

Abstract
Auranofin is an oral gold(I) compound, initially developed for the treatment of rheumatoid arthritis. Currently, Auranofin is under investigation for oncological application within a drug repurposing plan due to the relevant antineoplastic activity observed both in vitro and in vivo tumor models. In this review, we analysed studies in which Auranofin was used as a single drug or in combination with other molecules to enhance their anticancer activity or to overcome chemoresistance. The analysis of different targets/pathways affected by this drug in different cancer types has allowed us to highlight several interesting targets and effects of Auranofin besides the already well‐known inhibition of thioredoxin reductase. Among these targets, inhibitory‐κB kinase, deubiquitinates, protein kinase C iota have been frequently suggested. To rationalize the effects of Auranofin by a system biology‐like approach, we exploited transcriptomic data obtained from a wide range of cell models, extrapolating the data deposited in the Connectivity Maps website and we attempted to provide a general conclusion and discussed the major points that need further investigation.
Keywords: Auranofin, cancer, drug resistance, NF‐kB inhibition, proteasome inhibition, thioredoxin reductase inhibition
Abbreviations
- 17AAG
17‐Allylamino‐17‐demethoxygeldanamycin
- 2‐DG
2‐deoxy‐d‐glucose
- 5‐FU
5‐fluorouracil
- AAL
acute lymphoblastic leukemia
- ADE
adenanthin
- AF
auranofin
- AKT
serine/threonine kinase 1
- ANXA5
annexin A5
- APL
acute Promyelocytic Leukemia
- ATRA
all‐trans‐retinoic acid
- BAX
Bcl‐2 associated X‐protein
- BCP‐ALL
B‐cell acute lymphoblastic leukemia
- BRCA
BReast CAncer gene
- BSO
buthionine sulfoximine
- CCL
chronic lymphocytic leukemia
- CE
celecoxib
- cHL
classical Hodgkin lymphoma
- CMAP
connectivity Maps
- CML
chronic myelogenous leukemia
- DLBCL
diffuse large B‐cell lymphoma
- DR4/5
death receptor 4/5
- DTX
docetaxel
- DUBs
deubiquitinases
- EGFR
epidermal growth factor receptor
- ENZ
enzalutamide
- ERK
extracellular signal‐regulated kinases
- FDA
Food and Drug Administration
- FOXO3
Forkhead Box O3
- GCLC
Glutamate‐Cysteine Ligase Catalytic Subunit
- GSH
glutathione
- GSR
glutathione‐disulfide reductase
- Her2
receptor tyrosine‐protein kinase erbB‐2
- HER‐2
human epidermal growth factor receptor 2
- HNSCC
neck squamous cell carcinoma
- HO‐1
heme oxygenase‐1
- IAP
inhibitor of apoptosis proteins
- IkB
Inhibitor of NF‐κB
- IKK
IkB kinase
- IL‐6
interleukin 6
- IM
Imatinib mesylate
- JAK2
tyrosine‐protein kinase Janus Kinase 2
- JNK
c‐Jun N‐terminal kinase
- l‐ASC
l‐ascorbate
- MAPK
mitogen‐activated protein kinase
- MCL
mantle cell lymphoma
- Mcl‐1
myeloid‐cell leukemia 1
- MEK
MAPK/ERK kinase
- MLL
mixed lineage leukemia
- MM
multiple myeloma
- mTOR
mammalian target of rapamycin
- MUC4
mucin 4
- NAC
N‐acetyl‐l‐cysteine
- NF‐κB
nuclear factor kappa‐light‐chain‐enhancer of activated B cells
- NONO
non‐POU domain‐containing octamer‐binding
- Nrf2
nuclear factor erythroid 2–related factor 2
- NSCLC
non‐small cell lung cancer
- OC
ovarian cancer
- PAI‐2
plasminogen activator inhibitor
- PAK1
p21 activate kinase
- PB1
Phox and Bem 1
- PD‐L1
programmed death‐ligand 1
- PI3K
phosphatidylinositol‐3‐kinase
- PKCiota
protein kinase C iota
- PL
piperlongumine
- PRDXI
peroxiredoxin I
- Prx3
peroxiredoxin3
- PTEN
phosphatase and tensin homolog protein
- PTGR1
prostaglandin reductase 1
- ROS
reactive oxygen species
- SCLC
small cell lung carcinoma
- SeC
selenocysteine
- Smac
second mitochondria‐derived activator of caspases
- SP
side‐population
- STAT3
signal transducer and activator of transcription 3
- TAK1
transforming growth factor β‐activated kinase 1
- TIC
tumor‐initiating cell
- TNBC
triple negative tumors
- TP53
mutated or
- Trx
thioredoxin
- TrXR
thioredoxin reductase
- TUSC2
tumor suppressor candidate 2
- VDAC
voltage‐dependent anion‐selective channel protein
- XCT
cystine/glutamate transporter
- YAP1
yes‐associated protein 1
- ZnPP IX
zinc protoporphyrin IX
- γ‐GCL
γ‐glutamylcysteine ligase
1. INTRODUCTION
Auranofin (AF) is a linear gold(I) complex bearing triethylphosphine and thioglucose tetraacetate as ligands (Figure 1). AF has been adopted in clinical use since 1985 for the treatment of rheumatoid arthritis. It is an orally administered drug and is considered safe for human use due to the well‐known toxicity profile. After oral administration, 15%–25% of the drug can be detected in plasma and the serum albumin has been found to carry about 80%–95% of gold circulating in the blood. 1 , 2 The pharmacokinetics of AF has been extensively described by Chaffman et al. 3 The mean pick of AF concentration, following a single 6 mg dose, was attained after 102–120 min and it ranged between of 0.066 and 0.23 µg/ml (corresponding to a molar concentration of 0.1–0.34 µM). Steady‐state plasma gold concentrations following at least 12 weeks of 2–9 mg daily dose of AF were 0.20–1.0 µg/ml, respectively corresponding to 0.3–1.5 µM. More recently in a Phase I human study, where AF was used as an antiparasitic agent at the dose of 7 mg daily (dose recommended in rheumatoid arthritis therapy), gold level in plasma of treated subjects for over a week reached 1–1.5 µM. 4 The triethyl‐phosphine ligand is lipophilic, conferring membrane solubility to the complex. Once the compound has entered into the cells, the thiol ligand reacts with thiol and selenol groups, with which it forms stable and irreversible adducts. 5 In mammals most of the effects caused by this drug are thought to be related to its inhibitory activity against the thioredoxin reductase enzymes: TrxR1 and TrxR2. 6 , 7 , 8 , 9 , 10 The thioredoxin reductase enzymes play an important role in multiple intracellular processes including DNA synthesis, transcriptional regulation, cell growth and resistance to oxidative agents that induce oxidative stress and apoptosis. An increase of the thioredoxin reductase/thioredoxin (TrxR/Trx) system has been reported in many tumors compared to normal tissues and the ability of certain cancer cells to maintain a highly reduced intracellular environment is correlated with tumor aggressiveness and drug resistance. Furthermore, high expression level of TrxR/Trx system directly correlates with poor prognosis in a variety of cancers including lung cancer 11 and breast cancer. 12 Indeed, this system plays a pivotal role in maintaining the reduced and active state of several proteins including phosphatase and tensin homolog (PTEN) that negatively regulates phosphatidylinositol‐3‐kinase/AKT Serine/Threonine Kinase 1 (PI3K/Akt) pathway which is positively involved cell survival. 13 Several elegant reviews have already discussed the role of TrxR/Trx system under physiological and pathological conditions. 14 , 15 Other areas of research for the antiproliferative activity of AF focus on the inhibition of signal transducer and activator of transcription 3 (STAT3), 16 , 17 , 18 nuclear factor‐kappa B (NF‐κB) 18 , 19 and protein kinase C iota (PKCiota) 20 , 21 signaling. Furthermore, proteasome inhibition and activation of forkhead box O3 (FOXO3) were also reported as possible mechanisms involved in AF cytotoxicity. 22 Based on this evidence, AF is today object of important drug repurposing plans in anticancer therapy with encouraging results, as summarized in this review. In this context, results obtained by research studies have already been translated into clinical trials to develop new alternative therapeutic strategies. Indeed, despite enormous advances in cancer research and treatment, there are still some types of cancer for which there is no specific therapy and hence, chemotherapy and radiation therapy remain pillars of cancer treatment. Furthermore, in these cases patients often respond only partially to therapy and develop resistance over long‐term treatment. Hence, the development of combination therapies may be useful to eradicate cancer cells that are unresponsive to monotherapy. Up to now, Phase I and II studies on chronic lymphocytic leukemia (NCT01419691) and a pilot study on ovarian cancer (NCT01747798) have been completed, while other clinical trials with AF as monotherapy or in combination with other drugs are ongoing on glioblastoma (NCT02770378), on lung cancer (NCT01737502), on ovarian cancer (NCT03456700). AF is also under investigation for use in infective diseases (NCT02736968 for Giardia protozoa, NCT02961829 for HIV, and NCT02968927 for tuberculosis).
Figure 1.
Chemical structure of Auranofin
In this review we have reviewed articles that use AF (alone or in combination with other molecules) as an anticancer drug. We focused first on the effect of AF on different types of cancer cells and/or animal models and then we analysed and integrated the results by a system biology‐like approach, with the data available on Connectivity Map website (https://clue.io/cmap). Finally, we attempted to extrapolate general conclusion and discussed the major points that need further investigation. In Table 1 we reported AF effect and mechanism of action on different cancer models (cell lines and/or animal model). Most of the articles are commented on throughout the text.
Table 1.
Summary of the studies focusing on AF repurposing as single drug or in combination
| AF as single drug or in combination | AF concentration and time of treatmenta | Cell lines | In vivo experiment | Mechanisms/pathways involved | Referencesb |
|---|---|---|---|---|---|
| Multiple myeloma | |||||
| AF + NF‐kB inhibitor SN‐50 | 0.05–0.1 µM AF for 6, 12, 24 h | U266, RPMI8226, IM‐9 cell lines and primary cell lines | Inhibition of IL‐6 induced‐JAK/STAT pathway and NF‐kB activity | 18 | |
| AF + PX‐12 (inhibitor of Trx1) | 0–8 µM AF for 24 h (EC50 = 0.05 µM at 24) | RPMI8226, U266 ‐resistant to bortezomib | Oxidative stress (Trx system) | 31 | |
| AF | 0–8 µM AF for 24 h | RPMI8226, U266 ‐resistant to bortezomib | Oxidative stress (Trx system) and NF‐kB signaling pathway (NF‐kb p65) | 32 | |
| AF + HO‐1 inhibitor Zinc Protoporphyrin IX | 0–4 µM AF for 24 h (EC50 (24 h) = 0.48 µM for RPMI8226, 0.46 µM for U266, and 0.457 µM for OPM2 cells) | RPMI8226, U266, OPM2 and PBMCs | HO‐1 expression through the Nrf2 signaling | 35 | |
| AF + Prima‐1 Met (APR‐246) | HMCLs = 0.25–2 µM AF for 2 days; (EC50 = 0.4 µM). Primary cells = 50 or 500 nM AF for 24 h. | 7 HMCLs with a wild‐type TP53 status (MDN, NCI‐H929, NAN9, NAN11, XG3, XG6, XG7), 8 HMCLs with a missense TP53 mutation (JIM3, KMS12PE, LP1, NAN10, OPM2, U266, XG2, XG5) and 3 HMCLs with a TP53 indel leading to the lack of mRNA and/or protein expression (JJN3, L363, NAN7). Primary cell lines | Oxidative stress (ROS/GSH pathway) | 40 | |
| AF + ([Au(d2pype)2]Cl) | 0.25‐4 µM AF for 24 h Mice = AF 5 mg/kg, (5 times a week, for 2 weeks). | RPMI8226, U266 and JJN3 myeloma cells | human RPMI8226 xenograft model | Oxidative stress (Trx system) | 42 |
| AF + seventy‐six FDA‐approved oncology drugs and emerging therapeutics | MM cell lines = 0.01–10 µM AF for 24 and 72 h. Primary cells = 0.01–10 µM AF for 24 h (EC50 below 100 nM). | 25 multiple myeloma (MM) and 15 non‐Hodgkin's lymphoma cell lines and in 113 primary MM samples | Drug direct screening | 43 | |
| Leukemia | |||||
| AF | AF 3–7.5 mg/kg, every fourth day, daily, and twice daily | C57BL x DBA/2 F, mice inoculated with the lymphocytic leukemia P388 | 44 | ||
| AF | AF 1.5–96 mg/kg/daily for days 1–5. Optimal activity with 12 mg/kg/daily for days 1–5. | C57BL x DBA/2 F, mice inoculated with the lymphocytic leukemia P388 | 45 | ||
| AF + all‐trans retinoic acid (ATRA) | 0.1–1 µM AF for 4 days or 24 h. 0.3 µM AF and 5 nM ATRA | NB4 cell | Oxidative stress (Trx system) | 48 | |
| AF | 0.5‐2 µM AF for 1–12 h | HL‐60 cell | p38 MAPK activation | 52 | |
| AF + all‐trans retinoic acid (ATRA) and 1,25(OH)2 vit D3 | NB4 cells and APL blasts = 0.3–0.5 µM AF for 4–5 days. HL‐60 cells = 0.5 µM AF and 3 nM 1,25(OH)2 vit D3 in combination for 3 days. | NB4 cells and HL‐60, APL blasts from leukemia patients | Enhancing histone acetylation | 49 | |
| AF | Cells = 0.125‐4 µM AF for 24 h; 1 µM for 24 and 48 h. Mice = 10 mg/kg (5 days/week for 2 weeks) | Human chronic B‐cell leukemia, MEC‐1 cells, primary CD19 + CLL cells | TCL‐1 transgenic mice genotype (Tcl1‐g:p53 ‐/‐) | Oxidative stress (Trx system) and Endoplasmic Reticulum Stress | 53 |
| AF | Cells = 0.5‐2 µM AF for 6,12,24 and 48 h. Mice = AF 7 mg/kg/day for 12 days | KBM5 (Bcr‐Abl wild‐type) and KBM5‐T315I (Bcr‐Abl‐T315I) Chronic myelogenous leukemia cells | mouse imatinib‐resistant xenograft models | Bcr‐Abl signaling and proteasome‐dependent caspase activation | 57 |
| AF + [Au(d2pype)2]Cl | 0.25–4 µM AF for 24 and 48 h | K562 and KU812 CML cell lines sensitive and resistant to imatinib | Bcr‐Abl signaling and Trx system | 60 | |
| AF + Erastin and BSO | 0.1–2 µM AF for 24 h | Human T‐ALL (Jurkat, Molt‐4) and precursor (pre)‐B‐ALL | Oxidative stress pathways | 63 | |
| AF + adenanthin | BCP‐ALL cell lines = 0.0625–2 µM AF for 48 h. Primary leukemic BCP‐ALL blasts = 0.0156–2 µM AF for 92 h. Normal PBMC = 0.0125–2 µM AF for 4 days. Mice = AF 10 mg/kg, once daily for 3 weeks | Human BCP‐ALL cell lines (697, REH; SEM; SD1; BV17;, SUP‐B15) and B‐cell lymphoma cell line RL, primary BCP‐ALL blasts or their primografts | patient‐derived xenograft model | Oxidative stress pathways | 65 |
| AF + sorafenib | EC50 = 0.65 µM in Cells = MV11 and 0.71 µM in MV‐11R (sorafenib resistant) Mice = 10 mg/ml AF, 10 mg/ml sorafenib | MV‐11, MV‐11R | MV‐11R and MV‐11 xenograft | TrxR3 inhibition | 66 |
| Lymphoma | |||||
| AF | 0.5–4 µM AF for 30–40 min and 6, 8, 24 h | Jurkat T‐lymphoma and U937 monocytic cell lines | 67 | ||
| AF + doxorubicin, cisplatin, and gemcitabine | Cells = 0.1‐10 µM AF for 24 or 48 h. Mice = 10 mg/kg, 3 times a week | L‐1236, L‐428, KM‐H2, HDLM‐2, and L‐540 cHL‐derived cell lines; L‐540 gemcitabine‐resistant and HDLM‐2 brentuximab resistant cells | L‐540 gemcitabine resistant–derived tumor xenografts, | Oxidative stress pathways | 68 |
| AF + BSO | 0.1–3 µM AF for 24 h. 100 nM AF and 5 µM BSO in combination for 18 h | human DLBCL cell lines, SUD‐HL6 and OCI‐LY10, the MCL cell lines, Rec‐1 and Granta, and the MM cell lines, U266 and KMS‐12‐PE, primary MCL‐derived cells | TrxR and NF‐κB signaling | 69 | |
| AF + l‐ascorbate (l‐ASC) | Cells = 0.125–0.6 µM AF for 48 h. 0.5 µM AF and 200µM l‐ASC in combination for 1–6 h. Mice= AF (2.5 mg/kg once daily) and l‐ASC (3 g/kg twice daily) from day 2 to 7 and day 10 to 15 after cell inoculation | primary chronic lymphocytic leukemia B‐cells | murine B‐cell lymphoma model | 70 | |
| AF | Cells = 0.075‐5 µM for 72 h (EC50 assay). PDX model = 50 mg/kg, daily for 21 consecutive days after 3 days of tumor engraftment | OCI‐Ly8, OCI‐Ly7, and Su‐DHL‐10; OCL‐Ly3, OCI‐Ly10, U2932, TMD8, and HBL‐1); Z‐138, JVM‐2, Mino, Maver‐1, Jeko‐1, and Jeko‐R | TP53‐mutated DLBCL PDX model | TrxR/Trx system | 71 |
| Breast cancer | |||||
| AF + BSO + 2DG and 17AAG | 0.5 µM AF, 0.5 µM 17AAG, 20 mM 2DG, 1 mM BSO for 24 h | MDA‐MB 231 and SUM159 | Oxidative stress (GSH and Trx oxidation) | 72 | |
| AF + 2DG + DHEA | 1 µM AF, 24 h; 20 mM 2DG, 24–48 h; DHEA, 300 µM, 24–48 h | MDA‐MB23 | Oxidative stress | 73 | |
| AF | 1 µM AF, 12 h (for STAT3 inhibition) | MDA‐MB 231 | Inhibition of STAT3 phosphorylation through ROS‐dependent mechanism | 17 | |
| AF + Vitamin C | Cells =6 µM AF, 1 h Mice = AF 10 mg/kg+ Vitamin C 4 g/kg; AF 5 mg/kg + VitaminC 4 g/kg or AF | MDA‐MB‐231 | MDA‐MB‐231 xenograft | Oxidative stress | 74 |
| AF + Vitamin C + menadione | 05–1 µM AF for 24 h 3, 24 μM Menadione 50, 100 μM l‐Acs | MDA‐MB‐231, HCC1806 | Oxidative stress, inhibition of PRDXI activity | 75 | |
| AF | Cells = EC50 values of 1.5 μM (24 h) and 0.41 μM (48 h) Mice = 6 mg/kg/day for 21 days | MCF‐7 | MCF7 xenograft | Inhibition of DUBs | 76 |
| AF + anti PD‐L1 antibodies | EC50 = 0.5–2 µM depending on TNBC cell lines Mice = 5 mg/kg, 14 days | SUM159 and MDA‐MB‐231 | 4T1.2 model, MDA‐MB‐231 xenograft and patient‐derived tumor xenograft | Oxidative stress increased, increased EGFR and ERK1/2 phosphorylation, decreased STAT3 phosphorylation | 77 |
| AF + BSO and radiation | Cells = 0.25–0.5 μM AF for 1–3 h, 0.1 mM BSOfor 24 h Mice = BSO 450 mg/kg and AF 1.7 mg/kg 2 h, before irradiation | MDA‐MB 231 and SUM159 | MDA‐MB‐231 xenograft | Oxidative stress | 79 |
| AF | 0.1–5 μM AF | MDA‐MB‐231 | MDA‐MB‐231 xenograft 4T1.2 model | NONO expression inhibition | 80 |
| AF + mesupron (Urokinase‐type plasminogen activator inhibitor) | EC50 (24 h) = 0.7 μM on MDA‐MB; 231 and 0.2 μM on MCF‐7; 10 μM mesupron + 0.125 μM AF | MDA‐MB‐231, MCF‐7 | Oxidative stress, downregulation of Akt phosphorylation | 145 | |
| AF + Nutlin‐3a | 0.5 μM AF, 2.5 μM Nutlin‐3a | MCF4, MDA‐MB 231 | Oxidative stress | 146 | |
| AF + R428 (Axl inhibitor) | EC50 (48 h) = 0.6 μM on MDA‐MB‐231 and 0.47 μM on MCF‐7 2.5 μM R428 + 0.5 μM AF | MDA‐MB‐231, MCF‐7 cells | Oxidative stress | 147 | |
| Colon and gastric cancer | |||||
| AF + 5Z‐7‐oxozeaenol | Cells = 500 nM AF pretreatment, then 5 µM 5Z‐7‐oxozeaenol. Mice = 1.6 mg/kg AF and 15 mg/kg 5Z‐7‐oxozeaenol for 5 consecutive days, followed by 2 days of AF only, and then 4 more consecutive days of combination treatment. | HCT116 and SW620 | SW620 xenografts | Oxidative stress (Trx and GSH systems) | 82 |
| AF + celecoxib | Cells =1 µM AF; 1 µM AF + 10 µM celecoxib in combination for 24, 48 h. Mice = AF 10 mg/kg; CE 20 mg/kg; CE 60 mg/kg; AF 10 mg/kg + CE 20 mg/kg; AF 10 mg/kg + CE 60 mg/kg | DLD‐1, HCT116, and HT‐29 | DLD‐1 xenografts | Oxidative stress (Trx system) | 83 |
| AF + 5‐FU | Cells = 5–10 μM 5‐FU + 0.4 μM AF, 24 h Mice = 50 mg/kg/5‐FU once every 4 days, +6 mg/kg/d AF | 5‐FU‐resistant SW620 and HCT‐8 | SW620/5‐FU xenografts | FoxO3‐activation and TR1‐inhibition | 84 |
| AF | Organoids = 2 µM AF. Mice = 10 mg/kg three times a week for 3 weeks | CT26 and IEC6 | Human colon organoid, CTC26 xenografts | RE stress, UPR response mediated apoptosis | 86 |
| AF + piperlongumine (PL) | EC50 (24 h) = 2.3 μM AF in BC‐823, 1.8 μM in SGC‐7901 and 2.7 μM KATO III Mice = 2 mg/Kg/day AF + 4 mg/Kg/day PL | BGC‐823, SGC‐7901 and KATO III | SGC‐7901 xenografts | ROS mediated RE stress | 87 |
| AF | 0.5 μM AF, 72 h | SW480 and HCT116 | PKC iota inhibition | 148 | |
| Lung cancer | |||||
| AF + BSO + carboplatin | Cells = 0.5 μM AF for H292, 5 μM AF for A459 100 μM BSO, 20 mM 2DG, 2 μM carboplatin for clonogenic survival Mice = 15 mg/kg carboplatin, 450 mg/Kg BSO, 1.6 mg/g AF for 6 days | A549, H292 | A549 tumor xenografts | Oxidative stress (Trx system) | 89 |
| AF + SeC | Cells = 6 µM AF, 6 h after SeC 8 µM, 24 h Mice = 5 mg/kg SeC+2 mg/kg AF, 8 doses. | A549 | A549 tumor xenograft | Down regulation of ERK and Akt phosphorylation, p38 and JNK phosphorylation doesn't affected | 90 |
| AF + MK2206 (Akt inhibitor) | Cells = 0.1 µM AF, 1‐3 µM MK2206 Mice = 25 mg/kg+ MK2206 5 mg/kg AF | HCC193 and HI993 | H1993 tumor xenografts nude mice | ROS induction and JNK activation | 92 |
| AF | Cells = 0.5 µM, 12‐24 h Mice = 10 mg/kg/day | HCC366, Calu3 | Calu3 tumor xenograft | PI3K/Akt/mTOR pathway and NRF2‐mediated oxidative stress response inhibition | 93 |
| AF and TUSC2 gene delivered by nanovesicles + erlotinib | Cells = 0.5–0.6 μM AF, 1 μM erlotinib, 72 h Mice = 10 mg/kg AF five times per week, for two weeks, 30 mg/kg Erlotinib orally feed daily with a total of 8 times | Calu‐3, Calu‐6 and H522 | EGFR TUSC2‐deficient human H1299 cells xenograft | NRF2‐mediated oxidative stress response | 94 |
| AF + adryamicin | EC50(24 h) = 4 µM AF Several different combination Mice = AF 10 mg/kg, and ADM 5 mg/kg, once a week for 6 week | A549 and NCI‐H460 (these cell lines contained an elevate percentage of SP cells) | A549 tumor xenograft | Selective inhibition of SP cell growth by ROS dependent mechanism; ATP depletion through direct hexokinase inhibition | 95 |
| AF | Cells = 0.5 μM AF for 12‐48 h Mice = 10 mg/kg/day | A459 knocked out of GSR | Patient‐derived xenografts | Oxidative stress | 97 |
| AF + ibrutinib (EGFR inhibitor) | Cells = 0.25 µM AF, 0.1–0.3 µM ibrutinib, 24‐48 h Mice = 5 mg/kg AF + 25 mg/kg ibrutinib | EGFR wild‐type NSCLC cell lines (Calu3 and H460) EGFR mutant lines, sensitive and resistant to ibrutinib | H1975 tumor xenograft | Akt inhibition | 98 |
| AF + IPA3 (PAK1 inhibitor) | 15 µM AF for 6 h IPA3 5 µM Mice = 10 mg/kg AF + 10 mg/kg IPA3 | HCC827 (EGFR mutated), H23 (KRAS mutated) and H520 (PAK1 overexpression) | HCC827, H23, or H520 xenograft nude mice | PKCι signaling inhibition | 99 |
| AF + cisplatin | Cells = 1 µM AF; 1 µM cisplatin, 4 h Mice = 10 mg/kg/day AF + 3 mg/kg/day cisplatin | H69 and H196 (cisplatin resistant) | H69 xenograft nude mice | ROS induced apoptosis | 100 |
| AF + KU55933 (Ataxia ‐telangiectasicia mutated protein, ATM ibhibitor) | AF 2 µM in A549; 10 µM in MLF + 10 µM ATM, 4 h | A549, MLF | Oxidative stress | 149 | |
| AF | 3–4 µM at 24 h | Several different cell lines | Mitochondrial damage and apoptosis | 150 | |
| Ovarian cancer | |||||
| AF | EC50(24 h) = 1.4 µM (2008), 1.03 µM (C13) | cisplatin sensitive (2008) and its cisplatin‐resistant variant (C13) | Oxidative stress | 102 | |
| AF + selenite | 1 µM AF, 0.5 µM selenite for 48 h | cisplatin sensitive (2008) and its cisplatin‐resistant variant (C13) | Redox unbalance | 103 | |
| AF | 0.5 µMAF for 24 h | A2780 | Variation of amount of proteins involved in protein degradation and redox homeostasis (proteomic study) | 104 | |
| AF | 0.5 µM AF for 24 h | A2780 cisplatin‐resistant | Variation of amount of proteins involved in protein degradation and redox homeostasis (proteomic study) | 105 | |
| AF | EC50 (ES2 TICs) = 0.192 µM, 5 days Mice = 12 mg/kg, 6 days | ES2 and Skov3 cultured in stem cell medium and developing tumorigenic TIC (tumor‐initiating cell) phenotype | ovarian cancer orthotopic mouse model | PKCι signaling inhibition | 106 |
| AF | 0.1 µM AF for48 h | SCOV3 | Caspase‐3‐mediated apoptosis in FOXO3‐dependent manner | 22 | |
| AF + MUC4 silencing | 0.025 µM AF for 72 h | SKOV3 | Inhibition of Her2/Akt/FOXO3 pathway | 110 | |
| AF + BRCA1 silencing | 2 µM AF for 18 h | SKOV3, OVCAR5 | Double strand brakes in ROS dependent manner | 112 | |
| AF + HSP90 inhibitor (AUY92) | 1 µM AF, 0.1 µM AUY92, 24 h | A1847, A2780, OVCAR8, OVCAR4, PEO4, SKOV3 | DUBs inhibition | 113 | |
| AF | Cells = 0.7 µM, 48 h Mice = 15 mg/kg three times a week for 2 weeks | A2780 | A2780 orthotopic and subcutaneous xenograft mice | TrxR inhibition | 114 |
| Other cancers | |||||
| Hepatoma | |||||
| AF | 2 µM, 6 h | HEPG2 | Inhibition of IL‐6 mediated activation of JAK1‐STAT3 pathway | 16 | |
| AF + Disulfiram | 10 μM DSF, 0.2 μM AF or their combination for 24 h | SMMC‐ 7721 and HepG2 | HepG2 or SMMC‐7721 xenografts | proteasome inhibition, induction of ER stress | 115 |
| AF + Sorafenib | Different concentration depending on cell lines. Mice = 6 mg/kg/d AF for 7 days | Huh7, MHCC97L, Hep3B, HepG2, PLC/PRF/5) | Hydrodynamic tail‐vein (HDTV) injections of CRISPR‐Cas9‐KO p53 and PTEN plasmids in C57BL/6 N mice | TrxR1 inhibition | 117 |
| AF + morin | 1 µM AF,100‐200 µM morin, 24 h | Hep3B | Extrinsic and intrinsic apoptosis pathways | 118 | |
| AF + sulforaphane | 0.5, 1, 1.5, and 2 μM AF or 2.5, 5, 7.5, and 10 μM sulforaphane for 24 h | HepG3 | PI3K/Akt Signaling Pathway | 119 | |
| AF | EC50 = 0.43 (24 h) and 0.17 μM (48 h) Mice = 6 mg/kg/day for 21 days | HEPG2 | HEPG2 xenograft | DUBs inhibition | 76 |
| Prostate cancer | |||||
| AF | 2.5 µM AF for 24 h | PC‐3 | induction of Annexin A5 expression | 121 | |
| AF | 1 µM AF for 24 h | PC‐3 | Annexin A5 mediated ihibition of COX‐3 | 122 | |
| AF | 0.5–1 µM for 24 h | PC‐3 | Annexin A5 mediated ihibition of PAI‐2 | 123 | |
| AF | Cells = 1 µM AF for 24 h Mice = 6 mg/kg/d | LNcap and 22RV1 | 22RV1 xenograft | Inhibition of DUBs and androgen receptor degradation | 125 |
| AF + enzalutamide (ENZ) and docetaxel (DTX) | Cells = 2 µM AF for 12 h Mice = 5 mg/kg/d AF 10 mg/kg/d ENZ5 10 mg/kg/d, 5 days per week DTX once a week | cells resistant to DTX and androgen‐deprivation therapy | R1‐DDR (cells resistant to DTX and ENZ) xenograft | decrease the AR3‐E2F1 axis | 126 |
| AF | EC50 = 2.18 µM and 0.64 µM PNANC‐1 nutrient sufficient and deprived Mice = 12.5 mg/kg/5 times weekly for 5 weeks | PANC‐1 | xenograft models of human PSN‐1 | TrxR1 inhibition | 127 |
| AF + BSO + 2DG and 17AAG | 0.5 µM AF for 24 h, 17AAG 20 mM 2DG 15 mM, BSO 1 mM for 24 | PC‐3 | Oxidative stress | 72 | |
| AF | 1 µM AF for 24 h, 20 mM 2DG for 24–48 h, 300 µM DHEA for 24–48 h | PC3, DU145 | Oxidative stress | 73 | |
| Head and neck squamous cell carcinoma | |||||
| AF + BSO | Cells = 0.5 µM AF + 1 mM BSO Mice = 400 mg/kg/d BSO + mg/kg/d AF for 10 day | FaDu, Cal‐27 and SCC‐25 | Cal‐27 xenografts | Oxidative stress | 128 |
| AF + BSO | Cells = 0.1–0.5 μM AF + 5–25 μM BSO Mice: 2 mg/Kg/d AF + 450 mg/kg/d BSO | Several cell lines | HN3‐cisplatin resistant xenografts | Oxidative stress | 129 |
| Osteosarcome and Ewing sarcome | |||||
| AF | Cells = EC50(48 h) = 0.7 µM for KHOS, 1.3 µM for MG‐63 Mice = 0.1 mg/kg AF + 2.5 mg/kg vorinostat), or + 0.1 mg/kg rapamycin 5 days per week for 3 weeks | KHOS/NP and MG‐63 | KHOS/NP xenograft | Apoptosis induction | 130 |
| AF + ganetespib | Mice = 12 mg/kg through AF once a day for 5 days per week, and 150 mg/kg ganetespib once weekly | Several cell lines of Ewing sarcome | A673 xenografts | 131 | |
| Melanoma | |||||
| AF | Cells = 1–4 µM for 2 h 0.0125–0.2 µM for 2 h EC50(2 h) = 6.5 µM in stationary cell population; 7 µM in logarithmic cell population Mice = 2–24 mg/kg/daily on days 1 through 5. | B16 melanoma cell lines | C57BL x DBA/2 F, mice inoculated with B16 melanoma cells | 45 | |
| AF + MJ25 (2‐{[2‐(1,3‐benzothiazol‐2‐ylsulfonyl)ethyl]thio}−1,3‐benzoxazole) | 0.1–5 µM AF for 6, 12, 24 h | human melanoma cell line ARN8 stably transfected with RGCΔFosLacZ and its parental cell line A375; HT‐144 melanoma cells and several others cell lines | TrxR inhibition | 132 | |
| AF | 0.25–10 µM AF for 48 h (most potent anti‐melanogenic activity =1 µM) | B16F10 and MNT‑1 melanoma cells | Melanogenesis inhibition through by different mechanisms, including reduction of intracellular tyrosinase enzyme activity, reduction of cAMP levels and increase of immature melanosomes. | 133 | |
| Cervical cancer | |||||
| AF | EC50 = 2 µM AF for 24 h | HeLa | Oxidative stress | 134 | |
| AF + 2‐DG + BSO | Cells = 0.050 µM AF, 10 mM 2‐DG, 500 mM BSO for 24 h Mice = 400 mg/kg 2‐DG, 200 mg/kg BSO, and 1.5 mg/kg AF 3 times per week, over a period of 35 days. | SiHa, Caski, C33A, ME180 | SiHa and Caski xenografts | Oxidative stress | 135 |
Note: The principal mechanisms or pathways proposed as potential targets are reported.
AF concentrations, as single drug or in combination, used for determination of EC50 (if calculated in the study) or in key experiments, such as apoptosis and cytotoxicity determination, are reported.
References include also studies not specifically described in the text.
The bibliographic research was limited to studies published from 1980 to 2020 and found in PubMed and Google Scholar databases. Keywords used to retrieve documents were “Auranofin and cancer.” In PubMed the option “Best match” was applied. The word “auranofin” had to be cited in the title, in the abstract or among keywords. Studies performed with auranofin analogues, but not auranofin, were not included.
2. AF REPURPOSING IN CANCER THERAPY
2.1. Blood cancers
AF has been shown to exert a significant anticancer activity on several different types of blood cancer cells, namely myeloma, leukemia, and lymphoma. Indeed, as reported in the introduction, AF is currently in phase II clinical trials for the treatment of chronic lymphocytic leukemia.
2.1.1. Multiple myeloma (MM)
MM is the second most common hematological malignancy after non‐Hodgkin's lymphoma, and is characterized by the presence of differentiated plasma cells (PCs) in the bone marrow. 23 Standard therapies for MM are based on a combination of proteasome inhibitors (bortezomib and carfilzomib) and immunomodulatory agents (thalidomide and lenalidomide). 24 , 25 , 26 However, due to acquired drug resistance and the heterogeneity of myeloma cells, MM remains incurable. Therefore, new therapeutic approaches are needed to overcome drug resistance and improve clinical outcomes in the treatment of MM.
The first study dealing with the potential use of AF to treat MM was carried out by Nakaya et al. 18 They selected this gold(I) compound based on its well‐known anti‐inflammatory and immunosuppressive properties through the inactivation of NF‐κB signaling. 19 , 27 It has been reported that chemoresistance in MM is associated with constitutive activation of NF‐κB and tyrosine‐protein kinase Janus Kinase 2 (JAK2)/STAT3 signaling. 28 Consequently, Nakaya et al. examined whether AF could exert anticancer activity through the inhibition of these signaling pathways. They carried out the analysis on the human myeloma cell line U266 confirming some of the experiments also in the RPMI8226 and IM‐9 cell lines as well as in cells isolated from bone marrow samples of three patients. The results pointed out the ability of 0.05 µM AF, after 24 h of treatment, to arrest cell cycle in subG1 phase followed by apoptosis via both extrinsic and intrinsic pathways. Apoptosis was achieved by the inhibition of interleukin‐6 (IL‐6) induced‐JAK/STAT3 pathway and the downregulation of the antiapoptotic induced myeloid leukemia cell differentiation protein (Mcl‐1), one of the key proteins for survival of myeloma cells. 29 , 30 Furthermore, Nakaya et al. demonstrated that the combinatory treatment with 0.05 µM AF and the specific NF‐κB inhibitor SN‐50 potentiates the downregulation of Mcl‐1 and consequently the apoptotic effect. Raninga et al. showed for the first time a positive correlation between myeloma growth, chemoresistance and high Trx1 and TrxR1 expression levels. 31 They used AF as a selective inhibitor of TrxR1 and the compound PX‐12 to target Trx1. AF (1–4 µM) and PX‐12 treatment disrupted intracellular redox homeostasis in myeloma cells (RPMI8226 and U266) leading to cell growth arrest and activation of the apoptotic pathway. Therefore, they proposed to consider the inhibition of Trx1 and TrxR1 as a potential novel strategy to treat drug‐resistant MM. In a subsequent study Raninga et al. sought to analyze in more depth the role of TrxR1 in the acquisition of bortezomib (first proteasome inhibitor to be approved by the FDA) resistance in MM. 32 It is known that hypoxia is a key factor in the acquisition of bortezomib resistance even if the molecular mechanisms involved are still to be elucidated. 33 , 34 The authors first showed, by using the gene expression data set (GSE4581), that higher TrxR1 levels were associated to acquired drug resistance and to decreased myeloma patient survival. Second, they found an overexpression of TrxR1 in hypoxic myeloma cells RPMI8226 and U266 resistant to bortezomib, suggesting a possible involvement of this enzyme in the hypoxia‐induced bortezomib resistance. Thus, they used AF (1–4 µM) as a selective TrxR1 inhibitor in hypoxic myeloma cells. The results pointed out that TrxR1 inhibition overcame hypoxia‐induced bortezomib resistance. Accordingly, they demonstrated that hypoxia increased NF‐κB subunit p65 nuclear protein levels and that AF‐induced TrxR1 inhibition, decreased both NF‐κB p65 protein and messenger RNA (mRNA) levels as well as mRNA levels of downstream NF‐κB regulated genes, Survivin and Cyclin D1. Based on these data, the authors proposed TrxR1 inhibitors, such as AF, as single drug or in combination therapy to bypass bortezomib resistance and to increase myeloma patient survival. In the same year Raninga and colleagues investigated a possible crosstalk between TrxR and heme oxygenase‐1 (HO‐1) in multiple myeloma cell lines RPMI8226, U266, and OPM2, aiming to demonstrate the need to target multiple antioxidant systems. 35 HO‐1 participates in hemoglobin degradation and catalyzes the conversion of intracellular heme into biliverdin, free iron, and carbon monoxide. 36 In turn, biliverdin is reduced by biliverdin reductase to bilirubin which has anti‐inflammatory, antioxidative, and antiapoptosis properties. 37 HO‐1 expression and activity has been found increased in several types of cancer implying its potential involvement in chemoresistance. Moreover, HO‐1 expression is regulated by many transcription factors including NF‐κB 38 and nuclear factor erythroid 2‐related factor 2 (Nrf2) 39 but its regulation in myeloma cells is unclear. The obtained results pointed out that TrxR inhibition by a sublethal concentration of AF (1 µM) induced HO‐1 expression through the Nrf2 signaling pathway without leading to apoptosis. The study demonstrated that this increase was a compensatory effect to the decrease of TrxR activity and protected cells from undergoing apoptosis. Indeed, only a combinatory use of HO‐1 inhibitor Zinc Protoporphyrin IX (ZnPP IX) and a sublethal AF dose triggered apoptosis in myeloma cells. The idea of a combinatory use of AF in chemoresistant MM treatment was evaluated by Tessoulin and colleagues. 40 The aim of the study was to overcome chemoresistance in p53 or Bax/Bak mutated myeloma cells by targeting oxidative stress. Prima‐1Met (APR‐246) is a compound able to interact with mutated p53 protein restoring its tumor‐suppressor function and triggering cell death in various cancer types. Since Prima‐1Met has been shown to target TrxR1, 41 the authors investigated whether AF could display the same anticancer activity on a panel of 18 myeloma cell lines and in 10 primary cells from patients with multiple myeloma or plasma cell leukemia. The results confirmed the ability of both AF (0.4 µM) and Prima‐1Met to overcome resistance to apoptosis in p53 mutated MM cells inducing ROS dependent cell death. Besides, both compounds showed antiproliferative activity in myeloma cells resistant to the Bcl2‐specific BH3‐mimetic venetoclax, inducing apoptosis in a Bak/Bax‐independent manner.
The in vivo anticancer activity of AF was assessed in a human RPMI8226 xenograft model by Sze et al. 42 They demonstrated that AF was able to arrest tumor growth and induce apoptosis through TrxR inhibition.
Overall, these studies have strengthened the idea of a possible clinical use of AF. Indeed, very recently, AF, at a concentration ranging from 0.01 to 10 µM, was evaluated by Bonolo de Campos et al. in a “Direct Drug” screening with other 76 FDA‐approved oncology drugs and emerging therapeutics in 25 MM and 15 non‐Hodgkin's lymphoma cell lines and in 113 primary MM samples. 43 The aim of the study was to correlate the drug sensitivities to clinical phenotype, genetic mutation, and transcriptional profiles to favor individualized therapeutic approaches in MM and to support clinical trials. AF turned out to have the broadest cytotoxicity in primary MM among with proteasome inhibitors, dinaciclib, selinexor, venetoclax, and histone deacetylating agents.
2.1.2. Leukemia
In the last 40 years, several in vivo and in vitro studies have been published on AF as a potential anticancer agent for leukemia treatment. The first in vivo investigations were published in 1981 and 1985; these studies demonstrated the ability of this gold(I) compound to increase the life span of C57BL × DBA/2 F mice inoculated with the lymphocytic leukemia P388 cell line. 44 , 45 Later, the cytotoxic activity of AF was analyzed in acute promyelocytic leukemia (APL) model cells. This type of cancer is characterized by a block of differentiation and an accumulation of promyelocytic cells. All‐trans‐retinoic acid (ATRA) was utilized in the treatment of APL with a 90%–95% complete remission rate of patients. 46 High doses of ATRA can restart cell differentiation even if 30% of patients relapse within 4–5 years. 47 Several studies suggested a potential therapeutic benefit in treating APL with a combination of AF and a low dose of ATRA 48 or 1,25(OH)2 vitamin D3 49 (another differentiation‐inducing agent 50 thus overcoming the harmful side effects caused by high doses of these drugs (i.e., ATRA resistance in relapsed APL patients and the hypercalcemic side effect and incomplete cell differentiation with 1,25(OH)2 vitamin D3 51 ). In particular, Park et al. deciphered the molecular mechanism responsible for AF‐induced apoptosis demonstrating that AF (2 µM) proapoptotic effects in human HL‐60 cells depended on p38 mitogen‐activated protein kinase (MAPK) signaling activation. 52 ROS induction by AF was proposed as a mechanism of p38 MAPK activation, since N‐acetyl‐l‐cysteine (NAC) inhibited p38 MAPK phosphorylation, preventing apoptosis.
Fiskus et al. attempted to provide rationale to AF repurposing for chronic lymphocytic leukemia (CLL) therapy. 53 Standard therapy is based on myelotoxic chemoimmunotherapy. 54 However, in case of relapse a different treatment is required as standard therapy is no longer effective. This type of cancer was suitable for AF repurposing because CLL cells have intrinsically higher levels of ROS. 55 The authors demonstrated for the first time thatAF 1 µM triggered lethal oxidative stress through TrxR inhibition and ER stress response in cultured and patient derived primary CLL cells, including those with genetic features associated with poor clinical outcome. AF anticancer activity was also proved to be efficient in TCL‐1 transgenic mice, an in vivo model of CCL. 56
Chen et al. 57 evaluated AF on chronic myelogenous leukemia (CML) cell and animal models, as an alternative therapy to overcome acquired resistance to Imatinib mesylate (IM), a drug resistance mainly due to mutation in Bcr‐Abl. 58 , 59 They demonstrated that AF had antiproliferative activity in both Bcr‐Abl wild‐type and resistant Bcr‐Abl‐T315I CML cell lines as well as in mouse IM‐resistant xenograft models, thus illustrating AF ability to overcome drug resistance. AF‐induced apoptosis depended on both Bcr/Abl signaling downregulation and proteasome‐dependent caspase activation, while ROS were not involved in cell death process. In a subsequent study, Clapper et al. demonstrated that in imatinib resistant CML cells the TrxR/Trx system was upregulated. 60 The inhibition of the TrxR enzyme by AF and the gold compound [Au(d2pype)2]Cl decreased Bcr‐Abl and proto‐oncogene c‐myc protein expression leading to apoptosis also in IM resistant CML cells. The obtained results highlighted the effectiveness of TrxR targeting in overcoming IM resistance as well as the crosstalk between the TrxR system and Bcr‐Abl signaling pathway.
Acute lymphoblastic leukemia (AAL) is the most frequent neoplasm in childhood 61 and, as various other cancers, it is characterized by an unbalanced redox homeostasis. 62 For this reason, Haß et al. 63 selected different ROS inducers to sensitize ALL cells to therapeutic treatment based on the second mitochondria‐derived activator of caspases (Smac) mimetic LCL161 that antagonizes inhibitor of Apoptosis (IAP) proteins. They used AF alone or in combination with LCL161 to target TrxR, Erastin to inhibit the cystine/glutamate transporter XCT required for GSH production and buthionine sulfoximine (BSO) as specific inhibitor of γ‐glutamylcysteine ligase (γ‐GCL). 64 The results pointed out the effectiveness of this approach in being able to prime ALL cells for LCL161‐induced cell death. The thioredoxin system was also evaluated as a novel therapeutic target against B‐cell acute lymphoblastic leukemia (BCP‐ALL) by Fidyt et al. 65 This blood cancer is characterized by genetic heterogeneity and abnormal expansion of immature B cells. The currently available chemotherapy is effective in most cases. However, patients harboring certain genetic lesions, such as MLL rearrangements or Bcr‐Abl1 fusion, are resistant and need novel therapeutic approaches. The authors used AF and adenanthin (ADE), to inhibit the thioredoxin system. The results showed that these compounds triggered oxidative stress and ER stress leading to cell death, both in BCP‐ALL cell lines, pediatric and adult BCP‐ALL primary cells, including primary cells cocultured with bone marrow‐derived stem cells. AF efficacy was also demonstrated in vivo using a patient‐derived xenograft model. Recently Liu et al. identified a mitochondrial isoform of TrxR3 as a new target of AF. 66 The authors found that TrxR3 was overexpressed in myelomonocytic leukemia MV4‐11 cells resistant to sorafenib (MV4‐11R) in comparison to parental cell line and furthermore, patients with high‐risk acute myeloid leukemia showed high expression of this enzyme and worse survival in comparison to low‐risk patients. They demonstrated, both in vitro and in vivo models, that TrxR3 high expression conferred sorafenib resistance and that AF was able to overcome resistance.
2.1.3. Lymphoma
To our knowledge, the first study carried out on AF anticancer activity on lymphoma cell lines was published by Cox et al. 67 They demonstrated that AF treatment activated apoptotic signaling through a Bcl‐2 homologous antagonist/(Bax/Bak)‐dependent mechanism associated to TrxR inhibition and peroxiredoxin3 (Prx3) oxidation in Jurkat T‐lymphoma and U937 monocytic cell lines. Celegato et al. investigated repurposing AF for refractory classical Hodgkin lymphoma (cHL). 68 Its antitumour activity was demonstrated both in vitro and in vivo tumor models. AF (0.1–10 µM) has showed antiproliferative effects in L‐1236, L‐428, KM‐H2, HDLM‐2, and L‐540 cHL‐derived cell lines, in L‐540 gemcitabine‐resistant and HDLM‐2 brentuximab resistant cells. Furthermore, AF showed a synergistic activity with three chemotherapeutic drugs widely used in cHL treatment such as doxorubicin, cisplatin, and gemcitabine. This cytotoxicity was also demonstrated in L‐540 gemcitabine resistant–derived tumor xenograft. The proposed mechanism of action encompassed the inhibition of TrxR, ROS increase, and apoptotic pathway activation.
ROS‐induced apoptosis was also proposed as main mechanism in other studies where AF was used alone or in combination with other pro‐oxidant compound as l‐ascorbate, used at pharmacological concentration of 200 µM, and BSO. 69 , 70 Finally, Wang et al. repurposed AF to treat TP53‐mutated or (PTEN) deleted refractory B‐cell lymphoma. 71 In several refractory mantle cell lymphoma (MCL) and diffuse large B‐cell lymphoma (DLBCL) models, they demonstrated that AF inhibited TrxR, increased ROS production and apoptosis. Its cytotoxicity was mainly effective in TP53‐mutated or PTEN‐deleted lymphomas strengthening the hypothesis of AF repurposing for treatment of TP53‐mutated or PTEN‐deleted refractory B‐cell lymphoma.
2.2. Breast cancer
Breast cancer is the most common female cancer and the second most common cause of cancer death in women. Up to 20% of breast cancers are “triple negative” tumors (TNBC) as the three most common types of receptors known to fuel most breast cancer growth—estrogen, progesterone, and the receptor tyrosine‐protein kinase (HER‐2/neu) gene—are not present in cancer cells. TNBC are not treatable with antihormone therapy, thus chemotherapy and radiation therapy represent the only possible form of treatment. Unfortunately, most patients develop resistance to these interventions and there is an unmet clinical need to find novel targeted and combination therapies.
Several studies have recently investigated the ability of AF to counteract breast cancer both in vitro and in vivo. In most of them, AF has been used in combination with at least one other drug to increase oxidative stress and induce cell death. Scarbrough et al. reported that simultaneous targeting of both GSH with BSO and TrxR with AF (0.5 µM), enhanced chemo‐sensitization to 17‐Allylamino‐17‐demethoxygeldanamycin (17AAG) (an experimental chemotherapeutic agent believed to increase oxygen free radicals) and 2‐deoxy‐d‐glucose (2DG) combined treatment in MDA‐MB 23 and SUM159 cell lines. 72 The same research group also proved that inhibition of TrxR by AF (1 µM) potentiated cancer cells clonogenic killing induced by inhibiting both glycolysis (with 2‐DG) and pentose phosphate cycle (with dehydroepiandrosterone DHEA). 73 Similar results were also obtained using PC‐3 prostate cancer cell lines 73 Kim et al. demonstrated that AF (0.5–1 µM) was able to inhibit cell proliferation and cell growth on soft agar in MDA‐MB 23 through inhibition of STAT3 phosphorylation, in a ROS‐dependent fashion. 17 Other two studies on triple negative cell lines indicated that 0.5 µM AF in combination with vitamin C (used as prooxidant) or with vitamin C plus menadione induced cell death mostly through the generation of oxidative stress, strengthening the unbalance of the redox homeostasis as main mechanism of AF action. 74 , 75 On the other hand, in 2014 Liu et al. reported for the first time that proteasome inhibition, and not ROS production, was required for AF‐induced cytotoxicity and apoptosis both in vitro and in vivo model. 76 They demonstrated that AF inhibited the activity of deubiquitinases (DUBs) associated to 19S regulatory subunit of proteasome in MCF‐7 and in HepG2 (hepatoma cell line). This caused and accumulation of ubiquitinated proteins that was already detected after 3 h of 0.5 µM AF treatment indicating the proteasome inhibition occurred early and at low dose of the drug.
Very recently, AF was used in combination with an antiprogrammed death‐1 ligand (PD‐L1) antibodies to target this key immune checkpoint. 77 In fact, the immune system plays a critical role in controlling neoplastic transformation and progression and targeting immune checkpoints constitutes a valid possibility to treat tumors refractory to conventional therapies. One of these checkpoints is represented by programmed death‐1 (PD‐1) receptor and PD‐L1. PD‐1 receptor is expressed on the surface of T‐cells and displays immunoinhibitory activity. Cancer cells express PD‐L1 on their surface and interact with cytotoxic T‐cells, inhibiting their function and evading host immunosurveillance. In several cancers, PD‐L1 expression increases in response to chemotherapy and leads to drug resistance; in triple negative breast cancer it is overexpressed in approximately 20%–30% of patients and is associated with poor prognosis. 78 The authors demonstrated that AF induced cell death in different breast cancer cell lines (with IC50 values ranging between 0.5 and 2 µM in TNBC cells) and impaired the growth of TNBC‐derived spheroids. AF treatment exerted significant in vivo antitumour activity in multiple TNBC models including the syngenic 4T1.2 model, MDA‐MB‐231 xenograft and patient‐derived tumor xenograft. Interestingly the authors showed that AF alone failed to completely eliminate tumors in all in vivo models due to AF‐induced increased PD‐L1 expression through the ERK1/2‐myc axis. This observation led to the rationale of the implementation of a combined AF and anti‐PD‐L1 antibodies treatment. Combined treatment synergistically impaired the growth of 4T1.2 primary tumors, overcoming the AF‐induced increased PD‐L1 expression.
In addition, Rodman et al. have shown that AF is also capable of potentiating the response to radiation therapy both in vivo and in vitro. 79 They demonstrated that combined inhibition of GSH synthesis with BSO and TrxR with AF‐enhanced cancer cell clonogenic killing and radiation responses via a mechanism that could be inhibited by N‐acetylcysteine (NAC), thus depending on thiol redox state. Furthermore, pre‐treating breast cancer stem cells with AF sensitized them to radiation. Finally, in vitro results were confirmed by in vivo experiments showing that in human breast cancer xenografts, combined administration of AF and BSO before radiation significantly increased survival rate and decreased the number of cancer stem cells. Very recently, Kim et al. found that the RNA binding protein NONO was bounded to STAT3 mRNA and to STAT3 protein, increasing its stability, transcriptional activity and contributing to STAT3 oncogenic function. 80 Using a high‐throughput drug screening, they revealed that AF was a potential inhibitor of the expression of NONO, suggesting a new way in which AF could carry out its antitumour activity.
2.3. Colon cancer and gastric cancer
Colon cancer is one of main cancer types that lead to cancer‐related death worldwide. 81 Chemotherapy is an important treatment approach for this pathology; however, patients have few treatment options after two cycles of therapy. Hence, the discovery of new treatments for colon cancer care is mandatory. AF (0.5 µM) was combined with 5Z‐7‐oxozeaenol (5 µM), a molecule able to inhibit the transforming growth factor β‐activated kinase 1 (TAK1), leading to colon carcinoma cell death both in vivo and in vitro. 82 The inhibition of TrxR activity and the increased Trx oxidation were proposed to be responsible for the enhancement of sensitization of cancer cells to 5Z‐7‐oxozeaenol. Moreover, AF has been used in combination with the anti‐inflammatory drug celecoxib (CE) in different colon carcinoma cell lines. 83 The findings show that AF/CE combination greatly enhanced AF therapeutic efficacy both in vitro and in vivo. Drug mixture generated oxidative stress that inhibited hexokinase, induced TrxR oxidation and modifies mitochondrial redox state. Particularly in mitochondria, AF/CE promoted the oxidation of TrxR2 and the degradation of the cytochrome c oxidase subunit II, a component of complex IV of the electron transport chain. As consequence, mitochondrial respiration and ATP production resulted inhibited.
5‐FU is widely used in chemotherapy for colon cancer, but drug resistance limits its clinical effectiveness. In a recent study, Liu et al. found that nuclear FOXO3 was decreased in 5‐FU‐resistant SW620 and HCT‐8 (SW620/5‐FU and HCT‐8/5‐FU) cells, and that overexpressing FOXO3 could significantly reverse 5‐FU resistance. 84 FOXO3 is a transcription factor with key role in cell cycle, apoptosis, aging regulation, and tumor suppression. Several protein kinases (IkB kinase, Akt, MAP) phosphorylate FOXO3, promoting its translocation from the nucleus to the cytosol where it is degraded via the ubiquitin‐proteasome pathway. 85 Its inhibition leads to tumor development and progression, suggesting that FOXO3 reactivation could be a promising strategy to develop anticancer therapeutic drugs. The authors demonstrated that TrxR1 was the key protein participating in the regulation of FOXO3‐mediated 5‐FU resistance. Thus, they used AF as FOXO3 agonist and TrxR1 inhibitor to overcome 5‐FU resistance both in vitro and in vivo models.
The possible beneficial effect of AF during radiation treatment has been analysed in subcutaneous CT26 colon tumors, in normal gastrointestinal epithelium of mice and in human organoids formed using malignant and nonmalignant tissues from the same patient. 86 Indeed, one of the disadvantages in the use of radiotherapy in colon carcinoma therapy is the elevated radio sensitivity of normal intestinal epithelium. The results showed that AF pretreatment prevented radiation toxicity in normal cells in all these models. In fact, AF caused cell cycle arrest and p53/p21 pathway activation, leading to reduction of the irradiation‐induced DNA damages and increasing cell survival. On the other hand, in malignant cells AF inhibited proteasome activity and induced endoplasmic reticulum stress/unfolded protein response, activating apoptosis.
Concerning gastric cancer, Peng et al. demonstrated that in gastric cell lines, the natural compound piperlongumine (PL), known to induce ROS production, enhanced AF cytotoxicity. 87 In particular, 4 µM AF induced ROS increase, ER stress and mitochondrial dysfunction leading to apoptosis. The combination of AF and PL resulted in an increase of cell death in vitro and in a reduction of tumor size in vivo.
2.4. Lung cancer
Lung cancer is the leading cause of cancer‐related deaths in men and women. 88 In particular, non‐small cell lung cancer (NSCLC) accounts for about 85% of all lung cancers and has a poor prognosis with a 5‐year survival rate of 17%. Currently common therapies for NSCLC include platinum‐based chemotherapy with concurrent radiotherapy but cancer cell resistance limits treatment utility.
Fath et al. investigated AF efficacy in combination with 2DG and carboplatin in both in vitro and in vivo models. 89 First, the authors demonstrated that 2DG enhanced carboplatin‐induced clonogenic cell killing and that this effect was almost abolished by NAC, indicating that 2DG plus carboplatin effect in lung cancer cells was mediated by disruptions of thiol metabolism. Second, they showed that inhibiting both GSH synthesis with BSO and TrxR activity with AF resulted in the clonogenic killing of A459 and H292 cell lines (with AF 5 and 0.5 µM, respectively), while single drug treatment had no effect on A459 cell line and low effect on H292. These results represented the rationale to treat cancer cells with a combination of AF + BSO and 2DG + carboplatin. As expected, this treatment caused a decrease of the clonogenic survival of 2DG + carboplatin treated A549 cells as well as increasing thiol oxidation that was inhibited by NAC. Most importantly, in A549 xenograft mice, the combination of AF + BSO + carboplatin resulted in a highly significant decrease in tumor growth rate when compared to control and to treatment with BSO + AF or carboplatin alone.
Fan et al. demonstrated that selenocysteine (SeC) acted as a natural inhibitor of TrxR1 in vitro and in vivo to enhance AF‐induced human lung cancer cells killing through ROS‐mediated apoptosis and inactivation of Mitogen‐Activated Protein Kinase 1 (Erk) and Akt pathways. 90 Also, in this study, treatment of A459 with AF alone (6 µM) was not able to induce cell death, but pretreatment with SeC resulted in a severe cell death increase. The data were also confirmed in vivo: indeed, A459, xenograft growth in nude mice was more effectively inhibited by combined treatment with SeC and AF than with AF alone.
PI3K/Akt pathway plays a pivotal role in cancer cell growth and survival and inhibition of this pathway may represent a tool in cancer therapy. It has been reported that a large subset of NSCLC cells does not respond to Akt inhibitors, 91 thus Dai et al. used a genome‐wide siRNA library screening to identify synthetic lethality loci with Akt inhibitor MK2206. 92 Among these loci, they identified the TrxR1gene and demonstrated that inhibiting TrxR1 with siRNA or AF, sensitized lung cancer cell lines resistant to MK2206 to treatment with this drug. The combined treatment with AF 0.1 µM induced apoptosis by ROS activation of mitogen‐activated protein kinase 8 (JNK). Furthermore, in H1993 xenograft nude mice, treatment with MK2206 or AF as single drugs had no effect on tumor growth, but their combination significantly inhibited tumor growth and significantly prolonged animal survival. The same research group also investigated the cytotoxicity of AF alone in 10 different NSCLC cell lines and they found that AF sensitivity ranged from less than 1 µM to up to 2 µM. 93 They demonstrated that sensitive cells presented lower expression of TrxR1 than resistant cell lines. The authors selected representative sensitive (Calu3 and HCC366) and resistant (A549) cell lines for further experiments. In the sensitive cell lines, a high‐throughput protein profiling analysis of 214 proteins and phosphoproteins was performed and several key nodes in the PI3K/Akt/mTOR pathway resulted repressed. Since the transient overexpression of the TrxR1 protein partially reversed this repression, they postulated that TrxR1 may participate in the regulation of PI3K/Akt/mTOR pathway and that high TrxR1 levels can attenuate AF‐induced inhibition of the PI3K/Akt/mTOR pathway. The activity of AF was confirmed in vivo in Calu3 xenograft nude mice. The same group also proved that the antitumour activity of a combination of TUSC2 gene (tumor suppressor candidate 2) delivered by nanovesicles and erlotinib (epidermal growth factor receptor [EGFR] inhibitor) was enhanced by AF both in vitro and in vivo. 94
Hou et al. investigated the effect of AF on cancer stem cell side‐population (SP). 95 SP represents a subpopulation of stem‐like cancer cells that have a pivotal role in drug resistance due to their high expression of the ATP‐binding cassette transporter ABCG2 involved in drug export. The authors found that A549 cell line, contained a considerable amount of SP and AF could effectively deplete SP cells by increasing ROS generation. Interestingly, they proved that AF inhibited the glycolytic enzyme hexokinase leading to ATP depletion and decreasing the activity of ABCG2 pump. Furthermore, the synergistic effect of AF and the chemotherapeutic agent adriamycin was proved both in vivo and in vitro.
Trx/TrxR and glutathione/glutathione‐disulfide reductase (GSH/GSR) are the most relevant cellular antioxidant systems and functional deficiency in one of these systems renders cells dependent on the other system for survival. 96 Thus, it has been hypothesized that cells harboring a compromised GSH/GSR system could be more sensitive to TrxR inhibition. Yan et al. found that GSR gene is delete in 6% of lung adenocarcinoma. To investigate whether this deletion could sensitize cells to AF treatment, the authors evaluated AF sensitivity in several cell lines with different expression of genes involved in GSH homeostasis and in AF resistant A459 cell line in which GSR gene was knocked out or silenced. In all these conditions the authors demonstrated that the alteration of GSH homeostasis was correlated with increased sensitivity to AF treatment. 97 Moreover, they also demonstrated in vivo activity of AF in two lung cancer patient‐derived xenograft models with relatively low levels of GSR and glutamate‐cysteine ligase catalytic subunit expression.
Activating EGF mutations represent a driven force in lung tumor progression and limit the therapy based on EGFR inhibitors. It is interesting to note that AF (0.25 µM) enhanced ibrutinib activity in EGFR mutant non‐small cell lung cancer cells by inhibiting the expression or phosphorylation of multiple key nodes in Akt/mTOR and MEK/ERK pathways. 98 Ito et al. proved that AF in combination with IPA‐3, an inhibitor of p21 activate kinase (PAK1) which is in turn regulated by PKCiota, had a highly synergistic effect in EGFR or KRAS mutant adenocarcinoma and squamous cell carcinoma cell lines and decreased tumor volume in mice models. 99 In these cell lines the combination of IPA‐3 and AF abrogated the expression and/or phosphorylation of serine/threonine‐protein kinase PAK1, PKCiota, ERK 1/2, mTOR, Akt, and transcriptional coactivator YAP1 proteins.
Liu et al. developed a high‐throughput drug screen strategy to identify new drugs that can synergistically enhance antitumour efficiency of cisplatin in chemoresistant small cell lung carcinoma (SCLC) cells. 100 Among the FDA‐approved drug library of 1092 compounds they found that AF was able to overcome cisplatin resistance. In this study the authors demonstrated that AF synergized with cisplatin resulting in ROS overproduction, which in turn led to mitochondrial dysfunction and DNA damage in SCLC. The combination resulted in significant reduction of tumor growth also in vivo.
2.5. Ovarian cancer (OC)
OC is the most lethal gynecological disease and is characterized by heterogeneity, high risk of relapse and development of platinum resistance that result in poor prognosis. The cornerstone of treatment is maximal‐effort surgical cytoreduction combined with cytotoxic chemotherapy. An estimated 80%–85% of patients with OC who achieve full remission following first‐line platinum‐based therapy will develop recurrent disease and median survival for these patients ranges from 12 months to 24 months. 101 Except for bevacizumab and poly ADP‐ribose polymerase inhibitors, approved for BRCA1/2 mutated patients and for platinum‐sensitive patients without BRCA 1/2 mutation, there are few other options for women with platinum‐resistant OC.
The first articles that report a cytotoxic activity of AF on ovarian cancer cells were published by Marzano 102 and Rigobello. 103 In these studies, the authors demonstrated that AF could overcome cisplatin resistance in the cisplatin‐resistant variant (C13*) of the sensitive 2008 cell line. AF, by inhibiting TrxR activity, induced ROS mediated activation of apoptosis. The authors also explored the effect of AF (1 µM) in combination with selenite that is a powerful pro‐oxidant compound at moderate to high concentrations. Selenite pretreatment induced a dramatic increase in AF cytotoxicity in both resistant and sensitive cells. Cytotoxic AF properties have been investigated by 2D‐gel proteomic approach. 104 , 105 These studies, performed on A2780 sensitive and cisplatin‐ resistant (A2780R) cells showed that AF (0.5 µM) affected the proteins involved in redox balance (peroxiredoxin 1 and 6, thioredoxin‐like 1 protein) and in protein degradation.
In 2013, Wang and collaborators found a new AF target: the PKCiota protein a member of the protein kinase C family. 106 The authors cultured ES2 and SKOV3 ovarian cell lines in stem cell medium to obtain cells with tumorigenic tumor initiating cell (TIC) properties (TICs cells). TIC properties include expression of stem‐related genes, clonal expansion, enhanced transformed growth, resistance to chemotherapy. These characteristics resemble properties of ovarian cancer stem cells that play a key role in relapse following systemic chemotherapy. 107 These authors demonstrated that ovarian TICs require PKCiota for clonal expansion, enhanced anchorage‐independent‐growth, and tumor initiation in vivo. Pharmacologic inhibition of PKCiota with AF blocks the TIC phenotype in vitro and TIC‐mediated tumor formation in vivo. PKCiota inhibition was achieved with AF 1 µM treatment for 24 h. This inhibition was probably direct, since it has been previously demonstrated that two analogues of AF, namely aurothiomalate and aurothioglucose, interact with a critical cysteine residue within the PKCiota PB1 (Phox and Bem 1) domain leading to inhibition of oncogenic PKCiota signaling. 108
Two other interesting articles focused on the inhibition of FOXO3 pathway. Park et al. showed that AF at 0.1 µM for 48 h downregulated the expression of IkB‐kinase, one of the kinase responsible of FOXO3 phosphorylation 109 and induced the nuclear translocation of FOXO3 in SKOV3 cells that are p53 null. 22 Furthermore, FOXO3 silencing caused a decrease in apoptosis indicating that in this cell line, AF induced apoptosis through FOXO3. The same research group reported that the anticancer activity of AF via FOXO3 regulation could be enhanced by inhibition of the expression of mucin 4 (MUC4). 110 MUC4 is a membrane glycoprotein, it is overexpressed in several cancers and promotes tumor formation, tumor aggressiveness, and poor outcomes in various types of epithelial carcinomas, including ovarian cancer. 111 AF combined with MUC4 silencing decreased the expression of receptor tyrosine‐protein kinase erbB‐2 (Her2) and Akt phosphorylation, leading to inhibition of the Her2/Akt/FOXO3 pathway and thus inhibiting FOXO3 translocation from nucleus to cytosol.
It is well known that BRCA 1/2 mutation predisposes women to increased risks of developing ovarian and breast cancers. Oommen et al. demonstrated that in OVCAR5 and SKOV3 cells, BRCA1 silencing increased AF cytotoxicity and that AF‐induced DNA double strand brakes in a ROS‐dependent manner, thus demonstrating that BRCA1 deficiency renders tumor cells more sensitive to AF. 112 Hyter et al. tested the cytotoxicity of AF alone or in combination with HSP90 inhibitors in ten ovarian cancer cell lines founding that there were remarkable differences in AF sensitivity in different lines. 113 They utilized A1847 and PEO4 as representative cell lines for the sensitive and resistant groups respectively and they demonstrated a synergic effect of AF and the HSP90 inhibitor AUY922. They used the most effective synergic concentration to evaluate apoptosis, protein ubiquitination and expression level of Nrf2, a transcription factor that binds to the antioxidant response element of several detoxifying genes. They also created multigene signatures linked to sensitivity or resistance and sought out the prevalence of these genes in publicly available TCGA databases. This approach pointed out that the resistant cell lines were more like clinical samples and were more appropriate models. Recently Marzo et al. analysed the in vitro and in vivo properties of AF and an AF analogue, the iodido(triethylphosphine)gold(I) complex (Et3PAuI) in which the thiosugar ligand was simply replaced by one iodide ligand. 114 The two compounds exerted similar cytotoxic and proapoptotic effect on A2780 human ovarian cancer cells in vitro. However, when evaluated in a preclinical orthotopic model of ovarian cancer, Et3PAuI produced a far superior anticancer action than AF, inducing almost complete tumor remission. This result confirmed that the thiosugar ligand is not mechanistically indispensable and that the [Et3PAu]+ moiety is likely to be the true pharmacophore.
2.6. Other cancers
The literature contains several other studies concerning the use of AF as a cytotoxic and potentially antitumor agent in other cancer types. Taken together they confirm the possible broad spectrum of application of this drug.
Concerning hepatoma, Kim et al. reported that 2 μM AF was able to inhibit IL‐6 activation of JAK1‐STAT3 pathway in a ROS‐independent fashion. 16 Huang et al. found that AF (0.2 μM) in combination with Disulfiram (an aldehyde dehydrogenase, in clinical use to treat alcoholism) synergistically induced proteasome‐associated DUBs inhibition, reticulum endoplasmic (RE) stress and apoptosis in HepG2 and SMMC‐7721 cell lines. 115 They also proved that this mechanism was ROS‐independent since scavenging of ROS by 100 µM Vitamin C failed to block cell death. As already shown for other cancer types, also in hepatocellular carcinoma TrxR1 expression positively correlated with advanced clinical staging and poorer patient survival. 116 Thus, inhibition of TrxR1 represents a valid tool to counteract cancer progression. Lee et al. demonstrated that AF treatment sensitized several hepatoma cells lines toward conventional therapeutic Sorafenib and furthermore, AF alone was able to inhibit tumor growth in vivo. 117 Hwang‐Bo et al. demonstrated that 1 µM AF was able to induce oxidative stress in Hep3B cells and that the natural compounds morin and sulforaphane enhanced its anticancer activity. 118 , 119 They showed that cotreatment with morin induced activation of both extrinsic and intrinsic pathways of apoptosis by upregulating death receptors DR4 and DR5 and by modulating Bcl2 family proteins. Furthermore, AF plus sulforaphane displayed a synergic induction of ROS‐mediated PI3K/Akt signaling.
Annexin A5 (ANXA5) has a pivotal role in mediating cytotoxicity, apoptosis, and anti‐inflammatory effects. 120 Park et al. suggested that, in PC3 prostate cancer cell lines, AF (2.5 µM) induced ANXA5 expression and translocation into mitochondria where it possible participated in VDAC oligomerization and activation of caspase‐3. 121 More recently, the same group demonstrated that AF suppressed cyclooxygenase 2 (COX‐2) expression through ANXA5 induction, thus inhibiting COX‐2 mediated inflammation. 122 In PC‐3 cells, induction of ANXA5 by AF was also proposed to enhance apoptosis by suppressing the expression of plasminogen activator inhibitor (PAI‐2), 123 a protein involved in tumor vascularization, cell migration, and tumor metastasis. 124
Androgen receptor is often overexpressed in prostate cancer and is strongly linked to tumor growth and progression, thus, its targeting with competitive inhibitors or its down regulation is crucial for therapy. In this context, Liu et al. proved that AF (1 µM) promoted androgen receptor protein degradation and inhibited androgen receptor transcription in two androgen receptor‐positive cell lines, LNcap and 22RV1. 125 Xu et al. demonstrated that the combination of enzalutamide (ENZ) and docetaxel (DTX) effectively inhibited the growth of prostate cancer cells that are resistant to either single drug. 126 Thus, the authors generated a cell line resistant to both DTX and ENZ and demonstrated that AF could overcome resistance by inhibiting the growth of these drug‐resistant prostate cancer cells both in vitro and in vivo.
In prostate cancer as well as in other cancers, some regions of tumor microenvironments are often nutrient‐deprived and hypoxic because of aberrant cell proliferation and insufficient vascularization. Recently, Onodera et al. found that inhibitors of the redox system displayed preferential cytotoxicity to cancer cells in nutrient‐deprived conditions. 127 AF treatment in PANC‐1 cells showed preferential cytotoxicity under nutrient‐deprived compared with nutrient‐sufficient condition with an EC50 of 0.64 and 2.18, respectively.
In literature there are only two studies concerning AF in treatment of head and neck squamous cell carcinoma (HNSCC). 128 , 129 Both were based on the prooxidative activity of AF. Overall, they demonstrated that inhibition of the Nrf2 pathway in addition to inhibition of γ ‐GCL and TrxR could effectively eliminate resistant cells.
The high‐throughput screen of FDA approved drugs was used to discover new therapies for osteosarcoma and Ewing sarcoma. 130 , 131 In both cases AF emerged as a possible alternative to traditional treatments both as a single drug and in combination with rapamycin or vorinostat (for osteosarcoma) and with ganetespib, an HSP90 inhibitor (for Ewing sarcoma).
Few papers dealing with AF and melanoma emerged from Pubmed research. To the best of our knowledge, AF cytotoxic effects were examined in a melanoma cell line (B16 melanoma cells) for the first time by Mirabelli et al. in 1985. 45 Subsequently, Sachweh and colleagues proposed the inhibition of TrxR1 through compounds such as AF, as a possible therapy for malignant melanoma. 132 This type of cancer is the most dangerous type of skin cancer due to drug resistance and relapse following treatment and, in most cases, it is characterized by the inactivation of the tumor suppressor p53 through dysregulation of upstream pathways. In the attempt to find a new melanoma therapeutic strategy, the authors screened for compounds that can reactivate p53 in melanoma cells. They identify the nongenotoxic compound MJ25 (2‐[2‐(1,3‐benzothiazol‐2‐ ylsulfonyl)ethyl]thio}−1,3‐benzoxazole) and proved that MJ25 could inhibit TrxR1. Thus they demonstrated that AF displayed effects comparable with MJ25 on several melanoma cells lines, confirming TrxR inhibition as a potential therapeutic strategy.
The AF efficacy has also been investigated in melanogenesis; Goenka et al. found that nontoxic concentrations of AF (0.25–1 µM) showed antimelanogenic activity in a dose‐dependent manner both in mouse B16F10 and MNT‐1 human melanoma cells lines, by inhibiting intracellular tyrosinase activity, decreasing cAMP levels and increasing immature melanosomes. 133
Lastly, AF‐induced cell death via oxidative stress also in HeLa cervical cancer cells and was used in combination with 2‐DG and BSO (inhibitors of glycolysis and GSH synthesis, respectively) to treat highly glycolytic cervical cancer cell lines that were resistant to standard therapy with cisplatin plus pelvic irradiation. 134 , 135
3. DOSE‐DEPENDENT EFFECTS OF AURANOFIN THROUGH A SYSTEM BIOLOGY‐LIKE APPROACH
In the first part of this review, we reported an update on AF effects according to clinical applications in different cancers. Reporting the results obtained from different cell models, with different concentrations and exposition times, generates a fragmented vision of the AF mechanism of action. Thus, in this section we have summarized the major effects of AF in cancer cells by revisiting a large data set of transcriptomic data obtained with a wide range of cell models exposed to AF. The data are deposited in the connectivity maps (CMAP) website (https://clue.io/cmap) 136 that, using transcriptional expression data (mRNA transcript abundance of 978 “landmark” genes), allows to probe the relationships between drugs and many cellular models of diseases. In practice, changes in gene expression in response to a genetic perturbation (knockdown or overexpression of a gene) or treatments with small molecules (perturbagenes) are compared by similarity to all perturbation signatures of the database (more than 1 million of available profiles) obtaining a “tau score” which allows to predict the effects of a perturbagene. CMAP is equipped with all the bioinformatic tools and transcriptomic data sets to compare the effects of different drug dosages and exposition times in many cancer cell lines.
Searching for the connectivities of AF with other “perturbagenes,” CMAP performs its prediction using the gene expression profiles of nine cell lines (A375, A549, HCC515, HEPG2, HT29, MCF7, PC3, HA1E, and VCAP) exposed for 6 h at 10 µM of AF which is generally considered a high dose. The highest AF connection is with the NF‐κB inhibition (Figure 2A). The NF‐κB inhibition effects of AF have long been known. Jeon et al. were the first to show that NF‐κB was inhibited with 5–10 µM of AF treatment for 4 h in lipopolysaccharide stimulated macrophages blocking IκB kinase (IKK) activity. 19 , 27 Later, Nakaya et al. reported that 0.05 µM of AF inhibited NF‐κB DNA binding and decreased NF‐κB nuclear protein level in U266 multiple myeloma cells. 18 Moreover, Saei et al. recently proposed a proteomic based strategy to find candidate protein targets of AF, at 3 µM drug dose concentration, in HCT116 colon cancer cells. 137 Also in this case, NF‐κB resulted to be the most probable gold target while TrxR1 was only in third position.
Figure 2.
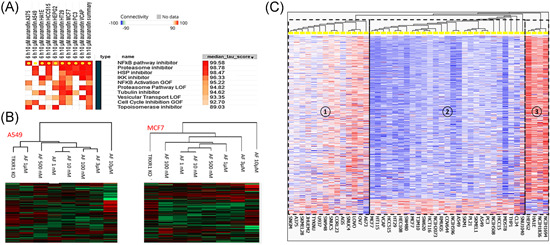
(A) Output of the connectivity map of AF realized with the transcriptomic data of nine cancer cell lines exposed to 10 µM of AF for 6 h. The median tau score is calculated from the tau score of each cell line. Only the “perturbagene” classes with tau score greater than 90 were reported. (B) Hierarchical clustering of the transcription profiles of A549 and MCF7 cell lines exposed to different AF concentrations together with the transcription profile of TrxR1‐silenced cells. (C) Hierarchical clustering of the transcription profiles of 45 cancer cell lines exposed to 10 µM of AF for 6 h. AF, Auranofin [Color figure can be viewed at wileyonlinelibrary.com]
The second AF correlation predicted by CMAP is with proteasome inhibitor family. Inhibition of DUBs by AF (0.5 µM for 3 h) was firstly proposed by Liu et al. in HEPG2 and MCF7 cells. 76 Later the same research group confirmed this finding in vitro and in vivo models of prostate cancer. 115
On the other hand, in a recent proteomic study Zhang et al. compared the proteome profiles of HCT116 colon cancer cells treated with AF, with the proteasome inhibitor bortezomib and with the 19S DUBs inhibitor b‐AP15. 138 They showed that AF dependent DUBs and proteasome inhibition only occurred at high dose of AF (>2.5 µM). Furthermore, AF (used at 1 and 5 µM concentration) induced a proteomic expression profile distinct from that of bortezomib and of AP15, both of which induced similar responses. Thus, AF‐dependent proteasome inhibition was considered an off‐target effect occurring at high dose of AF. Indeed, in the same study, monitoring the level of 1 HO‐1, considered a readout of Nrf2 activation, they observed that oxidative stress induced by TrxR1 inhibition occurred at lower concentration of AF (0.25 µM) compared to HCT116 cells IC50 (1.5 µM) and for this reason it should be considered the dominant mechanism underlying AF cytotoxicity. In our opinion, to try to interpret this set of sometimes conflicting results it should be considered that the cell type specific genetic background could also play a key role in determining the primary AF targets. This is confirmed by the comparison of the transcript expression profiles between different AF concentrations in two cancer cell lines and the effects of TrxR1 silencing, available in the CMAP data set. For example, A549 cells (lung cancer) exposed to 1 µM AF generate effects very similar to TrxR silencing whereas, MCF7 cells (breast cancer) exposed to different concentration of AF (1 nM to 10 µM) show low correlations with TrxR1 silencing (Figure 2B). Furthermore, considering the hierarchical clustering of the CMAP transcriptomic profiles of 45 cancer cell lines exposed to 10 µM AF for 6 h, three major clusters can be distinguished (Figure 2C). Cluster 1 is characterized by 14 cell lines showing very heterogeneous response to the treatment. Cluster 2 consists of 27 cell lines showing predominant transcript downregulation mostly linked to the cell cycle. Cluster 3 consists of four cell lines showing predominant transcript upregulation belonging to proliferative pathways such as PI3K–Akt and Jak–STAT3. Overall, these data confirm both dose and cell type‐dependent effects of AF and can provide a key to the interpretation of the different results reported in the studies summarized in previous paragraphs.
4. CONCLUSION AND PERSPECTIVES
Based on the studies performed on different cancer models, several mechanisms have been proposed to explain how AF alone or in combination with other drugs can exert its anticancer activity. These mechanisms can be partially overlapping and tumor and/or AF dosage dependent. As summarized in Table 1, AF EC50 after 24–48 h ranges between 0.5 and 2 µM in most of the cells. Most of the experiments were performed using AF in this range concentration for 12–24 h h. In some studies, AF was used at higher concentration (6–10 µM) for a shorter amount of time (2–6 h). These different approaches could lead to different pathway activations and complicate overall data interpretation as discussed in the previous paragraph. Since in humans, the plasma steady‐state physiological range of AF is 1–1.5 µM, 3 , 4 it is reasonable to hypothesize that in vivo, targets inhibited by low concentrations of AF have greater relevance.
In conclusion the main direct and indirect targets of AF are shown in Figure 3 and, in particularly, a direct interaction has been shown for the following proteins:
-
TrxR inhibition
The direct inhibition of TrxR was demonstrated for the first time in 1998 and up to date it represents the most proposed mechanism. 6 In most articles, the inhibition of TrxR1 and the consequent increase of ROS has been proposed as the primary way by which AF induces cytotoxicity. In this context, it should be also mentioned that TrxR1 inhibition can influence the redox state of several proteins controlled by the TrxR/Trx system and therefore the ultimate effector of AF action may result difficult to identify. 13 , 27 , 57 Another point to be considered is that not all cancers have the same expression level of this enzyme and thus, AF sensitivity may depend on this feature. In fact, several studies have shown that cell lines where TrxR1 expression levels were higher were also more resistant to AF cytotoxic activity. 31
-
IκB kinase
IκB kinase was proposed as direct AF target by Jeon et al. in LPS‐stimulated macrophages. The same authors also demonstrated that AF binds Cys179 of IκB kinase, thus blocking its activity. 19 , 27 IκB kinase activity is strictly related to activation of NF‐κB. Indeed, IκB kinase phosphorylates IκB protein that binds NF‐κB and sequestrates it in the cytosol in an inactive state. Once phosphorylated, IκB dissociates from NF‐κB, that migrates in the nucleus where it carries out its transcriptional activity. 139 In this review we have described several studies pointing out a reduction of NF‐κB activity upon AF treatment. This can impact on cancer progression, since NF‐κB regulates the expression of a plethora of genes involved in inflammation, oxidative stress and cell survival. In particular, since the correlation between tumor aggressiveness and inflammation has been extensively proved, the anti‐inflammatory properties of AF could be relevant if used in combination with other antitumour drugs. 140 Interestingly, IκB is not the only substrate of IκB kinase, indeed Hu et al. found that it also phosphorylates the tumor suppressor FOXO3 that once phosphorylated, translocates to the cytosol and becomes inactive. 109 Thus, IκB kinase inhibition by AF can evoke various responses whose importance for the cytotoxicity of the drug could depend on the specific cell type and, in vivo, on the tumor microenvironment.
-
PKCiota
PKCiota was indicated as AF target in 2013 by Wang et al. 20 A possible direct interaction has been proposed since two analogues of AF, namely aurothiomalate and aurothioglucose, inhibit the enzyme, by binding a critical cysteine residue. 108 Amplification of the PKCiota gene and elevated PKCiota mRNA level are observed in several cancers, that is, human primary lung squamous cell carcinomas, serous epithelial ovarian cancer, and cervical cancer. 141 Furthermore, high PKCiota expression level is associated with decreased survival in patients with NSCLC, ovarian, bile duct, and prostate tumors, suggesting its possible role as prognostic marker. 142 In consideration of all these aspects the AF inhibitory role on this kinase needs further investigation especially in cancer with high expression of this enzyme.
-
Hexokinase
The study of Hou et al. demonstrated that the purified enzyme hexokinase was inhibited by AF at 4–6 μM and that in vivo cellular glucose uptake, lactate and ATP production were also significantly suppressed by the same dose. 95 Most cancer cells express high levels of hexokinase and this is implicated in the accelerated glycolytic flux. 143 Furthermore, hexokinase (in particular the isoform II) can bind to mitochondria, where it plays additional functions such as cell death inhibition and autophagy regulation. 144 Thus, targeting hexokinase is considered a promising antitumour strategy, but further evidence is needed to understand the possible involvement of AF especially if used at more physiological doses.
-
Proteasome‐associated DUBs
In 2014 Liu et al. demonstrated that the proteasome associated deubiquitinases UCHL5 and USP14 of the 16S regulatory subunit are inhibited by AF both in vitro and in vivo and that this inhibition played a key role in AF cytotoxicity. 76 , 125 As discussed in previous paragraph, Zhang et al. considered DUBs an off‐target effect occurring at high dose of AF. 138 Further studies are needed to better understand dose‐dependent and/or cell type dependent inhibition of these targets.
All the other proteins summarized in Figure 3 resulted affected by AF treatment but, as fa as we know, no evidence was provided regarding a direct interaction. It is reasonable to assume that their modulation is related to the inhibition of the direct targets previously described, but in several cases the signaling cascade has not been clarified. Furthermore, more than one mechanism can contribute to the ultimate effect of the drug.
Overall, the studies described in this review support the use of AF in combination with the drugs normally used in the therapy of specific cancer, as a valuable aid to overcome resistance to chemotherapy and/or enhance the effectiveness of conventional therapies. Further efforts are needed to understand the effective mechanism through which AF performs its action on different types of cancer in relation to specific genome background and protein pattern expression. The studies reported in this review confirm the growing interest in AF drug repurposing and provide the basis for further insights that can support the translation of research in this field into clinical trials.
Figure 3.
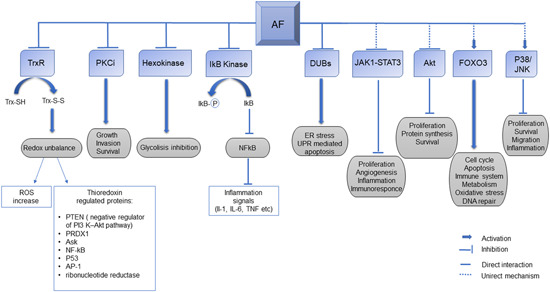
Overview of the main targets and the main signaling pathways affected by auranofin [Color figure can be viewed at wileyonlinelibrary.com]
ACKNOWLEDGEMENTS
Open access funding provided by Universita degli Studi di Firenz within the CRUI‐CARE Agreement.
Gamberi T, Chiappetta G, Fiaschi T, Modesti A, Sorbi F, Magherini F. Upgrade of an old drug: Auranofin in innovative cancer therapies to overcome drug resistance and to increase drug effectiveness. Med Res Rev. 2022;42:1111‐1146. 10.1002/med.21872
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in PubMed.
REFERENCES
- 1. Kean WF, Kean IRL, Buchanan WW. Auranofin. Rheumatology. 1997;36(5):560‐572. 10.1093/rheumatology/36.5.560 [DOI] [PubMed] [Google Scholar]
- 2. Kean WF, Kean IRL. Clinical pharmacology of gold. Inflammopharmacology. 2008;16(3):112‐125. 10.1007/s10787-007-0021-x [DOI] [PubMed] [Google Scholar]
- 3. Chaffman M, Brogden RN, Heel RC, Speight TM, Avery GS. Auranofin: a preliminary review of its pharmacological properties and therapeutic use in rheumatoid arthritis. Drugs. 1984;27(5):378‐424. 10.2165/00003495-198427050-00002 [DOI] [PubMed] [Google Scholar]
- 4. Capparelli EV, Bricker‐Ford R, Rogers MJ, McKerrow JH, Reed SL. Phase I clinical trial results of auranofin, a novel antiparasitic agent. Antimicrob Agents Chemother. 2016;61(1):e01947‐16. 10.1128/AAC.01947-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Messori L, Marcon G. Gold complexes in the treatment of rheumatoid arthritis. Met Ions Biol Syst. 2004;41:279‐304. [PubMed] [Google Scholar]
- 6. Gromer S, Arscott LD, Williams CH, Schirmer RH, Becker K. Human placenta thioredoxin reductase. J Biol Chem. 1998;273(32):20096‐20101. 10.1074/jbc.273.32.20096 [DOI] [PubMed] [Google Scholar]
- 7. Rackham O, Shearwood AM, Thyer R, et al. Substrate and inhibitor specificities differ between human cytosolic and mitochondrial thioredoxin reductases: implications for development of specific inhibitors. Free Radic Biol Med. 2011;50(6):689‐699. 10.1016/j.freeradbiomed.2010.12.015 [DOI] [PubMed] [Google Scholar]
- 8. Ralph SJ, Nozuhur S, ALHulais RA, Rodríguez‐Enríquez S, Moreno‐Sánchez R. Repurposing drugs as pro‐oxidant redox modifiers to eliminate cancer stem cells and improve the treatment of advanced stage cancers. Med Res Rev. 2019;39(6):2397‐2426. 10.1002/med.21589 [DOI] [PubMed] [Google Scholar]
- 9. Scalcon V, Bindoli A, Rigobello MP. Significance of the mitochondrial thioredoxin reductase in cancer cells: an update on role, targets and inhibitors. Free Radic Biol Med. 2018;127(March):62‐79. 10.1016/j.freeradbiomed.2018.03.043 [DOI] [PubMed] [Google Scholar]
- 10. Ghareeb H, Metanis N. The Thioredoxin system: a promising target for cancer drug development. Chem—A Eur J. 2020;26(45):10175‐10184. 10.1002/chem.201905792 [DOI] [PubMed] [Google Scholar]
- 11. Soini Y, Kahlos K, Näpänkangas U, et al. Widespread expression of thioredoxin and thioredoxin reductase in non‐small cell lung carcinoma. Clin Cancer Res. 2001;7(6):1750‐1757. [PubMed] [Google Scholar]
- 12. Bhatia M, McGrath KL, Di Trapani G, et al. The thioredoxin system in breast cancer cell invasion and migration. Redox Biol. 2016;8:68‐78. 10.1016/j.redox.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y, Park J, Han SJ, et al. Redox regulation of tumor suppressor PTEN in cell signaling. Redox Biol. 2020;34(April):101553. 10.1016/j.redox.2020.101553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bian M, Fan R, Zhao S, Liu W. Targeting the thioredoxin system as a strategy for cancer therapy. J Med Chem. 2019;62(16):7309‐7321. 10.1021/acs.jmedchem.8b01595 [DOI] [PubMed] [Google Scholar]
- 15. Zhang J, Li X, Han X, Liu R, Fang J. Targeting the thioredoxin system for cancer therapy. Trends Pharmacol Sci. 2017;38(9):794‐808. 10.1016/j.tips.2017.06.001 [DOI] [PubMed] [Google Scholar]
- 16. Kim NH, Lee MY, Park SJ, Choi JS, Oh MK, Kim IS. Auranofin blocks interleukin‐6 signalling by inhibiting phosphorylation of JAK1 and STAT3. Immunology. 2007;122(4):607‐614. 10.1111/j.1365-2567.2007.02679.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim NH, Park HJ, Oh MK, Kim IS. Antiproliferative effect of gold(I) compound auranofin through inhibition of STAT3 and telomerase activity in MDA‐MB 231 human breast cancer cells. BMB Rep. 2013;46(1):59‐64. 10.5483/BMBRep.2013.46.1.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakaya A, Sagawa M, Muto A, Uchida H, Ikeda Y, Kizaki M. The gold compound auranofin induces apoptosis of human multiple myeloma cells through both down‐regulation of STAT3 and inhibition of NF‐κB activity. Leuk Res. 2011;35(2):243‐249. 10.1016/j.leukres.2010.05.011 [DOI] [PubMed] [Google Scholar]
- 19. Jeon K‐I, Jeong J‐Y, Jue D‐M. Thiol‐reactive metal compounds inhibit NF‐κB activation by blocking IκB kinase. J Immunol. 2000;164(11):5981‐5989. 10.4049/jimmunol.164.11.5981 [DOI] [PubMed] [Google Scholar]
- 20. Wang Y, Hill KS, Fields AP. PKCi maintains a tumor‐initiating cell phenotype that is required for ovarian tumorigenesis. Mol Cancer Res. 2013;11(12):1624‐1635. 10.1158/1541-7786.MCR-13-0371-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Regala RP, Thompson EA, Fields AP. Atypical protein kinase Cι expression and aurothiomalate sensitivity in human lung cancer cells. Cancer Res. 2008;68(14):5888‐5895. 10.1158/0008-5472.CAN-08-0438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park SH, Lee JH, Berek JS, Hu MCT. Auranofin displays anticancer activity against ovarian cancer cells through FOXO3 activation independent of p53. Int J Oncol. 2014;45(4):1691‐1698. 10.3892/ijo.2014.2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raab MS, Podar K, Breitkreutz I, Richardson PG, Anderson KC. Multiple myeloma. Lancet. 2009;374(9686):324‐339. 10.1016/S0140-6736(09)60221-X [DOI] [PubMed] [Google Scholar]
- 24. Singhal S, Mehta J, Desikan R, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341(21):1565‐1571. 10.1056/NEJM199911183412102 [DOI] [PubMed] [Google Scholar]
- 25. Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high‐dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352(24):2487‐2498. 10.1056/NEJMoa043445 [DOI] [PubMed] [Google Scholar]
- 26. Richardson PG, Blood E, Mitsiades CS, et al. A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood. 2006;108(10):3458‐3464. 10.1182/blood-2006-04-015909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jeon K, Byun M, Jue D. G old com pound auranofin inhibits IκB kinase (IKK) by m odifying Cys‐179 of IKK β subunit. Exp Mol Med. 2003;35(2):61‐66. [DOI] [PubMed] [Google Scholar]
- 28. Hu J, Hu WX. Targeting signaling pathways in multiple myeloma: pathogenesis and implication for treatments. Cancer Lett. 2018;414:214‐221. 10.1016/j.canlet.2017.11.020 [DOI] [PubMed] [Google Scholar]
- 29. Puthier D, Bataille R, Amiot M. IL‐6 up‐regulates Mcl‐1 in human myeloma cells through JAK/STAT rather than Ras/MAP kinase pathway. Eur J Immunol. 1999;29(12):3945‐3950. [DOI] [PubMed] [Google Scholar]
- 30. Report S. Mcl‐1 and Bcl‐x L are co‐regulated by IL‐6 in human myeloma cells. Br J Haematol. 1999;107:392‐395. [DOI] [PubMed] [Google Scholar]
- 31. Raninga PV, Trapani GDi, Vuckovic S, Bhatia M, Tonissen KF. Inhibition of thioredoxin 1 leads to apoptosis in drug‐resistant multiple myeloma. Oncotarget. 2015;6(17):15410‐15424. 10.18632/oncotarget.3795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Raninga PV, Di Trapani G, Vuckovic S, Tonissen KF. TrxR1 inhibition overcomes both hypoxia‐induced and acquired bortezomib resistance in multiple myeloma through NF‐кβ inhibition. Cell Cycle. 2016;15(4):559‐572. 10.1080/15384101.2015.1136038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Muz B, De La Puente P, Azab F, Luderer M, Azab AK. Hypoxia promotes stem cell‐like phenotype in multiple myeloma cells. Blood Cancer J. 2014;4(12):e262. 10.1038/bcj.2014.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu J, Handisides DR, Van Valckenborgh E, et al. Targeting the multiple myeloma hypoxic niche with TH‐302, a hypoxia‐activated prodrug. Blood. 2010;116(9):1524‐1527. 10.1182/blood-2010-02-269126 [DOI] [PubMed] [Google Scholar]
- 35. Raninga PV, Di Trapani G, Vuckovic S, Tonissen KF. Cross‐talk between two antioxidants, thioredoxin reductase and heme oxygenase‐1, and therapeutic implications for multiple myeloma. Redox Biol. 2016;8:175‐185. 10.1016/j.redox.2016.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2(10):2557‐2568. 10.1096/fasebj.2.10.3290025 [DOI] [PubMed] [Google Scholar]
- 37. Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235(4792):1043‐1046. 10.1126/science.3029864 [DOI] [PubMed] [Google Scholar]
- 38. Lavrovsky Y, Schwartzman ML, Levere RD, Kappas A, Abraham NG. Identification of binding sites for transcription factors NF‐κB and AP‐2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci USA. 1994;91(13):5987‐5991. 10.1073/pnas.91.13.5987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase‐1 gene. J Biol Chem. 1999;274(37):26071‐26078. 10.1074/jbc.274.37.26071 [DOI] [PubMed] [Google Scholar]
- 40. Tessoulin B, Descamps G, Dousset C, Amiot M, Pellat‐Deceunynck C. Targeting oxidative stress with auranofin or prima‐1Met to circumvent p53 or Bax/Bak deficiency in myeloma cells. Front Oncol. 2019;9(Mar):1‐7. 10.3389/fonc.2019.00128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peng X, Zhang MQ, Conserva F, et al. APR‐246/PRIMA‐1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013;4(10):1‐7. 10.1038/cddis.2013.417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sze JH, Raninga PV, Nakamura K, et al. Anticancer activity of a Gold(I) phosphine thioredoxin reductase inhibitor in multiple myeloma. Redox Biol. 2020;28(June 2019):101310. 10.1016/j.redox.2019.101310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bonolo de Campos C, Meurice N, Petit JL, et al. “Direct to Drug” screening as a precision medicine tool in multiple myeloma. Blood Cancer J. 2020;10(5):54. 10.1038/s41408-020-0320-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Simon TM, Kunishima DH, Vibert GJ, Lorber A. Screening trial with the coordinated gold compound auranofin using mouse lymphocytic leukemia p388. Cancer Res. 1981;41(1):97. [PubMed] [Google Scholar]
- 45. Mirabelli CK, Johnson RK, Sung CM, Faucette L, Muirhead K, Crooke ST. Evaluation of the in vivo antitumor activity and in vitro cytotoxic properties of auranofin, a coordinated gold compound, in murine tumor models. Cancer Res. 1985;45(1):32‐39. [PubMed] [Google Scholar]
- 46. Watts JM, Tallman MS. Acute promyelocytic leukemia: what is the new standard of care? Blood Rev. 2014;28(5):205‐212. 10.1016/j.blre.2014.07.001 [DOI] [PubMed] [Google Scholar]
- 47. Gallagher RE. Retinoic acid resistance in acute promyelocytic leukemia. Leukemia. 2002;16(10):1940‐1958. 10.1038/sj.leu.2402719 [DOI] [PubMed] [Google Scholar]
- 48. Kim IS, Jin JY, Lee IH, Park SJ. Auranofin induces apoptosis and when combined with retinoic acid enhances differentiation of acute promyelocytic leukaemia cells in vitro. Br J Pharmacol. 2004;142(4):749‐755. 10.1038/sj.bjp.0705708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Park SJ, Kim M, Kim NH, et al. Auranofin promotes retinoic acid‐ or dihydroxyvitamin D3‐mediated cell differentiation of promyelocytic leukaemia cells by increasing histone acetylation. Br J Pharmacol. 2008;154(6):1196‐1205. 10.1038/bjp.2008.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Abe E, Miyaura C, Sakagami H, et al. Differentiation of mouse myeloid leukemia cells induced by 1α,25‐dihydroxyvitamin D3. Proc Natl Acad Sci USA. 1981;78(8 I):4990‐4994. 10.1073/pnas.78.8.4990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kingsley EC, Durie BGM, Garewal HS. Acute promyelocytic leukemia. West J Med. 1987;146(3):322‐327. 10.5005/jp/books/12582_38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Park SJ, Kim IS. The role of p38 MAPK activation in auranofin‐induced apoptosis of human promyelocytic leukaemia HL‐60 cells. Br J Pharmacol. 2005;146(4):506‐513. 10.1038/sj.bjp.0706360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fiskus W, Saba N, Shen M, et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014;74(9):2520‐2532. 10.1158/0008-5472.CAN-13-2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gribben JG, O'Brien S. Update on therapy of chronic lymphocytic leukemia. J Clin Oncol. 2011;29(5):544‐550. 10.1200/JCO.2010.32.3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS‐generating anticancer agents. Blood. 2003;101(10):4098‐4104. 10.1182/blood-2002-08-2512 [DOI] [PubMed] [Google Scholar]
- 56. Bichi R, Shinton SA, Martin ES, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci USA. 2002;99(10):6955‐6960. 10.1073/pnas.102181599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen X, Shi X, Zhao C, et al. Anti‐rheumatic agent auranofin induced apoptosis in chronic myeloid leukemia cells resistant to imatinib through both Bcr/Abl‐dependent and ‐independent mechanisms. Oncotarget. 2014;5(19):9118‐9132. 10.18632/oncotarget.2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kantarjian HM, Talpaz M, Giles F, O'Brien S, Cortes J. New insights into the pathophysiology of chronic myeloid leukemia and imatinib resistance. Ann Intern Med. 2006;145(12):913‐923. 10.7326/0003-4819-145-12-200612190-00008 [DOI] [PubMed] [Google Scholar]
- 59. McGahon A, Bissonnette R, Schmitt M, Cotter KM, Green DR, Cotter TG. BCR‐ABL maintains resistance of chronic myelogenous leukemia cells to apoptotic cell death. Blood. 1994;83(5):1179‐1187. 10.1182/blood.v83.5.1179.bloodjournal8351179 [DOI] [PubMed] [Google Scholar]
- 60. Clapper E, Wang S, Raninga PV, Di Trapani G, Tonissen KF. Cross‐talk between Bcr‐abl and the thioredoxin system in chronic myeloid leukaemia: implications for CML treatment. Antioxidants. 2020;9(3). 10.3390/antiox9030207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Odenike OM, Michaelis LC, Stock W. Acute lymphoblastic leukemia. Oncol An Evidence‐Based Approach. 2006:1182‐1209. 10.1007/0-387-31056-8_64 [DOI] [Google Scholar]
- 62. Irwin ME, Rivera‐Del Valle N, Chandra J. Redox control of leukemia: from molecular mechanisms to therapeutic opportunities. Antioxidants Redox Signal. 2013;18(11):1349‐1383. 10.1089/ars.2011.4258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Haß C, Belz K, Schoeneberger H, Fulda S. Sensitization of acute lymphoblastic leukemia cells for LCL161‐induced cell death by targeting redox homeostasis. Biochem Pharmacol. 2016;105:14‐22. 10.1016/j.bcp.2016.01.004 [DOI] [PubMed] [Google Scholar]
- 64. Griffith OW. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J Biol Chem. 1982;257(22):13704‐13712. [PubMed] [Google Scholar]
- 65. Fidyt K, Pastorczak A, Goral A, et al. Targeting the thioredoxin system as a novel strategy against B‐cell acute lymphoblastic leukemia. Mol Oncol. 2019;13(5):1180‐1195. 10.1002/1878-0261.12476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu X, Zhang Y, Lu W, et al. Mitochondrial TXNRD3 confers drug resistance via redox‐mediated mechanism and is a potential therapeutic target in vivo. Redox Biol. 2020;36:101652. 10.1016/j.redox.2020.101652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cox AG, Brown KK, Arner ESJ, Hampton MB. The thioredoxin reductase inhibitor auranofin triggers apoptosis through a Bax/Bak‐dependent process that involves peroxiredoxin 3 oxidation. Biochem Pharmacol. 2008;76(9):1097‐1109. 10.1016/j.bcp.2008.08.021 [DOI] [PubMed] [Google Scholar]
- 68. Celegato M, Borghese C, Casagrande N, et al. Preclinical activity of the repurposed drug auranofin in classical Hodgkin lymphoma. Blood. 2015;126(11):1394‐1397. 10.1182/blood-2015-07-660365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Graczyk‐Jarzynka A, Goral A, Muchowicz A, et al. Inhibition of thioredoxin‐dependent H2O2 removal sensitizes malignant B‐cells to pharmacological ascorbate. Redox Biol. 2019;21(2018):101062. 10.1016/j.redox.2018.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kiebala M, Skalska J, Casulo C, et al. Dual targeting of the thioredoxin and glutathione antioxidant systems in malignant B cells: a novel synergistic therapeutic approach. Exp Hematol. 2015;43(2):89‐99. 10.1016/j.exphem.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang J, Wang J, Lopez E, et al. Repurposing auranofin to treat TP53‐mutated or PTEN‐deleted refractory B‐cell lymphoma. Blood Cancer J. 2019;9(12):95. 10.1038/s41408-019-0259-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Scarbrough PM, Mapuskar KA, Mattson DM, Gius D, Watson WH, Spitz DR. Simultaneous inhibition of glutathione‐ and thioredoxin‐dependent metabolism is necessary to potentiate 17AAG‐induced cancer cell killing via oxidative stress. Free Radic Biol Med. 2012;52(2):436‐443. 10.1016/j.freeradbiomed.2011.10.493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Li L, Fath MA, Scarbrough PM, Watson WH, Spitz DR. Combined inhibition of glycolysis, the pentose cycle, and thioredoxin metabolism selectively increases cytotoxicity and oxidative stress in human breast and prostate cancer. Redox Biol. 2015;4:127‐135. 10.1016/j.redox.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hatem E, Azzi S, El Banna N, et al. Auranofin/vitamin C: a novel drug combination targeting triple‐negative breast cancer. JNCI, J Natl Cancer Inst. 2018;111:1‐12. 10.1093/ije/djy149 [DOI] [Google Scholar]
- 75. Bajor M, Graczyk‐Jarzynka A, Marhelava K, et al. Triple combination of ascorbate, menadione and the inhibition of peroxiredoxin‐1 produces synergistic cytotoxic effects in triple‐negative breast cancer cells. Antioxidants. 2020;9(4):1‐15. 10.3390/antiox9040320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu N, Li X, Huang H, et al. Clinically used antirheumatic agent auranofin is a proteasomal deubiquitinase inhibitor and inhibits tumor growth. Oncotarget. 2014;5(14):5453‐5471. 10.18632/oncotarget.2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Raninga PV, Lee AC, Sinha D, et al. Therapeutic cooperation between auranofin, a thioredoxin reductase inhibitor and anti‐PD‐L1 antibody for treatment of triple‐negative breast cancer. Int J Cancer. 2020;146(1):123‐136. 10.1002/ijc.32410 [DOI] [PubMed] [Google Scholar]
- 78. Mittendorf EA, Philips AV, Meric‐Bernstam F, et al. PD‐L1 expression in triple‐negative breast cancer. Cancer Immunol Res. 2014;2(4):361‐370. 10.1158/2326-6066.CIR-13-0127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rodman SN, Spence JM, Ronnfeldt TJ, et al. Enhancement of radiation response in breast cancer stem cells by inhibition of thioredoxin‐ and glutathione‐dependent metabolism. Radiat Res. 2016;186(4):385‐395. 10.1667/RR14463.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kim SJ, Ju JS, Kang MH, et al. RNA‐binding protein NONO contributes to cancer cell growth and confers drug resistance as a theranostic target in TNBC. Theranostics. 2020;10(18):7974‐7992. 10.7150/thno.45037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67(3):177‐193. 10.3322/caac.21395 [DOI] [PubMed] [Google Scholar]
- 82. Hrabe JE, O'leary BR, Fath MA, et al. Disruption of thioredoxin metabolism enhances the toxicity of transforming growth factor β‐activated kinase 1 (TAK1) inhibition in KRAS‐mutated colon cancer cells. Redox Biol. 2015;5:319‐327. 10.1016/j.redox.2015.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Han Y, Chen P, Zhang Y, et al. Synergy between auranofin and celecoxib against colon cancer in vitro and in vivo through a novel redox‐mediated mechanism. Cancers. 2019;11(7):931. 10.3390/cancers11070931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Liu C, Zhao Y, Wang J, et al. FoxO3 reverses 5‐fluorouracil resistance in human colorectal cancer cells by inhibiting the Nrf2/TR1 signaling pathway. Cancer Lett. 2020;470:29‐42. 10.1016/j.canlet.2019.11.042 [DOI] [PubMed] [Google Scholar]
- 85. Liu Y, Ao X, Ding W, et al. Critical role of FOXO3a in carcinogenesis. Mol Cancer. 2018;17(1):1‐12. 10.1186/s12943-018-0856-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Nag D, Bhanja P, Riha R, et al. Auranofin protects intestine against radiation injury by modulating p53/p21 pathway and radiosensitizes human colon tumor. Clin Cancer Res. 2019;25(15):4791‐4807. 10.1158/1078-0432.CCR-18-2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zou P, Chen M, Ji J, et al. Auranofin induces apoptosis by ROS‐mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancer. Oncotarget. 2015;6(34):36505‐36521. www.impactjournals.com/oncotarget [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Global Burden of Disease Cancer Collaboration , Fitzmaurice C, Dicker D, Pain A, et al. Europe PMC funders group the global burden of cancer 2013. JAMA Oncol. 2015;1(January 2014):505‐527. 10.1001/jamaoncol.2015.0735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fath MA, Ahmad IM, Smith CJ, Spence J, Spitz DR. Enhancement of carboplatin‐mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Clin Cancer Res. 2011;17(19):6206‐6217. 10.1158/1078-0432.CCR-11-0736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Fan C, Zheng W, Fu X, Li X, Wong YS, Chen T. Enhancement of auranofin‐induced lung cancer cell apoptosis by selenocystine, a natural inhibitor of TrxR1 in vitro and in vivo. Cell Death Dis. 2014;5(4):e1191. 10.1038/cddis.2014.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Meng J, Dai B, Fang B, et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non‐ small cell lung cancer in vitro and in vivo. PLOS One. 2010;5(11):1‐10. 10.1371/journal.pone.0014124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dai B, Yoo SY, Bartholomeusz G, et al. KEAP1‐dependent synthetic lethality induced by AKT and TXNRD1 inhibitors in lung cancer. Cancer Res. 2013;73(17):5532‐5543. 10.1158/0008-5472.CAN-13-0712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Li H, Hu J, Wu S, et al. Auranofin‐mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non‐small cell lung cancer cells. Oncotarget. 2015;7(3):3548‐3558. doi:10.18632/oncotarget.6516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Xiaobo C, Majidi M, Feng M, et al. TUSC2(FUS1)‐erlotinib induced vulnerabilities in epidermal growth factor receptor(EGFR) wildtype non‐small cell lung cancer(NSCLC) targeted by the repurposed drug auranofin. Sci Rep. 2016;6(September):1‐10. 10.1038/srep35741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hou GX, Liu PP, Zhang S, et al. Elimination of stem‐like cancer cell side‐population by auranofin through modulation of ROS and glycolysis article. Cell Death Dis. 2018;9(2):89. 10.1038/s41419-017-0159-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mandal PK, Schneider M, Kölle P, et al. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res. 2010;70(22):9505‐9514. 10.1158/0008-5472.CAN-10-1509 [DOI] [PubMed] [Google Scholar]
- 97. Yan X, Zhang X, Wang L, et al. Inhibition of thioredoxin/thioredoxin reductase induces synthetic lethality in lung cancers with compromised glutathione homeostasis. Cancer Res. 2019;79(1):125‐132. 10.1158/0008-5472.CAN-18-1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hu J, Zhang H, Cao M, Wang L, Wu S, Fang B. Auranofin enhances ibrutinib's anticancer activity in EGFR‐mutant lung adenocarcinoma. Mol Cancer Ther. 2018;17(10):2156‐2163. 10.1158/1535-7163.MCT-17-1173 [DOI] [PubMed] [Google Scholar]
- 99. Ito M, Codony‐Servat C, Karachaliou N, Rosell R. Targeting PKCι‐PAK1 in EGFR‐mutation positive non‐small cell lung cancer. Transl Lung Cancer Res. 2019;8(5):667‐673. 10.21037/tlcr.2019.08.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Liu X, Wang W, Yin Y, et al. A high‐throughput drug screen identifies auranofin as a potential sensitizer of cisplatin in small cell lung cancer. Invest New Drugs. 2019;37(6):1166‐1176. 10.1007/s10637-019-00750-2 [DOI] [PubMed] [Google Scholar]
- 101. Sorbi F, Prosperi P, Villanucci A, Berti V, Sisti G, Fambrini M. Laparoscopic splenectomy as quaternary cytoreduction for isolated parenchymal splenic recurrence of epitelial tubo‐ovarian cancer: report of a case and literature review. Gynecol Obstet Invest. 2015;80(4):265‐271. 10.1159/000366476 [DOI] [PubMed] [Google Scholar]
- 102. Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello MP. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin‐resistant human ovarian cancer cells. Free Radic Biol Med. 2007;42(6):872‐881. 10.1016/j.freeradbiomed.2006.12.021 [DOI] [PubMed] [Google Scholar]
- 103. Rigobello MP, Gandin V, Folda A, et al. Treatment of human cancer cells with selenite or tellurite in combination with auranofin enhances cell death due to redox shift. Free Radic Biol Med. 2009;47(6):710‐721. 10.1016/j.freeradbiomed.2009.05.027 [DOI] [PubMed] [Google Scholar]
- 104. Magherini F, Modesti A, Bini L, et al. Exploring the biochemical mechanisms of cytotoxic gold compounds: a proteomic study. J Biol Inorg Chem. 2010;15(4):573‐582. 10.1007/s00775-010-0624-3 [DOI] [PubMed] [Google Scholar]
- 105. Guidi F, Landini I, Puglia M, et al. Proteomic analysis of ovarian cancer cell responses to cytotoxic gold compounds. Metallomics. 2012;4(3):307‐314. 10.1039/c2mt00083k [DOI] [PubMed] [Google Scholar]
- 106. Wang Y, Hill KS, Fields AP. PKCι maintains a tumor‐initiating cell phenotype that is required for ovarian tumorigenesis. Mol Cancer Res. 2013;11(12):1624‐1635. 10.1158/1541-7786.MCR-13-0371-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sabini C, Sorbi F, Cunnea P, Fotopoulou C. Ovarian cancer stem cells: ready for prime time? Arch Gynecol Obstet. 2020;301(4):895‐899. 10.1007/s00404-020-05510-9 [DOI] [PubMed] [Google Scholar]
- 108. Erdogan E, Lamark T, Stallings‐Mann M, et al. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ci. J Biol Chem. 2006;281(38):28450‐28459. 10.1074/jbc.M606054200 [DOI] [PubMed] [Google Scholar]
- 109. Hu MC, Lee DF, Xia W, et al. IκB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117(2):225‐237. 10.1016/S0092-8674(04)00302-2 [DOI] [PubMed] [Google Scholar]
- 110. Bae JS, Lee J, Park Y, et al. Attenuation of MUC4 potentiates the anticancer activity of auranofin via regulation of the Her2/Akt/FOXO3 pathway in ovarian cancer cells. Oncol Rep. 2017;38(4):2417‐2425. 10.3892/or.2017.5853 [DOI] [PubMed] [Google Scholar]
- 111. Singh AP, Chaturvedi P, Batra SK. Emerging roles of MUC4 in cancer: a novel target for diagnosis and therapy. Cancer Res. 2007;67(2):433‐436. 10.1158/0008-5472.CAN-06-3114 [DOI] [PubMed] [Google Scholar]
- 112. Oommen D, Yiannakis D, Jha AN. BRCA1 deficiency increases the sensitivity of ovarian cancer cells to auranofin. Mutat Res—Fundam Mol Mech Mutagen. 2016;784‐785:8‐15. 10.1016/j.mrfmmm.2015.11.002 [DOI] [PubMed] [Google Scholar]
- 113. Hyter S, Hirst J, Pathak H, et al. Developing a genetic signature to predict drug response in ovarian cancer. Oncotarget. 2018;9(19):14828‐14848. 10.18632/oncotarget.23663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Marzo T, Massai L, Pratesi A, et al. Replacement of the thiosugar of auranofin with iodide enhances the anticancer potency in a mouse model of ovarian cancer. ACS Med Chem Lett. 2019;10(4):656‐660. 10.1021/acsmedchemlett.9b00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Huang H, Liao Y, Liu N, et al. Two clinical drugs deubiquitinase inhibitor auranofin and aldehyde dehydrogenase inhibitor disulfiram trigger synergistic anti‐tumor effects in vitro and in vivo. Oncotarget. 2016;7(3):2796‐2808. 10.18632/oncotarget.6425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Fu B, Meng W, Zeng X, Zhao H, Liu W, Zhang T. TXNRD1 is an unfavorable prognostic factor for patients with hepatocellular carcinoma. BioMed Res Int. 2017;2017:4698167. 10.1155/2017/4698167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lee D, Xu IM, Chiu DK, et al. Induction of oxidative stress through inhibition of thioredoxin reductase 1 is an effective therapeutic approach for hepatocellular carcinoma. Hepatology. 2019;69(4):1768‐1786. 10.1002/hep.30467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hwang‐Bo H, Lee WS, Nagappan A, et al. Morin enhances auranofin anticancer activity by up‐regulation of DR4 and DR5 and modulation of Bcl‐2 through reactive oxygen species generation in Hep3B human hepatocellular carcinoma cells. Phyther Res. 2019;33(5):1384‐1393. 10.1002/ptr.6329 [DOI] [PubMed] [Google Scholar]
- 119. Hwangbo H, Kim SY, Lee H, et al. Auranofin enhances sulforaphane‐mediated apoptosis in hepatocellular carcinoma hep3b cells through inactivation of the pi3k/akt signaling pathway. Biomol Ther. 2020;28(5):443‐455. 10.4062/biomolther.2020.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Peng B, Guo C, Guan H, Liu S, Sun MZ. Annexin A5 as a potential marker in tumors. Clin Chim Acta. 2014;427:42‐48. 10.1016/j.cca.2013.09.048 [DOI] [PubMed] [Google Scholar]
- 121. Park N, Chun YJ. Auranofin promotes mitochondrial apoptosis by inducing annexin A5 expression and translocation in human prostate cancer cells. J Toxicol Environ Heal—Part A Curr Issues. 2014;77:1467‐1476. 10.1080/15287394.2014.955834 [DOI] [PubMed] [Google Scholar]
- 122. Baek HS, Park N, Kwon YJ, Ye DJ, Shin S, Chun YJ. Annexin A5 suppresses cyclooxygenase‐2 expression by downregulating the protein kinase C‐ζ‐nuclear factor‐κB signaling pathway in prostate cancer cells. Oncotarget. 2017;8(43):74263‐74275. 10.18632/oncotarget.19392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Shin DW, Kwon YJ, Ye DJ, Baek HS, Lee JE, Chun YJ. Auranofin suppresses plasminogen activator inhibitor‐2 expression through annexin A5 induction in human prostate cancer cells. Biomol Ther. 2017;25(2):177‐185. 10.4062/biomolther.2016.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kubala MH, DeClerck YA. The plasminogen activator inhibitor‐1 paradox in cancer: a mechanistic understanding. Cancer Metastasis Rev. 2019;38(3):483‐492. 10.1007/s10555-019-09806-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Liu N, Guo Z, Xia X, et al. Auranofin lethality to prostate cancer includes inhibition of proteasomal deubiquitinases and disrupted androgen receptor signaling. Eur J Pharmacol. 2019;846(January):1‐11. 10.1016/j.ejphar.2019.01.004 [DOI] [PubMed] [Google Scholar]
- 126. Xu J, Yang X, Deshmukh D, Chen H, Fang S, Qiu Y. The role of crosstalk between AR3 and E2F1 in drug resistance in prostate cancer cells. Cells. 2020;9(5):1094. 10.3390/cells9051094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Onodera T, Momose I, Adachi H, et al. Human pancreatic cancer cells under nutrient deprivation are vulnerable to redox system inhibition. J Biol Chem. 2020;295(49):16678‐16690. 10.1074/jbc.RA120.013893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Sobhakumari A, Love‐Homan L, Fletcher EVM, et al. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLOS One. 2012;7(10):2‐11. 10.1371/journal.pone.0048175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Roh JL, Jang H, Kim EH, Shin D. Targeting of the glutathione, thioredoxin, and Nrf2 antioxidant systems in head and neck cancer. Antioxidants Redox Signal. 2017;27(2):106‐114. 10.1089/ars.2016.6841 [DOI] [PubMed] [Google Scholar]
- 130. Parrales A, McDonald P, Ottomeyer M, et al. Comparative oncology approach to drug repurposing in osteosarcoma. PLOS One. 2018;13(3):1‐16. 10.1371/journal.pone.0194224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Pessetto ZY, Chen B, Alturkmani H, et al. In silico and in vitro drug screening identifies new therapeutic approaches for Ewing sarcoma. Oncotarget. 2017;8(3):4079‐4095. 10.18632/oncotarget.13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Sachweh MC, Stafford WC, Drummond CJ, et al. Redox effects and cytotoxic profiles of MJ25 and auranofin towards malignant melanoma cells. Oncotarget. 2015;6(18):16488‐16506. 10.18632/oncotarget.4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Goenka S, Simon SR. Organogold drug auranofin exhibits anti‐melanogenic activity in B16F10 and MNT‐1 melanoma cells. Arch Dermatol Res. 2020;312(3):213‐221. 10.1007/s00403-019-01974-1 [DOI] [PubMed] [Google Scholar]
- 134. You BR, Shin HR, Han BR, Kim SH, Park WH. Auranofin induces apoptosis and necrosis in HeLa cells via oxidative stress and glutathione depletion. Mol Med Rep. 2015;11(2):1428‐1434. 10.3892/mmr.2014.2830 [DOI] [PubMed] [Google Scholar]
- 135. Rashmi R, Huang X, Floberg JM, et al. Radioresistant cervical cancers are sensitive to inhibition of glycolysis and redox metabolism. Cancer Res. 2018;78(6):1392‐1403. 10.1158/0008-5472.CAN-17-2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Subramanian A, Narayan R, Corsello SM, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171(6):1437‐1452. 10.1016/j.cell.2017.10.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Saei AA, Gullberg H, Sabatier P, et al. Comprehensive chemical proteomics for target deconvolution of the redox active drug auranofin. Redox Biol. 2020;32(March):101491. 10.1016/j.redox.2020.101491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Zhang X, Selvaraju K, Saei AA, et al. Repurposing of auranofin: thioredoxin reductase remains a primary target of the drug. Biochimie. 2019;162:46‐54. 10.1016/j.biochi.2019.03.015 [DOI] [PubMed] [Google Scholar]
- 139. Mitchell S, Vargas J, Hoffmann A. Signaling via the NFκB system. Wiley Interdiscip Rev: Syst Biol Med. 2016;8(3):227‐241. 10.1002/wsbm.1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Diakos CI, Charles KA, McMillan DC, Clarke SJ. Cancer‐related inflammation and treatment effectiveness. Lancet Oncol. 2014;15(11):e493‐e503. 10.1016/S1470-2045(14)70263-3 [DOI] [PubMed] [Google Scholar]
- 141. Murray NR, Kalari KR, Fields AP. Protein kinase Cι expression and oncogenic signaling mechanisms in cancer. J Cell Physiol. 2011;226(4):879‐887. 10.1002/jcp.22463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Parker PJ, Justilien V, Riou P, Linch M, Fields AP. Atypical protein kinase Cι as a human oncogene and therapeutic target. Biochem Pharmacol. 2014;88(1):1‐11. 10.1016/j.bcp.2013.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer's double‐edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25(34):4777‐4786. 10.1038/sj.onc.1209603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Ciscato F, Ferrone L, Masgras I, Laquatra C, Rasola A. Hexokinase 2 in cancer: a prima donna playing multiple characters. Int J Mol Sci. 2021;22(9):4716. 10.3390/ijms22094716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Lee JE, Kwon YJ, Baek HS, et al. Synergistic induction of apoptosis by combination treatment with mesupron and auranofin in human breast cancer cells. Arch Pharm Res. 2017;40(6):746‐759. 10.1007/s12272-017-0923-0 [DOI] [PubMed] [Google Scholar]
- 146. Ye DJ, Kwon YJ, Baek HS, Cho E, Kwon TU, Chun YJ. Combination treatment with auranofin and nutlin‐3a induces synergistic cytotoxicity in breast cancer cells. J Toxicol Environ Health A. 2019;82(10):626‐637. 10.1080/15287394.2019.1635934 [DOI] [PubMed] [Google Scholar]
- 147. Ryu YS, Shin S, An HG, et al. Synergistic induction of apoptosis by the combination of an Axl inhibitor and auranofin in human breast cancer. Cells. Biomol Ther. 2020;28(5):473‐481. 10.4062/biomolther.2020.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Du GS, Qiu Y, Wang WS, et al. Knockdown on aPKC‐ι inhibits epithelial‐mesenchymal transition, migration and invasion of colorectal cancer cells through Rac1‐JNK pathway. Exp Mol Pathol. 2019;107:57‐67. 10.1016/j.yexmp.2018.11.007 [DOI] [PubMed] [Google Scholar]
- 149. Ehrenfeld V, Heusel JR, Fulda S, van Wijk SJL. ATM inhibition enhances Auranofin‐induced oxidative stress and cell death in lung cell lines. PLOS One. 2020;15(12):e0244060. 10.1371/journal.pone.0244060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Cui XY, Park SH, Park WH. Auranofin inhibits the proliferation of lung cancer cells via necrosis and caspase‑dependent apoptosis. Oncol Rep. 2020;44(6):2715‐2724. 10.3892/or.2020.7818 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available in PubMed.