

Summary
Background
Tofacitinib is an oral, small molecule Janus kinase inhibitor for the treatment of ulcerative colitis. We present final data from OCTAVE Open, an open‐label, long‐term extension study.
Aims
The primary objective of OCTAVE Open was to assess the safety and tolerability of long‐term tofacitinib in patients with ulcerative colitis; evaluating efficacy was a secondary objective.
Methods
Eligible patients included OCTAVE Induction 1&2 non‐responders and OCTAVE Sustain completers/treatment failures. Patients in remission at OCTAVE Open baseline received tofacitinib 5 mg b.d.; all others received 10 mg b.d. Incidence rates (unique patients with events/100 patient‐years) for adverse events of special interest were calculated; ≤7.0 years of observation. Efficacy endpoints derived from Mayo score were reported ≤36 months (last scheduled endoscopy visit).
Results
In OCTAVE Open, 769 of 944 patients (81.5%) initially received tofacitinib 10 mg b.d. Among all patients (2440.8 patient‐years of exposure), incidence rates (IRs; 95% confidence intervals) for deaths and adverse events of special interest were: deaths, 0.25 (0.09‐0.54); serious infections, 1.61 (1.14‐2.20); herpes zoster (non‐serious and serious), 3.16 (2.47‐3.97); opportunistic infections, 0.87 (0.54‐1.33); major adverse cardiovascular events, 0.16 (0.04‐0.42); malignancies (excluding non‐melanoma skin cancer), 1.03 (0.67‐1.52); non‐melanoma skin cancer, 0.75 (0.45‐1.19); deep vein thrombosis, 0.04 (0.00‐0.23); pulmonary embolism, 0.21 (0.07‐0.48). At Month 36, 66.9% and 40.3% showed clinical response, 64.6% and 37.1% had endoscopic improvement, and 58.9% and 33.7% maintained or achieved remission, with tofacitinib 5 and 10 mg b.d. respectively.
Conclusion
Tofacitinib demonstrated consistent safety up to 7.0 years. Data collected up to Month 36 support long‐term efficacy beyond the 52‐week maintenance study.

1. INTRODUCTION
Ulcerative colitis is a chronic, immune‐mediated, idiopathic inflammatory bowel disease that is characterised by mucosal inflammation in the rectum and colon. 1 , 2 The management of patients with moderate to severe ulcerative colitis often requires long‐term maintenance treatment, the aims of which are to achieve sustained steroid‐free remission, improve quality of life, reduce morbidity and prevent colorectal cancer. 2 , 3 , 4
Tofacitinib is an oral, small molecule Janus kinase inhibitor for the treatment of ulcerative colitis. The efficacy and safety of tofacitinib were demonstrated in a phase 2, double‐blind, placebo‐controlled, 8‐week induction study (NCT00787202), 5 two identical phase 3, randomised, double‐blind, placebo‐controlled, 8‐week induction studies (OCTAVE Induction 1 and 2; NCT01465763 and NCT01458951) and one phase 3, randomised, double‐blind, placebo‐controlled, 52‐week maintenance study (OCTAVE Sustain; NCT01458574). 6
The safety and efficacy of tofacitinib in patients with ulcerative colitis have been further evaluated in a phase 3, multicentre, open‐label, long‐term extension study (OCTAVE Open; ClinicalTrials.gov: NCT01470612). 7 Patients enrolled in OCTAVE Open received tofacitinib for up to 7.0 years. The primary objective of OCTAVE Open was to assess the safety and tolerability of long‐term tofacitinib therapy in patients with ulcerative colitis. A secondary objective was to evaluate the efficacy of long‐term tofacitinib therapy in patients with ulcerative colitis. Here, we report the final safety and efficacy data from OCTAVE Open.
2. MATERIALS AND METHODS
2.1. Patients, study design and treatments
OCTAVE Open was a phase 3, multicentre, open‐label, long‐term extension study for patients who had previously enrolled in the phase 3 induction (OCTAVE Induction 1 and 2) and/or maintenance (OCTAVE Sustain) studies (Figure 1). Duration of treatment varied by patient, depending on when they enrolled in the study and when the study completed. By design, treatment durations were not fixed. Full study design details for OCTAVE Induction 1 and 2 and OCTAVE Sustain have been reported previously. 6
FIGURE 1.

Overview of the tofacitinib phase 3 OCTAVE clinical programme. †Final complete efficacy assessment at Week 8/52. Treatment continued up to Week 9/53. ‡Clinical response in OCTAVE Induction 1 and 2 was defined as a decrease from induction study baseline total Mayo score of ≥3 points and ≥30%, with a decrease in rectal bleeding subscore of ≥1 point or an absolute rectal bleeding subscore of 0 or 1. §Remission was defined as a total Mayo score of ≤2, with no individual subscore >1 and a rectal bleeding subscore of 0. Adapted from Winthrop et al 19 (in accordance with the CC BY‐NC licence). b.d., twice daily; N, number of patients treated
Patients initially enrolled in OCTAVE Induction 1 or 2 were randomised to receive either tofacitinib 10 mg twice daily (b.d.) or placebo for 8 weeks. Patients with the clinical response at the end of OCTAVE Induction 1 or 2 were eligible to enrol in OCTAVE Sustain, a 52‐week maintenance study in which patients were randomised to receive tofacitinib 5 mg b.d., tofacitinib 10 mg b.d. or placebo. Clinical response was defined as a decrease from induction study baseline total Mayo score of ≥3 points and ≥30%, plus a decrease in rectal bleeding subscore of ≥1 point or an absolute rectal bleeding subscore of 0 or 1.
Patients were eligible to enrol in OCTAVE Open if they were non‐responders after completing OCTAVE Induction 1 or 2 (Week 8 data), or had completed (Week 52 data) or withdrawn early (after experiencing treatment failure) from OCTAVE Sustain. Treatment failure in OCTAVE Sustain was defined as an increase in total Mayo score of ≥3 points from baseline of OCTAVE Sustain, accompanied by an increase in rectal bleeding subscore of ≥1 point, and an increase of endoscopic subscore (centrally read) of ≥1 point (yielding an absolute endoscopic subscore of ≥2), after a minimum of 8 weeks of treatment in the study. The study data collected at the Week 8 visit for OCTAVE Induction 1 or 2, or the Week 52 visit or early termination visit of OCTAVE Sustain, was recorded as the baseline data for OCTAVE Open.
Patients who completed OCTAVE Induction 1 or 2 and were classified as non‐responders were eligible to receive tofacitinib 10 mg b.d. in OCTAVE Open. Patients in remission at Week 52 of OCTAVE Sustain were assigned to receive tofacitinib 5 mg b.d. in OCTAVE Open; remission was defined as a total Mayo score of ≤2 with no individual subscore >1, and a rectal bleeding subscore of 0 (for eligibility assessment, the central read of the Mayo endoscopic subscore was used to determine if a patient was in remission). Patients who completed OCTAVE Sustain not in remission, and patients who withdrew early from OCTAVE Sustain due to treatment failure, were eligible to receive tofacitinib 10 mg b.d. in OCTAVE Open.
At Month 2 of OCTAVE Open, all patients underwent endoscopy, and non‐responders (centrally read) from OCTAVE Induction 1 and 2 were mandated to withdraw if they continued to fail to demonstrate clinical response at this Month 2 assessment.
Patients were permitted to adjust their dose after 8 weeks of initial treatment in OCTAVE Open. For patients receiving tofacitinib 5 mg b.d., the dose could be adjusted to 10 mg b.d. following the loss of response documented by a local read of endoscopy. Loss of response was defined as an increase in total Mayo score of ≥2 points from baseline of OCTAVE Sustain, accompanied by an increase in rectal bleeding subscore of ≥1 point. For patients who met these criteria for loss of response, an endoscopy was performed to determine if they were experiencing a flare. Flare was defined as an increase in total Mayo score of ≥3 points from baseline value of OCTAVE Sustain, accompanied by an increase in rectal bleeding subscore of ≥1 point, and an increase of endoscopic subscore of ≥1 point (unless the endoscopic subscore was a “3” at baseline and remained a “3”), after a minimum of 8 weeks of treatment in OCTAVE Open. Once a patient was confirmed to meet the definition for flare, the investigator could increase the tofacitinib dose from 5 to 10 mg b.d., providing the patient did not have any of the risk factors for pulmonary embolism. For patients receiving tofacitinib 10 mg b.d., the dose could be adjusted to 5 mg b.d. if the patient met specified laboratory criteria, or if the patient was in remission or partial Mayo score remission at, or after, Month 24 (Methods S1). Following protocol amendment 11 (June 2019), patients receiving tofacitinib 10 mg b.d. were required to dose de‐escalate to 5 mg b.d. if pulmonary embolism risk factors were identified during the study. The risk factors for pulmonary embolism are provided in Methods S2.
Prior immunosuppressant use was defined as taking non‐biologic agents such as azathioprine, 6‐mercaptopurine, methotrexate, thioguanine, cyclosporine, tacrolimus and mycophenolic acid products, and leukapheresis, but excluding tumour necrosis factor inhibitor (TNFi) agents. Full details of permitted and prohibited concomitant medications in OCTAVE Open are provided in Methods S3. Dose modifications of concomitant oral 5‐aminosalicylates or sulfasalazine were permitted during OCTAVE Open. Patients who entered the study on corticosteroids were required to taper their corticosteroid dose to achieve steroid‐free status. However, if a patient could not tolerate tapering their corticosteroid dose, they were permitted to remain in the study provided their dose did not exceed 10 mg/day.
This analysis includes final safety and efficacy data for OCTAVE Open (NCT01470612). The study was funded by Pfizer Inc. The study protocol was designed by Pfizer Inc in collaboration with the principal academic investigators. The study protocol (available online with this article) was approved by the Institutional Review Board or independent ethics committee at each participating centre. All patients provided written informed consent. The study was conducted in compliance with the ethical principles derived from the Declaration of Helsinki and in compliance with all International Conference on Harmonization Good Clinical Practice Guidelines. The study was carried out at 252 sites across 31 countries (Methods S4) between October 2012 and August 2020 (first patient first visit: October 1, 2012; last patient last visit: August 6, 2020). The study was determined to be complete by the sponsor as it had met its objectives of characterising long‐term safety and tolerability.
2.2. Assessments
2.2.1. Safety
The primary objective of OCTAVE Open was to assess the safety and tolerability of long‐term tofacitinib therapy in patients with ulcerative colitis. Safety endpoints reported here include: the incidence of deaths; the incidence and severity of adverse events of special interest (serious infections, herpes zoster [non‐serious and serious; including events classified as opportunistic infections], opportunistic infections, malignancies excluding non‐melanoma skin cancer [NMSC], NMSC, major adverse cardiovascular events [MACE], gastrointestinal perforations, deep vein thrombosis and pulmonary embolism); hepatic events evaluated for drug‐induced liver injury; and changes from baseline in clinical laboratory values.
Adverse events and serious adverse events (defined as an adverse event that was life‐threatening, required inpatient hospitalisation or prolongation of existing hospitalisation, or resulted in a persistent or significant disability/incapacity, a congenital anomaly/birth defect or death) were coded using the Medical Dictionary for Regulatory Activities, Version 23.0.
Cardiovascular, malignancy, hepatic, opportunistic infection and gastrointestinal perforation events were adjudicated by independent specialist review committees. All biopsies of suspected malignancy were confirmed by a central expert pathologist. Herpes zoster events that were adjudicated as multidermatomal (defined as non‐adjacent or >2 adjacent dermatomes that were not considered disseminated) or disseminated (defined as any of: diffuse rash [>6 dermatomes], encephalitis, pneumonia or other non‐skin organ involvement) were classified as opportunistic infections. Gastrointestinal perforations excluded preferred terms of pilonidal cyst, perirectal abscess, rectal abscess, anal abscess, perineal abscess and any preferred terms containing the term fistula.
2.2.2. Efficacy
The secondary objective of OCTAVE Open was to evaluate the efficacy of long‐term tofacitinib therapy in patients with ulcerative colitis, and there was no primary efficacy endpoint. Endoscopy was only performed at Months 2, 12, 24 and 36. Secondary efficacy endpoints reported here include the proportions of patients with clinical response, with endoscopic improvement or in remission at Months 2, 12, 24 and 36. Endoscopic improvement was defined as a Mayo endoscopic subscore of 0 or 1 and referred to as “mucosal healing” in the OCTAVE Open protocol. Secondary endpoints also included partial Mayo score (PMS) remission over time. PMS remission was defined as a PMS ≤2, with no individual subscore >1.
2.3. Statistical analysis
Safety and efficacy analyses were based on initial treatment assignment to the tofacitinib 5 and 10 mg b.d. groups and performed on the full analysis set, defined as all patients who received at least one dose of study drug in OCTAVE Open. Proportions of adverse events, serious adverse events and severe adverse events were evaluated. Proportions and IRs (the number of unique patients with events per 100 patient‐years of follow‐up) of adverse events of special interest were evaluated, with 95% confidence intervals (95% CIs).
Efficacy endpoints were derived from the Mayo score per local read of endoscopic scores, with non‐responder imputation (NRI) for missing data at all visits, but with last observation carried forward (LOCF) after a patient advanced to a follow‐up study (RIVETING [NCT03281304] or Japan post‐marketing surveillance studies) up to the visit they would have reached if they had stayed in OCTAVE Open.
3. RESULTS
3.1. Patients
A total of 944 patients received at least one dose of tofacitinib (for up to 7.0 years; range, 1‐2561 days) upon entry into OCTAVE Open. Per protocol, patients in remission at Week 52 of OCTAVE Sustain were assigned to tofacitinib 5 mg b.d. in OCTAVE Open; patients who completed OCTAVE Sustain not in remission, patients who withdrew early from OCTAVE Sustain due to treatment failure, and patients who were non‐responders at Week 8 of OCTAVE Induction 1 or 2 were assigned to tofacitinib 10 mg b.d. In total, 18.5% and 81.5% were assigned to tofacitinib 5 mg b.d. and tofacitinib 10 mg b.d. groups respectively (Table 1). Overall patient disposition is provided in Figure S1. Although dose groups in this analysis were based on initial treatment assignment, patients were permitted to switch dose during OCTAVE Open. Of the 175 patients assigned to tofacitinib 5 mg b.d. in OCTAVE Open, 50 (28.6%) increased their dose to 10 mg b.d. during OCTAVE Open. Of the 769 patients who initially received tofacitinib 10 mg b.d. in OCTAVE Open, 98 (12.7%) dose de‐escalated to 5 mg b.d. during the study (including those who dose de‐escalated due to specified laboratory criteria, remission/PMS remission status or pulmonary embolism risk factors).
TABLE 1.
Baseline demographics and disease characteristics in OCTAVE Open
| Tofacitinib 5 mg b.d. (N = 175) | Tofacitinib 10 mg b.d. (N = 769) | Tofacitinib All (N = 944) | |
|---|---|---|---|
| Remission, n (%) a , b , c | 163 (93.1) d | 1 (0.1) e | 164 (17.4) |
| Total Mayo score, mean (SD) a , c | 1.2 (0.9) | 8.1 (2.3) | 6.8 (3.4) |
| Age (years), mean (SD) | 44.5 (14.6) | 40.5 (13.5) | 41.2 (13.8) |
| Female, n (%) | 79 (45.1) | 310 (40.3) | 389 (41.2) |
| Race, n (%) | |||
| White | 136 (77.7) | 615 (80.0) | 751 (79.6) |
| Asian | 25 (14.3) | 97 (12.6) | 122 (12.9) |
| Weight (kg), mean (SD) f , g | 75.6 (15.7) | 73.4 (16.4) | 73.8 (16.3) |
| BMI (kg/m2), mean (SD) f , h | 25.8 (5.2) | 24.8 (4.7) | 25.0 (4.8) |
| Disease duration (years), median (range) c | 6.6 (1.6‐32.8) | 6.5 (0.6‐42.9) | 6.5 (0.6‐42.9) |
| Corticosteroid use, n (%) c | 1 (0.6) i | 212 (27.6) | 213 (22.6) |
| 5‐ASA use, n (%) c | 133 (76.0) | 539 (70.1) | 672 (71.2) |
| Prior TNFi exposure, n (%) j | 74 (42.3) | 441 (57.3) | 515 (54.6) |
| Prior TNFi failure, n (%) j | 66 (37.7) | 425 (55.3) | 491 (52.0) |
| Prior immunosuppressant use, n (%) j , k | 116 (66.3) | 602 (78.3) | 718 (76.1) |
| Prior immunosuppressant failure, n (%) j , k | 110 (62.9) | 588 (76.5) | 698 (73.9) |
| Extent of disease, n (%) j , l | |||
| Proctosigmoiditis | 38 (21.8) | 98 (12.8) | 136 (14.5) |
| Left‐sided colitis | 56 (32.2) | 261 (34.0) | 317 (33.7) |
| Extensive colitis/pancolitis | 80 (46.0) | 407 (53.1) | 487 (51.8) |
| Proctitis m | 0 (0.0) | 1 (0.1) | 1 (0.1) |
Abbreviations: 5‐ASA, 5‐aminosalicylates; b.d., twice daily; BMI, body mass index; N, number of patients in the treatment group; n, number of patients within the given category; SD, standard deviation; TNFi, tumour necrosis factor inhibitor.
Assessment based on 767 and 942 evaluable patients receiving tofacitinib 10 mg b.d. and tofacitinib all respectively.
Based on centrally read endoscopic subscore.
Baseline and clinical characteristics data per data from baseline of OCTAVE Open.
Twelve patients not in remission at OCTAVE Open baseline were assigned to tofacitinib 5 mg b.d. as protocol deviations.
One patient in remission at OCTAVE Open baseline was assigned to tofacitinib 10 mg b.d. in error by the clinical trial site.
Assessment based on 765 and 940 evaluable patients receiving tofacitinib 10 mg b.d. and tofacitinib all respectively.
From the last patient visit in the previous study.
BMI was calculated using weight (from the last patient visit in the previous study) and height (from screening of the induction studies).
One patient was receiving prednisone 7.5 mg daily at baseline of OCTAVE Open.
Baseline and clinical characteristics data per data from baseline of induction studies.
Immunosuppressant use was defined as taking non‐biologic agents such as azathioprine, 6‐mercaptopurine, methotrexate, thioguanine, cyclosporine, tacrolimus and mycophenolic acid products and leukapheresis, but excludes TNFi agents.
Assessment based on 174, 767 and 941 evaluable patients receiving tofacitinib 5 mg b.d., tofacitinib 10 mg b.d. and tofacitinib all respectively.
One patient with proctitis was enrolled into the induction study as a protocol deviation.
Demographics were generally similar between tofacitinib 5 and 10 mg b.d. groups; however, consistent with patients assigned to tofacitinib 5 mg b.d. being in remission at the time of entry, disease characteristics at OCTAVE baseline tended to be more severe in patients assigned to the tofacitinib 10 mg b.d. group than in those assigned to the 5 mg b.d. group (Table 1). Mean (standard deviation [SD]) total Mayo score was higher in the tofacitinib 10 mg b.d. group (8.1 [SD 2.3]) than in the 5 mg b.d. group (1.2 [SD 0.9]), who, per protocol, were in remission on entering the study. At baseline, 0.6% of patients in the tofacitinib 5 mg b.d. group and 27.6% of patients in the 10 mg b.d. group were taking corticosteroids; per protocol, only patients who withdrew from OCTAVE Sustain due to treatment failure or were non‐responders from OCTAVE Induction 1 and 2 were permitted to take corticosteroids at baseline, and these patients were required to undergo corticosteroid tapering as described. Extent of disease was most commonly extensive colitis/pancolitis in both tofacitinib 5 and 10 mg b.d. groups. A higher proportion of patients in the tofacitinib 10 mg b.d. group (55.3%) had prior TNFi failure, compared with those in the tofacitinib 5 mg b.d. group (37.7%). The tofacitinib 10 mg b.d. group also had a higher proportion of patients with prior immunosuppressant failure (76.5% and 62.9% of patients in the tofacitinib 10 and 5 mg b.d. groups respectively).
3.2. Safety
3.2.1. Adverse events, serious adverse events and discontinuations
Patients in the tofacitinib 5 and 10 mg b.d. groups had median (range) study drug exposures of 1529 (36‐2422) and 668 (1‐2561) days respectively and total patient‐years of exposure of 643.7 and 1797.0 respectively (Table 2). Tofacitinib discontinuation occurred in 48.0% and 86.5% of patients in the tofacitinib 5 and 10 mg b.d. groups respectively; patient discontinuations over time are reported in Figure S2. Overall, the most frequent reason leading to discontinuation was insufficient clinical response, which included the adverse event of worsening ulcerative colitis (11.4% and 42.4% of patients in the tofacitinib 5 and 10 mg b.d. groups respectively) (Figure S1). Discontinuations due to insufficient clinical response prior to, or at Month 2, occurred in 0.0% and 22.4% of patients in the tofacitinib 5 and 10 mg b.d. groups respectively. At Month 2 of OCTAVE Open, non‐responders (centrally read) from OCTAVE Induction 1 and 2, all of whom were assigned to tofacitinib 10 mg b.d., were mandated to withdraw if they continued to fail to demonstrate clinical response at this assessment. A total of 140 (14.8%) patients discontinued to enrol in RIVETING, an ongoing, randomised, parallel‐group study designed to evaluate the efficacy and safety of tofacitinib in patients with ulcerative colitis who were in stable remission on 10 mg b.d. In Japan, 12 (1.3%) patients enrolled into post‐marketing surveillance and three (0.3%) discontinued the study following regulatory approval. Additional reasons for discontinuation are shown in Table S1.
TABLE 2.
Summary of safety in OCTAVE Open
| Tofacitinib 5 mg b.d. (N = 175) | Tofacitinib 10 mg b.d. (N = 769) a | Tofacitinib All (N = 944) | |
|---|---|---|---|
| Total PY of exposure | 643.7 | 1797.0 | 2440.8 |
| Treatment duration (days), median (range) | 1529 (36‐2422) | 668 (1‐2561) | – |
| Discontinuations, n (%) b | 84 (48.0) | 665 (86.5) | 749 (79.3) |
| Due to adverse event excluding worsening ulcerative colitis c | 20 (11.4) d | 80 (10.4) d | 100 (10.6) |
| Due to insufficient clinical response c | |||
| Prior to or at Month 2 | 0 (0.0) | 172 (22.4) | 172 (18.2) |
| OCTAVE Open final analysis (cumulative) | 20 (11.4) | 326 (42.4) | 346 (36.7) |
| Due to enrolment into another study/regulatory approval | |||
| Enrolled into RIVETING (NCT03281304) | 2 (1.1) e | 138 (17.9) | 140 (14.8) |
| Enrolled into post‐marketing surveillance (Japan only) | 8 (4.6) | 4 (0.5) | 12 (1.3) |
| Regulatory approval in Japan | 1 (0.6) | 2 (0.3) | 3 (0.3) |
| Dose adjustments, n (%) | |||
| Dose escalation | 50 (28.6) | — | — |
| Dose de‐escalation | — | 98 (12.7) | — |
| All‐causality treatment‐emergent adverse events, n (%) | |||
| Adverse events | 154 (88.0) | 626 (81.4) | 780 (82.6) |
| Serious adverse events | 39 (22.3) | 147 (19.1) | 186 (19.7) |
| Severe adverse events | 25 (14.3) | 104 (13.5) | 129 (13.7) |
| All‐causality treatment‐emergent adverse events, by preferred term occurring in ≥10% of patients in any treatment group, n (%) | |||
| Worsening ulcerative colitis | 47 (26.9) | 159 (20.7) | 206 (21.8) |
| Nasopharyngitis | 41 (23.4) | 157 (20.4) | 198 (21.0) |
| Blood creatine phosphokinase increased | 19 (10.9) | 85 (11.1) | 104 (11.0) |
| Upper respiratory tract infection | 19 (10.9) | 77 (10.0) | 96 (10.2) |
| Influenza | 23 (13.1) | 65 (8.5) | 88 (9.3) |
Abbreviations: b.d., twice daily; N, number of patients in the treatment group; n, number of unique patients with a particular adverse event; PY, patient‐years.
All patients underwent endoscopy at Month 2; induction non‐responders were mandated to discontinue if they failed to achieve clinical response by Month 2.
Other reasons for withdrawal from OCTAVE Open included: no longer willing to participate in study, pregnancy, protocol violation, lost to follow‐up, not meeting entrance criteria, adverse events not related to study drug, other.
Adverse events of worsening ulcerative colitis leading to discontinuation were designated as insufficient clinical response.
Related to study drug in 12 (6.9%) patients who received tofacitinib 5 mg b.d., and 46 (6.0%) patients who received tofacitinib 10 mg b.d.
Two patients in the 5 mg b.d. arm of OCTAVE Open were enrolled in RIVETING in error.
Treatment‐emergent adverse events and serious adverse events were reported in 88.0% and 22.3% of patients in the tofacitinib 5 mg b.d. group, and in 81.4% and 19.1% of patients in the tofacitinib 10 mg b.d. group respectively (Table 2). The most frequent treatment‐emergent adverse events, by preferred term (≥10% of patients in any treatment group), were “colitis ulcerative” (worsening ulcerative colitis), “nasopharyngitis,” “blood creatine phosphokinase increased,” “upper respiratory tract infection” and “influenza” (Table 2). The most frequent treatment‐emergent adverse events, by system organ class, were infections and infestations (428/944 patients [45.3%]) and gastrointestinal disorders (361/944 patients [38.2%]).
The most frequent serious adverse events by preferred term were “colitis ulcerative” (worsening ulcerative colitis; 42/944 [4.4%] patients), “condition aggravated” (34/944 [3.6%] patients), “disease progression” (5/944 [0.5%] patients), “herpes zoster” (5/944 [0.5%] patients) and “pulmonary embolism” (5/944 [0.5%] patients). All other serious adverse events by preferred term occurred in fewer than five patients.
3.2.2. Deaths
There were six deaths in the study, all in the tofacitinib 10 mg b.d. group (Table S2). Two patients died due to serious adverse events that occurred during the study. The causes of death were: hepatic angiosarcoma (the patient died from blood loss following a liver biopsy); and pulmonary embolism, in a patient with cholangiocarcinoma metastasised to the peritoneum. Four patients died due to post‐therapy serious adverse events. The causes of death were: metastatic adenocarcinoma; acute myeloid leukaemia; cardiac arrest (in the setting of malignant lung cancer and a pneumothorax post lung biopsy); and multiple organ dysfunction syndrome (in the setting of malignant melanoma). The IR for deaths in the tofacitinib 10 mg b.d. group was 0.33 (95% CI, 0.12‐0.73).
3.2.3. Adverse events of special interest
The IRs for adverse events of special interest, for the overall population and by tofacitinib dose (initial treatment assignment), are summarised in Table 3.
TABLE 3.
Proportions and IRs of deaths and adverse events of special interest in OCTAVE Open
| Tofacitinib 5 mg b.d. (N = 175; 643.7 PY) | Tofacitinib 10 mg b.d. (N = 769; 1797.0 PY) | Tofacitinib All (N = 944; 2440.8 PY) | ||||
|---|---|---|---|---|---|---|
| n (%) | IR a (95% CI) | n (%) | IR a (95% CI) | n (%) | IR a (95% CI) | |
| Deaths b | 0 (0.0) | 0.00 (0.00‐0.57) | 6 (0.8) | 0.33 (0.12‐0.73) | 6 (0.6) | 0.25 (0.09‐0.54) |
| Serious infections c | 8 (4.6) d | 1.25 (0.54‐2.46) | 31 (4.0) e | 1.74 (1.18‐2.47) | 39 (4.1) | 1.61 (1.14‐2.20) |
| Herpes zoster (non‐serious and serious) c , f | 13 (7.4) | 2.08 (1.11‐3.55) | 60 (7.8) g | 3.55 (2.71‐4.58) | 73 (7.7) | 3.16 (2.47‐3.97) |
| Serious herpes zoster c | 1 (0.6) | 0.16 (0.00‐0.87) | 6 (0.8) | 0.33 (0.12‐0.73) | 7 (0.7) | 0.29 (0.12‐0.59) |
| Opportunistic infections c , h , i , j | 4 (2.3) | 0.63 (0.17‐1.60) | 17 (2.2) | 0.96 (0.56‐1.53) | 21 (2.2) | 0.87 (0.54‐1.33) |
| Malignancies excluding NMSC b , h | 7 (4.0) | 1.09 (0.44‐2.25) | 18 (2.3) | 1.00 (0.60‐1.59) | 25 (2.6) | 1.03 (0.67‐1.52) |
| NMSC b , h | 6 (3.4) | 0.96 (0.35‐2.08) | 12 (1.6) | 0.68 (0.35‐1.19) | 18 (1.9) | 0.75 (0.45‐1.19) |
| MACE b , h | 2 (1.1) | 0.31 (0.04‐1.13) | 2 (0.3) | 0.11 (0.01‐0.40) | 4 (0.4) | 0.16 (0.04‐0.42) |
| Gastrointestinal perforations c , h , k | 1 (0.6) | 0.16 (0.00‐0.87) | 1 (0.1) | 0.06 (0.00‐0.31) | 2 (0.2) | 0.08 (0.01‐0.30) |
| Deep vein thrombosis c | 0 (0.0) | 0.00 (0.00‐0.57) | 1 (0.1) | 0.06 (0.00‐0.31) | 1 (0.1) | 0.04 (0.00‐0.23) |
| Pulmonary embolism c | 0 (0.0) | 0.00 (0.00‐0.57) | 5 (0.7) | 0.28 (0.09‐0.65) | 5 (0.5) | 0.21 (0.07‐0.48) |
b.d., twice daily; CI, confidence interval; IR, incidence rate; MACE, major adverse cardiovascular events; N, number of patients who received at least one dose of study drug; n, number of patients with the specified event within the given category; NMSC, non‐melanoma skin cancer; PY, patient‐years.
IRs were calculated as the number of unique patients with events per 100 PY.
All events, including those outside the 28‐day risk period, were included.
All events occurred within 28 days after the last dose of study drug.
Three events were reported as severe (number of events): complicated appendicitis (1), gastroenteritis norovirus (1), necrotising fasciitis (1).
Sixteen events were reported as severe in a total of 13 patients (number of events): appendicitis (3), arthritis bacterial (1), atypical pneumonia (1), herpes zoster (3), herpes zoster meningitis (1), mastoiditis (1), meningitis viral (1), ophthalmic herpes zoster (1), osteomyelitis (1), perirectal abscess (1) sinusitis (1), wound infection (1).
Includes all herpes zoster events, including those classified as opportunistic infections.
Five events were reported as severe.
Adjudicated events.
Excludes tuberculosis and herpes zoster with two adjacent dermatomes; only multidermatomal herpes zoster (events that were non‐adjacent or with more than two adjacent dermatomes that were not considered disseminated) and disseminated herpes zoster (defined as any of: diffuse rash [>6 dermatomes], encephalitis, pneumonia or other non‐skin organ involvement) were classified as opportunistic infections.
Non‐herpes zoster opportunistic infections included a pulmonary mycosis (invasive cryptococcosis) case in the tofacitinib 5 mg b.d. group, and one case each of histoplasmosis, cytomegalovirus (CMV) infection and CMV hepatitis in the tofacitinib 10 mg b.d. group.
Excludes preferred terms of pilonidal cyst, perirectal abscess, rectal abscess, anal abscess, perineal abscess and any preferred terms containing the term fistula.
Serious infections were reported in eight (4.6%) patients in the tofacitinib 5 mg b.d. group and 31 (4.0%) patients in the tofacitinib 10 mg b.d. group, with IRs of 1.25 (95% CI, 0.54‐2.46), 1.74 (95% CI, 1.18‐2.47) and 1.61 (95% CI, 1.14‐2.20) for patients in the tofacitinib 5 mg b.d., tofacitinib 10 mg b.d. and tofacitinib all groups respectively.
A total of 13 (7.4%) patients in the tofacitinib 5 mg b.d. group and 60 (7.8%) patients in the tofacitinib 10 mg b.d. group had herpes zoster (non‐serious and serious; including events classified as opportunistic infections), with IRs of 2.08 (95% CI, 1.11‐3.55), 3.55 (95% CI, 2.71‐4.58) and 3.16 (95% CI, 2.47‐3.97) for patients in the tofacitinib 5 mg b.d., tofacitinib 10 mg b.d. and tofacitinib all groups respectively. Of the 73 patients who had herpes zoster in the study, 7 (9.6%) had herpes zoster serious adverse events and 66 (90.4%) had non‐serious herpes zoster. In total, 12 patients had herpes zoster with two adjacent dermatomes (three patients in the tofacitinib 5 mg b.d. group and nine patients in the tofacitinib 10 mg b.d. group). Of the 17 patients with adjudicated herpes zoster opportunistic infection, 12 were multidermatomal (two patients in the tofacitinib 5 mg b.d. group and 10 patients in the tofacitinib 10 mg b.d. group) and five were disseminated herpes zoster (one patient in the tofacitinib 5 mg b.d. group and four patients in the tofacitinib 10 mg b.d. group [two of which were cases of cutaneous disseminated herpes zoster]).
Adjudicated opportunistic infections were reported in four (2.3%) patients in the tofacitinib 5 mg b.d. group and 17 (2.2%) patients in the tofacitinib 10 mg b.d. group, with IRs of 0.63 (95% CI, 0.17‐1.60), 0.96 (95% CI, 0.56‐1.53) and 0.87 (95% CI, 0.54‐1.33) for patients in the tofacitinib 5 mg b.d., tofacitinib 10 mg b.d. and tofacitinib all groups respectively. Of the 21 patients with opportunistic infections, most (17) were herpes zoster opportunistic infections. Non‐herpes zoster opportunistic infections included a pulmonary mycosis (cryptococcosis) case in the tofacitinib 5 mg b.d. group, and one case each of histoplasmosis, cytomegalovirus (CMV) infection and CMV hepatitis in the tofacitinib 10 mg b.d. group. There was no specific clustering of viral infections or viral opportunistic infections other than herpes zoster. There were no COVID‐19 infections reported in OCTAVE Open.
Adjudicated malignancies (excluding NMSC) were reported in seven (4.0%) patients in the tofacitinib 5 mg b.d. group and 18 (2.3%) patients in the tofacitinib 10 mg b.d. group, with IRs of 1.09 (95% CI, 0.44‐2.25), 1.00 (95% CI, 0.60‐1.59) and 1.03 (95% CI, 0.67‐1.52) for patients in the tofacitinib 5 mg b.d., tofacitinib 10 mg b.d. and tofacitinib all groups respectively. There were four patients with colorectal cancer, three with breast cancer, two with cervical cancer, two with cancer of the penis, two with soft tissue sarcoma, two with cholangiocarcinoma, two with non‐Hodgkin lymphoma, two with malignant melanoma and two with lung cancer; all other malignancy types were reported in one patient each (Table S3). IRs for adjudicated NMSC were 0.96 (95% CI, 0.35‐2.08), 0.68 (95% CI, 0.35‐1.19) and 0.75 (95% CI, 0.45‐1.19) for patients in the tofacitinib 5 mg b.d., tofacitinib 10 mg b.d. and tofacitinib all groups respectively. A total of 13 patients experienced basal cell carcinoma (five in the tofacitinib 5 mg b.d. group and eight in the tofacitinib 10 mg b.d. group); and nine patients experienced squamous cell carcinoma (three in the tofacitinib 5 mg b.d. group and six in the tofacitinib 10 mg b.d. group) (Table S4).
Adjudicated MACE were reported in two (1.1%) patients in the tofacitinib 5 mg b.d. group and two (0.3%) patients in the tofacitinib 10 mg b.d. group, with IRs of 0.31 (95% CI, 0.04‐1.13) for patients in the tofacitinib 5 mg b.d. group (a non‐fatal myocardial infarction and a non‐fatal cerebrovascular accident), and 0.11 (95% CI, 0.01‐0.40) for patients in the tofacitinib 10 mg b.d. group (a non‐fatal cerebrovascular accident and a fatal cardiac death) (Table S5). The IR for adjudicated MACE in the tofacitinib all group was 0.16 (95% CI, 0.04‐0.42).
There were two patients with gastrointestinal perforations: one (0.6%) in the tofacitinib 5 mg b.d. group and one (0.1%) in the tofacitinib 10 mg b.d. group. IRs for adjudicated gastrointestinal perforations were 0.16 (95% CI, 0.00‐0.87), 0.06 (95% CI, 0.00‐0.31) and 0.08 (95% CI, 0.01‐0.30) for patients in the tofacitinib 5 mg b.d., tofacitinib 10 mg b.d. and tofacitinib all groups respectively. The patient with gastrointestinal perforation treated with tofacitinib 5 mg b.d. had complicated appendicitis deemed unrelated to study drug, which occurred 10 days after enrolment into OCTAVE Open, and had previously received tofacitinib 5 mg b.d. during OCTAVE Sustain. The patient treated with tofacitinib 10 mg b.d. had a gastrointestinal perforation which occurred 44 days after the start of study treatment, and after colectomy and subsequent colonic tissue examination a diagnosis of non‐Hodgkin lymphoma was made; this patient was receiving concomitant corticosteroids at the time of the event.
In the tofacitinib 10 mg b.d. group, 1 (0.1%) patient had a deep vein thrombosis and 5 (0.7%) patients had pulmonary embolism, with IRs of 0.06 (95% CI, 0.00‐0.31) and 0.28 (95% CI, 0.09‐0.65) respectively. The patient with deep vein thrombosis was diagnosed following a long‐haul flight and management of an infected leg wound sustained in a recent motorbike accident. Of the five patients with pulmonary embolism, four had the following notable medical history: one with prior deep vein thrombosis and pulmonary embolism; one with phlebothrombosis and stroke; one was receiving combined oral contraceptives for dysfunctional uterine bleeding; and one had cholangiocarcinoma and metastases to the peritoneum, and pulmonary embolism was the cause of death. The fifth patient was aged 33 years and had no pre‐existing risk factors for pulmonary embolism; he was not receiving concomitant steroids, had a body mass index of 22.7 kg/m2 and his pulmonary embolism event occurred on day 2447 of OCTAVE Open. He also had a PMS of 0 for many years prior to the pulmonary embolism. No cases of deep vein thrombosis or pulmonary embolism occurred in patients in the tofacitinib 5 mg b.d. group. The IRs for deep vein thrombosis and pulmonary embolism in the tofacitinib all group were 0.04 (95% CI, 0.00‐0.23) and 0.21 (95% CI, 0.07‐0.48) respectively.
3.2.4. Hepatic injury
Overall, 26 patients had events (laboratory abnormality or adverse events) evaluated for drug‐induced liver injury by adjudication; seven (4.0%) patients had received tofacitinib 5 mg b.d. and 19 (2.5%) patients had received tofacitinib 10 mg b.d. (Table S6). No patients had events assessed as probable, highly likely or definite for drug‐induced liver injury. One patient treated with tofacitinib 5 mg b.d. had elevated alanine aminotransferase (ALT) of mild severity, and the event was assessed as possible drug‐induced liver injury. Four patients treated with tofacitinib 10 mg b.d. had possible drug‐induced liver injury; one patient had an increase in aspartate aminotransferase (AST)/ALT ≥5× the upper limit of normal (no corresponding adverse event reported), and three patients had an adverse event of increased AST/ALT. There were no confirmed Hy’s Law cases.
3.2.5. Laboratory parameters of special interest
Laboratory parameters of special interest in OCTAVE Open included: liver enzymes (AST and ALT); lipids (low‐density lipoprotein and high‐density lipoprotein); haemoglobin; absolute lymphocyte count; and absolute neutrophil count. Observations of laboratory values over time were based on data generated through Month 60 (Figure 2); after this time point the number of values was greatly decreased. No major changes from baseline of OCTAVE Open were observed in laboratory parameters up to Month 60 (Figure 2). The proportion of patients with AST or ALT values ≥3 times the upper limit of normal was numerically higher in the tofacitinib 5 mg b.d. group compared with the tofacitinib 10 mg b.d. group (Table 4).
FIGURE 2.
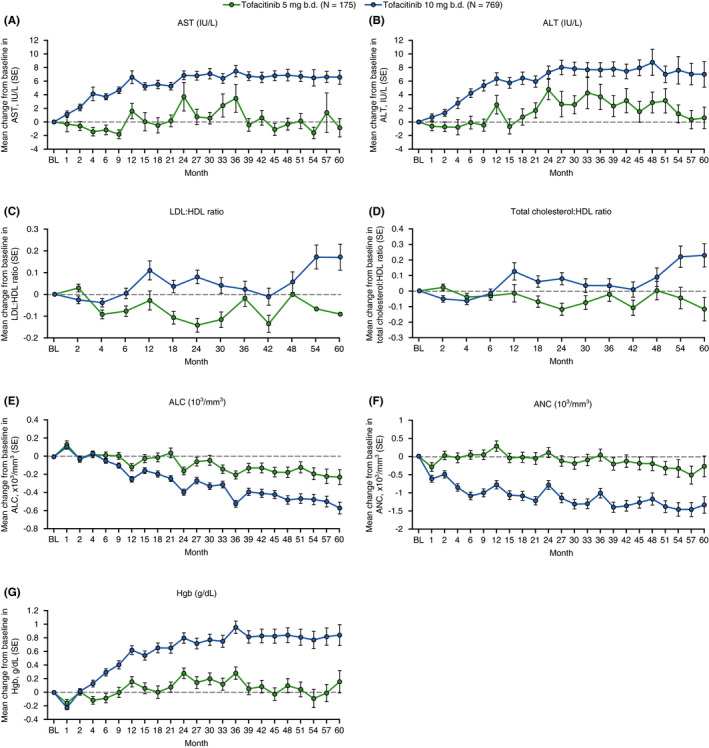
Mean change from baseline of OCTAVE Open over time up to Month 60 in AST, ALT, LDL:HDL ratio, total cholesterol:HDL ratio, ANC, ALC and Hgb. Values are mean ± SE. ALC, absolute lymphocyte count; ALT, alanine aminotransferase; ANC, absolute neutrophil count; AST, aspartate aminotransferase; b.d., twice daily; BL, baseline; Hgb, haemoglobin; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; N, number of evaluable patients; SE, standard error
TABLE 4.
Summary of liver function test values as multiples of the upper limit of normal and laboratory data meeting criteria for discontinuation in OCTAVE Open
| n (%) | Tofacitinib 5 mg b.d. (N = 175) | Tofacitinib 10 mg b.d. (N = 769) | Tofacitinib All (N = 944) |
|---|---|---|---|
| ALT | |||
| ≥1 ×ULN | 86 (49.1) | 211 (27.4) | 297 (31.5) |
| ≥2 ×ULN | 25 (14.3) | 52 (6.8) | 77 (8.2) |
| ≥3 ×ULN | 14 (8.0) | 19 (2.5) | 33 (3.5) |
| AST | |||
| ≥1 ×ULN | 72 (41.1) | 180 (23.4) | 252 (26.7) |
| ≥2 ×ULN | 22 (12.6) | 35 (4.6) | 57 (6.0) |
| ≥3 ×ULN | 10 (5.7) | 13 (1.7) | 23 (2.4) |
| Laboratory data for discontinuations | |||
| Two sequential Hgb <8.0 g/dL or >30% decrease from baseline | 0 (0.0) | 11 (1.4) | 11 (1.2) |
| Two sequential ANC <750 neutrophils/mm3 | 0 (0.0) | 1 (0.1) | 1 (0.1) |
| Two sequential ALC <500 lymphocytes/mm3 | 1 (0.6) | 14 (1.8) | 15 (1.6) |
| Two sequential creatine phosphokinase elevations >10 ×ULN a | 2 (1.1) | 4 (0.5) | 6 (0.6) |
| Two sequential AST or ALT elevations | |||
| ≥3 ×ULN with at least one total bilirubin value ≥2 ×ULN b | 1 (0.6) | 0 (0.0) | 1 (0.1) |
| Two sequential AST or ALT elevations | |||
| ≥5 ×ULN | 1 (0.6) | 1 (0.1) | 2 (0.2) |
Reference ranges for ULN parameters were dependent on patient sex and age.
Abbreviations: ALC, absolute lymphocyte count; ALT, alanine aminotransferase; ANC, absolute neutrophil count; AST, aspartate aminotransferase; b.d., twice daily; Hgb, haemoglobin; N, number of patients in the treatment group; n, number of patients meeting criteria; ULN, upper limit of normal.
Discontinuation was required unless the causality was known not to be medically serious (e.g. exercise induced).
Two sequential AST or ALT elevations ≥3 ×ULN alone was not a reason for discontinuation. Discontinuation was only required if accompanied by at least one total bilirubin value ≥2 ×ULN, or signs or symptoms consistent with hepatic injury.
For laboratory parameters of special interest, the proportions of patients meeting each discontinuation criteria were low (<2%) for each category in both the tofacitinib 5 and 10 mg b.d. groups (Table 4).
3.3. Efficacy
The proportions of patients who achieved clinical response, endoscopic improvement or remission at Months 2, 12, 24 and 36, “as observed” and with NRI‐LOCF, are shown in Figure 3. In the tofacitinib 5 mg b.d. group, the proportion of patients who maintained/achieved clinical response, endoscopic improvement or remission remained high at Months 2, 12, 24 and 36 when analysed as observed and with NRI‐LOCF. In the tofacitinib 10 mg b.d. group, 97.0% of patients had achieved clinical response, 86.3% achieved endoscopic improvement and 79.9% were in remission at Month 36 (as observed); with NRI‐LOCF, the proportions of patients with clinical response, endoscopic improvement or remission were generally maintained up to Month 36.
FIGURE 3.

Proportions of patients in (A) the tofacitinib 5 mg b.d. group, and (B) the tofacitinib 10 mg b.d. group, who achieved clinical response, endoscopic improvement or remission, ‘as observed’ and with NRI‐LOCF, in OCTAVE Open (FAS). Data are per local read of endoscopy. Patients in remission at the start of OCTAVE Open were assigned to tofacitinib 5 mg b.d.; all other patients were assigned to tofacitinib 10 mg b.d. b.d., twice daily; FAS, full analysis set; N1, number of patients in the specified category with non‐missing data; n, number of patients with the specified response within the given category; NRI‐LOCF, non‐responder imputation was applied after a patient discontinued and last observation carried forward imputation after a patient advanced to a subsequent study up to the visit they would have reached if they had stayed in the study. Non‐responder imputation was applied for intermittent missing data
The proportion of patients who were in PMS remission up to Month 60, ‘as observed’ and with NRI‐LOCF, are shown in Figure 4. Among patients that continued in OCTAVE Open (as observed), PMS remission was maintained over 60 months for the tofacitinib 5 mg b.d. group and increased through Month 60 for the 10 mg b.d. group; with NRI‐LOCF, the proportion of patients in PMS remission decreased over time, a trend which was more pronounced in the tofacitinib 5 mg b.d. group.
FIGURE 4.

Proportion of patients in (A) the tofacitinib 5 mg b.d. group, and (B) the tofacitinib 10 mg b.d. group, who achieved PMS remission, ‘as observed’ and with NRI‐LOCF, in OCTAVE Open (FAS). Patients in remission at the start of OCTAVE Open were assigned to tofacitinib 5 mg b.d.; all other patients were assigned to tofacitinib 10 mg b.d. b.d., twice daily; FAS, full analysis set; N1, number of patients in the specified category with non‐missing data; n, number of patients with the specified response within the given category; NRI‐LOCF, non‐responder imputation was applied after a patient discontinued and last observation carried forward imputation after a patient advanced to a subsequent study up to the visit they would have reached if they had stayed in the study. Non‐responder imputation was applied for intermittent missing data; PMS, partial Mayo score
4. DISCUSSION
As ulcerative colitis is a chronic disease, maintenance therapies need to be effective over long periods of time, and, ideally, treatments should not be associated with an increased risk of safety events. For example, while corticosteroids are an acknowledged induction therapy in patients with ulcerative colitis, 2 , 8 , 9 they are not recommended for maintenance therapy due to a lack of long‐term efficacy and their potential for side‐effects. 2 , 10 In contrast, TNFi therapies, including infliximab, adalimumab, golimumab, the anti‐α4β7 integrin antibody vedolizumab and the interleukin‐12/‐23 inhibitor ustekinumab, have been shown to be effective and well tolerated in long‐term extension trials. 11 , 12 , 13 , 14 , 15
This final analysis of OCTAVE Open, reporting data for up to 7.0 years of treatment with tofacitinib in patients with moderately to severely active ulcerative colitis, demonstrated that tofacitinib administered as 5 or 10 mg b.d. had a consistent safety profile during long‐term therapy for ulcerative colitis. The IRs for adverse events of special interest in this final analysis were similar to those from previous interim analyses of this study (November 2017 and May 2019 data cuts); serious infections, malignancies and venous thromboembolic events were infrequent and the safety profile was consistent with that observed in previous analyses from the OCTAVE clinical programme. 6 , 7 , 16 , 17 , 18 This study also demonstrated that tofacitinib 5 and 10 mg b.d. were effective as long‐term therapy for patients with ulcerative colitis, with efficacy being maintained for up to 36 months. In OCTAVE Open, the last scheduled endoscopy visit was at Month 36 and, beyond this, only PMS was evaluated.
Previous findings from the 52‐week maintenance study (OCTAVE Sustain) indicated that the IRs of herpes zoster (non‐serious and serious) were numerically higher with tofacitinib 10 mg b.d. (5.1% of patients; IR 6.64 [95% CI, 3.19‐12.22]) than with 5 mg b.d. (1.5% of patients; IR 2.05 [95% CI, 0.42‐6.00]). 6 , 19 Herpes zoster risk in the entire tofacitinib ulcerative colitis clinical programme has been evaluated previously; although there was an elevated risk of herpes zoster in patients receiving tofacitinib, the majority of cases were non‐serious, cutaneous and limited in their distribution. 19 Of note, the proportions and IRs of herpes zoster in pooled data from across the ulcerative colitis clinical programme were similar in the tofacitinib 10 mg b.d. (5.5% of patients; IR 4.25 [95% CI, 3.18‐5.56]) and 5 mg b.d. (6.5% of patients; IR 3.45 [95% CI, 1.78‐6.02]) groups. 19 These findings are consistent with the final analyses of OCTAVE Open, although it is important to note that dose dependency of IRs cannot be firmly concluded from the data presented here, as dose changes were permitted during the study. In OCTAVE Open, similar IRs for herpes zoster (non‐serious and serious) were seen in the tofacitinib 5 and 10 mg b.d. groups, and most (90.4%) herpes zoster cases were non‐serious. Importantly, the infection resolved without discontinuing tofacitinib therapy in the majority of cases. At baseline of OCTAVE Open, the majority of patients (81.5%) were assigned to and received tofacitinib 10 mg b.d., and the differences in duration of exposure (643.7 PY for tofacitinib 5 mg b.d. versus 1797.0 PY for tofacitinib 10 mg b.d.) between tofacitinib doses in OCTAVE Open should be taken into consideration when comparing the IRs between doses and studies.
With the exception of a higher IR for herpes zoster, the tofacitinib safety data observed in OCTAVE Open was similar to that of previously reported real‐world data for tofacitinib. 20 , 21 , 22 , 23 , 24 In a recent meta‐analysis of the real‐world effectiveness and safety of tofacitinib for moderate to severely active UC, which included data from 1162 patients in 17 studies, the herpes zoster IR was reported to be 6.9 (95% CI, 4.5‐9.3), 25 which is higher than the herpes zoster IR reported here (IR 3.16 [95% CI, 2.47‐3.97]). Of note, however, patients receiving tofacitinib in the real‐world settings described in this meta‐analysis had a higher rate of failure of biologic therapy, with nearly 90% of patients having prior biologic exposure and approximately two‐thirds having prior TNFi and vedolizumab failure, versus almost half of patients in the OCTAVE studies being naïve to biologic therapy. 25 Prior failure of biologic therapy is a known risk factor for herpes zoster in patients with UC. 26 In addition, with the exception of a higher IR for herpes zoster, the safety of tofacitinib in OCTAVE Open was similar to that reported for other approved UC therapies, including biologics. 27 The IRs of adverse events of special interest were generally similar between the tofacitinib 5 and 10 mg b.d. groups, although IRs for serious infections, all herpes zoster and pulmonary embolism were numerically higher in the 10 mg b.d. group than in the 5 mg b.d. group. All deep vein thrombosis and pulmonary embolism events were in the tofacitinib 10 mg b.d. group, although the IRs for deep vein thrombosis and pulmonary embolism were comparable with those reported for patients with ulcerative colitis in general. 28 , 29 , 30 Six deaths occurred during the study, all in the tofacitinib 10 mg b.d. group; one of the six deaths was assessed by the investigator to be related to study drug (a case of hepatic angiosarcoma). In OCTAVE Open, the four adjudicated MACE (two in the tofacitinib 5 mg b.d. group and two in the tofacitinib 10 mg b.d. group) occurred in patients aged ≥55 years, and three of the four patients had a prior medical history that included risk factors for cardiovascular disease. The higher number for some of these events in patients receiving tofacitinib 10 mg b.d. is not unexpected, considering the majority of patients (81.5%) were assigned to and received this dose at baseline of OCTAVE Open.
The safety profile of tofacitinib in OCTAVE Open was consistent with previously published long‐term studies of tofacitinib in patients with rheumatoid arthritis, active psoriatic arthritis or moderate to severe chronic plaque psoriasis. 31 , 32 , 33 , 34 It should be noted that co‐primary endpoint data from the recently completed ORAL Surveillance trial, a required post‐marketing surveillance study, showed an increased rate for MACE and malignancies (excluding NMSC) for tofacitinib relative to TNFi in rheumatoid arthritis patients aged ≥50 years with at least one additional cardiovascular risk factor, who were receiving methotrexate at study baseline. 35
Our findings show that the proportions of patients in the tofacitinib 5 mg b.d. group who achieved clinical response, endoscopic improvement or remission remained high throughout the study, whether data were analysed as NRI‐LOCF or observed. In contrast, for patients in the tofacitinib 10 mg b.d. group, the proportion of patients who achieved clinical response, endoscopic improvement or remission was higher when data were analysed as observed than as NRI‐LOCF. The lower proportion of patients achieving efficacy endpoints by NRI‐LOCF in the 10 mg b.d. group than in the 5 mg b.d. group may be due to the fact that, per protocol, patients who were in remission at Week 52 of OCTAVE Sustain were assigned to tofacitinib 5 mg b.d. upon entry into the open‐label, long‐term extension study. In contrast, patients who were initially assigned to tofacitinib 10 mg b.d. were either induction non‐responders, treatment failures in OCTAVE Sustain or patients who completed OCTAVE Sustain but did not meet the remission definition, indicating that this population of patients may be more refractory to treatment with tofacitinib.
The criteria for treatment assignment could also explain why discontinuations were more common in the tofacitinib 10 mg b.d. group than in the 5 mg b.d. group, with the most frequent reason for discontinuation being insufficient clinical response. Of note, non‐responders from OCTAVE Induction who continued to have non‐response at Month 2 of the open‐label, long‐term extension study were mandated to discontinue, with the reason for discontinuation termed “insufficient clinical response” to include worsening ulcerative colitis.
In this study, between‐group comparisons were limited by baseline differences between dose groups, and the fact that, due to study design, 81.5% of patients were assigned to and received tofacitinib 10 mg b.d. at baseline of the open‐label, long‐term extension study. Comparisons were also limited by the fact that dose groups were classified based on initial treatment assignment; patients were permitted to switch dose during the study, with no fixed treatment groups for comparison. Dose dependency of IRs could not be firmly concluded from the data presented here, as dose changes were permitted during the study. Finally, this was an open‐label study with no placebo or active comparator arm, and so does not allow direct comparison of tofacitinib with other treatments.
In conclusion, the safety profile of tofacitinib remained stable over long‐term treatment exposure (up to 7.0 years), with no increase in IRs compared with previous analyses of interim data from OCTAVE Open. 7 , 16 , 18 This study demonstrated that tofacitinib administered as 5 or 10 mg b.d. had an acceptable safety profile and was well tolerated during long‐term therapy for ulcerative colitis. Most adverse events were mild to moderate in severity and the safety findings are consistent with the established safety profile of tofacitinib. Efficacy was maintained with tofacitinib 5 and 10 mg b.d. up to 36 months beyond Week 52 of OCTAVE Sustain.
AUTHORSHIP
Guarantor of the article: X. Guo.
Author contributions: WJ Sandborn, S Danese, EV Loftus Jr. and J Panés were investigators for this study. N Lawendy, C Su and W Wang contributed to the conceptualisation of the study. N Lawendy, C Su and W Wang contributed to the methodology. N Lawendy, C Su, DT Judd, X Guo, I Modesto and W Wang contributed to data curation, project administration and validation. All authors contributed equally to formal analysis of the data and writing, reviewing and editing the manuscript. All authors approved the final version of the article, including the authorship list.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the patients, physicians and trial team involved in OCTAVE Open. Medical writing support, under the guidance of the authors, was provided by Chris Guise, PhD, CMC Connect, McCann Health Medical Communications and was funded by Pfizer Inc, New York, NY, USA in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med 2015;163:461–464). William J. Sandborn is supported in part by the National Institute of Diabetes and Digestive and Kidney Diseases‐funded San Diego Digestive Diseases Research Center (P30 DK120515). All authors approved the final version of the article, including the authorship list.
Declaration of personal interests: WJ Sandborn has received grant support, personal fees and non‐financial support from Pfizer Inc during the conduct of the study; research support from AbbVie, Abivax, Arena Pharmaceuticals, Boehringer Ingelheim, Celgene, Genentech, Gilead Sciences, GlaxoSmithKline, Janssen, Lilly, Pfizer Inc, Prometheus Biosciences, Seres Therapeutics, Shire, Takeda and Theravance Biopharma; consultancy fees from AbbVie, Abivax, AdMIRx, Alfasigma, Alimentiv (Robarts Clinical Trials, owned by Health Academic Research Trust [HART]), Alivio Therapeutics, Allakos, Amgen, Applied Molecular Transport, Arena Pharmaceuticals, Bausch Health (Salix), BeiGene, Bellatrix Pharmaceuticals, Boehringer Ingelheim, Boston Pharmaceuticals, Bristol‐Myers Squibb, Celgene, Celltrion, Cellularity, Cosmo Pharmaceuticals, Equillium, Escalier Biosciences, Forbion, Genentech/Roche, Gilead Sciences, Glenmark Pharmaceuticals, Gossamer Bio, Immunic (Vital Therapies), Index Pharmaceuticals, Intact Therapeutics, Janssen, Kyverna Therapeutics, Landos Biopharma, Lilly, Oppilan Pharma, Otsuka, Pandion Therapeutics, Pfizer Inc, Progenity, Prometheus Biosciences, Protagonist Therapeutics, Provention Bio, Reistone Biopharma, Seres Therapeutics, Shanghai Pharma Biotherapeutics, Shire, Shoreline Biosciences, Sublimity Therapeutics, Surrozen, Takeda, Theravance Biopharma, Thetis Pharmaceuticals, Tillotts Pharma, UCB, Vedanta Biosciences, Ventyx Biosciences, Vimalan Biosciences, Vivelix Pharmaceuticals, Vivreon Biosciences and Zealand Pharma; and has stock or stock options in Allakos, BeiGene, Gossamer Bio, Oppilan Pharma, Progenity, Prometheus Biosciences, Shoreline Biosciences, Ventyx Biosciences and Vimalan Biosciences. WJ Sandborn’s spouse has received consultancy fees from Iveric Bio and Oppilan Pharma; is an employee of Prometheus Biosciences; and has stock or stock options in Iveric Bio, Oppilan Pharma Progenity, Prometheus Biosciences, Ventyx Biosciences and Vimalan Biosciences. N Lawendy, C Su, DT Judd, X Guo, I Modesto and W Wang are all employees and stockholders of Pfizer Inc. S Danese has received consultancy fees from AbbVie, Allergan, Amgen, AstraZeneca, Biogen, Boehringer Ingelheim, Celgene, Celltrion, Ferring, Gilead Sciences, Hospira, Janssen, Johnson & Johnson, MSD, Mundipharma, Pfizer Inc, Roche, Sandoz, Takeda, TiGenix, UCB and Vifor. EV Loftus Jr. has received research support from AbbVie, Amgen, Bristol‐Myers Squibb, Celgene, Genentech, Gilead Sciences, Janssen, Pfizer Inc, Receptos, Robarts Clinical Trials, Takeda, Theravance and UCB; and consultancy fees from AbbVie, Amgen, Arena Pharmaceuticals, Bristol‐Myers Squibb, Boehringer Ingelheim, Calibr, Celgene, Lilly, Genentech, Gilead Sciences, Gossamer Bio, Iterative Scopes, Janssen, Lilly, Ono Pharma, Pfizer Inc, Sun Pharma, Takeda and UCB. A Hart has received consultancy fees from AbbVie, Allergan, Atlantic, Bristol‐Myers Squibb, Celltrion, Falk Pharma, Ferring, Janssen, MSD, Napp Pharmaceuticals, Pfizer Inc, Pharmacosmos, Shire and Takeda; lecture fees from AbbVie, Atlantic, Bristol‐Myers Squibb, Celltrion, Falk Pharma, Ferring, Janssen, MSD, Napp Pharmaceuticals, Pfizer Inc, Pharmacosmos, Shire and Takeda; other fees from Genentech (Global Steering Committee); and has served as an advisory board member for AbbVie, Atlantic, Bristol‐Myers Squibb, Celltrion, Falk Pharma, Ferring, Janssen, MSD, Napp Pharmaceuticals, Pfizer Inc, Pharmacosmos, Shire and Takeda. I Dotan has received personal fees from AbbVie, Abbott, Altman Research, Arena Pharmaceuticals, Athos, Cambridge Healthcare, Celgene/Bristol‐Myers Squibb, Celltrion, DSM, Falk Pharma, Ferring, Food Industries Organisation, Genentech/Roche, Gilead Sciences, Janssen, MSD, Neopharm, Nestlé, Pfizer Inc, Rafa Laboratories, Sandoz, Sangamo, Sublimity, Takeda and Wildbio. AOMC Damião has received personal fees from AbbVie, Ferring, Janssen, Takeda and Pfizer Inc. J Panés has received research support from AbbVie and Pfizer Inc; and personal fees from AbbVie, Arena Pharmaceuticals, Boehringer Ingelheim, Celgene, Celltrion, F. Hoffmann‐La Roche, Genentech, Gilead Sciences, GlaxoSmithKline, Immunic, Janssen, Origo, Pandion, Pfizer Inc, Progenity, Robarts Clinical Trials, Takeda, Theravance and Wassermann.
Sandborn WJ, Lawendy N, Danese S, et al. Safety and efficacy of tofacitinib for treatment of ulcerative colitis: final analysis of OCTAVE Open, an open‐label, long‐term extension study with up to 7.0 years of treatment. Aliment Pharmacol Ther. 2022;55:464–478. doi: 10.1111/apt.16712
The Handling Editor for this article was Dr Nicholas Kennedy, and it was accepted for publication after full peer‐review.
Funding information
OCTAVE Open was sponsored by Pfizer Inc. Medical writing support, under the guidance of the authors, was provided by Chris Guise, PhD, of CMC Connect, McCann Health Medical Communications and was funded by Pfizer Inc, New York, NY, USA in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med 2015;163:461–464).
DATA AVAILABILITY STATEMENT
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de‐identified participant data. See https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information.
REFERENCES
- 1. Ungaro R, Mehandru S, Allen PB, Peyrin‐Biroulet L, Colombel J‐F. Ulcerative colitis. Lancet. 2017;389:1756–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rubin DT, Ananthakrishnan AN, Siegel CA, Sauer BG, Long MD. ACG clinical guideline: ulcerative colitis in adults. Am J Gastroenterol. 2019;114:384–413. [DOI] [PubMed] [Google Scholar]
- 3. Harbord M, Eliakim R, Bettenworth D, et al. Third European evidence‐based consensus on diagnosis and management of ulcerative colitis. Part 2: current management. J Crohns Colitis. 2017;11:769–784. [DOI] [PubMed] [Google Scholar]
- 4. Kornbluth A, Sachar DB. Practice Parameters Committee of the American College of Gastroenterology. Ulcerative colitis practice guidelines in adults: American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol. 2010;105:501–523. [DOI] [PubMed] [Google Scholar]
- 5. Sandborn WJ, Ghosh S, Panes J, et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367:616–624. [DOI] [PubMed] [Google Scholar]
- 6. Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376:1723–1736. [DOI] [PubMed] [Google Scholar]
- 7. Lichtenstein GR, Loftus EV, Wei SC, et al. DOP61 Tofacitinib, an oral, small‐molecule Janus kinase inhibitor, in the treatment of ulcerative colitis: analysis of an open‐label, long‐term extension study with up to 5.9 years of treatment [abstract]. J Crohns Colitis. 2020;14(Suppl 1):S100–S101. Abstract DOP61. [Google Scholar]
- 8. Sandborn WJ, Travis S, Moro L, et al. Once‐daily budesonide MMX® extended‐release tablets induce remission in patients with mild to moderate ulcerative colitis: results from the CORE I study. Gastroenterology. 2012;143:1218–1226. [DOI] [PubMed] [Google Scholar]
- 9. Ford AC, Bernstein CN, Khan KJ, et al. Glucocorticosteroid therapy in inflammatory bowel disease: systematic review and meta‐analysis. Am J Gastroenterol. 2011;106:590–599; quiz 600. [DOI] [PubMed] [Google Scholar]
- 10. Bressler B, Marshall JK, Bernstein CN, et al. Clinical practice guidelines for the medical management of nonhospitalized ulcerative colitis: The Toronto consensus. Gastroenterology. 2015;148:1035–1058. [DOI] [PubMed] [Google Scholar]
- 11. Reinisch W, Sandborn WJ, Rutgeerts P, et al. Long‐term infliximab maintenance therapy for ulcerative colitis: the ACT‐1 and ‐2 extension studies. Inflamm Bowel Dis. 2012;18:201–211. [DOI] [PubMed] [Google Scholar]
- 12. Colombel J‐F, Sandborn WJ, Ghosh S, et al. Four‐year maintenance treatment with adalimumab in patients with moderately to severely active ulcerative colitis: data from ULTRA 1, 2, and 3. Am J Gastroenterol. 2014;109:1771–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reinisch W, Gibson PR, Sandborn WJ, et al. Long‐term benefit of golimumab for patients with moderately to severely active ulcerative colitis: results from the PURSUIT‐maintenance extension. J Crohns Colitis. 2018;12:1053–1066. [DOI] [PubMed] [Google Scholar]
- 14. Kaser A, James A. Long‐term effectiveness and safety of vedolizumab in patients with ulcerative colitis: 5‐year cumulative exposure of gemini 1 completers rolling into the gemini open‐label extension study [abstract]. Gut. 2017;66(Suppl. 2):A120. Abstract AODWE‐002. [Google Scholar]
- 15. Panaccione R, Danese S, Sandborn WJ, et al. Ustekinumab is effective and safe for ulcerative colitis through 2 years of maintenance therapy. Aliment Pharmacol Ther. 2020;52:1658–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lichtenstein GR, Loftus EV, Bloom S, et al. Tofacitinib, an oral Janus kinase inhibitor, in the treatment of ulcerative colitis: an interim analysis of an open‐label, long‐term extension study with up to 4.9 years of treatment [abstract]. Am J Gastroenterol. 2018;113(Suppl):S329; Abstract 571. [Google Scholar]
- 17. Sandborn WJ, Panés J, D’Haens GR, et al. Safety of tofacitinib for treatment of ulcerative colitis, based on 4.4 years of data from global clinical trials. Clin Gastroenterol Hepatol. 2019;17:1541–1550. [DOI] [PubMed] [Google Scholar]
- 18. Lichtenstein GR, Gary R, Loftus EV, et al. Tofacitinib, an oral Janus kinase inhibitor, in the treatment of ulcerative colitis: an interim analysis of an open‐label, long‐term extension study with up to 5.5 years of treatment [abstract]. United European Gastroenterol J 2019;7(Suppl):OP213. Abstract OP213. [Google Scholar]
- 19. Winthrop KL, Melmed GY, Vermeire S, et al. Herpes zoster infection in patients with ulcerative colitis receiving tofacitinib. Inflamm Bowel Dis. 2018;24:2258–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chaparro M, Garre A, Mesonero F, et al. Tofacitinib in ulcerative colitis: real‐world evidence from the ENEIDA registry. J Crohns Colitis. 2021;15:35–42. [DOI] [PubMed] [Google Scholar]
- 21. Biemans VBC, Sleutjes JAM, de Vries AC, et al. Tofacitinib for ulcerative colitis: results of the prospective Dutch Initiative on Crohn and Colitis (ICC) registry. Aliment Pharmacol Ther. 2020;51:880–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoffmann P, Globig A‐M, Thomann AK, et al. Tofacitinib in treatment‐refractory moderate to severe ulcerative colitis: real‐world experience from a retrospective multicenter observational study. J Clin Med. 2020;9:2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Honap S, Chee D, Chapman TP, et al. Real‐world effectiveness of tofacitinib for moderate to severe ulcerative colitis: a multicentre UK experience. J Crohns Colitis. 2020;14:1385–1393. [DOI] [PubMed] [Google Scholar]
- 24. Lair‐Mehiri L, Stefanescu C, Vaysse T, et al. Real‐world evidence of tofacitinib effectiveness and safety in patients with refractory ulcerative colitis. Dig Liver Dis. 2020;52:268–273. [DOI] [PubMed] [Google Scholar]
- 25. Taxonera C, Olivares D, Alba C. Real‐world effectiveness and safety of tofacitinib in patients with ulcerative colitis: systematic review with meta‐analysis. Inflamm Bowel Dis. 2021. 10.1093/ibd/izab011 [DOI] [PubMed] [Google Scholar]
- 26. Winthrop KL, Loftus EV, Baumgart DC, et al. Tofacitinib for the treatment of ulcerative colitis: analysis of infection rates from the ulcerative colitis clinical programme. J Crohns Colitis. 2021;15:914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Curtis JR, Regueiro M, Yun H, et al. Tofacitinib treatment safety in moderate to severe ulcerative colitis: comparison of observational population cohort data from the IBM MarketScan® administrative claims database with tofacitinib trial data. Inflamm Bowel Dis. 2021;27:1394–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weng M‐T, Park SH, Matsuoka K, et al. Incidence and risk factor analysis of thromboembolic events in East Asian patients with inflammatory bowel disease, a multinational collaborative study. Inflamm Bowel Dis. 2018;24:1791–1800. [DOI] [PubMed] [Google Scholar]
- 29. Kappelman MD, Horvath‐Puho E, Sandler RS, et al. Thromboembolic risk among Danish children and adults with inflammatory bowel diseases: a population‐based nationwide study. Gut. 2011;60:937–943. [DOI] [PubMed] [Google Scholar]
- 30. Bernstein CN, Blanchard JF, Houston DS, Wajda A. The incidence of deep venous thrombosis and pulmonary embolism among patients with inflammatory bowel disease: a population‐based cohort study. Thromb Haemost. 2001;85:430–434. [PubMed] [Google Scholar]
- 31. Valenzuela F, Korman NJ, Bissonnette R, et al. Tofacitinib in patients with moderate to severe chronic plaque psoriasis: long‐term safety and efficacy in an open‐label extension study. Br J Dermatol. 2018;179:853–862. [DOI] [PubMed] [Google Scholar]
- 32. Cohen SB, Tanaka Y, Mariette X, et al. Long‐term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: integrated analysis of data from the global clinical trials. Ann Rheum Dis. 2017;76:1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pfizer Inc. FDA Advisory Committee Meeting sNDA 203214 supplement 018 briefing document. 2018. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/GastrointestinalDrugsAdvisoryCommittee/UCM599514.pdf. Accessed August 8, 2021
- 34. Nash P, Coates LC, Kivitz AJ, et al. Safety and efficacy of tofacitinib in patients with active psoriatic arthritis: interim analysis of OPAL Balance, an open‐label, long‐term extension study. Rheumatol Ther. 2020;7:553–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pfizer Inc. Pfizer shares co‐primary endpoint results from post‐marketing required safety study of XELJANZ® (tofacitinib) in subjects with rheumatoid arthritis (RA) [press release]. 2021. https://www.pfizer.com/news/press‐release/press‐release‐detail/pfizer‐shares‐co‐primary‐endpoint‐results‐post‐marketing. Accessed August 8, 2021
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de‐identified participant data. See https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information.