

Abstract
Background and Aims
Poor response to ionizing radiation (IR) due to resistance remains a clinical challenge. Altered metabolism represents a defining characteristic of nearly all types of cancers. However, how radioresistance is linked to metabolic reprogramming remains elusive in hepatocellular carcinoma (HCC).
Approach and Results
Baseline radiation responsiveness of different HCC cells were identified and cells with acquired radio‐resistance were generated. By performing proteomics, metabolomics, metabolic flux, and other functional studies, we depicted a metabolic phenotype that mediates radiation resistance in HCC, whereby increased glucose flux leads to glucose addiction in radioresistant HCC cells and a corresponding increase in glycerophospholipids biosynthesis to enhance the levels of cardiolipin. Accumulation of cardiolipin dampens the effectiveness of IR by inhibiting cytochrome c release to initiate apoptosis. Mechanistically, mammalian target of rapamycin complex 1 (mTORC1) signaling‐mediated translational control of hypoxia inducible factor‐1α (HIF‐1α) and sterol regulatory element‐binding protein‐1 (SREBP1) remodels such metabolic cascade. Targeting mTORC1 or glucose to cardiolipin synthesis, in combination with IR, strongly diminishes tumor burden. Finally, activation of glucose metabolism predicts poor response to radiotherapy in cancer patients.
Conclusions
We demonstrate a link between radiation resistance and metabolic integration and suggest that metabolically dismantling the radioresistant features of tumors may provide potential combination approaches for radiotherapy in HCC.
Abbreviations
- 3Br‐PA
3‐bromopyruvate
- CL
cardiolipin
- CRLS1
CL synthase 1
- DHAP
dihydroxyacetone phosphate
- 4E‐BP
4E‐binding protein
- FBS
fetal bovine serum
- GPD1
glycerol‐3‐phosphate dehydrogenase 1
- GPL
glycerophospholipid
- GLUT1
glucose transporter 1
- G3P
glyceraldehyde‐3‐phosphate
- Glycerol‐3P
glycerol‐3‐phosphate
- HIF‐1α
hypoxia inducible factor‐1α
- HK2
hexokinase 2
- IR
ionizing radiation
- IR‐R
IR resistance
- LPIN2
lipin 2
- M‐HCl
minocycline hydrochloride
- mTORC1
mammalian target of rapamycin complex 1
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- PFKL
phosphofructokinase liver type
- PPARγ
peroxisome proliferator–activated receptor gamma
- RT
radiotherapy
- S6K
S6 kinase
- sh‐
short hairpin
- si‐
small interfering
- SCD
stearoyl‐CoA desaturase
- SREBP1
sterol regulatory element‐binding protein‐1
- TCA
tricarboxylic acid
- TPI1
triosephosphate isomerase 1
INTRODUCTION
HCC is one of the most prevalent malignancies, with dismal prognosis.[ 1 ] Radiotherapy (RT) is one of the few therapies with demonstrated clinical feasibility for patients with HCC, but its effectiveness has been constrained due to inherent or acquired resistance.[ 2 , 3 , 4 ] Recently, accumulating evidence has suggested a radiation resistance–abetting role of metabolic alterations in several cancer types.[ 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 ] Nevertheless, the liver acts as the metabolic hub of the body; whether and how reprogrammed metabolism regulates radiation responsiveness in HCC remains obscure.
To gain survival advantages, cancer cells flexibly co‐opt aberrant metabolic networks through a number of distinct as well as united avenues.[ 15 ] One of the central components of metabolic reprogramming in tumors is aerobic glycolysis (also known as the Warburg effect). Although glycolysis is less energy‐efficient in ATP production compared to aerobic respiration, the intermediary glucose metabolites can be funneled into anabolic pathways such as de novo lipid synthesis (lipogenesis), amino acid production and nucleotide synthesis, thus promoting proliferation and progression and suppressing apoptosis of tumor cells.[ 16 ] Hence, glucose often acts as a metabolic foundation of other metabolic pathways in tumors. Despite several studies reporting high rates of glycolysis imparting radioresistance, the precise fate of glucose and downstream metabolites and their roles in ionizing radiation (IR) responsiveness are yet to be fully explored.
IR mediates various forms of cancer cell death; among them, apoptosis is one of the main mechanisms of IR action. Damaged DNA and second messengers, such as ceramide and reactive oxygen species, can mediate IR‐induced apoptosis.[ 17 , 18 ] Recently, efficient DNA damage repair facilitated by activated nucleotide metabolism has gained much attention in the research field of radiation resistance.[ 5 , 8 , 19 ] However, how and what metabolites derived from altered metabolism are linked to IR‐induced apoptosis remain elusive. Here, by comparing the metabolic dependence of different HCC cells with intrinsic or acquired radioresistance, we delineate an integrated metabolic mechanism that inhibits apoptosis in driving radioresistance, using HCC as a model, and indicate a therapeutic strategy of disrupting metabolic dependencies in radiation sensitization.
MATERIALS AND METHODS
Cell culture and generation of sublines with acquired radioresistance
Cultured HCC cells were maintained in high‐glucose DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS) at 37°C in 5% CO2. The short tandem repeat reports of all cell lines used are provided in the Supporting Information. All cell experiments were performed within 10 passages after cell thawing.
MHCC97H and MHCC97L cells maintained in DMEM with 10% FBS were irradiated using a linear accelerator (Varian Clinac 23EX Linear Accelerator) in the Department of Radiation Oncology at Nanfang Hospital. Briefly, cells were plated at 70% confluence 24 h before IR. MHCC97H cells were subjected to 2 Gy IR daily for 25 fractions (taking weekends off), and MHCC97L cells were exposed to 8 Gy IR every 2 days for five fractions.[ 8 , 20 ] During the procedures, culture media were refed daily, and after 4 weeks of recovery time, cells were again exposed to 10 Gy IR one time (Figure 1B).
FIGURE 1.
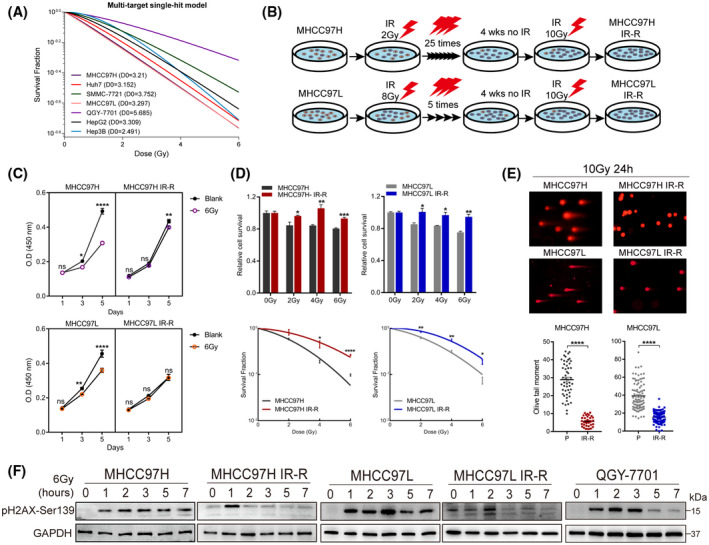
Response of HCC cell lines to IR and generation of radiation‐resistant sublines. (A) Surviving fractions of radiation colony formation of HCC cell lines exposed to the indicated doses of IR. (B) Schematic of the generation of sublines with acquired IR‐R. (C) Proliferation of control and IR‐R cells at 1, 3, or 5 days postexposure to 6 Gy IR by CCK8 assays. (D) Relative sensitivity of parental compared with IR‐R cells to increasing dose of IR as determined by MTT assays (72 h after IR) and colony formation assays. (E) Comet assays between parental and IR‐R cells postexposure to 10 Gy IR. (F) Western blots of pH2AX‐Ser139 levels in the indicated cell lines at 0, 1, 2, 3, 5, or 7 h following IR. Survival data were normalized to those of unirradiated control cells. Data are represented as mean ± SEM of at least three replicates. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Abbreviations: GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; OD, optical density; P, parental; ns, not significant; wks, weeks [Color figure can be viewed at wileyonlinelibrary.com]
Patient samples
Thirteen patients who underwent hepatectomy or ultrasonically guided liver biopsy within 1 year before RT from 2011 to 2019 were collected from the Department of Pathology at Nanfang Hospital. RT was given to biopsied lesions, metastases localized to the liver, or the intrahepatic vein in these patients. Post‐RT tumor samples from 5 patients were kindly provided by Professor Zhaochong Zeng at the Department of Radiation Oncology, Zhongshan Hospital, Fudan University, Shanghai.[ 21 ] Patients were given conventional (2 Gy per fraction) or hypofractionated (5‐8 Gy per fraction) IR, and total doses of gross tumor volume ranged 42–60 Gy. All patients received contrast‐enhanced CT or MRI before and after RT. Tumor response was assessed according to the Modified Response Evaluation Criteria in Solid Tumor.[ 22 ] Briefly, in assessments of tumors within 6 months after completion of RT, if reduction of tumor volume, loss of “fast‐in and fast‐out” phenomenon, or local tumor inactivation (no obvious enhancement in the arterial phase) was detected, these patients were defined as “response.” However, if tumor volume became larger or local recurrences or new lesions in the liver were observed, the patients were defined as “nonresponse.” Informed consent was obtained from all patients, and the procedures for human sample collection were approved by the Institute Research Medical Ethics Committee of Nanfang Hospital. Detailed clinical information is described in Table S3.
Xenograft Mouse studies
Athymic nude mice (BALB/c‐nu/nu, 4 weeks old) and C57BL6/N mice (4 weeks old), purchased from the animal center of Guangdong Province, were used for s.c. xenograft models. All mouse care and experiments were approved by the Institutional Animal Care and Use Committee of Nanfang Hospital, and all animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals. Radiation was given at a dose of 8 Gy for 2 or 3 consecutive days (8 Gy × 2 F or 8 Gy × 3 F). Only tumor locations were exposed to radiation, and the body was shielded with a lead plate. For drug combination studies, when tumors reaching a volume of ~100 mm3, mice were randomly distributed into the indicated groups. Rapamycin, 3‐bromopyruvate (3Br‐PA), ketoconazole, and MHY1485 were all purchased from Selleck; injected i.p. the day before IR; and maintained every 2 days at a concentration of 4 mg/kg, 5 mg/kg, 20 mg/kg, or 5 mg/kg, respectively. Glucose was dissolved in distilled water (5%) and administered for 2 days, followed by 1 day of normal drinking water. Tumor size was measured using digital vernier calipers, and tumor volumes were calculated by the formula 1/2 × Length × Width × Height.
Statistics
The results are presented as mean ± SEM. A two‐tailed, unpaired Student t test was used to compare the variables of two groups, and one‐way or two‐way ANOVA was performed for multigroup comparisons wherever appropriate. All statistical tests were performed using GraphPad Prism 7. Significant differences are indicated by *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Additional experimental procedures are provided in the Supporting Information.
RESULTS
Generation of radioresistant HCC cell lines
Colony formation assays with increasing doses of IR of HCC cell lines revealed variation in their radiosensitivity (Figure 1A; Figure S1A). Of note, QGY‐7701 was the most resistant line used. As the MHCC97H and MHCC97L cell lines were relatively sensitive to IR and weak in clonality, we then exposed them to conventional or hypofractionated IR, respectively, with the aim of generating sublines with acquired IR resistance (IR‐R) under different fraction schemes (Figure 1B). The resistance status of IR‐R sublines was determined by the Cell Counting Kit‐8 (CCK8) test, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) cytotoxicity assays, clonogenic survival, and neutral comet assays (Figure 1C–E; Figure S1B). Because low levels of phosphorylation of histone 2A (pH2AX‐Ser139) are indicative of effective DNA damage repair occurring in irradiated cells, we observed that the levels of pH2AX decreased faster with time in radioresistant cells than in MHCC97H and MHCC97L cells after exposure to IR (Figure 1F). Importantly, the increased radioresistance was found to be stable, with similar results obtained 10 passages after completion of the fractionated IR regimen.
Increased glucose metabolism fuels radioresistance of HCC cells
To explore the molecular mechanisms underlying resistance, we performed proteomics and observed that multiple metabolic pathways were significantly enriched in MHCC97H IR‐R cells compared to MHCC97H controls (Figure 2A; Figure S1C). Because cancer cells rely on nutrients that confer a survival advantage, we then cultured cells under glucose, glutamine, or serum deprivation conditions to examine the energy source of radioresistant cells; and nutrient‐rich conditions served as control. In agreement with recent reports indicating radio‐protective roles of glucose metabolism,[ 7 , 8 , 9 , 10 , 11 ] deprivation of glucose, but not glutamine or serum, promoted death of IR‐R cells (Figure 2B). As most cancer cells highly dependent on glucose are often forced into apoptosis when subjected to glucose blockade,[ 23 ] we observed that resistant cells demonstrated significant apoptosis and diminished survival under low‐glucose conditions (Figure 2C). Consistently, we found that IR‐R cells displayed higher glucose uptake than their parental controls, and the difference was larger in hypoxic conditions (Figure 2D). Expression of enzymes, like hexokinase 2 (HK2), glucose transporter 1 (GLUT1) solute carrier family 2 member 1 (SLC2A1), and phosphofructokinase liver type (PFKL), involved in glucose metabolism was also increased in IR‐R cells; and a combination of IR and low‐glucose culturing conditions synergistically led to much less clonogenic survival in IR‐R, but not in parental, cells (Figure 2E,F; Figure S1D). Of note, QGY‐7701 cells demonstrated consistent changes in glucose addiction (Figure S2). Together these findings establish that radioresistant HCC cells shared increased dependence on glucose.
FIGURE 2.
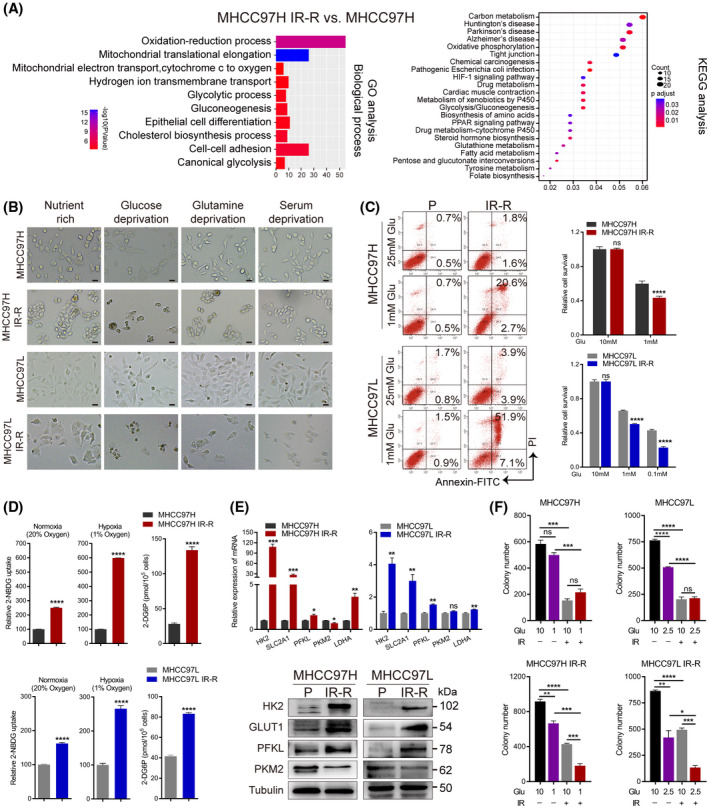
Increased glucose metabolism fuels radioresistance in HCC cells. (A) GO and KEGG enrichment analysis of the proteome profiles between MHCC97H parental and IR‐R cells. (B) Images of control and IR‐R cells cultured for 48 h in nutrient‐rich (10% FBS and 10 mM glucose), glucose‐deprived (10% FBS and 1 mM glucose), serum‐deprived (0.1% FBS and 10 mM glucose), or glutamine‐deprived (10% FBS and 10 mM glucose but no glutamine) DMEM. Scale bars, 100 μm. (C) Effect of glucose deprivation on apoptosis and cell survival as determined by annexin V and propidium iodide staining and MTT assays. Survival data were normalized to those of control cells cultured in 10 mM glucose. (D) Relative 2‐NBDG uptake (fluorescent glucose analogue) and 2‐deoxyglucose‐6‐phosphate content (reflects glucose analogue 2‐deoxyglucose uptake) in IR‐R and parental cells. Counts for 2‐NBDG uptake were normalized to the cell count in respective parental control cultured under normoxia or hypoxia. (E) Quantitative RT‐PCR and western blots of glycolytic genes. (F) Clonogenic survival of the indicated cells cultured in DMEM containing the indicated concentration of glucose with or without 6 Gy IR. Data are represented as mean ± SEM of at least three replicates. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Abbreviations: 2‐DG6P, 2‐deoxyglucose‐6‐phosphate; Glu, glucose; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; LDHA, lactate dehydrogenase A; 2‐NBDG, 2‐(N‐(7‐nitrobenz‐2‐oxa‐1,3‐diazol‐4‐yl) amino)‐2‐deoxy‐D‐glucose; ns, not significant; P, parental; PI, prodium iodide; PKM2, pyruvate kinase M2; SLC2A1, solute carrier family 2 member 1 [Color figure can be viewed at wileyonlinelibrary.com]
Enhanced glucose to cardiolipin synthesis contributes to repressed cytochrome c release in radioresistant HCC cells
To identify the metabolic fate of glucose in MHCC97H parental and IR‐R cells, we performed U‐13C glucose tracing with mass spectrometry. However, we found faster glucose flux into mainstream glycolytic metabolites and tricarboxylic acid (TCA) cycle–derived metabolites in MHCC97H cells than in IR‐R cells, while increased glucose carbon flux into branched metabolites, like serine and glycine, was observed in IR‐R cells. Combining our proteomic data and detection of higher HK2 activity but lower lactate production in IR‐R cells, we speculated whether the flexible assembly of glycolysis in IR‐R cells assigned intermediate metabolites to augment branched anabolism required combating radiation (Figure 3A; Figure S3A,B). We further performed untargeted metabolomics to identify the characteristically metabolic accumulation in IR‐R cells. Metabolic pathway and functional enrichment analyses demonstrated that glycerophospholipid (GPL) metabolism was among the most enriched pathways in MHCC97H IR‐R compared with control cells (Figure 3B). GPL synthesis efficiently links glucose and lipid metabolism. Its synthesis requires free fatty acids and the obligatory intermediary glycerol‐3‐phosphate, which can be generated by dehydrogenating dihydroxyacetone phosphate (DHAP), an intermediate metabolite of glycolysis. DHAP can either be converted to glyceraldehyde‐3‐phosphate (G3P) by triosephosphate isomerase 1 (TPI1) and return to the mainstream glycolysis, or be dehydrogenated by glycerol‐3‐phosphate dehydrogenase 1 (GPD1) to form glycerol‐3‐phosphate (Glycerol‐3P) and facilitate branched GPL synthesis.[ 24 ] Proteomic analyses showed that GPD1 expression was higher but TPI1 expression was lower in MHCC97H IR‐R cells than in parental controls, indicative of an activation of GPL synthesis (Figure 3A).
FIGURE 3.
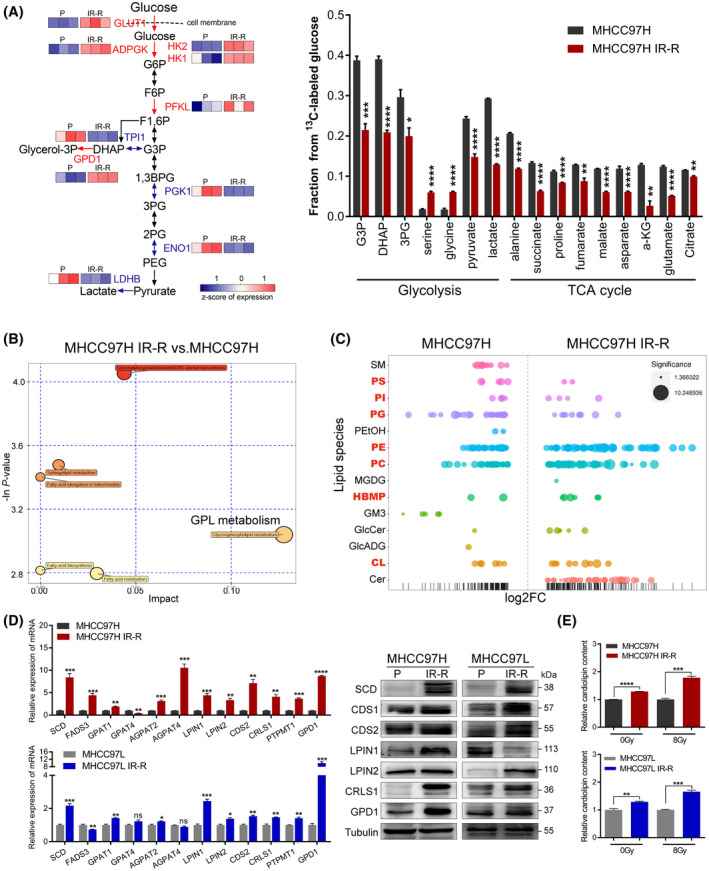
Increased glucose flux to CL anabolism in radioresistant HCC cells. (A) 13C‐labeled glycolytic and TCA metabolites as identified by gas chromatographic–mass spectrometric analysis and corresponding protein expression of indicated enzymes in glycolysis from proteomic analyses. Red indicates overexpressed enzymes in MHCC97H IR‐R cells, n = 3/group. (B) Metabolic pathway impact analysis of metabolites by Metaboanalyst 3.0 based on results of liquid chromatography–tandem mass spectrometry–based untargeted metabolomics, n = 3/group. (C) Relative expression levels of up‐regulated and down‐regulated lipid species displayed as log2 fold change in MHCC97H IR‐R compared to MHCC97H cells. Each spot represents a species of lipids, and the spot size indicates significance. Red indicates species of GPLs, n = 6/group. (D) Quantitative RT‐PCR and western blots of genes involved in CL synthesis. (E) Relative CL content in cells under basal conditions or at 24 h after 8 Gy IR as determined by ELISA. Data were calculated relative to respective untreated or 8 Gy‐treated controls. Data are represented as mean ± SEM of at least three replicates. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Abbreviations: ADPGK, ADP‐dependent glucokinase; AGPAT2/4, 1‐acylglycerol‐3‐phosphate O‐acyltransferase 2/4; a‐KG, alpha‐ketoglutarate; 1,3BPG, 1,3‐bisphosphoglyceric acid; CDS1/2, cytidine diphosphate–diacylglycerol synthase 1/2; Cer, ceramide; ENO1, enolase 1; FADS3, fatty acid desaturase 3; FC, fold change; F6P, fructose‐6‐phosphate; F1,6P, fructose‐1,6‐bisphosphate; GlcADG, glucuronosyldiacylglycerol; GlcCer, glucosylceramide; GM3, ganglioside monosialic acid 3; G6P, glucose‐6‐phosphate; GPAT1/4, glycerol‐3‐phosphate acyltransferase 1/4; HBMP, human bone morphogenetic protein ; LPIN1/2, lipin 1/2; MGDG, monogalactosyldiacylglycerol; P, parental; PC, polycarbonate; PE, phosphatidylethanolamine; PEG, polyethylene glycol; PEtOH, phosphatidylethanol; PG, phosphatidylglycerol; 2PG/3PG, 2/3‐phosphoglyceric acid; PGK1, phosphoglycerate kinase 1; PI, phosphatidylinositol; PS, phosphatidylserine; PTPMT1, protein tyrosine phosphatase mitochondrial 1; SM, sphingomyelin [Color figure can be viewed at wileyonlinelibrary.com]
Among kinds of GPLs, cardiolipins (CLs) are structurally unique phospholipids synthesized exclusively in mitochondria.[ 25 ] Early studies demonstrated that increased CL levels could strengthen the membrane binding of cytochrome c, thus rendering cells more likely to survive when challenged with apoptotic stimulus.[ 26 , 27 ] As cytochrome c–mediated apoptosis is one of the major death modes caused by IR, we asked whether CL synthesis was responsible for radioresistance. We first performed lipidomics to identify the global changes of the lipid profile in MHCC97H parental and IR‐R cells. The results revealed that lipids that markedly accumulated in IR‐R cells were mainly composed of GPLs. Importantly, the species of phosphatidylglycerol (PG; lipids consumed by CL synthase 1 [CRLS1], which catalyzes the final step of CL synthesis) decreased, while species of CLs increased in IR‐R cells compared to parental cells, implying enhancements of CL synthesis (Figure 3C; Figure S3C). Consistently, proteomic analyses, quantitative RT‐PCR, and western blot also identified up‐regulations of major genes, like stearoyl‐CoA desaturase (SCD), lipin 2 (LPIN2), CRLS1, etc., involved in CL synthesis in IR‐R and QGY‐7701 cells in comparison with the controls, along with higher cellular CL content and huge accumulation of lipid droplets in IR‐R cells (Figure 3D,E; Figures S2D and S3D,E).
Next, we studied the potential role of cytochrome c. We used minocycline hydrochloride (M‐HCl), a small molecule that inhibits cytochrome c release, to treat cells.[ 28 ] We detected an increase of cytoplasmic localized cytochrome c in parental cells after IR, and M‐HCl treatment significantly restored IR‐induced cell apoptosis and cytochrome c release in these cells, while no such alterations or restorative effects were observed in IR‐R and QGY‐7701 cells, indicating that cytochrome c release was blocked in radioresistant HCC cells to some extent (Figure 4A; Figure S3F). To further identify whether this block was CLs‐dependent, we generated small interfering (si‐) RNA against CRLS1 to pretreat resistant cells. The results revealed that repression of CRLS1 in tested cells could correspondingly promote cytochrome c release, decrease cellular CL content, and robustly increase cell death upon IR treatment (Figure 4B; Figure S4A,B). Collectively, these data demonstrate that integration of glucose and CL anabolism mediates radiation resistance by modulating cytochrome c extrusion in HCC cells.
FIGURE 4.
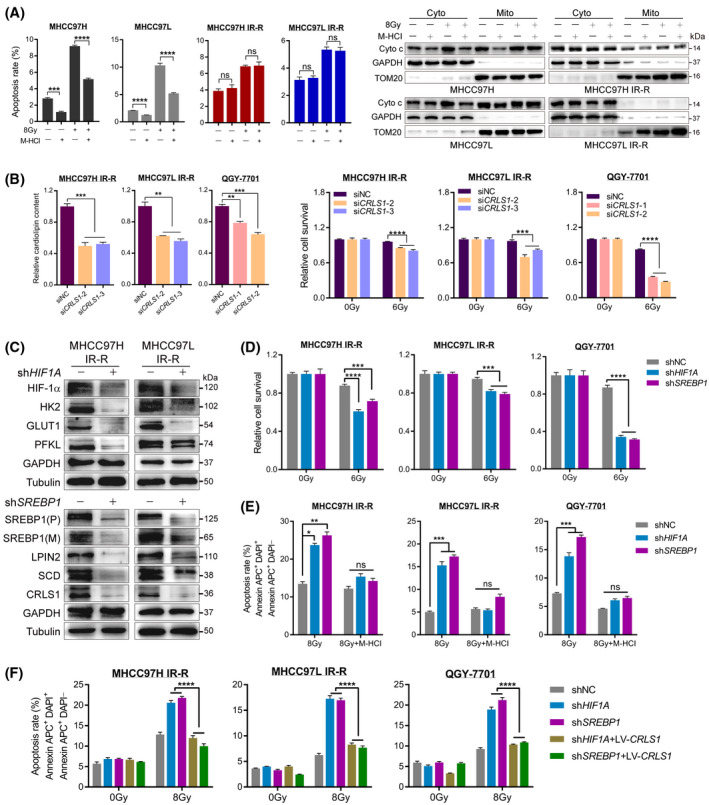
HIF‐1α and SREBP1 mediating increased glucose to CL anabolism represses cytochrome c release in radioresistant HCC cells. (A) Restorative effect of M‐HCl (20 μM, 48 h) on IR‐induced apoptosis and cytochrome c release in the indicated cell lines. Glyceraldehyde 3‐phosphate dehydrogenase and translocase of outer mitochondrial membrane 20 were loaded as cytoplasmic and mitochondrial markers, respectively. (B) Relative CL content (left) and cell survival determined by MTT assays (right) of radioresistant cells transiently expressing either scrambled siRNA or siRNA against CRLS1 following IR treatment. (C) Western blot of downstream targets in IR‐R cells upon HIF1A or SREBP1 knockdown using shRNA. (D) Cell viability determined by MTT assays in resistant cells upon HIF1A or SREBP1 knockdown. (E,F) Restorative effect of M‐HCl (E) or CRLS1 overexpression (F) on IR‐induced apoptosis in radioresistant cell lines with HIF1A or SREBP1 knockdown. Survival data were normalized to those of unirradiated control cells. Data are represented as mean ± SEM of at least three replicates. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Abbreviations: Cyto, cytoplasmic; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; DAPI, 4‐6‐diamidino‐2‐phenylindole; (M), mature; Mito, mitochondrial; ns, not significant; (P), precursor; TOM20, translocase of outer mitochondrial membrane 20 [Color figure can be viewed at wileyonlinelibrary.com]
Mammalian target of rapamycin complex 1 mediates up‐regulation of hypoxia‐inducible factor 1α and sterol regulatory element–binding protein 1, which are the metabolic master regulators in radioresistant HCC cells
The coordinated metabolic network in response to IR in HCC cells raised an intriguing possibility of a well‐integrated mechanism of action. Because our previous data showed that hypoxia further widened the difference of glucose absorption between IR‐R and parental cells, along with hypoxia inducible factor‐1 (HIF‐1) signaling being enriched in IR‐R cells, we wondered whether the glucose addiction of resistant cells was HIF‐1α‐dependent. Meanwhile, when transcriptionally regulating glucose anabolism, HIF‐1α is particularly indicative of mammalian target of rapamycin complex 1 (mTORC1) signaling activation, and mTORC1 signaling is essential to stimulate sterol regulatory element–binding protein 1 (SREBP1) or peroxisome proliferator–activated receptor‐gamma (PPARγ) to regulate lipogenesis.[ 29 ] Interestingly, we found that basal protein expression of HIF‐1α, SREBP1, and primary downstream effectors of mTORC1 were significantly up‐regulated, while rictor (indicative of mTORC2 activation) and PPARγ were down‐regulated in radioresistant cells compared with their respective controls (Figure S4C).
To investigate their roles, we first generated IR‐R and QGY‐7701 cells with stable knockdown of HIF‐1α or SREBP1. The results revealed that resistant cells with HIF‐1α or SREBP1 knockdown suppressed downstream metabolic targets involved in glucose metabolism or CL synthesis, respectively (Figure 4C; Figure S4D,E). Functionally, repression of either HIF‐1α or SREBP1 robustly diminished radioresistance, decreased cellular CL content, and accelerated IR‐induced cytochrome c release of resistant HCC cells (Figure 4D; Figure S4F,G). However, treatment with M‐HCl to inhibit cytochrome c liberation or ectopically overexpressed CRLS1 in tested cells to restore CL pools efficiently attenuated apoptosis that had been induced by exposure to IR in radioresistant HCC cells with either HIF‐1α or SREBP1 knockdown (Figure 4E,F; Figure S5).
To further examine the functions of mTORC1 signaling, we first used two inhibitors, BEZ235 and rapamycin, to pharmacologically inhibit mTORC1 activation. The results revealed that mTORC1 inhibition significantly reduced downstream metabolic targets and phenotypes in resistant cells, along with accelerated cytochrome c release upon IR treatment (Figure S6). Next, we examined if mTORC1 inhibition could abrogate radiation resistance. The results showed that the combination of rapamycin and IR sharply enhanced apoptosis in resistant cells, while no synergy was observed in MHCC97H and MHCC97L cells (Figure 5A; Figure S7A). Additionally, M‐HCl treatment or CRLS1 overexpression in radioresistant cells robustly restored the elimination of clonogenic survival induced by rapamycin plus IR (Figure 5B,C; Figure S7B). These data indicate a role of mTORC1 signaling in mediating IR‐R by modulating HIF‐1α, SREBP1, and their downstream targets in HCC cells.
FIGURE 5.
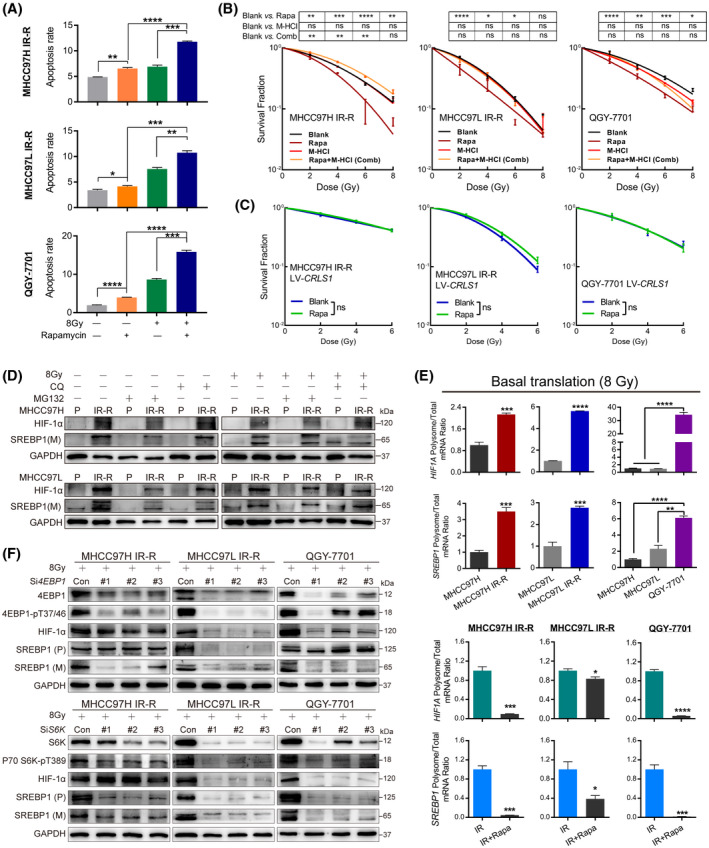
mTORC1‐mediated translation of HIF‐1α and SREBP1 drives IR‐R in HCC cells. (A) Synergy of rapamycin (2 μM, 48 h) with IR in radioresistant cells by apoptosis measurements. (B,C) Restorative effect of M‐HCl (10 μM) (B) or CRLS1 overexpression (C) on clonogenic survival of radioresistant cell lines treated with rapamycin (1 μM) and exposed to the indicated dose of IR. Survival data were normalized to respective unirradiated controls. (D) MG132 (20 μM, 24 h) or chloroquine (20 μM, 24 h) treatment on indicated cells with or without 8 Gy IR. (E) The ratio of HIF‐1α and SREBP1 mRNA loaded on polysomes to their total mRNA levels by quantitative RT‐PCR analysis in IR or IR plus rapamycin–treated conditions (2 μM, 24 h). Each value was normalized to tubulin loaded on the polysome and total tubulin expression. (F) Protein levels of HIF‐1α and SREBP1 in radioresistant cells with 4E‐BP1 or S6K knocked down by siRNA with 8 Gy IR. Data are represented as mean ± SEM of at least three replicates. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Abbreviations: Con, control; CQ, chloroquine; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; (M), mature; ns, not significant; P, parental; (P), precursor; Rapa, rapamycin [Color figure can be viewed at wileyonlinelibrary.com]
mTORC1 mediates radiation resistance of HCC cells by enhancing translation of HIF‐1α and SREBP1
Next, we sought to determine the mechanism of mTORC1 signaling in regulating HIF‐1α and SREBP1. We initially examined mRNA levels but failed to observe a significant difference, suggesting that mTORC1 functions at the posttranscriptional level (Figure S8A). As mTORC1 could regulate them by either increasing synthesis or suppressing degradation, to distinguish between these possibilities, we monitored HIF‐1α and SREBP1 half‐life in these cells but found that their decay rates were similar and that neither the proteasome inhibitor MG132 nor the lysosome inhibitor chloroquine reversed the protein discrepancy, suggesting a degradation‐independent mechanism (Figure 5D; Figure S8B,C). We next directly tested the translation of HIF‐1α and SREBP1 by testing their mRNA fractions in heavy polysome fractions, which indicates efficient translation.[ 30 ] In IR‐treated cells, we observed increased amounts of both HIF‐1α and SREBP1 mRNA loaded on the polysomes of radioresistant cells versus control cells, whereas rapamycin treatment significantly reduced polysome‐associated HIF‐1α and SREBP1 mRNA, while their total mRNA remained unaffected (Figure 5E; Figure S8D). These results suggest that mTORC1 signaling promoted the translation of both HIF‐1α and SREBP1 in radioresistant HCC cells.
Because mTORC1 stimulates translation by phosphorylating downstream targets including 4E‐binding proteins (4E‐BPs) and ribosomal protein S6 kinases (S6Ks),[ 31 ] we next generated siRNA against 4E‐BP1 and S6K to pretreat resistant HCC cells. Under normal culturing conditions, we found that knockdown of 4E‐BP1 only reduced protein expression of HIF‐1α, while S6K repression only led to SREBP1 suppression. However, when rechallenging these cells with IR, the results were far more complicated. In MHCC97H IR‐R cells, repression of 4E‐BP1 resulted in reduction of both HIF‐1α and SREBP1, while repression of S6K only suppressed SREBP1. In MHCC97L IR‐R and QGY‐7701 cells, knockdown of either 4E‐BP1 or S6K led to down‐regulation of both HIF‐1α and SREBP1 (Figure 5F; Figure S9A). Additionally, overexpression of HIF‐1α or SREBP1 efficiently restored downstream metabolic targets repressed by both 4E‐BP1 and S6K knockdown in these cells (Figure S9B). These data indicated that mTORC1 functions differently in regulating translation of HIF‐1α and SREBP1 between normal and IR‐treated conditions in radioresistant HCC cells.
Dismantling mTORC1‐mediated glucose to CL metabolism sensitizes tumors to IR in vivo
To assess whether the in vitro findings can be recapitulated in vivo, we first implanted HCC cells s.c. in athymic nude mice and randomly subjected them to control or IR (8 Gy × 2 F). The results revealed that radiation significantly diminished tumor volume in MHCC97H and MHCC97L tumor‐bearing mice, while a delay and slight reduction of tumor volume were observed in the MHCC97L IR‐R tumor‐bearing group (Figure 6A). Consistently, IR markedly injured cells, activated proapoptotic signaling, and repressed the proliferative index and mTORC1 signal in radiosensitive tumors; nevertheless, slight cell atypia and converse regulations of these targets were observed in MHCC97L IR‐R tumors compared to their respective blank controls (Figure 6B; Figure S10A).
FIGURE 6.
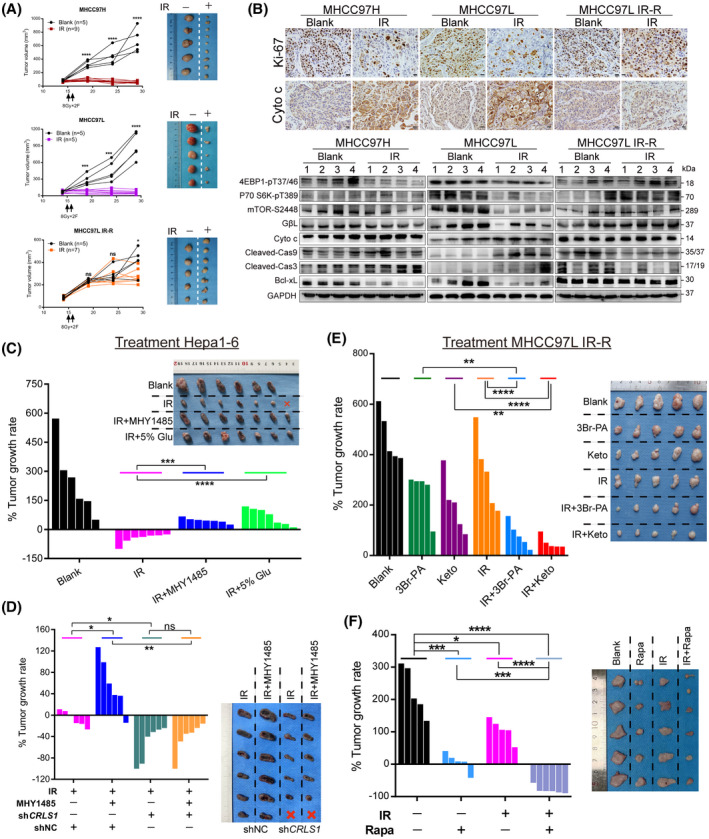
mTORC1 activation–mediated glucose to CL anabolism determines radiation sensitivity in vivo. (A,B) Tumor growth curves, tumor images, representative immunohistochemical staining, and western blots of s.c. xenograft models in nude mice with indicated cells and treatments. (C) Response of Hepa1‐6 xenografts in C57 mice treated with control, IR (8 Gy × 3 F), IR with MHY1485 (5 mg/kg), or IR with high‐glucose drinking (5%). (D) Response of Hepa1‐6 short hairpin RNA control (shNC) and shCRLS1 xenografts in C57 mice treated with IR (8 Gy × 2 F) or IR with MHY1485 (5 mg/kg). (E) Effect of cutting off glucose flux on radiation responsiveness in s.c. implanted MHCC97L IR‐R nude mice subjected to treatments with control, 3Br‐PA (5 mg/kg), ketoconazole (20 mg/kg), IR (8 Gy × 2 F), IR with 3Br‐PA, or IR with ketoconazole. (F) Response of MHCC97L IR‐R xenografts to treatment with control, rapamycin (4 mg/kg), IR (8 Gy × 2 F), or combination. Waterfall plot showing the percentage of tumor growth rate per individual mouse and tumor images upon necropsy presented in (C–F). Data are represented as mean ± SEM. Scale bars, 20 μm. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Abbreviations: Cas3/9, caspase 3/9; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; Glu, glucose; Keto, ketoconazole; ns, not significant; Rapa, rapamycin [Color figure can be viewed at wileyonlinelibrary.com]
Then, we examined whether a high‐glucose condition or mTORC1 activation would dampen IR effectiveness. We established s.c. tumors in C57BL/6 mice using Hepa1‐6 cells (murine HCC cells). Once the size of xenografts reached approximately 100–150 mm3, the mice were separated into the following four subgroups: (1) vehicle (PBS), (2) IR treatment (8 Gy × 3 F), (3) IR + drinking water with high glucose (5%), (4) IR + MHY1485 (mTORC1 agonist, 5 mg/kg). Compared with the blank control group, mice with engrafted tumors treated with IR had significant tumor shrinkage; however, either high‐glucose drinking or mTORC1 activation strongly counteracted RT efficacy (Figure 6C). To test if the radio‐protective effects of MHY1485 reversed when CL metabolism was inhibited, we performed knockdown of CRLS1 in Hepa1‐6 cells and established xenograft models with C57BL/6 mice, which were divided into IR (8 Gy × 2 F) and IR+MHY1485 groups. Compared with the corresponding short hairpin RNA control group, CRLS1 knockdown significantly sensitized tumors to IR and efficiently dampened the effects of MHY1485, along with decreased CL content in these tumors (Figure 6D; Figure S10C,D).
Leveraging this metabolic mechanism, we next sought to determine whether cutting off glucose flux or repressing mTORC1 activation could overcome IR‐R in vivo. To interrupt glucose flow, we used 3Br‐PA and ketoconazole[ 32 , 33 ] to target HK2, the first rate‐limiting enzyme of glycolysis, which was most significantly up‐regulated in radioresistant HCC cells. We s.c. inoculated MHCC97L IR‐R cells to nude mice. Once xenografts were about 100 mm3 in size, they were randomized to receive indicative treatments (Figure S10E). The results revealed that although treatment with either 3Br‐PA or ketoconazole could reduce tumor volume and cell proliferation compared to the control group, both combination therapies showed significantly synergistic powers in tumor suppression and apoptosis induction compared to the other subgroups (Figure 6E; Figure S10F–H). Next, we further examined the effects of rapamycin on IR response in the same MHCC97L IR‐R xenograft models. The results also showed that although rapamycin alone robustly repressed tumor growth, combination therapy synergistically shrunk the tumor volume, inhibited metabolic activation, and stimulated apoptosis in tumors (Figure 6F; Figure S11A,B). The single or combined treatments were all well tolerated (Supporting Figures S10B,F and S11C). Together, these data suggest that dismantling mTORC1‐mediated glucose to CL metabolism may serve as an adjuvant approach for RT in HCC.
Activated metabolism correlates with the response of cancer patients to RT
We further investigated the clinical relevance of metabolic activation to RT in patients with HCC. We obtained 13 pre‐RT and 5 post‐RT HCC samples to perform immunohistochemical staining. The results revealed that high expression of metabolic targets was robustly associated with poor response to RT, while samples from responders tended to show significant reduction in protein levels of these targets in tumor cells (Figure 7A–C). Moreover, using The Cancer Genome Atlas public database, we found that patients with combination of high expression of SLC2A1, HK2, and PFKL tended to have inferior overall survival after RT in several other cancer types (Figure S11D). However, expression of genes involved in CL synthesis failed to predict prognosis in these tumors (data not shown). Together, these data suggest that stimulation of glucose metabolism likely presents as a prevalent characteristic among patients with cancer and a dismal RT response, while activated CL biosynthesis might be a unique feature of HCC with radioresistance.
FIGURE 7.
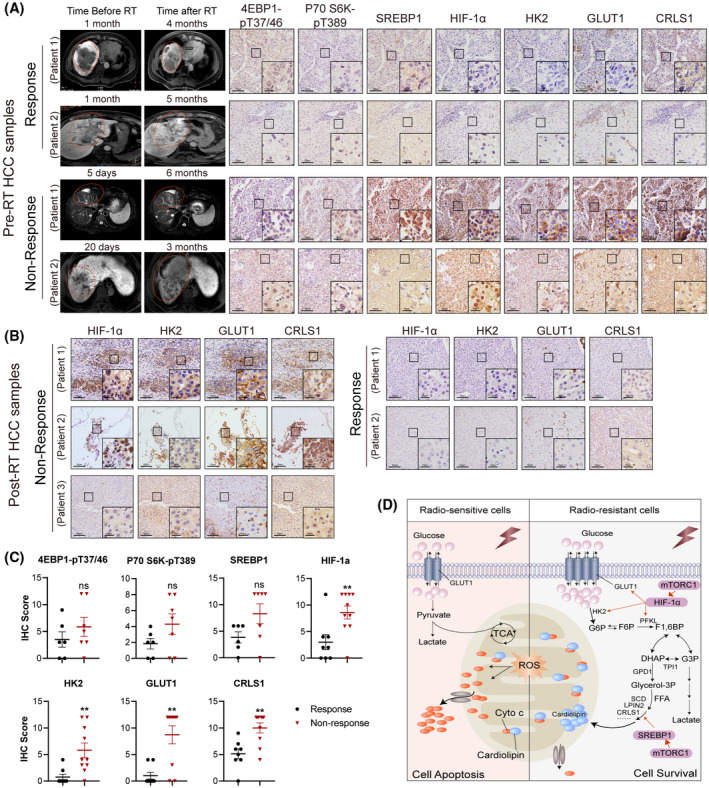
Activated metabolism correlates with response of patients with HCC to RT. (A) Representative images of indicated immunohistochemical staining of tumor samples from patients with HCC who received surgical resection or liver biopsy before RT. Matched MRIs from corresponding patients before and after RT are displayed. (B) Representative immunohistochemical staining of indicated targets in five tumor samples from patients with HCC who received surgical resection after RT. Scale bars, 50 μm/10 μm (inset). (C) Statistics of indicated targets between RT response and nonresponse in patients with HCC, as related to (A,B). (D) Working model depicting the mechanisms that drive glucose and CL anabolism underlying radioresistance of HCC cells. Abbreviations: FFA, free fatty acid; F6P, fructose‐6‐phosphate; F1,6P, fructose‐1,6‐bisphosphate; G6P, glucose‐6‐phosphate; IHC, immunohistochemistry; ROS, reactive oxygen species [Color figure can be viewed at wileyonlinelibrary.com]
DISCUSSION
RT is increasingly used in advanced HCC and has been reported to confer survival benefits[ 2 , 3 , 4 ]; nevertheless, radioresistance has been a major hurdle. Several mechanisms of radiation resistance encompassing different molecular pathways in HCC have been suggested,[ 10 , 34 , 35 , 36 , 37 ] but most previous studies mainly addressed one single gene in mediating resistance, ignored either intrinsic or acquired resistance to RT, and did not illustrate a general mechanism shared by different resistant backgrounds. To span beyond these restrictions, in the current study, we initially sought to characterize the inherent radiosensitivity of different HCC cells and generated sublines with acquired radioresistance through conventional fractions and hypofractions, with the aim of better delineating the mechanism underlying radioresistance encountered in clinical practice.
Several metabolic alterations have been reported to play roles in RT in cancers recently, such as highly activated glycolysis, enhanced lipogenesis or fatty acid β‐oxidation, and increased nucleotide metabolism.[ 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 ] Although therapy‐induced metabolic modifications of both cancer cells and their microenvironments have recently been the focus of attention, the influence of metabolic reprogramming on radiation response in HCC remains unclear. Here, we describe a survival mechanism triggered by IR in HCC cells that substantially contributes to both acquired and intrinsic radioresistance. In this context, radioresistant HCC cells were highly addicted to glucose, while glucose did not feed into mainstream glycolysis to produce pyruvate or lactate but rather fed into branched pathways for CL anabolism.
CLs are structurally unique GPLs synthesized exclusively in mitochondria, which rely on a series of metabolic enzymes, including 1‐acylglycerol‐3‐phosphate O‐acyltransferase, cytidine diphosphate–diacylglycerol synthase, phosphatidylglycerophosphate synthase 1, protein tyrosine phosphatase mitochondrial 1 (PTPMT1), and CRLS1.[ 25 ] Inhibition of CL biosynthesis was reported to be able to increase cytoplasmic release of cytochrome c, thus enhancing cytochrome c–mediated caspase activation and cell apoptosis.[ 26 , 38 , 39 , 40 ] Here, increased CL accumulation in radioresistant HCC cells also retarded the liberation of cytochrome c, thereby inhibiting IR‐induced apoptosis to acquire radiation resistance and a survival advantage. Ectopically manipulating CRLS1 expression efficiently altered IR responsiveness of HCC cells. Echoing our present study, the crucial roles of CLs in HCC have been unveiled recently. It has been reported that mTORC2 stimulated glucosylceramide and that CL synthesis robustly promotes HCC development.[ 25 ] Bao et al. identified that PTPMT1‐driven CL synthesis leads to hypoxic survival of HCC cells.[ 41 ] What is more, CRLS1 attaches PG to diacylglyceride, producing immature CL that is then remodeled in a series of reactions to generate mature CL species, and an elevated level of immature CL species, but not mature species, was reported to promote the progression of NAFLD to HCC.[ 42 ] Considering the direct survival advantages of CLs endowing cells, dissecting additional and mechanistic roles of CLs in HCC and exploring specific inhibitors targeting CL synthesis will be valuable areas of future investigation.
Hypoxia, which could stabilize HIF‐1α, has been linked with radioresistance in several cancer types. The main focus was on HIF‐1‐dependent regulation of glycolysis and the pentose phosphate pathway, which could increase the antioxidant capacity of tumors.[ 43 ] However, no studies have directly determined the control of combined HIF‐1α and SREBP1 on sequential glucose and lipid metabolism in radiation responsiveness. Here, our data demonstrated that radioresistant HCC cells had elevated expression of HIF‐1α as well as SREBP1, which serve in controlling glucose metabolism and CL synthesis, respectively. Coactivation of the two built a serial metabolic network to increase glucose flux into the CL biosynthetic pathway, and silencing either of them strongly sensitized radioresistant cells to IR. Tumors display activation of HIF‐1α under normoxia, often indicating hyperactivation of mTORC1 signaling[ 44 ]; and HIF‐1α and SREBP1 are two major downstream commanders of mTORC1 signaling in controlling anabolism.[ 45 ] We also observed a central role of mTORC1 activation in radioresistant HCC cells. Previous work identified that the mechanism by which mTORC1 regulates HIF‐1α and SREBP1 appears to be quite different. mTORC1 signaling could increase the transcription or translation of HIF‐1α,[ 29 , 46 ] while it activates SREBP1 mainly by transcriptional programming or promoting its nuclear translocation.[ 47 ] Interestingly, in our work, we found that mTORC1 functioned differently in enhancing translation of HIF‐1α and SREBP1 between normal and IR‐treated conditions through either 4E‐BP1 or S6K in radioresistant HCC cells. These findings provided additional insights into the pivotal contributions of mTORC1 to IR response in HCC cells, and it will be interesting to study this in future explorations.
Additionally, our data showed that high glucose or mTORC1 activation exhibited significant radiation‐abating effects in vivo. Cutting off glucose flux by targeting HK2, repressing CRLS1 expression, or impeding mTORC1 activation exerted a strong radiosensitizing effect in xenograft models. Because the HK2 inhibitor ketoconazole is Food and Drug Administration–approved for other indications and mTORC1 inhibitors, like rapamycin and everolimus, are currently in clinical trials for HCC,( 48 ) the barrier to clinical translation is relatively low. What is more, consistent with these preclinical results, stimulation of glucose to CL metabolism likely contributed to inferior RT response in patients with HCC, and glucose metabolism likely acted as a common feature among patients with cancer who have a dismal RT response. Therefore, the combination strategies documented here likely also have implications for other cancer types that depend on glucose for resisting IR.
In summary, our study deciphers an integration of glucose and CL anabolism, regulated by mTORC1/HIF‐1α/SREBP1 signaling, that mediates radiation resistance through inhibiting cytochrome c release in HCC cells with acquired as well as intrinsic radioresistance (Figure 7D). With the gradual blossoming of RT in HCC treatment, understanding the metabolic vulnerability in such a metabolic tumor and elaborating the underlying mechanism may help to determine more effective multimodality treatments for patients with HCC.
CONFLICT OF INTEREST
Nothing to disclose.
AUTHOR CONTRIBUTIONS
Yuan Fang, Li Liang, Yi Ding, and Dehua Wu conceived and designed the experiments. Yuan Fang, Yizhi Zhan, Yuwen Xie, Yaowei Zhang, and Keli Chen performed the experiments. Yizhi Zhan and Yuwen Xie analyzed the data. Shisuo Du, Yuhan Chen, Zhaochong Zeng, Yongjia Wang, Li Liang, Yi Ding, and Dehua Wu contributed radiation therapy equipment, materials, tumor tissues, and analysis tools. Yuan Fang, Li Liang, Yi Ding, and Dehua Wu wrote the paper.
Supporting information
Fang Y, Zhan Y, Xie Y, Du S, Chen Y, Zeng Z, et al. Integration of glucose and cardiolipin anabolism confers radiation resistance of HCC. Hepatology. 2022;75:1386–1401. 10.1002/hep.32177
Yuan Fang, Yizhi Zhan, and Yuwen Xie contributed equally.
Funding information
Supported by the National Natural Science Foundation of China (82073394, 82073342, 81872470, 81872399); the Guangdong Natural Science Funds for Distinguished Young Scholar (2015A030306015); the Pearl River Nova Program of Guangzhou, Guangdong Province (2014J2200015); the Excellent Young Teachers Program of Higher Education of Guangdong Province (YQ2015036); the Guangdong Program for Support of Top‐notch Young Professionals (2015TQ01R279); the Guangdong Provincial Regional Joint Fund‐Youth Fund Project (2020A1515110006); and the President Foundation of Nanfang Hospital, Southern Medical University (2020B012).
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2. Rajyaguru DJ, Borgert AJ, Smith AL, Thomes RM, Conway PD, Halfdanarson TR, et al. Radiofrequency ablation versus stereotactic body radiotherapy for localized hepatocellular carcinoma in nonsurgically managed patients: analysis of the national cancer database. J Clin Oncol. 2018;36:600–8. [DOI] [PubMed] [Google Scholar]
- 3. Wahl DR, Stenmark MH, Tao Y, Pollom EL, Caoili EM, Lawrence TS, et al. Outcomes after stereotactic body radiotherapy or radiofrequency ablation for hepatocellular carcinoma. J Clin Oncol. 2016;34:452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wei X, Jiang Y, Zhang X, Feng S, Zhou B, Ye X, et al. Neoadjuvant three‐dimensional conformal radiotherapy for resectable hepatocellular carcinoma with portal vein tumor thrombus: a randomized, open‐label, multicenter controlled study. J Clin Oncol. 2019;37:2141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou W, Yao Y, Scott AJ, Wilder‐Romans K, Dresser JJ, Werner CK, et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat Commun. 2020;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tan Z, Xiao L, Tang M, Bai F, Li J, Li L, et al. Targeting CPT1A‐mediated fatty acid oxidation sensitizes nasopharyngeal carcinoma to radiation therapy. Theranostics. 2018;8:2329–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rashmi R, Huang X, Floberg JM, Elhammali AE, McCormick ML, Patti GJ, et al. Radioresistant cervical cancers are sensitive to inhibition of glycolysis and redox metabolism. Cancer Res. 2018;78:1392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gunda V, Souchek J, Abrego J, Shukla SK, Goode GD, Vernucci E, et al. MUC1‐mediated metabolic alterations regulate response to radiotherapy in pancreatic cancer. Clin Cancer Res. 2017;23:5881–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen H, Hau E, Joshi S, Dilda PJ, McDonald KL. Sensitization of glioblastoma cells to irradiation by modulating the glucose metabolism. Mol Cancer Ther. 2015;14:1794–804. [DOI] [PubMed] [Google Scholar]
- 10. Yu L, Sun Y, Li J, Wang Y, Zhu Y, Shi Y, et al. Silencing the Girdin gene enhances radio‐sensitivity of hepatocellular carcinoma via suppression of glycolytic metabolism. J Exp Clin Cancer Res. 2017;36:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiao L, Hu Z‐Y, Dong X, Tan Z, Li W, Tang M, et al. Targeting Epstein‐Barr virus oncoprotein LMP1‐mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene. 2014;33:4568–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu Y, Murray‐Stewart T, Casero RAJ, Kagiampakis I, Jin L, Zhang J, et al. Targeting hexokinase 2 inhibition promotes radiosensitization in HPV16 E7‐induced cervical cancer and suppresses tumor growth. Int J Oncol. 2017;50:2011–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Singh D, Banerji AK, Dwarakanath BS, Tripathi RP, Gupta JP, Mathew TL, et al. Optimizing cancer radiotherapy with 2‐deoxy‐d‐glucose dose escalation studies in patients with glioblastoma multiforme. Strahlentherapie und Onkologie. 2005;181:507–14. [DOI] [PubMed] [Google Scholar]
- 14. Souchek JJ, Baine MJ, Lin C, Rachagani S, Gupta S, Kaur S, et al. Unbiased analysis of pancreatic cancer radiation resistance reveals cholesterol biosynthesis as a novel target for radiosensitisation. Br J Cancer. 2014;111:1139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 17. Kim BM, Hong Y, Lee S, Liu P, Lim JH, Lee YH, et al. Therapeutic implications for overcoming radiation resistance in cancer therapy. Int J Mol Sci. 2015;16:26880–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ferlini C, D’Amelio R, Scambia G. Apoptosis induced by ionizing radiation. The biological basis of radiosensitivity. Subcell Biochem. 2002;36:171–86. [PubMed] [Google Scholar]
- 19. Fu S, Li Z, Xiao L, Hu W, Zhang LU, Xie B, et al. Glutamine synthetase promotes radiation resistance via facilitating nucleotide metabolism and subsequent DNA damage repair. Cell Rep. 2019;28:1136–43.e4. [DOI] [PubMed] [Google Scholar]
- 20. Cheng JC, Bai A, Beckham TH, Marrison ST, Yount CL, Young K, et al. Radiation‐induced acid ceramidase confers prostate cancer resistance and tumor relapse. J Clin Invest. 2013;123:4344–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Du S, Chen G, Yuan B, Hu Y, Yang P, Chen Y, et al. DNA sensing and associated type 1 interferon signaling contributes to progression of radiation‐induced liver injury. Cell Mol Immunol. 2020:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 23. Flaveny C, Griffett K, El‐Gendy B‐D, Kazantzis M, Sengupta M, Amelio A, et al. Broad anti‐tumor activity of a small molecule that selectively targets the Warburg effect and lipogenesis. Cancer Cell. 2015;28:42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krishnan J, Suter M, Windak R, Krebs T, Felley A, Montessuit C, et al. Activation of a HIF1alpha–PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009;9:512–24. [DOI] [PubMed] [Google Scholar]
- 25. Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, Moes S, et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell. 2017;32:807–23.e12. [DOI] [PubMed] [Google Scholar]
- 26. Belikova NA, Jiang J, Tyurina YY, Zhao Q, Epperly MW, Greenberger J, et al. Cardiolipin‐specific peroxidase reactions of cytochrome c in mitochondria during irradiation‐induced apoptosis. Int J Radiat Oncol Biol Phys. 2007;69:176–86. [DOI] [PubMed] [Google Scholar]
- 27. Orrenius S, Zhivotovsky B. Cardiolipin oxidation sets cytochrome c free. Nat Chem Biol. 2005;1:188–9. [DOI] [PubMed] [Google Scholar]
- 28. Zhu S, Stavrovskaya IG, Drozda M, Kim BYS, Ona V, Li M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–8. [DOI] [PubMed] [Google Scholar]
- 29. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Su X, Yu Y, Zhong Y, Giannopoulou EG, Hu X, Liu H, et al. Interferon‐gamma regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat Immunol. 2015;16:838–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morita M, Gravel S‐P, Chénard V, Sikström K, Zheng L, Alain T, et al. MTORC1 controls mitochondrial activity and biogenesis through 4E‐BP‐dependent translational regulation. Cell Metab. 2013;18:698–711. [DOI] [PubMed] [Google Scholar]
- 32. Agnihotri S, Mansouri S, Burrell K, Li M, Mamatjan Y, Liu J, et al. Ketoconazole and posaconazole selectively target HK2‐expressing glioblastoma cells. Clin Cancer Res. 2019;25:844–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lis P, Dyląg M, Niedźwiecka K, Ko Y, Pedersen P, Goffeau A, et al. The HK2 dependent “Warburg effect” and mitochondrial oxidative phosphorylation in cancer: Targets for effective therapy with 3‐bromopyruvate. Molecules. 2016;21:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shao Y, Song X, Jiang W, Chen Y, Ning Z, Gu W, et al. MicroRNA‐621 acts as a tumor radiosensitizer by directly targeting SETDB1 in hepatocellular carcinoma. Mol Ther. 2019;27:355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Y, Zheng L, Ding Y, Li Q, Wang R, Liu T, et al. MiR‐20a induces cell radioresistance by activating the PTEN/PI3K/Akt signaling pathway in hepatocellular carcinoma. Int J Radiat Oncol Biol Phys. 2015;92:1132–40. [DOI] [PubMed] [Google Scholar]
- 36. Bamodu OA, Chang H‐L, Ong J‐R, Lee W‐H, Yeh C‐T, Tsai J‐T. Elevated PDK1 expression drives PI3K/AKT/MTOR signaling promotes radiation‐resistant and dedifferentiated phenotype of HCC. Cells. 2020;9(3):746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sun J, Zhu Z, Li W, Shen M, Cao C, Sun Q, et al. UBE2T‐regulated H2AX monoubiquitination induces hepatocellular carcinoma radioresistance by facilitating CHK1 activation. J Exp Clin Cancer Res. 2020;39:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS Lett. 2000;466:323–6. [DOI] [PubMed] [Google Scholar]
- 39. Potze L, Di Franco S, Grandela C, Pras‐Raves ML, Picavet DI, van Veen HA, et al. Betulinic acid induces a novel cell death pathway that depends on cardiolipin modification. Oncogene. 2016;35:427–37. [DOI] [PubMed] [Google Scholar]
- 40. Mashima T, Oh‐hara T, Sato S, Mochizuki M, Sugimoto Y, Yamazaki K, et al. p53‐defective tumors with a functional apoptosome‐mediated pathway: a new therapeutic target. J Natl Cancer Inst. 2005;97:765–77. [DOI] [PubMed] [Google Scholar]
- 41. Bao M‐R, Yang C, Tse A‐W, Wei L, Lee D, Zhang MS, et al. Genome‐wide CRISPR‐Cas9 knockout library screening identified PTPMT1 in cardiolipin synthesis is crucial to survival in hypoxia in liver cancer. Cell Rep. 2021;34:108676. [DOI] [PubMed] [Google Scholar]
- 42. Zhu Y, Zhang C, Xu F, Zhao M, Bergquist J, Yang C, et al. System biology analysis reveals the role of voltage‐dependent anion channel in mitochondrial dysfunction during non‐alcoholic fatty liver disease progression into hepatocellular carcinoma. Cancer Sci. 2020;111:4288–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meijer TWH, Kaanders JHAM, Span PN, Bussink J. Targeting hypoxia, HIF‐1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin Cancer Res. 2012;18:5585–94. [DOI] [PubMed] [Google Scholar]
- 44. De Berardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. He L, Gomes AP, Wang X, Yoon SO, Lee G, Nagiec MJ, et al. mTORC1 promotes metabolic reprogramming by the suppression of GSK3‐dependent Foxk1 phosphorylation. Mol Cell. 2018;70:949–60.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Snaebjornsson MT, Janaki‐Raman S, Schulze A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. 2020;31:62–76. [DOI] [PubMed] [Google Scholar]
- 48. Lu X, Paliogiannis P, Calvisi DF, Chen X. Role of the mammalian target of rapamycin pathway in liver cancer: from molecular genetics to targeted therapies. Hepatology. 2021;72(Suppl 1):49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials