b) taxa in Malus domestica (modern apple).
Table S1 Metadata table with the information regarding Malus accessions used in the present study.
Table S2 List of ITS sequences used to calculate distances among Malus species and the phylogenetic trees.
Table S3 Analysis of variance on the effect of domestication and Malus phylogeny (species) on fungal and bacterial diversity based on Shannon index.
Table S4 Results of statistical pairwise‐comparisons of fungal and bacterial diversity based on Shannon index and community composition based on Bray–Curtis dissimilarity index using adonis (˜PERMANOVA).
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
This work was funded equally by the European Union's Horizon2020 under ‘Nurturing excellence by means of cross‐border and cross‐sector mobility’ program for MSCA‐IF‐2018‐Individual Fellowships, grant agreement 844114 (AA) and by BARD, Israel‐US Binational Agricultural Research and Development Fund (IS‐5040‐17) awarded to SD and MW.
Contributor Information
Ahmed Abdelfattah, Email: ahmed.abdelfattah@tugraz.at.
Michael Wisniewski, Email: wisniewski@vt.edu.
Data availability
The datasets generated and/or analyzed during the current study are available in the ‘SRA NCBI’ repository, and can be accessed from the following link https://www.ncbi.nlm.nih.gov/bioproject/PRJNA702287. The code used in this study is available on zenodo and can be accessed from the following link https://doi.org/10.5281/zenodo.5578000.
References
- Abarenkov K, Henrik Nilsson R, Larsson K‐H, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T et al. 2010. The UNITE database for molecular identification of fungi – recent updates and future perspectives. New Phytologist 186: 281–285. [DOI] [PubMed] [Google Scholar]
- Abdelfattah A, Freilich S, Bartuv R, Zhimo VY, Kumar A, Biasi A, Salim S, Feygenberg O, Burchard E, Dardick C et al. 2021a. Global analysis of the apple fruit microbiome: are all apples the same? Environmental Microbiology 23: 6038–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelfattah A, Whitehead SR, Macarisin D, Liu J, Burchard E, Freilich S, Dardick C, Droby S, Wisniewski M. 2020. Effect of washing, waxing and low‐temperature storage on the postharvest microbiome of apple. Microorganisms 8: 944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelfattah A, Wisniewski M, Droby S, Schena L. 2016. Spatial and compositional variation in the fungal communities of organic and conventionally grown apple fruit at the consumer point‐of‐purchase. Horticulture Research 3: 16047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelfattah A, Wisniewski M, Schena L, Tack AJM. 2021b. Experimental evidence of microbial inheritance in plants and transmission routes from seed to phyllosphere and root. Environmental Microbiology 23: 2199–2214. [DOI] [PubMed] [Google Scholar]
- Abdullaeva Y, Ambika Manirajan B, Honermeier B, Schnell S, Cardinale M. 2020. Domestication affects the composition, diversity, and co‐occurrence of the cereal seed microbiota. Journal of Advanced Research 31: 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam E, Bernhart M, Müller H, Winkler J, Berg G. 2018. The Cucurbita pepo seed microbiome: genotype‐specific composition and implications for breeding. Plant and Soil 422: 35–49. [Google Scholar]
- Arbizu M. 2019. Pairwise multilevel comparison using Adonis. R package v.0.4. [WWW document] URL https://github.com/pmartinezarbizu/pairwiseAdonis. [Google Scholar]
- Ballester A‐R, Norelli J, Burchard E, Abdelfattah A, Levin E, González‐Candelas L, Droby S, Wisniewski M. 2017. Transcriptomic response of resistant (PI613981–Malus sieversii) and susceptible (“Royal Gala”) genotypes of apple to blue mold (Penicillium expansum) infection. Frontiers in Plant Science 8: 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga A, Orme CDL. 2012. betapart: an R package for the study of beta diversity. Methods in Ecology and Evolution 3: 808–812. [Google Scholar]
- Beilsmith K, Thoen MPM, Brachi B, Gloss AD, Khan MH, Bergelson J. 2019. Genome‐wide association studies on the phyllosphere microbiome: embracing complexity in host–microbe interactions. The Plant Journal 97: 164–181. [DOI] [PubMed] [Google Scholar]
- Berg G. 2009. Plant–microbe interactions promoting plant growth and health: perspectives for controlled use of microorganisms in agriculture. Applied Microbiology and Biotechnology 84: 11–18. [DOI] [PubMed] [Google Scholar]
- Berg G, Rybakova D, Fischer D, Cernava T, Vergès M‐C, Charles T, Chen X, Cocolin L, Eversole K, Corral GH et al. 2020. Microbiome definition re‐visited: old concepts and new challenges. Microbiome 8: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergna A, Cernava T, Rändler M, Grosch R, Zachow C, Berg G. 2018. Tomato seeds preferably transmit plant beneficial endophytes. Phytobiomes Journal 2: 183–193. [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al‐Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using Qiime 2. Nature Biotechnology 37: 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouffaud M‐L, Poirier M‐A, Muller D, Moënne‐Loccoz Y. 2014. Root microbiome relates to plant host evolution in maize and other Poaceae. Environmental Microbiology 16: 2804–2814. [DOI] [PubMed] [Google Scholar]
- Bray JR, Curtis JT. 1957. An ordination of the upland forest communities of southern Wisconsin. Ecological Monographs 27: 325–349. [Google Scholar]
- Brooks AW, Kohl KD, Brucker RM, van Opstal EJ, Bordenstein SR. 2016. Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biology 14: e2000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brucker RM, Bordenstein SR. 2013. The hologenomic basis of speciation: gut bacteria cause hybrid lethality in the genus Nasonia . Science 341: 667–669. [DOI] [PubMed] [Google Scholar]
- Brundrett MC. 2002. Coevolution of roots and mycorrhizas of land plants. New Phytologist 154: 275–304. [DOI] [PubMed] [Google Scholar]
- Bulgarelli D, Garrido‐Oter R, Münch Philipp C, Weiman A, Dröge J, Pan Y, McHardy Alice C, Schulze‐Lefert P. 2015. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host & Microbe 17: 392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. Dada2: high‐resolution sample inference from Illumina amplicon data. Nature Methods 13: 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinale M, Grube M, Erlacher A, Quehenberger J, Berg G. 2015. Bacterial networks and co‐occurrence relationships in the lettuce root microbiota. Environmental Microbiology 17: 239–252. [DOI] [PubMed] [Google Scholar]
- Chen X, Krug L, Yang H, Li H, Yang M, Berg G, Cernava T. 2020. Nicotiana tabacum seed endophytic communities share a common core structure and genotype‐specific signatures in diverging cultivars. Computational and Structural Biotechnology Journal 18: 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman‐Derr D, Desgarennes D, Fonseca‐Garcia C, Gross S, Clingenpeel S, Woyke T, North G, Visel A, Partida‐Martinez LP, Tringe SG. 2016. Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytologist 209: 798–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornille A, Giraud T, Smulders MJM, Roldán‐Ruiz I, Gladieux P. 2014. The domestication and evolutionary ecology of apples. Trends in Genetics 30: 57–65. [DOI] [PubMed] [Google Scholar]
- Cornille A, Gladieux P, Smulders MJM, Roldán‐Ruiz I, Laurens F, Le Cam B, Nersesyan A, Clavel J, Olonova M, Feugey L et al. 2012. New insight into the history of domesticated apple: secondary contribution of the European wild apple to the genome of cultivated varieties. PLoS Genetics 8: e1002703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Huntley RB, Zeng Q, Steven B. 2021. Temporal and spatial dynamics in the apple flower microbiome in the presence of the phytopathogen Erwinia amylovora . The ISME Journal 15: 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Hondt K, Kostic T, McDowell R, Eudes F, Singh BK, Sarkar S, Markakis M, Schelkle B, Maguin E, Sessitsch A. 2021. Microbiome innovations for a sustainable future. Nature Microbiology 6: 138–142. [DOI] [PubMed] [Google Scholar]
- Delaux P‐M, Schornack S. 2021. Plant evolution driven by interactions with symbiotic and pathogenic microbes. Science 371: eaba6605. [DOI] [PubMed] [Google Scholar]
- Deng S, Caddell DF, Xu G, Dahlen L, Washington L, Yang J, Coleman‐Derr D. 2021. Genome wide association study reveals plant loci controlling heritability of the rhizosphere microbiome. The ISME Journal 15: 3181–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan N, Bai Y, Sun H, Wang N, Ma Y, Li M, Wang X, Jiao C, Legall N, Mao L et al. 2017. Genome re‐sequencing reveals the history of apple and supports a two‐stage model for fruit enlargement. Nature Communications 8: 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favela A, Bohn MO, Kent AD. 2021. Maize germplasm chronosequence shows crop breeding history impacts recruitment of the rhizosphere microbiome. The ISME Journal 15: 2454–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French E, Kaplan I, Iyer‐Pascuzzi A, Nakatsu CH, Enders L. 2021. Emerging strategies for precision microbiome management in diverse agroecosystems. Nature Plants 7: 256–267. [DOI] [PubMed] [Google Scholar]
- Gundel PE, Rudgers JA, Ghersa CM. 2011. Incorporating the process of vertical transmission into understanding of host–symbiont dynamics. Oikos 120: 1121–1128. [Google Scholar]
- Hardoim PR, Van Overbeek LS, Berg G, Pirttilä AM, Compant S, Campisano A, Döring M, Sessitsch A. 2015. The hidden world within plants: ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiology and Molecular Biology Reviews 79: 293–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson S, de Cates C, Hodgson J, Morley NJ, Sutton BC, Gange AC. 2014. Vertical transmission of fungal endophytes is widespread in forbs. Ecology and Evolution 4: 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeksema JD. 2010. Ongoing coevolution in mycorrhizal interactions. New Phytologist 187: 286–300. [DOI] [PubMed] [Google Scholar]
- Jousset A, Bienhold C, Chatzinotas A, Gallien L, Gobet A, Kurm V, Küsel K, Rillig MC, Rivett DW, Salles JF et al. 2017. Where less may be more: how the rare biosphere pulls ecosystems strings. The ISME Journal 11: 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassambara A, Mundt F. 2020. factoextra: extract and visualize the results of multivariate data analyses. R package v.1.0.7. [WWW document] URL https://CRAN.R‐project.org/package=factoextra. [Google Scholar]
- Kiers ET, Hutton MG, Denison RF. 2007. Human selection and the relaxation of legume defences against ineffective rhizobia. Proceedings of the Royal Society B: Biological Sciences 274: 3119–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Lee KK, Jeon J, Harris WA, Lee Y‐H. 2020. Domestication of Oryza species eco‐evolutionarily shapes bacterial and fungal communities in rice seed. Microbiome 8: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, Bushman FD, Knight R, Kelley ST. 2011. Bayesian community‐wide culture‐independent microbial source tracking. Nature Methods 8: 761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köberl M, Müller H, Ramadan EM, Berg G. 2011. Desert farming benefits from microbial potential in arid soils and promotes diversity and plant health. PLoS ONE 6: e24452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krings M, Taylor TN, Hass H, Kerp H, Dotzler N, Hermsen EJ. 2007. Fungal endophytes in a 400‐million‐yr‐old land plant: infection pathways, spatial distribution, and host responses. New Phytologist 174: 648–657. [DOI] [PubMed] [Google Scholar]
- Kruskal WH, Wallis WA. 1952. Use of ranks in one‐criterion variance analysis. Journal of the American Statistical Association 47: 583–621. [Google Scholar]
- Kusstatscher P, Adam E, Wicaksono WA, Bernhart M, Olimi E, Müller H, Berg G. 2021a. Microbiome‐assisted breeding to understand cultivar‐dependent assembly in Cucurbita pepo . Frontiers in Plant Science 12: 642027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusstatscher P, Cernava T, Abdelfattah A, Gokul J, Korsten L, Berg G. 2021b. Microbiome approaches provide the key to biologically control postharvest pathogens and storability of fruits and vegetables. FEMS Microbiology Ecology 96. doi: 10.1093/femsec/fiaa1119. [DOI] [PubMed] [Google Scholar]
- Lahti L, Shetty S. 2017. Tools for microbiome analysis in R. microbiome R package 1.10.0. [WWW document] URL http://microbiome.github.com/microbiome. [Google Scholar]
- Leff JW, Lynch RC, Kane NC, Fierer N. 2017. Plant domestication and the assembly of bacterial and fungal communities associated with strains of the common sunflower, Helianthus annuus . New Phytologist 214: 412–423. [DOI] [PubMed] [Google Scholar]
- Li P, Xue Y, Shi J, Pan A, Tang X, Ming F. 2018. The response of dominant and rare taxa for fungal diversity within different root environments to the cultivation of Bt and conventional cotton varieties. Microbiome 6: 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limborg MT, Heeb P. 2018. Special issue: coevolution of hosts and their microbiome. Genes 9: 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Abdelfattah A, Norelli J, Burchard E, Schena L, Droby S, Wisniewski M. 2018. Apple endophytic microbiota of different rootstock/scion combinations suggests a genotype‐specific influence. Microbiome 6: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto H, Fan X, Wang Y, Kusstatscher P, Duan J, Wu S, Chen S, Qiao K, Wang Y, Ma B et al. 2021. Bacterial seed endophyte shapes disease resistance in rice. Nature Plants 7: 60–72. [DOI] [PubMed] [Google Scholar]
- Mazel F, Davis KM, Loudon A, Kwong WK, Groussin M, Parfrey LW. 2018. Is host filtering the main driver of phylosymbiosis across the tree of life? mSystems 3: e00097‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8: e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes LW, Raaijmakers JM, de Hollander M, Mendes R, Tsai SM. 2018. Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. The ISME Journal 12: 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutch LA, Young JPW. 2004. Diversity and specificity of Rhizobium leguminosarum biovar viciae on wild and cultivated legumes. Molecular Ecology 13: 2435–2444. [DOI] [PubMed] [Google Scholar]
- Oksanen J, Kindt R, Legendre P, O’Hara B, Stevens MHH, Oksanen MJ, Suggests M. 2007. The vegan package. Community ecology package 10: 631–637. [Google Scholar]
- Paulson JN, Stine OC, Bravo HC, Pop M. 2013. Differential abundance analysis for microbial marker‐gene surveys. Nature Methods 10: 1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer JA, Spor A, Koren O, Jin Z, Tringe SG, Dangl JL, Buckler ES, Ley RE. 2013. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proceedings of the National Academy of Sciences, USA 110: 6548–6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peres‐Neto PR, Jackson DA. 2001. How well do multivariate data sets match? The advantages of a Procrustean superimposition approach over the Mantel test. Oecologia 129: 169–178. [DOI] [PubMed] [Google Scholar]
- Pérez‐Jaramillo JE, Carrión VJ, de Hollander M, Raaijmakers JM. 2018. The wild side of plant microbiomes. Microbiome 6: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter SS, Sachs JL. 2020. Agriculture and the disruption of plant‐microbial symbiosis. Trends in Ecology & Evolution 35: 426–439. [DOI] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2012. The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Research 41: D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2020. R: a language and environment for statistical computing (v.4.1.1). Vienna, Austria: R Foundation for Statistical Computing. [WWW document] URL https://www.r‐project.org/. [Google Scholar]
- Ramirez KS, Knight CG, de Hollander M, Brearley FQ, Constantinides B, Cotton A, Creer SI, Crowther TW, Davison J, Delgado‐Baquerizo M et al. 2018. Detecting macroecological patterns in bacterial communities across independent studies of global soils. Nature Microbiology 3: 189–196. [DOI] [PubMed] [Google Scholar]
- Ravanbakhsh M, Kowalchuk GA, Jousset A. 2021. Targeted plant hologenome editing for plant trait enhancement. New Phytologist 229: 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell CW, Bouvaine S, Newell PD, Douglas AE. 2013. Shared metabolic pathways in a coevolved insect‐bacterial symbiosis. Applied and Environmental Microbiology 79: 6117–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu PK, Mishra S. 2021. Effect of hybridization on endophytes: the endo‐microbiome dynamics. Symbiosis 84: 369–377. [Google Scholar]
- Saikkonen K, Ion D, Gyllenberg M. 2002. The persistence of vertically transmitted fungi in grass metapopulations. Proceedings of the Royal Society of London. Series B: Biological Sciences 269: 1397–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. 1987. The neighbor‐joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4: 406–425. [DOI] [PubMed] [Google Scholar]
- Schlaeppi K, Dombrowski N, Oter RG, Loren V, van Themaat E, Schulze‐Lefert P. 2014. Quantitative divergence of the bacterial root microbiota in Arabidopsis thaliana relatives. Proceedings of the National Academy of Sciences, USA 111: 585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliep KP. 2010. phangorn: phylogenetic analysis in R. Bioinformatics 27: 592–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shade A, Jacques M‐A, Barret M. 2017. Ecological patterns of seed microbiome diversity, transmission, and assembly. Current Opinion in Microbiology 37: 15–22. [DOI] [PubMed] [Google Scholar]
- Shade A, McManus PS, Handelsman J. 2013. Unexpected diversity during community succession in the apple flower microbiome. mBio 4: e00602‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahzad R, Khan AL, Bilal S, Asaf S, Lee I‐J. 2018. What is there in seeds? Vertically transmitted endophytic resources for sustainable improvement in plant growth. Frontiers in Plant Science 9: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Nie J, Dong Y, Kuang L, Li Y, Zhang J. 2018. Compositional shifts in the surface fungal communities of apple fruits during cold storage. Postharvest Biology and Technology 144: 55–62. [Google Scholar]
- Singh J, Sun M, Cannon SB, Wu J, Khan A. 2021. An accumulation of genetic variation and selection across the disease‐related genes during apple domestication. Tree Genetics & Genomes 17: 29. [Google Scholar]
- Spor A, Roucou A, Mounier A, Bru D, Breuil M‐C, Fort F, Vile D, Roumet P, Philippot L, Violle C. 2020. Domestication‐driven changes in plant traits associated with changes in the assembly of the rhizosphere microbiota in tetraploid wheat. Scientific Reports 10: 12234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Janisiewicz WJ, Nichols B, Jurick Ii WM, Chen P. 2017. Composition of phenolic compounds in wild apple with multiple resistance mechanisms against postharvest blue mold decay. Postharvest Biology and Technology 127: 68–75. [Google Scholar]
- Tan J, Kerstetter JE, Turcotte MM. 2021. Eco‐evolutionary interaction between microbiome presence and rapid biofilm evolution determines plant host fitness. Nature Ecology & Evolution 5: 670–676. [DOI] [PubMed] [Google Scholar]
- Theis KR, Dheilly NM, Klassen JL, Brucker RM, Baines JF, Bosch TCG, Cryan JF, Gilbert SF, Goodnight CJ, Lloyd EA et al. 2016. Getting the hologenome concept right: an eco‐evolutionary framework for hosts and their microbiomes. mSystems 1: e00028‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent JB, Weiblen GD, May G. 2016. Host associations and beta diversity of fungal endophyte communities in New Guinea rainforest trees. Molecular Ecology 25: 825–841. [DOI] [PubMed] [Google Scholar]
- Volk GM. 2019. Temperate tree fruits of North America: M alus Mill., Prunus L., Diospyros L., and Asimina Adans. In: Greene SL, Williams KA, Khoury CK, Kantar MB, Marek LF, eds. North American crop wild relatives, vol. 2: important species. Cham, Switzerland: Springer International, 353–386. [Google Scholar]
- Volk GM, Chao CT, Norelli J, Brown SK, Fazio G, Peace C, McFerson J, Zhong G‐Y, Bretting P. 2015. The vulnerability of US apple (Malus) genetic resources. Genetic Resources and Crop Evolution 62: 765–794. [Google Scholar]
- Wagner MR. 2021. Prioritizing host phenotype to understand microbiome heritability in plants. New Phytologist 232: 502–509. [DOI] [PubMed] [Google Scholar]
- Wassermann B, Kusstatscher P, Berg G. 2019a. Microbiome response to hot water treatment and potential synergy with biological control on stored apples. Frontiers in Microbiology 10. doi: 10.3389/fmicb.2019.02502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassermann B, Müller H, Berg G. 2019b. An apple a day: which bacteria do we eat with organic and conventional apples? Frontiers in Microbiology 10: 1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TJ, Bruns T, Lee S, Taylor J. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, eds. PCR protocols: a guide to methods and applications, vol. 18. New York, NY, USA: Academic Press, 315–322. [Google Scholar]
- Whitehead SR, Poveda K. 2019. Resource allocation trade‐offs and the loss of chemical defences during apple domestication. Annals of Botany 123: 1029–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead SR, Wisniewski M, Droby S, Abdelfattah A, Freilich S, Mazzola M. 2021. The apple microbiome: structure, function, and manipulation for improved plant health. In: Korban SS, ed. The apple genome. Cham, Switzerland: Springer, 341–382. [Google Scholar]
- Yeoh YK, Dennis PG, Paungfoo‐Lonhienne C, Weber L, Brackin R, Ragan MA, Schmidt S, Hugenholtz P. 2017. Evolutionary conservation of a core root microbiome across plant phyla along a tropical soil chronosequence. Nature Communications 8: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Stacked bar charts showing the fungal (a) and bacterial (b) community composition of the most abundant taxa with a relative abundance > 0.1% across all the investigated Malus species.
Fig. S2 Microbial gene copy numbers in apple shoots determined by qPCR and the estimated beta diversity partitioning.
Fig. S3 Bar chart showing the estimated sources of the fungal (a) and bacterial (b) taxa in Malus domestica (modern apple).
Table S1 Metadata table with the information regarding Malus accessions used in the present study.
Table S2 List of ITS sequences used to calculate distances among Malus species and the phylogenetic trees.
Table S3 Analysis of variance on the effect of domestication and Malus phylogeny (species) on fungal and bacterial diversity based on Shannon index.
Table S4 Results of statistical pairwise‐comparisons of fungal and bacterial diversity based on Shannon index and community composition based on Bray–Curtis dissimilarity index using adonis (˜PERMANOVA).
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available in the ‘SRA NCBI’ repository, and can be accessed from the following link https://www.ncbi.nlm.nih.gov/bioproject/PRJNA702287. The code used in this study is available on zenodo and can be accessed from the following link https://doi.org/10.5281/zenodo.5578000.