

Abstract
Background
D‐dimer antigen is a heterogeneous mixture of fibrin degradation products that when present at high levels in plasma indicate ongoing coagulation and fibrinolysis. The heterogeneous nature of the target D‐dimer antigen and the variety of assay systems means that it is difficult to compare results from different methods.
Objectives
To identify a universally agreed D‐dimer standard that could help harmonize results from different methods.
Methods
A pool of patient plasma with high D‐dimer levels was freeze‐dried and investigated as a long‐term World Health Organization international standard for D‐dimer. Fibrin degradation products from clot lysis reactions were also freeze‐dried in various formulations and investigated in commutability studies with patient plasma.
Results
Problems of instability of D‐dimer plasma emerged suggesting loss of reactivity after freeze‐drying and storage at −20°C of 10%–18% per year. Freeze‐dried fibrin degradation products added to plasma were also unstable, but the sugar trehalose was found to improve stability. However, this preparation was not suitable as a standard in widely used assay platforms. Previous studies suggest fibrin degradation products are prone to structural rearrangements and amyloid formation, which may explain the instability of candidate D‐dimer standards.
Conclusions
The known difficulties of D‐dimer standardization are compounded by instability of D‐dimer antigen after freeze‐drying, described in this report. Fibrin degradation products added to plasma and stabilized by trehalose are not suitable as a standard for D‐dimer measurement harmonization. Trehalose stabilization of pooled patient plasma containing high D‐dimer levels may produce a useful standard, but this requires confirmation.
Keywords: amyloid, D‐dimer, fibrin degradation, protein aggregation, standardization
Essentials.
Results from D‐dimer assays may be harmonized by a suitable freeze‐dried plasma standard.
A candidate D‐dimer plasma standard was unstable, losing 10%–18% activity per year at −20°C.
Fibrin degradation products could be stabilized by freeze‐drying in plasma with added trehalose.
Further investigation of D‐dimer stability in clinical samples is warranted.
1. INTRODUCTION
Elevated plasma D‐dimer is a marker for ongoing coagulation and fibrinolysis. The antigen target in D‐dimer tests is actually a heterogeneous mixture of fibrin degradation products (FDP), of varying size, that are generated by plasmin following activation of the fibrinolytic system. 1 These fibrin fragments contain cross‐linked D domains, formed by activated factor XIII (FXIIIa), a transglutaminase produced by the action of thrombin on the zymogen precursor FXIII. 2 Early work on the identification and use of fibrin breakdown products as a diagnostic marker 3 , 4 were boosted by the introduction of monoclonal antibody technology 5 and subsequently led to the development of a large diagnostic testing industry. 6 However, common D‐dimer tests are characterized by high sensitivity but low specificity, 7 , 8 which means the results have good negative predictive value (NPV), but poor positive predictive value (PPV, a high rate of false positives). Therefore, D‐dimer testing in clinical practice is widely used to exclude thrombosis, but confirmatory testing is required if patient values exceed a predefined cut‐off. NPV can be improved by various algorithms such as the Wells’ score, which aim to reduce the likelihood of positive results in the population tested. Elevated D‐dimer levels are seen in a number of other pathological states, such as disseminated intravascular coagulation, cardiovascular disease, cancer, inflammatory disease, trauma, 9 and infections, including SARS‐Cov‐2, 10 , 11 and are used to monitor therapy in patients with previous thrombosis. 6 Measurement and interpretation of D‐dimer assay results are complicated by the poor PPV and the establishment of reliable cut‐off values to make clinical decisions.
Further problems in routine D‐dimer testing, reporting, and interpretation are the use of different units: D‐dimer units or fibrinogen equivalent units (FEU), both expressed as mass/volume, but which can be ng/ml or µg/ml or µg/L etc. 12 , 13 , 14 Different methods have different cut‐off values or recommend users define their own cut‐off values, sometimes with adjustment for age. 15 , 16 The diverse antibodies used in available methods react in different ways to the heterogeneous mixture of D‐dimer antigens in samples found in the various conditions in which elevated D‐dimer is of interest. 2 , 6 , 9 All these factors mean it is impossible to directly compare raw results from different methods, but it may be possible to harmonize results between methods by using a common standard. 17 , 18 A pooled plasma with elevated D‐dimers from a large number of patients might be suitable as a common reference material. 19 , 20 , 21 Alternatively, a mixture of FDP prepared from digested fibrin added to plasma may work, would be easier to produce reproducibly at scale, and may be more comparable with calibrators prepared by manufacturers. 20 , 22 The work described below reports attempts to generate a potential international standard for D‐dimer from pooled plasma or in vitro‐generated cross‐linked FDP, which might improve harmonization and reliability of results. However, a problem of instability of D‐dimer antigen in plasma after freeze‐drying was found, which further complicates development of reliable reference materials and harmonization of D‐dimer measurements. These observations also suggest the stability of D‐dimer antigen in clinical samples should be investigated.
2. MATERIALS AND METHODS
2.1. D‐dimer measurement
Several different methods were used and compared when studying D‐dimer in patient plasma or FDP added to plasma, including the Dade Behring Innovance D‐Dimer Assay (Siemens Healthcare); HemosIL D‐dimer HS on an ACL Top 500 (Instrumentation Laboratory SpA); Hyphen Zymutest DDimer (HYPHEN BioMed); Stago Asserachrom D‐DI (Diagnostica Stago SAS), Technoclone Technozyme D‐dimer ELISA (Technoclone). Hyphen and Stago methods were ELISA‐based and were performed using a Softmax M5 plate reader (Molecular Devices) and results were analyzed using GraphPad Prism Software (GraphPad Software). Additional details for methods and platforms used for commutability studies are described below.
2.2. Preparation of SS258
Patient plasma left over from routine testing and with high levels of D‐dimer was collected at the Royal Hallamshire Hospital UK and its use in standard development was approved by the National Institute for Biological Standard and Control (NIBSC) ethics committee. Thirty plasma pools were provided of 10–12 ml and D‐dimer in each pool was determined using the Dade Behring Innovance D‐dimer Assay. D‐dimer values for these samples ranged from approximately 3 to >20 µg/ml (FEU units) and gave a volume of 360 ml with an expected estimated overall value of approximately 6 µg/ml, using the Dade Behring Innovance method. The pooled plasma was chilled on ice and 1 M Hepes buffer, pH 7.4 was added to the plasma to a final concentration of 40 mM before the pool was dispensed in 0.5 ml aliquots into 3 ml ampoules for freeze‐drying over 4 days using eight steps from −50 to +25°C and 150 to 25 mTorr. Finally, ampoules were backfilled with nitrogen gas and temporarily stoppered before sealing using a Bausch Strobel sealer. This batch of ampoules is referred to as SS258.
2.3. Preparation of SS444 and SS523
FDP were generated in a such way to produce a heterogenous mixture of fibrin fragments, from a purified system. Clots were formed using purified fibrinogen and thrombin, which included FXIIIa, plasminogen activator, and plasminogen. Plasmin was generated from plasminogen by tissue plasminogen activator (tPA, code, 98/714; NIBSC) or urokinase plasminogen activator (code 11/184; NIBSC). All solutions were made up in 10 mM Hepes buffer pH 7.4, containing 0.15 M NaCl and 0.01% Tween 20. Clot lysis mixtures were prepared by mixing 0.2 ml of activator solution (containing thrombin and FXIIIa [code 02/170; NIBSC] plus tPA or urokinase [uPA]) with 1.0 ml of fibrinogen solution (also containing plasminogen). The final mixtures contained 4 nM thrombin (code 01/578, NIBSC) plus 1 IU/ml activated FXIIIa and 2.5 mg/ml fibrinogen (Calbiochem) and 0.22 µM plasminogen, either glu‐plasminogen (HYPHEN BioMed) or lys‐plasminogen (Immuno) with 2 nM tPA or 1 nM uPA. Clot lysis at 37°C was followed in four mixtures of tPA or uPA with glu‐ or lys‐plasminogen (abbreviated to TG, TL, UG, UL) for varying amounts of time and 1 ml samples were taken at four time points: the moment the clot collapsed, t0 (6–12 minutes), 2× t0, 60 min and 300 min. Reactions were stopped by cooling immediately on ice and adding concentrated aprotinin (Baxter) to a final concentration of 7 µM (see Longstaff and Locke 23 ) before adding to plasma and snap freezing for storage at −80°C. Assays on samples from these reaction mixtures were performed using the Technoclone Technozyme D‐dimer ELISA, the Hyphen Zymutest DDimer ELISA method, and the Stago Asserachrom D‐DI ELISA method and the HemosiIL D‐dimer HS latex automated method (ACL TOP). A larger pool of FDP in plasma was prepared by combining equal volumes of these different reaction mixtures into 100 volumes of buffer or plasma and additional excipients to prepare samples A, B, C, and D, as shown in Results. This set of freeze‐dried ampoules was labeled SS444 A to D. Sucrose and trehalose excipients were from Fluka Analytical. All these formulations were freeze‐dried in 0.5 ml aliquots as described above for SS258. A second batch of FDP in plasma +trehalose (the same as SS444 D) was prepared for commutability studies and is labeled batch SS523.
2.4. Thioflavin T assays
A selection of 10 World Health Organization (WHO) standards were used when available as part of accelerated degradation studies taking place at NIBSC. Standards were reconstituted as directed in their instructions for use and four‐point, 2‐fold dilution ranges prepared in phosphate‐buffered saline (PBS). A working solution of 50 µM Thioflavin T (ThT) was prepared in PBS and 175 µl of this solution mixed with 25 µl of the protein solution from the WHO standards in Corning Costar black microtiter plates (Corning Life Sciences). After 30 min incubation at room temperature, the fluorescence was read in an M5 fluorescent plate reader (Molecular Devices) using an excitation wavelength of 435 nm and emission wavelength of 485 nm. 24 A linear dose response was obtained, and results are presented for the highest reading (most concentrated protein) in Results, after subtraction of background in no‐protein blanks.
2.5. Commutability studies
All assays were conducted by Roche Diagnostics. Three different D‐dimer assay systems were used: Roche Tinaquant on a Cobas c501 platform; Stago STA LIA D‐dimer plus on a STA platform; Innovance D‐Dimer assay on Sysmex CS‐5100 platform. All methods included the same internal reference calibrators plus candidate standards SS258 and SS523. A selection of 54 patient plasma samples, labeled 1–54, containing varying levels of D‐dimer were included. Results are presented below as from anonymized methods A, B, and C, not necessarily in the order of methods listed above.
2.6. Degradation analysis
Degradation studies were performed by measuring the change in relative potency (D‐dimer antigen concentration) over time of freeze‐dried candidate standards stored at a range of temperatures, −70, −20, 4, 20, 37, 45°C. Degradation rates at each temperature were modeled using the linear form of the Arrhenius equation, Equation (1), by a specially designed computer program previously established at NIBSC. 25 , 26 , 27 This modeling assumes a first order decay of activity, which is reasonable from previous studies, 25 shown in Equation (2), or in the linearized form as Equation (3). Rate constants can also be calculated directly by fitting degradation data over time at each temperature using Equation (2) or (3) by non‐linear or linear regression, respectively. These calculations were performed using R. 28
The linear transformation of the Arrhenius equation:
| (1) |
First‐order reaction A→P
| (2) |
linear transformation:
| (3) |
3. RESULTS
A pool of plasma with a high level of a heterogeneous mixture of D‐dimer antigen was collected with the aim of preparing a batch of freeze‐dried plasma in ampoules that could be investigated as a candidate standard to be used to calibrate manufacturer or other working standards. The characteristics of this pool and the freeze‐dried material are shown in Table 1.
TABLE 1.
Characteristics of the freeze‐dried candidate standard SS258
| Plasma volume | 290 ml with 40 mM Hepes buffer |
|---|---|
| D‐dimer content | Estimated >6 µg/ml |
| Ampoules filled @ weight (CV) | 556 @ 0.51 g (0.72%) |
| Dry weight (CV) | 0.038 g (1.61%) |
| Residual moisture (CV) | 0.19 mg (13.5%) |
The pool was made up of many small pools of 5–12 ml of patient plasma with D‐dimer levels ranging between approximately 3 to >20 µg/ml. Hepes buffer, pH 7.4 was added to the plasma, as commonly done with WHO plasma standards before freeze‐drying. There was no prior indication that the D‐dimer antigen present in this freeze‐dried plasma was likely to be unstable, but stability studies, using accelerated degradation methods, must be performed on potential WHO International Standards before they can be established. Therefore, in line with usual practice, ampoules were stored at temperatures of −20, 4, 20, 37, and 45°C for extended periods so that the amount of activity loss could be determined, and predictions made of likely stability on long‐term storage at −20°C. The D‐dimer content of these ampoules was measured after 0.55, 1.24, 1.91, 2.47 years of storage. Unexpectedly, a significant loss of potency (measurable D‐dimer antigen) was observed, even at the earliest time points. Several different D‐dimer assay methods were included in these studies (Innovance D‐Dimer Assay, HemosIL D‐dimer HS, Zymutest DDimer, Stago Assechrom D‐DI), which gave similar results on the same samples. A bar chart summarizing the loss of activity for D‐dimer in SS258 over time at each storage temperature is shown in Figure 1.
FIGURE 1.
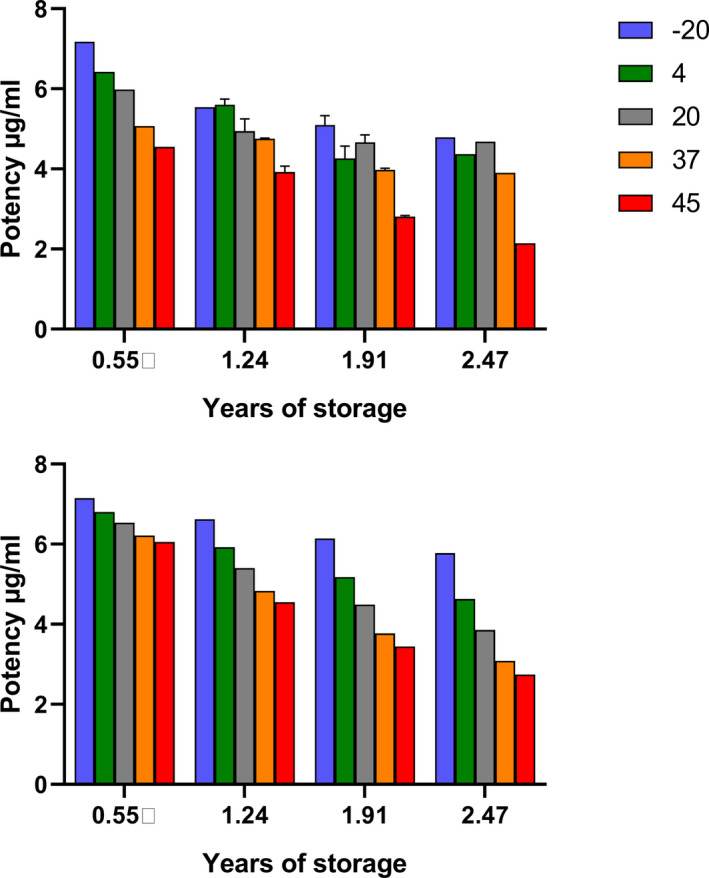
Estimates for D‐dimer concentrations over time in freeze‐fried plasma stored at elevated temperatures. A, Observed data for activity at each temperature of the time period of storage. B, Fitted data from the Degtest program assuming a first order reaction that obeys the Arrhenius equation
This degree of instability is much greater than seen in other freeze‐dried plasma standards prepared by NIBSC as WHO International Standards. Accelerated degradation results are analyzed by established methods 25 , 26 to calculate rate constants for loss of activity at each temperature investigated and to predict the loss of activity per year with confidence limits. However, the theoretical basis for these calculations assumes first order kinetics for the loss of activity, and that the reaction follows the Arrhenius equation (see Materials and Methods).
Results from the Degtest program 26 produced values for the rate constant for loss of activity at the temperatures shown, but the fitting deviated from Degtest predictions based on assumptions of first order reaction kinetics and validity of the Arrhenius equation for this system (compare Figure 1A and B). Other possible sources of error were the inclusion of different methods and the long time period of study, which meant that the lot number and calibrators of kits changed, or kits exceeded their stated shelf‐life. A summary of results from Degtest analysis is presented in Table 2. It is also possible to use time and activity data at each temperature and fit the results to linear equations for first order reaction kinetics (see Equation [3]). This analysis is also presented in Table 2 and high values for rate constants were also obtained by non‐linear fitting to Equation (2). It is striking that whichever analysis method was used there was a significant loss of activity of 10–18% after 1 year on storage at −20°C. In practice these results mean that this freeze‐dried plasma containing D‐dimer is not suitable as a WHO Standard due to this marked instability.
TABLE 2.
Fitted rate constant for loss of D‐dimer activity on storage at various temperatures
| Temperature °C | Degtest | First order | ||
|---|---|---|---|---|
| k year−1 | % loss | k year−1 | % loss | |
| −20 | 0.11 | 10.4 | 0.2 | 18.3 |
| 4 | 0.27 | 23.8 | 0.24 | 21.5 |
| 20 | 0.46 | 36.8 | 0.19 | 17.6 |
| 37 | 0.75 | 52.9 | 0.25 | 22 |
| 45 | 0.92 | 60.2 | 0.48 | 37.9 |
Rate constants were derived from fitting using the Degtest program or to a linearized first order reaction equation at each temperature.
3.1. Generation of FDP from fibrin
FXIIIa crosslinked FDP were made from clotted fibrinogen in the presence of tPA or uPA and glu‐ or lys‐plasminogen as described in Materials and Methods to give four reaction time courses designated as TG, TL, UG, UL where activator was tPA (T) or uPA (U) with glu‐plasminogen (G) or lys‐plasminogen (L). Samples were taken from each clot lysis mixture at the point of clot collapse, at t0= 6–12 min, 2 × t0, 60 min and 300 min, the reactions were stopped and FDP added to plasma. This approach was developed to generate a diverse mixture of FDP that might reflect the mixture of D‐dimer structures present in clinical samples from different disease conditions. D‐dimer values were measured in all these mixtures using the Technoclone Technozyme D‐dimer ELISA, the Hyphen Zymutest DDimer ELISA, the Stago Asserachrom D‐DI ELISA, and the HemosiIL D‐dimer HS latex automated method (ACL TOP) and results are shown in Figure 2.
FIGURE 2.
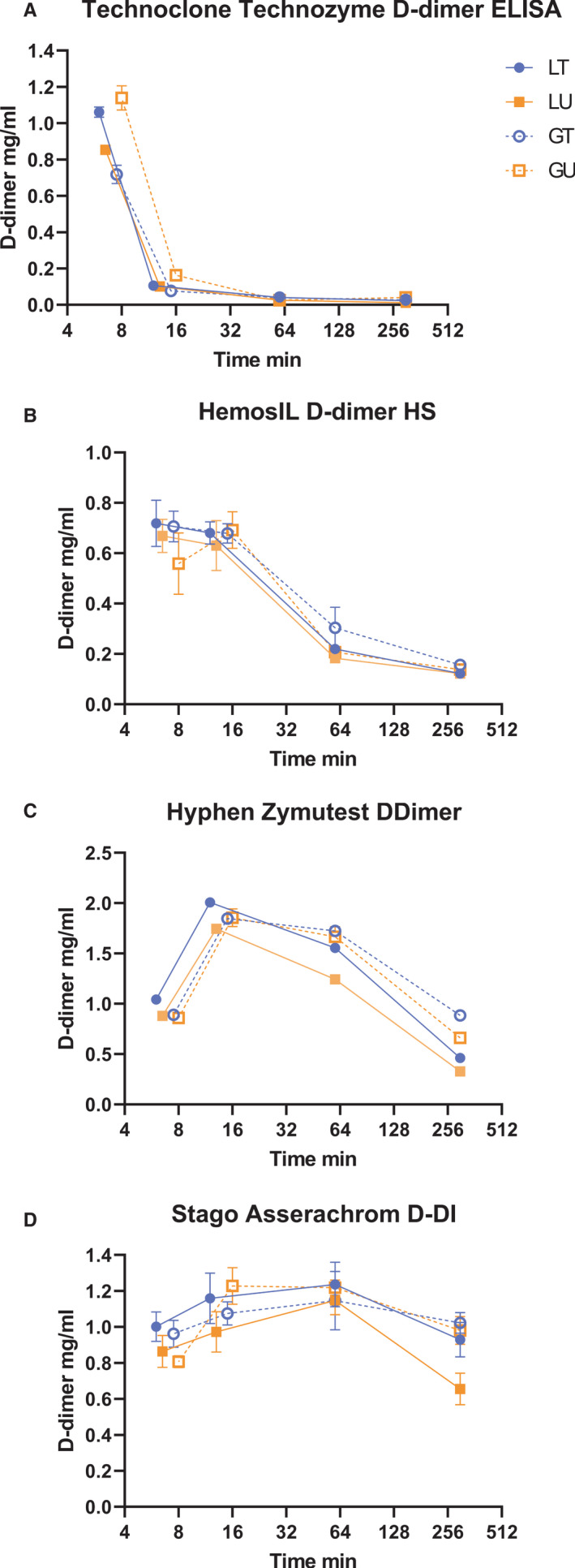
Analysis of fibrin degradation product (FDP) generation from clotting and lysis of fibrin over time using four methods. Concentrations of D‐dimer refer to the initial reaction mixture of 2.5 mg/ml fibrinogen clotted with thrombin in the presence of activated factor XIII (FXIIIa) and either tissue plasminogen activator (tPA; T) or urokinase (uPA; U) with glu‐plasminogen (G) or lys‐plasminogen (L). Time is presented on a log scale to see changes more easily at the four time points chosen
Results shown in Figure 2 indicate there was no clear difference between reaction mixtures with plasminogen activators tPA or uPA; or with glu‐ or lys‐plasminogen. However, this group of methods provides a clear indication of the different sensitivities observed for FDP generated over time corresponding to large sizes at early times and small FDP fragments at longer times. The Stago ELISA method gave a reasonably consistent response from 6 min to 5 hours of fibrin digestion, whereas other methods showed changing responses over the period of clot lysis. The absolute concentration of D‐dimer is not consistent across methods, peaking around 0.7 mg/ml for HemosIL, at the lower end and reaching around 2.0 mg/ml for the Hyphen Zymutest ELISA. These discrepancies highlight the lack of intermethod standardization. These results are corrected for dilutions into the different assay methods and so report D‐dimer formation in the clot lysis mixture which contained 2.5 mg/ml fibrinogen. The diverse results shown in Figure 2 emphasize the need for a heterogeneous mixture of D‐dimer fragments in any standard aimed at harmonizing different assay methods.
3.2. Stabilization of fibrin degradation products
In order to explore the stabilization of freeze‐dried FDP, experiments were performed on a mixture of 10 µg/ml FDP from the reactions used in Figure 2, which were added to four different formulations for freeze‐drying and stability testing, as shown in the legend to Figure 3. Subsequently, a second FDP formulation in plasma with added trehalose, the same as sample D of SS444, was prepared and this was labeled SS523.
FIGURE 3.
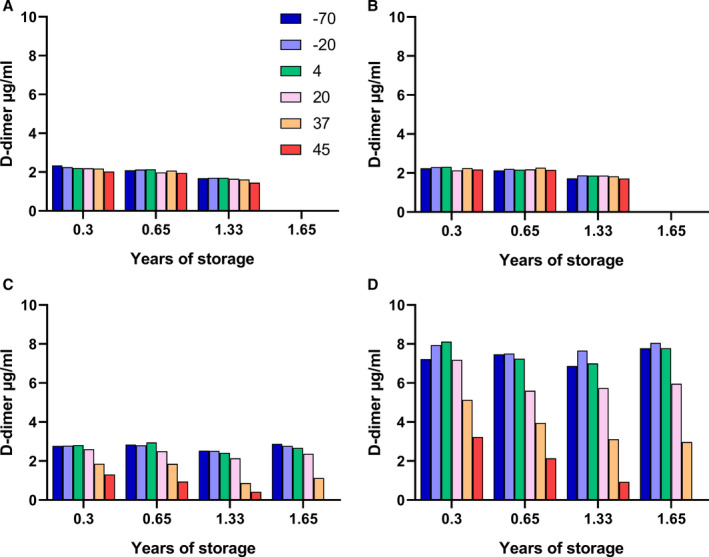
D‐dimer measurements over time in samples of SS444, freeze‐dried fibrin degradation product (FDP) in four formulations (A–D), stored at various temperatures at the times shown. The panels correspond to results for 10 µg/ml FDP added to 10 mM HEPES buffer, pH 7.4, containing 0.15 M NaCl, with additions: (A) 10 mg/ml trehalose, (B) 10 mg/ml sucrose, (C) plasma, and (D) plasma + 10 mg/ml trehalose. Note: no measurements were made after 1.33 years for samples A and B or the 45°C sample at 1.65 years because of insolubility
Immediately after freeze‐drying, samples were placed at elevated temperatures for storage and periodic measurement of D‐dimer antigen using the HemosIL kit method on the ACL TOP platform. Results are shown in Figure 3 for assays after 0.3, 0.65, 1.33, and 1.65 years of storage (or only up to 1.33 years for formulations A and B because there was little change between 0.3 and 1.33 years). It is apparent from the results shown in Figure 3 that there was an initial loss of activity in samples A‐C at all temperatures from −70 to +45°C. Maximum activity was close to 2.0–2.5 µg/ml or only 20%–25% of the expected value. Therefore, formulations A‐C with sugars, trehalose or sucrose, or with plasma and HEPES buffer do not give good recovery after freeze‐drying. Recovery was much better, at around 75%, in formulation D containing plasma, HEPES buffer, and 10 mg/ml trehalose. On extended storage there was further loss of activity in formulations C and D but only at higher temperatures, 20, 37, and 45°C.
This loss of activity in formulation D was explored further by analyzing the decay over time using the Degtest program. The analysis produced a set of decay constants and loss per year shown in Table 3, although the results are based only on changes in values seen at 20, 37, and 45°C. Good agreement between observed and predicted data, as percent of starting value, is also shown in Figure 4. There is obviously clustering of values around 100% (very little loss of activity), which come from the samples stored at −70, −20, and 4°C, but overall, the slope of 0.86 and intercept of 11.8%, R2 of 0.92 are satisfactory. Without the clustered values around 100% the linear fit is better, intercept = 1.3%, slope = 1.08, and R2 = 0.93, suggesting a good fit of model and data.
TABLE 3.
Derived rate constants and calculated % loss of activity per year for storage of SS444 formulation D at various temperatures
| Temperature | k (SE) year−1 | % Loss per year |
|---|---|---|
| −70 | 0 | 0 |
| −20 | 0.00016 (0.00012) | 0.016 |
| 4 | 0.008 (0.0033) | 0.801 |
| 20 | 0.076 (0.018) | 7.32 |
| 37 | 0.64 (0.043) | 47.4 |
| 45 | 1.62 (0.067) | 80.2 |
FIGURE 4.
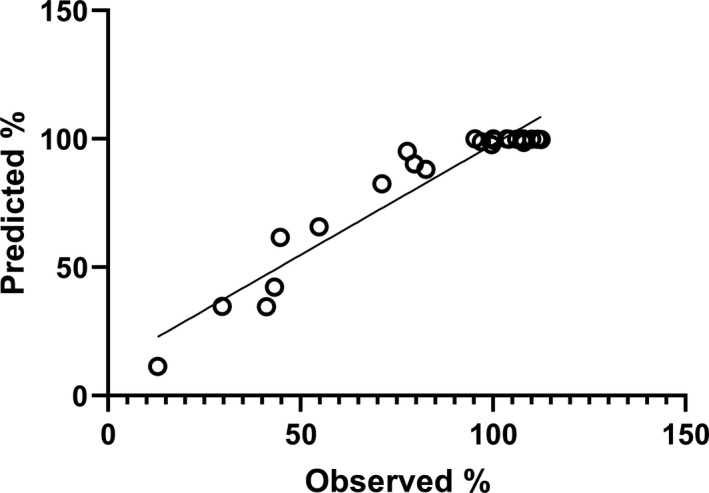
Observed versus predicted percent activity remaining in ampoules of SS444 stored at elevated temperatures over time
The conclusions from Table 2 are that this preparation is stable at temperatures below zero and could serve as a standard for long‐term use on this basis. However, it is not known whether the FDP in plasma in the presence of trehalose could act as a standard for plasma D‐dimer from patient plasma, which is explored below.
3.3. Commutability
A reference material (standard) is said to be commutable if the same results are obtained for clinical samples between methods that will use the reference material. Commutability studies are performed with clinical samples when new reference materials are being introduced, or when a new method is introduced. Therefore, in the case of D‐dimer standards it is important to investigate potential reference materials with patient samples in established methods. A series of such studies are shown in Figure 5.
FIGURE 5.
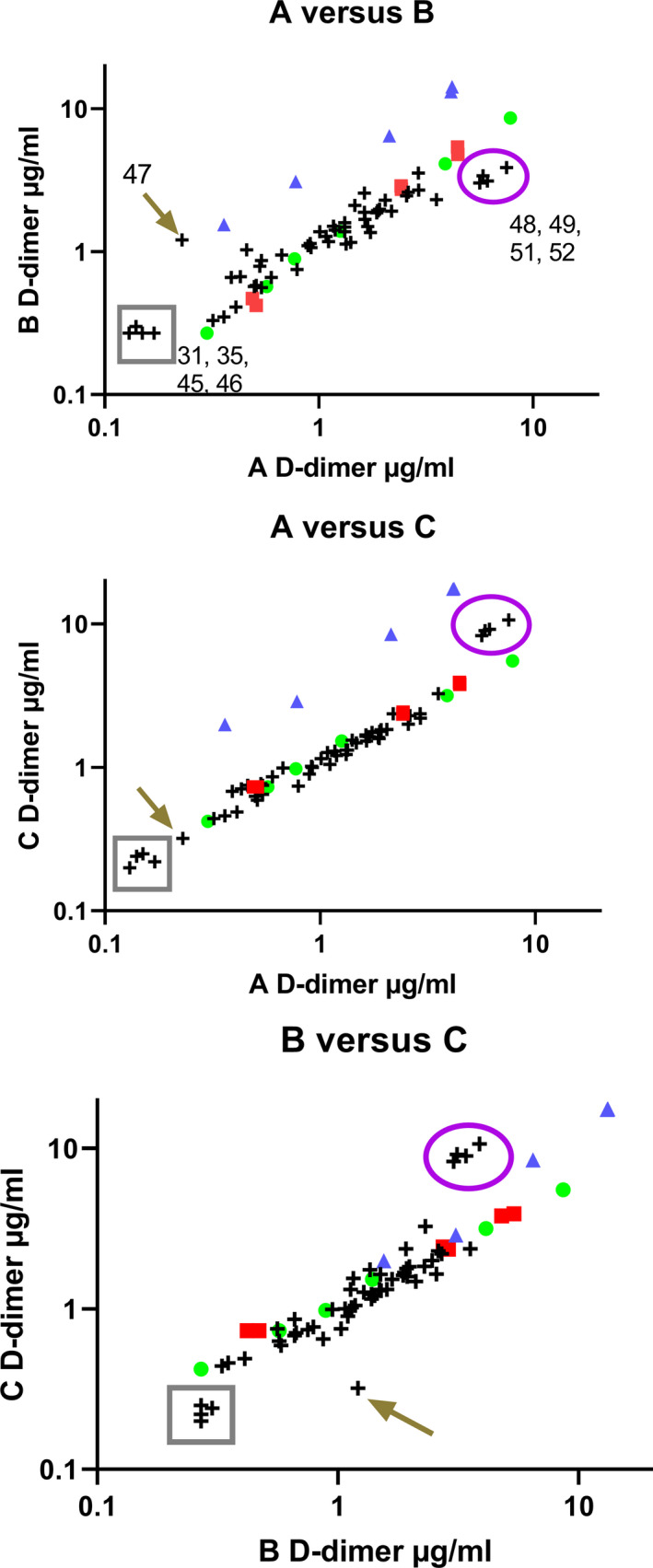
Correlation of results between methods A, B, and C as pairwise comparisons. Samples shown are an internal standard (green circle), and freeze‐dried candidate standards SS258 (red square) and SS523 (blue triangle) with 54 plasma samples (crosses) containing various levels of D‐dimer. Two groups of inconsistent samples are highlighted in the box (plasmas 31, 35, 45, 46) and in the oval (48, 49, 52, 53), along with sample 47 highlighted with the arrow. Log scales are used for presentation purposes
There was good agreement between the common standard (green circle) and the candidate standard SS258 (which had been stored for >5 years at −40°C at this point). Conversely, preparation SS523, FDP in plasma with trehalose, showed very different behavior between assays indicating that method A detected lower levels of D‐dimer in this sample than methods B and C. Optimal agreement between pairs of methods would lead to a slope of 1 and intercept of zero on linear plots, and in this series of experiments the slopes ranged from 0.52 to 1.65 for the internal standard, SS258, and plasmas. However, the slopes for candidate SS523 in the three‐method pairings shown in Figure 5 were 3.19, 4.20, and 1.38, for methods A versus B, A versus C, and B versus C, respectively. The distribution of plasma samples was generally consistent; however, there are several extreme samples with variable behavior between methods as shown circled in purple (plasma numbers 48, 49, 52, 53) or in the gray box (31, 35, 45, 46) and indicated by the arrow (47). These results suggest that overall the internal standard used in this series of testing performed in a broadly similar way to the freeze‐dried plasma SS258, but the mixture of FDP added to plasma and freeze‐dried in the presence of trehalose was not appropriate as a standard. Candidate standard SS523 is certainly not commutable between methods; and the individual plasma samples highlighted are also problematic between methods and all standards. These results suggest that no combination of methods or standards will be completely commutable for all D‐dimer plasma samples.
3.4. Mechanism of loss of D‐dimer antigen
It is possible that aggregation of D‐dimer fibrin fragments could explain the loss of D‐dimer signal over time as aggregation during fibrinolysis is known. 29 , 30 , 31 , 32 Fibrin aggregation during fibrinolysis is accompanied by cross beta sheet formation and amyloid development as demonstrated by the binding of ThT assessed by increased fluorescence. 33 , 34 However, it is not known whether this process can take place in freeze‐dried proteins. To explore this question, a selection of 10 freeze‐dried preparations that were undergoing accelerated degradation studies were investigated, providing 99 data points from various times of storage at temperatures between −70 and 45 °C. The results shown in Figure 6 clearly show a trend of increasing ThT fluorescent signal over baseline indicating some structural rearrangement and amyloid structure formation with increasing storage temperature. In most cases the major protein present in the samples was albumin either purified or in plasma, but there were also preparations of purified immunoglobulin or alpha‐1‐proteinase inhibitor. Therefore, it is entirely possible that freeze‐dried D‐dimer could undergo the same structural rearrangement and aggregation on storage after freeze‐drying, which would explain the loss of D‐dimer antigen response noted above. Conservative statistical analysis using Tukey’s method for comparing this family of estimates gave statistically significant differences for all pairwise comparisons above and including 20°C. Interestingly, if only samples that included trehalose in the freeze‐dried formulation were analyzed, the only statistically significant difference was for the −20 and 45°C pair.
FIGURE 6.
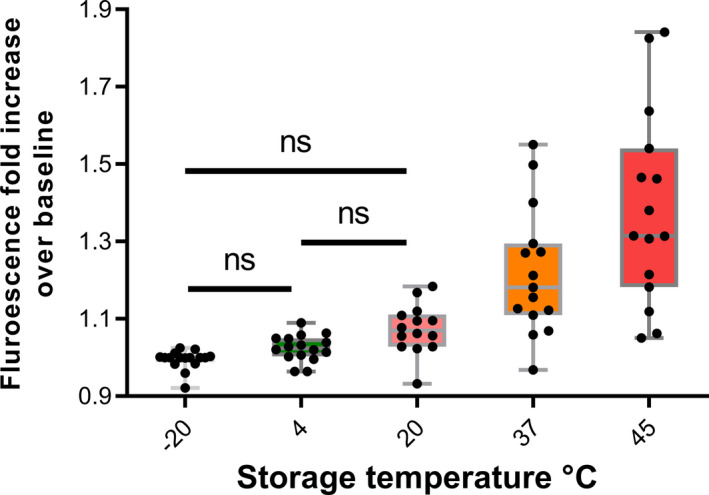
Development of cross‐beta amyloid structures in freeze‐dried proteins stored at elevated temperatures. Various proteins undergoing accelerated degradation studies were incubated with thioflavin T (ThT) and increasing fluorescence indicated the development of cross‐beta structures after storage at the higher temperatures. Tukey’s method for P‐value adjustment for a family of estimates was used to calculate significance levels of differences between results from each storage temperature. P‐values for all combinations were <0.05 where no ns is indicated
4. DISCUSSION
Freeze‐dried WHO international standards are generally very stable and have shelf lives of years to decades, and stability at −20°C must be confirmed by degradation studies (for example, the study to establish the 4th International Standard for Streptokinase found no loss of activity of standards in collaborative studies over a 30‐year period 35 ). The unusual instability of D‐dimer or FDP was manifested in two ways as shown in Figures 1 and 3. In the case of preparation SS258, the patient plasma with elevated D‐dimer, there was a rapid decline in measured D‐dimer at all temperatures during storage. The rate of loss was estimated to be 10%–18% per year, though these estimates rely on an assumption of simple first order kinetics, which may not be justified. In the case of SS444, the freeze‐dried FDP, there was a poor recovery of added D‐dimer antigen in formulations A–C compared to formulation D, which included plasma, Hepes buffer, and trehalose. Longer term, D‐dimer measurements were stable in sample D during storage at 4°C and below. It is not clear what the mechanism might be for the loss of D‐dimer antigen in these freeze‐dried samples, but based on previous studies it is known that FDPs have a tendency to aggregate, which could mask available D‐dimer antigen sites.
The propensity of fibrin(ogen) to aggregate, sometimes with the formation of amyloid structures, is well known. For example, there are a group of hereditary fibrinogen amyloidosis disorders associated with mutations that cause increases in β‐sheet regions and self‐assembly of fibrinogen with amyloid fibril deposition. 36 , 37 Other heterogeneous amyloid‐promoting molecules such as β‐amyloid 38 or serum amyloid A 39 can bind to fibrinogen and cause oligomerization with associated amyloid structure development. Aggregation or gelation of fibrinogen or FDP by histones has also been observed. 40 , 41 Fibrin polymerization catalyzed by thrombin does not normally result in development of β‐sheet from fibrinogen α‐helix, although stressed fibrin will show such secondary structure rearrangements. 42 , 43 However, aggregates formed by FDPs during fibrinolysis have been observed in many studies, 29 , 30 , 31 , 32 , 44 and in some cases these have been associated with development of β‐sheet amyloid structures that bind ThT. 33 , 34 Using a column clot perfusion system to generate and analyze FDPs from lysing clots, Walker and Nesheim 32 identified the structures and molecular weights of large fibrin fragments. Significantly, they also concluded that FDPs bound to fibrin with an affinity proportional to their mass. Therefore, the conclusion from these diverse studies using a variety of methods is that plasmin digestion of fibrin leads to rearrangement of the fibrin secondary structure and generation of large fragments with some amyloid structure. These fragments tend to aggregate, and the larger fragments have higher binding affinity. In the context of D‐dimer antigen measurement, it is plausible that this aggregation would lead to a loss of binding sites and reduced D‐dimer measurement. This type of association would explain the loss of D‐dimer signal over time in the case of SS258 and SS444 preparations. How association of D‐dimer fragments in patient samples might affect measurement in clinical samples is worthy of further investigation.
Results from Figure 3 suggest trehalose stabilizes D‐dimer measurements in plasma, at least on low temperature storage. If high molecular weight D‐dimer fragments are more prone to aggregation, 32 stabilization with trehalose may lead to an enrichment of these fragments in the freeze‐dried preparations SS523 and SS444 (D). This could explain the results in Figure 4 in which the FDP + plasma + trehalose preparation, SS523, behaved very differently to the other standards: the internal standard (a commercial preparation of unknown composition) and SS258 plasma, which were not formulated with trehalose. Alternatively, the FDP preparation that was formulated to contain a heterogeneous mixture of D‐dimer fragments may be unrepresentative of D‐dimer in clinical samples and hence unsuitable for this reason. In either case, it appears that a synthetic mixture of degraded fibrin products is not a straightforward option as a standard for D‐dimer assays. 20 The results shown in Figure 5 comparing the internal standard and SS258 in these three assay pairings suggest that SS258, D‐dimer plasma may be a useful standard for harmonization purposes. However, the freeze‐dried preparation of SS258 was more than 5 years old (stored at −40°C) by the time this set of experiments was performed, and based on the results of Figure 1, would be a different preparation from that immediately after freeze‐drying. Thus, although the results from Figure 5 suggest SS258 would be potentially a useful standard, the instability is not acceptable. Figure 6 includes data from 10 diverse freeze‐dried standards that indicate the development of amyloid‐like structures that bind ThT, particularly in samples stored at higher temperatures. As mentioned above, these structures are seen in fibrin 33 , 34 in solution, but these results suggest that structural rearrangements and aggregation can take place in freeze‐dried preparations. Previous studies on pharmaceutical protein solutions have suggested that protein misfolding with amyloid structure formation and aggregation is a general mechanism to explain immunogenicity in aged or stressed therapeutic drug preparations. 24 Our data may be the first time freeze‐dried proteins have been shown to generate these structures. Trehalose is widely used as a stabilizer for freeze‐dried proteins, but the mechanism of action is not fully understood. Nevertheless, it is interesting that trehalose has been shown to inhibit amyloid plaque formation in the case of β‐amyloid peptide that is associated with Alzheimer’s disease. 45
In summary, it has not so far been possible to make a standard for D‐dimer that would be useful in harmonizing results from different assay methods. The current work has highlighted a previously unforeseen problem of stability in freeze‐dried D‐dimer preparations, but also presents a potential solution to the problem using trehalose as a stabilizer. Further work is required to develop such a standard and prove its utility.
CONFLICTS OF INTEREST
The authors have no conflicts of interest.
AUTHOR CONTRIBUTIONS
CL conceived the work, performed experiments, and wrote the manuscript. SB performed experiments and helped with the manuscript.
ACKNOWLEDGMENTS
We are grateful for advice and support from chairs and members of the Fibrinolysis Subcommittee of the SSC/ISTH during these investigations. We are grateful to Dr. Steve Kitchen and Dr. Ian Jennings of the Sheffield Teaching Hospitals NHS Foundation Trust for providing D‐dimer plasma, expertise, and advice throughout the project; and Dr. Piet Meijer of the ECAT Foundation, NL for advice and guidance. We thank the Standards Processing Division of NIBSC for their work on preparing freeze‐dried candidate standards and Dr. Peter Rigsby of the Biostatistics Department, NIBSC for helpful discussions relating to data analysis and presentation. We are grateful to Dr. Norbert Gottschalk and his team at Roche Diagnostics, Penzberg, Germany for providing the results on commutability testing.
Bevan S, Longstaff C. Is it possible to make a common reference standard for D‐dimer measurements? Communication from the ISTH SSC Subcommittee on Fibrinolysis. J Thromb Haemost. 2022;20:498–507. doi: 10.1111/jth.15555
Manuscript handled by: Joost Meijers
Final decision: Joost Meijers, 11 October 2021
REFERENCES
- 1. Gaffney PJ, Edgell T, Creighton‐Kempsford LJ, Wheeler S, Tarelli E. Fibrin degradation product (FnDP) assays: analysis of standardization issues and target antigens in plasma. Br J Haematol. 1995;90:187‐194. [DOI] [PubMed] [Google Scholar]
- 2. Adam SS, Key NS, Greenberg CS. D‐dimer antigen: current concepts and future prospects. Blood. 2009;113:2878‐2887. [DOI] [PubMed] [Google Scholar]
- 3. Gaffney PJ, Lane DA, Kakkar VV, Brasher M. Characterisation of a soluble D dimer‐E complex in crosslinked fibrin digests. Thromb Res. 1975;7:89‐99. [DOI] [PubMed] [Google Scholar]
- 4. Whitaker AN, Rowe EA, Masci PP, Gaffney PJ. Identification of D dimer‐E complex in disseminated intravascular coagulation. Thromb Res. 1980;18:453‐459. [DOI] [PubMed] [Google Scholar]
- 5. Rylatt DB, Blake AS, Cottis LE, et al. An immunoassay for human D dimer using monoclonal antibodies. Thromb Res. 1983;31:767‐778. [DOI] [PubMed] [Google Scholar]
- 6. Linkins LA, Takach Lapner S. Review of D‐dimer testing: Good, Bad, and Ugly. Int J Lab Hematol. 2017;39(suppl 1):98‐103. [DOI] [PubMed] [Google Scholar]
- 7. Dempfle CE. Validation, calibration, and specificity of quantitative D‐dimer assays. Semin Vasc Med. 2005;5:315‐320. [DOI] [PubMed] [Google Scholar]
- 8. Schutgens RE, Esseboom EU, Haas FJ, Nieuwenhuis HK, Biesma DH. Usefulness of a semiquantitative D‐dimer test for the exclusion of deep venous thrombosis in outpatients. Am J Med. 2002;112:617‐621. [DOI] [PubMed] [Google Scholar]
- 9. Johnson ED, Schell JC, Rodgers GM. The D‐dimer assay. Am J Hematol. 2019;94:833‐839. [DOI] [PubMed] [Google Scholar]
- 10. Yu B, Li X, Chen J, et al. Evaluation of variation in D‐dimer levels among COVID‐19 and bacterial pneumonia: a retrospective analysis. J Thromb Thrombolysis. 2020;50:548‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang L, Yan X, Fan Q, et al. D‐dimer levels on admission to predict in‐hospital mortality in patients with Covid‐19. J Thromb Haemost. 2020;18:1324‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Olson JD, Cunningham MT, Higgins RA, Eby CS, Brandt JT. D‐dimer: simple test, tough problems. Arch Pathol Lab Med. 2013;137:1030‐1038. [DOI] [PubMed] [Google Scholar]
- 13. Thachil J, Longstaff C, Favaloro EJ, Lippi G, Urano T, Kim PY. Thrombosis SSCSoFotISo, Haemostasis. The need for accurate D‐dimer reporting in COVID‐19: Communication from the ISTH SSC on fibrinolysis. J Thromb Haemost. 2020;18:2408‐2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Longstaff C, Adcock D, Olson JD, et al. Harmonisation of D‐dimer ‐ A call for action. Thromb Res. 2016;137:219‐220. [DOI] [PubMed] [Google Scholar]
- 15. Favresse J, Lippi G, Roy PM, et al. D‐dimer: Preanalytical, analytical, postanalytical variables, and clinical applications. Crit Rev Clin Lab Sci. 2018;55:548‐577. [DOI] [PubMed] [Google Scholar]
- 16. Olson JD. D‐dimer: an overview of hemostasis and fibrinolysis, assays, and clinical applications. Adv Clin Chem. 2015;69:1‐46. [DOI] [PubMed] [Google Scholar]
- 17. Jennings I, Woods TA, Kitchen DP, Kitchen S, Walker ID. Laboratory D‐dimer measurement: improved agreement between methods through calibration. Thromb Haemost. 2007;98:1127‐1135. [DOI] [PubMed] [Google Scholar]
- 18. Meijer P, Haverkate F, Kluft C, de Moerloose P, Verbruggen B, Spannagl M. A model for the harmonisation of test results of different quantitative D‐dimer methods. Thromb Haemost. 2006;95:567‐572. [DOI] [PubMed] [Google Scholar]
- 19. Adema E, Gebert U. Pooled patient samples as reference material for D‐Dimer. Thromb Res. 1995;80:85‐88. [DOI] [PubMed] [Google Scholar]
- 20. Dempfle CE, Zips S, Ergul H, Heene DL. The fibrin assay comparison trial (FACT): correlation of soluble fibrin assays with D‐dimer. Thromb Haemost. 2001;86:1204‐1209. [PubMed] [Google Scholar]
- 21. Nieuwenhuizen W. A reference material for harmonisation of D‐dimer assays. Fibrinogen Subcommittee of the Scientific and Standardization Committee of the International Society of Thrombosis and Haemostasis. Thromb Haemost. 1997;77:1031‐1033. [PubMed] [Google Scholar]
- 22. Dempfle CE. D‐dimer: standardization versus harmonization. Thromb Haemost. 2006;95:399‐400. [DOI] [PubMed] [Google Scholar]
- 23. Longstaff C, Locke M. Increased urokinase and consumption of alpha2 ‐antiplasmin as an explanation for the loss of benefit of tranexamic acid after treatment delay. J Thromb Haemost. 2019;17:195‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maas C, Hermeling S, Bouma B, Jiskoot W, Gebbink MF. A role for protein misfolding in immunogenicity of biopharmaceuticals. J Biol Chem. 2007;282:2229‐2236. [DOI] [PubMed] [Google Scholar]
- 25. Kirkwood TB. Design and analysis of accelerated degradation tests for the stability of biological standards III. Principles of design. J Biol Stand. 1984;12:215‐224. [DOI] [PubMed] [Google Scholar]
- 26. Kirkwood TB, Tydeman MS. Design and analysis of accelerated degradation tests for the stability of biological standards II. A flexible computer program for data analysis. J Biol Stand. 1984;12:207‐214. [DOI] [PubMed] [Google Scholar]
- 27. Tydeman MS, Kirkwood TB. Design and analysis of accelerated degradation tests for the stability of biological standards I. Properties of maximum likelihood estimators. J Biol Stand. 1984;12:195‐206. [DOI] [PubMed] [Google Scholar]
- 28. R Development Core Team . R: A Language and Environment for Statistical Computing. 3.2.2 edn. R Foundation for Statistical Computing; 2019. https://www.R‐project.org/. [Google Scholar]
- 29. Diamond SL. Engineering design of optimal strategies for blood clot dissolution. Annu Rev Biomed Eng. 1999;1:427‐462. [DOI] [PubMed] [Google Scholar]
- 30. Sakharov DV, Nagelkerke JF, Rijken DC. Rearrangements of the fibrin network and spatial distribution of fibrinolytic components during plasma clot lysis. Study with confocal microscopy. J Biol Chem. 1996;271:2133‐2138. [DOI] [PubMed] [Google Scholar]
- 31. Veklich Y, Francis CW, White J, Weisel JW. Structural studies of fibrinolysis by electron microscopy. Blood. 1998;92:4721‐4729. [PubMed] [Google Scholar]
- 32. Walker JB, Nesheim ME. The molecular weights, mass distribution, chain composition, and structure of soluble fibrin degradation products released from a fibrin clot perfused with plasmin. J Biol Chem. 1999;274:5201‐5212. [DOI] [PubMed] [Google Scholar]
- 33. Gebbink MF. Tissue‐type plasminogen activator‐mediated plasminogen activation and contact activation, implications in and beyond haemostasis. J Thromb Haemost. 2011;9(suppl 1):174‐181. [DOI] [PubMed] [Google Scholar]
- 34. Longstaff C, Thelwell C, Williams SC, Silva MM, Szabo L, Kolev K. The interplay between tissue plasminogen activator domains and fibrin structures in the regulation of fibrinolysis: kinetic and microscopic studies. Blood. 2011;117:661‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Locke M, Rigsby P, Longstaff C, SSC Communication on behalf of the Fibrinolysis Subcommittee . An international collaborative study to establish the WHO 4th International Standard for Streptokinase: communication from the SSC of the ISTH. J Thromb Haemost. 2020;18:1501‐1505. [DOI] [PubMed] [Google Scholar]
- 36. Stangou AJ, Banner NR, Hendry BM, et al. Hereditary fibrinogen A alpha‐chain amyloidosis: phenotypic characterization of a systemic disease and the role of liver transplantation. Blood. 2010;115:2998‐3007. [DOI] [PubMed] [Google Scholar]
- 37. Vilar R, Fish RJ, Casini A, Neerman‐Arbez M. Fibrin(ogen) in human disease: both friend and foe. Haematologica. 2020;105:284‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ahn HJ, Zamolodchikov D, Cortes‐Canteli M, Norris EH, Glickman JF, Strickland S. Alzheimer's disease peptide beta‐amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci USA. 2010;107:21812‐21817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Page MJ, Thomson GJA, Nunes JM, et al. Serum amyloid A binds to fibrin(ogen), promoting fibrin amyloid formation. Sci Rep. 2019;9:3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gonias SL, Pasqua JJ, Greenberg C, Pizzo SV. Precipitation of fibrinogen, fibrinogen degradation products and fibrin monomer by histone H3. Thromb Res. 1985;39:97‐116. [DOI] [PubMed] [Google Scholar]
- 41. Locke M, Francis RJ, Tsaousi E, Longstaff C. Fibrinogen protects neutrophils from the cytotoxic effects of histones and delays neutrophil extracellular trap formation induced by ionomycin. Sci Rep. 2020;10:11694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kell DB, Pretorius E. Proteins behaving badly. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: lessons from and for blood clotting. Prog Biophys Mol Biol. 2017;123:16‐41. [DOI] [PubMed] [Google Scholar]
- 43. Litvinov RI, Faizullin DA, Zuev YF, Weisel JW. The α‐helix to β‐sheet transition in stretched and compressed hydrated fibrin clots. Biophys J. 2012;103:1020‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weisel JW, Veklich Y, Collet JP, Francis CW. Structural studies of fibrinolysis by electron and light microscopy. Thromb Haemost. 1999;82:277‐282. [PubMed] [Google Scholar]
- 45. Liu FF, Ji L, Dong XY, Sun Y. Molecular insight into the inhibition effect of trehalose on the nucleation and elongation of amyloid beta‐peptide oligomers. J Phys Chem B. 2009;113:11320‐11329. [DOI] [PubMed] [Google Scholar]