

Abstract
Background
Only a few studies have reported muscle imaging data on small cohorts of patients with myotonic dystrophy type 1 (DM1). We aimed to investigate the muscle involvement in a large cohort of patients in order to refine the pattern of muscle involvement, to better understand the pathophysiological mechanisms of muscle weakness, and to identify potential imaging biomarkers for disease activity and severity.
Methods
One hundred and thirty‐four DM1 patients underwent a cross‐sectional muscle magnetic resonance imaging (MRI) study. Short tau inversion recovery (STIR) and T1 sequences in the lower and upper body were analyzed. Fat replacement, muscle atrophy and STIR positivity were evaluated using three different scales. Correlations between MRI scores, clinical features and genetic background were investigated.
Results
The most frequent pattern of muscle involvement in T1 consisted of fat replacement of the tongue, sternocleidomastoideus, paraspinalis, gluteus minimus, distal quadriceps and gastrocnemius medialis. Degree of fat replacement at MRI correlated with clinical severity and disease duration, but not with CTG expansion. Fat replacement was also detected in milder/asymptomatic patients. More than 80% of patients had STIR‐positive signals in muscles. Most DM1 patients also showed a variable degree of muscle atrophy regardless of MRI signs of fat replacement. A subset of patients (20%) showed a ‘marbled’ muscle appearance.
Conclusions
Muscle MRI is a sensitive biomarker of disease severity alsofor the milder spectrum of disease. STIR hyperintensity seems to precede fat replacement in T1. Beyond fat replacement, STIR positivity, muscle atrophy and a ‘marbled’ appearance suggest further mechanisms of muscle wasting and weakness in DM1, representing additional outcome measures and therapeutic targets for forthcoming clinical trials.
Keywords: clinical trials, CTG, muscle atrophy and inflammation, muscle MRI, myotonic dystrophy type 1
We refined the pattern of muscle involvement in DM1 by upper and lower body muscle magnetic resonance imaging (MRI), identifying the most frequent pattern of fat replacement and confirming that muscle MRI is a sensitive biomarker of disease burden in DM1. We also observed: STIR‐positive muscles in 80% of patients preceding fat replacement, muscle atrophy in muscles unreplaced by fat, and progeroid muscle appearance supporting a premature muscle senescence. Our findings provide novel insights into the pathophysiological mechanisms of muscle wasting and weakness in DM1, and could represent additional outcome measures and therapeutic targets for forthcoming clinical trials.
INTRODUCTION
Myotonic dystrophy type 1 (DM1) (OMIM #160900) is the most frequent inherited muscle disease in adults [1, 2]. DM1 is a multisystem disease caused by an unstable trinucleotide (CTG) expansion in the DMPK gene, which ultimately leads to the production of toxic mRNA transcripts [3, 4]. Skeletal and cardiac muscles are primarily affected, causing myotonia associated with progressive muscular weakness and atrophy, and variable respiratory and cardiac involvement [5, 6, 7].
Muscle magnetic resonance imaging (MRI) represents the gold standard technique for a muscle imaging study, providing useful information for diagnostic purposes through the detection of disease‐specific patterns of muscle involvement in various neuromuscular conditions [8, 9]. Furthermore, muscle MRI has been considered a biomarker of disease progression [10, 11, 12], and also a potential non‐invasive tool to better understand the pathophysiology of specific muscle diseases [13, 14].
Several MRI studies, mainly including small cohorts of patients and focusing on the lower limbs, have assessed the pattern and severity of muscle involvement in DM1 [15, 16, 17, 18, 19]. Two other studies explored the muscle involvement of upper limbs but omitted head, neck and scapular girdle muscles [20, 21]. A recent study analyzed the fat fraction and atrophy in 20 lower extremity muscles of 33 DM1 patients by quantitative muscle MRI, suggesting it as a valuable biomarker for this disease [22].
In this framework we aimed to extensively investigate the muscle involvement in a large cohort of DM1 patients evaluated by muscle MRI, in order to refine the global pattern of muscle involvement and to better understand the pathophysiological mechanisms of muscle weakness in this disease. We also aimed to identify which characteristics could represent reliable imaging biomarkers for assessing disease activity and severity, representing a useful tool to monitor disease progression and treatment response for forthcoming clinical trials.
PATIENTS AND METHODS
Study design and patient cohort
This was a multicenter, retrospective, cross‐sectional study involving four neuromuscular centers in Rome. It was carried out in compliance with standards of local ethical committees, the Helsinki Declaration and the Good Clinical Practices; all patients (or legal representative in patients aged <18 years or in patients unable to give their consent) gave written informed consent.
We retrospectively reviewed muscle MRI in 134 DM1 patients performed for routine clinical‐radiological follow‐up purposes. All patients included in this study were in clinical follow‐up in the abovementioned centers and had the muscle MRI performed between July 2016 and December 2019. All patients received a complete neurological examination, including Muscular Impairment Rating Scale (MIRS), based on an extensive manual muscle test scored by the Medical Research Council (MRC) scale [23] as part of their clinical follow‐up, within 6 months from muscle MRI assessment. Patients were stratified in five clinical forms based on disease onset: (i) congenital (at birth), (ii) infantile (1–10 years), (iii) juvenile (11–20 years), (iv) adult (21–40 years) and (v) late onset (>40 years).
In all cases, the genetic diagnosis of DM1 had been performed on DNA extracted from peripheral leukocytes at the moment of clinical diagnosis. According to CTG expansion range, patients were also grouped in three expansion classes: E1 (CTG 50–150), E2 (CTG 151–1000) and E3 (CTG >1000).
Muscle MRI study
Muscle imaging was obtained by a 1.5T MRI device (Magnetom Espree, Siemens AG), following the already reported internal protocol in accordance with the international consensus recommendations [24, 25].
Non‐contrast images were obtained using T1‐TSE (turbo spin echo) and T2‐STIR sequences. Axial T1 sequence (TR/TE of 400/13 ms, thickness/gap 4 mm/0, 4 mm, field of view [FOV] 370 mm) was performed by two contiguous stacks to obtain an anatomic coverage from the skull base to the ankles. STIR axial images (TR/TE/TI 3000/35/160 ms) were also acquired with the same anatomical coverage of T1 sequences. For the upper body, T1 sequences also included coronal (TR/TE of 450/13 ms, thickness/gap 3.5 mm/0.35 mm, FOV 400 mm) and sagittal (TR/TE of 630/13 ms, thickness/gap 5 mm/0.5 mm, FOV 400 mm) acquisitions in order to better evaluate the neck and thoracic muscles. Three coronal stacks were obtained to assess anterior and posterior thoracic muscles and neck muscles. Slices were oriented along (i) the axis of the pectoralis major muscle for anterior thoracic muscles, (ii) parallel to the dorsal kyphosis for posterior thoracic muscles and (iii) the major axis of the neck for neck muscles. Sagittal stack was oriented to cover from one shoulder to the other. The duration of the MRI exam was approximately 50 min.
Thirty‐two couples of isolated or grouped muscles of lower body (lower limbs, pelvis, abdominal muscles and lumbar paraspinal) and sixteen couples (plus genioglossus) of upper body muscles (head, neck, scapular girdle and thoracic) were studied by axial, coronal and sagittal planes and evaluated throughout their whole extension. The following muscles were grouped and evaluated together because of the difficulty in evaluating them independently in most cases: obliquus abdominis externus/internus‐transversus abdominis; obturatorius externus/internus; extensor digitorum/hallucis longus; peroneus longus/brevis; tibialis posterior/flexor digitorum longus; pterygoideus internus/externus; cervical paraspinal muscles; thoracic paraspinal muscles; teres major/minor.
T1 sequences were used to for semi‐quantitative evaluation of fat replacement and muscle atrophy. Fat replacement was scored using a six‐point scale (Table S1, Supplementary Materials) expanded from the original Mercuri scale modified by Fisher [26] as follows: 0 = normal; 1 = minimal/initial replacement (one isolated area); 2 = <50% volume replacement; 3 = ≥50% replacement; 4 = complete replacement (≥99%); 5 = complete muscle atrophy, fat replacement not‐evaluable. We introduced the additional stage of 5 in the revised Mercuri scoring system to assign a T1 score to completely atrophic muscles, in order to better represent the clinical consequences of the Mercuri scale in DM1, even if no fat replacement was apparently present at MRI. Global T1 score was used to assess the severity of fat replacement ranging in lower body from 0 to 320 and in upper body from 0 to 165.
In addition, the degree of muscle atrophy was evaluated by a revised version of the scale reported by Brogna et al. [27] for lower limbs, considering posterior, medial and anterior compartments of the thigh and anterior and posterior compartments of the leg. Conversely, in the upper body, due to the difficulty of differentiating scapular girdle and neck muscles in compartments, we decided to assess muscle atrophy of four isolated muscles: sternocleidomastoideus, trapezius, pectoralis minor and major. The scoring system was based on a five‐point scale implemented from a previous study [27] (Table S2, Supplementary Materials).
We defined fat replacement as asymmetrical if we identified a difference of at least 2 points or more in T1‐weighted images between both sides.
Muscle hyperintensity in STIR sequences was evaluated with a binomial system (1: positive; 0: negative) regardless of the intensity and extension of the positive signal.
Scans were retrospectively and independently evaluated by three radiologists/neurologists with expertise in muscle imaging (T.T., M.G. and T.N.) who were blinded to the clinical data.
Statistical analysis
Clinical and genetic data for statistical analysis included gender, age at MRI, age at disease onset, clinical forms, disease duration, MIRS score, and CTG expansion range and classes. MRI parameters considered for statistical analysis included: T1 score of lower limbs, upper body or total, STIR score, atrophy score, asymmetry and atypical findings.
In a first stage we confirmed that all variables followed a normal distribution (Kolmorgonov−Smirnov test, p < 0.05) and used parametric statistic studies. Differences in continuous variables between groups were assessed using Student’s t‐test or ANOVA depending on the number of groups to be compared. Correlation between variables was studied using Pearson’s test, considering that a correlation was excellent if the correlation coefficient (ρ) was higher than 0.8, and good if it was between 0.6 and 0.8. Hierarchical analysis, using mean fat replacement as the value analyzed, and graphical representation as a heatmap was performed using R software (V.4.0.3). Statistical analyses were performed using IBM SPSS Statistics (V.27) (IBM).
RESULTS
Patient cohort
Some 160 patients were considered for this study. Eleven patients did not participate in the MRI study because they were cardiac pacemaker wearers, six were ruled out for not completing the MRI study because of claustrophobia and nine patients refused. Finally, 134 patients participated in the muscle MRI study. All patients completed the MRI study of the lower body, and 115 patients also completed the upper body muscle MRI study. Participants’ characteristics are summarized in Table S4 (Supplementary Materials).
Eighty‐one patients were male (60.5%) and 53 were female (39.5%). Five patients (3.7%) had a congenital form of DM1, 13 (9.7%%) infantile, 36 (26.9%) juvenile, 51 (38.1%) adult and 29 (21.6%) late‐onset forms. Median age at MRI was 47 (range 12–81) years and average disease duration from disease onset to MRI was 16.5 years, ranging from 0 (asymptomatic patients) to 46 years. At the time of the muscle MRI, MIRS was = 1 (asymptomatic) for 10 patients (7%); for 35 patients (26%) it was = 2 (minimal signs); for 55 patients (41%) it was = 3 (distal weakness), for 33 patients (24%) it was = 4 (mild‐moderate proximal weakness) and for only 1 patient (<1%) it was = 5 (severe proximal weakness). CTG expansion ranged from 50 to >1500: 28 patients (20%) belonged to class E1, 97 (71%) to class E2 and 9 patients (6%) to class E3.
Muscle MRI study
T1 sequences
Lower body muscles were generally more affected than upper body. T1 score in the lower body was higher than that in the upper body in 90.4% of cases, with an overall mean T1 score nearly double in the lower than in the upper body (1.47 vs. 0.75, respectively). Four patients had completely normal MRI (global T1 score = 0). Frequency and severity of T1 changes in each muscle are shown in Figure 1.
FIGURE 1.
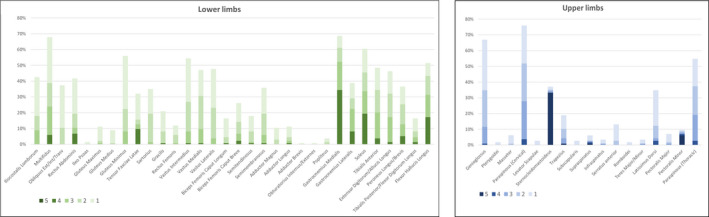
Frequency and severity of T1 changes in muscles showing the most frequently (higher columns) and severely (darker color) affected muscles in the lower limbs (left panel) and upper body (right panel)
At the abdomen and pelvis, the most affected muscles were multifidus and gluteus minimus, followed by rectus abdominis and iliocostalis lumborum. Notably, the iliopsoas muscle was spared until the late‐end stage of disease in almost all patients (97.7%).
At the thigh, the vasti were the most affected muscles and showed a distal‐proximal gradient of fat replacement (Figure 2). The posterior compartment of the thigh was less frequently affected with semimembranosus and biceps femoris (both heads) showing a milder involvement regardless of the overall severity. Gracilis and sartorius were also less affected than quadriceps, but, similarly, showed a distal‐proximal gradient of fibro‐fatty replacement.
FIGURE 2.
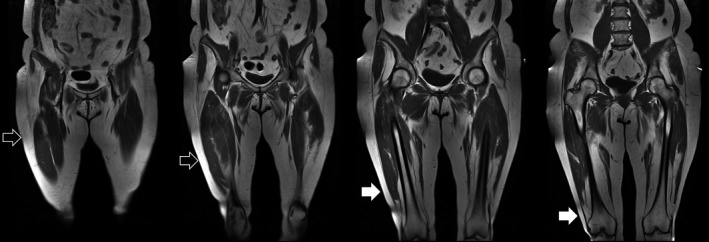
Distal‐proximal gradient of the quadriceps: quadriceps’ vasti showing a distal‐proximal gradient of fatty replacement. The proximal part of the muscle is spared (empty arrows) while the distal part is replaced (white arrows)
In the leg, gastrocnemius medialis was the most frequently and severely involved muscle, followed by soleus, gastrocnemius lateralis, flexor hallucis longus and tibialis anterior. Tibialis posterior was the most frequently spared muscle.
Overall, legs were more severely affected than thighs and pelvis. The most frequent pattern of muscle involvement detected in lower limbs was multifidus (>gluteus minimus> iliocostalis and rectus abdominis), vasti (distal‐proximal gradient), medial gastrocnemius (>soleus> flexor hallucis longus>tibialis anterior=gastrocnemius lateralis) (Figure 3, Table S3, Supplementary Materials).
FIGURE 3.
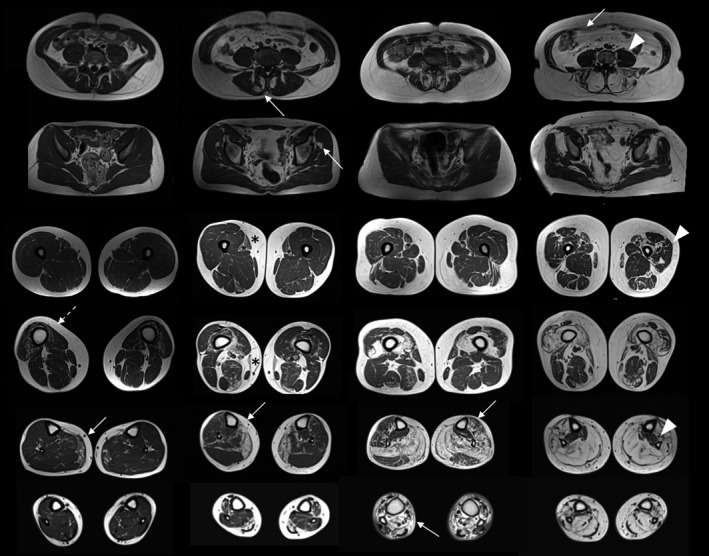
Pattern of muscle involvement in the lower limbs. Magnetic resonance imaging scans from four patients, from milder (left) to severer affected (right), showing the most frequent pattern of muscle involvement in the lower limbs (arrows): gastrocnemius and distal quadriceps (vasti) followed by glueteus minimus and soleus. Rectus abdominis, flexor hallucis longus, tibialis anterior and gastrocnemius lateralis are mildly affected. Psoas, proximal quadriceps (vasti) and tibialis posterior are spared until the late‐end stage of disease (arrowheads). Note the atrophy of quadriceps regardless of overall severity (milder patient, outlined arrow). Note the distal‐proximal gradient of fibro‐fatty replacement of sartorius (asterisk)
All these findings are summarized in the heatmap of the lower limbs (Figure 4). According to global T1 score, nine patients (6.7%) had a normal muscle MRI (T1 score = 0), whereas 24 (18%) had a total T1 score ≤2. Gastrocnemius medialis is one of the earliest affected muscles in DM1, therefore it could represent a good candidate for monitoring the onset of disease and its progression, whereas the distal‐proximal progression of fat replacement in the quadriceps could represent a useful biomarker in more advanced stages, since its frequent but uncomplete involvement until the late‐end stages of disease (only one patient had a score of 4 in one of three vasti). For this reason, even if vasti are frequently affected also in milder patients, and in moderate‐severe patients usually show a complete fat replacement in the distal part of the muscle (score 4), the proximal muscle bulk is frequently spared, resulting in an average global score of 2 or 3. Conversely, other muscles showed a homogeneous mild‐to‐moderate (score 1–2) fat replacement and sometimes (e.g., semimembranosus) it occurs regardless the overall severity of T1 changes, suggesting that both the timing of involvement and the rate of progression may differ between different muscles, as observed by quantitative muscle testing evaluations [28, 29, 30]. The most frequently affected muscles in the upper body were genioglossus, paraspinalis (both cervical and thoracic), trapezius and latissimus dorsi, followed by serratus anterior and supraspinatus (Figure 5). Eleven patients (11/115, 9.5%) had higher global T1 scores in the upper body than the lower body but, in most cases, it was due to sternocleidomastoid atrophy.
FIGURE 4.
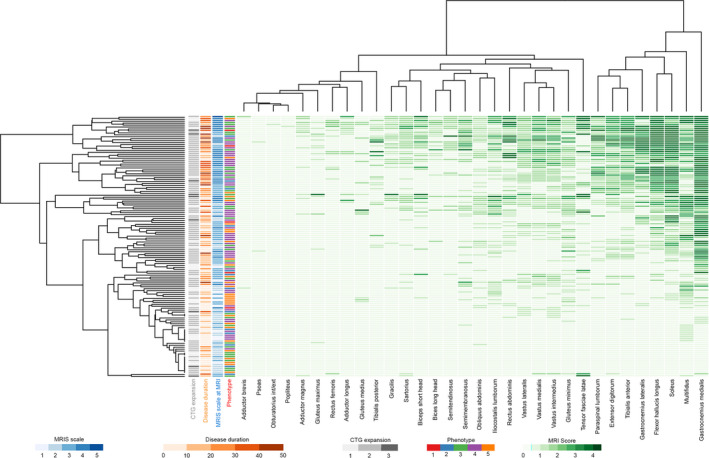
Heatmap of lower limbs. Hierarchical clustering matching severity of muscle involvement in each muscle (apsis) and overall T1 score (ordinat) in all patients. Each column represents one muscle and each line represents one patient. Darker green rectangles represent a higher T1‐Mercuri score. On the right side of the heatmap are located the most severely affected muscles (higher global T1 score), while in the upper part of the panel there are the more severe patients (higher global T1 score). Most of the more severely affected muscles are leg muscles (right side). In the middle of the panel are located muscles with moderate fat replacement as the result (1) of the average score between the distal‐replaced (score of 3–4) and proximal‐spared part (score 0–1) of the muscle (e.g., vasti) or (2) of a moderate but homogeneous fat replacement along the whole muscle bulk (e.g., semimembranosus). On the left side are the most spared muscles even in more severe patients (psoas, adductor brevis, obturatori, popliteus). Among patients, in the lower part of the panel there are patients with minimal or absent muscle involvement at magnetic resonance imaging (MRI) and just above there are those patients with minimal MRI alterations. Note that some patients with Muscular Impairment Rating Scale (MIRS) of 3 do not show MRI changes (probably underestimated because of a lacking upper limb study). Conversely, patients with MIRS 1 or 2 (milder‐asymptomatic spectrum of disease) can show muscle MRI alterations. The T1 score correlates with MIRS and disease duration but only weakly with CTG expansion
FIGURE 5.
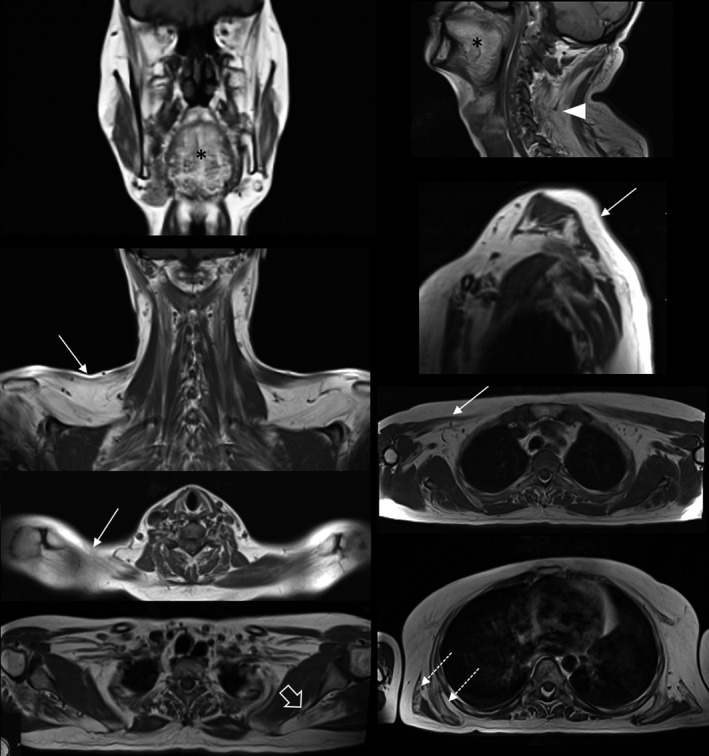
Muscle involvement in upper body. Set of images showing the most frequently affected muscles: tongue (asterisk), trapezius (arrows), paraspinalis (arrowhead), supraspinatus (open arrow), latissimus dorsi and serratus anterior (outlined arrows).
Overall, the fat replacement estimated by global T1 score showed a significant correlation with clinical severity assessed by MIRS (ρ = 0.65, p < 0.001) and with disease duration (ρ = 0.5, p < 0.001), but only a weak correlation with CTG range (ρ = 0.25, p = 0.002) (Figure 6).
FIGURE 6.
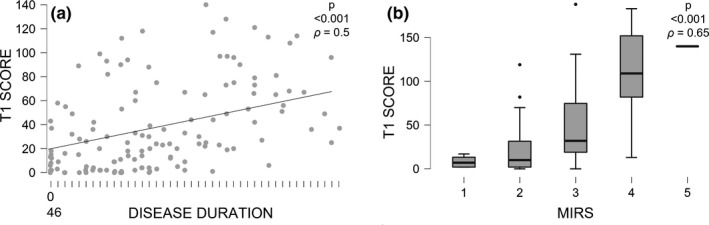
Fat replacement correlation with Muscular Impairment Rating Scale (MIRS) and disease duration. Severity of muscle involvement in lower body (higher global T1 score) is associated with longer disease duration (a). Global T1 score (upper and lower body) is also associated with worst clinical severity assessed by MIRS (b). Note that also the milder spectrum of disease (MIRS 1 and 2) shows some degree of fibro‐fatty replacement. Note also the possible underestimation of median global T1 score in the group of MIRS 3
STIR sequences
One‐hundred and thirteen patients (84.4%) had at least one STIR‐positive muscle in the lower limbs: the most frequent STIR‐positive muscles were vastus lateralis and medialis (15% of patients) in the thighs, flexor hallucis longus (17%), tibialis anterior (15%) and gastrocnemii (11%) in the legs. In general, STIR‐positive signals surrounded the fibro‐fatty replaced muscle detected in T1 sequences and showed a distal‐proximal gradient in the legs and vasti (Figure 7).
FIGURE 7.
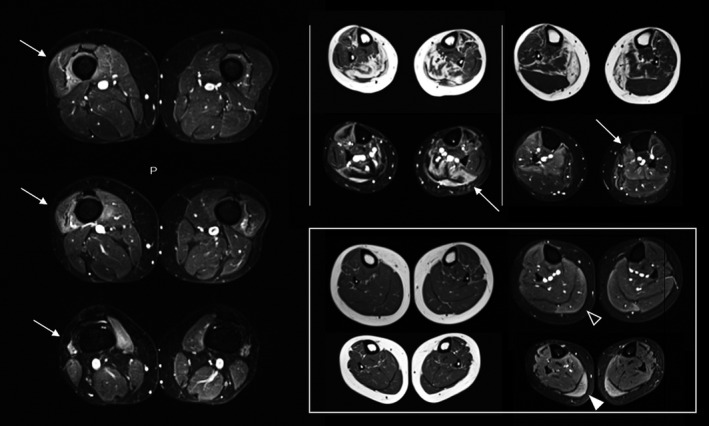
Short tau inversion recovery (STIR) findings. Blended hyperintensity of distal part of quadriceps’ vasti (left column, arrows). T1‐STIR matched images of the lower legs showing STIR‐positive signal in the residual areas of muscles (upper middle) or transitional area from fatty replacement to spared muscles (upper right). Distal‐proximal gradient of STIR positivity in T1‐negative gastrocnemius medialis (square, distal white arrowhead and proximal open arrowhead)
Conversely, both pelvis and upper body muscles were only rarely affected in STIR.
Interestingly, one‐third (3/9) of patients with negative muscle MRI by T1 sequences (T1 score = 0) showed >2 STIR positive muscles, and adding patients with very mild muscle MRI changes (global T1 score ≤2), the percentage of patients with >2 STIR‐positive muscles resulted in higher percentages (45%, 10/23 cases). Conversely, the patients with total negative STIR sequences (21, 15.6%) had a median T1 score of 2 (range 0–118, average 15.4).
STIR positivity correlated with the degree of fat replacement estimated by global T1 score (ρ = 0.55, p<0.001) and, as also observed in T1, with clinical severity by MIRS (ρ = 0.48, p < 0.001), disease duration (ρ = 0.42, p < 0.001) and weakly with the average CTG value (ρ = 0.23, p < 0.006).
Muscle atrophy
In contrast to T1 or STIR MRI changes, muscle atrophy was most frequently observed in the upper body than in lower limbs (66.9% and 16.4% of patients, respectively). In the lower limbs it mainly affected the anterior thighs compartment, particularly the distal part of the quadriceps, and more frequently both anterior and posterior compartments of the legs.
In the lower limbs muscle atrophy was nearly always not complete, and frequently showed some degree of fatty replacement. Conversely, in the upper body, muscle atrophy was more frequently complete and was observed also in normal T1 muscles. The most frequent (67%) and severely atrophic muscle was the sternocleidomastoideus which showed a complete atrophy in almost half of cases. Pectoralis minor and trapezius also showed partial muscle atrophy, regardless of fat replacement, in a lower number of patients (16.6% and 12.2%, respectively) (Figure 8A).
FIGURE 8.
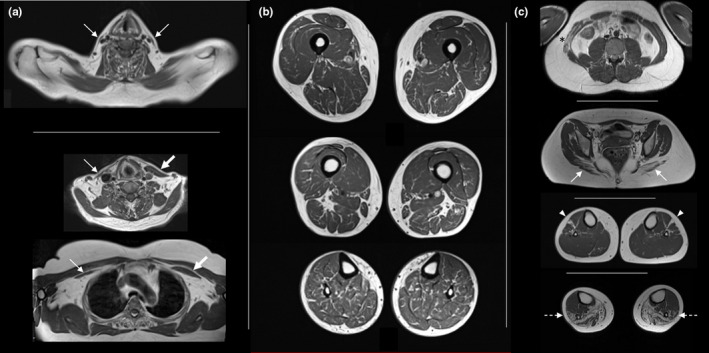
Muscle atrophy, ‘marbled’ appearance and atypical findings. (a) Bilateral atrophy of sternocleidomastoideus (top). Asymmetrical atrophy of sternocleidomastoideus (middle) and pectoralis minor (bottom). (b) Diffuse ‘marble’ texture of muscle tissue in lower limbs. (c) Atypical findings in different patients: transversus abdominis more affected than rectus abdominis with asymmetrical involvement (asterisk), gluteus maximus more affected than gluteus minimus (arrows), isolated involvement of extensor digitorum longus (arrowheads), peroneal muscles more affecter than tibialis anterior (outlined arrows)
Sternocleidomastoid atrophy at MRI correlated strongly with global fibro‐fatty replacement in T1 sequences (ρ = 0.6, p<0.001) and moderately with clinical severity (ρ = 0.44, p < 0.001) and disease duration (ρ = 0.42, p < 0.001). A weaker correlation was also observed with STIR positivity (ρ = 0.25, p < 0.008).
Atypical findings
A subset of patients (20%) showed a diffuse ‘marbled’ muscle appearance in the lower limbs detected by T1 sequences, regardless of global T1 score, STIR positivity, clinical severity, disease duration and genetic background. This peculiar aspect was slightly more frequent in older DM1 patients (ρ = 0.22, p < 0.009).
Muscle involvement in DM1 is typically bilateral and symmetric as observed in the large majority of patients, both clinically and through MRI studies. Accordingly, an asymmetric muscle involvement was observed in less than 1% of the couple of muscles evaluated (Figure 8b).
Atypical patients not following the classical pattern of muscle replacement were also observed. In these cases, certain muscles, typically mildly or later affected, were earlier and sometimes selectively replaced, while the most frequently affected muscles in the whole cohort were spared in these patients (Figure 8c).
DISCUSSION
Growing evidence supports the use of MRI in the diagnostic workup of various muscle diseases due to its ability to define the pattern of muscle involvement [8, 31, 32, 33]. Furthermore, muscle MRI may also represent a useful biomarker for disease severity and progression and can provide important information on disease pathophysiology [34, 35]. Because of its very suggestive clinical presentation, muscle MRI is usually not needed for diagnosis of DM1, but it could still be useful for deep phenotyping and better understanding of the pathophysiology of the disease [34].
Based on these assumptions, we have investigated MRI characteristics in a large cohort of DM1 patients. We observed that T1 and STIR hyperintensity as well as muscle atrophy occur in the muscles of DM1 patients. All these MRI parameters correlated with disease severity (as evaluated by MIRS grade) and disease duration, leading us to hypothesize that they may represent valuable biomarkers of the clinical impairment. We also observed that muscle MRI detected changes even in clinically spared muscles, revealing a subclinical involvement also in the mildest disease spectrum (asymptomatic or pauci‐symptomatic patients with MIRS 1–2).
About 80% of DM1 patients showed a predominant pattern of muscle involvement, characterized by involvement of tongue, paraspinalis, vasti and medial gastrocnemius, associated with atrophy of sternocleidomastoideus. Interestingly, the vasti and gastrocnemius medialis showed a distal‐proximal gradient of fat replacement. Considering patterns of muscle involvement, although not necessary for differential diagnosis in most cases, its recognition has some similarities with other muscle diseases that is worth mentioning. Tongue and paraspinalis are early and frequently affected in late‐onset Pompe disease (LOPD) [36] which can also show a similar pattern, but in LOPD quadriceps shows a prominent involvement of vastus intermedius, and gastrocnemius medialis is usually affected later in the disease course when other thigh muscles (semimembranosus and bicep femoris long head) become affected [12]. As well, tongue, paraspinalis and gluteal muscles are frequently affected in oculopharyngeal muscular dystrophy (OPMD), but with a prominent involvement of the soleus in the leg instead of gastrocnemius medialis [37].
Distal‐proximal gradient in the quadriceps with gastrocnemius medialis involvement is also typically observed in inclusion body myositis (IBM) [38], while the opposite proximo‐distal gradient in the quadriceps has been observed in sarcoglycanopathies and Pompe disease [36, 39, 40]. The prominent involvement of gastrocnemius medialis has been observed in many diseases including dystrophinopathy, calpainopathy, dysferlinopathy, laminopathies, RYR1‐related myopathies and tubular aggregate myopathies [41, 42, 43, 44, 45, 46].
Concerning STIR sequences, we observed that STIR hyperintensity frequently occurred in the transitional areas located between normal muscle and fat replacement, and was most evident in muscles with distal‐proximal gradient. Moreover, some patients without fat replacement (normal T1 study) showed altered STIR signal in the distal part of some muscles with a distal‐proximal gradient. These findings suggest that STIR positivity precedes the fat replacement process. This is also supported by a STIR‐T1 correspondence in the majority of muscles in the thigh and leg (Video 1). Evidence that STIR‐positive signal precedes the muscle degeneration has previously been reported in both acquired and genetic myopathies, such as idiopathic inflammatory myopathies and facioscapulohumeral muscular dystrophy [47, 48, 49] where STIR positivity corresponded to muscle edema and, more specifically, to inflammation [14]. To date, there is no evidence of an inflammatory process behind muscle degeneration in DM1 muscle biopsies. Nevertheless, our results suggest that an ‘active’ process, reflected by STIR positivity, may precede fat replacement, as described in other muscular dystrophies. On this basis, STIR hyperintensity could be used as a biomarker of disease activity in DM1 and could be a candidate as an outcome measure in forthcoming clinical trials.
VIDEO 1.
T1‐STIR (short tau inversion recovery) matching. First slide, T1 features: compartmental heatmaps of fat replacement (left panel), fat replacement in thighs (right, top) and unreplaced proximal‐distal legs (right, bottom). Second slide: STIR features: compartmental heatmaps of STIR‐positive muscles (left panel), STIR positivity in thighs (right, top) and legs (right, bottom). Note that STIR positivity occurs in the same areas of the heatmap with overall higher T1 scores in the thighs and legs, and the STIR‐positive signal is evident in still unreplaced muscle areas (Mercuri score <4) (thighs) and in still unreplaced muscles with distal‐proximal gradient (legs), suggesting that STIR positivity precedes the fat replacement
Muscle atrophy was another of the main findings in our study and could represent another important mechanism of muscle weakness in DM1. Classically, muscle atrophy is a consequence of disuse, or represents the last end‐stage of fat replacement. However, we observed that many muscles in the upper body were atrophic but not replaced by fat. This is the case for sternocleidomastoideus, which was the most frequently atrophic muscle, but without evidence of T1 hyperintensity in many cases. Atrophy of sternocleidomastoideus is a well‐known characteristic of the clinical phenotype in DM1, and the evidence that it is frequently not due to fat replacement suggests additional pathophysiological mechanisms for muscle wasting in DM1. Several explanations could be considered such as (i) embryological misdevelopment in some muscles leading to small hypoplastic muscles possibly due to the aberrant expression of fetal protein isoforms or insufficient regeneration of myofibers due to ineffective satellite cells turnover, (ii) abnormal muscle growth during childhood, (iii) prolonged mechanic stress (e.g., improper disuse/inactivity) or even (iv) premature loss of muscle volume as observed in aging processes leading to sarcopenia [50].
Finally, one‐fifth of patients presented a diffuse ‘marbled’ muscular appearance in the lower limbs as is sometimes observed in the elderly. This feature was detected in a minority of patients regardless of the overall severity of fat replacement (in both mild and severe patients) and in both spared or more affected muscles. This feature did not show any association with specific clinical, genetic or MRI characteristics. As expected, it was slightly more frequent in older patients [51]. Nevertheless it has been observed also in some young patients (age <30 years). This ‘marbled’ appearance of muscles could be related to a premature muscle senescence occurring in patients affected by DM1, in the context of the ‘progeroid’ process also reported in other affected tissues [52, 53].
This study has some limitations. First, the lack of correlation between MRI and genetic background likely depends on (i) the occurrence of somatic mosaicism (variable size of CTG‐expansions among different tissues) and (ii) triplet repeat expansion instability, which tends to increase over time. This last issue appears particularly relevant considering that the genetic diagnosis and MRI study were performed at different times in most patients included in this study. Second, our MRI protocol did not include the evaluation of the upper limbs, leading to a potential underestimation of the overall T1 score. This consideration especially concerns the moderate spectrum of disease, particularly patients with MIRS 3, having a predominant distal weakness of the upper limbs. This could also explain why in our study lower limbs (legs and thighs) showed more frequently STIR‐positive muscles than pelvis and upper body (proximal muscles more spared by disease). Third, the most severe spectrum of disease was not adequately represented in our sample (only one patient had MIRS = 5) because of contraindications (presence of cardiac devices) or low compliance with MRI examination (severe muscle disability, respiratory involvement with difficulty staying in a supine position for a long time). Fourth, we only used the MIRS scale to assess the disease severity; even if it has been widely used for many years, it has some limitations and reflects only approximatively the general disability of patients. Fifth, we used the manual muscle testing, scored by the MRC, to assess muscular weakness instead of the Quantitative Manual Testing (QMT); even if QMT is more accurate, it is more time consuming and in the routine clinical evaluation the Manual Muscle Testing (MMT) scale is more commonly used to evaluate outpatients, reserving QMT only for patients enrolled in clinical trials in our centers. Sixth, the muscle MRI study did not include quantitative acquisitions as Dixon or T2 relaxation time mapping for fat replacement or muscle edema, respectively. Finally, we did not include longitudinal MRI data, which represent the gold standard for proving disease progression over time. Nevertheless, because disease duration (time from symptom onset) correlated with global T1 score (degree of fat replacement) (p < 0.001), it is possible to assume that the muscles affected in the earliest stage of disease (in patients with low disease duration) represent effectively the earliest affected muscles, whereas those affected in the later stages represent the last affected muscles. In fact, the earliest affected muscles are also always affected in more severe patients with longer disease duration. This ‘natural history approach’ has also been argued in other cross‐sectional studies of muscle MRI with strong correlation between disease duration and severity of fat replacement at muscle MRI [43]. In our study it is also supported by the evidence that STIR positivity frequently occurs in the transitional areas between normal and replaced muscles, and that STIR positivity occurs in T1‐negative muscle expected to be the next affected muscles. In this way, STIR hyperintensity represents a useful biomarker for disease activity in DM1 and the severity of fat replacement in T1 represents the disease progression in the natural history of the disease.
Longitudinal follow‐up studies of muscle MRI in DM1 are warranted to confirm the data that have emerged from the present study.
In conclusion, muscle MRI data collected in the present study provided important insights for DM1. Muscle MRI is a valuable biomarker of disease severity also in the mildest spectrum of disease; also STIR positivity may represent one of the best candidates for an outcome measure for disease activity and treatment efficacy in forthcoming clinical trials. STIR positivity, muscle atrophy and ‘marbled’ muscle appearance, in addition to fat replacement, suggest a composite pathophysiological mechanism for muscle wasting and weakness in DM1, and could represent additional therapeutic targets in this disease. Overall, evidence acquired in this study paves the way for further studies aimed at better elucidating the pathophysiological mechanisms of muscle degeneration and could lead to the development of new treatment strategies.
CONFLICT OF INTEREST
The authors report no competing interests.
AUTHOR CONTRIBUTIONS
Matteo Garibaldi: Conceptualization (lead); Data curation (lead); Formal analysis (lead); Investigation (lead); Methodology (lead); Project administration (lead); Supervision (lead); Visualization (lead); Writing‐original draft (lead); Writing‐review & editing (lead). Tommaso Nicoletti: Conceptualization (supporting); Data curation (lead); Formal analysis (equal); Methodology (supporting); Project administration (equal); Supervision (supporting); Validation (supporting); Writing‐original draft (supporting); Writing‐review & editing (supporting). Elisabetta Bucci: Data curation (equal); Investigation (supporting); Writing‐review & editing (supporting). Laura Fionda: Data curation (equal); Writing‐review & editing (equal). Luca Leonardi: Data curation (supporting); Formal analysis (supporting); Writing‐review & editing (supporting). Stefania Morino: Data curation (supporting); Writing‐review & editing (supporting). Laura Tufano: Data curation (supporting); Project administration (supporting); Writing‐review & editing (supporting). Girolamo Alfieri: Data curation (supporting); Writing‐review & editing (supporting). Antonio Lauletta: Data curation (supporting); Writing‐review & editing (supporting). Gioia Merlonghi: Data curation (supporting); Writing‐review & editing (supporting). Alessia Perna: Data curation (supporting); Writing‐review & editing (supporting). Salvatore Rossi: Data curation (supporting); Writing‐review & editing (supporting). Jorge Alonso Perez: Data curation (supporting). Enzo Ricci: Data curation (supporting); Methodology (supporting); Writing‐review & editing (supporting). Tommaso Tartaglione: Data curation (lead); Formal analysis (equal); Methodology (equal); Project administration (equal); Writing‐review & editing (supporting). Antonio Petrucci: Data curation (equal); Project administration (supporting); Writing‐review & editing (supporting). Elena Maria Pennisi: Data curation (equal); Project administration (equal); Writing‐review & editing (supporting). Marco Salvetti: Supervision (equal); Writing‐review & editing (supporting). Gary Cutter: Formal analysis (lead); Methodology (equal); Supervision (supporting); Writing‐review & editing (supporting). Jordi Díaz‐Manera: Data curation (supporting); Formal analysis (equal); Investigation (equal); Methodology (equal); Project administration (equal); Software (lead); Visualization (supporting); Writing‐review & editing (supporting). Gabriella Silvestri: Data curation (lead); Formal analysis (equal); Investigation (equal); Methodology (equal); Project administration (equal); Supervision (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Giovanni Antonini: Conceptualization (supporting); Data curation (equal); Formal analysis (equal); Investigation (equal); Methodology (equal); Project administration (supporting); Supervision (supporting); Writing‐original draft (supporting); Writing‐review & editing (supporting).
Supporting information
ACKNOWLEDGMENTS
The authors acknowledge support from the Associazione Italiana Miologia (AIM) and Associazione Italiana Sistema Nervoso Periferico (ASNP). S.R. received a fellowship partially granted by the A.I. Vi.P.S. Onlus (Associazione Italiana Vivere la Paraparesi Spastica). Open Access funding provided by Universita degli Studi di Roma La Sapienza within the CRUI‐CARE Agreement. [Correction added on 20 May 2022, after first online publication: CRUI funding statement has been added.]
Garibaldi M, Nicoletti T, Bucci E, et al. Muscle magnetic resonance imaging in myotonic dystrophy type 1 (DM1): Refining muscle involvement and implications for clinical trials. Eur J Neurol.2022;29:843–854. doi: 10.1111/ene.15174
Gabriella Silvestri and Giovanni Antonini contributed equally to this work.
Funding information
This study was awarded a grant from “Giornata Malattie Neuromuscolari 2018” (GMN2018)
DATA AVAILABILITY STATEMENT
Anonymized data from this study not published in this article are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Turner C, Hilton‐Jones D. The myotonic dystrophies: diagnosis and management. J Neurol Neurosurg Psychiatry. 2010;81(4):358‐367. 10.1136/jnnp.2008.158261 [DOI] [PubMed] [Google Scholar]
- 2. Yotova V, Labuda D, Zietkiewicz E, et al. Anatomy of a founder effect: myotonic dystrophy in Northeastern Quebec. Hum Genet. 2005;117(2‐3):177‐187. 10.1007/s00439-005-1298-8 [DOI] [PubMed] [Google Scholar]
- 3. Miller JW, Urbinati CR, Teng‐Umnuay P, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19(17):4439‐4448. 10.1093/emboj/19.17.4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68(4):799‐808. 10.1016/0092-8674(92)90154-5 [DOI] [PubMed] [Google Scholar]
- 5. Bucci E, Testa M, Licchelli L, et al. A 34‐year longitudinal study on long‐term cardiac outcomes in DM1 patients with normal ECG at baseline at an Italian clinical centre. J Neurol. 2018;265(4):885‐895. 10.1007/s00415-018-8773-3 [DOI] [PubMed] [Google Scholar]
- 6. Garibaldi M, Lauletta A, Bucci E, et al. Gender effect on cardiac involvement in myotonic dystrophy type 1. Eur J Neurol. 2021;28(4):1366‐1374. 10.1111/ene.14665 [DOI] [PubMed] [Google Scholar]
- 7. Rossi S, Della Marca G, Ricci M, et al. Prevalence and predictor factors of respiratory impairment in a large cohort of patients with Myotonic Dystrophy type 1 (DM1): a retrospective, cross sectional study. J Neurol Sci. 2019;399:118‐124. 10.1016/j.jns.2019.02.012 [DOI] [PubMed] [Google Scholar]
- 8. Díaz‐Manera J, Llauger J, Gallardo E, Illa I. Muscle MRI in muscular dystrophies. Acta Myol. 2015;34(2–3):95‐108. [PMC free article] [PubMed] [Google Scholar]
- 9. Warman‐Chardon J, Diaz‐Manera J, Tasca G, Straub V, MRI workshop study group . 247th ENMC International Workshop: muscle magnetic resonance imaging ‐ implementing muscle MRI as a diagnostic tool for rare genetic myopathy cohorts. Hoofddorp, The Netherlands, September 2019. Neuromuscul Disord. 2020;30(11):938‐947. 10.1016/j.nmd.2020.08.360 [DOI] [PubMed] [Google Scholar]
- 10. Barnard AM, Willcocks RJ, Triplett WT, et al. MR biomarkers predict clinical function in Duchenne muscular dystrophy. Neurology. 2020;94(9):e897‐e909. 10.1212/WNL.0000000000009012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Willis TA, Hollingsworth KG, Coombs A, et al. Quantitative muscle MRI as an assessment tool for monitoring disease progression in LGMD2I: a multicentre longitudinal study. PLoS One. 2013;8(8):e70993. 10.1371/journal.pone.0070993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Figueroa‐Bonaparte S, Llauger J, Segovia S, et al. Quantitative muscle MRI to follow up late onset Pompe patients: a prospective study. Sci Rep. 2018;8(1):10898. 10.1038/s41598-018-29170-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kinali M, Arechavala‐Gomeza V, Cirak S, et al. Muscle histology vs MRI in Duchenne muscular dystrophy. Neurology. 2011;76(4):346‐353. 10.1212/WNL.0b013e318208811f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lassche S, Küsters B, Heerschap A, et al. Correlation between quantitative MRI and muscle histopathology in muscle biopsies from healthy controls and patients with IBM, FSHD and OPMD. J Neuromuscul Dis. 2020;7(4):495‐504. 10.3233/JND-200543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Castillo J, Pumar JM, Rodríguez JR, et al. Magnetic resonance imaging of muscles in myotonic dystrophy. Eur J Radiol. 1993;17(3):141‐144. 10.1016/0720-048x(93)90091-z [DOI] [PubMed] [Google Scholar]
- 16. Stramare R, Beltrame V, Dal Borgo R, et al. MRI in the assessment of muscular pathology: a comparison between limb‐girdle muscular dystrophies, hyaline body myopathies and myotonic dystrophies. Radiol Med. 2010;115(4):585‐599. 10.1007/s11547-010-0531-2 [DOI] [PubMed] [Google Scholar]
- 17. Kornblum C, Lutterbey G, Bogdanow M, et al. Distinct neuromuscular phenotypes in myotonic dystrophy types 1 and 2: a whole body highfield MRI study. J Neurol. 2006;253(6):753‐761. 10.1007/s00415-006-0111-5 [DOI] [PubMed] [Google Scholar]
- 18. Park D, Lee S‐H, Shin J‐H, Park J‐S. Lower limb muscle magnetic resonance imaging in myotonic dystrophy type 1 correlates with the six‐minute walk test and CTG repeats. Neuromuscul Disord. 2018;28(1):29‐37. 10.1016/j.nmd.2017.08.005 [DOI] [PubMed] [Google Scholar]
- 19. Song J, Fu J, Ma M, et al. Lower limb muscle magnetic resonance imaging in Chinese patients with myotonic dystrophy type 1. Neurol Res. 2020;42(2):170‐177. 10.1080/01616412.2020.1716494 [DOI] [PubMed] [Google Scholar]
- 20. Hayashi K, Hamano T, Kawamura Y, et al. Muscle MRI of the upper extremity in the myotonic dystrophy type 1. Eur Neurol. 2016;76(1–2):87‐94. 10.1159/000448328 [DOI] [PubMed] [Google Scholar]
- 21. Sugie K, Sugie M, Taoka T, et al. Characteristic MRI findings of upper limb muscle involvement in myotonic dystrophy type 1. PLoS One. 2015;10(4):e0125051. 10.1371/journal.pone.0125051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heskamp L, van Nimwegen M, Ploegmakers MJ, et al. Lower extremity muscle pathology in myotonic dystrophy type 1 assessed by quantitative MRI. Neurology. 2019;92(24):e2803‐e2814. 10.1212/WNL.0000000000007648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mathieu J, Boivin H, Meunier D, Gaudreault M, Bégin P. Assessment of a disease‐specific muscular impairment rating scale in myotonic dystrophy. Neurology. 2001;56(3):336‐340. 10.1212/wnl.56.3.336 [DOI] [PubMed] [Google Scholar]
- 24. Garibaldi M, Tasca G, Diaz‐Manera J, et al. Muscle MRI in neutral lipid storage disease (NLSD). J Neurol. 2017;264(7):1334‐1342. 10.1007/s00415-017-8498-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Warman Chardon J, Díaz‐Manera J, Tasca G, et al. MYO‐MRI diagnostic protocols in genetic myopathies. Neuromuscul Disord. 2019;29(11):827‐841. 10.1016/j.nmd.2019.08.011 [DOI] [PubMed] [Google Scholar]
- 26. Fischer D, Kley RA, Strach K, et al. Distinct muscle imaging patterns in myofibrillar myopathies. Neurology. 2008;71(10):758‐765. 10.1212/01.wnl.0000324927.28817.9b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brogna C, Cristiano L, Verdolotti T, et al. MRI patterns of muscle involvement in type 2 and 3 spinal muscular atrophy patients. J Neurol. 2020;267(4):898‐912. 10.1007/s00415-019-09646-w [DOI] [PubMed] [Google Scholar]
- 28. Gagnon C, Petitclerc É, Kierkegaard M, Mathieu J, Duchesne É, Hébert LJ. A 9‐year follow‐up study of quantitative muscle strength changes in myotonic dystrophy type 1. J Neurol. 2018;265(7):1698‐1705. 10.1007/s00415-018-8898-4 [DOI] [PubMed] [Google Scholar]
- 29. Roussel M‐P, Fiset M‐M, Gauthier L, et al. Assessment of muscular strength and functional capacity in the juvenile and adult myotonic dystrophy type 1 population: a 3‐year follow‐up study. J Neurol. 2021;268(11):4221‐4237. 10.1007/s00415-021-10533-6 [DOI] [PubMed] [Google Scholar]
- 30. Hammarén E, Kjellby‐Wendt G, Lindberg C. Muscle force, balance and falls in muscular impaired individuals with myotonic dystrophy type 1: a five‐year prospective cohort study. Neuromuscul Disord. 2015;25(2):141‐148. 10.1016/j.nmd.2014.11.004 [DOI] [PubMed] [Google Scholar]
- 31. Mercuri E, Jungbluth H, Muntoni F. Muscle imaging in clinical practice: diagnostic value of muscle magnetic resonance imaging in inherited neuromuscular disorders. Curr Opin Neurol. 2005;18(5):526‐537. 10.1097/01.wco.0000183947.01362.fe [DOI] [PubMed] [Google Scholar]
- 32. Carlier R‐Y, Quijano‐Roy S. Myoimaging in congenital myopathies. Semin Pediatr Neurol. 2019;29:30‐43. 10.1016/j.spen.2019.03.019 [DOI] [PubMed] [Google Scholar]
- 33. Verdú‐Díaz J, Alonso‐Pérez J, Nuñez‐Peralta C, et al. Accuracy of a machine learning muscle MRI‐based tool for the diagnosis of muscular dystrophies. Neurology. 2020;94(10):e1094‐e1102. 10.1212/WNL.0000000000009068 [DOI] [PubMed] [Google Scholar]
- 34. Nuñez‐Peralta C, Alonso‐Pérez J, Díaz‐Manera J. The increasing role of muscle MRI to monitor changes over time in untreated and treated muscle diseases. Curr Opin Neurol. 2020;33(5):611‐620. 10.1097/WCO.0000000000000851 [DOI] [PubMed] [Google Scholar]
- 35. Meyerspeer M, Boesch C, Cameron D, et al. 31 P magnetic resonance spectroscopy in skeletal muscle: experts’ consensus recommendations. NMR Biomed. Published online February 10, 2021;34:e4246. 10.1002/nbm.4246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Díaz‐Manera J, Walter G, Straub V. Skeletal muscle magnetic resonance imaging in Pompe disease. Muscle Nerve. Published online November 6, 2020;63(5):640–650. 10.1002/mus.27099 [DOI] [PubMed] [Google Scholar]
- 37. Alonso‐Jimenez A, Kroon RHMJM, Alejaldre‐Monforte A, et al. Muscle MRI in a large cohort of patients with oculopharyngeal muscular dystrophy. J Neurol Neurosurg Psychiatry. 2019;90(5):576‐585. 10.1136/jnnp-2018-319578 [DOI] [PubMed] [Google Scholar]
- 38. Tasca G, Monforte M, De Fino C, Kley RA, Ricci E, Mirabella M. Magnetic resonance imaging pattern recognition in sporadic inclusion‐body myositis. Muscle Nerve. 2015;52(6):956‐962. 10.1002/mus.24661 [DOI] [PubMed] [Google Scholar]
- 39. Tasca G, Monforte M, Díaz‐Manera J, et al. MRI in sarcoglycanopathies: a large international cohort study. J Neurol Neurosurg Psychiatry. 2018;89(1):72‐77. 10.1136/jnnp-2017-316736 [DOI] [PubMed] [Google Scholar]
- 40. Alonso‐Pérez J, González‐Quereda L, Bello L, et al. New genotype‐phenotype correlations in a large European cohort of patients with sarcoglycanopathy. Brain. 2020;143(9):2696‐2708. 10.1093/brain/awaa228 [DOI] [PubMed] [Google Scholar]
- 41. Barp A, Bello L, Caumo L, et al. Muscle MRI and functional outcome measures in Becker muscular dystrophy. Sci Rep. 2017;7(1):16060. 10.1038/s41598-017-16170-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barp A, Laforet P, Bello L, et al. European muscle MRI study in limb girdle muscular dystrophy type R1/2A (LGMDR1/LGMD2A). J Neurol. 2020;267(1):45‐56. 10.1007/s00415-019-09539-y [DOI] [PubMed] [Google Scholar]
- 43. Diaz‐Manera J, Fernandez‐Torron R, LLauger J, et al. Muscle MRI in patients with dysferlinopathy: pattern recognition and implications for clinical trials. J Neurol Neurosurg Psychiatry. 2018;89(10):1071‐1081. 10.1136/jnnp-2017-317488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Garibaldi M, Fattori F, Riva B, et al. A novel gain‐of‐function mutation in ORAI1 causes late‐onset tubular aggregate myopathy and congenital miosis. Clin Genet. 2017;91(5):780‐786. 10.1111/cge.12888 [DOI] [PubMed] [Google Scholar]
- 45. GóMez‐Andrés D, Díaz‐Manera J, Alejaldre A, et al. Muscle imaging in laminopathies: synthesis study identifies meaningful muscles for follow‐up. Muscle Nerve. 2018;58(6):812‐817. 10.1002/mus.26312 [DOI] [PubMed] [Google Scholar]
- 46. Jokela M, Tasca G, Vihola A, et al. An unusual ryanodine receptor 1 (RYR1) phenotype: mild calf‐predominant myopathy. Neurology. 2019;92(14):e1600‐e1609. 10.1212/WNL.0000000000007246 [DOI] [PubMed] [Google Scholar]
- 47. Renard D, Taieb G, Garibaldi M, et al. Inflammatory facioscapulohumeral muscular dystrophy type 2 in 18p deletion syndrome. Am J Med Genet A. 2018;176(8):1760‐1763. 10.1002/ajmg.a.38843 [DOI] [PubMed] [Google Scholar]
- 48. Tasca G, Pescatori M, Monforte M, et al. Different molecular signatures in magnetic resonance imaging‐staged facioscapulohumeral muscular dystrophy muscles. PLoS One. 2012;7(6):e38779. 10.1371/journal.pone.0038779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pinal‐Fernandez I, Casal‐Dominguez M, Carrino JA, et al. Thigh muscle MRI in immune‐mediated necrotising myopathy: extensive oedema, early muscle damage and role of anti‐SRP autoantibodies as a marker of severity. Ann Rheum Dis. 2017;76(4):681‐687. 10.1136/annrheumdis-2016-210198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Palmio J, Udd B. Borderlines between sarcopenia and mild late‐onset muscle disease. Front Aging Neurosci. 2014;6:267. 10.3389/fnagi.2014.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yamada Y. Muscle mass, quality, and composition changes during atrophy and sarcopenia. Adv Exp Med Biol. 2018;1088:47‐72. 10.1007/978-981-13-1435-3_3 [DOI] [PubMed] [Google Scholar]
- 52. Azzabou N, Hogrel J‐Y, Carlier PG. NMR based biomarkers to study age‐related changes in the human quadriceps. Exp Gerontol. 2015;70:54‐60. 10.1016/j.exger.2015.06.015 [DOI] [PubMed] [Google Scholar]
- 53. Hogrel J‐Y, Barnouin Y, Azzabou N, et al. NMR imaging estimates of muscle volume and intramuscular fat infiltration in the thigh: variations with muscle, gender, and age. Age (Dordr). 2015;37(3):9798. 10.1007/s11357-015-9798-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Anonymized data from this study not published in this article are available from the corresponding author upon reasonable request.