

Abstract
Medical device (MD) is a broad term that encompasses products ranging from, for example, examination gloves to digital dermoscopy systems; all of which are regulated by a new regulatory framework in the EU from May 2021. The new Medical Device Regulation (MDR) (Regulation EU 2017/745) will have a significant effect on suppliers of MD and will have subsequent effects also for dermatologists and other clinicians. Medical device software and apps are reclassified leading to more stringent requirements on documentation within, e.g. clinical evidence, as well as regulatory authority control. The changes will likely have positive effects on quality, to the benefit of patients. There will, however, be implications affecting the availability and support of existing devices and the introduction of new devices, as well as a likely price increase due to the higher costs for suppliers. Dermatologists, other clinicians and administrators need to be aware of the effects of MDR to ensure that existing devices and new purchases can be used as planned. Specifically, clinicians need to be aware of the following: (i) improved quality of MD and follow‐up of incidents can be expected. (ii) Only ‘non‐significant’ updates will be permitted after May 2021 to many existing systems and devices unless approved under the new MDR. (iii) Existing devices that do not achieve approval under the new regulation will no longer be manufactured after May 2024. (iv) New products and methods will take longer time to be approved and available. (v) Prices will likely increase. (vi) Suppliers of products that do not fulfil the new regulation will disappear, and the availability of consumables, spare parts or upgrades might be discontinued. (vii) A trend to oligopoly may appear in the market. It is therefore important to check with your suppliers as to how and when they will adhere to the new MDR regulation.
Introduction
Almost every product used by clinicians in managing patients or medical procedures is considered a ‘medical device’. This covers everything from facemasks and gowns to dermatoscopes and digital dermoscopy systems. Medical devices (MD) placed on the market within the EU need to have a CE mark and have been regulated through EU directives called Medical Device Directive (MDD) and Active Implantable Medical Device Directive (AIMDD) for many years. 1 Following various incidents such as the PIP breast implant scandal in France 2 and the metal hip prosthesis issue, the EU decided to improve safety and control by strengthening the regulation of medical devices. 3 The result is a new set of industry regulations that came into effect in the EU in May 2021 (Regulation EU 2017/745). 4 The new regulation, called MDR (Medical Device Regulation), tightens the control mechanisms for medical devices, including medical device software (which includes apps), and will have a substantial impact on medical device manufacturers and distributors and will in turn also have an effect on dermatologists, supporting clinicians and other staff and to the final beneficiaries, the patients.
Also, in vitro diagnostic devices will be affected by a new regulation called In Vitro Diagnostic Medical Device Regulation (IVDR) that will replace the current In Vitro Diagnostic Medical Device Directive (IVDD) in May 2022. This article focuses, however, on the new MDR regulation and its consequences.
The availability of new methods and devices and the improvement of existing medical devices will both be affected. Clinicians should be aware of these changes to make informed decisions when considering new devices and when evaluating suppliers of medical equipment. One of the key goals of the MDR regulation is to increase patient safety by improving the quality, safety and performance of medical devices. 5 , 6 , 7 Many devices today are seen to have insufficient clinical evidence of safety and efficacy, partly because they were assigned a low‐risk classification under the previous MDD, and thus had limited controls of clinical validation of safety and efficacy. This will change under the new MDR regulation, and many devices will be ‘up‐classified’ to a higher risk classification under MDR.
The requirements for medical device approval have been tightened, and the process has become more standardized. This affects how clinical trials and performance tests are conducted, and even basic aspects such as the identification and traceability of products. In the long run, this aims to improve patient management and benefit patient safety.
Methods
The key points of the new MD regulation were determined and analysed to identify the main issues related to MD certification and classification and to identify the information that dermatologists should request from the manufacturers of MD.
Definition of MD in the EU
The definition of a medical device is very broad and covers in principle almost all devices and materials that are used for diagnostic or therapeutic purposes, except where the principal function is based on pharmacological, immunological or metabolic means.
The definition of medical devices in MDR Article 2 (abbreviated) is any article (also including software products) intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes:
Diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease.
Diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability.
Investigation, replacement or modification of the anatomy or of a physiological or pathological process or state.
Providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations, and which does not achieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means.
This definition includes everything from plasters, skin biopsy punches and disposable gloves to phototherapy lamps, software apps, diagnostic devices, pacemakers and radiation systems. Medical devices, including software, must have a medical purpose in order to qualify as a medical device. This includes apps for mobile phones that provide a therapeutic recommendation or suggest a diagnosis. 8
Aesthetic, wellness or other devices which do not fall under the above definition are generally not seen as medical devices. However, devices that are similar in function and risk profile to products intended for medical use are also covered by the new regulation, such as dermal fillers, liposuction or lasers for skin resurfacing.
Classification of medical devices
Medical devices, including all medical device software, are classified according to their risk level (see Chapter 5 in Regulation (EU 2017/745)), 3 and the regulatory requirements increase with the risk level (Fig. 1). Class I devices are subject to the least regulatory control and are not intended to help support or sustain life or be substantially important in preventing impairment to human health, whereas Class III devices are ‘high risk’ and are usually those that support or sustain human life.
Figure 1.
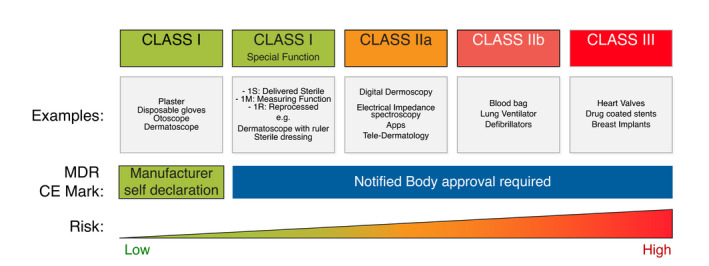
Classes of medical devices in the new regulation, examples, requirements and risk.
With the new MDR regulation, many products will be reclassified to a higher classification level, which increases the requirements the suppliers and the products must fulfil to be approved and sold to clinicians.
Consequences of the new MDR in dermatology
In the previous legislation, under the MDD, devices in Class I had a very broad scope – the same set of rules covered almost all devices within dermatology – from examination gloves to total body photography and digital dermoscopy systems. Many software‐based systems, for example, are today classified by the manufacturer as a Class I ‘documentation only system’, and so have the lowest level of control. 8 Systems, however, that claim to assist with diagnosis are classified as Class II products in the new regulatory framework.
Classification of many systems will change under MDR. Any software system that ‘provides information contributing to a diagnosis’, such as tele‐dermatology apps or digital dermoscopy, will be reclassified from Class I to Class IIa or higher. This has major consequences for the manufacturers and distributors of such devices – they must comply with multiple new regulations and the certification process usually takes 2–3 years.
Diagnostic devices will require robust clinical evaluations, usually implying backing from their own, solid clinical data. This is a major change in the enforcement of the legislation designed to ensure that devices perform as claimed. It means that many companies will need to perform new clinical trials to support their existing or any new claims. Furthermore, the clinical data that are to be used as evidence to support safety and performance need to be generated in studies that are shown to be reliably and robustly designed and run.
All medical devices will be registered in a common database named European Database of Medical Devices (EUDAMED). 9 This will provide the possibility for authorities to monitor, e.g. all reported incidents for a device. The database will to certain extent also be available for clinicians and the public to search for information about devices.
In addition, there are complex requirements regarding manufacturer quality management processes – everything from the documentation of the technical development process and testing to the control of production processes by vendors are highly controlled. Authorities perform regular inspections to ensure that MDR requirements and responsibilities are met (though less so for Class I products). After approval, the manufacturer needs to provide regular ‘postmarket’ evaluations and reports.
Certification process of MD
To achieve CE marking for a product under MDR, there are several requirements that need to be fulfilled and documented by the manufacturer. The requirements involve items such as a quality management system, extensive technical documentation of the development and manufacturing for a product, risk management for the product in clinical use, clinical evaluation that shows the safety and performance of the product and that the benefits of using the device outweigh any risks. In addition, for all devices higher than Class I, a notified body will perform reviews of the documentation as well as regular inspections of the manufacturer.
Where accordance with MDR has been demonstrated, following specific conformity assessment procedures, manufacturers shall provide an EU declaration of conformity to show that requirements in MDR are fulfilled.
Manufacturers also must plan and perform ‘postmarket surveillance’ activities, which involve follow‐up after the device is put on the market – such as postmarket clinical studies – to verify that safety and performance criteria are met in normal clinical practice.
The clinical evaluation, risk assessment and the postmarket surveillance activities as well as a benefit–risk analysis must be updated on a regular basis after the product is on the market to maintain the manufacturer’s certification and the CE mark.
Consequences of MDR that dermatologists should understand
Clinicians can expect to see both positive and negative aspects of this regulation change. These will primarily be indirect as it is the medical device manufacturers and distributors that need to comply with the new stricter rules. In general, clinicians can expect that product quality will improve, claims on performance will be better substantiated and new releases/updates more controlled, all of which will benefit the patient. However, there will be consequences regarding the availability of some devices, delays or barriers to introducing new products as well as price increases. 10
The expected consequences of the MDR are as follows:
-
Improved quality and adherence to safety and performance claims from manufacturers.
Devices will need to be thoroughly tested and validated in clinical studies regarding safety and efficacy prior to being made available to clinicians, and all devices except Class I will have to be authorized by a notified body. This means that clinicians can feel more secure that devices actually perform as specified.
However, MDR does not as yet require detailed labelling of ingredients on the packaging of MD’s, which may result in unnecessary irritant or allergic reactions to patients if these ingredients had been listed on the label. 11
-
Improved follow‐up of potential issues with devices in clinical use.
Because there are more stringent requirements on follow‐up of potential incidents, complaints or limitations of a medical device, an improved level of control and follow‐up of quality or safety issues can be expected. This applies for all medical device suppliers no matter which classification or what product they supply. This information has previously been often difficult to obtain, but under MDR, certain information such as serious incident reports and field service corrective actions is to be made available in the EUDAMED database.
-
Only non‐significant upgrades to software and hardware permitted from May 2021.
Medical devices already on the market may continue to be manufactured with a CE mark under the previous MDD regulation until May 2024 and sold until May 2025. However, if the products do not carry a CE mark under the new MDR, they will be essentially ‘frozen’ as they were in May 2021. In other words, no significant changes 12 are allowed until the device and the manufacturer or supplier are approved according to the new MDR regulation. The risk is that manufacturers decide not to continue updating or supporting products because the work and cost involved to get them approved under MDR is too high.
-
Existing devices with a CE mark under MDD that are not re‐approved under MDR can no longer be manufactured after May 2024 and some suppliers will disappear.
Since the burden of the new requirements is significant, time consuming and costly, some manufacturers may either ‘give up’ or not succeed in fulfilling the requirements. This is not only due to the effort and costs of updating procedures and technical documentation but also due to the challenge of providing supporting clinical evidence for existing product performance claims and safety, especially if it involves new clinical studies.
-
New products and methods will take longer to reach the market and clinical use.
It will most likely take significantly longer for new methods and devices to be released onto the market, since CE marking under the new MDR regulation is much more burdensome compared with MDD. This is due to the many new requirements for documentation of safety and performance and the need for evidence from clinical evaluations with the device in question.
-
Prices will likely increase.
Regulatory improvements always come with a price tag. In the case of the new MDR regulation, it can be quite substantial as it brings fundamental changes to the requirements suppliers must manage. So higher costs for the manufacturers will almost certainly also increase the prices for medical devices.
-
A trend to oligopoly may appear in the market.
The stricter requirements in MDR may lead many innovators and innovative start‐ups to disappear or give up in the quest to search for innovative solutions. This may cause a trend towards the creation of an oligopoly, where market is dominated by a smaller group of larger companies with the implications this may have for access to novel products and methods.
Special considerations for MDR in artificial intelligence (AI)‐based systems
AI image analysis systems are an example of a type of system that is directly impacted by MDR. This is not specifically because they are based on AI, but more because they usually provide diagnostic guidance and being software‐based is now much more clearly covered by the regulations. The new regulations aim to ensure that systems that contribute to diagnosis (and in turn therapy) are accurate and reliable.
AI systems that provide ‘risk scores’ or classify/diagnose lesions in any way become Class IIa devices (or higher) and are subject to much tougher regulation. They will also require comprehensive own clinical data that need to be assessed and approved by a third party, that is a MDR accredited notified body. As of May 2021, only one diagnostic system within dermatology had received approval under MDR. 13
Effects on current MD’s
Devices with an existing approval under MDD may still be sold during an interim period until late May 2025, provided that no significant changes 12 are made to the product. This will limit the introduction of new product features and updates, and especially significant software updates and new clinical analyses or indications.
Certain new requirements in the MDR, such as monitoring of safety issues on products placed on the market, inclusion in the EUDAMED database, etc., are mandatory even if the manufacturer holds an approval under MDD during the interim period.
Information to be provided by manufacturers
For devices already in use in a practice that do not get ‘updated’, there is generally nothing to worry about. However, for software‐based products, for example, it is a good idea to request written confirmation from the supplier that the devices are correctly classified, and will be approved under MDR (and in what timeframe).
If dermatologists are considering buying a new product, then they need to make sure that the product and any accessories will remain available after May 2021 and under what class. If the product will definitely remain a Class I product under MDR (i.e. it provides no analysis or diagnostic interpretation), there is no need for concern. However, if the product does supply any type of diagnostic information, it will likely end up reclassified to a higher risk class, so make sure the supplier will fulfil requirements and be approved under MDR. If not, there is no guarantee that it will be possible to update the system in future. It is likely that you can still use it as is, but it is unclear how support and updates will be managed.
Suppliers or products that do not fulfil the new regulation will sooner or later disappear from the market, and then, the availability of consumables, spare parts or upgrades might be discontinued.
So, it is important that you check with your suppliers how and when they will adhere to the new MDR regulation.
Checklist for dermatologists
Finally, we propose a checklist for dermatologists to ensure your MD fulfils the necessary safety and performance requirements required by MDR, with the following points: If considering a new MD for a practice or clinic:
-
Verify that the device has a CE mark under the new MDR.
Prior to purchase, check that the device has the MD symbol showing it is implemented under MDR, or have your supplier provide you with a ‘Declaration of Conformity’ (DoC) to MDR for the product, and in case, the device is not a Class I also a ‘EC certificate’ for the manufacturer.
-
If the device has no CE mark under MDR:
Have the supplier provide assurance and time plan for MDR certification as well as information on how updates will be handled in the interim period.
If the device does not receive CE mark under MDR, you must have option to return the device.
If evaluating an existing MD in a practice or clinic:
Ask the supplier to provide assurance and a time plan for MDR certification.
If assurance is not provided, consider replacing the device with a device that has (or will obtain) a CE mark under MDR.
Conclusions
The new Medical Device Regulation (MDR) will have a significant impact on medical devices in Europe. It will provide benefits for patients because it requires improved quality and adherence to safety and performance claims from manufacturers. However, the updated classification of devices combined with stricter requirements on manufacturers will have impact on the future availability, upgradeability and price of devices. This needs to be considered by clinicians, both for their existing devices and when purchasing new devices.
Conflicts of interest
All of the authors have submitted separate COI forms. The authors have no conflict of interest and received no specific funding for this work. The authors of this article represent both academia, clinicians, regulatory expertise and industry.
Funding source
None declared.
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- 1. European commission . Harmonised standards for Medical devices. https://ec.europa.eu/growth/single‐market/european‐standards/harmonised‐standards/medical‐devices_en
- 2. Donawa . Gray: The breast implant scandal and European Medical Device Regulation. https://www.donawa.com/wp‐content/uploads/2019/06/Breast‐implants‐GMP‐Review‐Apr12.pdf
- 3. Majety RPD, Katru S, Veluchuri JP, Juturi RKR. New era in medical device regulations in the European Union. Pharmaceut Reg Affairs 2021; 10: 234. [Google Scholar]
- 4. EU Medical Device Regulation 2017/745 (MDR: https://eur‐lex.europa.eu/legal‐content/EN/TXT/PDF/?uri=CELEX:32017R0745
- 5. Thienpont E, Quaglio G, Karapiperis T, Kjaersgaard‐Andersen P. Guest editorial: new medical device regulation in Europe: a collaborative effort of stakeholders to improve patient safety. Clin Orthop Relat Res 2020; 478: 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Migliore A. On the new regulation of medical devices in Europe. Expert Rev Med Devices 2017; 14: 921–923. [DOI] [PubMed] [Google Scholar]
- 7. Martelli N, Eskenazy D, Déan C et al. New european regulation for medical devices: what is changing? Cardiovasc Intervent Radiol 2019; 42: 1272–1278. [DOI] [PubMed] [Google Scholar]
- 8. Freeman K, Dinnes J, Chuchu N et al. Algorithm based smartphone apps to assess risk of skin cancer in adults: systematic review of diagnostic accuracy studies. BMJ 2020; 10: m127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. and each medical device will have a Unique Device Identifier (UDI). https://ec.europa.eu/tools/eudamed/#/screen/home
- 10. Fink M, Akra B. Regulatory clearance: how are outcome measurements critical? Injury 2020; 51(Suppl 2): S67–S70. [DOI] [PubMed] [Google Scholar]
- 11. Herman A, Uter W, Rustemeyer T et al. EAACI Position statement: The need for EU legislation to require disclosure and labelling of the composition of medical devices. JEADV 2021;35:1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. MDCG 2020‐3, March 2020. https://ec.europa.eu/health/sites/default/files/md_sector/docs/md_MDCG_guidance_significant_changes_annexes_en.pdf
- 13. Malvehy J, Hauschild A, Curiel‐Lewandrowski C et al. Clinical performance of the Nevisense system in cutaneous melanoma detection: an international, multicentre, prospective and blinded clinical trial on efficacy and safety. Br J Dermatol 2014; 171: 1099–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.