

Abstract
Background and Aims
HEV infection is the most common cause of liver inflammation, but the pathogenic mechanisms remain largely unclear. We aim to explore whether HEV infection activates inflammasomes, crosstalk with antiviral interferon response, and the potential of therapeutic targeting.
Approach and Results
We measured IL‐1β secretion, the hallmark of inflammasome activation, in serum of HEV‐infected patients and rabbits, and in cultured macrophage cell lines and primary monocyte‐derived macrophages. We found that genotypes 3 and 4 HEV infection in rabbits elevated IL‐1β production. A profound increase of IL‐1β secretion was further observed in HEV‐infected patients (1,733 ± 1,234 pg/mL; n = 70) compared to healthy persons (731 ± 701 pg/mL; n = 70). Given that macrophages are the drivers of inflammatory response, we found that inoculation with infectious HEV particles robustly triggered NOD‐like receptor family pyrin domain‐containing 3 (NLRP3) inflammasome activation in primary macrophages and macrophage cell lines. We further revealed that the ORF2 capsid protein and the formed integral viral particles are responsible for activating inflammasome response. We also identified NF‐κB signaling activation as a key upstream event of HEV‐induced NLRP3 inflammasome response. Interestingly, inflammasome activation antagonizes interferon response to facilitate viral replication in macrophages. Pharmacological inhibitors and clinically used steroids can effectively target inflammasome activation. Combining steroids with ribavirin simultaneously inhibits HEV and inflammasome response without cross‐interference.
Conclusions
HEV infection strongly activates NLRP3 inflammasome activation in macrophages, which regulates host innate defense and pathogenesis. Therapeutic targeting of NLRP3, in particular when combined with antiviral agents, represents a viable option for treating severe HEV infection.
Abbreviations
- ALP
alkaline phosphatase
- Casp‐1
caspase‐1
- gRNA
genomic RNA
- GTs
genotypes
- H&E
hematoxylin‐eosin
- IFIT1
IFN‐induced protein with tetratricopeptide repeats 1
- IFN
interferon
- ISG
IFN‐stimulated gene
- ISRE
interferon‐stimulated response element
- JAK
Janus kinase
- LPS
lipopolysaccharides
- MX1
MX dynamin‐like GTPase 1
- NLRP3
NOD‐like receptor family pyrin domain‐containing 3
- OASL
2′‐5′‐oligoadenylate synthetase‐like
- PBMCs
peripheral blood mononuclear cells
- PMA
phorbol 12‐myristate 13‐acetate
- RIG‐I
retinoic‐acid–inducible gene I
- STAT1
signal transducer and activator of transcription 1
- UV
ultraviolet
- wpi
weeks postinoculation
INTRODUCTION
HEV is a nonenveloped single‐stranded RNA virus, encoding three proteins, including ORF1, ‐2, and ‐3. Although only one serotype, there are eight genotypes (GTs) and at least GTs 1, 2, 3, 4, and 7 have been reported to infect humans.[ 1 ] HEV infection is usually asymptomatic and self‐limiting in healthy persons. However, acute infection with GT1 HEV in pregnant women may lead to severe liver inflammation with high mortality. In immunocompromised organ transplant recipients, exposure to GT3 or ‐4 HEV bears the high risk of developing chronic hepatitis E with accelerated progression to liver fibrosis and cirrhosis.[ 2 ]
HEV‐infection–caused liver diseases are intricately related to dysregulation of immune responses.[ 3 ] Hepatic macrophages, consisting of liver resident Kupffer cells and monocyte‐derived macrophages, play a central role in immunosurveillance of the homeostatic liver and in initiating inflammatory response when encountering pathogen invasion. In acute hepatitis E patients, the frequency of monocytes‐macrophages and dendritic cells are increased compared to healthy controls. However, the phagocytic activity of macrophages is significantly impaired in HEV‐infected pregnant women who develop acute liver failure compared to those who do not.[ 4 ] Moreover, several proinflammatory cytokines, including interferon (IFN)‐γ, TNF‐ɑ, IL‐10, and IL‐18, have been reported to be increased and associated with adverse outcomes in HEV patients.[ 5 , 6 ]
Inflammasome activation is a hallmark for the pathogenesis and progression of many inflammatory diseases.[ 7 ] In human macrophages, inflammasomes are cytosolic sensors that detect pathogens and subsequently trigger maturation and release of several proinflammatory cytokines, in particular IL‐1β.[ 8 ] Currently, the best‐characterized inflammasome is the NOD‐like receptor family pyrin domain‐containing 3 (NLRP3) inflammasome. Full activation of the NLRP3 inflammasome usually requires two signals. The priming signal is triggered by various pathogen‐associated molecular patterns or damage‐associated molecular patterns, which induce the synthesis of pro‐IL‐1β and NLRP3 through activation of NF‐κB. The second signal triggers assembly into the NLRP3 inflammasome complex, leading to caspase‐1 autoactivation, cleavage of pro‐IL‐1β, and release of the mature IL‐1β.[ 9 , 10 ]
IFN and inflammatory responses constitute two major arms of innate immunity. IFN response is the first line of defense against many viral infections. Pathogens are recognized by different pattern recognition receptors, including Toll‐like receptors and retinoic‐acid–inducible gene I (RIG‐I)‐like receptors, and subsequently trigger IFN production. Secreted IFN binds to their cognate receptors on the membrane of cells to induce hundreds of interferon‐stimulated genes (ISGs) through the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway that eventually counteract viral infections.[ 11 , 12 ] We previously have demonstrated that HEV infection is capable of activating IFN response in experimental models and in the liver of acute HEV patients.[ 13 ] However, the field remains open for study on whether HEV infection activates the inflammasome and the possibility of interacting with antiviral IFN response.
This study explored in vivo evidence of inflammasome activation in HEV‐infected rabbits and patients by measuring IL‐1β secretion in serum, a hallmark of inflammasome activation. We next investigated the mechanism of action, crosstalk with antiviral IFN response, and potential of therapeutic targeting in cultured human macrophages.
PATIENTS AND METHODS
Patient materials
Serum of HEV‐infected patients (n = 70) and healthy persons (n = 70) were retrieved at The Fifth Medical Center of Chinese PLA General Hospital (Beijing, China). Concentrations of IL‐1β were measured by ELISA. Four liver biopsies from patients diagnosed with acute or chronic hepatitis E were stained with anti‐HEV ORF2 antibody and hematoxylin‐eosin (H&E). One biopsy from a hepatic hemangioma patient without HEV infection served as a control. This study was approved by the medical ethical committee of the Fifth Medical Center of Chinese PLA General Hospital. Informed consent was waived by IBR.
HEV infection in rabbits
White rabbits were inoculated with GT3 (GenBank Accession: JX109834) and GT4 (GenBank Accession: MT993748) HEV as described.[ 14 ] Blood samples were collected preinoculation and every 2 weeks after for measuring IL‐1β. Viral titers were quantified in collected fecal samples by qRT‐PCR. Liver tissues were stained with H&E. The animal study was approved by the Committee of Laboratory Animal Welfare and Ethics, Peking University Health Science Center, China.
Cell culture
Human monocytic cell lines (THP‐1, HL60, and U937) were cultured in RPMI 1640 medium (ThermoFisherScientific, Waltham, MA, USA), complemented with 10% (v/v) inactivated fetal bovine serum (FBS) with 100 IU/mL of penicillin and 100 mg/mL of streptomycin. For macrophage differentiation, THP‐1 and HL60 cells were treated with 15 and 40 ng/mL of phorbol 12‐myristate 13‐acetate (PMA) at 37°C for 48 hours, respectively. Then, cells were cultured for another 24 hours without PMA.
Primary macrophages were differentiated from monocytes as described.[ 15 ] Briefly, peripheral blood mononuclear cells (PBMCs) were isolated from healthy donors (Sanquin, The Netherlands) by Ficoll density gradient centrifugation. Monocytes were isolated from PBMCs through plastic adherence in Iscove’s modified Dulbecco’s medium (IMDM), supplemented with 2% human serum. Obtained monocytes were cultured in IMDM (with ultraglutamine), complemented with 8% (v/v) inactivated FBS, 1% penicillin/streptomycin, and 50 ng/mL of macrophage colony‐stimulating factor, for 7 days to generate macrophages. Expression of monocyte (day 0) markers and macrophage (day 7) markers was analyzed by flow cytometry. All samples were measured on a FACSCanto II flow cytometer (BD Biosciences) and analyzed using FlowJo software (version 10.6.1; Tree Star, Inc.).
For the IFN‐stimulated response element (ISRE) reporter model (Huh7‐ISRE‐Luc), human hepatic Huh7 cells were transduced with a lentiviral transcriptional reporter system expressing the firefly luciferase gene driven by a promoter containing multiple ISRE promoter elements (SBI Systems Biosciences, Mountain View, CA, USA). Luciferase activity represents ISRE promoter activation. Huh7‐HCV‐luciferase cells contained a subgenomic HCV bicistronic replicon (1389/NS3‐3V/LucUbiNeo‐ET) and were maintained with 250 μg/mL of G418. All cells were maintained at 37°C in a 5% CO2 atmosphere.
HEV models
GTs 1, 3, and 7 full‐length, and GT3 subgenomic, HEV genomic RNA (gRNA) were produced by using the Ambion mMESSAGE nMACHINE in vitro RNA transcription kit. The p6 subgenomic HEV replicon (p6‐Luc) contains a Gaussia princeps luciferase reporter gene.
Viral replication was tested in THP‐1 cells electroporated with full‐length and subgenomic RNA. HL60 and U937 cells were electroporated with full‐length HEV gRNA or inoculated with infectious virus particles to monitor the level of viral replication. Infectious HEV particles were produced from Huh7 cells eletroporated with GT3 HEV RNA.
Coculture of macrophages with Huh7 cells harboring HEV
THP‐1 cells were treated with 15 ng/mL of PMA at 37°C for 48 hours. Then, cells were cultured for another 24 hours without PMA. Coculture of THP‐1 macrophages with Huh7‐p6 or Huh7‐p6‐Luc cells was established at a ratio of 1:4, mimicking the relative percentages of these cell populations in the human liver.
Statistical analysis
GraphPad Prism software (version 7; GraphPad Software Inc., La Jolla, CA) was used for data analysis, using a Mann‐Whitney U test. All results are presented as mean ± SD. p values <0.05 (single asterisks in figures) were considered statistically significant, whereas p values <0.01 (double asterisks), 0.001 (triple asterisks), and 0.0001 (four asterisks) were considered highly significant. Correlation was evaluated using Pearson’s correlation coefficient.
Additional materials and methods are presented in the Supporting Information.
RESULTS
HEV infection significantly elevates IL‐1β secretion, the hallmark of inflammasome activation, in rabbits and patients
To search in vivo evidence on whether HEV infection activates inflammasome response, we initially tested a rabbit model inoculated with GT3 or ‐4 HEV (Figure 1A). Successful infection was confirmed by virus shedding in fecal samples (Figure 1B,D) and histology of liver tissues showing evidence of inflammatory cells and lymphocytes infiltration (Figure 1A). The dynamics of IL‐1β secretion and HEV shedding were analyzed for each inoculated rabbit over time (Figure S1A‐D). IL‐1β level in the serum of rabbits inoculated with GT3 HEV peaked at 6 weeks postinoculation (wpi), although it was not statistically significant (Figure 1C). This was confirmed in GT4 HEV‐inoculated rabbits. In particular, at 4 wpi, there was a significant elevation of IL‐1β level in serum (n = 9; p < 0.01; Figure 1E).
FIGURE 1.
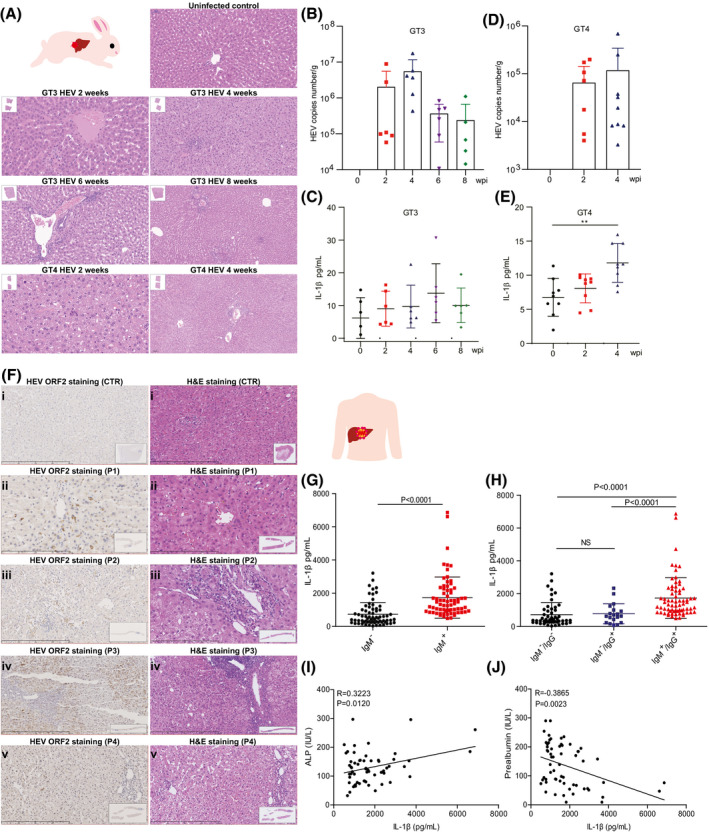
HEV infection significantly elevates IL‐1β secretion, the hallmark of inflammasome activation, in rabbits and patients. (A) Rabbits were inoculated with GT3 and ‐4 HEV. Histology showing liver inflammation in infected, but not in the control, animals. (B,C) Shedding of GT3 HEV into feces and secretion of IL‐1β into blood were measured at 0, 2, 4, 6, or 8 wpi. (D,E) Shedding of GT4 HEV and secretion of IL‐1β were measured at 0, 2, and 4 wpi. (F) i is control tissue, which was derived from a hepatic hemangioma patient without HEV infection. i showed no clear liver injury. ii, iii, iv, and v tissues were from 4 different HEV‐infected patients, respectively, showing different histological/pathological features. ii shows apparent cholestasis in hepatic bile capillary with pigment‐containing granulocytophagous cells. iii shows infiltration of inflammatory cells in the portal area. iv shows an enlarged portal area with abundant inflammatory cells with a tendency of fibrosis. v shows some punctate necrosis and infiltration of a small amount of inflammatory cells in the portal area. (G) IL‐1β levels in serum of patients (IgM positive; n = 70) and healthy persons (IgM negative; n = 70) were determined by ELISA. (H) IL‐1β levels in serum of patients (IgM positive; n = 70) and in healthy controls who never encountered HEV (IgM−/IgG−; n = 19) or who were historically exposed to HEV (IgM−/IgG+; n = 51). Correlations between ALP (I) or prealbumin (J) and IL‐1β were analyzed. Data are means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Abbreviations: CTR, control; NS, not significant (Mann‐Whitney U test); P, patient
Next, we evaluated an HEV patient cohort who were admitted to the hospital because of complications and matched with healthy controls. Immunohistochemical staining showed positivity of HEV ORF2 expression in hepatocytes, and histology showed evidence of liver inflammation (Figure 1F). Stratifying by anti‐HEV IgM status, we found significantly higher levels of IL‐1β in the serum of HEV‐infected patients (1,733 ± 1,234 pg/mL; n = 70) compared to healthy persons (731 ± 701 pg/mL; n = 70; Figure 1G). We further divided healthy persons into two groups: never encountered HEV (IgM−/IgG−; n = 51) or historically exposed to HEV (IgM−/IgG+; n = 19). We found no significant difference in IL‐1β level between these two subgroups, further supporting that only ongoing HEV infection elevated IL‐1β secretion (Figure 1H). In addition, IL‐1β level in serum was not affected by sex and age (Figure S1E,F).
We next analyzed the potential association of HEV‐induced IL‐1β secretion with biomarkers related to liver functions and diseases (Table S1). We found that alkaline phosphatase (ALP) was positively (Figure 1I), whereas prealbumin was negatively (Figure 1J), correlated with IL‐1β production. Taken together, our results from rabbits and patients strongly indicated inflammasome activation triggered by HEV infection, which provoked us to further study the mechanism.
Inoculation of HEV activates NLRP3 inflammasome in human primary macrophages and macrophage cell lines
Monocytes and macrophages are the key drivers of pathological inflammation. A recent study has reported that HEV is capable of infecting human monocytes and macrophages.[ 16 ] We found that the undifferentiated human THP‐1 monocyte cell line supports the replication of HEV by using the GT3 subgenomic (p6‐luc) and full‐length HEV (p6) models (Figure S2A‐C). Naïve Huh7 cells inoculated with conditioned cell‐culture medium from HEV‐infected THP‐1 cells resulted in active infection shown at viral RNA and protein levels (Figure S2D,E). Infection was also reached in other myeloid cell lines, including HL60 and U937 cells (Figure S2F‐H). IFN‐ɑ treatment inhibited HEV infection in THP‐1 and HL60 cells (Figure S2I‐M), but appeared to have facilitated viral replication in U937 cells with different patterns of downstream ISG induction (Figure S2N,O).
We next tested THP‐1 and HL60 macrophages differentiated by PMA treatment and confirmed their susceptibility to HEV infection (Figure S2P,Q). We further established that primary human macrophages differentiated from monocytes using isolated PBMCs (Figure 2A). We have characterized their phenotype by flow cytometry analysis of surface marker expression (Figure S2R). Consistently, human primary macrophages also moderately supported viral replication upon inoculation of HEV particles (Figure S2S). Collectively, human monocytes and macrophages appear to support the full life cycle of HEV infection, although at a low level of viral replication. However, inoculation of HEV failed to induce IL‐1β secretion in undifferentiated THP‐1 monocytes (Figure S2T,U).
FIGURE 2.
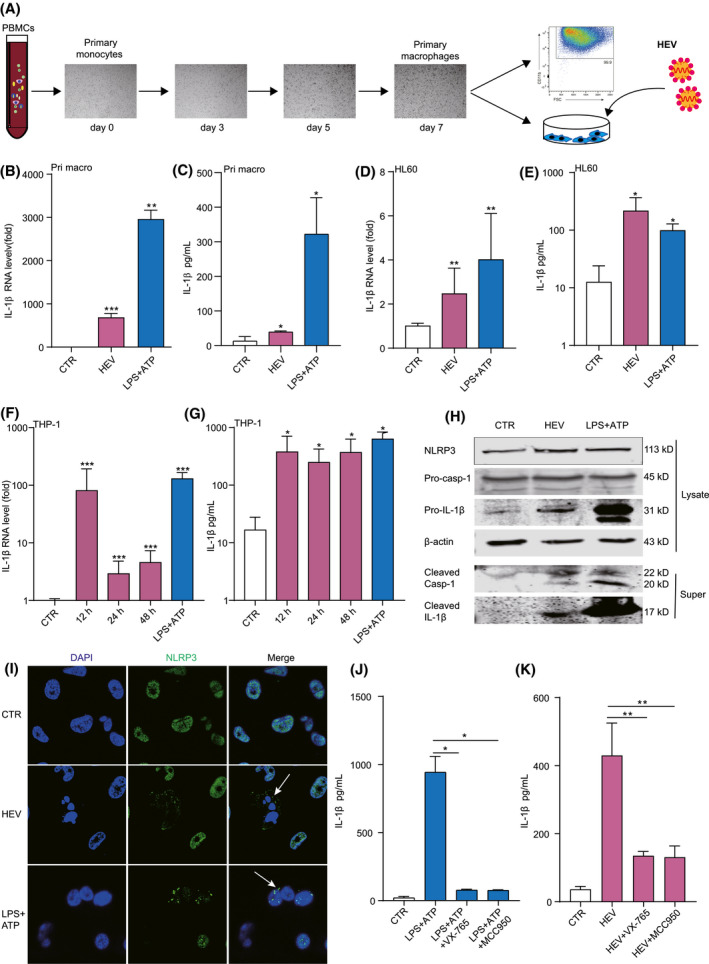
Inoculation of HEV activates NLRP3 inflammasome in human primary macrophages and macrophage cell lines. (A) Schematic illustration of generating primary macrophages from PBMC‐derived monocytes. Primary macrophages and HL60 macrophages were inoculated with HEV for 24 hours or treated with LPS (400 ng/mL) for 6 hours, followed by ATP (5 mM) for 40 minutes. IL‐1β mRNA (B,D) and protein (C,E) levels were quantified by qRT‐PCR (n = 4‐6) and ELISA (n = 4‐6), respectively. THP‐1 macrophages were inoculated with HEV for 12, 24, or 48 hours or treated with LPS (400 ng/mL) for 6 hours, followed by ATP (5 mM) for 40 minutes. IL‐1β mRNA (F) and protein (G) levels were quantified by qRT‐PCR (n = 8) and ELISA (n = 4), respectively. (H) Mature IL‐1β and cleaved Casp‐1 in supernatant or pro‐IL‐1β, pro‐Casp‐1, and NLRP3 in lysates were determined by western blotting. (I) THP‐1 macrophages were inoculated with HEV for 24 hours or treated with LPS (400 ng/mL) for 6 hours, followed by ATP (5 mM) for 40 minutes. Subcellular localizations of NLRP3 (green) and the nucleus marker, DAPI (blue), were examined under confocal microscopy. (J) THP‐1 macrophages were pretreated with 50 µM of Casp‐1 inhibitor (VX‐765) or 10 µM of NLRP3 inhibitor (MCC950) for 2 hours and then were treated with LPS (400 ng/mL) or LPS (400 ng/mL) plus 50 µM of Casp‐1 inhibitor (VX‐765) or 10 µM of NLRP3 inhibitor (MCC950) for 6 hours, followed by ATP (5 mM) for 40 minutes. IL‐1β level in supernatant was quantified by ELISA (n = 4). (K) THP‐1 macrophages were pretreated with 50 µM of Casp‐1 inhibitor (VX‐765) or 10 µM of NLRP3 inhibitor (MCC950) for 2 hours and then were incubated with HEV plus 50 µM of Casp‐1 inhibitor (VX‐765) or 10 µM of NLRP3 inhibitor (MCC950) for 24 hours. IL‐1β level in supernatant was quantified by ELISA (n = 4). NLRP3 inflammasome is marked by an arrow. Data were normalized to the control (CTR; set as 1). Data are means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Abbreviations: NS, not significant (Mann‐Whitney U test); Super, supernatant
We thus investigated whether HEV can activate inflammasome response in monocyte‐derived human primary macrophages (Figure 2A). We found that HEV inoculation significantly activated IL‐1β mRNA expression (Figure 2B) and protein secretion (Figure 2C), although to a substantially lower level, as compared to the positive control treated with lipopolysaccharides (LPS) together with ATP. Effective activation of IL‐1β mRNA expression (Figure 2D) and protein secretion (Figure 2E) were observed in the HL60 macrophage cell line. Finally, we demonstrated a robust induction of IL‐1β mRNA expression (Figure 2F) and IL‐1β production (Figure 2G) in THP‐1 macrophages upon HEV inoculation, comparable to that of LPS stimulation. Activation of both IL‐1β mRNA and protein was rapid and reached a plateau at 12 hours postinoculation. Concentrations of released IL‐1β in the supernatant of THP‐1 macrophages were 378 ± 285 pg/mL at 12 hours, 294 ± 179 pg/mL 24 hours, and 369 ± 225 pg/mL at 48 hours (Figure 2G). Western blotting confirmed the secretion of matured IL‐1β into cell supernatant (Figure 2H). Intracellular protein expression of the inflammasome effector, caspase‐1 (Casp‐1), was dramatically activated by HEV infection, and cleaved Casp‐1 protein was detected in cell supernatant (Figure 2H). Importantly, we observed NLRP3 inflammasome assembly triggered by HEV (Figure 2I). This activation by LPS or HEV could be specifically and profoundly blocked by treatment with MCC950 (NLRP3 inhibitor) or VX‐765 (Casp‐1 inhibitor; Figure 2J,K).
Taken together, both monocytes and macrophages moderately supported HEV infection, but macrophage is likely a key cell type for HEV‐targeted NLRP3 inflammasome activation.
HEV gRNA only moderately activates inflammasome response
To dissect which viral components are responsible for inflammasome response, we initially tested whether HEV gRNA is a potential inflammasome stimulus. RNA was produced by in vitro transcription of linearized HEV plasmid constructs. Transfection of GT3 HEV RNA into THP‐1 macrophages significantly induced IL‐1β mRNA transcription (Figure 3A) and secretion of mature IL‐1β (Figure 3B,C). This was further confirmed by transfection of GT1 and ‐7 HEV RNA (Figure 3D‐F), suggesting a pangenotype effect (Figure 3G). Consistently, all three genotypes of HEV RNA induced the expression of several proinflammatory cytokines in THP‐1 cells (Figure S3A‐C). In addition, inoculation of primary macrophages with HEV particles also induced the expression of proinflammatory cytokines (Figure S3D). However, level of IL‐1β production was much lower by HEV‐RNA transfection (30‐60 pg/mL) compared to that of inoculating HEV particles (300‐500 pg/mL; Figures 2G and 3H), suggesting that HEV genome per se is likely not the main activator of inflammasome response.
FIGURE 3.
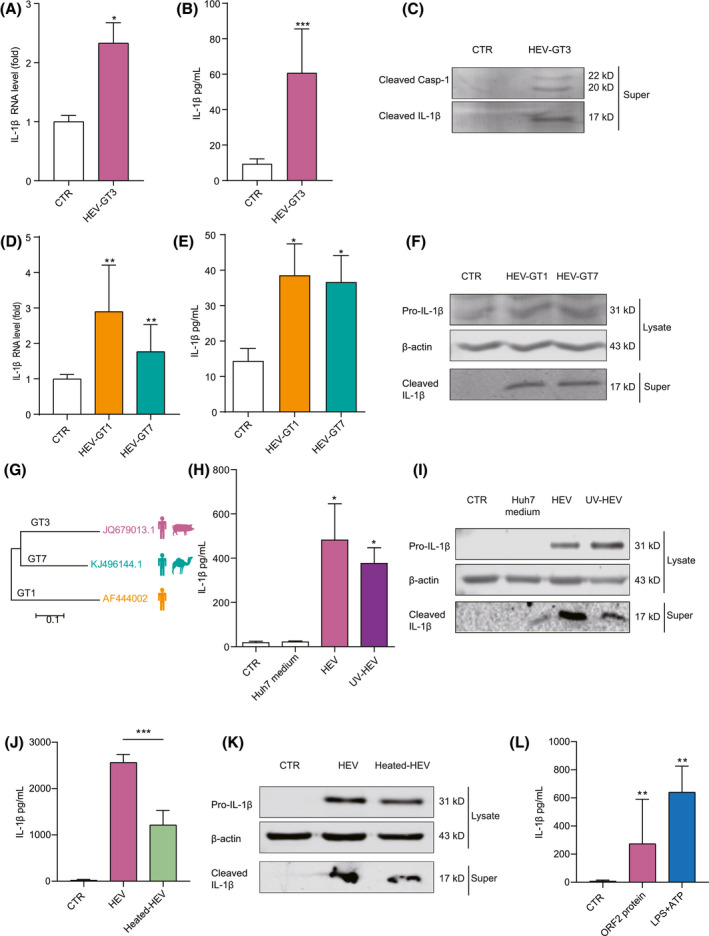
Integrity of HEV particle and ORF2 protein, but not HEV gRNA, potently activates inflammasome response. THP‐1 macrophages were transfected with GT3 HEV for 24 hours, and IL‐1β mRNA (A) was quantified by qRT‐PCR (n = 4). IL‐1β protein level (B) in supernatant was quantified by ELISA (n = 4), and (C) mature IL‐1β and cleaved Casp‐1 in supernatant were determined by western blotting. THP‐1 macrophages were transfected with GT1 and ‐7 HEV for 24 hours. IL‐1β mRNA (D) was quantified by qRT‐PCR (n = 6), and IL‐1β protein in supernatant (E) was quantified by ELISA (n = 6). (F) Mature IL‐1β in supernatant or pro‐IL‐1β in lysates were determined by western blotting. (G) Phylogenetic tree of HEV complete genome sequences. Three representative HEV strains (GenBank Accession Numbers: AF444002, JQ679013, and KJ496144) used in this study are indicated. THP‐1 macrophages were inoculated with infectious HEV and UV‐ and heat‐inactivated HEV. (H,J) IL‐1β protein in supernatant was quantified by ELISA (n = 6). (I,K) Mature IL‐1β in supernatant and pro‐IL‐1β in lysates were determined by western blotting. (L) THP‐1 macrophages were incubated with 50 µg/mL of purified recombinant ORF2 protein, and IL‐1β was measured by ELISA after 24 hours (n = 6). Data were normalized to the control (CTR; set as 1). Data are means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. Abbreviations: NS, not significant (Mann‐Whitney U test); Super, supernatant
The ORF2 capsid protein and the integral HEV particle in particular robustly activate inflammasome response
We explored whether ultraviolet (UV)‐inactivated HEV particles that have lost infectivity in macrophages (Figure S3E) can trigger inflammasome response. Surprisingly, exposure of UV‐inactivated HEV largely retained the capacity of inducing IL‐1β production in THP‐1 macrophages, comparable to that of inoculating infectious HEV particles (Figure 3H,I). UV irradiation damages viral RNA to prevent viral replication, but preserves the integrity of the immunological epitopes. In contrast, heat treatment can inactivate viruses by denaturing the secondary structures of proteins and thus is expected to disrupt the integrity of HEV particles.
Next, we perfumed heat inactivation of cell‐culture–produced infectious HEV particles and confirmed the loss of their infectivity (Figure S3F). We found that heat‐inactivated HEV remained capable of inducing lL‐1β production, but had lost >50% of potency (Figure 3J,K). These results suggest that integrity of the physical structure of the HEV particle is essential, but the disrupted capsid proteins may retain some activity in triggering inflammasome activation.
Finally, we used purified recombinant ORF2 protein produced by yeast.[ 17 ] Incubation of THP‐1 macrophages with ORF2 protein effectively triggered IL‐1β mRNA expression (Figure S3G) and IL‐1β protein secretion (Figure 3L). These results collectively indicate that the ORF2 capsid protein and the integral HEV particle in particular are robust activators of inflammasome response.
NF‐κB signaling activation by HEV is essential for NLRP3 inflammasome response
NF‐κB, a family of inducible transcription factors, regulates a large number of genes involved in immune and inflammatory responses. To determine whether the NF‐κB pathway is activated by HEV infection, THP‐1 macrophages were inoculated with HEV particles. HEV infection induced nuclear translocation of RelA, a hallmark of NF‐κB activation, comparable to the positive control of TNF‐ɑ treatment (Figure 4A). In general, IL‐1β activation is regulated by two signals, including pro‐IL‐1β mRNA transcription regulated by NF‐κB and the processing of IL‐1β mediated by NLRP3 inflammasome.[ 18 ] We have confirmed that HEV triggers NLRP3 inflammasome assembly in human macrophages (Figure 2I,K).
FIGURE 4.
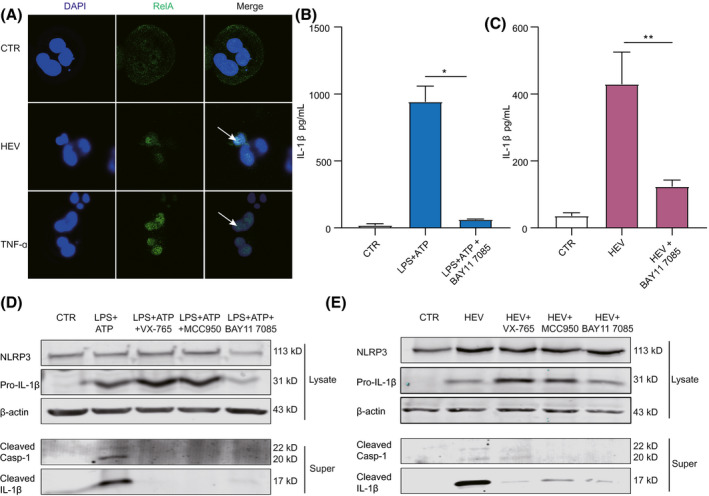
NF‐κB signaling activation by HEV is essential for NLRP3 inflammasome response. (A) THP‐1 macrophages were inoculated with HEV or treated with TNF‐ɑ (100 ng/mL) for 24 hours. Subcellular localizations of NF‐κB (green) and the nucleus marker, DAPI (blue), were examined under confocal microscopy. (B) THP‐1 macrophages were pretreated with 10 µM of NF‐κB inhibitor (BAY11 7085) for 2 hours and then treated with LPS (400 ng/ml) or LPS (400 ng/mL) plus 10 µM of NF‐κB inhibitor (BAY11 7085) for 6 hours, followed by ATP (5 mM) for 40 minutes. IL‐1β level in supernatant was quantified by ELISA (n = 4). (C) THP‐1 macrophages were pretreated with 10 µM of NF‐κB inhibitor (BAY11 7085) for 2 hours and then inoculated with HEV plus 10 µM of NF‐κB inhibitor (BAY11 7085) for 24 hours. IL‐1β level in supernatant was quantified by ELISA (n = 4). (D) Mature IL‐1β and cleaved Casp‐1 in supernatant or pro‐IL‐1β and NLRP3 in lysates were determined after LPS plus NF‐κB inhibitor treatment by western blotting. (E) Mature IL‐1β and cleaved Casp‐1 in supernatant or pro‐IL‐1β and NLRP3 in lysates were determined after HEV infection plus NF‐κB inhibitor by western blotting. NF‐κB activation is marked by an arrow. Data were normalized to the control (CTR; set as 1). Data are means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; Abbreviations: NS, not significant (Mann‐Whitney U test); Super, supernatant
To assess the biological functions of NF‐κB on HEV‐induced NLRP3 inflammasome response, we used a pharmacological inhibitor of NF‐κB (BAY11 7085). In the positive control treated with LPS, the induction of IL‐1β production, IL‐1β maturation, and Casp‐1 cleavage were effectively blocked by this inhibitor (Figure 4B,D). Similar effects were observed in THP‐1 macrophages infected with HEV (Figure 4C,E). For example, the concentration of IL‐1β in supernatant treated with BAY11 7085 was significantly reduced to 124 ± 15 pg/mL, compared to 422 ± 84 pg/mL in HEV‐infected cells without BAY11 7085 treatment (Figure 4C). Overall, the potency of the NF‐κB inhibitor in inhibiting NLRP3 inflammasome activation is comparable to that of MCC950 or VX‐765 (Figures 2J,K and 4B‐E). Of note, these inhibitors alone did not affect the basal level of IL‐1β secretion and cell viability (Figure S4A‐C). These results collectively suggest that NF‐κB activation is likely a key upstream event priming HEV‐triggered NLRP3 inflammasome response.
HEV RNA triggers robust antiviral IFN response in macrophages
Inflammasome activation and IFN response have been reported to cooperatively orchestrate viral and bacterial infections.[ 19 , 20 ] We have previously demonstrated that HEV potently induces IFN response in hepatocytes of the patient liver, and viral RNA is the main inducer of IFN response.[ 13 ] It is interesting to investigate whether HEV also activates IFN response in macrophages and whether there is crosstalk with inflammasome response.
We found that inoculation of primary macrophages with HEV particles significantly, but moderately, induced the expression of IFN‐β (Figure 5A) and downstream several antiviral ISGs (e.g., STAT1, IFN‐induced protein with tetratricopeptide repeats 1 [IFIT1], and 2′‐5′‐oligoadenylate synthetase‐like [OASL]; Figure 5B). In contrast, transfection of in vitro transcribed GT3 HEV RNA in THP‐1 macrophages resulted in a robust induction of type 1 (IFN‐ɑ and IFN‐β) and type 3 IFN (IFN‐λ1 and IFN‐λ2) genes (Figure 5C and Figure S5A) and ISG expression, including STAT1, IFIT1, myxovirus resistance gene A, melanoma differentiation‐associated gene 5 (MDA5), and RIG‐I (Figure 5C and Figure S5A,B).
FIGURE 5.
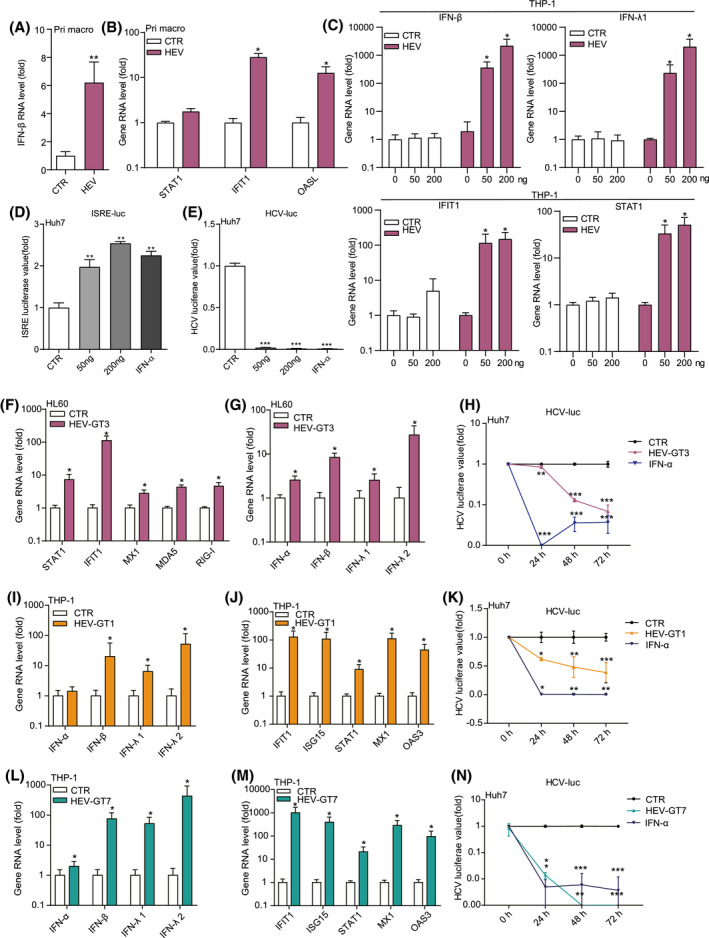
HEV RNA triggers robust antiviral IFN response in macrophages. Primary macrophages were inoculated with HEV for 24 hours. IFN‐β (A) and ISG (B) gene expression was quantified by qRT‐PCR (n = 4‐6). THP‐1 macrophage cells were transfected with GT3 HEV RNA (50 or 200 ng/well in 48‐well plates). Expression levels of IFNs and ISGs (C) were quantified at 48 hours (n = 4). (D) ISRE luciferase value and (E) HCV luciferase value were measured at 48 hours after the treatment of conditioned medium from THP‐1 cells transfected with GT3 HEV RNA (n = 6). HL60 macrophage cells were transfected with GT3 HEV RNA. Expression levels of ISGs (F) and IFNs (G) were quantified at 48 hours (n = 5). (H) HCV luciferase value was measured at 48 hours after the treatment of conditioned medium from HL60 cells transfected with GT3 HEV RNA (n = 7). THP‐1 macrophage cells were transfected with and GT1 or ‐7 HEV RNA. Expression levels of IFNs (I,L) and ISGs (J,M) were quantified at 48 hours (n = 8). HCV luciferase value was measured at 48 hours after the treatment of conditioned medium from THP‐1 cells transfected with GT1 (K) or GT7 (N) HEV RNA (n = 4). Data were normalized to the control (CTR; set as 1). Data are means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. Abbreviations: NS, not significant (Mann‐Whitney U test); OAS3, 2′‐5′‐oligoadenylate synthetase 3
To examine whether functional IFNs are produced, we collected conditioned medium from the transfected cells (supernatant; Figure S5C) and performed an ISRE‐based IFN reporter assay and a highly IFN‐sensitive HCV replicon. We found that conditioned medium treatment strongly increased ISRE luciferase activity (Figure 5D) and inhibited HCV replication (Figure 5E). Similar results were observed in HL60 macrophages (Figure 5F‐H). Furthermore, GT1 and ‐7 HEV RNA also effectively triggered IFN response (Figure 5I‐N). These results demonstrated that HEV gRNA, irrespective of genotype, can robustly trigger antiviral IFN response in macrophages.
Reciprocal antagonism between HEV‐induced inflammasome activation and antiviral IFN response
Given that the JAK‐STAT pathway mediates IFN response to induce ISG expression,[ 21 ] treatment with JAK inhibitor 1 almost completely blocked HEV‐induced STAT1 phosphorylation (Figure 6A) and ISG expression (Figure 6A,B and Figure S6A) in THP‐1 macrophages. Expression of IFN genes was also largely demolished (Figure 6C and Figure S6B). Treatment with the recombinant viral 136R/Y136 protein, an IFN receptor inhibitor, also profoundly inhibited the expression of HEV‐induced IFNs and ISGs (Figure 6D,E and Figure S6C,D).
FIGURE 6.
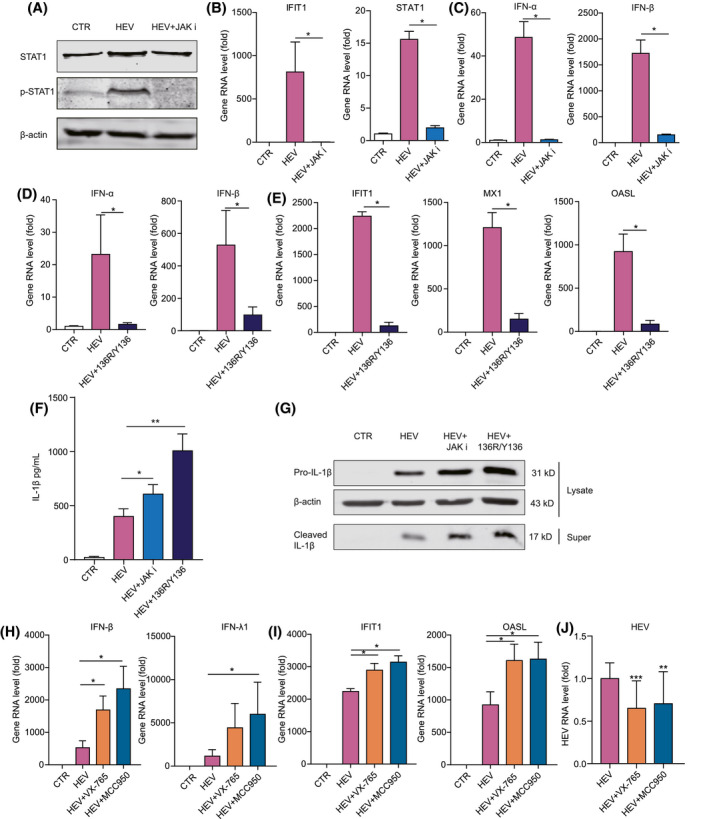
Crosstalk between HEV‐induced inflammasome activation and antiviral IFN response. (A) THP‐1 macrophage cells were transfected with GT3 HEV RNA or GT3 HEV RNA plus JAK inhibitor 1 for 24 hours. Protein expression of STAT1 and p‐STAT1 was determined by western blotting. THP‐1 macrophages were transfected with GT3 HEV RNA or GT3 HEV RNA plus JAK inhibitor 1 for 24 hours, and expression levels of ISGs (B) and IFNs (C) were quantified at 24 hours by qRT‐PCR (n = 4). THP‐1 macrophages were transfected with GT3 HEV RNA or GT3 HEV RNA plus recombinant viral 136R/Y136 for 24 hours, and expression levels of IFNs (D) and ISGs (E) were quantified at 24 hours (n = 4). THP‐1 macrophages were infected with HEV or HEV plus JAK inhibitor 1 (10 µM) or recombinant viral 136R/Y136 (1,000 ng/mL) for 24 hours. IL‐1β level in supernatant was quantified by ELISA (n = 4) (F), and mature IL‐1β in supernatant and pro‐IL‐1β in cell lysates were quantified by western blotting (G). THP‐1 macrophages were inoculated with HEV or HEV with 50 µM of Casp‐1 inhibitor (VX‐765) or 10 µM of NLRP3 inhibitor (MCC950) for 24 hours. Expression levels of IFNs (H) and ISGs (I) were quantified by qRT‐PCR (n = 4). (J) HEV‐RNA levels were quantified by qRT‐PCR (n = 18). Data were normalized to the control (CTR; set as 1). Data are means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. Abbreviations: NS, not significant (Mann‐Whitney U test); p‐STAT1, phosphorylated STAT1; Super, supernatant
Interestingly, blocking IFN response enhanced HEV‐induced IL‐1β production and activation (Figure 6F,G). Conversely, blocking inflammasome response by Casp‐1 inhibitor (VX‐765) and NLRP3 inhibitor (MCC950) significantly increased HEV‐induced type 1 (IFN‐ɑ and IFN‐β) and type 3 (IFN‐λ1 and IFN‐λ2) IFN gene expression in THP‐1 macrophages (Figure 6H and Figure S6E). Consistently, the induction of ISGs, including IFIT1, MX dynamin‐like GTPase 1 (MX1), and OASL, was also markedly augmented (Figure 6I and Figure S6F). This may explain why treatment with inflammasome inhibitors significantly (although moderately) inhibited HEV replication in THP‐1 macrophages (Figure 6J). These results suggest an active crosstalk between the HEV‐induced inflammasome and IFN response, showing reciprocal antagonism.
Therapeutic targeting NLRP3 inflammasome and combining with antiviral therapy
Because of pathological hyperinflammation in severe acute HEV infection, we hypothesize that antiviral therapy alone is insufficient, whereas combining targeted anti‐inflammatory treatment may be necessary. We demonstrated that a preclinical pharmacological inhibitor effectively inhibited HEV‐induced NLRP3 inflammasome activation (Figure 2K). However, identifying inflammasome inhibitors from U.S. Food and Drug Administration–approved medications would facilitate expedited clinical application. Steroids, the classical anti‐inflammatory drugs, have been shown to exert clinical benefits in severe COVID‐19 patients.[ 22 ] Interestingly, we found that both dexamethasone and prednisone significantly attenuated HEV‐induced IL‐1β production and secretion in macrophages (Figure 7A‐C), suggesting targeting of inflammasome activation.
FIGURE 7.
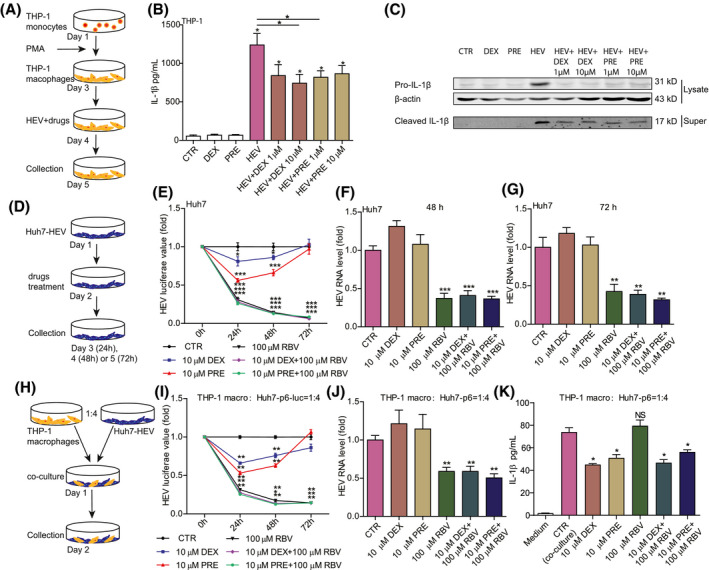
Therapeutic targeting NLRP3 inflammasome and combining with ribavirin treatment. (A) Schematic illustration of differentiation of THP‐1 monocytes to macrophages and drug treatment. THP‐1 macrophages were incubated with HEV particles and 1 or 10 µM of dexamethasone (DEX) or 1 or 10 µM of prednisone (PRE) for 24 hours. (B) IL‐1β protein levels were quantified by ELISA (n = 6), and (C) mature IL‐1β in supernatant and pro‐IL‐1β in lysates were measured by western blotting. (D) Schematic illustration of DEX or PRE treatment in Huh7‐p6 and Huh7‐p6‐luc HEV models. Huh7‐p6‐luc or Huh7‐p6 cells were treated with 10 µM of DEX, 10 µM of PRE, 100 µM of ribavirin, 10 µM of DEX plus 100 µM of ribavirin, or 10 µM of PRE plus 100 µM of ribavirin for 24, 48, and 72 hours. (E) HEV luciferase values were measured. HEV‐RNA levels were quantified at 48 (F) and 72 (G) hours by qRT‐PCR (n = 6‐8). (H) Schematic illustration of the coculture system of Huh7‐based HEV cells with THP‐1 macrophages. The coculture was treated with 10 µM of DEX, 10 µM of PRE, 100 µM of ribavirin, 10 µM of DEX plus 100 µM of ribavirin, or 10 µM of PRE plus 100 µM of ribavirin for 24, 48, and 72 hours. (I) HEV luciferase values were measured. HEV‐RNA levels were quantified at 48 hours (J) by qRT‐PCR (n = 4‐6), and IL‐1β levels (K) were measured by ELISA (n = 4). Data were normalized to the control (CTR; set as 1). Data are means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. Abbreviations: NS, not significant (Mann‐Whitney U test); Super, supernatant
Given that hepatocytes are the primary targets of HEV infection, we investigated the effects of steroids in two Huh7‐based HEV models: the subgenomic replicon and infectious model (Figure 7D). Overall, treatment with dexamethasone or prednisone had minimal effects on HEV replication and did not interfere with the antiviral activity of ribavirin (Figure 7E‐G), supporting the applicability of combing inflammasome inhibitor with antiviral therapy.
Finally, we established a coculture system of Huh7 with THP‐1 macrophages for assessing combination therapy (Figure 7H). In this system, the combination of dexamethasone or prednisone with ribavirin simultaneously inhibited viral replication (Figure 7I,J) and HEV‐induced IL‐1β production (Figure 7K), without evidence of cross‐interference or affecting cell viability (Figure S7). Ribavirin significantly inhibited HEV replication (Figure 7I,J) without affecting the steroid‐mediated inhibition of IL‐1β production (Figure 7K). Conversely, steroids did not compromise the anti‐HEV activity of ribavirin (Figure 7I,J). These results demonstrated a proof of concept of combining an antiviral agent with an inflammasome inhibitor to simultaneously inhibit infection and inflammation.
DISCUSSION
This study has provided compelling evidence that HEV infection, in rabbits and patients, activates inflammasome response. We further demonstrated macrophage as a key cell type in initiating the NLRP3 inflammasome in response to HEV infection. We detailed and characterized the mechanism of actions and demonstrated a proof of concept of therapeutic targeting of HEV‐triggered inflammasome activation.
In healthy persons, HEV infection is usually asymptomatic and self‐limiting without evidence of causing inflammation. In contrast, acute hepatitis E in pregnant women can manifest severe liver diseases featured by massive inflammatory response. In HEV‐infected pregnant women, higher levels of proinflammatory cytokines, including TNF‐α, IL‐6, and IFN‐γ, are associated with adverse pregnancy outcome.[ 23 ] In this study, we observed histological evidence of liver inflammation in HEV‐infected rabbits and hospitalized HEV patients. Importantly, HEV infection resulted in a dramatic elevation of IL‐1β, particularly in HEV patients. In these patients, IL‐1β level was positively corrected with ALP, but negatively associated with prealbumin, suggesting that inflammasome activation may contribute to poor liver functions and more‐advanced disease stages. Our patients, although not pregnant women, have developed complications by the infection. The disease severity of our patients is likely very mild compared to that in HEV‐infected pregnant women. We thus speculate that the level of inflammasome activation in HEV‐infected pregnant women could be much more robust.
Monocytes and macrophages in the liver play a key role in inflammasome‐mediated defense against viral or bacterial infections.[ 24 ] Macrophages are the major cell type initiating inflammasome response in the liver. Gram‐negative bacteria and their products and LPS entering into the liver by the gut‐liver axis can trigger macrophage activation, leading to the production of a large amount of inflammatory cytokines through inflammasome activation. In HCV‐infected patients, IL‐1β production by macrophages confers liver inflammation through HCV‐induced inflammasome signaling. The production of a variety of cytokines, such as IL‐6, IL‐8, and IL‐1β, in particular is associated with HCV infection and liver disease severity.[ 25 ] Although macrophages are not permissible to HCV infection, HCV can indirectly trigger NLRP3 inflammasome response and IL‐1β production in human macrophages.[ 26 ] This is mainly through the phagocytosis of HCV particles or viral components by macrophages.[ 26 , 27 ] In HEV‐infected pregnant women, increased frequencies of monocytes‐macrophages and dendritic cells have been observed.[ 4 ] We and others have shown that both human monocytes and macrophages are permissive to HEV infection.[ 16 ] We found that HEV infection robustly activates NLRP3 inflammasome in human primary macrophages and macrophage cell lines. Because of the distinct susceptibility to HCV and HEV infections, the mechanisms in activating the inflammasome in macrophages could be very different by these two hepatotropic viruses.
Upon entry into host cells, HEV particles release their viral genome to initiate the replication process. In this study, we demonstrated that direct transfection of HEV gRNA, irrespective of genotypes, can moderately activate the inflammasome in macrophages. HCV gRNA has also been reported to activate NLRP3 inflammasome in human myeloid cells,[ 28 ] despite that HCV RNA is not capable of replicating in these cells. Although both HCV and Zika virus are from the Flaviviridae viral family, Zika virus RNA is not involved in NLRP3 inflammasome activation and IL‐1β secretion.[ 29 ] We think HEV RNA is not the main activator of the inflammasome, because inoculation of infectious virus particles elicited much stronger activation.
More interestingly, inoculation with UV‐inactivated HEV particles exerted a comparable level of inflammasome activation. UV irradiation damages the HEV genome to prevent replication, but preserves the integrity of viral particles. This suggests that exposure to the viral particle rather than active infection is required for inflammasome activation in macrophages. When the viral structure was damaged by heat inactivation, we found a dramatic reduction, but a retained ~45% activity, in triggering IL‐1β production. This indicates that the disrupted capsid proteins are likely also capable of activating the inflammasome. Indeed, incubation of macrophages with recombinant ORF2 protein effectively activates IL‐1β gene transcription and protein secretion. These findings indicate that the ORF2 capsid protein and especially the integrity of HEV particles are the main stimulus of inflammasome response in macrophages.
In the canonical model of inflammasome response, two distinct signals are required for activation and release of IL‐1β.[ 18 ] The first signal triggered by pathogens drives translocation of NF‐κB into the nucleus to induce production of pro‐IL‐1β. The second signal subsequently activates NLRP3, the adapter molecule apoptosis‐associated speck‐like protein containing a C‐terminal caspase recruitment domain, and procaspase‐1 inflammasome assembly to promote cleavage of procaspase‐1 and secretion of IL‐1β. We found that HEV infection in macrophages induced NF‐κB activation. Importantly, IL‐1β maturation and production induced by HEV infection can be effectively blocked by a pharmacological NF‐κB inhibitor (Figure 4), suggesting that NF‐κB activation likely acts as an upstream event in HEV‐induced NLRP3 inflammasome activation. NF‐κB has been long proposed as a target for treating many diseases.[ 30 ] A variety of preclinical compounds are being explored for targeting this pathway, which may bear relevance in treating pathological inflammation for HEV patients based on findings in this study, but future research is required to further explore this approach.
Concurrent activation of inflammasome and IFN responses has been reported during viral infection.[ 31 ] We previously have reported evidence of IFN response in hepatocytes of HEV‐infected patients.[ 13 ] Consistent with our previous findings in liver cell lines,[ 13 ] we found that HEV gRNA robustly activates antiviral IFN response, whereas activation by inoculation of HEV particles is very moderate. In contrast, the HEV particle compared to gRNA is highly potent in activating the NLRP3 inflammasome. Thus, different viral components appear to be responsible for activating IFN and inflammasome responses, respectively, although simultaneously.
Crosstalk between inflammasome and IFN responses has been described in many diseases.[ 32 ] We here revealed mutual antagonism between inflammasome and IFN response during HEV infection. This likely explains why treatment with inhibitors targeting the NLRP3 inflammasome machinery significantly, although moderately, inhibited HEV replication in macrophages. A suppressive effect of IFN on IL‐1β production has been reported in Mycobacterium tuberculosis–infected macrophages.[ 33 ] In multiple sclerosis patients treated with IFN‐β, monocytes produced much less IL‐1β compared to that from healthy donors.[ 34 ] Thus, our results largely agree with previous evidence that inflammasome and IFN responses mutually counteract,[ 35 ] but this is likely pathogen and context dependent.
Therapeutic development has primarily focused on treating chronic hepatitis E by inhibiting the virus with antiviral agent, such as ribavirin,[ 36 ] which is very effective in the majority of patients. However, treating severe acute HEV infection (e.g., in pregnant women) remains an unexplored territory. We hypothesize that an antiviral agent alone is insufficient for these patients, because of massive inflammatory response. There is an interesting lesson learned from treating severe COVID‐19 patients with steroids, which significantly reduce mortality.[ 22 ] The pathology of severe COVID‐19 is closely related to intense, rapid activation of the NLRP3 inflammasome pathway in monocytes and macrophages.[ 37 ] We therefore repurposed steroids, and, excitingly, both dexamethasone or prednisone significantly inhibited HEV‐induced inflammasome response. A previous study has demonstrated that dexamethasone can inhibit pro‐IL‐1β translation.[ 38 ] This appears to support our results, but the detailed mechanisms of how steroids inhibit inflammasome remain to be further clarified. We think that treating with an inflammasome inhibitor alone is not sufficient, given that it does not inhibit, but may even promote, viral replication, thus bearing the risk of developing chronic infection as observed in organ transplant patients receiving immunosuppressants.[ 39 ] In this study, by using a coculture model of HEV‐infected liver cells with macrophages, we demonstrated a proof of concept of combing steroids with ribavirin to simultaneously inhibit viral replication and inflammasome activation without cross‐interference. Our approach appears to be supported by a recent case who developed severe neurological complications with multiple disseminated inflammatory lesions in the central nervous system by acute HEV infection. Interestingly, this patient was recovered by a combination of anti‐inflammatory (methylprednisolone) and antiviral (ribavirin) treatment.[ 40 ]
The potency of steroids in inhibiting the inflammasome is rather suboptimal, as shown in our experimental models, urging the need of developing more specific and potent inflammasome inhibitors. A recent study has discovered that nucleoside reverse‐transcriptase inhibitors, which are widely used for treating HIV‐1 or hepatitis B, have anti‐inflammasome properties.[ 41 ] Several biologicals and many small molecules targeting the inflammasome are at preclinical and clinical evaluation. We thus expect that effective anti‐inflammasome medications could soon be available in the clinic.
In summary, this study convincingly demonstrated activation of the NLRP3 inflammasome in macrophages upon HEV infection. We comprehensively characterized the mode of action and crosstalk between inflammasome and IFN response, and explored the potential of therapeutic targeting. These findings are essential for a better understanding HEV‐host interactions and the pathogenic mechanisms of HEV infection. Our proposed strategy of combining an inflammasome inhibitor with an antiviral agent provides a viable option for treating severe HEV infections (e.g., in pregnant women), as well as many other severe viral infections such as COVID‐19.
CONFLICT OF INTEREST
Nothing to report.
AUTHOR CONTRIBUTIONS
Yang Li, Peifa Yu, Jiaye Liu, Yunlong Li, Pengfei Li, Amy L. Kessler, Jingyi Shu, Yining Wang, Zhaochao Liang, Sonja I. Buschow, and Ling Wang performed experiments and/or performed data analysis. Yijin Wang, Xiaoyan Liu, and Aixia Liu collected and analyzed patient samples and information. Zhongren Ma, Maikel P. Peppelenbosch, Marco J. Bruno, Robert A. de Man, and Ling Wang discussed the project and provided key resources. Qiuwei Pan, Yijin Wang, and Lin Wang supervised the project. Yang Li and Qiuwei Pan wrote the manuscript, and the final manuscript was revised and approved by all the authors.
Supporting information
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Ruud Delwel (Department of Hematology, Erasmus University Medical Center, Rotterdam, The Netherlands) for generously providing THP‐1 and U937 cell lines and Dr. Gwenny Fuhler (Department of Gastroenterology and Hepatology, Erasmus University Medical Center, Rotterdam, The Netherlands) for providing the HL60 cell line. We also thank Dr. Suzanne U. Emerson (National Institute of Allergy and Infectious Diseases, Bethesda, Maryland, USA) for generously providing GT1 and GT3 HEV plasmids to generate subgenomic and full‐length HEV genomic RNA; Dr. Tian‐Cheng Li (National Institute of Infectious Diseases, Tokyo, Japan) for providing the plasmid containing the full‐length dromedary camel HEV genome (GenBank Accession: KJ496144); and Dr. Rasa Petraitytė‐Burneikienė (Vilnius University, Lithuania) and Dr. Rainer Günter Ulrich (Federal Research Institute for Animal Health, Greifswald, Germany) for providing purified recombinant HEV ORF2 protein.
Li Y, Yu P, Kessler AL, Shu J, Liu X, Liang Z, et al. Hepatitis E virus infection activates NOD‐like receptor family pyrin domain‐containing 3 inflammasome antagonizing interferon response but therapeutically targetable. Hepatology.2022;75:196–212. 10.1002/hep.32114
Funding information
Supported by a VIDI grant (No. 91719300) from the Netherlands Organisation for Scientific Research (NWO; to Q. Pan) and the China Scholarship Council for funding Ph.D. fellowship (to Yang Li; No. 201703250073)
Contributor Information
Lin Wang, Email: lin_wang@pku.edu.cn, Email: wangyj3@sustech.edu.cn, Email: q.pan@erasmusmc.nl.
Yijin Wang, Email: wangyj3@sustech.edu.cn.
Qiuwei Pan, Email: q.pan@erasmusmc.nl.
REFERENCES
- 1. Zhou JH, Li XR, Lan X, Han SY, Wang YN, Hu Y, et al. The genetic divergences of codon usage shed new lights on transmission of hepatitis E virus from swine to human. Infect Genet Evol. 2019;68:23–9. [DOI] [PubMed] [Google Scholar]
- 2. Kamar N, Bendall R, Legrand‐Abravanel F, Xia NS, Ijaz S, Izopet J, et al. Hepatitis E. Lancet. 2012;379:2477–88. [DOI] [PubMed] [Google Scholar]
- 3. Heymann F, Tacke F. Immunology in the liver—from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13:88–110. [DOI] [PubMed] [Google Scholar]
- 4. Sehgal R, Patra S, David P, Vyas A, Khanam A, Hissar S, et al. Impaired monocyte‐macrophage functions and defective Toll‐like receptor signaling in hepatitis E virus‐infected pregnant women with acute liver failure. Hepatology. 2015;62:1683–96. [DOI] [PubMed] [Google Scholar]
- 5. Debes JD, Groothuismink ZMA, Doukas M, de Man RA, Boonstra A. Immune dissociation during acute hepatitis E infection. Int J Infect Dis. 2019;87:39–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kumar A, Devi SG, Kar P, Agarwal S, Husain SA, Gupta RK, et al. Association of cytokines in hepatitis E with pregnancy outcome. Cytokine. 2014;65:95–104. [DOI] [PubMed] [Google Scholar]
- 7. Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol. 2012;57:642–54. [DOI] [PubMed] [Google Scholar]
- 8. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–65. [DOI] [PubMed] [Google Scholar]
- 9. Franchi L, Muñoz‐Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13:325–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 2015;25:308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schneider WM, Chevillotte MD, Rice CM. Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang W, Wang Y, Qu C, Wang S, Zhou J, Cao W, et al. The RNA genome of hepatitis E virus robustly triggers an antiviral interferon response. Hepatology. 2018;67:2096–112. [DOI] [PubMed] [Google Scholar]
- 14. Li S, He Q, Yan LI, Li M, Liang Z, Shu J, et al. Infectivity and pathogenicity of different hepatitis E virus genotypes/subtypes in rabbit model. Emerg Microbes Infect. 2020;9:2697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Graaff P, Berrevoets C, Rӧsch C, Schols HA, Verhoef K, Wichers HJ, et al. Curdlan, zymosan and a yeast‐derived β‐glucan reshape tumor‐associated macrophages into producers of inflammatory chemo‐attractants. Cancer Immunol Immunother. 2021;70:547–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sayed IM, Seddik MI, Gaber MA, Saber SH, Mandour SA, El‐Mokhtar MA. Replication of hepatitis E virus (HEV) in primary human‐derived monocytes and macrophages in vitro. Vaccines (Basel). 2020;8:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kubickova B, Schenk JA, Ramm F, Markuškienė K, Reetz J, Dremsek P, et al. A broadly cross‐reactive monoclonal antibody against hepatitis E virus capsid antigen. Appl Microbiol Biotechnol. 2021;105:4957–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32. [DOI] [PubMed] [Google Scholar]
- 19. Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, von Messling V, et al. Type I IFN triggers RIG‐I/TLR3/NLRP3‐dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 2013;9:e1003256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J Exp Med. 2007;204:987–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang W, Xu L, Su J, Peppelenbosch MP, Pan Q. Transcriptional regulation of antiviral interferon‐stimulated genes. Trends Microbiol. 2017;25:573–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. RECOVERY Collaborative Group ; Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, et al. Dexamethasone in hospitalized patients with Covid‐19. N Engl J Med. 2021;384:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Devi SG, Kumar A, Kar P, Husain SA, Sharma S. Association of pregnancy outcome with cytokine gene polymorphisms in HEV infection during pregnancy. J Med Virol. 2014;86:1366–76. [DOI] [PubMed] [Google Scholar]
- 24. Serti E, Werner JM, Chattergoon M, Cox AL, Lohmann V, Rehermann B. Monocytes activate natural killer cells via inflammasome‐induced interleukin 18 in response to hepatitis C virus replication. Gastroenterology. 2014;147:209 – 20.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Negash AA, Ramos HJ, Crochet N, Lau DTY, Doehle B, Papic N, et al. IL‐1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013;9:e1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shrivastava S, Mukherjee A, Ray R, Ray RB. Hepatitis C virus induces interleukin‐1β (IL‐1β)/IL‐18 in circulatory and resident liver macrophages. J Virol. 2013;87:12284–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marukian S, Jones CT, Andrus L, Evans MJ, Ritola KD, Charles ED, et al. Cell culture‐produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology. 2008;48:1843–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen W, Xu Y, Li H, Tao W, Xiang YU, Huang B, et al. HCV genomic RNA activates the NLRP3 inflammasome in human myeloid cells. PLoS One. 2014;9:e84953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang W, Li G, Wu D, Luo Z, Pan P, Tian M, et al. Zika virus infection induces host inflammatory responses by facilitating NLRP3 inflammasome assembly and interleukin‐1β secretion. Nat Commun. 2018;9:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baig MS, Roy A, Saqib U, Rajpoot S, Srivastava M, Naim A, et al. Repurposing thioridazine (TDZ) as an anti‐inflammatory agent. Sci Rep. 2018;8:12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dubois H, Sorgeloos F, Sarvestani ST, Martens L, Saeys Y, Mackenzie JM, et al. Nlrp3 inflammasome activation and Gasdermin D‐driven pyroptosis are immunopathogenic upon gastrointestinal norovirus infection. PLoS Pathog. 2019;15:e1007709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Labzin LI, Lauterbach MA, Latz E. Interferons and inflammasomes: cooperation and counterregulation in disease. J Allergy Clin Immunol. 2016;138:37–46. [DOI] [PubMed] [Google Scholar]
- 33. Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer‐Barber KD, et al. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL‐1β production in human macrophages. J Immunol. 2011;187:2540–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, et al. Type I interferon inhibits interleukin‐1 production and inflammasome activation. Immunity. 2011;34:213–23. [DOI] [PubMed] [Google Scholar]
- 35. Burke TP, Engström P, Chavez RA, Fonbuena JA, Vance RE, Welch MD. Inflammasome‐mediated antagonism of type I interferon enhances Rickettsia pathogenesis. Nat Microbiol. 2020;5:688–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. European Association for the Study of the Liver . EASL Clinical Practice Guidelines on hepatitis E virus infection. J Hepatol. 2018;68:1256–71. [DOI] [PubMed] [Google Scholar]
- 37. Merad M, Martin JC. Author correction: pathological inflammation in patients with COVID‐19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20:448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kern JA, Lamb RJ, Reed JC, Daniele RP, Nowell PC. Dexamethasone inhibition of interleukin 1 beta production by human monocytes. Posttranscriptional mechanisms. J Clin Invest. 1988;81:237‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Y, Metselaar HJ, Peppelenbosch MP, Pan Q. Chronic hepatitis E in solid‐organ transplantation: the key implications of immunosuppressants. Curr Opin Infect Dis. 2014;27:303–8. [DOI] [PubMed] [Google Scholar]
- 40. Rahmig J, Grey A, Berning M, Schaefer J, Lesser M, Reichmann H, et al. Disseminated inflammation of the central nervous system associated with acute hepatitis E: a case report. BMC Neurol. 2020;20:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ambati J, Magagnoli J, Leung H, Wang SB, Andrews CA, Fu D, et al. Repurposing anti‐inflammasome NRTIs for improving insulin sensitivity and reducing type 2 diabetes development. Nat Commun. 2020;11:4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material