

Abstract
Background and Aims
Lipoprotein Z (LP‐Z) is an abnormal free cholesterol (FC)–enriched LDL‐like particle discovered from patients with cholestatic liver disease. This study aims to define the diagnostic value of LP‐Z in alcohol‐associated hepatitis (AH) and interrogate the biology behind its formation.
Approach and Results
We measured serum levels of LP‐Z using nuclear magnetic resonance spectroscopy, a well‐established clinical assay. Serum levels of LP‐Z were significantly elevated in four AH cohorts compared with control groups, including heavy drinkers and patients with cirrhosis. We defined a Z‐index, calculated by the ratio of LP‐Z to total apolipoprotein B–containing lipoproteins, representing the degree of deviation from normal VLDL metabolism. A high Z‐index was associated with 90‐day mortality independent from the Model for End‐Stage Liver Disease (MELD) and provided added prognosticative value. Both a Z‐index ≤ 0.6 and a decline of Z‐index by ≥0.1 in 2 weeks predicted 90‐day survival. RNA‐sequencing analyses of liver tissues demonstrated an inverse association in the expression of enzymes responsible for the extrahepatic conversion of VLDL to LDL and AH disease severity, which was further confirmed by the measurement of serum enzyme activity. To evaluate whether the FC in LP‐Z could contribute to the pathogenesis of AH, we found significantly altered FC levels in liver explant of patients with AH. Furthermore, FC in reconstituted LP‐Z particles caused direct toxicity to human hepatocytes in a concentration‐dependent manner, supporting a pathogenic role of FC in LP‐Z.
Conclusions
Impaired lipoprotein metabolism in AH leads to the accumulation of LP‐Z in the circulation, which is hepatotoxic from excessive FC. A Z‐index ≤ 0.6 predicts 90‐day survival independent from conventional biomarkers for disease prognostication.
Abbreviations
- AH
alcohol‐associated hepatitis
- ALD
alcohol‐associated liver disease
- ASH
alcohol‐associated steatohepatitis
- AUROC
area under the receiver operating characteristic curve
- BIDMC
Beth Israel Deaconess Medical Center
- CE
cholesteryl ester
- DF
Maddrey’s discriminant function
- ELAD
Extracorporeal Liver Assist Device
- FC
free cholesterol
- HL
hepatic lipase
- InTEAM
Integrated Approaches for Identifying Molecular Targets
- IRB
institutional review board
- LCAT
lecithin cholesterol acyltransferase
- LIPC
lipase C hepatic type
- LP‐Z
lipoprotein Z
- MELD
Model for End‐Stage Liver Disease
- NMR
nuclear magnetic resonance
- rLP‐Z
LP‐Z‐like emulsion particles
- ROC
receiver operating characteristic
- SM
sphingomyelin
- TG
triglyceride
- TREAT
Translational Research and Evolving Alcoholic Hepatitis Treatment
INTRODUCTION
Over the last decade, many countries have seen a rise of alcohol‐associated liver disease (ALD) as a significant cause of death.[ 1 ] Alcohol‐associated hepatitis (AH), the most precarious form of ALD, carries a mortality rate of up to 50% in its severe form.[ 2 , 3 ] The pathogenesis of AH is poorly understood, and current AH treatment is limited to abstinence, nutritional support, and selected use of corticosteroids.[ 4 ] ALD has become the leading cause for liver transplant in the United States and Europe in the post–hepatitis C era.[ 5 , 6 , 7 ] Currently, patients with AH compete with other wait‐listed patients for liver transplant based on the Model for End‐Stage Liver Disease, a score validated largely for decompensated cirrhosis.
We recently identified an abnormal lipoprotein among individuals with high bilirubin using nuclear magnetic resonance (NMR) and named this particle lipoprotein Z (LP‐Z).[ 8 ] LP‐Z is an LDL‐like particle that carries a higher content of triglyceride (TG) and free cholesterol (FC), but a reduced amount of cholesteryl ester (CE) compared with LDL. The liver orchestrates lipoprotein metabolism. Not only does the liver produce major precursors of circulating lipoproteins, including VLDL and HDL, but the liver is also the primary source of circulating enzymes responsible for extrahepatic lipoprotein metabolism, such as lecithin cholesterol acyltransferase (LCAT) and hepatic lipase (HL), also known as lipase C hepatic type (LIPC). LCAT catalyzes the transfer of fatty acyl chains from phosphatidylcholine to FC to generate hydrophobic CE, whereas HL converts hydrophobic TG to diacylglycerol and free fatty acids.[ 9 ] We found that patient samples containing high levels of LP‐Z most frequently carried a diagnosis of ALD.[ 8 ] The current study examines the specificity of LP‐Z in diagnosing AH, the mechanism for its accumulation, and the diagnostic and therapeutic implications of LP‐Z for patients with AH.
PATIENTS AND METHODS
Patient cohorts
Beth Israel Deaconess Medical Center (BIDMC) AH registry was a single‐center prospective observational study that started in 2018. The study enrolled 39 patients with a clinical diagnosis of AH from the inpatient liver service based on standard criteria including a history of heavy alcohol consumption (3+ drinks for women and 4+ drinks for men), signs of liver injury (aspartate aminotransferase [AST] > 50 IU/L and AST/alanine aminotransferase [ALT] > 1.5) and liver failure with elevated bilirubin (>3 mg/dL).[ 10 ] Patients with an alternative diagnosis contributing to liver failure were excluded from the study. Patients considered for liver transplant were not excluded. Blood samples were obtained at the time of enrollment. BIDMC NAFLD registry started in 2009 and recruited patients with a diagnosis of NAFLD based on liver biopsy as described previously.[ 11 ] Patients with other forms of chronic liver disease or alcohol consumption of more than 20 g per day were excluded. Outpatient cirrhosis registry was an ongoing prospective study of patients with stage 3 and above fibrosis based on histology or noninvasive fibrosis staging. Inpatient alcohol‐associated cirrhosis cohort was derived from an ongoing study of patients with decompensated cirrhosis admitted for hospitalization at BIDMC. Laboratory tests and serum in these studies were also collected at the time of enrollment. The BIDMC transplant registry, established in 2015, recruited patients undergoing solid organ transplant for all causes. Subjects with liver transplant for cirrhosis were included in the study. Blood samples were collected from patients on the day of the transplant. All studies were approved by the BIDMC institutional review board (IRB) and were conducted in accordance with the Helsinki declaration of 1975, as revised in 1983. All participants consented to the study at enrollment.
The Extracorporeal Liver Assist Device (ELAD) study of AH (VTI‐208) (NCT01471028) was a randomized open‐label multicenter clinical trial sponsored by Vital Therapy to evaluate the safety and efficacy of the ELAD devices in treating severe AH. The study enrolled subjects aged 18–50 with a clinical diagnosis of AH who met the inclusion criteria of international normalized ratio ≤2.5, creatinine <1.3 mg/dL, total bilirubin ≥16 mg/dL, and MELD score <30. A total of 91 subjects (49 control, 42 ELAD) with baseline samples available for testing were included in our study. Inasmuch as ELAD treatment did not change the patient outcome or LP‐Z levels, both the treatment and control arms in ELAD were combined. Ethylene diamine tetraacetic acid (EDTA) plasma samples were obtained on days 0 and 14. The study was approved by the IRB of participating institutes, and all participants signed informed consent before enrollment.
The Translational Research and Evolving Alcoholic Hepatitis Treatment (TREAT) (NCT02172898) was an observational study of patients with well‐characterized AH (n = 196) and matched heavy drinkers without liver disease (n = 169) sponsored by the National Institute of Alcohol Abuse and Alcoholism (NIAAA) from September 2012 through June 2018.[ 12 ] The study was conducted at Indiana University, Mayo Clinic, and Virginia Commonwealth University. Study participants were evaluated at baseline and then at 6 and 12 months for clinical and survival data and biosample acquisition. This study was approved by the IRBs at the respective institutions, and all participants signed an informed consent form before enrollment.
Integrated Approaches for Identifying Molecular Targets (InTEAM) is an international NIAAA‐sponsored consortium to study the pathophysiology of AH (NCT02075918).[ 13 ] We included 20 patients with AH with liver biopsy in this study. The diagnosis of AH in all patients was based on the recently described criteria.[ 10 ] The following subjects and tissues samples were included in the transcriptomics analysis: early alcohol‐associated steatohepatitis (ASH, n = 12), patients with histologically confirmed AH who were biopsied before any treatment (AH, n = 18), and explants from patients with AH who underwent early transplantation (n = 11). Groups of controls include nondiseased human livers (n = 10), NAFLD without alcohol abuse (n = 9), noncirrhotic HCV infection (n = 10), and compensated HCV‐related cirrhosis (n = 9). Patients with malignancies were excluded from the study. All patients gave written informed consent. The research protocols were approved by the local ethics committees and by the central IRB of the University of North Carolina at Chapel Hill.
Analyses in the study included all subjects in each cohort, unless otherwise specified for the identification of a subgroup within a cohort.
Quantification of LP‐Z by NMR spectroscopy
Lipoprotein profiling by NMR LipoProfile analysis is in routine clinical use by primary care clinicians and cardiologists.[ 14 ] NMR LipoProfile spectra in this study were collected at 400 MHz on either NMR Profiler or Vantera Clinical Analyzer platforms. Both EDTA plasma and serum are suitable specimens for this analysis, and freeze‐thaw cycles have minimal impact on lipoprotein quantification.[ 15 ] LP‐Z particle concentrations were measured using the LP4 algorithm as previously described.[ 8 ] In short, a one‐dimensional proton NMR spectrum of each sample was acquired, and the broad lipid methyl group signal envelope interrogated by deconvolution to quantify the separate contributions made to it by different‐size lipoprotein particle subclasses, including LP‐Z.
Agarose gel electrophoresis
Serum samples were loaded on Sebia Hydragel 15/30 lipoprotein (e) agarose gels (Sebia, Inc.) and run at 100 V for 1 hour in Barbital buffer (Millipore Sigma) in a Titan gel electrophoresis chamber (Helena Laboratories). Gels were fixed and stained with Sudan Black (Millipore Sigma) to detect neutral lipids or freshly prepared filipin (Millipore Sigma) to detect FC using previously described methods.[ 16 ]
Lipoprotein isolation
Two methods were used to isolate LDL or LP‐Z lipoproteins. To prepare samples for cryo‐electron microscopy, plasma samples were centrifuged at 10,000 x g for 5 min and loaded on a Superose 6 10/300 GL column (Millipore Sigma) through an AKTA–fast protein liquid chromatography system (Millipore Sigma) at room temperature. Same fractions corresponding to 9.5–12 ml elution volume in PBS were pooled and stored at 4°C. For lipidomics analysis, 120‐µl plasma samples were first centrifuged at 90,000 rpm at 4°C in a TLA‐120.2 rotor (Beckman) for 3 hours. The lower half of the sample was separated and mixed with 60 µl of potassium bromide at 1.12 g/ml and centrifuged at 90,000 rpm, 4°C for 18 h. After centrifugation, the top half of the sample containing LDL and LP‐Z (1.006–1.063 g/ml) was collected for lipidomics. For cell toxicity assay, we pooled five plasma samples from patients with AH with high Z‐index (0.89 ± 0.02), and five samples with a Z‐index of 0. After density gradient centrifugation, the LDL and LP‐Z fractions were dialyzed extensively against PBS, concentrated 10‐fold, filtered against a 0.22‐µm filtering device, and stored for cell treatment.
Transcriptomics analysis
RNA extraction, sequencing, and bioinformatic analysis of the InTEAM study were described previously.[ 13 ] We identified genes relevant to lipoprotein metabolism including apolipoprotein A1 (APOA1), APOA2, APOA4, APOA5, APOC1, APOC2, APOC3, APOE, hepatocyte nuclear factor 4 (HNF4A), cholesteryl ester transfer protein (CETP) , LCAT, lipase C hepatic type (LIPC), fatty acid–binding protein 1 (FABP1), microsomal triglyceride transfer protein (MTTP), diacylglycerol acyltransferase (DGAT1), DGAT2, sterol O‐acyltransferase (SOAT1 ), SOAT2, LDL receptor (LDLR), and LDLR‐related protein (LRP). The levels of mRNA were plotted by transcripts per kBp million in the following groups: normal control, ASH, mild–moderate AH, severe AH 90‐day survivors, severe AH 90‐day nonsurvivors, AH explant, HCV, NAFLD, and HCV cirrhosis.
Statistical analysis
We first compared the levels of LP‐Z in four cohorts of AH and four control groups using ANOVA and post‐test pairwise comparison using Bonferroni’s multiple tests. The Z‐index is calculated as the ratio between particle concentrations of LP‐Z and total apolipoprotein B (apoB)–containing lipoproteins (i.e., VLDL and LDL), where total LDL concentration includes LP‐Z.
We used the ELAD study as a derivative cohort to examine the relationship between serum LP‐Z concentration, Z‐index, and outcomes of patients with AH at 90 days and 1 year. Cox proportional hazard models were used after confirming proportional hazard tests. To compare the predictive value of the Z‐index to current standard scores for prognostication, Z‐index as a continuous variable in increments of 0.1 (0–10) was first compared with MELD, Maddrey’s discriminant function (DF), and their components as continuous variables. The regression was then adjusted for MELD and DF in multivariate models to compare the relative associations. Receiver operating characteristic (ROC) curves of Z‐index were plotted for 90‐day mortality to evaluate the performance of the Z‐index and to determine cutoffs that optimize sensitivity and specificity. The goal was to identify a cutoff with either high sensitivity or specificity, to maximize clinical utility. We also aimed to set a cutoff point that was convenient to use. We examined Z‐index cutoffs with 0.1 intervals and chose a cutoff of 0.6 that maximizes the specificity for survival. Kaplan‐Meier’s survival analysis was performed using a binary cutoff of Z‐index at 0.6, and p‐value was calculated using the log‐rank test. The same analyses were performed in two validation cohorts of the TREAT study and the BIDMC AH registry. To evaluate whether changes of Z‐index carried prognostic information, we calculated the delta Z‐index between day 14 and baseline in ELAD (n = 81). Patients without day 14 samples were not included in the study. We compared the 90‐day survival from day 14 between those with a decrease of day‐14 Z‐index by more than 0.1 to their counterparts by Kaplan‐Meier survival analysis. To compare the predictive value of Z‐index and MELD, we pooled all AH data and compared the ROC of Z‐index, MELD, and a regression model using Z‐index and MELD using the DeLong’s test. All statistical analyses were performed using STATA version 14.
Additional information on cryo‐electron microscopy, fluorescent microscopy imaging and data analysis, untargeted lipoprotein lipidomics, enzyme activity measurements, cell culture, and preparation of reconstituted LP‐Z like emulsion particles are available in the Supporting Material and Methods.
RESULTS
Circulating LP‐Z particles in patients with AH
Patients with AH exhibit an atypical pattern of circulating lipoproteins. When analyzed by agarose gel electrophoresis, a typical plasma sample from a healthy subject has three bands, corresponding to HDL, VLDL and LDL respectively, as stained by Sudan black for neutral lipids (Figure 1A). In contrast, the plasma of patients with AH demonstrated only one band with a migration pattern similar to that of LDL. This LDL‐sized band was enriched with FC, which can be stained by filipin, a fluorescent dye (Figure 1A). LP‐Z, like LDL, carries one copy of apoB (Figure 1B).[ 8 ] But unlike LDL, LP‐Z is enriched with TG instead of CE in its lipid core, and carries a significant amount of FC on the surface.[ 8 ] Isolated LP‐Z showed a morphology similar to LDL by cryo‐electron microscopy imaging (Figure 1C, Figure S1). LP‐Z particles had a mean size of 21.5 ± 1.8 nm, smaller and more homogenous than LDL particles at 24.4 ± 5.2 nm (p < 0.001) (Figure 1D). LDL particles in side views showed characteristic stripes, a result of layered CE packing at a low temperature.[ 17 ] This was not seen in LP‐Z, in keeping with a deficiency of CE (Figure 1C, Figure S1). Lipidomics analysis of isolated LP‐Z and LDL particles confirmed these features, in which the LP‐Z‐enriched sample had higher levels of TG (21.0% vs. 14.4%, p = 0.003), but lower levels of CE (0.7% vs. 3.4%, p < 0.001) compared with LDL‐enriched samples (Figure 1E). In addition, LP‐Z‐enriched samples had lower levels of sphingomyelin, lysophosphatidylcholine, lysodimethylphosphatidyl‐ethanolamine, and phosphatidic acid (Table S1).
FIGURE 1.
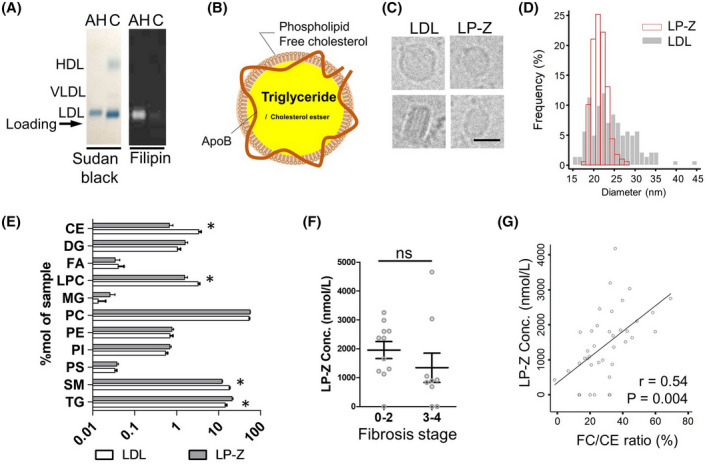
Characteristics of lipoprotein Z (LP‐Z) in alcohol‐associated hepatitis (AH). (A) Comparison of serum lipoproteins from patients with AH and healthy control (C) analyzed by agarose gel electrophoresis. Sudan black staining of neutral lipids (triglyceride [TG] and cholesteryl ester [CE]) highlights LDL, HDL, and VLDL. Arrow indicates the reference position of sample loading. Filipin staining of free cholesterol (FC) on the same samples shown on the right. (B) Diagram of the composition of LP‐Z. (C) Representative cryo‐EM images of LDL and LP‐Z in different views. Scale bar equals 20 nm. (D) Histograms of the size of LDL and LP‐Z measured from cryo‐EM images. (E) Comparison of lipid composition between LP‐Z and LDL measured in %mol. Asterisks indicates p value < 0.005 by t test. (F) Comparison of LP‐Z in AH with advanced cirrhosis and early cirrhosis in a subcohort of Integrated Approaches for Identifying Molecular Targets (InTEAM). (G) Relationship between free cholesterol (FC)/CE ratio and LP‐Z concentration demonstrating a linear correlation. P value calculated by linear regression
Elevated circulating levels of LP‐Z are specific to AH
We measured lipoprotein concentrations in the plasma using NMR in four cohorts of AH: an AH registry at BIDMC (n = 39), the ELAD clinical trial (n = 91), patients with AH in the TREAT consortium (n = 196), and subjects with liver biopsy from the InTEAM consortium (n = 20) (Table 1). Although AH is a clinical diagnosis, most patients in these cohorts (79%–90%) meet the American Gastroenterology Association (AGA)‐recommended laboratory criteria for AH clinical trials. This was compared to five control cohorts: a registry of biopsy‐confirmed NAFLD without cirrhosis (n = 178), an outpatient cirrhosis registry (n = 114), a liver transplant registry (n = 10), heavy drinkers without AH in the TREAT consortium (n = 169), and an inpatient alcohol‐associated cirrhosis cohort (Tables S2–S6). In four cohorts of AH, the mean levels of circulating LP‐Z were similar (p > 0.05), and significantly higher than those from the control groups (p < 0.001) (Figure 2). In the TREAT consortium, the heavy drinker controls and similar demographics, body mass index, comorbidities, and even higher mean Alcohol Use Disorder Identification Test (AUDIT score (27.5 ± 7.5 vs. 24.0 ± 8.7, p < 0.001) than patients with AH (Table S4). The significantly higher occurrence of LP‐Z in patients with AH indicated that alcohol consumption alone was not sufficient to cause LP‐Z accumulation. Among those patients admitted for decompensation from alcohol‐associated cirrhosis, LP‐Z was detected in 5 of 6 patients who were actively drinking within 3 months of admission, and 3 of 11 subjects who were not actively drinking (Table S6). These observations suggested that LP‐Z accumulation in AH was not driven by isolated hepatic steatosis, heavy drinking, or cirrhosis pathologies presented in control subjects. The level of LP‐Z was similar in early fibrosis (stage 0–2) and advanced fibrosis (stages 3 or 4) among patients in InTEAM with liver biopsy (Figure 1F). The serum FC to CE ratio was 0.41 in patients with AH compared to 0.15 in NAFLD (p < 0.001) (Table S7). The FC/CE ratio in AH was linearly associated with the concentration of LP‐Z (Pearson correlation 0.54, p = 0.004) (Figure 1G).
TABLE 1.
Description of AH Cohorts
| BIDMC AH registry (n = 39) | ELAD (n = 91) | InTEAM (n = 20) | TREAT | ||
|---|---|---|---|---|---|
| Heavy Drinker (n = 169) | AH (n = 196) | ||||
| Study description | A prospective single‐center observational study of AH; subjects were consecutively recruited from the hospitalized patients at BIDMC | A phase 3, randomized, open‐label, multicenter clinical trial of ELAD (VTL‐308); subjects with baseline samples were included in this study | A multicenter prospective observational study aimed to identify molecular targets in AH; subjects with liver biopsy were included in this study | A multicenter observational case‐control study comparing subjects with heavy drinking and AH | |
| Age | <35: 14 (36%) | <35: 31 (34%) | <35: 2 (10%) | <35: 44 (26%) | <35: 39 (20%) |
| ≥35: 25 (64%) | ≥35: 60 (66%) | ≥35: 18 (90%) | ≥35: 125 (74%) | ≥35: 157 (80%) | |
| Gender (% F) | 36% | 39% | 45% | 35% | 20% |
| Meeting AGA laboratory criteria for AH trials (%)a | 82.1% | 79.1% | 90.0% | — | 80.1% |
| MELD (median, IQR) | 28 (24, 34) | 27 (24, 30) | 21 (19, 26) | 7 (6, 8) | 23 (18, 27) |
| DF (median, IQR) | 58 (42, 97) | 47 (30, 67) | 19 (13, 36) | 0.5 (0.3, 1) | 41 (19, 61) |
| Total bilirubin (mg/dL, mean ± SD) | 18.7 ± 11.5 | 23.7 ± 8.7 | 13.1 ± 7.7 | 0.6 ± 0.6 | 14.2 ± 11.0 |
| Triglyceride (mg/dL, mean ± SD) | 101 ± 65 | 100 ± 54 | 137 ± 88 | 175 ± 122 | 127 ± 78 |
| LP‐Z (nmol/L) (mean ± SD) | 1440 ± 996 | 1295 ± 943 | 1680 ± 1256 | 76 ± 317 | 1611 ± 1296 |
| Z‐index (mean ± SD) | 0.63 ± 0.30 | 0.58 ± 0.25 | 0.63 ± 0.32 | 0.04 ± 0.11 | 0.56 ± 0.32 |
Abbreviations: F, female; and IQR, interquartile range.
FIGURE 2.
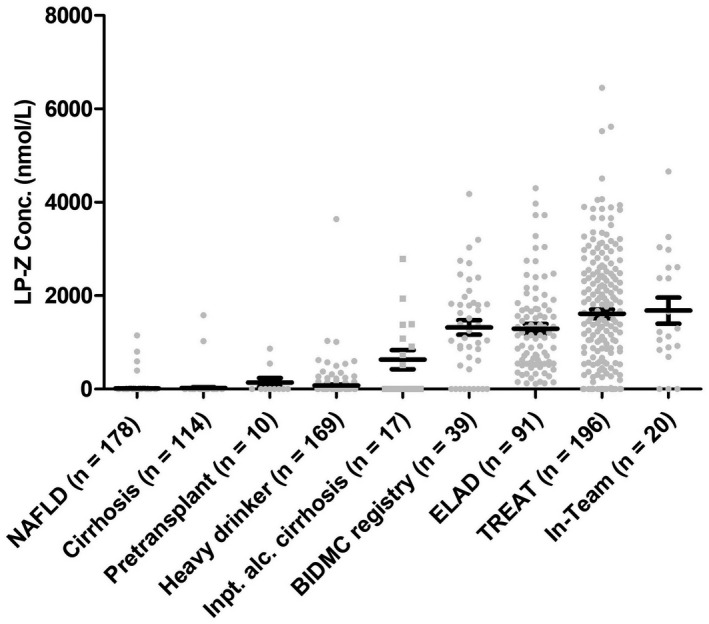
Accumulation of LP‐Z in patients with AH. Comparison of LP‐Z levels from four AH cohorts: Beth Israel Deaconess Medical Center (BIDMC) AH registry, Extracorporeal Liver Assist Device (ELAD) trial, Translational Research and Evolving Alcoholic Hepatitis Treatment (TREAT) AH arm, InTEAM subcohort, and five control cohorts: NAFLD, outpatient cirrhosis registry, pretransplant registry, inpatient decompensated alcohol‐associated cirrhosis, and heavy drinker arm from TREAT
Z‐index predicts 90‐day survival in AH
The marked elevation of circulating LP‐Z in AH suggested that LP‐Z might predict clinical outcomes in AH. In the ELAD cohort, patients with high baseline LP‐Z showed no significant differences in labs of liver function, prognostication indices, or clinical outcome (Table S8), nor was LP‐Z concentration associated with mortality at 90 days or 1 year in Cox proportional hazard models. As LP‐Z is structurally similar to LDL and has derived from VLDL like LDL, we next examined whether the proportion of LP‐Z among apoB containing lipoproteins (VLDL, LDL,s and LP‐Z) could be predictive of clinical outcomes. This ratio was named the Z‐index and ranged from 0 to 1. Every 0.1‐unit increase of the Z‐index was associated with a HR of 1.6 (95% CI 1.2–2.1, p = 0.001) at 90 days and HR of 1.3 (95% CI 1.1–1.5, p = 0.009) at 1 year (Table 2). Likely in part due to sample homogeneity in a clinical trial, MELD and DF were not predictive of 90‐day mortality in ELAD. The Z‐index remained significant after adjustment for either MELD or DF in predicting 90‐day mortality (Table 2). To find a cutoff for the Z‐index that would be clinically informative, we examined the predictive values of the Z‐index at 0.1‐unit intervals and concluded that the Z‐index was more useful in predicting survival than mortality. Among the 38 patients with a baseline Z‐index ≤ 0.6, 36 individuals (95%) survived at 90 days despite a median MELD of 25 at baseline. A Z‐index ≤ 0.6 had a specificity of 91% and a positive predictive value of 95% in predicting 90‐day survival. Conversely, most of the mortality occurred when the baseline Z‐index was >0.6, with a log‐rank p value of 0.0004 (Figure 3A, Table S9). We also examined whether changes in Z‐index carried prognostic value. On day 14, those with the Z‐index decreased by 0.1 unit or more from baseline had a significantly higher rate of 90‐day survival compared to their counterparts, with no significant decreases in the Z‐index (Figure 3B). This indicated that repeated measurements of the Z‐index could inform the trajectory of patients during treatment.
TABLE 2.
Predictive Values of Z‐index, MELD, and DF for 90‐Day Mortality in the ELAD Cohort
| HR | 95% CI | P Value | |
|---|---|---|---|
| Univariate analysis | |||
| Z‐index a | 1.61 | 1.23–2.12 | 0.001 |
| Creatinine | 1.55 | 1.11–2.16 | 0.01 |
| Bilirubin | 1.01 | 0.97–1.06 | 0.6 |
| INR | 0.67 | 0.24–1.82 | 0.4 |
| MELD | 1.07 | 0.97–1.19 | 0.2 |
| DF | 1.01 | 0.99–1.03 | 0.5 |
| Multivariate analysis | |||
| Z‐index (model 1) b | 1.60 | 1.21–2.11 | 0.001 |
| Z‐index (model 2) c | 1.62 | 1.23–2.14 | 0.001 |
Abbreviation: INR, international normalized ratio.
Z‐index in the unit of 0.1 (0–10) used in Cox proportional hazard regression.
Model 1 adjusted for MELD.
Model 2 adjusted for DF.
FIGURE 3.
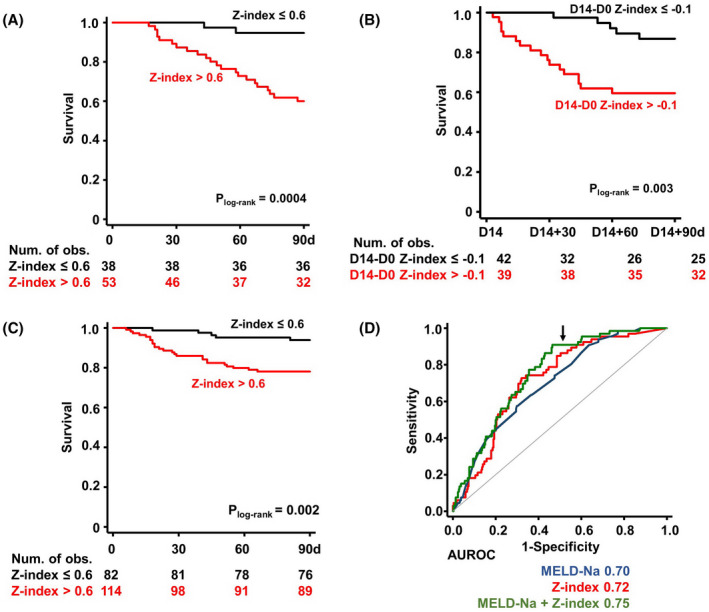
Z‐index predicts the 90‐day outcome in AH. (A) Kaplan‐Meier curves of 90‐day survival in ELAD categorized by baseline Z‐index cutoff at 0.6 (n = 91). Study subjects include both ELAD treatment and control arms. Baseline samples before ELAD treatment were used to calculate the Z‐index. (B) Kaplan‐Meier curves of 90‐day survival in ELAD categorized by the cutoff of changes in Z‐index by −0.1 on day 14 (n = 81). (C) Kaplan‐Meier curves of 90‐day survival in TREAT categorized by baseline Z‐index cutoff at 0.6 (n = 196). At‐risk tables are shown in the bottom of each panel. P values calculated by log‐rank tests. (D) Comparison of receiver operating characteristic (ROC) curves of Model for End‐Stage Liver Disease–Sodium (MELD‐Na), Z‐index, and MELD‐Na + Z‐index in predicting 90‐day mortality. Areas under the ROC (AUROCs) are provided under the plot. Arrow indicates specificity of 0.5
The predictive value of the Z‐index was validated in two additional cohorts: patients with AH in the TREAT consortium and the BIDMC AH registry. In Cox proportional hazard analysis, every 0.1‐unit increase of the Z‐index was associated with a HR of 1.3 (95% CI 1.1–1.6, p = 0.001) in predicting 90‐day mortality. A Z‐index ≤ 0.6 significantly predicted survival in Kaplan‐Meier analysis (log‐rank p value = 0.002) (Figure 3C) and has a specificity of 83.3% in predicting 90‐day survival with a positive predictive value of 93.9% (Table S9). In the BIDMC AH registry, 13 patients had a Z‐index ≤ 0.6 at enrollment, and mortality occurred only in 1 patient (7.7%) within 90 days. This translated to a similar specificity of 85% and a positive predictive value of 92.3%. Of note, in the BIDMC registry, 5 patients received a liver transplant, only 2 of whom had a baseline Z‐index more than 0.6.
We compared the performance of Z‐index against existing scores to prognosticate outcomes in AH. Most current prognostic scores of AH outcome use composite indices calculated by multiple measurements of liver and organ failure, whereas the Z‐index is a singular measurement of altered lipoprotein metabolism. In the TREAT consortium, the Z‐index as well as MELD, DF, and ABIC[ 18 ] demonstrated robust association with 90‐day mortality using Cox proportional hazard models (Table S10). We compared the performance of Z‐index against existing scores to prognosticate outcomes in AH. Most current prognostic scores of AH outcome use composite indices calculated by multiple measurements of liver and organ failure, whereas the Z‐index is a singular measurement of altered lipoprotein metabolism. In the TREAT consortium, the Z‐index, as well as MELD, DF and ABIC, demonstrated robust association with 90‐day mortality using Cox proportional hazard models with pooled data of all four cohorts, the area under the receiver operating characteristic curve (AUROC) of the Z‐index and MELD in predicting 90‐mortality was 0.72 versus 0.70, respectively (Figure 3D). A regression model that combined MELD and Z‐index further increased the AUROC to 0.75. The ROC curve shape of the Z‐index and combined model differed from that of MELD by exhibiting better performance in sensitivity, in keeping with its utility in predicting survival. At a specificity of 0.5, Z‐index improved the sensitivity of predicting 90‐day mortality from MELD at 0.75 to 0.85, whereas the combined model improved the sensitivity further to 0.91.
LP‐Z results from impaired extrahepatic lipoprotein metabolism in AH
We next investigated the mechanism of LP‐Z accumulation in AH. The liver regulates lipoprotein metabolism by producing key apolipoproteins, such as apoB and apolipoprotein A‐I, and circulating enzymes (LCAT and HL) that convert VLDL to LDL (Figure 4A). To examine the expression of lipoprotein‐related genes in AH, we compared the mRNA levels in liver biopsy and explant tissues from patients with AH to liver biopsies from control subjects as previously reported.[ 13 ] RNA‐sequencing data suggested that AH was associated with a significant down‐regulation of genes involved in lipoprotein metabolism, including APOA1, LIPC (HL), and LCAT (Figure 4B, Figure S2). Further analysis segregating patients with AH with an increasing degree of disease severity showed a gradual decline in LCAT and LIPC expression, but not changes in APOB expression (Figure 4C). Other down‐regulated genes included APOA2, APOA5, APOC1, APOC3, CETP, MTTP, and DGAT2, whereas up‐regulated genes included SOAT1, LDLR, and LRP1 (Figure S2). To validate these findings, we measured the LCAT and lipase activities in the serum of patients with AH and found that they were significantly lower than those in NAFLD controls (Figure 4D). When these enzyme activities were compared with Z‐index, we found that subjects with Z‐index >0.6 had lower lipase but similar LCAT activity compared to those with Z index ≤ 0.6 (Figure 4E). Together, these observations support the premise that the down‐regulation and functional deficiency of LCAT and LIPC is at least partially responsible for the rise of the Z‐index in AH.
FIGURE 4.
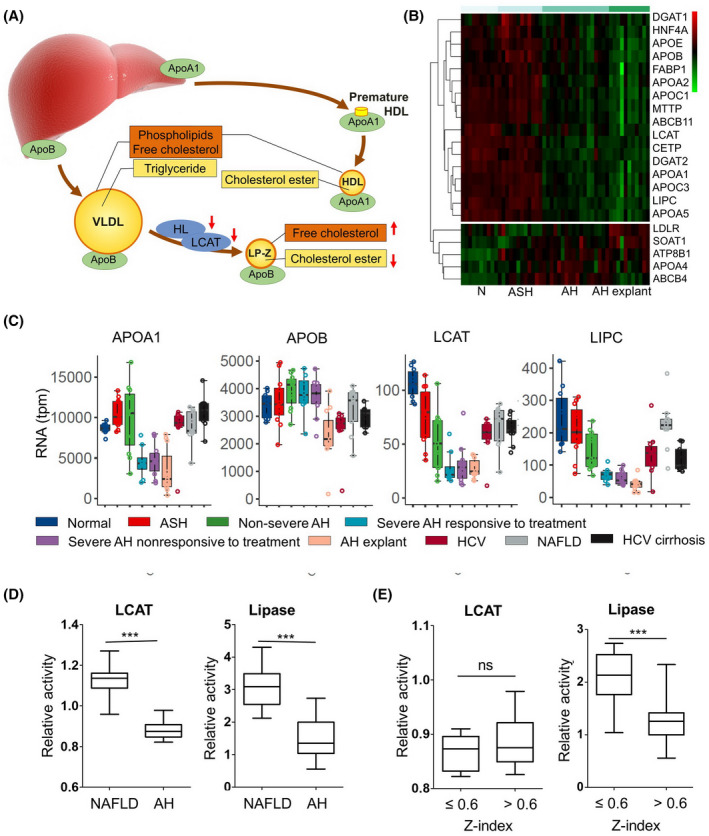
Mechanism for the accumulation of LP‐Z in AH. (A) A diagram of lipoprotein metabolism highlights liver‐derived apolipoproteins and enzymes responsible for the extrahepatic lipoprotein metabolism: lecithin cholesterol acyltransferase (LCAT) and hepatic lipase (HL) (i.e., lipase C hepatic type [LIPC]). (B) Heat map of genes responsible for lipoprotein metabolism from liver biopsies of healthy controls (N), early alcohol‐associated steatohepatitis (eASH), AH, and explants from patients with AH who underwent liver transplant. (C) Box plot comparisons of APOA1, APOB, LCAT, LIPC (HL), and CETP among healthy controls, eASH, mild–moderate AH, severe AH 90‐day survivors, severe AH 90‐day nonsurvivors, AH explant, HCV, NAFLD, and compensated HCV cirrhosis. (D) Serum activities of LCAT and total lipase among patients with AH (n = 39) and NAFLD controls (n = 40). (E) Serum activities of LCAT and total lipase among individuals with Z‐index ≤ 0.6 (n = 13) and Z‐index >0.6 (n = 26). P values calculated by a two‐sided t tests. ***p value < 0.001. Abbreviations: APOA1, apolipoprotein A1; APOB, apolipoprotein B; CETP, cholesteryl ester transfer protein
LP‐Z contributes to FC‐medicated hepatocellular toxicity in AH
High levels of FC are cytotoxic.[ 19 ] Compared with LDL, a key characteristic of LP‐Z is its high concentration of FC, suggesting a potential pathogenic role in AH. We compared the distribution and amount of FC in cryo‐preserved tissue from liver explants of patients with AH who underwent transplant and in control liver tissues without AH. In the liver tissue without AH, FC was observed in discontinuous clusters in the plasma membrane and intracellular membranous organelles as stained by filipin (arrows, Figure 5A). FC content in hepatocytes of controls was homogeneous within the tissue and across subjects (Figure 5B). In AH, FC content was heterogenous both from cells‐to‐cells within the same tissue and across patients with AH. Some hepatocytes in AH explant liver demonstrated intense FC staining within a significant portion of the cytoplasm as demonstrated in heatmap reconstruction of five serial focus stacks of images, suggesting a state of FC overload in these cells (Figure 5A, bottom panels). To test whether LP‐Z in AH could be toxic to hepatocytes, we purified lipoproteins at LDL density from patients with high Z‐index (0.89 ± 0.02), and compared to LDL from patients with Z‐indices of zero. LP‐Z at serum concentration showed significantly higher toxicity than LDL to HepG2 cells in vitro (Figure 5C). We then tested whether FC in LP‐Z was the cause of this hepatotoxicity. We synthesized LP‐Z‐like emulsion particles (rLP‐Z) that reproduced the structure of LP‐Z (Figure S3). To track the uptake of FC by human hepatocytes, rLP‐Z was green fluorescence–labeled with Topfluor FC. When rLP‐Z was added to human hepatocytes in culture, in 2 hours FC from rLP‐Z (green) entered hepatocytes and almost completely overlapped with the filipin signal that stained the entire intracellular cholesterol pool (blue) (Figure 5D). In 6 hours, significant cell death was noted in human hepatocytes, as measured by the propidium iodide uptake assay (Figure 5E). We compared the toxicity of rLP‐Z with various FC concentrations to test the direct relationship between FC content and cytotoxicity using cultured HepG2. Exposure to rLP‐Z at a particle concentration similar to those observed in patients with AH induced FC concentration–dependent cell death, reaching its maximum in 6 hours (Figure 5F). This observation supports FC‐mediated hepatocellular toxicity by LP‐Z.
FIGURE 5.
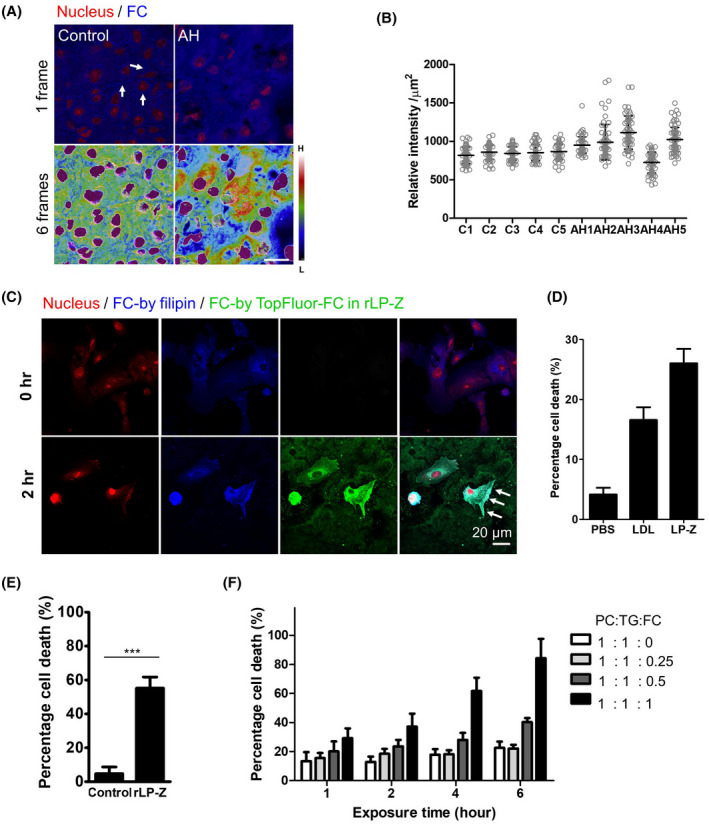
LP‐Z may exacerbate FC‐mediated hepatotoxicity. (A) Confocal microscopy images of explants from patients with AH compared with non‐AH controls. Samples were cryo‐preserved without formalin fixation to preserve lipids. Top panels: Arrows highlight segregated FC in membranous structures stained by filipin. Bottom panels: Heatmap of five Z‐stack series of FC signals. Scale bar represents 10 µm. (B) Hepatocellular FC concentrations in AH and controls. The relative intracellular FC concentration was measured by filipin intensity of 60 5‐µm‐diameter circular areas from 10 representative hepatocytes on 6 unedited micrographs in 5 AH explants (AH1–5) and 5 controls (C1–5). (C) Toxicity of LP‐Z and LDL in HepG2 cells. LP‐Z and LDL are purified from pooled plasma of patients with AH based on the Z‐index (0.89 ± 0.02 for LP‐Z vs. 0 for LDL). (D) Confocal microscopy images of FC uptake from reconstituted LP‐Z‐like emulsion particles (rLP‐Z) by human hepatocytes. rLP‐Z was prepared with TopFluor FC tracer (green) at 33 mg/dL FC and 1:1:0.5 PC:TG:FC ratio. Arrows indicate an expansion of the plasma membrane with a focally high concentration of FC. Scale bar represents 20 µm. In (A)–(C), filipin was used to stain FC, and propidium iodide (PI) to stain nucleic acids. (E) Toxicity of rLP‐Z in human hepatocytes. rLP‐Z concentration was the same as (D). (F) Impact of FC on rLP‐Z‐induced hepatocellular toxicity. rLP‐Z was prepared with 67 mg/dL TG, 67 mg/dL PC, and 0‐67 mg/dL FC with indicated ratio. In (C), (E) and (F), cell viability was measured by PI uptake assay with 0.1% Triton‐X100 as positive controls (100% cell death). Error bars represent standard error. **p < 0.01; ***p < 0.001
DISCUSSION
We report a distinctive elevation of circulating LP‐Z in AH. This recently identified abnormal lipoprotein evades detection by a conventional lipid panel, but can be readily detected and quantified by NMR.[ 8 ] We devised a Z‐index based on the ratio of LP‐Z to total apoB‐containing lipoproteins, and demonstrated that it predicted 90‐day survival among patients with AH independent of their MELD or DF. Together, these observations suggest the potential clinical utility of LP‐Z for the risk stratification of patients with AH and provide insights into the role of abnormal lipoprotein metabolism in the pathophysiology of AH.
Proposed diagnostic utility of LP‐Z and Z‐index
We showed that the accumulation of LP‐Z is specific to AH in comparison to other forms of liver disease, including cirrhosis and heavy drinkers without liver damage. LP‐Z was found in 2 of 114 patients with advanced fibrosis or cirrhosis, 2 of 10 pretransplant patients, and 3 of 11 patients with decompensated alcohol‐associated cirrhosis who had maintained sobriety. LP‐Z‐like lipoproteins were first described in obstructive jaundice by Kostner et al. in 1976.[ 20 ] Sabesin subsequently reported abnormal lipoproteins in 4 patients with AH with decreased CE, increased FC, and suggested that functional LCAT deficiency could have explained the observation.[ 21 ] Unfortunately, this unique anomaly of lipoprotein metabolism in AH was not further investigated over the next 40 years, largely due to the difficulty of its measurement. Because rapid quantitation of LP‐Z can now be routinely assessed clinically by NMR lipoprotein profiling, evaluation of its diagnostic value is now possible.
Our data provided evidence that LP‐Z measurement can improve the diagnosis and prognostication of AH outcomes. A significantly elevated serum level of LP‐Z is strongly supportive of a diagnosis of AH when other competing diagnoses are present (Figure 1F). As shown in Figure 2A,C, a low Z‐index less than 0.6 robustly predicts 90‐day survival in AH independent from the MELD, the current standard in liver transplant candidate selection. Compared with MELD, which includes parameters indicative of liver failure and hemodynamic decompensation, the Z‐index reflects changes in lipoprotein metabolism, a process previously not well‐studied in liver failure. Although the Z‐index AUROC is comparable to that of MELD, it differs in the shape of the ROC curves, indicating complementary utility. The Z‐index offered superior accuracy in predicting survival (sensitivity of mortality) to MELD (Figure 2D). The predictive power of the Z‐index was greater for short‐term outcomes than long‐term outcomes, in keeping with the complexity of factors contributing to the long‐term survival of patients with AH. A decline in the Z‐index, when measured over time is predictive of a favorable outcome. Because NMR lipoprotein profiling is in widespread clinical use, calculation of the Z‐index can be obtained expediently at a low cost. An increasing number of transplant centers have started to offer early liver transplant for patients with severe alcohol‐associated hepatitis that fail medical therapy.[ 22 , 23 ] The Z‐index could be useful in the risk stratification of patients with AH with high MELD scores, particularly among potential candidates for urgent liver transplantation.
Implication of LP‐Z in AH pathogenesis
The LP‐Z concentration itself is not directly correlated with AH outcome, but instead, the Z‐index, the ratio between LP‐Z and apoB‐containing lipoproteins, is prognostic. This observation of LP‐Z in AH may have important biological implications. The circulating concentration of LP‐Z is determined by the rate of VLDL production, VLDL‐to‐LP‐Z conversion, and LP‐Z clearance. LP‐Z is likely cleared by the LDLR, a process occurring in both hepatocytes and non‐hepatic cells. Meanwhile, the Z‐index reflects specifically the impairment in VLDL‐to‐LDL metabolism. Extrahepatic VLDL metabolism is driven by key enzymes produced by the liver, such as LCAT and HL. The expression of these enzymes was better correlated with AH disease severity compared with the expression of apoB, the structural component of VLDL (Figure 3C). Argemi et al. recently reported that a transcriptional hallmark of AH was defective HNF4α‐dependent gene expression.[ 13 ] HNF4α is a master transcription factor for hepatocyte differentiation, which regulates genes responsible for liver function, including lipoprotein metabolism (Figure S2).[ 24 , 25 , 26 , 27 , 28 ] The dysregulation of HNF4α leads to the dedifferentiation of hepatocytes and clinical presentation of liver failure. RNA‐sequencing analyses suggest that both LCAT and LIPC mRNA levels were associated with the dysregulation of HNF4α transcription and disease severity of AH. This finding needs to be validated by targeted mRNA quantification in future studies. To this extent, the Z‐index may reflect the extent of hepatocellular dedifferentiation with subsequent impairment of lipoprotein metabolism, a key feature of AH.
The biology of LP‐Z also suggests a state of impaired cholesterol metabolism. The FC‐to‐phospholipid ratio is tightly regulated in the cell membrane in order to prevent the accumulation of FC, which is known to cause cellular injury at high levels.[ 19 ] LP‐Z particles in AH carry a high level of FC, and as shown here, rLP‐Z at concentrations observed in AH can cause direct cytotoxicity to human hepatocytes in vitro (Figure 4D,E). We found that hepatocytes from AH explanted livers sometimes have significantly increased FC content distributed throughout the cell (Figure 4A,B). The up‐regulation of LDLR and LRP1 as AH becomes more severe could increase the uptake of LP‐Z into hepatocytes (Figure S2). Furthermore, an altered microbiome in AH may contribute to the impairment in cholesterol metabolism and exacerbate cholesterol‐mediated toxicity.[ 29 , 30 ]
Contextual considerations and unresolved questions
Our study showed a frequent accumulation of LP‐Z in AH, but has not sufficiently ruled out its presence in other liver diseases. It remains to be seen whether LP‐Z accumulation occurs in biliary diseases such as primary biliary cholangitis, primary sclerosing cholangitis, or acute liver failure. The Z‐index bears a moderate prognostic value with AUROC < 0.8. Although it is not intended to replace MELD or other existing prognostic scores, it can provide additional value due to its unique biology that is not captured by conventional indices. Our study used multiple existing cohorts, each of which provided unique strength, but also carried specific limitations. The case mix in these studies is likely to be different from real‐world clinical practice. Future prospective studies are required to validate the prognostic value of the Z‐index against other indices in prospective studies. Recently, cytokeratin 18 has been shown to predict outcomes in AH.[ 31 ] It will be interesting to compare the performance of Z‐index with CK18. Future studies may also address the impact of nutritional status and subsequent therapy as well as corticosteroid treatment on LP‐Z levels, and whether Z‐index could prognosticate responses to medical therapy. Our data suggested that cholesterol toxicity is a pivotal pathogenic factor in AH. We have not fully explored other lipid species that are differentially present in LP‐Z compared with LDL. The role of lipid toxicity in AH needs to be evaluated in animal models, or ex vivo human‐derived multicellular models, such as organoids or liver‐in‐chip. Finally, because the potential pathogenic role of cholesterol toxicity in the pathogenesis of AH, it is conceivable that cholesterol‐lowering medications, such as statins, may have beneficial effects in these patients, either preventing AH or influencing disease severity. Further mechanistic and clinical investigations should be performed to address this question.
In conclusion, we show that hepatic lipoprotein and cholesterol metabolism is profoundly impaired in AH, leading to the accumulation of abnormal LP‐Z particles in the circulation of patients with AH. LP‐Z exerts direct, FC‐mediated hepatotoxicity in vitro and may contribute to the pathogenesis of AH. The Z‐index, based on serum levels of LP‐Z, strongly predicts 90‐day survival in AH and could be used as a risk‐stratification tool in the management of patients with AH.
CONFLICT OF INTEREST
Dr. Jiang consults for Olix and received grants from Pfizer and Gilead. Dr. Sugahara is employed by Phoenixbio. Dr. Curry consults and received grants from Gilead and Sonic Incytes. He consults for Mallinckrodt. Dr. Szabo consults for Durect, Evive, Novartis, Pandion, Merck, Quest, Surrozen, Zomagen, and Pfizer. She advises Terra Firma and owns stock in Glympse. Dr. Saito consults for EA Pharma and Ajinomot. He received grants from Phoenixbio. Dr. Connelly is employed, owns stock, and holds intellectual property rights with Labcorp. Dr. Sanyal consults and received grants from Conatus, Gilead, Mallinckrodt, Immuron, Boehringer Ingelheim, Novartis, Bristol‐Myers Squibb, Merck, Eli Lilly, Novo Nordisk, Fractyl, Siemens, Madrigal, Inventiva, and Covance. He consults and owns stock in Genfit. He consults for Intercept, Pfizer, Salix, Galectin, Hemoshear, Terns, Albireo, Sanofi, Janssen, Takeda, Northsea, AMRA, Perspectum, Poxel, 89 Bio, AstraZeneca, NGM, Amgen, Regeneron, Genentech, Roche, Prosciento, Histoindex, Path AI, and Biocellvia. He received grants from Echosens‐Sandhill, Owl, and Second Genome. He owns stock in Exhalenz, Sanyal Bio, Durect, Indalo, Tiziana, and Rivus. He received royalties from Elsevier and UptoDate.
ETHICS APPROVAL STATEMENT
The study has been approved by the Institutional Review Board at Beth Israel Deaconess Medical Center for ethnics approval (IRB 2018P000055).
AUTHOR CONTRIBUTIONS
Data acquisition and analysis: K.H., M.C.P.M., J.A., E.V., I.S., E.B., L.L., G.S., H.D., K.D., S.T., H.C., H.H., Y.Y., J.V.B., M.V.C., K.M., A.P.G., S.N., S.I.B., L.H.A., Z.F., M.L., M.C., K.J.M., R.M., and Z.G.J. Study concept: M.A.C., N.P.C., R.B., and Z.G.J. Funding obtainment: A.J.S., J.D.O., T.S., M.A.C., N.P.C., R.B., and Z.G.J. Manuscript draft and revision: K.H., M.C.P.M., J.A., E.V., E.B., Y.V.P., N.A., G.S., A.J.S., T.S., M.A.C., N.P.C., R.B., and Z.G.J.
Supporting information
ACKNOWLEDGMENT
The authors thank Dr. Zhaoli Sun at the Department of Surgery, John Hopkins Medical Center, for providing human liver explant from patients with AH and control tissues; Dr. Robert Brocia at Roar Biomedical for enzyme activity measurements; Dr. C. James McKnight at the Department of Physiology and Biophysics, Boston University School of Medicine, for assistance in the isolation of lipoproteins; investigators at Vital Therapies for providing samples of patients with AH; investigators and colleagues at LabCorp for the testing of patient samples; Ms. Susan McDermott for the assistance with clinical studies; and Dr. J. Thomas Lamont for the critical review in the preparation of this manuscript at Division of Gastroenterology and Hepatology, Beth Israel Deaconess Medical Center.
Hu K, Perez‐Matos MC, Argemi J, Vilar‐Gomez E, Shalaurova I, Bullitt E, et al. Lipoprotein Z, a hepatotoxic lipoprotein, predicts outcome in alcohol‐associated hepatitis. Hepatology. 2022;75:968–982. doi: 10.1002/hep.32203
Kunpeng Hu, Maria C. Perez‐Matos, Josepmaria Argemi, and Eduardo Vilar‐Gomez contributed equally to this work.
Funding information
The Alan Hofmann Clinical and Translational Research Award from the American Associations for the Study of Liver Diseases (AASLD) and Gilead Research Scholar Award from Gilead Science (to Z.G.J.); research grants from the University Hospital of Lausanne; the Novartis Foundation for Medical‐Biological Research and the Gottfried und Julia Bangerter‐Rhyner Foundation (to J.V.B.); an Advanced/Transplant Hepatology Award from AASLD (to Z.F.); a PhoenixBio Research Grant (to T.S.); Irving W. and Charlotte F. Rabb Award (to Z.G.J. and Y.V.P.), a research grant from PSC Partners Seeking a Cure Canada (to Y.V.P.); National Institute of Diabetes and Digestive and Kidney Disease grant (K08DK115883 to Z.G.J., K23DK083439 to M.L., and DK10596 to A.J.S.); National Institute of Alcohol Abuse and Alcoholism grants (U01AA021908 and U01AA02082 to R.B., 1U01AA026979 to A.J.S., and 5U01AA021883‐04, 5U01AA021891‐04, 5U01AA021788‐04, and 5U01AA021840‐04 to N.P.C.); and National Center for Advancing Translational Sciences (UL1TR002649 to A.J.S.).
Contributor Information
Ramon Bataller, Email: bataller@pitt.edu.
Z. Gordon Jiang, Email: zgjiang@bidmc.harvard.edu.
REFERENCES
- 1. Tapper EB, Parikh ND. Mortality due to cirrhosis and liver cancer in the United States, 1999–2016: observational study. BMJ. 2018;362:k2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mathurin P, Bataller R. Trends in the management and burden of alcoholic liver disease. J Hepatol. 2015;62:S38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liangpunsakul S. Clinical characteristics and mortality of hospitalized alcoholic hepatitis patients in the United States. J Clin Gastroenterol. 2011;45:714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thursz MR, Richardson P, Allison M, Austin A, Bowers M, Day CP, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med. 2015;372:1619–28. [DOI] [PubMed] [Google Scholar]
- 5. Goldberg D, Ditah IC, Saeian K, Lalehzari M, Aronsohn A, Gorospe EC, et al. Changes in the prevalence of hepatitis C virus infection, nonalcoholic steatohepatitis, and alcoholic liver disease among patients with cirrhosis or liver failure on the waitlist for liver transplantation. Gastroenterology. 2017;152:1090–9.e1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belli LS, Perricone G, Adam R, Cortesi PA, Strazzabosco M, Facchetti R, et al. Impact of DAAs on liver transplantation: major effects on the evolution of indications and results. An ELITA study based on the ELTR registry. J Hepatol. 2018;69:810–7. [DOI] [PubMed] [Google Scholar]
- 7. Lee BP, Im GY, Rice JP, Weinberg E, Hsu C, Fix OK, et al. Underestimation of Liver transplantation for alcoholic hepatitis in the national transplant database. Liver Transpl. 2019;25:706–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bedi S, Garcia E, Jeyarajah EJ, Shalaurova I, Perez‐Matos MC, Jiang ZG, et al. Characterization of LP‐Z lipoprotein particles and quantification in subjects with liver disease using a newly developed NMR‐based assay. J Clin Med. 2020;9:2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rousset X, Vaisman B, Amar M, Sethi AA, Remaley AT. Lecithin: cholesterol acyltransferase—from biochemistry to role in cardiovascular disease. Curr Opin Endocrinol Diabetes Obes. 2009;16:163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crabb DW, Bataller R, Chalasani NP, Kamath PS, Lucey M, Mathurin P, et al. Standard definitions and common data elements for clinical trials in patients with alcoholic hepatitis: recommendation from the NIAAA alcoholic hepatitis consortia. Gastroenterology. 2016;150:785–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang ZG, Tapper EB, Connelly MA, Pimentel CFMG, Feldbrügge L, Kim M, et al. Steatohepatitis and liver fibrosis are predicted by the characteristics of very low density lipoprotein in nonalcoholic fatty liver disease. Liver Int. 2016;36:1213–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lourens S, Sunjaya DB, Singal A, Liangpunsakul S, Puri P, Sanyal A, et al. Acute alcoholic hepatitis: natural history and predictors of mortality using a multicenter prospective study. Mayo Clin Proc Innov Qual Outcomes. 2017;1:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Argemi J, Latasa MU, Atkinson SR, Blokhin IO, Massey V, Gue JP, et al. Defective HNF4alpha‐dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat Commun. 2019;10:3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jeyarajah EJ, Cromwell WC, Otvos JD. Lipoprotein particle analysis by nuclear magnetic resonance spectroscopy. Clin Lab Med. 2006;26:847–70. [DOI] [PubMed] [Google Scholar]
- 15. Matyus SP, Braun PJ, Wolak‐Dinsmore J, Jeyarajah EJ, Shalaurova I, Xu Y, et al. NMR measurement of LDL particle number using the Vantera Clinical Analyzer. Clin Biochem. 2014;47:203–10. [DOI] [PubMed] [Google Scholar]
- 16. Lefevre M. Localization of lipoprotein unesterified cholesterol in nondenaturing gradient gels with filipin. J Lipid Res. 1988;29:815–8. [PubMed] [Google Scholar]
- 17. Liu Y, Luo D, Atkinson D. Human LDL core cholesterol ester packing: three‐dimensional image reconstruction and SAXS simulation studies. J Lipid Res. 2011;52:256–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dominguez M, Rincón D, Abraldes JG, Miquel R, Colmenero J, Bellot P, et al. A new scoring system for prognostic stratification of patients with alcoholic hepatitis. Am J Gastroenterol. 2008;103(11):2747–56. [DOI] [PubMed] [Google Scholar]
- 19. Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J Clin Invest. 2002;110:905–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kostner GM, Laggner P, Prexl HJ, Holasek A. Investigation of the abnormal low‐density lipoproteins occurring in patients with obstructive jaundice. Biochem J. 1976;157:401–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sabesin SM, Hawkins HL, Kuiken L, Ragland JB. Abnormal plasma lipoproteins and lecithin‐cholesterol acyltransferase deficiency in alcoholic liver disease. Gastroenterology. 1977;72:510–8. [PubMed] [Google Scholar]
- 22. Mathurin P, Moreno C, Samuel D, Dumortier J, Salleron J, Durand F, et al. Early liver transplantation for severe alcoholic hepatitis. N Engl J Med. 2011;365:1790–800. [DOI] [PubMed] [Google Scholar]
- 23. Lee BP, Mehta N, Platt L, Gurakar A, Rice JP, Lucey MR, et al. Outcomes of early liver transplantation for patients with severe alcoholic hepatitis. Gastroenterology. 2018;155:422–30.e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Duncan SA, Nagy A, Chan W. Murine gastrulation requires HNF‐4 regulated gene expression in the visceral endoderm: tetraploid rescue of Hnf‐4(‐/‐) embryos. Development. 1997;124:279–87. [DOI] [PubMed] [Google Scholar]
- 25. Inoue Y, Peters LL, Yim SH, Inoue J, Gonzalez FJ. Role of hepatocyte nuclear factor 4alpha in control of blood coagulation factor gene expression. J Mol Med. 2006;84:334–44. [DOI] [PubMed] [Google Scholar]
- 26. Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol. 2001;21:1393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li J, Ning G, Duncan SA. Mammalian hepatocyte differentiation requires the transcription factor HNF‐4alpha. Genes Dev. 2000;14:464–74. [PMC free article] [PubMed] [Google Scholar]
- 28. Inoue Y, Hayhurst GP, Inoue J, Mori M, Gonzalez FJ. Defective ureagenesis in mice carrying a liver‐specific disruption of hepatocyte nuclear factor 4alpha (HNF4alpha). HNF4alpha regulates ornithine transcarbamylase in vivo. J Biol Chem. 2002;277:25257–65. [DOI] [PubMed] [Google Scholar]
- 29. Ghazalpour A, Cespedes I, Bennett BJ, Allayee H. Expanding role of gut microbiota in lipid metabolism. Curr Opin Lipidol. 2016;27:141–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duan YI, Llorente C, Lang S, Brandl K, Chu H, Jiang LU, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. 2019;575:505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Atkinson SR, Grove JI, Liebig S, Astbury S, Vergis N, Goldin R, et al. In severe alcoholic hepatitis, serum keratin‐18 fragments are diagnostic, prognostic, and theragnostic biomarkers. Am J Gastroenterol. 2020;115:1857–68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials