

Abstract
Background and Aims
Hepatic fibrosis secondary to HCV infection can lead to cirrhosis and hepatic decompensation. Sustained virologic response (SVR) is possible with direct‐acting antiviral drug regimens; however, patients with advanced fibrosis have an increased risk for HCC. Heat shock protein 47 (HSP47), a key collagen chaperone, has been implicated in fibrosis development. We evaluated the efficacy and safety of BMS‐986263, a lipid nanoparticle delivering small interfering RNA designed to degrade HSP47 mRNA, for the treatment of advanced fibrosis.
Approach and Results
NCT03420768 was a Phase 2, randomized (1:1:2), placebo‐controlled trial conducted at a hepatology clinic in the United States. Patients with HCV‐SVR (for ≥ 1 year) and advanced fibrosis received once‐weekly i.v. infusions of placebo or BMS‐986263 (45 or 90 mg) for 12 weeks. The primary endpoint was ≥ 1 METAVIR stage improvement at Week 12; key secondary endpoints included Ishak score improvement, pharmacokinetics, fibrosis biomarkers, and safety. All 61 patients completed treatment, and 2/15 (13%, placebo), 3/18 (17%, 45 mg), and 6/28 (21%, 90 mg) had METAVIR improvements of ≥ 1 stage at Week 12. Five patients in the 90‐mg arm had Ishak improvements by ≥ 2 stages. BMS‐986263 plasma concentrations increased in a generally dose‐proportional fashion between BMS‐986263 doses, with no notable accumulation with weekly dosing. All adverse events (AEs) were mild or moderate in intensity; most treatment‐related AEs were infusion‐related reactions in the BMS‐986263 arms. At baseline, collagen levels were low, indicating low levels of fibrogenesis in these patients.
Conclusions
In patients with HCV‐SVR, BMS‐986263 administration was generally well tolerated through Week 36 and resulted in METAVIR and Ishak score improvements. Further evaluation of BMS‐986263 in patients with active fibrogenesis is warranted.
Abbreviations
- ADA
anti‐drug antibody
- AE
adverse event
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- C3M
matrix metalloproteinase–derived collagen type III–specific neo‐epitope peptide
- CPA
collagen proportionate area
- DPD
di‐retinamide‐PEG‐di‐retinamide
- HSC
hepatic stellate cell
- HSP47
heat shock protein 47
- INR
international normalized ratio
- IRR
infusion‐related reaction
- LNP
lipid nanoparticle
- MRE
magnetic resonance elastography
- PIIINP
N‐terminal propeptide of type III procollagen
- PK
pharmacokinetics
- PRO‐C3
N‐terminal propeptide of type III collagen
- qFIB
quantitative assessment of liver fibrosis
- QW
once weekly
- siRNA
small interfering RNA
- SVR
sustained viral response
- TIMP‐1
tissue inhibitor of matrix metalloproteinases 1
- ULN
upper limit of normal
INTRODUCTION
Hepatic fibrosis can result from a variety of etiologies including chronic HCV infection and NAFLD/NASH.[ 1 ] Patients with hepatic fibrosis have a higher risk of adverse clinical outcomes over time, such as cirrhosis, hepatic decompensation, and HCC.[ 2 ] Additionally, mortality risk increases as fibrosis progresses such that patients with advanced fibrosis and cirrhosis have the highest all‐cause and liver‐related mortality risk.[ 2 , 3 , 4 ] In recent years, direct‐acting antiviral regimens have become standard treatment for chronic HCV infection, and they enable sustained virologic response (SVR) rates > 95% with 8‐ to 12‐week regimens, even in patients with advanced fibrosis.[ 5 , 6 ] However, despite HCV‐SVR, adults with cirrhosis have an increased HCC risk, and regular surveillance testing is advised.[ 7 ] With no currently approved therapies for advanced hepatic fibrosis, there is an urgent need for treatments that can stabilize or reverse fibrotic disease progression.
Two key factors in fibrosis development, collagen dysregulation and imbalanced extracellular matrix (ECM) remodeling, are therapeutic targets that have been evaluated in preclinical and clinical studies.[ 8 , 9 , 10 , 11 , 12 , 13 , 14 ] Collagen is a vital component of the ECM, and heat shock protein 47 (HSP47) is a molecular chaperone that binds triple‐helical procollagen in the endoplasmic reticulum to prevent inappropriate collagen unfolding or aggregation.[ 9 ] In preclinical models, HSP47 mRNA and protein expression are markedly increased in fibrotic tissue, suggesting a role for HSP47 in collagen accumulation and fibrosis progression.[ 15 , 16 , 17 ] It is thought that HSP47 mRNA silencing through small interfering ribonucleic acids (siRNAs) may reduce fibrosis by hepatic stellate cells (HSCs),[ 9 , 10 , 12 , 18 ] which produce excess collagen following liver injury.[ 14 , 19 ]
BMS‐986263 is a retinoid‐conjugated lipid nanoparticle (LNP) containing HSP47 siRNA. The LNP contains a critical lipid targeting agent, di‐retinamide‐PEG‐di‐retinamide (DPD), which facilitates direction of the LNP to HSCs. There, retinoid‐containing moieties on the LNP bind retinol‐binding protein (RBP) expressed on HSCs; in vitro studies that show strong binding of the DPD‐containing LNP to human RBP support the mechanism of DPD‐mediated LNP uptake by liver HSCs.[ 12 ] Upon intracellular release, HSP47 siRNA recruits the RNA‐induced silencing complex, which degrades HSP47 mRNA and prevents HSP47 protein translation.[ 13 , 20 ] Thus, BMS‐986263‐mediated HSP47 mRNA inhibition may reduce or reverse liver fibrosis by disrupting collagen formation and promoting HSC apoptosis, respectively.[ 21 ]
To date, BMS‐986263 has been studied in healthy volunteers (Phase 1) and in patients with advanced hepatic fibrosis secondary to NASH or HCV (Phases 1b and 1b/2).[ 13 , 22 , 23 ] BMS‐986263 administration once weekly (QW) or twice weekly for 3–5 weeks was generally well tolerated with linear pharmacokinetics (PK) and resulted in biopsy‐assessed histopathological improvements in patients with advanced fibrosis. Here, we present the results from a Phase 2 study to evaluate the safety, tolerability, PK, and pharmacodynamics of BMS‐986263 administered QW for 12 weeks in patients with advanced hepatic fibrosis secondary to HCV.
PATIENTS AND METHODS
Study design
NCT03420768 was a Phase 2, randomized, double‐blind, placebo‐controlled, parallel‐group study in adults with advanced hepatic fibrosis due to HCV who had achieved SVR (HCV‐SVR; Figure 1). The study was conducted at The Texas Liver Institute (San Antonio, TX). Eligible patients had documented SVR (i.e., no detectable serum HCV RNA at least 12 weeks after completing antiviral therapy) for at least 1 year and advanced hepatic fibrosis (METAVIR F3 or F4) according to local pathology interpretation of liver tissue obtained within 8 weeks of screening. Exclusion criteria included Child‐Pugh score > 6, Model for End‐Stage Liver Disease score > 12, or evidence of HCC based on serum concentration of alpha‐fetoprotein or imaging at screening. Additionally, patients were ineligible if they had a history of hepatic decompensation or transplantation, albumin < 3.5 g/dL, international normalized ratio (INR) greater than the upper limit of normal (ULN), or total bilirubin > ULN. The minimum allowable platelet count was initially 140,000/µL; however, to account for the frequency of thrombocytopenia in patients with cirrhosis, the protocol was amended toward the end of enrollment to lower the platelet count threshold to < 100,000/µL.
FIGURE 1.
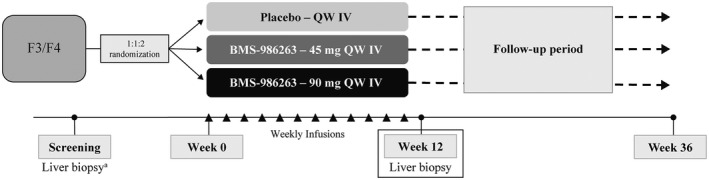
NCT03420768 study design. Eligible patients were randomized 1:1:2 to receive placebo or BMS‐986263 (45 mg or 90 mg) i.v. QW for 12 weeks. aLiver biopsies were performed within 8 weeks prior to or during the screening period
Randomization using interactive response technology followed a computer‐generated block scheme that stratified patients by METAVIR fibrosis stage (F3 vs. F4). Assignments were made 1:1:2 to receive placebo (0.3–0.6 mL intralipid in 100 mL 5% dextrose solution) or BMS‐986263 (45 or 90 mg diluted in 0.9% saline to ~100 mL) infused i.v. QW for 12 weeks (Weeks 0–11; Figure 1). Pretreatment with antihistamines or hydrocortisone was allowed at the investigator’s discretion to reduce potential for infusion‐related reactions (IRRs). Protocol amendments were made to evaluate and determine the optimal BMS‐986263 infusion rate. Liver biopsies were performed at Week 12, and safety was monitored through Week 36.
Study endpoints
The primary endpoint was the proportion of patients with ≥ 1 METAVIR stage improvement in hepatic fibrosis at Week 12 as determined by histopathological interpretation of liver tissue obtained from biopsy. Key secondary endpoints were the proportion of patients with ≥ 2 Ishak stage improvement, BMS‐986263 plasma concentrations, and safety at Week 36. Exploratory endpoints included changes from baseline to Week 12 in the following parameters: HSP47 mRNA and HSP47 protein in liver, fibrosis assessed by dual‐photon microscopy (qFibrosis [qFIB]), serum biomarkers of collagen formation (N‐terminal propeptide of type III collagen [PRO‐C3]), collagen degradation (matrix metalloproteinase–derived collagen type III–specific neo‐epitope peptide [C3M] and tissue inhibitor of matrix metalloproteinase [TIMP‐1]), and collagen turnover (N‐terminal propeptide of type III procollagen [PIIINP]).
Study assessments
Baseline liver histology was analyzed by a local pathologist to identify eligible study patients (i.e., those with METAVIR F3 and F4 fibrosis). Per the study protocol, the baseline biopsy specimens were subsequently reassessed by a blinded central pathologist (Z.G.) to enable comparison with Week 12 biopsy specimens. Morphometric quantification of hepatic collagen in each biopsy specimen was performed on 4‐µm‐thick sections stained with picrosirius red; digital images of the sections were acquired using an Aperio ScanScope XT (Leica Biosystems, Vista, CA) at ×20 magnification. Collagen proportionate area (CPA) was defined as the percentage of the tissue section that was positive for red staining as determined by the Aperio Positive Pixel Count Algorithm. qFIB, a stain‐free, fully automated, quantitative tissue imaging tool, was also used to assess fibrosis changes between baseline and Week 12 using 4‐µm‐thick tissue sections and second‐harmonic generation/two‐photon excitation fluorescence microscopy.[ 24 ] Baseline blinded serum samples were analyzed for circulating concentrations of collagen and fibrosis biomarkers; data were stratified by METAVIR fibrosis stage. PRO‐C3 and C3M were measured with competitive ELISAs developed and performed by Nordic Bioscience (Herlev, Denmark). PIIINP was measured using an ELISA developed by CisBio (Codolet, France) and performed by Nordic Bioscience, and TIMP‐1 was measured using Luminex technology (Myriad RBM, Austin, TX).
Magnetic resonance elastography (MRE) and FibroScan were used to assess liver stiffness at baseline as a surrogate marker of liver fibrosis. All MRE images were analyzed by the central imaging facility. HSP47 protein was measured in 4‐µm liver tissue sections by immunohistochemistry (Mosaic Laboratories, Lake Forest, CA), and HSP47 mRNA was measured in 5‐ to 10‐mm2 tissue sections using the HTG EdgeSeq system (HTG Molecular Diagnostics, Inc., Tucson, AZ) in liver tissue collected at baseline and Week 12. Blood PK samples were collected preinfusion, midinfusion, and postinfusion at Weeks 0, 1, 4, 6, and 11; and additional postdose samples were collected 1 and 3 weeks following the Week 11 dose. siRNA PK analysis was evaluated with a validated hybridization ELISA method that used complementary capture and detection oligonucleotides.
All treatment‐emergent adverse events (TEAEs) were recorded through Week 36 and classified using MedDRA (v21.1). Bone mineral density (BMD) was measured by dual‐energy X‐ray absorptiometry (DXA) at baseline and Week 36 by the central imaging facility. Antidrug antibody (ADA) responses to BMS‐986263 were measured in plasma using a three‐tier screening, confirmatory, and titer detection method where BMS‐986263 served as a capture and detection reagent for ADAs.[ 25 ] For these analyses, preinfusion samples were collected from each patient at Weeks 1, 2, 4, and 12.
Statistical analyses
The planned study sample size was 60 patients randomized into three arms; this population estimate was not based on formal statistical justification. Efficacy and safety analyses included all randomized patients who received at least one dose of study treatment. Biomarker and PK analyses included all patients who had received at least one dose of study treatment with at least one posttreatment biomarker measurement and available concentration–time data, respectively. For the primary efficacy endpoint, a 95% exact CI and ORs were used to estimate differences in the BMS‐986263 versus placebo arms; those with a missing Week 12 histology score were considered to have no fibrosis improvement. The secondary efficacy endpoint was analyzed similarly to the primary endpoint except that patients without a Week 12 histology score were excluded. Safety analyses included all reported TEAEs through Week 36, which were listed and summarized by system organ class, preferred term, and arm. All exploratory endpoints were summarized using descriptive statistics by arm and study day, with change from baseline or percentage change from baseline reported as appropriate. Secondary and exploratory endpoints were not adjusted for multiplicity. Statistical Analysis Software (SAS), version 9.4, was used for statistical analyses and tabulations.
Oversight
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the institutional review board or independent ethics committee. All patients gave written informed consent prior to study participation, and an external data monitoring committee monitored safety throughout the study.
RESULTS
Study population
The study was conducted between February 14, 2018, and May 28, 2019. All 61 patients who were enrolled and randomized 1:1:2 to receive placebo (n = 15), 45 mg BMS‐986263 (n = 18), or 90 mg BMS‐986263 (n = 28) completed the treatment and follow‐up periods. Most patients were White (92%) or Hispanic (53%), and the mean age was 60 years (Table 1). For all patients, the median time since SVR was 38 months; the 90‐mg BMS‐986263 arm had a median time of 46 months since SVR compared with 31 months for placebo and 32 months for the 45‐mg BMS‐986263 arm.
TABLE 1.
Baseline patient demographics and characteristics
| Parameters | Placebo QW (n = 15) | 45 mg BMS‐986263 QW (n = 18) | 90 mg BMS‐986263 QW (n = 28) | Total (N = 61) |
|---|---|---|---|---|
| Demographics | ||||
| Age, mean (SD), years | 61.8 (6.5) | 60.2 (7.5) | 59.8 (8.2) | 60.4 (7.5) |
| Female, n (%) | 8 (53) | 7 (39) | 12 (43) | 27 (44) |
| White, n (%) | 13 (87) | 17 (94) | 26 (93) | 56 (92) |
| Hispanic, n (%) | 6 (40) | 13 (72) | 13 (46) | 32 (53) |
| Characteristics | ||||
| Time since SVR, median (range), months | 31 (15–70) | 32 (15–58) | 46 (14–66) a | 37 (14–70) a |
| BMI, mean (SD), kg/m2 | 27.4 (4.2) | 30.0 (3.4) | 29.8 (3.7) | 29.3 (3.8) |
| T2DM, n (%) | 2 (13) | 4 (22) | 8 (29) | 14 (23) |
Abbreviations: BMI, body mass index; T2DM, type 2 diabetes mellitus.
Two patients had a missing SVR date.
All baseline fibrosis scores of enrolled patients were METAVIR F3 or F4 as assessed by the local pathologist; however, the central pathologist staged 32/61 (52%) of the baseline tissue samples as F1–F2 (Table 2). Most discrepancies between the local and central readings were related to patients staged as F3 at screening. Results from additional fibrosis measures for patients grouped by central pathologist–assigned baseline fibrosis scores are shown in Figure S1. Almost all baseline biopsy specimens were ≥ 15 mm in length (placebo, 14/15, 93%; 45 mg BMS‐986263, 16/18, 89%; 90 mg BMS‐986263, 24/28, 86%) with ≥ 11 portal areas (58/61, 95%; Figure S2). Similarly, most Week 12 biopsy specimens were ≥ 15 mm in length (placebo, 15/15, 100%; 45 mg BMS‐986263, 18/18, 100%; 90 mg BMS‐986263, 21/28; 75%) with ≥ 11 portal areas (59/61, 97%). One patient (90‐mg arm) had an inadequate Week 12 tissue sample. For the overall study population, baseline liver histology analysis generally revealed low levels of collagen and thin septa. Across METAVIR stages, median PRO‐C3 and PIIINP concentrations were not appreciably different at baseline (Figure S3); however, median C3M and TIMP‐1 values increased as fibrosis stage increased (Figure S4). Fibrosis parameters and liver‐related laboratory values were similar across arms. The overall study population had preserved hepatic synthetic capacity, as evidenced by normal albumin and INR values.
TABLE 2.
Baseline fibrosis stage by central read, liver biopsy tissue characteristics, and laboratory values
| Parameters | Placebo QW (n = 15) | 45 mg BMS‐986263 QW (n = 18) | 90 mg BMS‐986263 QW (n = 28) | Total (N = 61) |
|---|---|---|---|---|
| METAVIR fibrosis stage, n (%) | ||||
| F1 | 1 (7) | 4 (22) | 3 (11) | 8 (13) |
| F2 | 5 (33) | 8 (44) | 11 (39) | 24 (39) |
| F3 | 7 (47) | 4 (22) | 8 (29) | 19 (31) |
| F4 | 2 (13) | 2 (11) | 6 (21) | 10 (16) |
| Liver biopsy tissue characteristics | ||||
| Tissue length, mean (SD), mm | 20.1 (3.6) | 19.6 (4.0) | 18.3 (3.7) | 19.1 (3.8) |
| ≥ 20 mm, n (%) | 11 (73) | 11 (61) | 10 (36) | 32 (52) |
| ≥ 15 mm, n (%) | 14 (93) | 16 (89) | 24 (86) | 54 (89) |
| ≥ 11 portal areas, n (%) | 15 (100) | 17 (94) | 26 (93) | 58 (95) |
| Fibrosis parameters, mean (SD) | ||||
| CPA (%) | 3.49 (2.05) | 3.36 (2.47) | 3.04 (1.97) | 3.25 (2.12) |
| α‐SMA (%) | 4.71 (4.93) | 4.01 (3.92) | 4.44 (4.12) | N/A |
| Fibrosis‐4 index | 1.70 (0.61) | 1.81 (0.85) | 1.46 (0.42) | 1.62 (0.63) |
| PRO‐C3, ng/mL | 11.14 (3.89) | 12.57 (3.39) | 12.20 (3.79) | 12.05 (3.68) |
| PIIINP, ng/mL | 7.27 (2.24) | 7.27 (1.80) | 7.08 (1.78) | 7.19 (1.88) |
| TIMP‐1, ng/mL | 105.3 (23.1) | 122.7 (54.1) | 122.1 (41.2) | 118.2 (42.1) |
| C3M, ng/mL | 11.7 (2.4) | 11.2 (2.1) | 11.8 (2.4) | 11.57 (2.29) |
| MRE, kPa | 3.89 (1.88) | 3.49 (1.03) | 3.83 (0.97) | 3.74 (1.26) |
| FibroScan, kPa | 11.95 (6.94) | 8.61 (3.73) | 11.90 (6.03) | 10.94 (5.82) |
| Liver‐related laboratory values, mean (SD) | ||||
| ALT, U/L | 18.6 (6.1) | 32.1 (18.4) | 24.9 (9.1) | 25.5 (12.9) |
| AST, U/L | 20.9 (6.9) | 28.4 (10.0) | 23.2 (6.6) | 24.2 (8.2) |
| Alkaline phosphatase, IU/L | 76.3 (23.5) | 86.0 (25.4) | 71.1 (23.4) | 76.8 (24.5) |
| Platelets, ×109/L | 186.0 (26.6) | 191.2 (49.2) | 201.1 (42.9) | 194.5 (41.5) |
| Albumin, mg/dL | 4590 (280) | 4610 (397) | 4610 (336) | 4610 (338) |
| Total bilirubin, mg/dL | 0.48 (0.16) | 0.55 (0.19) | 0.56 (0.23) | 0.54 (0.20) |
| INR | 1.09 (0.09) | 1.10 (0.10) | 1.07 (0.08) | 1.08 (0.09) |
| Sodium, mmol/L | 141.1 (2.2) | 139.6 (3.4) | 139.7 (2.6) | 140.0 (2.8) |
| Serum creatinine, µmol/L | 70.0 (15.2) | 77.2 (18.6) | 78.1 (16.5) | 75.8 (16.9) |
Abbreviations: α‐SMA, alpha‐smooth muscle actin; N/A, not available.
Change in METAVIR stage at Week 12
Reduction in ≥ 1 METAVIR stage from baseline to Week 12 was observed for 2/15 (13%), 3/18 (17%), and 6/28 (21%) patients in the placebo, 45‐mg BMS‐986263, and 90‐mg BMS‐986263 arms, respectively (Figure 2). The ORs for response compared with placebo were 1.30 (95% CI, 0.13–17.70; 45‐mg arm) and 1.77 (95% CI, 0.26–20.23; 90‐mg arm). When analyzed by baseline METAVIR stage, 4/8 (50%) F4 patients treated with either dose of BMS‐986263 had fibrosis improvement. Further analysis of Week 12 METAVIR improvement according to central pathologist−assigned baseline METAVIR stage is shown in Tables S1 and S2. Of the patients who achieved ≥ 1 METAVIR stage improvement at Week 12, 9/11 (82%) also showed an improvement in qFIB score, including 6/6 (100%) patients in the 90‐mg BMS‐986263 arm (Figure S5). A summary of the concordance of METAVIR and qFIB responses at Week 12 is shown in Table S3a.
FIGURE 2.
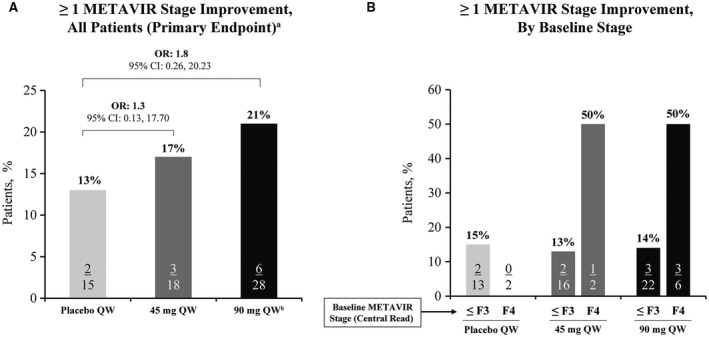
METAVIR stage change from baseline to Week 12. Patients with ≥ 1 METAVIR stage improvement at Week 12 stratified by (A) arm or (B) baseline fibrosis stage. aOR and difference in response rates are shown for each arm compared with placebo. bOne F2 patient had an inadequate Week 12 biopsy specimen and was considered to have no fibrosis improvement
Change in Ishak Score at Week 12
The secondary endpoint, a ≥ 2 Ishak score improvement from baseline to Week 12, was observed in 5 patients from the 90‐mg BMS‐986263 arm (Figure 3). Most (3/5) had cirrhosis at baseline (i.e., an Ishak score of 5–6). Of patients who achieved ≥ 2 stage improvement in Ishak score, all 5 also had an improvement in fibrosis as determined by qFIB (Figure S6). A summary of the concordance of Ishak and qFIB responses at Week 12 is shown in Table S3b.
FIGURE 3.

Ishak score change from baseline to Week 12. Patients with ≥ 2 Ishak score improvement at Week 12 stratified by study arm and baseline fibrosis stage. aOne F2 patient had an inadequate Week 12 biopsy specimen and was excluded from the analysis
Target Engagement
From baseline to Week 12, liver HSP47 mRNA levels were reduced in 2/15 (13%), 8/18 (44%), and 20/28 (71%) patients in the placebo, 45‐mg BMS‐986263, and 90‐mg BMS‐986263 arms, respectively (Figure 4A). The median HSP47 mRNA percentage change (range) was 13.7% (−19.3% to 51.8%) for the placebo arm, −1.2% (−37.3% to 57.6%) for the 45‐mg BMS‐986263 arm, and −5.9% (−29.9% to 22.6%) for the 90‐mg BMS‐986263 arm (Figure 4B). For HSP47 protein, the median percentage change from baseline to Week 12 (range) was 23.5% (−33.9% to 225.4%) for the placebo arm, −11.3% (−56.5% to 122.3%) for the 45‐mg BMS‐986263 arm, and −10.1% (−55.8% to 202.9%) for the 90‐mg BMS‐986263 arm (Figure 4C).
FIGURE 4.
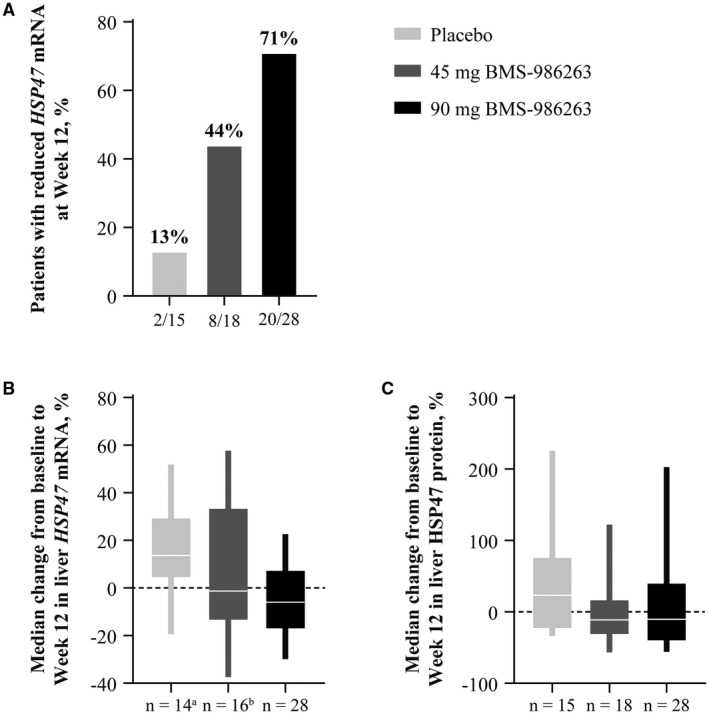
Liver HSP47 mRNA and protein change from baseline to Week 12. (A) Percentage of patients per arm with reduced HSP47 mRNA at Week 12. (B,C) Median percentage change in HSP47 mRNA (B) and HSP47 protein (C). aMissing data, n = 1. bMissing data, n = 2
PK
In patients who received either BMS‐986263 dose, plasma concentrations increased during the infusion course and were higher at the end of the infusion compared with midinfusion. BMS‐986263 plasma concentrations decreased rapidly after the end of infusion. In general, dose‐proportional plasma concentration increases were observed between the 45‐mg and 90‐mg BMS‐986263 arms. No apparent BMS‐986263 plasma accumulation occurred with weekly dosing based on similar peak and trough concentrations observed across weekly infusions (Figure 5).
FIGURE 5.
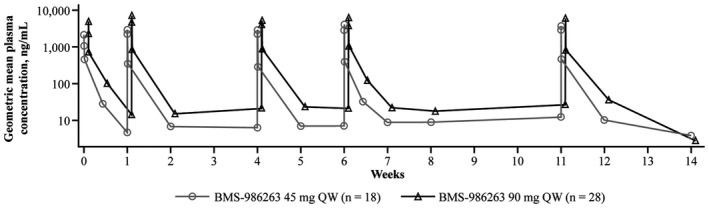
Geometric mean BMS‐986263 plasma concentration. Figure shows the mean concentration time profile following administration of all infusions in the study. PK samples were collected at preinfusion, midinfusion, and postinfusion at Weeks 0, 1, 4, 6, and 11. Trough PK samples were collected preinfusion at Weeks 2, 5, 7, and 8. Samples during a dosing interval were collected at 72 h postinfusion following the doses at Weeks 0 and 6. Additional postinfusion PK samples were collected at 1 and 3 weeks following the Week 11 dose
Safety
By Week 36, 37/61 (61%) patients experienced at least one adverse event (AE), all of which were mild or moderate in intensity (Table 3). Twenty‐five patients (41%) reported AEs that were considered possibly BMS‐986263‐related; patients in either BMS‐986263 arm had a higher frequency of BMS‐986263‐related AEs compared with those who received placebo. The most common BMS‐986263‐related AE was IRR, reported by 21/61 (34%) patients; all events were mild to moderate in intensity. The most common symptom associated with IRRs was back pain and/or spasms, reported by 17/21 patients. There were no reported events of anaphylaxis. Sixteen patients (45‐mg BMS‐986263 arm [n = 3], 90‐mg BMS‐986263 arm [n = 13]) had at least one temporary infusion interruption due to an IRR; despite this, all patients completed all scheduled infusions. There were no reported BMS‐986263‐related serious AEs, discontinuations due to AEs, or deaths. Additionally, there were no reported events of hepatic decompensation or laboratory changes indicative of potential DILI. At Week 36, no clinically meaningful changes were observed in BMD by DXA (data not shown).
TABLE 3.
Safety summary
| Patients, n (%) | Placebo QW (n = 15) | 45 mg QW BMS‐986263 (n = 18) | 90 mg QW BMS‐986263 (n = 28) | Total (N = 61) |
|---|---|---|---|---|
| ≥ 1 AE | 8 (53) | 10 (56) | 19 (68) | 37 (61) |
| Any treatment‐related AE | 3 (20) | 7 (39) | 15 (54) | 25 (41) |
| Infusion‐related reaction | 0 | 6 (33) | 15 (54) | 21 (34) |
| Headache | 1 (7) | 2 (11) | 0 | 3 (5) |
| Anxiety | 1 (7) | 0 | 1 (4) | 2 (3) |
| Back pain | 0 | 0 | 2 (7) | 2 (3) |
| Any severe AE | 0 | 0 | 1 (4) a | 1 (2) |
| AEs of special interest | ||||
| Infusion‐related reaction | ||||
| Mild | 0 | 2 (11) | 2 (7) | 4 (7) |
| Moderate | 0 | 4 (22) | 13 (46) | 17 (28) |
| Severe | 0 | 0 | 0 | 0 |
| Liver safety | ||||
| ALT and/or AST elevation >3× ULN | 0 | 2 (11) | 1 (4) | 3 (5) |
| HCC | 0 | 2 (11) | 1 (4) | 3 (5) b |
Ankle fracture at Week 10, considered unrelated to study drug.
In the posttreatment follow‐up period, HCC was detected in 3 patients through surveillance.
During the study, 3 patients (45‐mg BMS‐986263 arm [n = 2], 90‐mg BMS‐986263 arm [n = 1]) had elevated alanine aminotransferase (ALT) or aspartate aminotransferase (AST) concentrations > 3× ULN. One patient had elevated ALT (67 U/L) at baseline. For the other 2 patients, transaminitis occurred in the setting of elevated creatine phosphokinase (6,832 U/L) or in the setting of an AE of bronchitis for which azithromycin and hydrocortisone were administered. For all 3 patients, the elevations were transient, not associated with symptoms or other liver‐related laboratory abnormalities, and considered unrelated to BMS‐986263. During routine surveillance in the posttreatment follow‐up period, HCC was detected in 3 patients; during the treatment period, 2 patients received 45 mg BMS‐986263 and 1 received 90 mg BMS‐986263 (Table S4).
BMS‐986263 immunogenicity analyses are summarized in Table S5. At baseline, preexisting antibodies to BMS‐986263 were identified in 1 patient each in the placebo and 90‐mg BMS‐986263 arms. During treatment, 2 patients (11%) in the 45‐mg BMS‐986263 arm developed ADAs; the first patient tested positive at Weeks 2 and 4, and the second patient tested positive at Week 1. ADAs were not detected at subsequent time points in either patient in the 45‐mg arm. In the 90‐mg BMS‐986263 arm, ADAs were detected in the same patient who was positive at baseline at Weeks 2 and 4 at the same titer.
DISCUSSION
In patients with hepatic fibrosis, the risk of hepatic decompensation, HCC, and mortality increases with advancing fibrosis stage.[ 2 , 3 , 4 ] Though studies suggest that fibrosis reversal may improve clinical outcomes for patients with HCV,[ 26 ] the HCC rate in patients with cirrhosis pre‐HCV‐SVR and/or post‐HCV‐SVR can be as high as 2.3%–12.5%.[ 27 , 28 ] It is critical to continue study of this patient population to determine who is at highest risk of disease progression and might experience the greatest benefit from antifibrotic therapy. In this Phase 2, placebo‐controlled, proof‐of‐concept study of patients with HCV‐SVR, 12‐week treatment with BMS‐986263 improved liver fibrosis in some patients, including those with cirrhosis. The limitations of the trial included a small sample size from one study site and challenges associated with the enrolled patient population; however, a number of key findings were made that will inform the design of future studies with BMS‐986263.
The choice to enroll patients with HCV‐SVR in this study was made in an attempt to study the antifibrotic effects of BMS‐986263 in a well‐defined population of patients with advanced liver fibrosis more than 1 year post‐SVR. The overall population of patients with HCV‐SVR is sizeable and does not have ongoing drivers of inflammation. Within this study population, patients had a median time of 3 years of HCV‐SVR prior to the study. Though the 90‐mg BMS‐986263 arm had a longer median time since SVR compared with the other arms, it is unlikely that this difference impacted the study’s results, which are based mainly on comparisons between baseline and the end of treatment at Week 12 among the three treatment groups.
Eligible patients were those with advanced fibrosis, staged as METAVIR F3 or F4 by the local pathologist at screening. In this single‐center study, trial eligibility was determined based on fibrosis staging of pretreatment biopsy tissue by a local pathologist, while efficacy was determined by a central pathologist who performed fibrosis staging of pretreatment and posttreatment biopsy tissue. Ideally, the highly experienced expert central pathologist would have performed all fibrosis staging in this trial; however, local pathology reading of baseline fibrosis was done in order to screen patients in the timeliest fashion. Surprisingly, though the biopsy tissue quality was robust in terms of adequate length and portal tract inclusion, 50% of patients who were staged as F3 or F4 by the local pathologist were subsequently staged as F1 or F2 by the central pathologist. The staging performed by the central pathologist was supported by several noninvasive tests, such as MRE and Fibrosis‐4 performed at baseline, as well as by the remarkably thin bands of collagen, which were the predominant findings in these post‐SVR biopsy specimens. Together, these results suggest that the central pathologist’s staging was more accurate and, thus, that the study population inadvertently included some patients with relatively mild fibrosis. Staging of fibrosis regression in patients post‐SVR is not well characterized; liver biopsies are rarely performed on patients after they have achieved HCV‐SVR, and thus, there are no comprehensive data sets that describe changes in fibrosis > 3 years post‐SVR. The results from this trial imply that fibrosis staging post‐SVR is complex and may lead to greater interreader variability in fibrosis staging than might normally be expected in patients with fibrotic liver disease, particularly in cases where a reader does not routinely perform fibrosis staging for patients with HCV‐SVR. Thus, a comprehensive review to standardize post‐SVR fibrosis assessment would be useful to the field.
Exploratory assessments of collagen content and fibrosis biomarkers revealed that the patients with HCV‐SVR in this study had lower CPA than what has been previously reported for patients with chronic HCV infection[ 29 ] or NASH[ 30 ] but similar to that of patients with post‐HCV‐SVR fibrosis regression.[ 31 ] Independent of METAVIR stage, median PRO‐C3 concentrations for all arms were within the range of those observed in healthy adults,[ 32 ] who would be expected to have low levels of fibrogenesis. This suggests that, despite the diagnosis of advanced fibrosis, these patients with HCV‐SVR had low levels of fibrogenesis and that some fibrosis improvements occurred after SVR, prior to the baseline liver biopsy for this study. At baseline, patients with higher METAVIR stages had higher serum concentrations of C3M and TIMP‐1 compared to patients with earlier stages of fibrosis. Although the biomarker ranges overlapped across METAVIR stages, these data suggest higher levels of fibrolysis in patients who have higher versus lower fibrosis scores, as assessed by the central pathologist in this study. By silencing HSP47 mRNA, BMS‐986263 decreases fibrogenesis by reducing mature collagen formation; therefore, it is reasonable to hypothesize that BMS‐986263 will have greater efficacy in patients with high levels of active fibrogenesis compared to those with low levels, which explains the modest efficacy observed in patients with HCV‐SVR in this study. This is supported by an earlier Phase 1b study (NCT02227459) of BMS‐986263 in patients with advanced hepatic fibrosis in which patients with active fibrogenesis in the context of NASH experienced greater clinical benefit than did patients with HCV‐SVR.[ 13 ]
The trial was designed to include a 12‐week treatment duration to assess the antifibrotic effect of BMS‐986263, which was considered sufficient for this proof‐of‐concept study based on preclinical experiments[ 12 ] and the Phase 1b study (NCT02227459) in which patients with advanced hepatic fibrosis received 5 weeks of BMS‐986263 treatment.[ 13 ] Compared with placebo, treatment with BMS‐986263 demonstrated target engagement by reducing liver HSP47 mRNA levels and decreasing liver HSP47 protein levels at Week 12. However, maximal target engagement may not have been evident in the posttreatment liver tissue samples because the biopsy collection was approximately 7 days after the last BMS‐986263 dose. Notably, improvements in fibrosis were observed in some patients after BMS‐986263 treatment. At Week 12, nearly 20% of patients who received BMS‐986263, compared with 13% of patients who received placebo, had fibrosis improvement that was consistent with a decrease in METAVIR score by at least one stage. Additionally, half of patients in the 90‐mg BMS‐986263 group with cirrhosis at baseline had improved by at least one METAVIR stage. When Ishak score improvement was measured in this population, only patients in the 90‐mg BMS‐986263 group (n = 5) had achieved at least a two‐stage improvement, and 3 of these patients had cirrhosis at baseline. In addition to pathologist interpretation, an automated tool, qFIB, was used to perform an exploratory analysis of fibrosis changes in unstained liver biopsy specimens. Of the patients with METAVIR and Ishak score improvements, 82% or 100% of patients, respectively, were also shown to have improved fibrosis as assessed by qFIB. These results are in agreement with other studies that suggest a potential utility for qFIB as a supplemental means to stage fibrosis in biopsy tissue.[ 24 ] However, while published studies have linked fibrosis staging interpreted by pathologists to clinical outcomes,[ 3 ] no such data exist yet for qFIB.
BMS‐986263 treatment for 12 weeks at 45 and 90 mg QW was generally safe and well tolerated and exhibited dose‐proportional increases in exposure and no notable accumulation of BMS‐986263 plasma concentrations with weekly dosing. Although the frequency of AEs was higher in the BMS‐986263 treatment groups compared with placebo, all reported AEs were mild or moderate in intensity, and the majority were IRRs that did not result in treatment discontinuation. During the study, no patients showed evidence of potential DILI or hepatic decompensation. HCC was detected in 3 patients during the follow‐up period; these events were not deemed to be related to BMS‐986263 treatment and were likely a reflection of the increased HCC risk in this patient population that persists even after successful viral eradication.[ 13 , 27 , 34 ] BMS‐986263 immunogenicity analyses revealed a low rate and titer of ADAs. Only 2 patients in the 45‐mg dose group exhibited ADAs up to Week 4, and no patients in the 90‐mg dose group were positive for ADAs relative to baseline. Together, the early, low‐level ADA response, lack of ADA persistence in the 45‐mg arm, and absence of ADAs in the 90‐mg arm relative to baseline suggest that BMS‐986263 has low immunogenicity.
In summary, in this Phase 2 study of patients with HCV‐SVR, 12‐week BMS‐986263 administered through weekly i.v. infusion improved METAVIR and Ishak scores, demonstrated target engagement, and was generally well tolerated through Week 36. Further evaluation of BMS‐986263 in patients with advanced liver fibrosis, particularly those with active fibrogenesis, is warranted. A Phase 2 study to evaluate the safety and efficacy of BMS‐986263 in adults with NASH and compensated cirrhosis is ongoing (NCT04267393).
CONFLICT OF INTEREST
Dr. Lawitz consults for and received grants from Metacrine, Boehringer Ingelheim, and Bristol Myers Squibb. He received grants from 89Bio, AbbVie, Akcea, Arena, Celgene, Conatus, Durect, Enanta, Enyo, Galectin, Galmed, Genfit, Gilead, Hanmi, Intercept, Madrigal, Novartis, Novo Nordisk, Octeta, and Zydus. Dr. Shevell was an employee of Bristol Myers Squibb when the study was performed and owns company stock. Dr. Tirucherai is employed by, owns stock in, and holds intellectual property rights with Bristol Myers Squibb. Dr. Du is employed by and owns stock in Bristol Myers Squibb. Dr. Karsdal is employed by and owns stock in Nordic Bioscience. Dr. Nielsen is employed by and owns stock in Nordic Bioscience. M. Karsdal and M. Nielsen are among the original inventors and patent holders of assays to measure PRO‐C3 and C3M. Dr. Charles is employed by, owns stock in, and holds intellectual property rights with Bristol Myers Squibb. W. Chen was an employee of Bristol Myers Squibb at the time of this study and may hold company stock. U. Kavita was an employee of Bristol Myers Squibb at the time of this study and may hold company stock. A. Coste has no disclosures to report. F. Poordad has participated in speakers’ bureaus for and received grants from Gilead Sciences and Intercept Pharmaceuticals. Z. Goodman has received grants from Alexion Pharmaceuticals, Allergan, Conatus Pharmaceuticals, Exalenz Bioscience, Galactin Therapeutics, Gilead Sciences, and Intercept Pharmaceuticals.
AUTHOR CONTRIBUTION
Eric J. Lawitz, Diane E. Shevell, Giridhar S. Tirucherai, Shuyan Du, Morten Karsdal, Zachary Goodman, and Edgar D. Charles contributed to the design of the study protocol. Eric J. Lawitz, Warner Chen, Uma Kavita, Angie Coste, Fred Poordad, Morten Karsdal, Mette Nielsen, and Zachary Goodman participated in data acquisition. Edgar D. Charles analyzed the study data, had access to all data, and can vouch for the integrity of the data analyses. All authors were involved in data interpretation and manuscript review.
CLINICAL TRIAL NUMBER
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Joel Myers and Arkendu Chatterjee of Bristol Myers Squibb for their critical review and guidance in the development of the manuscript, George Klinger of Bristol Myers Squibb for serving as the clinical operations lead for the HSP47 clinical development program, Ratna Revankar for biostatistics support, and Jamuna Karkhanis of PRA HealthSciences for serving as the medical monitor for this trial. Medical writing support was provided by Kendall Foote, Ph.D., and Amanda Martin, Ph.D., of Medical Expressions (Chicago, IL) and was funded by Bristol Myers Squibb. The Bristol Myers Squibb policy on data sharing may be found at https://www.bms.com/researchers‐and‐partners/independent‐research/data‐sharing‐request‐process.html.
Lawitz EJ, Shevell DE, Tirucherai GS, Du S, Chen W, Kavita U, et al. BMS‐986263 in patients with advanced hepatic fibrosis: 36‐week results from a randomized, placebo‐controlled phase 2 trial. Hepatology. 2022;75:912–923. 10.1002/hep.32181
SEE EDITORIAL ON PAGE xxx
Funding information
Bristol Myers Squibb
REFERENCES
- 1. Weiskirchen R, Weiskirchen S, Tacke F. Recent advances in understanding liver fibrosis: bridging basic science and individualized treatment concepts. F1000Res 2018;7:921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vilar‐Gomez E, Calzadilla‐Bertot L, Wai‐Sun Wong V, Castellanos M, Aller‐de la Fuente R, Metwally M, et al. Fibrosis severity as a determinant of cause‐specific mortality in patients with advanced nonalcoholic fatty liver disease: a multi‐national cohort study. Gastroenterology. 2018;155:443–57.e17. [DOI] [PubMed] [Google Scholar]
- 3. Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta‐analysis. Hepatology. 2017;65:1557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xu F, Moorman AC, Tong X, Gordon SC, Rupp LB, Lu M, et al. All‐cause mortality and progression risks to hepatic decompensation and hepatocellular carcinoma in patients infected with hepatitis C virus. Clin Infect Dis. 2016;62:289–97. [DOI] [PubMed] [Google Scholar]
- 5. Curry MP, Tapper EB, Bacon B, Dieterich D, Flamm SL, Guest L, et al. Effectiveness of 8‐ or 12‐weeks of ledipasvir and sofosbuvir in real‐world treatment‐naive, genotype 1 hepatitis C infected patients. Aliment Pharmacol Ther. 2017;46:540–8. [DOI] [PubMed] [Google Scholar]
- 6. American Association for the Study of Liver Diseases , Infectious Diseases Society of America . When and in whom to initiate HCV therapy. HCV guidance: recommendations for testing, managing, and treating hepatitis C. Available at: https://www.hcvguidelines.org/evaluate/when‐whom. Accessed 25 Nov 2020. [DOI] [PMC free article] [PubMed]
- 7. American Association for the Study of Liver Diseases, Infectious Diseases Society of America . Monitoring patients who are starting HCV treatment, are on treatment, or have completed therapy. HCV guidance: recommendations for testing, managing, and treating hepatitis C. Available at: https://www.hcvguidelines.org/evaluate/monitoring. Accessed 25 Nov 2020.
- 8. Karsdal MA, Daniels SJ, Holm Nielsen S, Bager C, Rasmussen DGK, Loomba R, et al. Collagen biology and non‐invasive biomarkers of liver fibrosis. Liver Int. 2020;40:736–50. [DOI] [PubMed] [Google Scholar]
- 9. Ito S, Nagata K. Roles of the endoplasmic reticulum‐resident, collagen‐specific molecular chaperone Hsp47 in vertebrate cells and human disease. J Biol Chem. 2019;294:2133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roderfeld M. Matrix metalloproteinase functions in hepatic injury and fibrosis. Matrix Biol. 2018;68–69:452–62. [DOI] [PubMed] [Google Scholar]
- 11. Parsons CJ, Bradford BU, Pan CQ, Cheung E, Schauer M, Knorr A, et al. Antifibrotic effects of a tissue inhibitor of metalloproteinase‐1 antibody on established liver fibrosis in rats. Hepatology. 2004;40:1106–15. [DOI] [PubMed] [Google Scholar]
- 12. Sato Y, Murase K, Kato J, Kobune M, Sato T, Kawano Y, et al. Resolution of liver cirrhosis using vitamin A–coupled liposomes to deliver siRNA against a collagen‐specific chaperone. Nat Biotechnol. 2008;26:431–42. [DOI] [PubMed] [Google Scholar]
- 13. Lawitz E, Balabanska RI, Charles ED, Friedman SL, Gutierrez JA, Niitsu Y, et al. Clinical phase 1b/2 study results for safety, pharmacokinetics, and efficacy of ND‐L02‐s0201, a novel targeted lipid nanoparticle (LNP) delivering HSP47 siRNA for the treatment of patients with advanced liver fibrosis [Abstract]. Hepatology. 2017;66:237A. [Google Scholar]
- 14. Soule B, Tirucherai G, Kavita U, Kundu S, Christian R. Safety, tolerability, and pharmacokinetics of BMS‐986263/NDL02‐s0201, a novel targeted lipid nanoparticle delivering HSP47 siRNA, in healthy participants: a randomised, placebo‐controlled, double‐blind, phase 1 study [Abstract]. J Hepatol. 2018;68:S112. [Google Scholar]
- 15. Masuda H, Fukumoto M, Hirayoshi K, Nagata K. Coexpression of the collagen‐binding stress protein HSP47 gene and the alpha 1(I) and alpha 1(III) collagen genes in carbon tetrachloride‐induced rat liver fibrosis. J Clin Invest. 1994;94:2481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Razzaque MS, Hossain MA, Kohno S, Taguchi T. Bleomycin‐induced pulmonary fibrosis in rat is associated with increased expression of collagen‐binding heat shock protein (HSP) 47. Virchows Arch. 1998;432:455–60. [DOI] [PubMed] [Google Scholar]
- 17. Honzawa Y, Nakase H, Shiokawa M, Yoshino T, Imaeda H, Matsuura M, et al. Involvement of interleukin‐17A‐induced expression of heat shock protein 47 in intestinal fibrosis in Crohn's disease. Gut. 2014;63:1902–12. [DOI] [PubMed] [Google Scholar]
- 18. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Friedman SL. Targeting siRNA to arrest fibrosis. Nat Biotechnol. 2008;26:399–400. [DOI] [PubMed] [Google Scholar]
- 20. Tam C, Wong JH, Cheung RCF, Zuo T, Ng TB. Therapeutic potentials of short interfering RNAs. Appl Microbiol Biotechnol. 2017;101:7091–111. [DOI] [PubMed] [Google Scholar]
- 21. Arthur MJ. Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology. 2002;122:1525–8. [DOI] [PubMed] [Google Scholar]
- 22. Soule B, Charles ED, Tirucherai G, Roodt F, Kavita U, Revankar R, et al. Safety, pharmacokinetics, and immunogenicity of BMS‐986263/ND‐L02‐s0201 in healthy Japanese and non‐Japanese participants. [Abstract] Hepatology. 2018;68:739A. [Google Scholar]
- 23. Sakamoto N, Ogawa K, Suda G, Morikawa K, Sho T, Nakai M, et al. Clinical phase 1b study results for safety, pharmacokinetics and efficacy of ND‐L02‐s0201, a novel targeted lipid nanoparticle delivering HSP47 SIRNA for the treatment of Japanese patients with advanced liver fibrosis. [Abstract] J Hepatol. 2018;68:S242. [Google Scholar]
- 24. Wang Y, Vincent R, Yang J, Asgharpour A, Liang X, Idowu MO, et al. Dual‐photon microscopy‐based quantitation of fibrosis‐related parameters (q‐FP) to model disease progression in steatohepatitis. Hepatology. 2017;65:1891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kavita U, Miller W, Ji QC, Pillutla RC. A fit‐for‐purpose method for the detection of human antibodies to surface‐exposed components of BMS‐986263, a lipid nanoparticle–based drug product containing a siRNA drug substance. AAPS J. 2019;21:92. [DOI] [PubMed] [Google Scholar]
- 26. Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. 2015;64:830–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. George SL, Bacon BR, Brunt EM, Mihindukulasuriya KL, Hoffmann J, Di Bisceglie AM. Clinical, virologic, histologic, and biochemical outcomes after successful HCV therapy: a 5‐year follow‐up of 150 patients. Hepatology. 2009;49:729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ioannou GN, Beste LA, Green PK, Singal AG, Tapper EB, Waljee AK, et al. Increased risk for hepatocellular carcinoma persists up to 10 years after HCV eradication in patients with baseline cirrhosis or high FIB‐4 scores. Gastroenterology. 2019;157:1264–78.e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen S‐H, Peng C‐Y, Chiang I‐P, Lai H‐C, Lee C‐J, Su W‐P, et al. Comparison of collagen proportionate areas in liver fibrosis quantification between chronic hepatitis B and C. Medicine (Baltimore). 2016;95:e4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harrison SA, Abdelmalek MF, Caldwell S, Shiffman ML, Diehl AM, Ghalib R, et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology. 2018;155:1140–53. [DOI] [PubMed] [Google Scholar]
- 31. Pan JJ, Bao F, Du E, Skillin C, Frenette CT, Waalen J, et al. Morphometry confirms fibrosis regression from sustained virologic response to direct‐acting antivirals for hepatitis C. Hepatol Commun. 2018;2:1320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Daniels SJ, Erhardtsen E, Kehlet SN, Leeming DJ, Shevell D, Rasmussen DGK, et al. Establishing the stability, precision, potential influencing factors and reference ranges of a biomarker of liver fibrosis, PRO‐C3, in healthy subjects, obese, and NAFLD patients [Abstract]. Hepatology. 2020;72:911A. [Google Scholar]
- 33. Tachi Y, Hirai T, Miyata A, Ohara K, Iida T, Ishizu Y, et al. Progressive fibrosis significantly correlates with hepatocellular carcinoma in patients with a sustained virological response. Hepatol Res. 2015;45:238–46. [DOI] [PubMed] [Google Scholar]
- 34. Kanwal F, Kramer J, Asch SM, Chayanupatkul M, Cao Y, El‐Serag HB. Risk of hepatocellular cancer in HCV patients treated with direct‐acting antiviral agents. Gastroenterology. 2017;153:996–1005.e1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material