

Abstract
Many Drosophila species differ widely in their distributions and climate niches, making them excellent subjects for evolutionary genomic studies. Here, we have developed a database of high‐quality assemblies for 46 Drosophila species and one closely related Zaprionus. Fifteen of the genomes were newly sequenced, and 20 were improved with additional sequencing. New or improved annotations were generated for all 47 species, assisted by new transcriptomes for 19. Phylogenomic analyses of these data resolved several previously ambiguous relationships, especially in the melanogaster species group. However, it also revealed significant phylogenetic incongruence among genes, mainly in the form of incomplete lineage sorting in the subgenus Sophophora but also including asymmetric introgression in the subgenus Drosophila. Using the phylogeny as a framework and taking into account these incongruences, we then screened the data for genome‐wide signals of adaptation to different climatic niches. First, phylostratigraphy revealed relatively high rates of recent novel gene gain in three temperate pseudoobscura and five desert‐adapted cactophilic mulleri subgroup species. Second, we found differing ratios of nonsynonymous to synonymous substitutions in several hundred orthologues between climate generalists and specialists, with trends for significantly higher ratios for those in tropical and lower ratios for those in temperate‐continental specialists respectively than those in the climate generalists. Finally, resequencing natural populations of 13 species revealed tropics‐restricted species generally had smaller population sizes, lower genome diversity and more deleterious mutations than the more widespread species. We conclude that adaptation to different climates in the genus Drosophila has been associated with large‐scale and multifaceted genomic changes.
Keywords: climate adaptation, Drosophila, incomplete lineage sorting, introgression, phylogenomics, phylostratigraphy
1. INTRODUCTION
The genus Drosophila contains about 1600 species (taxodros.uzh.ch) covering a wide range of ecological niches, making it a rich resource for comparative studies. The first 12 Drosophila genomes were published by Clark et al. (2007) and many more have since been released. Comparative genomic analyses on some of these species have investigated aspects of the biology of the group, such as cactophilic adaptation (Guillen et al., 2014; Rane et al., 2019), genome evolution (Garrigan et al., 2012; Hu et al., 2013; Palmieri et al., 2014; Richards et al., 2005; Sanchez‐Flores et al., 2016), lifespan evolution (Fonseca et al., 2013), sex chromosome evolution (Zhou & Bachtrog, 2015), species invasion (Ometto et al., 2013) and speciation (Nolte et al., 2013), and climate adaptation (Parker et al., 2018; Rane et al., 2019). However, most of these comparisons have been limited by their focus on just a few closely related species in one or two species groups, rather than spanning a broad evolutionary range across subgenera.
Comparative genomic studies on such a broad scale require a well resolved phylogeny. However, despite numerous phylogenetic studies of Drosophila species, many relationships among them remain controversial and the genus also now appears to be paraphyletic to several other genera (Finet et al., 2021; van der Linde & Houle, 2008; van der Linde et al., 2010; O'Grady & DeSalle, 2018). Major uncertainties occur within each of the three major nominate Drosophila subgenera (Drosophila, Sophophora, and Dorsilopha), and even within the much studied melanogaster species group in the subgenus Sophophora. Incomplete lineage sorting (ILS, the evolutionary process where ancestral polymorphisms are randomly fixed in descendent lineages) has been implicated at several nodes in the melanogaster, eugracilis, takahashii and suzukii subgroups of this group (Pollard et al., 2006; Rosenfeld et al., 2012; Wong et al., 2007). And introgression of genes between species subsequent to natural hybridisation events has been reported in the simulans (Garrigan et al., 2012) and yakuba (Turissini & Matute, 2017) clades within the melanogaster subgroup. However, the overall contribution of ILS and introgression to phylogenetic ambiguities across the genus remains unclear.
Another prerequisite for comparative genomic analyses is detailed knowledge of species differences in traits underpinning their ecological differences. One of the best understood sets of traits in Drosophila in this respect involves resistance to extremes of temperature and humidity. Different species can be found in four of the five major Köppen climate groups (Kottek et al., 2006), and these resistances have been shown to influence the distribution of many of the species (Kellermann et al., 2009; Parratt et al., 2021). The widespread species that thrive in a variety of climatic conditions have high resistance to climatic stresses while species with restricted distributions tend to have lower resistance levels (Kellermann, Loeschcke, et al., 2012; Kellermann, Overgaard, et al., 2012).
Complementing such interspecific studies, selection experiments, geographic comparisons and strain comparisons have indicated high levels of genetic variability for these traits within some species (Hoffmann et al., 2003). Further, studies following gene expression changes during stress responses (Koniger & Grath, 2018; Sørensen et al., 2007), allele frequency changes during selection (Telonis‐Scott et al., 2012, 2016) and differences among populations or species with different geographical distributions (Parker et al., 2018) have identified sets of candidate genes and biochemical networks which may be responsible for the variation. A core set of 45 genes enriched for stimulus, stress and defense response have been related to desiccation resistance in population studies (Telonis‐Scott et al., 2016) and about 250 candidate genes show accelerated divergence between species in cold and warm regions (Parker et al., 2018). Several specific genes have also been functionally validated, for example nan and trp in sensing humidity (Liu et al., 2007), capa in the regulation of cellular ion and water homeostasis (Terhzaz et al., 2015), and Tps1 and Treh in regulation of body water homeostasis (Yoshida et al., 2016). Resistance to different climate stresses may also be correlated (Liu et al., 2007), such as cross‐resistance to cold and desiccation (Terhzaz et al., 2015).
Here, we compared the genomes of 46 species from the genus Drosophila and one from the paraphyletic lineage Zaprionus. Fifteen of these are published here for the first time and 20 published previously are improved with additional data. We also present population resequencing data for several of them. We constructed a well resolved phylogeny for the genus but also found high levels of ILS and/or introgression in some lineages. We then used phylostratigraphy, climate niche analysis and various tests of selection to screen for genomic signatures for species occupying different climate niches.
2. MATERIALS AND METHODS
2.1. Sequencing and assembly
Table S1 provides details of the stocks of the 15 species which were sequenced de novo, the 20 species whose published assemblies were improved with additional sequencing, and the 13 species for which individuals were resequenced for the population genomic analyses.
Multiple paired‐end libraries covering a range of insert sizes (250 base pairs [bp], 500 bp, 800 bp, 2 kbp, 5 kbp and 10 kbp) were prepared for each of the 15 species which were sequenced de novo (Table S2). An Illumina HiSeq 2000 platform was used to sequence 49 bp paired‐ends for large insert size libraries (≥2 kb) and 100 bp or 150 bp paired‐ends for short insert size libraries (<2 kb). Reads were discarded if more than 33% (short insert libraries) or 80% (large insert libraries) of their bases had Phred scores lower than seven, or more than 10% of their bases were scored as “N”. Paired‐end reads were also discarded if both reads were identical or overlapped by more than 10 bp. Genomes were assembled by platanus (Kajitani et al., 2014). We first constructed contigs from the reads from the short insert libraries using parameters set at “‐k 19 ‐s 10 ‐u 0.2 ‐d 0.6”. Reads from the long insert libraries were then aligned to the contigs and scaffolds constructed using the “‐u 0.2” parameter setting. Finally, gaps were closed using all the paired‐end reads and the default parameters. Assembly qualities were evaluated using busco (Simão et al., 2015) with the “arthropoda” database (https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi) as reference (Table S3).
We collected the genome assemblies for the other 32 species from NCBI and improved 20 of them with additional sequences from two paired‐end libraries with insert sizes of 500 bp. We closed the gaps of these assemblies with the additional data using the krskgf software Zhang et al. (2014) and gapcloser (Luo et al., 2012). Note that the genome which we attribute to D. mercatorum herein was originally published by us as that of the closely related D. repleta. However, we subsequently found that two mitochondrial sequences of this (Australian) stock had a 99.85% match with those of (American and Asian) D. mercatorum but only an 89.25% match with (American) D. repleta. Morphological comparisons of our stock to images of (American) type specimens also then showed a better match to D. mercatorum than D. repleta. Furthermore, a previous report of notional D. repleta in Australia described mating incompatibility between the Australian taxon and American D. repleta (Humphrey, 1974). For all these reasons we assume our sequenced stock is not D. repleta and probably is D. mercatorum herein. However, confirmation of the latter will require further taxonomic work, particularly as previous publications have never reported D. mercatorum in Australia, despite intensive study of Drosophilids in some of its favoured Opuntia cactus habitats here (Barker et al., 2005; Barker & Starmer, 1982).
We also sequenced mixed life stage transcriptomes of 19 species on the Illumina HiSeq 2000 platform to assist gene annotation (Table S1). Fifteen of them were each sequenced from a single strand specific library (dUTP) and the other three from a standard library with insert size 200 bp. In total, we obtained about 2–4 Gb of raw reads for each species. Three types of reads were filtered out; those in which N constituted more than 10% of bases, low quality reads that had 40 bases with Q20 less than or equal to seven, and reads with adapter contamination (note that the RNA‐seq data was only used to assist the annotation and any batch effects should not have affected this use.)
About 20 flies (except for one species for which only six were available) were resequenced from 13 of the 47 species above (Table S1). For each individual a library with an insert size of about 500 bp was made, from which we generated ~18 M paired‐end reads about 150 bp in length (20× coverage) on an Illumina HiSeq 4000 platform. Reads were quality filtered using soapnuke (parameters “‐Q 2 ‐G ‐l 10 ‐q 0.2 ‐d ‐5 1”) (Chen et al., 2018) so as to retain only reads with Phred scores >10 for 80% of the bases. Reads were then mapped to the corresponding genome assemblies using the mem algorithm in bwa (Li, 2013) with default parameters, and masking duplicates with picard (http://broadinstitute.github.io/picard/).
2.2. Genome annotation
Transposable elements (TEs) were annotated in all 47 genomes using a combination of both homologue‐ and de novo‐based approaches. For the former, we used repeatmasker and proteinmask (Tarailo Graovac & Chen, 2009) to identify known TEs at the DNA and protein levels. For the de novo‐based method, we first used piler (Edgar & Myers, 2005), ltr_finder (Xu & Wang, 2007) and repeatscout (Price et al., 2005) to construct de novo repeat libraries and then used repeatmasker to identify the TEs in those libraries. We also searched for tandem repeats using tandem repeats finder (Benson, 1999). The proportions of total genome sizes occupied by various classes of repeats were then calculated. The correlation between log10‐transformed TE content and genome size was also calculated after accounting for the phylogeny using the phylogenetic generalized least squares (PGLS) function, with branch lengths transformed using the maximum likelihood of lambda, in the caper package (https://cran.r‐project.org).
We used Ensembl (release‐84) to annotate protein coding genes in the gap‐closed D. melanogaster genomes. We transferred the Ensembl gene set from the downloaded assembly to the gap‐closed one after whole genome alignment using lastz (Harris, 2007). The models for all except seven of the 13,918 genes in the downloaded assembly were recovered in identical form in the gap‐closed one. Application of genewise (Birney et al., 2004) recovered models for five of the other seven which were very close to the originals. No model was predicted for the other two, both on chromosome 3R, which were therefore discarded.
Annotation of protein coding genes in most of the other species was conducted using a custom pipeline based on a combination of homology and de novo approaches and assisted by 24 previously published transcriptomes and the 19 we generated above (Table S1). Homology‐based annotation was carried out using gene models from D. melanogaster (Ensembl release 84), D. mojavensis (Flybase: GCA_000005175) and D. pseudoobscura (Flybase: GCA_000001765). Putative locations for seed proteins were identified on each target genome using blastp (BLASTall 2.2.23) with parameters “‐e 1e‐5”. These locations were then passed to genewise (version 2.2.0) for accurate spliced alignments and annotation. These predictions were then aggregated, to prioritise the D. melanogaster‐based annotation or a better alternative from the other two seed species if they had more complete length or higher genewise score. De novo predictions were also made using augustus (Stanke et al., 2006) and the repeat masked genome, only retaining predicted models that did not overlap with any of the models from the aggregated homology set. In species where transcriptome data was available, cleaned reads were aligned to the reference using star (STAR‐STAR_2.4.0i) and assembled into transcripts by cufflinks (version 2.0.2, parameter “‐I 50000 ‐u”) (Trapnell et al., 2012). Open reading frame (ORF) of the assembled transcripts were predicted using Hidden Markov Models trained on aggregated genewise models. The ORFs and transcripts were translated into protein sequences and compared to aggregated homology and de novo gene sets, and evidences were combined where a gene model and UTRs could be identified and consolidated into a complete gene.
2.3. Phylogenetics
We used two data sets for our phylogenetic analyses, the first of which was a whole genome alignment across all 47 species. To develop this, pairwise whole genome alignments were carried out between D. melanogaster and each of the other 46 species using lastz (with parameter settings “E = 30 K = 2200 L = 4000 H = 2000 Y = 3400 –scores =birdMatrix”), and Chain/Net (Abay et al., 2019; with parameter settings for the axtchain program of “–minScore = 1000, ‐linearGap = loose”). Chains unlikely to be netted by chainnet were captured by chainprenet and the remainder were netted by chainnet. Nets were converted into maf format for further analysis. Multiple hits in lastz were filtering down to retain only the one with the highest score. Multiway alignments across the 47 species were then generated by multiz (Blanchette et al., 2004). Segments with more than five species missing were removed from the alignment. Over‐aligned sequences (Zhang et al., 2014) were also removed using a script (Zhang et al., 2014) which scans for and removes regions of ≥36 bp that have <55% sequence identity to all the other species after allowing for gaps. The segments retained were then realigned using mafft (https://mafft.cbrc.jp) with the parameter “‐‐maxiterate 1000 –localpair”. The final alignment, containing 37,389,162 sites including gaps, was then concatenated.
The second data set only used the coding sequences (CDSs) of orthologous genes. One‐to‐one orthologues were identified between D. melanogaster and each of the other species if they met both reciprocal best blastp hit (RBH) with blastp (e value <1e‐5) and synteny criteria. For the latter, any RBH gene pair colinear with another RBH pair within five genes up‐ or downstream was considered as shared synteny. Orthologous pairs from the different comparisons were merged. Genes present in fewer than 42 species were then deleted, leaving a final data set of 8550 orthologues. The corresponding amino acid sequences were first aligned using sate (http://phylo.bio.ku.edu/software/sate) plus prank (Loytynoja, 2014) and a method for removing poorly aligned sites (Zhang et al., 2014). The remaining sites were then realigned using sate plus mafft and transferred back to CDS alignments. All CDS alignments were concatenated into a data set containing 18,793,872 sites.
We used both concatenation and coalescent approaches to infer species trees from each of the two data sets. We used the examl (Kozlov et al., 2015) package with both the GAMMA and PSR models for the concatenation analyses and astral (Zhang et al., 2018) packages for the coalescent analyses. Each data set was run 100 times under both “–m PSR” and “‐m GAMMA” in the examl package and we chose the tree with highest likelihood for each data set for the GAMMA model. For the PSR model, we calculated the likelihood of the 100 trees generated under the GAMMA model to identity the best ML tree because likelihood values under the PSR model are meaningless. For the coalescent analyses, the whole genome alignment was cut into 37,386 windows of length 1000 bp. A tree was constructed for each window using raxml (Stamatakis, 2014) under the GAMMA model and all trees were then parsed to astral to get the consensus species tree (Figure S1).
Divergence times were calculated using ~60,000 bp of sites randomly extracted from the whole genome alignments by MCMCtree in the paml package (Yang & Rannala, 2006). MCMCtree needs fossil records or biogeographic data for calibration and there are no such records for any of the nodes in our tree. However, estimates have been made for three of our nodes by Tamura et al. (2004) and Russo et al. (2013) based on fossil calibrations for other nodes in their trees, we used the time estimates for these nodes from the literature in our dating analyses. We set the lower and upper bounds of the nodes as 2*σ of their estimations. This gave us calibration time ranges of 33–76 Mya for the node separating the melanogaster and obscura groups, 20–32 Mya for the node separating the virilis and repleta groups, and 39–87 Mya for the node separating the subgenera Drosophila and Sophophora.
We used two methods, discovista (Zhang et al., 2018) and quibl (Edelman et al., 2019), to scan for ILS and introgression in the trees generated from our whole genome alignments. Individual trees were calculated for 1 kbp windows of aligned sequence at least 5 kbp apart (to reduce the probability of physical linkage), omitting windows with gaps at more than 40% of sites. The quibl analyses required an outgroup, so we treated the two subgenera separately for that analysis, using D. mojavensis as the outgroup for subgenus Sophophora, and D. melanogaster as the outgroup for subgenus Drosophila (and using the corresponding branch lengths from the full tree in the two sub‐trees). More than 100 runs were performed for each subgenus, and from each run 500 individual trees were selected at random from the 6754 and 5431 individual trees generated by the Sophophora and Drosophila analyses, respectively. discovista (but not quibl) was also run on trees generated from the orthologue data set.
discovista calculates the average frequency of each of the three possible arrangements at each branch (with respect to an outgroup) across all the analysed windows. ILS and/or introgression are suggested if the combined frequencies of the two less common, that is, discordant, arrangements are high. ILS is suggested if the relative frequencies of the two discordant arrangements are similar to one another. Introgression is suggested to the extent that their relative frequencies differ. quibl then takes a statistical approach to infer the proportions of ILS and introgression. It distinguishes between the two based on the distribution of the internal branch lengths of individual trees in a particular topology. Specifically, it compares two models, one with ILS only and one with a mixture of ILS and introgression, using the Bayesian information criterion test (BIC). We used the threshold of △BIC = 10 as recommended to select the preferred model (Edelman et al., 2019).
2.4. Phylostratigraphy
Phylostratigraphy uses blastp‐scored sequence similarity to estimate the taxonomic distribution and thence the minimal age of every protein coding gene. The NCBI nonredundant database is queried with the protein sequence corresponding to each gene to detect the most distant species in which a sufficiently similar sequence is present. It then posits that the gene is at least as old as the age of the common ancestor (Domazet‐Loso et al., 2007, 2017). The method is specifically designed to identify genes arising by noncopying mechanisms which thus have the potential to encode proteins with novel domains (Carvunis et al., 2012). Conservatively, it classifies genes as being as old as the most ancient parts of the proteins they encode, and duplicate copies as being as old as their parent genes.
In our case we evaluated the ages of all proteins at least 40 amino acids long and screened each of them against the entire NCBI nonredundant (NR) database by blastp and hmmer, using a permissive e‐value threshold of 10−3 and allowing a maximum number of 200,000 hits. This process, plus the fact that our annotations were done on repeat‐masked genomes, follows recent best practice and is specifically designed to reduce the risk of underestimating the ages of short genes (Arendsee et al., 2019; Domazet‐Loso et al., 2017; Hanschen et al., 2016; Kim et al., 2019; Vakirlis et al., 2020). The timing of lineage divergence events was estimated with TimeTree (Kumar et al., 2017) for the taxonomic branching before the divergence of drosophilids. Within drosophilids, we used the timing provided by our own analyses above.
We then counted the number of genes in each species originating in each of 34 phylostrata (PS) represented in our data, and also aggregated those counts into four broad evolutionary eras encompassing the phylostrata. The ancient era encompassed PS 1–8 (cellular organisms to Ecdysozoa, 4290–743 Mya), the middle era PS 9‐18 (Panarthropoda to Holometabola, 743–325 Mya), the young era PS 19‐25 (Diptera to Acalyptratae, 325–132 Mya), and the newest PS 26‐last (132 Mya‐present). For some analyses we also normalised the numbers of genes originating in each PS or era by the duration of the PS/era. The age of the genes assigned to each PS (PS midpoint) was defined as the average of the age of the taxonomic node defining that PS and the age of the node defining the previous PS.
We also calculated correlations between the numbers of genes arising in different phylostrata across species and with phylogenetic distance and three ecological characteristics of the species. To do this we constructed matrices of inter‐species absolute distances in each parameter across the 47 species. Estimates of the three ecological parameters, drying power of air, precipitation and temperature, were based on distributional data for each species assembled from Taxodros (www.taxodros.uzh.ch) and museum records and verified by taxonomy experts. Climate data for each species’ distribution were then extracted from worldclim V. 1.4 (Hijmans et al., 2005) and drying power of air calculated following Kellermann et al. (2009). Mean values per species were used. Mantel correlations were calculated using mantel.rtest, as implemented in the ade4 package (Dray & Dufour, 2007). When correlating distances based on number of genes per phylostratum, we additionally used a partial Mantel test and added a third distance matrix based on the total number of genes. For this we used the function mantel.partial implemented in the vegan package (Oksanen et al., 2019).
We also modelled the data on the age profiles of the genes in each species against a range of possible mathematical functions in order to better understand the underlying processes. To varying extents the profiles for all 47 species showed disproportionately high numbers of genes that arose in the most recent and ancient eras (see below) and for each of the species we found that this pattern was best explained by a simple linear combination of a growing and a decaying exponential function of time. We therefore used this function to generate two plots for each of the 47 species: (1) the number of genes vs. phylostratum midpoint, and (2) the ratio of the number of genes to phylostratum duration vs. phylostratum midpoint. For both (1) and (2), we fitted curves of the form f(t) = a*exp(‐b*t) + c*exp(d*t), where a, b, c, and d are positive parameters and time t is measured in Mya (t = 0 corresponding to the present, and large t to ancient time). The parameters a and c capture gene numbers as they arise close to the present (a), and had accumulated in the distant past (c). The exponents b and d capture the decrease in the number of novel genes moving backwards in time from relatively high numbers in the newest PS (−b) and the increase in the number of ancient genes in the most ancient PS (+d). For each species, best fit choices of the parameters a, b, c, and d were generated using the Marquardt‐Levenberg algorithm implemented using our own computer code as well as the freely distributed gnuplot software package. The half‐life of novel gene decay is simply obtained from curve (1) as ln2/b, and the time when the number of genes has its lowest value is calculated directly from this curve as the point when the derivative of the function f(t) is zero.
All statistical analysis for the phylostratigraphy was done in Excel, StatPlus, Prism and R. Skew and kurtosis were calculated and plots created in R. The R package phylo.heatmap (Revell, 2012) was also used to generate some figures.
2.5. Nonsynonymous substitution rates
Three subsets of species defined on the basis of their climate niches were identified by matching their distributions against the Köppen climate classifications. Two of the subsets contained different climate specialists as defined by their presence in just the tropical and temperate plus continental Köppen climate classes respectively, and the third comprised generalists that were present in all four classes covered by Drosophila. For each of the two specialist‐versus‐generalist comparisons, groups of orthologues containing genes from all species included in that comparison and the outgroup Scaptodrosophila lebanonensis (Vicoso & Bachtrog, 2015) were aligned by prank and filtered for poorly aligned sites using gblocks (Castresana, 2000). We used 9143 orthologues in the tropical vs. generalist comparison and 9520 in the temperate‐continental vs. generalist comparison. In each comparison the distribution of the ratio of nonsynonymous to synonymous substitutions, dN/dS, was assessed for goodness of fit against three models by codeml in paml V4.9h (Yang, 1997). The three models were: no difference between species (H0), differences between the two subsets compared (H1), and differences between species unrelated to subsets (i.e., a free ratio model; H2). χ2‐derived p‐values from likelihood ratio tests adjusted for a false discovery rate cutoff of 0.05 were used to compare the goodness of fit of the different models. Genes for which the model of subset differences gave a significantly better fit for H1 than H0 and no significantly better fit for H2 than H1 were considered candidates for involvement in the ecological differences between the respective specialist subset and the generalist subset. GO terms enriched among these candidates were found using clusterprofiler (Yu et al., 2012) and the org. Dm.eg.db database (version 3.13).
2.6. Population genomics
The samples of the 13 species that were resequenced were each obtained from a single locality. Variant calling followed the two‐step pipeline recommended by gatk Best Practice (https://gatk.broadinstitute.org/hc/en‐us). First, we called haplotypes for each individual using the gatk HaplotypeCaller command with a minimum phred‐scaled confidence threshold of 30. Then we combined the calls for all individuals in each species into a final VCF file using gatk GenotypeGVCF. Only biallelic variants with QD >2.0, FS <60.0, MQ <40.0, MQRankSum > −12.5 and ReadPosRankSum > −8.0 were retained, using the filtering command gatk VariantFiltration.
Nucleotide diversity (Pi) was calculated for each 10 kb‐window across each species’ genome using vcftools (Danecek et al., 2011). Then the correlation of Pi with niche position was calculated across species, correcting for phylogenetic position using the PGLS function in caper and the phylogeny and divergence times obtained above. Niche position, calculated using the niche function in the R package ade4, was defined as the distance from the mean of the multivariate climate data for the known distribution of each species to the mean for all species in the analysis (Dolédec et al., 2000). The distributional and climatic data were obtained as per the previous section. Given the large differences between species in sampling density, the species distribution model for each species incorporated a bias layer, which was generated by using the sampling points for all 47 species to create an overall point density layer in arcgis. This layer would be heavily influenced by the cosmopolitan species, and therefore should be a reasonable estimation of differences in sampling effort.
To calculate population size we first generated the site frequency spectrum (sfs) using angsd (Korneliussen et al., 2014) and then estimated per site Watterson theta (θ w) according to θ w = k/an , where k is the number of segregating sites and an is the (n–1)th harmonic number. Then the effective population size was estimated according to θ w = (4N e μ)*nsite, where μ is the mutation rate per site per generation calculated for four‐fold degenerate sites with phylofit (https://rdrr.io), and nsite is the number of all sites in the sfs.
We then tested whether the number of genes appearing at each phylostratum estimated above was correlated with N e. To account for nonindependence of species due to phylogenetic inertia we used phylogenetic generalized least squares regression (pgls). Pgls was conducted using the gls function in the ape package (Paradis & Schliep, 2019) independently in each PS and using the total number of genes assigned to the PS as a covariate in the form Genes in PS ~ N e + Total number of genes. The tree was rooted at the midpoint using the function midpoint.root and Pagel’s lambda estimated using the phylosig function, both implemented in phytools (Revell, 2012). The estimated lambda for N e was 0.87. We ran pgls in two modes; in one we let pgls estimate the lambda initialising the value at 1 and in the other we considered a Brownian Motion model equivalent to a lambda fixed at 1. Values of p were corrected using the Benjamini‐Hochberg correction and considered significant when p‐adjusted was <.05.
Finally, we carried out two tests comparing the influence of natural selection on the nucleotide diversity seen in each of the resequenced species. The first, based on the Grantham scores of non‐synonymous substitutions in their coding regions (Grantham, 1974), evaluated the proportions of deleterious mutations in each genome on the basis of the level of physicochemical dissimilarity between the alternative amino acids encoded by those substitutions, with values >150 being classed as deleterious. The second test was based on the neutrality index NITG, which was calculated according to Stoletzki and Eyre‐Walker (2011). NITG is a relative measure which in essence compares the proportion of nonsynonymous to synonymous polymorphisms within a species (Pn and Ps respectively) to the proportion of nonsynonymous to synonymous divergence between that species and other closely related species (Dn and Ds, respectively). For each species, we chose three or four closely related species as outgroup species. Amino acids encoded by orthologous genes in each species under test were first aligned with those in each of its outgroup species using prank and then translated into CDS alignments with gaps removed. Then Dn, Ds, Pn and Ps were counted using script in Nolte et al. (2013) and NITG calculated as:
where i refers to the ith gene. An NITG value of unity signifies neutral evolution, with deviations above or below one indicating various forms of selective difference, either within the target species or between it and the outgroup species.
3. RESULTS
3.1. New and improved genome assemblies and annotations
The 15 newly sequenced species represent a combination of restricted and widespread species from different parts of the Drosophila phylogeny (Table S1). They include rainforest‐restricted and more widespread montium subgroup species, cactophilic and cosmopolitan repleta group species and climate restricted and cosmopolitan immigrans group species, plus representatives of other lineages. All but one (D. ironensis) of these 15 species were inbred to varying degrees under laboratory conditions to reduce heterozygosity and facilitate assembly. An average of 150× coverage sequencing data was obtained for each species (Table S2). The scaffold N50 was larger than 1 Mb for 13 of the resulting assemblies, and around 500 kb for D. serrata and D. pseudoananassae. busco analysis identified more than 97% genes as complete and <1% genes as missing for most of the species (Table S3).
We also generated 9 Gb/50× coverage of additional resequencing data (from two paired‐end libraries with 500 bp insert size) for 20 of the previously sequenced species whose assemblies were less complete than the others (Tables S1–S3). The additional data were used to close gaps and on average increased contig N50 size by a third (Table S4). Even for the benchmark species D. melanogaster the gap length was reduced from 1.15 to 0.89 Mbp. On average across the 47 species in the data set, scaffold N50 was around 8.5 Mb and busco gene recovery was 98.6% in total and 97.8% for complete genes only.
Genome size varied substantially across species, ranging from 115 Mbp for D. navojoa to 252 Mbp for D. albomicans, with an average of 166 Mbp (Table S3). Genome size was strongly correlated (r = .8, p = 2.02e‐11) with TE content, which ranged from 8.2% for D. mauritiana to 45.6% for D. ananassae (Table S5). Overall, the most abundant TEs were LTRs (long terminal repeat elements), followed by DNA transposons and LINEs (long interspersed nuclear elements). Protein coding genes were annotated through a combination of homology‐ and de novo‐based approaches, assisted by previously published transcriptomes for 24 species and 2–4 Gb mixed life stage transcriptomes generated herein for 19 others. The number of predicted genes for each species ranged from 10,758 for D. navojoa to 17,530 for D. albomicans (Table S5). The gene number variation is partially due to quality differences of the genome assemblies and the lack of transcriptome data for annotation in some species.
Substitution rates were estimated across the phylogeny below from the whole genome alignments (Table S6). Species in the repleta‐virilis radiation (4.79 × 10−3 substitutions per site per million years) had the highest rate, more than twice that of species in the obscura group (1.9 × 10−3 substitutions per site per million years). The rates were positively correlated (r = .90, p = .034) with the number of species per group (taxodros.uzh.ch as at 2019) if the ananassae group was excluded from the analysis. The rate for the latter was higher than would be expected from its relatively low species number, but this group may contain many cryptic species given that three new species have only recently been described (McEvey & Schiffer, 2015). Correlations between substitution and net speciation rates have also been observed in birds and reptiles (Eo & DeWoody, 2010; Lanfear et al., 2010).
3.2. Evolutionary history of the Drosophila
We applied the concatenation (examl (Kozlov et al., 2015)) and coalescent (astral; (Zhang et al., 2018)) approaches to each of two data sets to reconstruct the phylogenetic tree of the 47 species. One data set, obtained using multiz (Blanchette et al., 2004), comprised 36 Mb of whole genome alignments across all species, covering both coding and non‐coding regions. The other, obtained using D. melanogaster as reference and the reciprocal best hit method, comprised 20 Mb of CDSs for 8550 sets of orthologous genes across all species, allowing no more than five species to be missing each gene.
The examl trees obtained from both data sets had congruent topologies for all except two adjacent nodes, involving D. mercatorum and D. hydei, as representatives of the mercatorum and hydei subgroups of the repleta species group in subgenus Drosophila (Figures S1 and S2). Previous studies had also failed to reach a consensus on the placement of these two subgroups (Durando et al., 2000; Tatarenkov & Ayala, 2001). examl CDS trees had D. mercatorum diverging first under both GAMMA and PSR models, as did those from the whole genome alignment with PSR but not with GAMMA, which instead had D. hydei diverging first. astral trees using the two data sets, which were otherwise concordant with the examl trees, also conflicted here, with the CDS trees supporting D. mercatorum diverging first and the whole genome trees supporting D. hydei diverging first (Figure S2). We therefore calculated the site concordance factor (sCF) (Minh et al., 2020), that is, the proportion of sites that support each topology for the branch in question using the two data sets, finding more sites in both data sets supported D. mercatorum diverging first (Table S7). We used this topology in the analyses below.
Our phylogenies (Figure 1) agree with the generally accepted view that the genus Drosophila is paraphyletic to other genera and can be divided into two large clades (Finet et al., 2021; van der Linde et al., 2010; O'Grady & DeSalle, 2018), one including subgenus Sophophora and genus Lordiphosa, and the other containing subgenus Drosophila, plus various other small lineages, including subgenus Dorsilopha and genus Zaprionus. However, our data resolved several previously ambiguous or unknown relationships within these two large clades.
FIGURE 1.
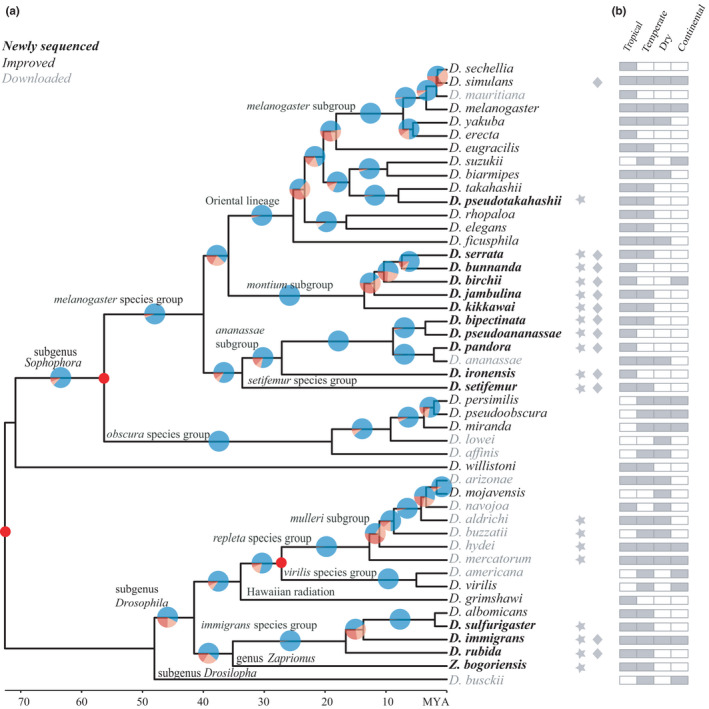
Phylogeny and climate niches of the 47 species. (a) Phylogenomic analyses. Species with names in bold are newly sequenced, those in black are improved by additional sequences generated in this study (see Table S4 for details) and those in grey are as previously published. The species for which we added transcriptome data are indicated with stars and those for which we resequenced multiple individuals are indicated with rhombuses. Pie chart areas for each node show the proportions of the three possible topologies for the corresponding branch, with blue denoting the most common topology (i.e., the species tree, as shown) and red and orange the two alternatives (i.e., with each of the two daughter lineages in the species tree as the outgroup instead). The three nodes indicated with red dots are those for which dates had been estimated by Tamura et al. (2004). (b) Climate zones occupied by each species according to the Köppen classification are presented on the right, with grey denoting presence in that environment
Within the clade containing the subgenera Drosophila and Dorsilopha and genus Zaprionus, our trees showed D. busckii was the outgroup to all other species. Earlier studies have placed this species as an outgroup of both Sophophora and Drosophila (Pitnick et al., 1995) or closest to either the immigrans–tripunctata (Zhou & Bachtrog, 2015) or Hawaiian‐repleta radiations (Finet et al., 2021) within the Drosophila. We placed Z. bogoriensis, the only Zaprionus species in our data set, together with the immigrans–tripunctata radiation, which concurs with three earlier studies (Finet et al., 2021; O'Grady & DeSalle, 2018; Yassin, 2013) that grouped at least some Zaprionus species with that radiation. Within the immigrans‐tripunctata radiation, we placed D. rubida as an outgroup to both D. immigrans and D. albomicans/D. sulfurigaster, whereas one earlier phylogeny paired D. rubida with D. immigrans as a sister to D. albomicans/D. sulfurigaster (Finet et al., 2021). Consistent with some (Finet et al., 2021; O'Grady & DeSalle, 2018; Remsen & O'Grady, 2002; Yassin, 2013) but not all (Grimaldi, 1990) other studies, we found the Hawaiian Drosophila (represented by D. grimshawi) were nested closest to the virilis‐repleta grouping, forming a sister lineage to the immigrans–tripunctata radiation.
Traditional taxonomy divided the other large clade, subgenus Sophophora, into three groups, the neotropical willistoni, “Old World” obscura and melanogaster groups. Our trees agreed with the generally accepted topology that has willistoni as the outgroup to the other two. Results for the five obscura group species we analysed also concurred with the topology for those species found in previous studies (Barrio & Ayala, 1997; van der Linde & Houle, 2008). However, our placement of the 25 melanogaster group species we analysed was at variance with previous work on this group (Da Lage et al., 2007; Finet et al., 2021; Kopp, 2006; Kopp & True, 2002; Yang et al., 2004, 2012), most of which was based on sequences for just one or a few genes.
The 190 species (taxodros.uzh.ch) in the melanogaster group have traditionally been divided into 12 subgroups, nine of them included in our study (the other three, flavohirta, longissimia and denticulata, seldom being described). These nine subgroups have in turn generally been divided into three monophyletic clades, one comprising the ananassae subgroup, one the montium subgroup and the third encompassing the other seven subgroups (sometimes referred to as the Oriental lineage; Figure 1) (Seetharam & Stuart, 2013). Our analysis suggested that ananassae is the outgroup to the other two, in agreement with most previous studies except one based on sequences for a single gene which put montium as the outgroup (Yang et al., 2004).
However, we also found that the melanogaster species group is paraphyletic, putting the setifemur group closest to the ananassae subgroup with high support in all our trees. The phylogenetics of the setifemur group have not previously been discussed but the fima clade has previously been placed in a similar position (Kopp et al., 2019). Given our trees also put the montium subgroup as an outgroup to both the setifemur group and ananassae subgroup, we support the proposal (Da Lage et al., 2007) to elevate both the ananassae and montium subgroups to the level of species groups.
The relationships we found within the ananassae subgroup concurred with the few previous analyses of this clade (van der Linde et al., 2010) but provided the first evidence on the positions of D. pandora and D. ironensis. We found D. pandora is closest to D. ananassae in the ananassae complex, and D. ironensis to be an outgroup to both the ananassae and bipectinata complexes.
All our trees showed D. kikkawai to result from the earliest split of the montium subgroup, followed by D. jambulina, then D. birchii and finally D. serrata and D. bunnanda (Figure S1). This ordering agreed with one earlier study (Finet et al., 2021) but differed from another which found D. jambulina split first (Da Lage et al., 2007) and still others that placed D. jambulina closer to D. serrata than to D. birchii (van der Linde & Houle, 2008) or placed D. jambulina closer to D. birchii than to the serrata clade (Pissios & Scouras, 1993). The early split of D. kikkawai is also consistent with a recent analysis of the montium group based on 60 genes (Conner et al., 2021) which clearly separates D. jambulina from D. serrata and relatives, although D. jambulina in that phylogeny does not split earlier than the serrata group of species.
There has been little clarity on several subgroup relationships in the Oriental lineage until now (Finet et al., 2021; Kopp, 2006; Yang et al., 2004, 2012). It is generally agreed that the elegans and rhopaloa subgroups formed a pair that is an outgroup to the takahashii, eugracilis, melanogaster and suzukii subgroups but the position of ficusphila was less clear. All our trees put ficusphila as the outgroup to other subgroups in the Oriental lineage, in agreement with some previous trees (Finet et al., 2021; Kopp & True, 2002; Schawaroch, 2002), but not others which either put the elegans‐rhopaloa clade as the outgroup (Da Lage et al., 2007; Kopp, 2006; van der Linde et al., 2010) or combined the elegans, rhopaloa and ficusphila subgroups (Seetharam & Stuart, 2013). The relationships among the takahashii, eugracilis, melanogaster and suzukii subgroups was also uncertain. The most comprehensive studies either had takahashii closest to suzukii and this pair plus eugracilis and melanogaster in a polytomy (van der Linde & Houle, 2008) or had takahashii/suzukii closest to eugracilis with melanogaster as the outgroup (Finet et al., 2021; van der Linde et al., 2010). Our trees paired takahashii with suzukii and eugracilis with melanogaster.
3.3. Prevalence of introgression and incomplete lineage sorting
We used the discovista package (Zhang et al., 2018) to evaluate the extent of gene tree discordance with the species tree across the phylogeny in Figure 1. We found over one third of the internal branches had a combined frequency of the two alternative topologies higher than 20% and for 12 of these it was higher than 40% (Figure 1a, Figure S3). Most of the previously controversial nodes had relatively high discordance levels. There was an overall negative correlation between branch length and the frequency of alternative topologies (Figure 2a), which is consistent with widespread ILS, as shorter speciation times tend to increase the probability of ILS (Pamilo & Nei, 1988). However, while most branches had a similar frequency for the two alternative topologies, seven of the nodes with >20% discordance showed a frequency difference between the two alternatives of greater than 20%, implying the presence of introgression (Figure S4). Notably the controversial node in the repleta group showed a higher frequency of the alternative tree placing D. mercatorum and the mulleri subgroup (represented by D. buzzatii) together than that putting D. mercatorum and D. hydei together (42%–24%) but the frequency of the latter was still quite high. This suggests a mix of introgression and ILS.
FIGURE 2.
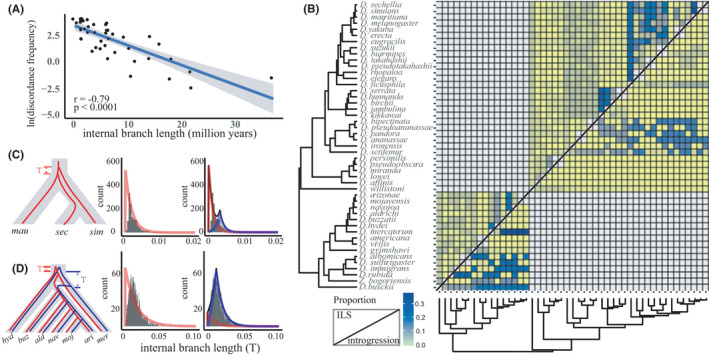
Causes of gene tree and species tree discordance. (a) Discordance frequency calculated by discovista as a function of branch length in our Drosophila species tree. The grey shaded region indicates the 95% confidence interval from a linear model (“lm” function in R). (b) Species pairwise metrics for the proportions of ILS (above diagonal) and introgression (below diagonal) estimated by quibl. More blue denotes a higher value. (c) Left, ILS is the only factor causing the closer genomic relationship between D. mauritiana and D. sechellia. The grey background indicates the species tree and the red line indicates the ILS genealogy. “T” is the internal branch length under ILS. Middle and right panels show the distribution of the internal branch lengths supporting the closer relationship of D. mauritiana and D. sechellia, which fitted better with the model assuming only ILS (middle, average of Bayesian information criteria (BIC) = –1815.24) than it did with the model assuming a mixture of ILS and introgression (right, average of BIC = –1776.57). The blue line denotes the inferred distribution of introgression, the red line denotes ILS and the black line their combination. The histograms show the distribution of internal branch lengths of individual trees supporting a closer relationship of D. mauritiana and D. sechellia. mau, mauritiana; sim, simulans; sec, sechellia. (d) Left, both ILS and introgression occurred during the evolution of the repleta group. The grey background indicates the species tree, the blue line denotes the genealogy of introgression and the red lines denotes that of by ILS. “T” is the internal branch length for the corresponding genealogies. The middle and right panels show that the distribution of the internal branch lengths supporting the closer relationship of D. mercatorum and the ancestor of the five mulleri subgroup species fitted better with a model assuming a mixture of ILS and introgression (right, average of BIC = –1415.26) than it did with a model assuming ILS only (middle, average of BIC = –1359.66). The histograms show the distribution of internal branch lengths of individual trees supporting a closer relationship of D. mercatorum and the ancestor of the five mulleri subgroup species. hyd, hydei; buz, buzzatii; ald, aldrichi; nav, navojoa; moj, mojavensis; ari, arizonae; mer, mercatorum
We then used the statistical approach in quibl (Edelman et al., 2019) to infer the proportions of ILS and hybridisation for each internal branch. Overall, the combined frequency of ILS and introgression events estimated by QulBL was similar to the frequency of gene tree discordance calculated by discovista (Figure S4B). However, the QulBL analysis revealed differences between the two subgenera in the prevalences of ILS and introgression.
In subgenus Sophophora, quibl found low levels of both ILS and introgression in the ananassae subgroup and obscura group (nodes 19–28 in Figures S3, S4) but identified ILS as the primary cause of phylogenetic discordances in nodes of the Oriental lineage and montium subgroup (Figure 2b, Figures S3 and S4). For example, while ILS and hybridisation have both been proposed previously in the node (node 1) separating D. simulans and D. sechellia (Garrigan et al., 2012; Pease & Hahn, 2013), our analyses suggested all of the discordant loci in this node were attributable to ILS (Figure 2c). We also observed high ILS and low introgression in several previously controversial branches in the Oriental lineage (e.g., nodes 6, 9, 10, 12) and montium subgroup (e.g., nodes 15 and 16) (Da Lage et al., 2007; Kopp, 2006; van der Linde et al., 2010; Russo et al., 2013; Yang et al., 2004, 2012; Yassin et al., 2016). The only major exception to this trend in this subgenus involved the node (node 18) separating the Oriental lineage from the montium subgroup, where more introgression signal was detected than ILS (Figure 2b).
In contrast to the situation in subgenus Sophophora, introgression was a major contributor to several of the higher gene tree discordances in subgenus Drosophila (Figure 2b). This included some relatively deep branches involving splits among the genus Zaprionus, immigrans species group and virilis‐repleta radiation, suggesting a long history of introgression in the subgenus. Notably, we detected high levels of both ILS and introgression in the problematic branches (node 34) between D. mercatorum, D. hydei and the mulleri subgroup. In particular, high levels of introgression (36% of total loci) were detected between D. mercatorum and the mulleri subgroup (Figure 2b,d and Figure S5).
3.4. Trajectories of novel gene flux differ between lineages
To investigate the link between gene gain and loss events and lineage‐specific adaptations in the Drosophila phylogeny, we determined the taxonomic distribution of genes and thence their minimal evolutionary ages by phylostratigraphy (Domazet‐Loso et al., 2007; Tautz & Domazet‐Loso, 2011). The ages of the genes in our species cover 32–36 phylostrata (PS) from four successive evolutionary eras, spanning the origin of cellular organisms through to the divergence of D. melanogaster. On average across the 47 species, we classified the majority, 76 ± 0.3%, as deriving from the ancient era (PS 1–8, up to the origin of the Panarthropoda), with 7.0 ± 0.04% from the middle (PS9‐18, up to the Holometabola), 8.2 ± 0.08% from the young (PS19‐25, up to the Acalyptratae) and 8.8 ± 0.3% from the newest era (PS 26‐last, up to the present) (Figure 3a,b, Figure S6A).
FIGURE 3.
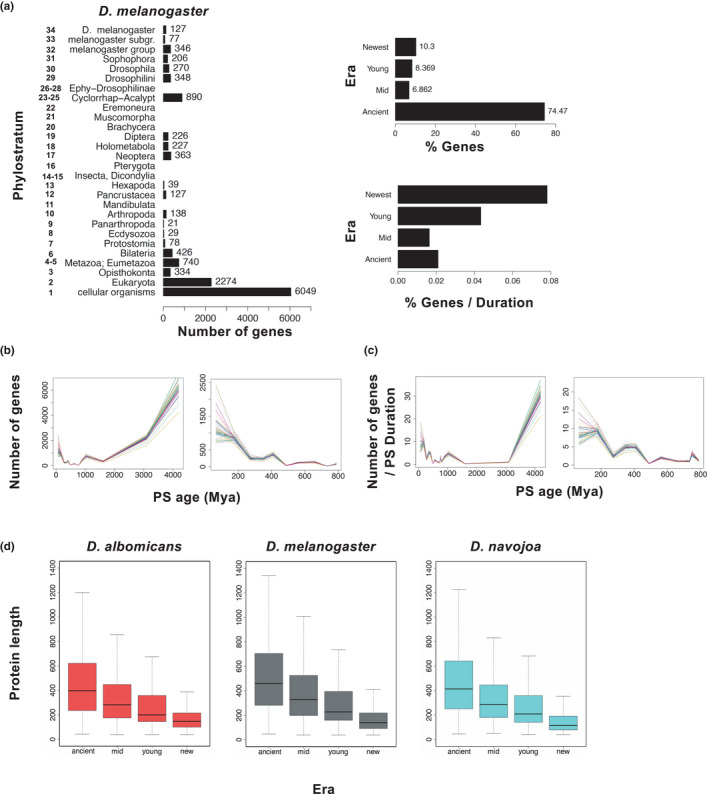
Temporal dynamics of novel gene gain and loss. (a) Raw numbers of novel genes in D. melanogaster estimated to have arisen in the 34 phylostrata (left) and four eras (top right), and the number of these genes arising in the four eras normalised for the (very different) durations of the eras (lower right). (b) Plots of the absolute numbers of genes estimated to have arisen in each phylostratum for each of the 47 species up to 4290 Ma (left) and zoomed in to 800 Ma (right). (c) Plots of the phylostratum duration‐normalised numbers of genes estimated to have arisen in each phylostratum for each of the 47 species up to 4290 Ma (left) and zoomed in to 800 Ma (right). Each species in (b) and (c) is represented by a different colour (same in each plot). See Figures S6 and S7 for further details. (d) Average lengths of proteins encoded by genes originating in the different evolutionary eras in three species from phylogenetically diverse lineages
However, the older eras are much longer than the younger ones (see Materials and Methods) and after correcting for their respective durations, we found the rate of gene acquisition was highest in the newest era (Figure 3a,c, Figure S6B). The same patterns were evident when the data were analysed in terms of phylostrata rather than eras (Figure 3a, Figure S7) and in broad terms they also recapitulate the pattern previously reported for genes in D. melanogaster and, indeed, some other animals (Domazet‐Loso et al., 2017; Neme & Tautz, 2013; Tautz & Domazet‐Loso, 2011). The disproportionately high numbers of novel genes that arose in the recent past may in part be because many of the genes arising in earlier eras subsequently disappeared, whereas there was less time for genes arising more recently to be lost.
There was significant variation in the age distribution of genes between species, with the differences greatest for genes originating in the newest and ancient eras (Figure 3b,c, Figure S8). The highest numbers of genes acquired in the newest era (PS 26‐last, c.f. the diversification of Drosophila species, which spans PS 30‐last) were largely concentrated in two lineages respectively containing three pseudoobscura subgroup species each occupying the same three (temperate, continental and dry) Köppen climate classes (14.3 ± 0.9%) and the five desert‐adapted cactophilic mulleri subgroup species (11.3 ± 0.7%). On the other hand, five of the six species with the lowest numbers of new genes arising in the newest era (D. ironensis, D. willistoni, D. grimshawi, D. albomicans and D. busckii; 6.2 ± 0.5%) were phylogenetically widely separated and variously climate specialists (each just covering one or two Köppen classes). The five cosmopolitan generalists (each covering all four Köppen classes) varied in new gene numbers, depending on their lineage; they were low in the immigrans group species D. immigrans (5.5%), relatively high in the melanogaster subgroup species D. melanogaster and D. simulans (10.3% and 9.6%), and intermediate in the repleta group species D. hydei and D. mercatorum (7.9% and 8.5%).
Notably the species differences in the numbers of genes retained from the ancient era reflected the opposite trends. For example, the numbers were relatively low in the three pseudoobscura (70.7 ± 1.0%) and the five cactophilic mulleri (73.6 ± 0.7%) subgroup species and relatively high in the five phylogenetically well separated climate specialists above (77.8 ± 0.5%). In fact, calculation of matrices of species’ pairwise differences in the numbers of genes arising in each era revealed that those for the ancient and newest eras were quite close mirror images of each other, with the pseudoobscura and mulleri lineages standing out most in both cases (Figure S9) and little obvious signal in the matrices for the middle and young eras. This suggests some form of trade‐off between the fates of recently arisen and ancient genes. However, it seems unlikely that this was a direct effect, because proteins encoded by genes retained from the newest era were on average substantially smaller than those from the ancient era (Figure 3d).
We also calculated the difference matrices for the species’ phylogenetic distances and three features of their ecological niches, namely temperature, precipitation and drying power of air (Figure S10) and examined their correlations with the corresponding matrices for the numbers of genes acquired in each phylostratum and era (either raw numbers or numbers normalized to genome size, total number of genes or phylogenetic distance) (Figures S11 and S12). We found that the differences in numbers of genes arising in the newest era had low but significant correlations with differences in phylogenetic distance (r = .18), drying power of air (r = .24), temperature (r = 0.21) and precipitation (r = .17) (controlling for differences in the total number of genes as a covariate). No significant correlation was evident for genes arising in the other eras. Thus, species that were most divergent in terms of their phylogenetic distance and climate‐linked ecological niches also differed most in the numbers of genes acquired in the most recent era.
Finally, we modelled the temporal dynamics of the appearance of new genes in each species, using a linear combination of a growing exponential function and a decaying exponential function in each case (Figures S13 and S14, Table S8). The models were chosen with the minimal number of parameters necessary for capturing gene emergence rates across all the PS, and data fitting was performed using the Marquardt‐Levenberg algorithm (see Materials and Methods). The modelled trajectories showed similar levels of species differences as the primary data. Notwithstanding those differences, the parameter estimates for the models for the 47 species (Table S8) enabled us to calculate average values of 97 My (interquartile range (IQR) 91–104 My) for the half‐life of novel genes, and 589 Ma (IQR: 574–603 Ma) for the time when the number of novel genes was at a minimum.
3.5. Nonsynonymous substitution rates vary with climate niches
We screened for genome‐wide differences in nonsynonymous substitution rates among three subsets of species defined on the basis of their Köppen climate classifications. One subset contained six species (D. bunnanda, D. grimshawi, D. ironensis, D. mauritiana, D. pandora and D. pseudoananassae) restricted to tropical climates. Another contained four species (D. americana, D. busckii, D. suzukii, and D. virilis) which each inhabited both temperate and continental but not tropical or dry climates, and the third contained five generalist species (D. hydei, D. immigrans, D. melanogaster, D. mercatorum and D. simulans) which were each found in all four of these climate zones. We then compared the dN/dS ratios of orthologous genes from each of the tropical and temperate‐continental subsets against those in the generalist subset using the PAML package and orthologues from Scaptodrosophila lebanonensis as an outgroup.
We found 460 genes with significantly different dN/dS values between the tropical specialists and the generalists among 9143 orthologous pairs tested, and 506 genes with significantly different dN/dS values between the temperate‐continental specialists and the generalists among 9520 orthologous pairs tested (Table S9). Given the high levels of gene tree incongruence seen in some lineages above, we then checked whether these differences remained if we used gene‐ rather than species‐trees as guide trees for PAML in the analysis (see Materials and Methods). We found that three of the 460 and seven of the 506 genes that had shown significant dN/dS in the two comparisons above did not also do so when using the respective gene trees as guides (Table S9). So only those 457 and 499 genes which yielded significant differences using both types of guide tree were retained for further analysis.
Notably, 87 genes appeared in both the tropical specialist vs. generalist and temperate‐continental specialist comparisons, far more than expected just by chance (p << .001 for each comparison; Table S9). Despite that overlap, the distributions of values across all the genes involved in the significant dN/dS differences also differed significantly between the two comparisons. Notwithstanding many individual exceptions, dN/dS values were generally higher in the temperate‐continental specialists than the generalists but lower in the tropical specialists than the generalists (Figure 4).
FIGURE 4.
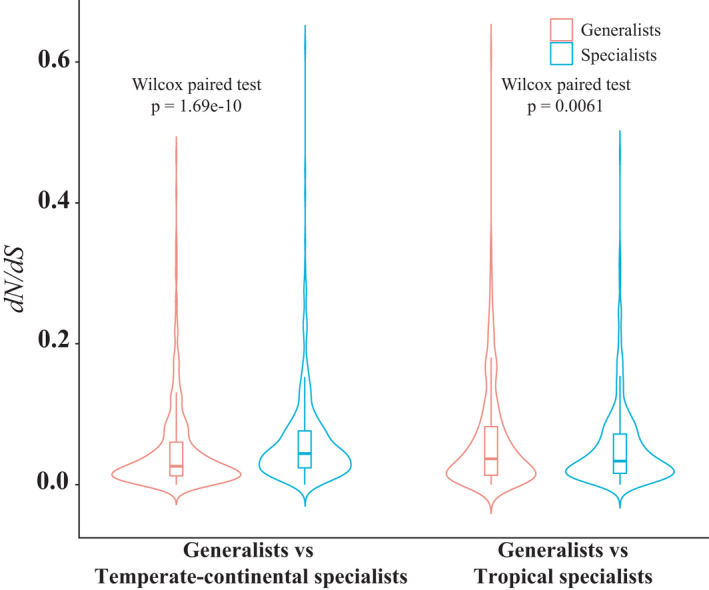
Distributions of dN/dS values differing between generalists and specialists. The 499 and 457 genes differing in the comparisons of dN/dS values between the generalists and temperate‐continental and tropical specialists respectively are listed in Table S9
Four gene ontology (GO) terms were enriched among the genes showing significant differences between the tropical and generalist species, and eight GO terms were enriched among the genes differing between the temperate‐continental and generalist species (Table S10; Figure S15A). About half the enriched terms in each comparison involved membrane and transmembrane transport activities, although only one term, “identical protein binding”, was enriched in both. Most of the enriched terms did not individually show significant directional trends in their respective comparisons but one in the temperate‐continental vs. generalist comparison did show a significant difference in the direction expected from the overall difference seen in all 499 genes involved in that comparison (Figure S15B). Twenty‐five of the 32 genes in this term, “inorganic molecular entity transmembrane transporter activity” showed higher dN/dS values in the temperate‐continental than generalist species (Figure S15B).
None of the genes in this particular GO term have previously been implicated in climate stress tolerance in Drosophila but five genes not assigned to this GO term which were previously implicated in such tolerance in D. melanogaster showed significant dN/dS differences in one or both of the comparisons. Four of these were significant in the temperate‐continental vs. generalist comparison and, consistent with the overall trend, all had higher dN/dS in the former than the latter. These were Gadd45, whose expression increases in the nervous system under exposure to hyperthermia (Moskalev et al., 2012; 0.043 vs. 0.012, q‐value = 0.026), Nan, which is involved in detecting reduced humidity in air (Liu et al., 2007; 0.021 vs. 0.006, q‐value = 0.002), thawb, knockdown of which causes insensitivity to thermal nociception (Honjo et al., 2016; 0.061 vs. 0.027, q‐value = 0.002) and CG13510, the expression of which increases under cold hardening (Qin et al., 2005; 0.073 vs. 0.044, q‐value = 0.03). Gadd45 again and Treh, which regulates trehalose metabolism and is essential for water homeostasis and desiccation resistance (Yoshida et al., 2016), also differed in dN/dS between the tropical specialists and generalists, but in both these cases the direction of the difference went against the overall trend in this comparison for values to be higher in the generalists than tropical specialists (0.026 vs. 0.049, q‐value = 0.045 and 0.065 vs. 0.108, q‐value = 0.049).
3.6. Population genomic differences between generalists and tropics‐restricted specialists
To investigate whether and how genome‐wide levels of polymorphism might differ between climate specialist vs. generalist species, we then resequenced multiple individuals from 13 species in Australia (Table S1). These species occupied a variety of climate niches as defined by Köppen climate classes, although all included the tropics in their distributions (Figure 1). However, their climate niches fell into two nonoverlapping groups on the basis of the outlying mean indices (OMIs) of their distributions, that is, the distance from the mean of the multivariate climate data for the distribution of each species to the mean for all species in the analysis (Table S11 and see also Materials and Methods). Eight species with OMIs ranging from 6.19 to 10.46 were essentially tropics‐restricted specialists living in monsoonal Australia and in some cases also in southeast Asia. The other five species, with OMIs ranging from 0.16 to 3.67, were much more widespread. They included the cosmopolitan generalists D. simulans and D. immigrans, D. kikkawai which is semi‐cosmopolitan across tropical and temperate Asia and Australia (Ranga et al., 2017), and D. setifemur and D. serrata, which occur in both tropical and temperate regions of eastern Australia. About 20 individuals for each species were sequenced (except for D. kikkawai where only six were available) on a HiSeq 4000 platform to a mean depth of ~20‐fold for each individual.
We found no obvious difference in the number of SNPs between the climatically widespread and tropics‐restricted specialist species (Table S12) but the average nucleotide diversity (Pi) was generally higher in the widespread than specialist species (Figure 5a). Moreover, their Pi values were significantly negatively correlated with their niche positions as defined by their OMIs, whether or not phylogenetic non‐independence was taken into account (using the pgls function in the R package caper) (Figure 5b). Two species with relatively extreme niches, D. rubida and D. pseudoananassae, strongly supported this trend but another species with a relatively extreme niche, D. bunnanda, was an outlier against the trend. The correlation remained significant if all three of these outlier species were omitted from the analysis (r = –.80, p = .004 for phylogenetic nonindependence). D. rubida and D. pseudoananassae were collected near the boundaries of their niches but so were D. jambulina, D. bipectinata and D. ironensis (Table S12). Our findings suggest that the species with tropics‐restricted climate niches have lower levels of nucleotide diversity than more widespread species.
FIGURE 5.
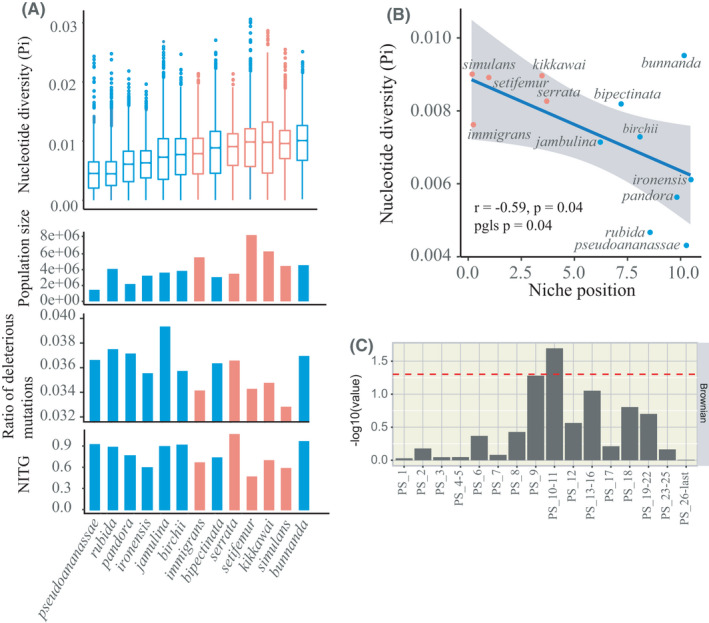
Population genomic statistics for tropics‐restricted specialists (blue) vs. widespread generalists (red). (a) Top two panels: The average overall nucleotide diversity (Pi) and effective population size (N e) were generally larger in the generalists than the tropics‐restricted specialists. Bottom two panels: The proportion of deleterious mutations and neutrality index NITG were generally smaller in the generalists than the tropics‐restricted specialists. (b) Pi was significantly negatively correlated with niche position. Confidence limits were calculated using the lm function in R. The generalists are shown in red and the specialists in blue in all panels in (a) and (b). (c) Significance of the correlation between effective population size and gene numbers across the 13 species for different phylostrata. Significance was determined using PGLS methods and assuming a Brownian motion model as detailed in the Materials and Methods (see also Figure S16 legend)
We calculated the effective population size (N e) of each species from the nucleotide diversity data (Figure 5a, Table S13), finding a range from a high N e of ~8.4 × 106 for the widespread D. setifemur down to ~1.4 × 106 for the tropics‐restricted D. pseudoananassae. We have not found any previous reports of N e estimates for these 13 species, but they are broadly comparable with the estimates of 1.8 × 106 and 1.4 × 106 previously obtained for D. suzukii and D. melanogaster (noting that the latter was based on just two isofemale lines; Adrion et al., 2014; Keightley et al., 2014). In our analysis we found the values for four of the five widespread species were amongst the five highest, possibly reflecting less exposure of widespread species to stochastic fluctuations. The exception was D. serrata, which occupies both tropical and temperate niches in eastern Australia and has an N e of just 3.5 × 106. The result for this species is considered further below.
We also examined the correlation of N e with the number of extant genes originating in each phylostratum. No correlation was found with the numbers of genes arising in the most recent phylostrata. However population size was found to be positively correlated (nominal pgls p < .05) with the number of genes originating in PS 10–11, that is, during the appearance of panarthropods (Figure 5c, Figure S16). Notably this relationship was very largely due to the widespread species (Figure S16). Thus, the differences in the numbers of genes retained in those species which originated in that period are associated with their population sizes, suggesting the genes retained from that period influence the niches which the species can now occupy.
Four of the five widespread species also had the lowest proportions of deleterious mutations as judged from the Grantham scores for the physicochemical effects of the amino acid differences encoded by nonsynonymous SNPs in their coding regions (Grantham, 1974), with D. serrata again the one exception (Figure 5a, Table S13). Intriguingly, a similar pattern was found in the values for the neutrality index NITG (Figure 5a, Table S14), even though it is an independent test based on population genetic rather than physicochemical criteria (see Materials and Methods). Thus NITG was on average lower across the five widespread species (0.71 ± 0.10) than among the eight tropics‐restricted ones (0.85 ± 0.04) (p < .05 on the Wilcoxon rank sum test (the “Wilcox test” in R); Figure 5a, Table S14), and once again D. serrata went against the trend, in this case with the highest NITG (1.08). The difference between the widespread and restricted species could indicate more effective selection against deleterious variants within the former, which would be consistent with the Grantham scores and some theory (Whitlock & Bürger, 2004), although other explanations involving stronger divergent selection between those species and their outgroup relatives are also possible.
The low Ne and high Grantham and NITG scores for D. serrata may in part reflect the high numbers of chromosome inversion polymorphisms in this species (Stocker et al., 2004), which would decrease effective population size and protect large blocks of physically linked deleterious recessive variants via recombination suppression and heterozygote advantage (Albornoz & Dominguez, 1994; Schwander et al., 2014). Also possibly relevant here is that this species has only recently expanded southwards into more temperate climates and its range is still limited by its response to cold periods (Jenkins & Hoffmann, 1999).
Overall, our resequencing data suggest a combination of smaller population sizes, less nucleotide diversity but more deleterious mutations may characterise the tropics‐restricted species.
4. DISCUSSION
4.1. A robust Drosophila phylogeny despite widespread ILS and introgression
The phylogenies we constructed using both the whole genome and orthologue‐specific data sets with the concatenation and coalescent methods showed a high level of concordance and disentangled several previously ambiguous relationships in the Drosophila genus. They confirmed mounting evidence for the paraphyly of the genus and found additional paraphylies in at least two major lineages within it. These results suggested that the subgenus Drosophila should also include subgenus Dorsilopha, and at least part of the genus Zaprionus. Within the subgenus Sophophora, they placed the setifemur group as sister to the ananassae subgroup, and the montium subgroup as sister to both setifemur and ananassae. Our phylogenies also resolved previously ambiguous relationships in the melanogaster group of subgenus Sophophora, revealing sister relationships between the takahashii and suzukii subgroups and between the eugracilis and melanogaster subgroups, and also between those two pairs. We therefore support the proposals (van der Linde & Houle, 2008; van der Linde et al., 2010; O'Grady & DeSalle, 2018; O'Grady & Markow, 2009) to overhaul the taxonomy of some areas of both major subgenera.
The one relationship which remained problematic in our phylogenies concerns the repleta group and mercatorum, hydei and mulleri subgroups within subgenus Drosophila. On balance our trees provide strong support for a closer relationship between hydei and mulleri, with mercatorum as the outgroup, but there is also significant support for hydei as the outgroup in our various analyses and data sets. Our discovista and quibl analyses suggest the ambiguity is due in good part to significant levels of both ILS and introgression, especially introgression between mercatorum and mulleri. We conclude that the divergences among the three lineages, regardless of the order of events, occurred within a short evolutionary time period and involved considerable ILS and introgression. This is intriguing given the evidence for high levels of gene duplication and positive selection associated with the uptake of cactus host use in these lineages and subsequent specialisation on cactus hosts in the mulleri subgroup (Rane et al., 2019).
In fact, we found high levels of ILS and/or introgression in several lineages of our phylogeny. In subgenus Sophophora, ILS was particularly common in the nominal melanogaster group, where two clusters of three adjacent nodes with high ILS probably explain why previous phylogenies, generally based on limited numbers of genes, have generally failed to concur on the relationships among the ficusphila, takahashii, suzukii, eugracilis and melanogaster subgroups in one case (Finet et al., 2021; Kopp, 2006; Kopp & True, 2002; van der Linde & Houle, 2008; van der Linde et al., 2010; Schawaroch, 2002; Seetharam & Stuart, 2013; Wong et al., 2007; Yang et al., 2004, 2012), and D. kikkawai, D. jambulina and the serrata clade in the montium subgroup in the other case (Da Lage et al., 2007; Finet et al., 2021; van der Linde & Houle, 2008).
Some authors have previously considered the possibility that ILS and/or introgression might have contributed to phylogenetic incongruences in the intensively studied melanogaster subgroup. ILS was suggested by the observation that more accurately inferred trees were obtained within the D. simulans clade when sequences were used from low‐recombination regions, where levels of ILS are expected to be low (Pease & Hahn, 2013). On the other hand, some hybridisation was proposed between D. simulans and D. mauritiana on the basis of mtDNA sequence differences (Ballard, 2000; Nunes et al., 2010), and between the cosmopolitan D. simulans and the island endemics D. mauritiana and D. sechellia on the basis of whole genome alignments (Garrigan et al., 2012). However, ILS was not considered in either of those studies and neither used the topology‐based discovista and quibl methods used here. Our results for other branches in the subgroup concur with previous work, in that we found <10% of ILS and very little hybridisation in the D. melanogaster‐D. simulans‐D. sechellia (Rosenfeld et al., 2012) and D. melanogaster‐D. yakuba‐D. erecta (Turissini & Matute, 2017; Wong et al., 2007) clades. Interestingly, some work on these clades suggests higher levels of introgression, some of it apparently adaptive, in mitochondrial than nuclear genomes (Bachtrog et al., 2006; Ballard, 2000; Llopart et al., 2005, 2014).
While ILS was generally more common than introgression in the subgenus Sophophora, we found introgression was relatively more frequent in subgenus Drosophila, where it contributed strongly to phylogenetic incongruences involving the base of the subgenus, the Zaprionus split and the immigrans–tripunctata radiation, in addition to the repleta‐hydei‐mulleri ambiguity noted above. We do not know the reason for this difference between the subgenera, but we note that the one case where introgression made a similarly large contribution to discordance (>20% of all gene trees) in the Sophophora was a relatively deep node, where the Oriental lineage and montium subgroup bifurcated.
The prevalence of ILS and introgression across the phylogeny has significant implications for studies of Drosophila trait and genome evolution. Many studies in the area to date have ignored the heightened risk of hemiplasy (a false pattern of convergence) (Guerrero & Hahn, 2018) due to high ILS and introgression. This happens because both processes increase the probability that the evolutionary history of a trait or gene of interest differs from the species tree. This difference is often interpreted in terms of multiple origins of the trait or gene, whereas ILS or introgression could in fact explain it with a single origin. The original Drosophila 12‐species genome study (Clark et al., 2007) found a substantial portion of Drosophila‐specific homologous genes required more than one gain or loss event to explain their evolution on a fixed species tree. Instead, our results suggest this pattern might at least in part represent hemiplasy caused by ILS. Future studies of the evolution of specific genes or traits, in this genus at least, should use hemiplasy‐aware methods (Hibbins et al., 2020; Wu et al., 2018) for reconstructing the evolutionary process.
4.2. Evolutionary and ecological genetic differences between specialists and generalists
While the ILS and hybridisation events may have impacted our phylostratigraphical analysis, it would have actually worked against our finding of higher novel gene numbers per unit time originating in the most recent era/phylostrata, because its tendency to disperse genes across more taxa compared to simple vertical transmission would make genes appear older than they actually are. In any event the effects of the ILS and hybridisation events would generally be relatively small, given that most ILS/introgression occurs in relatively closely related lineages compared to the timeframes considered in most of the phylostratigraphy. Even the pattern of species differences we found in comparisons which were within the shorter timeframes of the most recent phylostrata were not explicable in terms of ILS or introgression. For example, lineages such as the Oriental clade and the branches leading to the cactophilic mulleri subgroup species had some of the highest levels of discordance but did not have the relatively low rates of recent novel gene gains expected under ILS scenarios (Figures 1 and 2, Figure S8).
The analysis of genes’ evolutionary ages identified two clades with particularly high rates of novel gene production in the recent era, namely three sister species in the pseudoobscura subgroup (D. pseudoobscura, D. persimilis and D. miranda) and the five cactophilic species in the mulleri subgroup (D. aldrichi, D. arizonae, D. buzzatii, D. mojavensis and D. navojoa). The three pseudoobscura species are unique among the 47 analysed in occupying the temperate, dry and continental but not tropical Köppen classes. Whether their Köppen profile is directly relevant is unclear; other species like D. immigrans have broader climate niches but much lower recent rates of novel gene acquisition (Figure 1, Figures S6–S8). Instead, the relatively high rates of recent novel gene gain in the pseudoobscura subgroup may relate to their unusually broad host range, which includes rotting fruits, decaying vegetation, slime fluxes and fungi (Powell, 1997). The high recent rate of novel gene gain in the cactophilic repleta species may relate in part to their climate profiles, which show some variation between the species, but all include hot dry environments (Figure 1), although the demands of their relatively toxic cactus hosts may also have required significant numbers of new genes and functions (Matzkin, 2014; Oliveira et al., 2012). The high rate of novel gene gains in this lineage concurs with evidence for high rates of gene duplication and positive selection early in their move to desert environments and cactus hosts, although those rates subsequently declined to low levels as the species became more specialised to their climate and host niches (Rane et al., 2019).
Five of the six species with the lowest rates of recent novel gene gain (D. ironensis, D. willistoni, D. grimshawi, D. albomicans and D. busckii) were phylogenetically widely distributed and each was only found in one or two Köppen classes (Figure 1). However, the sixth (D. immigrans) was a cosmopolitan generalist. Overall, the five cosmopolitan climate generalists were relatively variable in their recent rates of gene gain, with D. melanogaster and D. simulans having relatively high rates and D. hydei and D. mercatorum intermediate rates (Figure 1, Figures S6–S8). Noting that D. melanogaster and D. simulans lie in the highly speciose melanogaster group and Oriental clade, and that the newest era (over which recent gene gains were calculated) encompasses the whole genus, relatively high rates of recent gene gains may be characteristic of lineages that have undergone more recent speciation, while lower rates may be found in species that have occupied broad niches for longer timeframes.
Interestingly, the pattern of species differences in recent gene gains was a close mirror image of the pattern of species differences in genes retained from the ancient era, possibly suggesting some form of trade‐off between the two sorts of genes. Given the size differences between the peptides/proteins encoded by the two classes of genes, this is unlikely to be a direct like‐for‐like effect. Unfortunately, little is yet known about the specific functions of recently acquired genes in any organism, although evidence for Drosophila indicates a significant number rapidly become important for fitness (Chen et al., 2010; Xia et al., 2020) and some have been implicated in male reproductive function (Begun et al., 2007).
Our dN/dS calculations identified several hundred orthologous genes which were under different levels of selective constraint in comparisons of climate generalists with either the tropical or temperate‐continental specialist species. Five of the genes distinguishing one or other (and in one case both) types of specialists from the generalists have functionally validated roles in climate stress responses in D. melanogaster. There was significant overlap between the genes distinguishing the two types of specialists from the generalists, suggesting many of the same genes are involved in adaptation to the different niches involved in the comparisons. Consistent with this, GO term enrichment analysis showed membrane and transmembrane transport processes were implicated in both sets of differences Importantly, however, the genes responded in different ways to the different environments; whereas there was a significant trend for the tropical specialists to have lower dN/dS values than the generalists, the reverse was true for the temperate‐continental specialists. The dN/dS data thus suggest that adaptation to different climate niches involves differing responses in quite large numbers of genes.
Additionally, our population resequencing analyses on a subset of the species revealed higher levels of nucleotide variation but more effective selection against deleterious mutations in the widespread species. We did not have resequencing data for the temperate‐continental specialists but some of the differences between the tropics‐restricted and widespread species at least are consistent with well‐established population and evolutionary genetic processes. In line with our observations, widespread species would be expected to maintain high levels of nucleotide variation due to ongoing gene flow, high effective population sizes and ongoing heterogeneous selection pressures (Star & Spencer, 2013; Willi et al., 2006). On the other hand, species that have become restricted to a relatively narrow niche may experience DNA decay and loss of function of some previously important genes, further limiting their capacity to expand (Dworkin & Jones, 2009; Hoffmann & Willi, 2008; Ostrowski et al., 2007). Intriguingly, we found that the population size differences between species were themselves associated with differences in the numbers of genes which they retained that originated in the phylostratum overlapping the origin of panarthropods. Overall, our population resequencing results suggest links between several aspects of genomic variation and climate specialism vs. generalism.
AUTHOR CONTRIBUTIONS
Guojie Zhang, Carla M. Sgrò, John G. Oakeshott and Ary A. Hoffmann designed the research. Zijun Xiong, Fang Li, Zimai Li, Stephen Pearce, Kerensa McElroy and Rahul V. Rane assembled and annotated the genomes. Fang Li and Jiawei Chen performed the phylogenomics, ILS and introgression and population genetics analysis. Renee A. Catullo provided the ecological data. Philippa C. Griffin, Michele Schiffer, Stephen Pearce, Kerensa McElroy, Ann Stocker, Jennifer Shirriffs, Fiona Cockerell, Chris Coppin developed insect and sequencing resources. Victor Luria, Amir Karger, John W. Cain, Jessica A. Weber, Gabriel Santpere and Marc W. Kirschner performed the phylostratigraphy analysis and modelling of gene flux. Fang Li wrote the first draft of the manuscript and Victor Luria, Rahul V. Rane, Renee A. Catullo, Siu Fai Lee, Ary A. Hoffmann, John G. Oakeshott and Guojie Zhang in particular contributed to revising the manuscript.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
We thank Madeleine Gane and Mark Schutze for assistance in species collection and inbreeding programs, Lea Rako and Kelly Richardson for assistance with stock maintenance and inbreeding, Lars Jermiin for advice on the phylogenomics and Shane McEvey for help in accessing collection records for the niche calculations. We are also grateful for the insights of three anonymous reviewers of an earlier version of this manuscript. The work was supported by Australia’s Science and Industry Endowment Fund and the Australian Research Council and their Laureate Fellowship Scheme. This work was also supported by Strategic Priority Research Program of the Chinese Academy of Sciences (XDB31020000), International Partnership Program of Chinese Academy of Sciences (No. 152453KYSB20170002), Carlsberg foundation (CF16‐0663), Villum Foundation (No. 25900) to G.Z. The project that gave rise to some of the results received the support of a fellowship from “la Caixa” Foundation (ID 100010434, to G.S.). The fellowship code is LCF/BQ/PI19/11690010. G.S. is also supported by Ministerio de Ciencia e Innovación, Spain (PID2019‐104700GA‐I00). V.L. and M.W.K. acknowledge funding support from the National Institutes of Health (NIH grants R01 HD073104 and R01 HD091846, to M.W.K.). V.L. thanks Anne O’Donnell‐Luria for discussions and also for support from the William Randolph Hearst Fund Award and a Boston Children’s Hospital Career Development Fellowship (to A.O.L.).
APPENDIX 1. GENOME ASSEMBLIES
Were whole genome shotgun libraries sequenced at high coverage for the target species (provide in terms of "X coverage ")?
Yes, an average of 150X for each de novo sequenced species.
Did the study generate a pan‐genome assembly from several individual samples?
No.
Were ‘long‐read’ libraries sequenced and included in the genome assembly (specify type and coverage)?
Yes, we sequenced libraries with insert size of 2000, 5000 and 10,000 bp for each do novo sequenced species with total coverage about 80X.
What are the basic assembly statistics: genome size, percent assembled, # contigs, contig N50, # scaffolds, scaffold N50.
These statistics were provided in Table S3.
Was mapping of some form (genetic, physical, optical) incorporated to order scaffolds?
No.
Were scaffolds anchored to chromosome positions, and if so, what proportion of the genome is anchored to chromosomes?
No.
Were analyses included that assess the quality of the genome assembly (GenomeScope, or busco)?
Yes, busco was used to assess genome assembly quality.
Were RNAseq libraries sequenced to assemble transcriptomes and annotate genes?
Yes, RNAseq libraries were sequenced, and transcriptome data were used to improve genes annotation.
Is the genome assembly publicly available through a web‐based genome browser?
No.
How is the genome assembly in this manuscript useful for broader research in the field of molecular ecology?
The 15 de novo assemblies and 20 improved genomes of Drosophila species in this manuscript provide a large volume of genomic resources for researchers working on a variety of species in the genus Drosophila.
Li, F. , Rane, R. V. , Luria, V. , Xiong, Z. , Chen, J. , Li, Z. , Catullo, R. A. , Griffin, P. C. , Schiffer, M. , Pearce, S. , Lee, S. F. , McElroy, K. , Stocker, A. , Shirriffs, J. , Cockerell, F. , Coppin, C. , Sgrò, C. M. , Karger, A. , Cain, J. W. , … Zhang, G. (2022). Phylogenomic analyses of the genus Drosophila reveals genomic signals of climate adaptation. Molecular Ecology Resources, 22, 1559–1581. 10.1111/1755-0998.13561
Contributor Information
Ary A. Hoffmann, Email: ary@unimelb.edu.au.
John G. Oakeshott, Email: john.oakeshott@csiro.au.
Guojie Zhang, Email: guojie.zhang@bio.ku.dk.
DATA AVAILABILITY STATEMENT
The raw and processed sequence data underlying this article have been made available in NCBI under the project accession number PRJNA736147. Files for the phylogeny code, coding and peptide sequences used for phylogenetic analyses, and the orthologue table for dN/dS calculations are available in figshare https://doi.org/10.6084/m9.figshare.14747817 (Li et al., 2021)
REFERENCES13561
- Adrion, J. R. , Kousathanas, A. , Pascual, M. , Burrack, H. J. , Haddad, N. M. , Bergland, A. O. , Machado, H. , Sackton, T. B. , Schlenke, T. A. , Watada, M. , Wegmann, D. , & Singh, N. D. (2014). Drosophila suzukii: The genetic footprint of a recent, worldwide invasion. Molecular Biology and Evolution, 31(12), 3148–3163. 10.1093/molbev/msu246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albornoz, J. , & Dominguez, A. (1994). Inversion polymorphism and accumulation of lethals in selected lines of Drosophila melanogaster . Heredity, 73(1), 92–97. 10.1038/hdy.1994.103 [DOI] [PubMed] [Google Scholar]
- Arendsee, Z. , Li, J. , Singh, U. , Seetharam, A. , Dorman, K. , & Wurtele, E. S. (2019). phylostratr: A framework for phylostratigraphy. Bioinformatics, 35(19), 3617–3627. 10.1093/bioinformatics/btz171 [DOI] [PubMed] [Google Scholar]
- Bachtrog, D. , Thornton, K. , Clark, A. , & Andolfatto, P. (2006). Extensive introgression of mitochondrial DNA relative to nuclear genes in the Drosophila yakuba species group. Evolution, 60(2), 292–302. 10.1111/j.0014-3820.2006.tb01107.x [DOI] [PubMed] [Google Scholar]
- Ballard, J. W. O. (2000). When one is not enough: Introgression of mitochondrial DNA in Drosophila . Molecular Biology and Evolution, 17(7), 1126–1130. 10.1093/oxfordjournals.molbev.a026394 [DOI] [PubMed] [Google Scholar]
- Barker, J. S. F. , Krebs, R. A. , & Davies, H. I. (2005). Geographical distributions, relative abundance and coexistence of Drosophila aldrichi and Drosophila buzzatii in Australia. Austral Ecology, 30(5), 546–557. 10.1111/j.1442-9993.2005.01470.x [DOI] [Google Scholar]
- Barker, J. S. F. , & Starmer, W. T. (1982). Ecological genetics and evolution: The cactus‐yeast‐Drosophila model system. Academic Press. [Google Scholar]
- Barrio, E. , & Ayala, F. J. (1997). Evolution of the Drosophila obscura species group inferred from the Gpdh and Sod genes. Molecular Phylogenetics and Evolution, 7(1), 79–93. 10.1006/mpev.1996.0375 [DOI] [PubMed] [Google Scholar]
- Begun, D. J. , Lindfors, H. A. , Kern, A. D. , & Jones, C. D. (2007). Evidence for de novo evolution of testis‐expressed genes in the Drosophila yakuba/Drosophila erecta clade. Genetics, 176(2), 1131–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, G. (1999). Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Research, 27(2), 573–580. 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney, E. , Clamp, M. , & Durbin, R. (2004). GeneWise and genomewise. Genome Research, 14(5), 988–995. 10.1101/gr.1865504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette, M. , Kent, W. J. , Riemer, C. , Elnitski, L. , Smit, A. F. A. , Roskin, K. M. , Baertsch, R. , Rosenbloom, K. , Clawson, H. , Green, E. D. , Haussler, D. , & Miller, W. (2004). Aligning multiple genomic sequences with the threaded blockset aligner. Genome Research, 14(4), 708–715. 10.1101/gr.1933104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvunis, A. R. , Rolland, T. , Wapinski, I. , Calderwood, M. A. , Yildirim, M. A. , Simonis, N. , Charloteaux, B. , Hidalgo, C. A. , Barbette, J. , Santhanam, B. , Brar, G. A. , Weissman, J. S. , Regev, A. , Thierry‐Mieg, N. , Cusick, M. E. , & Vidal, M. (2012). Proto‐genes and de novo gene birth. Nature, 487(7407), 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution, 17(4), 540–552. 10.1093/oxfordjournals.molbev.a026334 [DOI] [PubMed] [Google Scholar]
- Chen, S. , Zhang, Y. E. , & Long, M. (2010). New genes in Drosophila quickly become essential. Science, 330(6011), 1682–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Chen, Y. , Shi, C. , Huang, Z. , Zhang, Y. , Li, S. , Li, Y. , Ye, J. , Yu, C. , Li, Z. , Zhang, X. , Wang, J. , Yang, H. , Fang, L. , & Chen, Q. (2018). SOAPnuke: A MapReduce acceleration‐supported software for integrated quality control and preprocessing of high‐throughput sequencing data. Gigascience, 7(1), 1–6. 10.1093/gigascience/gix120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, A. G. , Eisen, M. B. , Smith, D. R. , Bergman, C. M. , Oliver, B. , Markow, T. A. , Kaufman, T. C. , Kellis, M. , Gelbart, W. , Iyer, V. N. et al (2007). Evolution of genes and genomes on the Drosophila phylogeny. Nature, 450(7167), 203–218. [DOI] [PubMed] [Google Scholar]
- Conner, W. R. , Delaney, E. K. , Bronski, M. J. , Ginsberg, P. S. , Wheeler, T. B. , Richardson, K. M. , Peckenpaugh, B. , Kim, K. J. , Watada, M. , Hoffmann, A. A. , Eisen, M. B. , Kopp, A. , Cooper, B. S. , & Turelli, M. (2021). A phylogeny for the Drosophila montium species group: A model clade for comparative analyses. Molecular Phylogenetics and Evolution, 158, 107061. 10.1016/j.ympev.2020.107061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Lage, J. L. , Kergoat, G. J. , Maczkowiak, F. , Silvain, J. F. , Cariou, M. L. , & Lachaise, D. (2007). A phylogeny of Drosophilidae using the Amyrel gene: Questioning the Drosophila melanogaster species group boundaries. Journal of Zoological Systematics and Evolutionary Research, 45(1), 47–63. 10.1111/j.1439-0469.2006.00389.x [DOI] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , Handsaker, R. E. , Lunter, G. , Marth, G. T. , Sherry, S. T. , McVean, G. , & Durbin, R. (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolédec, S. , Chessel, D. , & Gimaret‐Carpentier, C. (2000). Niche separation in community analysis: A new method. Ecology, 81(10), 2914–2927. 10.1890/0012-9658(2000)081.2914:NSICAA.2.0.CO;2 [DOI] [Google Scholar]
- Domazet‐Loso, T. , Brajkovic, J. , & Tautz, D. (2007). A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends in Genetics, 23(11), 533–539. 10.1016/j.tig.2007.08.014 [DOI] [PubMed] [Google Scholar]
- Domazet‐Loso, T. , Carvunis, A. R. , Alba, M. M. , Sestak, M. S. , Bakaric, R. , Neme, R. , & Tautz, D. (2017). No evidence for phylostratigraphic bias impacting inferences on patterns of gene emergence and evolution. Molecular Biology and Evolution, 34(4), 843–856. 10.1093/molbev/msw284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray, S. , & Dufour, A.‐B. (2007). The ade4 package: Implementing the duality diagram for ecologists. Journal of Statistical Software, 22(4), 1–20. [Google Scholar]
- Durando, C. M. , Baker, R. H. , Etges, W. J. , Heed, W. B. , Wasserman, M. , & DeSalle, R. (2000). Phylogenetic analysis of the repleta species group of the genus Drosophila using multiple sources of characters. Molecular Phylogenetics and Evolution, 16(2), 296–307. 10.1006/mpev.2000.0824 [DOI] [PubMed] [Google Scholar]
- Dworkin, I. , & Jones, C. D. (2009). Genetic changes accompanying the evolution of host specialization in Drosophila sechellia . Genetics, 181(2), 721–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman, N. B. , Frandsen, P. B. , Miyagi, M. , Clavijo, B. , Davey, J. , Dikow, R. B. , García‐Accinelli, G. , Van Belleghem, S. M. , Patterson, N. , & Neafsey, D. E. J. S. (2019). Genomic architecture and introgression shape a butterfly radiation. Science, 366(6465), 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. , & Myers, E. W. (2005). PILER: Identification and classification of genomic repeats. Bioinformatics, 21, I152–I158. 10.1093/bioinformatics/bti1003 [DOI] [PubMed] [Google Scholar]
- Eo, S. H. , & DeWoody, J. A. (2010). Evolutionary rates of mitochondrial genomes correspond to diversification rates and to contemporary species richness in birds and reptiles. Proceedings of the Royal Society B, 277(1700), 3587–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , Rane, R. V. , Luria, V. , Xiong, Z. , Chen, J. , Li, Z. , Catullo, R. A. , Griffin, P. C. , Schiffer, M. , Pearce, S. , Lee, S. F. , McElroy, K. , Stocker, A. , Shirriffs, J. , Cockerell, F. , Coppin, C. , Sgrò, C. M. , Karger, A. , Cain, J. W. , … Zhang, G. (2021). Phylogenomic analyses of the genus Drosophila reveals genomic signals of climate adaptation. NCBI, PRJNA736147. Figshare. 10.6084/m9.figshare.14747817 [DOI] [PMC free article] [PubMed]
- Finet, C. , Kassner, V. A. , Carvalho, A. B. , Chung, H. , Day, J. P. , Day, S. , Delaney, E. K. , De Ré, F. C. , Dufour, H. D. , Dupim, E. , Izumitani, H. F. , Gautério, T. B. , Justen, J. , Katoh, T. , Kopp, A. , Koshikawa, S. , Longdon, B. , Loreto, E. L. , Nunes, M. D. S. , … Marlétaz, F. (2021). DrosoPhyla: Resources for drosophilid phylogeny and systematics. Genome Biology and Evolution, 13(8), evab179. 10.1093/gbe/evab179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca, N. A. , Morales‐Hojas, R. , Reis, M. , Rocha, H. , Vieira, C. P. , Nolte, V. , Schlötterer, C. , & Vieira, J. (2013). Drosophila americana as a model species for comparative studies on the molecular basis of phenotypic variation. Genome Biology and Evolution, 5(4), 661–679. 10.1093/gbe/evt037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigan, D. , Kingan, S. B. , Geneva, A. J. , Andolfatto, P. , Clark, A. G. , Thornton, K. R. , & Presgraves, D. C. (2012). Genome sequencing reveals complex speciation in the Drosophila simulans clade. Genome Research, 22(8), 1499–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham, R. (1974). Amino acid difference formula to help explain protein evolution. Science, 185(4154), 862–864. [DOI] [PubMed] [Google Scholar]
- Grimaldi, D. A. (1990). A phylogenetic, revised classification of genera in the Drosophilidae (Diptera). Bulletin of the American Museum of Natural History, 197, 1–139. [Google Scholar]
- Guerrero, R. F. , & Hahn, M. W. (2018). Quantifying the risk of hemiplasy in phylogenetic inference. Proceedings of the National Academy of Sciences of the United States of America, 115(50), 12787–12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillén, Y. , Rius, N. , Delprat, A. , Williford, A. , Muyas, F. , Puig, M. , Casillas, S. , Ràmia, M. , Egea, R. , Negre, B. , Mir, G. , Camps, J. , Moncunill, V. , Ruiz‐Ruano, F. J. , Cabrero, J. , de Lima, L. G. , Dias, G. B. , Ruiz, J. C. , Kapusta, A. , … Ruiz, A. (2014). Genomics of ecological adaptation in cactophilic Drosophila . Genome Biology and Evolution, 7(1), 349–366. 10.1093/gbe/evu291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanschen, E. R. , Marriage, T. N. , Ferris, P. J. , Hamaji, T. , Toyoda, A. , Fujiyama, A. , Neme, R. , Noguchi, H. , Minakuchi, Y. , Suzuki, M. , Kawai‐Toyooka, H. , Smith, D. R. , Sparks, H. , Anderson, J. , Bakarić, R. , Luria, V. , Karger, A. , Kirschner, M. W. , Durand, P. M. , … Olson, B. J. S. C. (2016). The Gonium pectorale genome demonstrates co‐option of cell cycle regulation during the evolution of multicellularity. Nature Communications, 7(1), 1–10. 10.1038/ncomms11370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, R. S. (2007). Improved pairwise alignmnet of genomic DNA. Pennsylvania State University. [Google Scholar]
- Hibbins, M. S. , Gibson, M. J. , & Hahn, M. W. (2020). Determining the probability of hemiplasy in the presence of incomplete lineage sorting and introgression. eLife, 9, e63753. 10.7554/eLife.63753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijmans, R. J. , Cameron, S. E. , Parra, J. L. , Jones, P. G. , & Jarvis, A. (2005). Very high resolution interpolated climate surfaces for global land areas. International Journal of Climatology, 25(15), 1965–1978. 10.1002/joc.1276 [DOI] [Google Scholar]
- Hoffmann, A. A. , Sørensen, J. G. , & Loeschcke, V. (2003). Adaptation of Drosophila to temperature extremes: Bringing together quantitative and molecular approaches. Journal of Thermal Biology, 28(3), 175–216. 10.1016/S0306-4565(02)00057-8 [DOI] [Google Scholar]
- Hoffmann, A. A. , & Willi, Y. (2008). Detecting genetic responses to environmental change. Nature Reviews Genetics, 9(6), 421–432. 10.1038/nrg2339 [DOI] [PubMed] [Google Scholar]
- Honjo, K. , Mauthner, S. E. , Wang, Y. , Skene, J. P. , & Tracey, W. D. Jr (2016). Nociceptor‐enriched genes required for normal thermal nociception. Cell Reports, 16(2), 295–303. 10.1016/j.celrep.2016.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, T. T. , Eisen, M. B. , Thornton, K. R. , & Andolfatto, P. (2013). A second‐generation assembly of the Drosophila simulans genome provides new insights into patterns of lineage‐specific divergence. Genome Research, 23(1), 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey, C. M. (1974). Genetic isolation among six strains of Drosophila repleta from the eastern United States, Central America, Hawaii, and Australia. Georgia Institute of Technology. [Google Scholar]
- Jenkins, N. L. , & Hoffmann, A. A. (1999). Limits to the southern border of Drosophila serrata: Cold resistance, heritable variation, and trade‐offs. Evolution, 53(6), 1823–1834. [DOI] [PubMed] [Google Scholar]
- Kajitani, R. , Toshimoto, K. , Noguchi, H. , Toyoda, A. , Ogura, Y. , Okuno, M. , Yabana, M. , Harada, M. , Nagayasu, E. , Maruyama, H. , Kohara, Y. , Fujiyama, A. , Hayashi, T. , & Itoh, T. (2014). Efficient de novo assembly of highly heterozygous genomes from whole‐genome shotgun short reads. Genome Research, 24(8), 1384–1395. 10.1101/gr.170720.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D. , Ness, R. W. , Halligan, D. L. , & Haddrill, P. R. (2014). Estimation of the spontaneous mutation rate per nucleotide site in a Drosophila melanogaster full‐sib family. Genetics, 196(1), 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellermann, V. , Loeschcke, V. , Hoffmann, A. A. , Kristensen, T. N. , Flojgaard, C. , David, J. R. , Svenning, J. C. , & Overgaard, J. (2012). Phylogenetic constraints in key functional traits behind species’ climate niches: Patterns of desiccation and cold resistance across 95 Drosophila species. Evolution, 66(11), 3377–3389. [DOI] [PubMed] [Google Scholar]
- Kellermann, V. , Overgaard, J. , Hoffmann, A. A. , Flojgaard, C. , Svenning, J. C. , & Loeschcke, V. (2012). Upper thermal limits of Drosophila are linked to species distributions and strongly constrained phylogenetically. Proceedings of the National Academy of Sciences of the United States of America, 109(40), 16228–16233. 10.1073/pnas.1207553109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellermann, V. , van Heerwaarden, B. , Sgrò, C. M. , & Hoffmann, A. A. (2009). Fundamental evolutionary limits in ecological traits drive Drosophila species distributions. Science, 325(5945), 1244–1246. [DOI] [PubMed] [Google Scholar]
- Kim, H.‐M. , Weber, J. A. , Lee, N. , Park, S. G. , Cho, Y. S. , Bhak, Y. , Lee, N. , Jeon, Y. , Jeon, S. , & Luria, V. (2019). The genome of the giant Nomura’s jellyfish sheds light on the early evolution of active predation. BMC Biology, 17(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koniger, A. , & Grath, S. (2018). Transcriptome analysis reveals candidate genes for cold tolerance in Drosophila ananassae . Genes (Basel), 9(12), 624. 10.3390/genes9120624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp, A. (2006). Basal relationships in the Drosophila melanogaster species group. Molecular Phylogenetics and Evolution, 39(3), 787–798. 10.1016/j.ympev.2006.01.029 [DOI] [PubMed] [Google Scholar]
- Kopp, A. , Barmina, O. , & Prigent, S. R. (2019). Phylogenetic position of the Drosophila fima and dentissima lineages, and the status of the D. melanogaster species group. Molecular Phylogenetics and Evolution, 139, 106543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp, A. , & True, J. R. (2002). Phylogeny of the oriental Drosophila melanogaster species group: A multilocus reconstruction. Systematic Biology, 51(5), 786–805. 10.1080/10635150290102410 [DOI] [PubMed] [Google Scholar]
- Korneliussen, T. S. , Albrechtsen, A. , & Nielsen, R. (2014). ANGSD: Analysis of next generation sequencing data. BMC Bioinformatics, 15(1), 356. 10.1186/s12859-014-0356-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottek, M. , Grieser, J. , Beck, C. , Rudolf, B. , & Rubel, F. (2006). World map of the Köppen‐Geiger climate classification updated. Meteorologische Zeitschrift, 15(3), 259–263. 10.1127/0941-2948/2006/0130 [DOI] [Google Scholar]
- Kozlov, A. M. , Aberer, A. J. , & Stamatakis, A. (2015). ExaML version 3: A tool for phylogenomic analyses on supercomputers. Bioinformatics, 31(15), 2577–2579. 10.1093/bioinformatics/btv184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Suleski, M. , & Hedges, S. B. (2017). TimeTree: A resource for timelines, timetrees, and divergence times. Molecular Biology and Evolution, 34(7), 1812–1819. 10.1093/molbev/msx116 [DOI] [PubMed] [Google Scholar]
- Lanfear, R. , Ho, S. Y. , Love, D. , & Bromham, L. (2010). Mutation rate is linked to diversification in birds. Proceedings of the National Academy of Sciences of the United States of America, 107(47), 20423–20428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv preprint arXiv:13033997.
- Liu, L. , Li, Y. , Wang, R. , Yin, C. , Dong, Q. , Hing, H. , Kim, C. , & Welsh, M. J. (2007). Drosophila hygrosensation requires the TRP channels water witch and nanchung. Nature, 450(7167), 294. [DOI] [PubMed] [Google Scholar]
- Llopart, A. , Herrig, D. , Brud, E. , & Stecklein, Z. (2014). Sequential adaptive introgression of the mitochondrial genome in Drosophila yakuba and Drosophila santomea . Molecular Ecology, 23(5), 1124–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llopart, A. , Lachaise, D. , & Coyne, J. A. (2005). Multilocus analysis of introgression between two sympatric sister species of Drosophila: Drosophila yakuba and D. santomea . Genetics, 171(1), 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loytynoja, A. (2014). Phylogeny‐aware alignment with PRANK. Methods in Molecular Biology, 1079, 155–170. [DOI] [PubMed] [Google Scholar]
- Luo, R. , Liu, B. , Xie, Y. , Li, Z. , Huang, W. , Yuan, J. , He, G. , Chen, Y. , Pan, Q. I. , Liu, Y. , Tang, J. , Wu, G. , Zhang, H. , Shi, Y. , Liu, Y. , Yu, C. , Wang, B. O. , Lu, Y. , Han, C. , … Wang, J. (2012). SOAPdenovo2: An empirically improved memory‐efficient short‐read de novo assembler. Gigascience, 1(1), 18. 10.1186/2047-217X-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzkin, L. M. (2014). Ecological genomics of host shifts in Drosophila mojavensis . Ecological Genomics, 781, 233–247. [DOI] [PubMed] [Google Scholar]
- McEvey, S. F. , & Schiffer, M. (2015). New species in the Drosophila ananassae subgroup from Northern Australia, New Guinea and the South Pacific (Diptera: Drosophilidae), with historical overview. Records of the Australian Museum, 67(5), 129–161. 10.3853/j.2201-4349.67.2015.1651 [DOI] [Google Scholar]
- Minh, B. Q. , Hahn, M. W. , & Lanfear, R. (2020). New methods to calculate concordance factors for phylogenomic datasets. Molecular Biology and Evolution, 37(9), 2727–2733. 10.1093/molbev/msaa106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskalev, A. , Plyusnina, E. , Shaposhnikov, M. , Shilova, L. , Kazachenok, A. , & Zhavoronkov, A. (2012). The role of D‐GADD45 in oxidative, thermal and genotoxic stress resistance. Cell Cycle, 11(22), 4222–4241. 10.4161/cc.22545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neme, R. , & Tautz, D. (2013). Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution. BMC Genomics, 14(1), 1–13. 10.1186/1471-2164-14-117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte, V. , Pandey, R. V. , Kofler, R. , & Schlötterer, C. (2013). Genome‐wide patterns of natural variation reveal strong selective sweeps and ongoing genomic conflict in Drosophila mauritiana . Genome Research, 23(1), 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes, M. D. , Wengel, P. O. , Kreissl, M. , & Schlötterer, C . (2010). Multiple hybridization events between Drosophila simulans and Drosophila mauritiana are supported by mtDNA introgression. Molecular Ecology, 19(21), 4695–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Grady, P. M. , & DeSalle, R. (2018). Phylogeny of the genus Drosophila . Genetics, 209(1), 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Grady, P. M. , & Markow, T. A. (2009). Phylogenetic taxonomy in Drosophila: Problems and prospects. Fly (Austin), 3(1), 10–14. 10.4161/fly.3.1.7748 [DOI] [PubMed] [Google Scholar]
- Oksanen, J. , Blanchet, F. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , Minchin, P. , O’Hara, R. , Simpson, G. , & Solymos, P. (2019). vegan: Community ecology package. R package version 2.5. 4. 2019.
- Oliveira, D. C. S. G. , Almeida, F. C. , O'Grady, P. M. , Armella, M. A. , DeSalle, R. , & Etges, W. J. (2012). Monophyly, divergence times, and evolution of host plant use inferred from a revised phylogeny of the Drosophila repleta species group. Molecular Phylogenetics and Evolution, 64(3), 533–544. 10.1016/j.ympev.2012.05.012 [DOI] [PubMed] [Google Scholar]
- Ometto, L. , Cestaro, A. , Ramasamy, S. , Grassi, A. , Revadi, S. , Siozios, S. , Moretto, M. , Fontana, P. , Varotto, C. , Pisani, D. , Dekker, T. , Wrobel, N. , Viola, R. , Pertot, I. , Cavalieri, D. , Blaxter, M. , Anfora, G. , & Rota‐Stabelli, O. (2013). Linking genomics and ecology to investigate the complex evolution of an invasive Drosophila pest. Genome Biology and Evolution, 5(4), 745–757. 10.1093/gbe/evt034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski, E. A. , Ofria, C. , & Lenski, R. E. (2007). Ecological specialization and adaptive decay in digital organisms. American Naturalist, 169(1), E1–E20. 10.1086/510211 [DOI] [PubMed] [Google Scholar]
- Palmieri, N. , Kosiol, C. , & Schlotterer, C. (2014). The life cycle of Drosophila orphan genes. eLife, 3, e01311. 10.7554/eLife.01311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamilo, P. , & Nei, M. (1988). Relationships between gene trees and species trees. Molecular Biology and Evolution, 5(5), 568–583. [DOI] [PubMed] [Google Scholar]
- Paradis, E. , & Schliep, K. (2019). ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics, 35(3), 526–528. 10.1093/bioinformatics/bty633 [DOI] [PubMed] [Google Scholar]
- Parker, D. J. , Wiberg, R. A. W. , Trivedi, U. , Tyukmaeva, V. I. , Gharbi, K. , Butlin, R. K. , Hoikkala, A. , Kankare, M. , & Ritchie, M. G. (2018). Inter and intraspecific genomic divergence in Drosophila montana shows evidence for cold adaptation. Genome Biol Evol, 10(8), 2086–2101. 10.1093/gbe/evy147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parratt, S. R. , Walsh, B. S. , Metelmann, S. , White, N. , Manser, A. , Bretman, A. J. , Hoffmann, A. A. , Snook, R. R. , & Price, T. A. (2021). Temperatures that sterilize males better match global species distributions than lethal temperatures. Nature Climate Change, 11(6), 481–484. 10.1038/s41558-021-01047-0 [DOI] [Google Scholar]
- Pease, J. B. , & Hahn, M. W. (2013). More accurate phylogenies inferred from low‐recombination regions in the presence of incomplete lineage sorting. Evolution, 67(8), 2376–2384. 10.1111/evo.12118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pissios, P. , & Scouras, Z. G. (1993). Mitochondrial DNA evolution in the Montium‐species subgroup of Drosophila . Molecular Biology and Evolution, 10(2), 375–382. [DOI] [PubMed] [Google Scholar]
- Pitnick, S. , Markow, T. A. , & Spicer, G. S. (1995). Delayed male maturity is a cost of producing large sperm in Drosophila . Proceedings of the National Academy of Sciences of the United States of America, 92(23), 10614–10618. 10.1073/pnas.92.23.10614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard, D. A. , Iyer, V. N. , Moses, A. M. , & Eisen, M. B. (2006). Widespread discordance of gene trees with species tree in Drosophila: Evidence for incomplete lineage sorting. PLoS Genetics, 2(10), 1634–1647. 10.1371/journal.pgen.0020173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, J. R. (1997). Progress and prospects in evolutionary biology: The Drosophila model. Oxford University Press. [Google Scholar]
- Price, A. L. , Jones, N. C. , & Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics, 21(Suppl 1), i351–358. 10.1093/bioinformatics/bti1018 [DOI] [PubMed] [Google Scholar]
- Qin, W. , Neal, S. J. , Robertson, R. M. , Westwood, J. T. , & Walker, V. K. (2005). Cold hardening and transcriptional change in Drosophila melanogaster . Insect Molecular Biology, 14(6), 607–613. 10.1111/j.1365-2583.2005.00589.x [DOI] [PubMed] [Google Scholar]
- Rane, R. V. , Pearce, S. L. , Li, F. , Coppin, C. , Schiffer, M. , Shirriffs, J. , Sgrò, C. M. , Griffin, P. C. , Zhang, G. , Lee, S. F. , Hoffmann, A. A. , & Oakeshott, J. G. (2019). Genomic changes associated with adaptation to arid environments in cactophilic Drosophila species. BMC Genomics, 20(1), 52. 10.1186/s12864-018-5413-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranga, P. , Prakash, R. , & Mrinal, N. (2017). Sibling Drosophila species (Drosophila leontia and Drosophila kikkawai) show divergence for thermotolerance along a latitudinal gradient. Evolutionary Ecology, 31(1), 93–117. 10.1007/s10682-016-9880-1 [DOI] [Google Scholar]
- Remsen, J. , & O'Grady, P. (2002). Phylogeny of Drosophilinae (Diptera: Drosophilidae), with comments on combined analysis and character support. Molecular Phylogenetics and Evolution, 24(2), 249–264. 10.1016/S1055-7903(02)00226-9 [DOI] [PubMed] [Google Scholar]
- Revell, L. J. (2012). phytools: An R package for phylogenetic comparative biology (and other things). Methods in Ecology and Evolution, 3(2), 217–223. 10.1111/j.2041-210X.2011.00169.x [DOI] [Google Scholar]
- Richards, S. , Liu, Y. , Bettencourt, B. R. , Hradecky, P. , Letovsky, S. , Nielsen, R. , Thornton, K. , Hubisz, M. J. , Chen, R. , & Meisel, R. P. (2005). Comparative genome sequencing of Drosophila pseudoobscura: Chromosomal, gene, and cis‐element evolution. Genome Research, 15(1), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld, J. A. , Payne, A. , & DeSalle, R. (2012). Random roots and lineage sorting. Molecular Phylogenetics and Evolution, 64(1), 12–20. 10.1016/j.ympev.2012.02.029 [DOI] [PubMed] [Google Scholar]
- Russo, C. A. , Mello, B. , Frazão, A. , & Voloch, C. M. (2013). Phylogenetic analysis and a time tree for a large drosophilid data set (Diptera: Drosophilidae). Zoological Journal of the Linnean Society, 169(4), 765–775. [Google Scholar]
- Sanchez‐Flores, A. , Peñaloza, F. , Carpinteyro‐Ponce, J. , Nazario‐Yepiz, N. , Abreu‐Goodger, C. , Machado, C. A. , & Markow, T. A. (2016). Genome evolution in three species of cactophilic Drosophila . G3: Genes, Genomes, Genetics, 6(10), 3097–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schawaroch, V. (2002). Phylogeny of a paradigm lineage: The Drosophila melanogaster species group (Diptera : Drosophilidae). Biological Journal of the Linnean Society, 76(1), 21–37. 10.1111/j.1095-8312.2002.tb01711.x [DOI] [Google Scholar]
- Schwander, T. , Libbrecht, R. , & Keller, L. J. C. B. (2014). Supergenes and complex phenotypes. Current Biology, 24(7), R288–R294. 10.1016/j.cub.2014.01.056 [DOI] [PubMed] [Google Scholar]
- Seetharam, A. S. , & Stuart, G. W. (2013). Whole genome phylogeny for 21 Drosophila species using predicted 2b‐RAD fragments. PeerJ, 1, e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão, F. A. , Waterhouse, R. M. , Ioannidis, P. , Kriventseva, E. V. , & Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31(19), 3210–3212. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- Sørensen, J. G. , Nielsen, M. M. , & Loeschcke, V. (2007). Gene expression profile analysis of Drosophila melanogaster selected for resistance to environmental stressors. Journal of Evolutionary Biology, 20(4), 1624–1636. 10.1111/j.1420-9101.2007.01326.x [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30(9), 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Tzvetkova, A. , & Morgenstern, B. (2006). AUGUSTUS at EGASP: Using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biology, 7(Suppl 1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Star, B. , & Spencer, H. G. (2013). Effects of genetic drift and gene flow on the selective maintenance of genetic variation. Genetics, 194(1), 235–244. 10.1534/genetics.113.149781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker, A. J. , Foley, B. , & Hoffmann, A. (2004). Inversion frequencies in Drosophila serrata along an eastern Australian transect. Genome, 47(6), 1144–1153. [DOI] [PubMed] [Google Scholar]
- Stoletzki, N. , & Eyre‐Walker, A. (2011). Estimation of the neutrality index. Molecular Biology and Evolution, 28(1), 63–70. 10.1093/molbev/msq249 [DOI] [PubMed] [Google Scholar]
- Tamura, K. , Subramanian, S. , & Kumar, S. (2004). Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Molecular Biology and Evolution, 21(1), 36–44. 10.1093/molbev/msg236 [DOI] [PubMed] [Google Scholar]
- Tarailo Graovac, M. , & Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics. Chapter, 4, Unit 4.10. 10.1002/0471250953.bi0410s25 [DOI] [PubMed] [Google Scholar]
- Tatarenkov, A. , & Ayala, F. J. (2001). Phylogenetic relationships among species groups of the virilis–repleta radiation of Drosophila . Molecular Phylogenetics and Evolution, 21(2), 327–331. 10.1006/mpev.2001.1002 [DOI] [PubMed] [Google Scholar]
- Tautz, D. , & Domazet‐Loso, T. (2011). The evolutionary origin of orphan genes. Nature Reviews Genetics, 12(10), 692–702. 10.1038/nrg3053 [DOI] [PubMed] [Google Scholar]
- Telonis‐Scott, M. , Gane, M. , DeGaris, S. , Sgrò, C. M. , & Hoffmann, A. A. (2012). High resolution mapping of candidate alleles for desiccation resistance in Drosophila melanogaster under selection. Molecular Biology and Evolution, 29(5), 1335–1351. 10.1093/molbev/msr294 [DOI] [PubMed] [Google Scholar]
- Telonis‐Scott, M. , Sgrò, C. M. , Hoffmann, A. A. , & Griffin, P. C. (2016). Cross‐study comparison reveals common genomic, network, and functional signatures of desiccation resistance in Drosophila melanogaster . Molecular Biology and Evolution, 33(4), 1053–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhzaz, S. , Teets, N. M. , Cabrero, P. , Henderson, L. , Ritchie, M. G. , Nachman, R. J. , Dow, J. A. , Denlinger, D. L. , & Davies, S.‐A. (2015). Insect capa neuropeptides impact desiccation and cold tolerance. Proceedings of the National Academy of Sciences of the United States of America, 112(9), 2882–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Roberts, A. , Goff, L. , Pertea, G. , Kim, D. , Kelley, D. R. , Pimentel, H. , Salzberg, S. L. , Rinn, J. L. , & Pachter, L. (2012). Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nature Protocols, 7(3), 562–578. 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turissini, D. A. , & Matute, D. R. (2017). Fine scale mapping of genomic introgressions within the Drosophila yakuba clade. PLoS Genetics, 13(9), e1006971. 10.1371/journal.pgen.1006971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakirlis, N. , Acar, O. , Hsu, B. , Castilho Coelho, N. , Van Oss, S. B. , Wacholder, A. , Medetgul‐Ernar, K. , Bowman, R. W. , Hines, C. P. , Iannotta, J. , Parikh, S. B. , McLysaght, A. , Camacho, C. J. , O’Donnell, A. F. , Ideker, T. , & Carvunis, A.‐R. (2020). De novo emergence of adaptive membrane proteins from thymine‐rich genomic sequences. Nature Communications, 11(1), 1–18. 10.1038/s41467-020-14500-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linde, K. , & Houle, D. (2008). A supertree analysis and literature review of the genus Drosophila and closely related genera (Diptera, Drosophilidae). Insect Systematics & Evolution, 39(3), 241–267. 10.1163/187631208788784237 [DOI] [Google Scholar]
- van der Linde, K. , Houle, D. , Spicer, G. S. , & Steppan, S. J. (2010). A supermatrix‐based molecular phylogeny of the family Drosophilidae. Genetical Research, 92(1), 25–38. 10.1017/S001667231000008X [DOI] [PubMed] [Google Scholar]
- Vicoso, B. , & Bachtrog, D. (2015). Numerous transitions of sex chromosomes in Diptera. PLoS Biology, 13(4), e1002078. 10.1371/journal.pbio.1002078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock, M. C. & Bürger, R. (2004). Fixation of new mutations in small populations. In Ferrie`re R., Dieckmann U. & Couvet D. (Eds.), Evolutionary conservation biology (pp. 155 –170. Cambridge, UK: Cambridge University Press. [Google Scholar]
- Willi, Y. , Van Buskirk, J. , & Hoffmann, A. A. (2006). Limits to the adaptive potential of small populations. Annual Review of Ecology Evolution and Systematics, 37, 433–458. 10.1146/annurev.ecolsys.37.091305.110145 [DOI] [Google Scholar]
- Wong, A. , Jensen, J. D. , Pool, J. E. , & Aquadro, C. F. (2007). Phylogenetic incongruence in the Drosophila melanogaster species group. Molecular Phylogenetics and Evolution, 43(3), 1138–1150. 10.1016/j.ympev.2006.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, M. , Kostyun, J. L. , Hahn, M. W. , & Moyle, L. C. (2018). Dissecting the basis of novel trait evolution in a radiation with widespread phylogenetic discordance. Molecular Ecology, 27(16), 3301–3316. 10.1111/mec.14780 [DOI] [PubMed] [Google Scholar]
- Xia, S. , VanKuren, N. W. , Chen, C. , Zhang, L. , Kemkemer, C. , Shao, Y. , Jia, H. , Lee, U. , Advani, A. S. , & Gschwend, A. (2020). New genes in drosophila quickly evolved essential functions in viability during development. bioRxiv.
- Xu, Z. , & Wang, H. (2007). LTR_FINDER: An efficient tool for the prediction of full‐length LTR retrotransposons. Nucleic Acids Research, 35, W265–W268. 10.1093/nar/gkm286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Hou, Z. C. , Qian, Y. H. , Kang, H. , & Zeng, Q. T. (2012). Increasing the data size to accurately reconstruct the phylogenetic relationships between nine subgroups of the Drosophila melanogaster species group (Drosophilidae, Diptera). Molecular Phylogenetics and Evolution, 62(1), 214–223. 10.1016/j.ympev.2011.09.018 [DOI] [PubMed] [Google Scholar]
- Yang, Y. , Zhang, Y. P. , Qian, Y. H. , & Zeng, Q. T. (2004). Phylogenetic relationships of Drosophila melanogaster species group deduced from spacer regions of histone gene H2A–H2B. Molecular Phylogenetics and Evolution, 30(2), 336–343. 10.1016/S1055-7903(03)00212-4 [DOI] [PubMed] [Google Scholar]
- Yang, Z. H. (1997). PAML: A program package for phylogenetic analysis by maximum likelihood. Computer Applications in the Biosciences, 13(5), 555–556. 10.1093/bioinformatics/13.5.555 [DOI] [PubMed] [Google Scholar]
- Yang, Z. , & Rannala, B. (2006). Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Molecular Biology and Evolution, 23(1), 212–226. 10.1093/molbev/msj024 [DOI] [PubMed] [Google Scholar]
- Yassin, A. (2013). Phylogenetic classification of the Drosophilidae rondani (Diptera): The role of morphology in the postgenomic era. Systematic Entomology, 38(2), 349–364. 10.1111/j.1365-3113.2012.00665.x [DOI] [Google Scholar]
- Yassin, A. , Delaney, E. K. , Reddiex, A. J. , Seher, T. D. , Bastide, H. , Appleton, N. C. , Lack, J. B. , David, J. R. , Chenoweth, S. F. , Pool, J. E. , & Kopp, A. (2016). The pdm3 locus is a hotspot for recurrent evolution of female‐limited color dimorphism in Drosophila . Current Biology, 26(18), 2412–2422. 10.1016/j.cub.2016.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, M. , Matsuda, H. , Kubo, H. , & Nishimura, T. (2016). Molecular characterization of Tps1 and Treh genes in Drosophila and their role in body water homeostasis. Scientific Reports, 6, 30582. 10.1038/srep30582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, G. C. , Wang, L. G. , Han, Y. Y. , & He, Q. Y. (2012). clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS: A Journal of Integrative Biology, 16(5), 284–287. 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Rabiee, M. , Sayyari, E. , & Mirarab, S . (2018). ASTRAL‐III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics, 19(6), 15–30. 10.1186/s12859-018-2129-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G. , Li, B. , Li, C. , Gilbert, M. T. P. , Jarvis, E. D. , Wang, J. , Consortium AG (2014). Comparative genomic data of the Avian Phylogenomics Project. Gigascience, 3(1), 26. 10.1186/2047-217X-3-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Q. , & Bachtrog, D. (2015). Ancestral chromatin configuration constrains chromatin evolution on differentiating sex chromosomes in Drosophila . PLoS Genetics, 11(6), e1005331. 10.1371/journal.pgen.1005331 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Li, F. , Rane, R. V. , Luria, V. , Xiong, Z. , Chen, J. , Li, Z. , Catullo, R. A. , Griffin, P. C. , Schiffer, M. , Pearce, S. , Lee, S. F. , McElroy, K. , Stocker, A. , Shirriffs, J. , Cockerell, F. , Coppin, C. , Sgrò, C. M. , Karger, A. , Cain, J. W. , … Zhang, G. (2021). Phylogenomic analyses of the genus Drosophila reveals genomic signals of climate adaptation. NCBI, PRJNA736147. Figshare. 10.6084/m9.figshare.14747817 [DOI] [PMC free article] [PubMed]
Supplementary Materials
Supplementary Material
Data Availability Statement
The raw and processed sequence data underlying this article have been made available in NCBI under the project accession number PRJNA736147. Files for the phylogeny code, coding and peptide sequences used for phylogenetic analyses, and the orthologue table for dN/dS calculations are available in figshare https://doi.org/10.6084/m9.figshare.14747817 (Li et al., 2021)