

Abstract
Efficient synthetic approaches for the incorporation of nitrogen into polyaromatic compounds (PACs) in different patterns as stabilising moiety for π‐extended systems and modification tool for optoelectronic properties remain a challenge until today. Herein, we developed a new versatile pathway to napthyridine‐based PACs as non‐symmetric and regioisomeric pendant to pyrazine‐based PACs. A combination of a gold‐catalysed synthesis of 2‐aminoquinolines and the development of an in situ desulfonation and condensation of these precursors are the key steps of the protocol. The shape and type of attached functional groups of the PACs can be designed in a late stage of the overall synthetic procedure by the chosen anthranile and backbone of the ynamide introduced in the gold‐catalysed step. Single‐crystal X‐ray diffraction and the investigation of electronic properties of the compounds show the influence of the attached substituents. All naphthyridine‐based PACs show halochromic behaviour implying their use as highly sensitive proton sensor in non‐protic solvents.
Keywords: gold catalysis, N-containing polyaromatic compounds, proton sensing, sensors, synthetic methods
A potent modular two‐step approach to naphthyridine‐based polyaromatic compounds was achieved through gold catalysis followed by acid‐mediated condensation and in situ desulfonation. Besides the halochromic behaviour of all synthesised π‐extended systems, one example shows outstanding sensitivity in detecting protons in non‐protic solvents and allows a quantitative analysis in the low μm area.
Introduction
During the past decades, in the development and synthesis of suitable molecules for electronic devices, [1] the isosteric substitution of carbon atoms by nitrogen in polyaromatic carbon‐based structures was established as a tool to significantly modify the material properties. This incorporation of nitrogen into π‐conjugated systems leads to an energetic decrease of the electronic states and often to a different crystal packing induced by C−H⋅⋅⋅N interactions in comparison to the carbon‐based analogues. [2] Due to the low‐lying LUMOs, [3] this often results in a stabilisation against moisture and oxidation. Furthermore, a changed semiconducting behaviour, based on both electronic effects as well as the effect of the altering of crystal packings, [4] is observed. In addition, the donating properties of the nitrogen lone pair opens up several versatile applications, such as proton [5] and anion sensors, [6] sensors for cationic metal species, [7] and as chelating ligands for metal complexes. [8]
A large part of N‐containing polyaromatic compounds (N‐PACs) is based on acene‐related structures with pyrazine backbones. This can be attributed to straightforward syntheses that are based on condensation methods [9] or palladium‐catalysed cross‐couplings. [10] This leads to a wide variety of symmetric replacements of carbon units by nitrogen atoms in already known systems as well as the access to yet unknown even larger acene systems. [11] Examples for N‐containing PACs, showing a non‐symmetric substitution of carbon units by nitrogen, such as a pyridine moiety, are less common [12] and the introduction of the corresponding naphthyridine units has rarely been investigated, mostly due to a lack of efficient synthetic approaches. Only a few examples exist, [13] although PACs with naphthyridine backbones are potentially highly interesting regioisomeric systems to quinoxaline‐based PACs with respect to their different packing and electronic properties. [2a]
Unlike in the symmetric approach used for the synthesis of pyrazine‐substituted N‐PACs, a condensation approach towards the regioisomeric naphthyridine pendants requires a pre‐oriented 2‐aminoquinoline system connected to its backbone, which moreover needs an easily applicable and versatile synthetic approach as well. In recent times gold catalysis proved to be a valuable tool for the synthesis of polyaromatic compounds and the construction of extended π‐systems for materials science. [14] In this context, we recently published the synthesis of carbazole‐based systems suitable for organic field‐effect transistors (OFET) [15] and the C−H annulation of biaryls as route to acridine systems. [16] In addition, we published a gold‐catalysed synthesis of quinolines from propargyl silyl ethers [17] as a direct and functional‐group‐tolerant access to formyl‐substituted quinolines and 2‐aminoquinolines. Attracted by the potential of the attached formyl unit as synthetic handle in combination with established acid‐mediated condensation methods to acridines, [18] we envisioned that we could apply these 2‐aminoquinolines as potential precursors for naphthyridine systems (Scheme 1). However, the application of known acid‐mediated condensations would require a deprotection of the sulfonamide protection group, which requires harsh conditions and an accompanying low functional group tolerance in an extra step. [19] To overcome this problem, we developed an acid‐mediated cyclisation and deprotection of the sulfonamide group in one step, which in combination with gold catalysis delivers a convenient and versatile access to the rarely investigated substance class of N‐PACs with naphthyridine backbones.
Scheme 1.
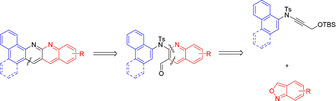
Synthetic approach for the synthesis of N‐PACs with naphthyridine subunits.
Results and Discussion
Synthesis of N‐PACs with Naphthyridine Backbones (5 a–h)
As during the synthesis of large π‐extended systems stability issues play an important role, we started our project with 4 a as model substrate for the condensation to a π‐extended naphthyridine system. Our aim was to avoid any problems during the later characterisation which might arise from oxidation or decomposition of the products (Scheme 2). For this reason, the applied phenanthrene backbone was used first in order to provide higher stability of the product by the introduction of more Clar sextets into the final target. In an initial experiment, we applied our earlier reported optimised conditions [17] for the gold‐catalysed step to 4 a from ynamide 1. But only 25 % of 4 a were obtained, which can be rationalised by the steric interaction between the bulky phenanthrene unit and the IPr‐ligand. By changing the catalyst to the less bulky gold(III) catalyst PicAuCl2 (3), we were able to enhance the yield to 75 %. With these adjusted reaction conditions, we then synthesised compounds 4 a to 4 h as depicted in Scheme 3. The reactions of electron‐deficient anthranils 2 b–d and 2 f gave moderate yields of the corresponding quinolines 4 b–d and 4 f (51–65 %) in comparison to an unsubstituted (2 a) and an electron‐rich (methoxy‐substituted) anthranil (2 e; 75 and 89 %) which confirmed the reactivity trend observed in earlier reports. [17] Even the synthesis of interesting starting materials like 4 g from π‐extended anthranils was possible, which gives access to potential precursors for larger π‐extended systems. The replacement of the phenanthrene moiety with other extended aromatic systems like naphthalene (4 h) was also possible. In this special case IPrAuCl/AgNTf2 turned out to be the more efficient catalyst system (31 % yield) than the routinely used PicAuCl2 (15 %).
Scheme 2.
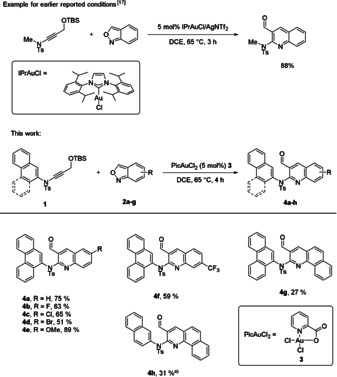
Example for earlier reported conditions and the reaction conditions used in this work: Synthesis of compounds 4 a–4 h, reaction conditions: 1 (0.2 mmol), anthranile 2 a–2 g (0.3 mmol) and 5 mol % of catalyst in 1 mL DCE at 65 °C for 4 hours. a) IPrAuCl (5 mol %) and AgNTf2 (5 mol %) were used as catalyst.
Scheme 3.
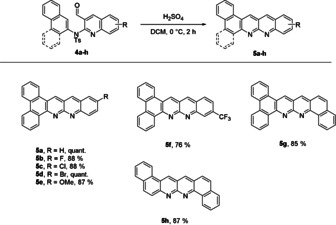
Synthesis of different naphthyridine‐condensed polyaromatic compounds 5 a–5 h. Reaction conditions: 4 a–4 h in DCM (1 mL of solvent per 1 mg of quinoline 4 a–4 h), 0 °C, dropwise addition of H2SO4 (1 vol% acid), 2 h.
The direct condensation and deprotection of sulfonamide 4 a to 5 a was at first achieved with trifluoromethane sulfonic acid (TfOH) in dry DCE under nitrogen atmosphere in quantitative yield. We observed the same efficiency and yield under open flask conditions by using sulfuric acid in DCM as solvent (Scheme 3). Typically, a similar type of condensation reactions to acridines requires high temperatures and/or the acid as solvent,[ 18a , 18d ] but interestingly for yet unknown reasons in this case, the cyclisation to the condensed ring system already occurred under mild conditions and yields after basic workup were generally on a high level (76 % for the electron‐deficient naphthyridine 5 f to quantitative for 5 a and 5 d). This two‐step protocol consisting of a gold‐catalysed approach to quinolines and the subsequent acid‐mediated condensation to π‐extended naphthyridine systems enables a remarkably simple way to a little‐noticed substance class of N‐PACs from ynamides.
Synthesis of Large π‐Extended Systems (11 a–d)
To demonstrate the versatility of this reaction sequence, a bidirectional approach towards the synthesis of large π‐extended systems was conducted. The synthesis of the large acene systems with pyrene backbones 11 a to 11 d is depicted in Scheme 4. To enable substitution of the K‐region of pyrene 6 the nucleophilic positions were sterically blocked by tert‐butyl groups via Friedel–Crafts alkylation. [20] Acetyl groups were introduced in position 4 and 9 by Friedel–Crafts acylation followed by the reduction to the corresponding benzylic alcohols 7. A transformation of 7 to the diaminopyrene 8 was then carried out by following a procedure reported by Liu et al. [21] Noteworthy, this molecule and its derivatives are rarely used in literature, even though regarding to its symmetry it can be regarded as a highly interesting building block for the synthesis of pyrene‐based structures for materials science. Difficult accessibility by earlier approaches hampered its utilisation so far, [22] and we believe that our scalable synthetic approach, with higher yield in comparison (45 % over 4 steps vs. 15 %) and the easier access to only a single regioisomer, might pave the way for future applications of this building block. After tosylation of the diamine with tosylchloride, ynamide 9 was prepared in analogy to the synthesis of 1 (see Supporting Information). The gold‐catalysed synthesis of quinolines 10 a–d from anthranils 2 a and 2 h–j, bearing different alkyl chain tethers to increase solubility, was achieved in good yields up to 81 %. In a next step they were converted through condensation to the extended π‐systems 11 a to 11 d in high yields up to 91 %.
Scheme 4.
Synthesis of 11 a–d. a) tert‐Butylchloride, AlCl3, DCM, rt, 85 %, b) AcCl, AlCl3, DCM, rt, 1 h, 60 %, c) NaBH4, MeOH/DCM, rt, 2.5 h, quant., d) TsCl, pyridine, 0 °C–rt, 4 h, 83 %, e) Cs2CO3, phenyl((trimethylsilyl)ethynyl)iodonium triflate, DMF/DCM, rt, 4 h, 68 %, f) n‐BuLi, paraformaldehyde, −78 °C–rt, 3 h 68 %, g) TBSCl, 1H‐Imidazole, DCM, rt, 2 h, 86 %.
Crystal Structure and Packing
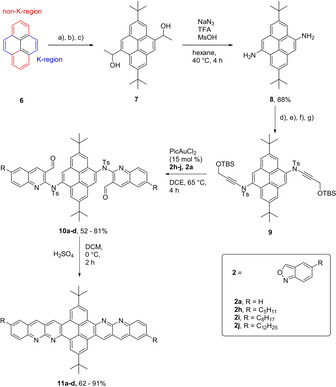
Crystal structures and packings were obtained for compounds 11 d (Figure 1 a), 5 a (Figure 1 c) and 5 c (Supporting Information, Figure S1a). [23] 11 d packs in a slipped stack mode with a plain distance of 3.31 Å and 7.28 Å. Compound 5 a packs under dimer formation in a herringbone‐type arrangement with a monomer–monomer distance of 3.29 Å and with a distance of the C−H‐arene interaction of 2.87 Å. 5 c arranges one‐dimensionally in a dimer formation with an interplane distance of 3.37 Å (see Supporting Info Figure S 1b). The formation of dimeric arrangements in the structure pattern indicates the direct effect on the crystal packing through a substitution of nitrogen into a polyaromatic structure. The C−H⋅⋅⋅N interaction induces a preferred order, which is in line with observations from other examples in the literature. [4] Introduction of substituents onto the polyaromatic backbone has a big influence on the packing pattern and can induce, together with the weak C−H⋅⋅⋅N interactions, formation of different crystal packings dominated by π‐stacking or C−H⋅⋅⋅arene interactions.
Figure 1.
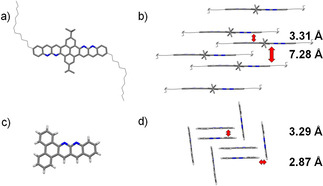
a) Solid‐state molecular structure of 11 d, b) packing of 11 d in a slipped stack mode, with an interplane distance of 3.31 Å, c) solid‐state molecular structure of 5 a, d) packing of 5 a, in a herringbone mode, with an interplane distance of 3.29 Å and a distance of the C−H interaction of 2.87 Å. [23]
Electronic Properties
The introduction of different functional groups on the acene part of the molecules was investigated with DFT calculations using the B3LYP functional and the 6‐311G basis set, in combination with measurements of UV/Vis spectra, fluorescence spectra, and cyclovoltammetry (for details and spectra see Supporting Information). The HOMO–LUMO energy gaps were extracted from the onset of the bathochromic absorption bands and LUMO energies were estimated from the reduction potentials of the cyclovoltammetry spectra using the formula E LUMO=−(E red+4.8) eV. [24] The HOMO energy was then calculated from the extracted HOMO–LUMO energy gap of the UV/Vis spectra and the estimated LUMO energies were calculated from the cyclovoltammetry spectra. A comparison of the resulting values is shown in Table 1.
Table 1.
Optoelectronic, electrochemical and quantum chemical data of naphthyridines 5 a–h and 11 a–d.
|
Compound |
E gap (UV) [eV][a] |
E red (CV) [eV][b] |
E LUMO (CV) [eV][c] |
E HOMO [eV][d] |
E HOMO (calc.) [eV][e] |
E LUMO (calc.) [eV][e] |
|---|---|---|---|---|---|---|
|
5 a |
2.65 |
−1.58 |
−3.22 |
−5.87 |
−5.95 |
−2.80 |
|
5 b |
2.71 |
−1.56 |
−3.24 |
−5.95 |
−6.05 |
−2.92 |
|
5 c |
2.67 |
−1.50 |
−3.30 |
−5.97 |
−6.08 |
−2.95 |
|
5 d |
2.68 |
−1.49 |
−3.31 |
−5.99 |
−6.05 |
−2.93 |
|
5 e |
2.65 |
−1.67 |
−3.13 |
−5.78 |
−5.75 |
−2.79 |
|
5 f |
2.68 |
−1.43 |
−3.37 |
−6.05 |
−6.16 |
−3.11 |
|
5 g |
2.75 |
−1.67 |
−3.13 |
−5.88 |
−5.95 |
−2.70 |
|
5 h |
2.69 |
−1.68 |
−3.12 |
−5.81 |
−5.91 |
−2.75 |
|
11 a |
2.67 |
–[f] |
– |
– |
−5.89 |
−2.83 |
|
11 b |
2.65 |
–[f] |
– |
– |
−5.79 |
−2.79 |
|
11 c |
2.67 |
–[f] |
– |
– |
– |
– |
|
11 d |
2.67 |
–[f] |
– |
– |
– |
– |
[a] The optical gap (E gap) was extracted from the onset of the bathochromic absorption band in the absorption spectrum. [b] The cyclovoltammetric reduction (E red) potentials were measured with ferrocene as internal standard. [c] The LUMO energies (E LUMO) were estimated with the formula E LUMO= −(E red+4.8) eV. [24] [d] E HOMO=E LUMO−E gap. [e] Calculated with the B3LYP functional and the 6‐311G basis set. [f] Could not be measured due to low solubility.
The optical gaps do not differ a lot from each other, even the large acene systems 11 a–d show a similar energy gap to the small naphthyridine systems 5 a–h ranging from 2.65–2.71 eV. An introduction of electron‐withdrawing or ‐donating groups or annulation of ring systems (in our cases phenanthroline, naphthalene, and pyrene) has little to no effect on the energy difference of HOMO and LUMO, which indicates that this energy difference is more likely influenced by the number of linear fused aromatic rings as well as the positioning and absolute number of the nitrogen substitutions in the carbon backbone, which corresponds well with the literature.[ 2a , 25 ] In the emission spectra, a backbone‐ and substituent‐dependent stokes shift is instead visible (see Supporting Information, Figure S5). Whereas the length of the alkyl chain on the large acene systems 11 a–d does not play a big role on the stokes shift of the emission spectrum, a change of the arrangement of the aromatic rings leads to a smaller emission band and the introduction of strong electron‐donating and ‐withdrawing groups shifts the emission spectra to a bathochromic direction. In contrast to the missing change in the energy gap, the HOMO and LUMO energies considered individually depend on the introduction of different substituents. Considering the cyclovoltammetry data (Supporting Information, Figure S2 and Figure S3), compound 5 f with a strong electron‐withdrawing CF3‐group has a first reduction potential of −1.43 V and therefore an estimated LUMO of −3.37 eV, whereas compound 5 e with a strong electron‐donating methoxy group, for example, has a first reduction potential of −1.67 V and therefore an estimated LUMO of −3.13 eV. DFT calculations are consistent with the experimentally measured and estimated values. As illustrated in Figure 2, the frontier orbitals are delocalised over the acene core of the molecule, implying that a substitution on the linear aromatic system would influence both HOMO and LUMO energy equally leading to only small changes in the energy difference of HOMO and LUMO.
Figure 2.
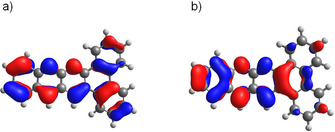
Frontier orbitals of 5 a. a) HOMO of 5 a, b) LUMO of 5 a.
Acid‐Induced Change of UV/Vis and Fluorescence
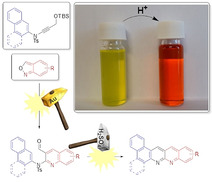
Mixing acid with all of the nitrogen‐substituted PACs 5 a–h and 11 a–d induced changes in the absorption and fluorescence spectra. Protonation of the nitrogen lone pairs caused a red shift of the whole spectrum and the colour of the solutions changed from yellow to red. For a quantitative investigation of the formation of the new species, 11 c was chosen due to its high extinction coefficient of 44 713 l mol−1 cm−1 in the visible area (445 nm) of the absorption spectrum. Titration of methane sulfonic acid in 0.5 equivalent steps to a 2.61 μm solution in chloroform is depicted as normalised absorption spectra in Figure 3 a. By adding acid, a formation of a new absorption maximum at 469 nm is observed with the appearance of an isosbestic point at 451 nm which indicates a direct formation of a new species from 11 c. The emission with two maxima at 478 nm and 508 nm decreases as shown in Figure 4 a with the increase of acid concentration, whereas a new weak emission band of the protonated species with a maximum at 601 nm appears. The sensitivity of the proton detection in this setup ranges between around 1 and 10 μm as shown through plotting the increase of the absorption at 469 nm (Figure 3 b) and the decrease of the emission at 478 nm (Figure 4 b). The linear area of the diagram curves allows a quantitative analysis of the proton concentration in the range of 0 to 5 μm, which makes compound 11 c a highly sensitive proton sensor with potential applications in, for example, detecting low doses of γ‐radiation with halogenated solvents. [26]
Figure 3.
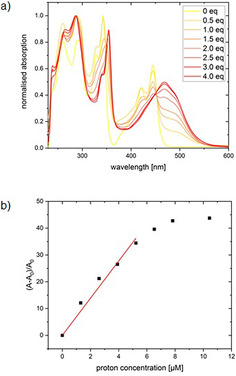
a) Normalised absorption spectra of 11 c in CHCl3 (2.61 μm) and change of the absorption bands by addition of MeSO3H in 0.5 equiv steps, b) relative increase of absorption of 11 c in CHCl3 as function of the concentration of MeSO3H. The data points from 0–5 μm proton concentration were linear fitted (R2=0.991).
Figure 4.
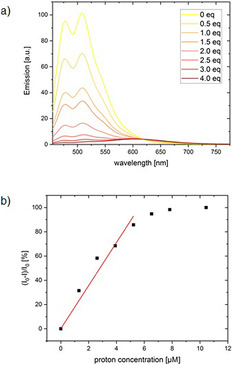
a) Change of the emission of 11 c (2.61 μM) in CHCl3 with addition of MeSO3H in 0.5 equiv steps, b) relative decrease of the emission of 11 c in CHCl3 as function of the concentration of MeSO3H. The data points from 0–5 μm proton concentration were linear fitted (R2=0.984).
Conclusion
We developed a versatile strategy for the synthesis of π‐extended napthyridine‐based polyaromatic compounds through a minor modification of the reaction conditions of the gold‐catalysed [27] synthesis of 2‐aminoquinolines from ynamides and a newly developed method of a one‐step condensation and desulfonisation. The versatility through the introduction of different substituents in the late stage of the overall synthesis and the generally high yields of the last step allowed a structure–property study regarding electronic properties and packing in the crystal structure. The introduction of substituents on structures with the same backbone has an equal effect on the HOMO and LUMO energies individually and therefore shows little to no effect on the HOMO–LUMO gap. The addition of substituents influences the packing pattern with a high impact and is in competition with the influence of the C−H⋅⋅⋅N interaction. Furthermore, the first naphthyridine‐based π‐extended system with eight linear annulated rings was synthesised, showing the great advantage and versatility of this modular synthetic approach. All synthesised naphthyridines show halochromic behaviour by addition of acid and can be used, as shown in the case of a large π‐extended system, as highly sensitive proton sensor in non‐protic solvents.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors acknowledge support by the state of Baden‐Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/467‐1 FUGG (JUSTUS cluster). The authors are grateful for funding by the DFG (SFB 1249 N‐Heteropolyzyklen als Funktionsmaterialien). Open Access funding enabled and organized by Projekt DEAL.
F. Stuck, M. C. Dietl, M. Meißner, F. Sebastian, M. Rudolph, F. Rominger, P. Krämer, A. S. K. Hashmi, Angew. Chem. Int. Ed. 2022, 61, e202114277.
References
- 1.
- 1a. Ameri T., Lia N., Brabecab C. J., Energy Environ. Sci. 2013, 6, 2390–2413; [Google Scholar]
- 1b. Burghard M., Klauk H., Kern K., Adv. Mater. 2009, 21, 2586–2600; [DOI] [PubMed] [Google Scholar]
- 1c. Kordt P., Holst J. J. M. v. d., Helwi M. A., Kowalsky W., May F., Badinski A., Lennartz C., Andrienko D., Adv. Funct. Mater. 2015, 25, 1955–1971; [Google Scholar]
- 1d. Lüssem B., Riede M., Leo K., Phys. Status Solidi A 2013, 210, 9–43; [Google Scholar]
- 1e. Pron A., Gawrys P., Zagorska M., Djuradoa D., Demadrillea R., Chem. Soc. Rev. 2010, 39, 2577–2632; [DOI] [PubMed] [Google Scholar]
- 1f. Roncali J., Leriche P., Blanchard P., Adv. Mater. 2014, 26, 3821–3838. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Winkler M., Houk K. N., J. Am. Chem. Soc. 2007, 129, 1805–1815; [DOI] [PubMed] [Google Scholar]
- 2b. He Z., Mao R., Liu D., Miao Q., Org. Lett. 2012, 14, 4190–4193; [DOI] [PubMed] [Google Scholar]
- 2c. Fleischhauer J., Zahn S., Beckert R., Grummt U.-W., Birckner E., Görls H., Chem. Eur. J. 2012, 18, 4549–4557. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Liang Z., Tang Q., Xu J., Miao Q., Adv. Mater. 2011, 23, 1535–1539; [DOI] [PubMed] [Google Scholar]
- 3b. Lindner B. D., Engelhart J. U., Tverskoy O., Appleton A. L., Rominger F., Peters A., Himmel H.-J., Bunz U. H. F., Angew. Chem. Int. Ed. 2011, 50, 8588–8591; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8747–8750; [Google Scholar]
- 3c. Naibi Lakshminarayana A., Ong A., Chi C., J. Mater. Chem. C 2018, 6, 3551–3563. [Google Scholar]
- 4.
- 4a. Hisaki I., Ji Q., Takahashi K., Tohnai N., Nakamura T., Cryst. Growth Des. 2020, 20, 3190–3198; [Google Scholar]
- 4b. Klues M., Witte G., CrystEngComm 2018, 20, 63–74; [Google Scholar]
- 4c. Liang Z., Tang Q., Mao R., Liu D., Xu J., Miao Q., Adv. Mater. 2011, 23, 5514–5518; [DOI] [PubMed] [Google Scholar]
- 4d. Tang Q., Liang Z., Liu J., Xu J., Miao Q., Chem. Commun. 2010, 46, 2977–2979; [DOI] [PubMed] [Google Scholar]
- 4e. Maly K. E., Cryst. Growth Des. 2011, 11, 5628–5633; [Google Scholar]
- 4f. Xin Q., Duhm S., Hosoumi S., Ueno N., Tao X.-t., Kera S., J. Phys. Chem. C 2011, 115, 15502–15508; [Google Scholar]
- 4g. Black H. T., Perepichka D. F., Angew. Chem. Int. Ed. 2014, 53, 2138–2142; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2170–2174. [Google Scholar]
- 5.
- 5a. Schneider J. A., Perepichka D. F., J. Mater. Chem. C 2016, 4, 7269–7276; [Google Scholar]
- 5b. He B., Dai J., Zherebetskyy D., Chen T. L., Zhang B. A., Teat S. J., Zhang Q., Wang L., Liu Y., Chem. Sci. 2015, 6, 3180–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Gu P.-Y., Gao J., Wanga C., Zhang Q., RSC Adv. 2015, 5, 80307–80310; [Google Scholar]
- 6b. Gu P.-Y., Wang Z., Zhang Q., J. Mater. Chem. B 2016, 4, 7060–7074. [DOI] [PubMed] [Google Scholar]
- 7. Li J., Chen S., Zhang P., Wang Z., Long G., Ganguly R., Li Y., Zhang Q., Chem. Asian J. 2016, 11, 136–140. [DOI] [PubMed] [Google Scholar]
- 8. Xiang H., Cheng J., Ma X., Zhou X., Chruma J. J., Chem. Soc. Rev. 2013, 42, 6128–6185. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Bunz U. H. F., Engelhart J. U., Lindner B. D., Schaffroth M., Angew. Chem. Int. Ed. 2013, 52, 3810–3821; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3898–3910; [Google Scholar]
- 9b. Kulisic N., More S., Mateo-Alonso A., Chem. Commun. 2011, 47, 514–516; [DOI] [PubMed] [Google Scholar]
- 9c. Mateo-Alonso A., Kulisic N., Valenti G., Marcaccio M., Paolucci F., Prato M., Chem. Asian J. 2010, 5, 482–485; [DOI] [PubMed] [Google Scholar]
- 9d. Bunz U. H. F., Acc. Chem. Res. 2015, 48, 1676–1686; [DOI] [PubMed] [Google Scholar]
- 9e. Kohl B., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2015, 54, 6051–6056; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6149–6154; [Google Scholar]
- 9f. Gao B., Wang M., Cheng Y., Wang L., Jing X., Wang a. F., J. Am. Chem. Soc. 2008, 130, 8297–8306. [DOI] [PubMed] [Google Scholar]
- 10. Bunz U. H. F., Engelhart J. U., Chem. Eur. J. 2016, 22, 4680–4689. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Jester S.-S., Sigmund E., Röck L. M., Höger S., Angew. Chem. Int. Ed. 2012, 51, 8555–8559; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8683–8687; [Google Scholar]
- 11b. Ahrens L., Hahn S., Rominger F., Freudenberg J., Bunz U. H. F., Chem. Eur. J. 2019, 25, 14522–14526; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Endres A. H., Schaffroth M., Paulus F., Reiss H., Wadepohl H., Rominger F., Krämer R., Bunz U. H. F., J. Am. Chem. Soc. 2016, 138, 1792–1795. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Liu Y.-Y., Song C.-L., Zeng W.-J., Zhou K.-G., Shi Z.-F., Ma C.-B., Yang F., Zhang H.-L., Gong X., J. Am. Chem. Soc. 2010, 132, 16349–16351; [DOI] [PubMed] [Google Scholar]
- 12b. Song C.-L., Ma C.-B., Yang F., Zeng W.-J., Zhang H.-L., Gong X., Org. Lett. 2011, 13, 2880–2883; [DOI] [PubMed] [Google Scholar]
- 12c. Wiesner T., Ahrens L., Rominger F., Freudenberg J., Bunz U. H. F., Chem. Eur. J. 2021, 27, 4553–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Ukwitegetse N., Saris P. J. G., Sommer J. R., Haiges R. M., Djurovich P. I., Thompson M. E., Chem. Eur. J. 2019, 25, 1472–1475; [DOI] [PubMed] [Google Scholar]
- 13b. Irgashev R. A., Demina N. S., Kazin N. A., Rusinov G. L., Tetrahedron Lett. 2019, 60, 1135–1138; [Google Scholar]
- 13c. Sampathkumar N., Kumar N. V., Rajendran S. P., Synth. Commun. 2004, 34, 2019–2024. [Google Scholar]
- 14. Hendrich C. M., Sekine K., Koshikawa T., Tanaka K., Hashmi A. S. K., Chem. Rev. 2021, 121, 9113–9163. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Hendrich C. M., Bongartz L. M., Hoffmann M. T., Zschieschang U., Borchert J. W., Sauter D., Krämer P., Rominger F., Mulks F. F., Rudolph M., Dreuw A., Klauk H., Hashmi A. S. K., Adv. Synth. Catal. 2021, 363, 549–557; [Google Scholar]
- 15b. Hendrich C. M., Hannibal V. D., Eberle L., Hertwig L. E., Zschieschang U., Rominger F., Rudolph M., Klauk H., Hashmi A. S. K., Adv. Synth. Catal. 2021, 363, 1401–1407. [Google Scholar]
- 16. Zeng Z., Jin H., Sekine K., Rudolph M., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2018, 57, 6935–6939; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7051–7056. [Google Scholar]
- 17. Jin H., Tian B., Song X., Xie J., Rudolph M., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2016, 55, 12688–12692; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12880–12884. [Google Scholar]
- 18.
- 18a. Li J., Tan E., Keller N., Chen Y.-H., Zehetmaier P. M., Jakowetz A. C., Bein T., Knochel P., J. Am. Chem. Soc. 2019, 141, 98–103; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Shimo S., Takahashi K., Iwasawa N., Chem. Eur. J. 2019, 25, 3790–3794; [DOI] [PubMed] [Google Scholar]
- 18c. Zeghada S., Bentabed-Ababsa G., Mongin O., Erb W., Picot L., Thiéry V., Roisnel T., Dorcet V., Mongin F., Tetrahedron 2020, 76, 131435; [Google Scholar]
- 18d. Kuninobu Y., Tatsuzaki T., Matsuki T., Takai K., J. Org. Chem. 2011, 76, 7005–7009. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Sabitha G., Reddy B. V. S., Abraham S., Yadav J. S., Tetrahedron Lett. 1999, 40, 1569–1570; [Google Scholar]
- 19b. Javorskis T., Orentas E., J. Org. Chem. 2017, 82, 13423–13439; [DOI] [PubMed] [Google Scholar]
- 19c. Vedejs E., Lin S., J. Org. Chem. 1994, 59, 1602–1603; [Google Scholar]
- 19d. Nandi P., Redko M. Y., Petersen K., Dye J. L., Lefenfeld M., Vogt P. F., Jackson J. E., Org. Lett. 2008, 10, 5441–5444. [DOI] [PubMed] [Google Scholar]
- 20. Wu Z.-H., Huang Z.-T., Guo R.-X., Sun C.-L., Chen L.-C., Sun B., Shi Z.-F., Shao X., Li H., Zhang H.-L., Angew. Chem. Int. Ed. 2017, 56, 13031–13035; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13211–13215. [Google Scholar]
- 21. Liu J., Qiu X., Huang X., Luo X., Zhang C., Wei J., Pan J., Liang Y., Zhu Y., Qin Q., Song S., Jiao N., Nat. Chem. 2019, 11, 71–77. [DOI] [PubMed] [Google Scholar]
- 22. Ji L., Krummenacher I., Friedrich A., Lorbach A., Haehnel M., Edkins K., Braunschweig H., Marder T. B., J. Org. Chem. 2018, 83, 3599–3606. [DOI] [PubMed] [Google Scholar]
- 23.Deposition numbers 2103412 (5 a), 2103413 (5 c), 2103414 (5 e), 2103415 (5 h), 2103416 (11 d), and 2103417 (17) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 24. Cardona C. M., Li W., Kaifer A. E., Stockdale D., Bazan G. C., Adv. Mater. 2011, 23, 2367–2371. [DOI] [PubMed] [Google Scholar]
- 25. Ruiz-Morales Y., J. Phys. Chem. A 2002, 106, 11283–11308. [Google Scholar]
- 26. Han J.-M., Xu M., Wang B., Wu N., Yang X., Yang H., Salter B. J., Zang L., J. Am. Chem. Soc. 2014, 136, 5090–5096. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Hashmi A. S. K., Chem. Rev. 2007, 107, 3180–3211; [DOI] [PubMed] [Google Scholar]
- 27b. Hashmi A. S. K., Bührle M., Salathé R., Bats J. W., Adv. Synth. Catal. 2008, 350, 2059–2064; [Google Scholar]
- 27c. Hashmi A. S. K., Blanco M. C., Eur. J. Org. Chem. 2006, 4340–4342. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information