

Abstract
Aims
GSK3358699 is a mononuclear myeloid‐targeted bromodomain and extra‐terminal domain (BET) family inhibitor which demonstrates immunomodulatory effects in vitro. This phase 1, randomized, first‐in‐human study evaluated the safety, pharmacokinetics, and pharmacodynamics of GSK3358699 in healthy male participants (NCT03426995).
Methods
Part A (N = 23) included three dose‐escalating periods of 1‐40 mg of GSK3358699 or placebo in two cohorts in a single ascending‐dose crossover design. Part C (N = 25) was planned as an initial dose of 10 mg of GSK3358699 or placebo daily for 14 days followed by selected doses in four sequential cohorts.
Results
In part A, exposure to GSK3358699 and its metabolite GSK3206944 generally increased with increasing doses. The median initial half‐life ranged from 0.7 to 1.1 (GSK3358699) and 2.1 to 2.9 (GSK3206944) hours after a single dose of 1‐40 mg. GSK3206944 concentrations in monocytes were quantifiable at 1‐hour post‐dose following 10 mg of GSK3358699 and 1 and 4 hours post‐dose following 20‐40 mg. Mean predicted percentage inhibition of ex vivo lipopolysaccharide‐induced monocyte chemoattractant protein (MCP)‐1 reached 75% with 40 mg of GSK3358699. GSK3358699 did not inhibit interleukin (IL)‐6 and tumour necrosis factor (TNF).
The most common adverse event (AE) was headache. Four AEs of nonsustained ventricular tachycardia were observed across parts A and C. One serious AE of atrial fibrillation (part C) required hospitalization.
Conclusions
Single doses of GSK3358699 are generally well tolerated with significant metabolite concentrations detected in target cells. A complete assessment of pharmacodynamics was limited by assay variability. A causal relationship could not be excluded for cardiac‐related AEs, resulting in an inability to identify a suitable repeat‐dose regimen and study termination.
Keywords: bromodomain and extra‐terminal domain, epigenetic reader protein, GSK3358699, myeloid, pharmacodynamics, pharmacokinetics
1. What is already known about this subject
Bromodomain and extra‐terminal domain (BET) proteins may contribute to inflammatory disease by maintaining or upregulating the expression of genes that drive pathological disease mechanisms.
BET inhibitors suppress inflammatory processes in animal models.
Clinical development of pan‐BET inhibitors has been hindered by dose‐limiting toxicities including thrombocytopenia, gastrointestinal symptoms and QTc prolongation.
What this study adds
GSK3358699 is an esterase‐sensitive motif‐containing BET inhibitor that preferentially targets mononuclear‐myeloid cells.
Targeted BET inhibition with GSK3358699 largely mitigated the common clinical toxicities observed with pan‐BET inhibitors and single doses were generally well tolerated in healthy adult males.
Repeat dosing of GSK3358699 was associated with clinically significant cardiac adverse events.
1. INTRODUCTION
The bromodomain and extra‐terminal domain (BET) family of proteins regulate gene expression at an epigenetic level 1 , 2 and may contribute to inflammatory and fibrotic disease by maintaining or upregulating the expression of proinflammatory genes that drive pathological disease mechanisms. 1 In animal models, BET inhibitors have been found to suppress the proinflammatory mechanisms that underlie several immune‐inflammatory diseases. 3 , 4 , 5 , 6 However, clinical development of pan‐BET inhibitors has been hindered by dose‐limiting toxicities, primarily thrombocytopenia and gastrointestinal symptoms. 7
GSK3358699 was developed to explore the therapeutic potential of a BET inhibitor that preferentially targets mononuclear‐myeloid cells, including the potential to mitigate the preclinical and clinical toxicities observed with pan‐BET inhibition. GSK3358699 incorporates within its structure an esterase‐sensitive ester motif (ESM) that allows preferential delivery of a pharmacologically active acid metabolite, GSK3206944, to cells that express human carboxyesterase‐1 (CES‐1), namely cells of the mononuclear‐myeloid lineage, including monocytes, macrophages and dendritic cells. 8 GSK3358699 is hydrolysed intracellularly by CES‐1 to form the acid metabolite GSK3206944, which is selectively generated and, by virtue of its physicochemical properties, is retained in mononuclear‐myeloid cells. Both GSK3358699 and GSK3206944 are potent BET inhibitors. Thus, GSK3358699 was postulated to have potential therapeutic benefits in diseases where myeloid cells are important, such as rheumatoid arthritis, psoriasis, fibrotic liver disease and inflammatory bowel disease.
Preclinical studies support the therapeutic potential of GSK3358699 for the treatment of inflammatory diseases. GSK3358699 demonstrated potent immunomodulatory effects in ex vivo experiments with human whole blood and in blood samples from patients with rheumatoid arthritis. In addition, GSK3358699 inhibited joint inflammation and bone erosion in a mouse model of arthritis and decreased lipopolysaccharide (LPS)‐induced cytokine production in cynomolgus monkeys.
In this first‐in‐human study, we evaluated the safety and tolerability of single and multiple ascending doses of GSK3358699 in healthy male participants. The pharmacokinetic and pharmacodynamic profile of GSK3358699 and the metabolite GSK3206944 and quantification of GSK3206944 in monocytes following dosing of GSK3358699 were also evaluated.
2. METHODS
The study was approved by a national ethics committee and was conducted according to the recommendations of Good Clinical Practice and the Declaration of Helsinki. All participants provided written informed consent before the performance of any study procedures.
2.1. Study design
In this phase 1, randomized, double‐blind (sponsor open), planned three‐part study, we evaluated the safety, pharmacokinetics and pharmacodynamics of GSK3358699 treatment in healthy males ( Clinicaltrials.gov identifier: NCT03426995). Study participants were randomized to a treatment group using validated internal software centrally controlled by Registration and Medication Ordering System Next Generation (RAMOS NG). Participants and study site staff were blinded to the allocation of GSK3358699 or matching placebo.
In part A of the study, two interlocking cohorts of participants (cohorts 1 and 2) received GSK3358699 in a single ascending‐dose crossover design during three dose‐escalating treatment periods (Figure 1). The decision to escalate to the next dosing level was made by the Dose Escalation Committee (DEC). Dose‐escalation‐stopping criteria are described in the Supplementary Information . Based on available preclinical and clinical data, 1‐45 mg was selected as the planned dose range. However, after data from the first two treatment periods was reviewed by the DEC, the maximum dose level in treatment period 3 was changed to 40 mg for cohort 1 and 30 mg for cohort 2. Each participant in part A received a maximum of two single ascending doses of GSK3358699 and one dose of placebo. At each of the six dose levels, GSK3358699 and placebo were administered in a 2:1 ratio within each treatment period according to the randomization schedule. On day 1, one of the two sentinel participants received GSK3358699 and the other received placebo. On establishing adequate safety in the sentinel participants, after approximately 48 hours, the remaining participants in the cohort were dosed. There were at least 14 days between each dosing level in cohorts 1 and 2, resulting in a ≥28‐day washout period between dosing in each treatment period for each cohort. Part A of the study also included a fourth treatment period where participants were treated with GSK3358699 or placebo followed by an in vivo LPS (cohort 1) or granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) challenge (cohort 2) with concurrent cantharidin‐induced blisters. The dose of GSK3358699 selected for this treatment period was 25 mg based on analysis of safety, pharmacokinetic and pharmacodynamic data available to the DEC from the previous part A single ascending‐dose treatment periods. The exploratory results from this fourth period will be reported elsewhere.
FIGURE 1.
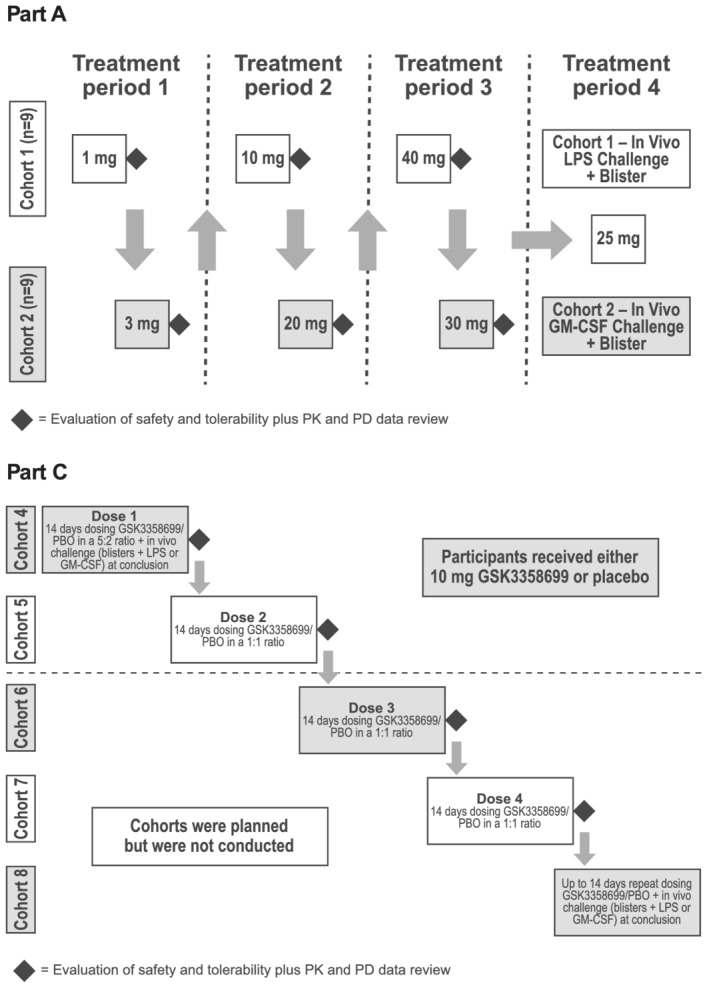
Dosing schematic. In part A of the study, two interlocking cohorts of participants (cohorts 1 and 2) received GSK3358699 in a single ascending‐dose crossover design during three dose‐escalating treatment periods (treatment periods 1‐3). Each participant received a maximum of two single ascending oral doses of GSK3358699 (1, 3, 10, 20, 40 or 30 mg) and one dose of placebo. In treatment period 4, participants were treated with 25 mg of GSK3358699 or placebo followed by an in vivo LPS (cohort 1) or GM‐CSF challenge (cohort 2). Part C of the study was planned to be a repeat‐dose design in sequential cohorts with participants randomized to either 10 mg of GSK3358699 or placebo daily for 14 days. However, the study was terminated during cohort 5 and cohorts 6‐8 were not conducted. GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; LPS, lipopolysaccharide; PBO, placebo; PD, pharmacodynamic; PK, pharmacokinetic
Part B, which was planned to evaluate the food effect of GSK3358699, did not occur.
Part C of the study was originally planned as a repeat‐dose design in four sequential cohorts (cohorts 4‐7; Figure 1). Participants in cohort 4 were treated with either 10 mg of GSK3358699 or placebo daily for 14 days in a 5:2 ratio according to the randomization schedule. The 10 mg of GSK3358699 dose was selected based on data obtained from the dose‐escalation treatment periods (treatment periods 1‐3) in part A of the study. On day 14, it was originally planned that, similar to part A, LPS or GM‐CSF in vivo challenges with concurrent cantharidin‐induced blisters would be investigated at each dose of GSK3358699.
Following a temporary halt to investigate cardiac‐related adverse events (AEs) during cohort 4, the study design was amended for cohorts 5‐7. Each cohort was to receive one daily dose of either GSK3358699 or placebo for 14 days in a 1:1 ratio to allow for collection of safety data in balanced treatment groups (Figure 1). Participants in cohort 5 were treated with 10 mg of GSK3358699. Sentinel dosing with staggering of the first two participants was implemented as described for part A. The planned in vivo challenges were not included in the revised design for cohorts 5‐7. Instead, cohort 8 was planned, which would include investigation of these immunological challenges after up to 14 days of repeat GSK3358699 dosing, but cohort 8 was not conducted. The study was terminated during cohort 5.
2.2. Participants
Healthy males aged 18‐56 years were enrolled in the study. Study participants were required to have a body weight ≥50 kg and a body mass index within the range of 18.5‐35.0 kg/m2. Participants were ineligible for the study if they had a chronic history of pancreatitis, diabetes mellitus/glucose intolerance, cardiac disease, gastrointestinal disease, liver disease, clinically significant renal disease or clinically significant respiratory disease. Participants with an electrocardiogram (ECG) QT interval corrected for heart rate using Fridericia's formula (QTcF) of >450 milliseconds (ms) based on triplicate ECGs or a family history of premature cardiovascular disease were also excluded. After the temporary halt during cohort 4, participants with nonsustained ventricular tachycardia (NSVT) and any clinically relevant abnormality on the screening ECG were also not eligible for the study. Full inclusion and exclusion criteria are described in the Supplementary Information .
2.3. Study assessments and procedures
The primary objective of the study was to assess the safety and tolerability of GSK3358699, including evaluation of AEs, laboratory safety data (clinical chemistry, haematology, urinalysis), vital signs, cardiac telemetry and 12‐lead ECGs. Additional objectives included evaluation of the systemic pharmacokinetic profile of GSK3358699 and the metabolite GSK3206944, the intracellular pharmacokinetic profile of GSK3206944 in monocytes and the pharmacodynamic profile measured by the extent of target engagement (eg, inhibition of cytokine production) in the blood after ex vivo LPS treatment.
Samples for pharmacokinetic analyses were collected on day 1 of treatment periods 1‐4 of part A and days 1 and 14 of part C at pre‐dose and 15 minutes, 30 minutes and 1, 2, 4, 6, 8, 12, 24 and 48 hours post‐dose (except 48‐hour samples were not collected on day 1 of part C). During part C, samples were also collected pre‐dose on days 4, 8 and 12. Plasma pharmacokinetic parameters were analysed for GSK3358699 and GSK3206944 using a validated analytical method based on protein precipitation, followed by high‐performance liquid chromatography tandem mass spectrometry. The lower limit of quantification (LLQ) was 0.1 ng/mL and the higher limit of quantification (HLQ) was 100 ng/mL.
Blood samples for intracellular pharmacokinetic analyses of GSK3206944 were collected on day 1 of treatment periods 1‐3 of part A at 1, 4, 8, 24 and 48 hours post‐dose. During treatment period 4 of part A, samples were collected on day 1 at 4 and 24 hours post‐dose. During part C, samples were collected at 1, 4 and 8 hours post‐dose on days 1 and 14, and also at 24 and 48 hours post‐dose on day 14. Samples were also collected pre‐dose on days 4, 8 and 12. Monocytes were isolated directly from the blood samples using cluster of differentiation 14 magnetic beads and lysed using mammalian protein extraction reagent (M‐PER) buffer. Intracellular pharmacokinetic parameters for GSK3206944 were analysed using the same methods as for the plasma pharmacokinetic parameters. The LLQ and HLQ for intracellular pharmacokinetic analysis were 0.2 and 100 ng/mL, respectively.
Blood samples for ex vivo LPS pharmacodynamic analyses during treatment periods 1‐3 of part A were collected the day prior to treatment and on day 1 at pre‐dose and 1, 4, 8, 12, 24 and 48 hours post‐dose. During part C, samples were collected the day prior to treatment and on days 1 and 14 at pre‐dose, and 1, 4 and 8 hours post‐dose. On day 14, samples were also collected 24 and 48 hours post‐dose. Additional samples during part C were collected pre‐dose on days 2, 4, 8 and 12. In part A, 1 mL blood samples were collected into an LPS TruCulture tube (Myriad RBM, Austin, TX, USA) and a null (no LPS) TruCulture tube. All samples were incubated for approximately 22 hours at 37 °C. The cellular and soluble contents were separated, and the soluble fraction analysed for concentrations of monocyte chemoattractant protein (MCP)‐1, interleukin (IL)‐6 and tumour necrosis factor (TNF)‐α using the Meso Scale Discovery platform (Meso Scale Diagnostics, LLC, Rockville, MD, USA).
2.4. Statistical analyses
The sample size was based on feasibility and no formal hypotheses were tested. The safety and pharmacodynamic endpoints were analysed in all randomized participants who received at least one dose of study treatment (safety population). The pharmacokinetic parameters were measured in all participants in the safety population who received an active dose of the study drug and for whom pharmacokinetic samples were obtained and analysed (pharmacokinetic population). An estimation approach was used to quantify the single‐dose pharmacokinetics for each dose level studied and to assess pharmacokinetic parameters following 14 days repeat dosing relative to single dosing.
Safety, pharmacokinetic and pharmacodynamic assessments were summarized descriptively. Pharmacokinetic parameters were calculated by standard noncompartmental analysis according to current working practices and using WinNonlin software (Certara, Princeton, NJ, USA). The pharmacokinetic parameters analysed were area under the concentration‐time curve (AUC) from time zero to the time of the last quantifiable concentration (AUC(0‐t)), AUC extrapolated to infinity (AUC(0‐∞)), AUC from time zero to 24 hours post‐dose (AUC(0‐24)), AUC from time zero to the end of the dosing period (AUC(0‐tau)), the maximum observed concentration (C max), the time to reach C max (T max), the apparent terminal half‐life (t 1/2 terminal), actual initial half‐life (t 1/2 initial), accumulation ratio between one single dose (day 1) and the repeat dose (day 14; R 0) and steady state ratio (R S).
3. RESULTS
3.1. Study population
Part A included 23 participants, 17 of whom completed part A (Figure 2). Part C included 25 participants (11 placebo, 14 GSK3358699), but only four participants (two placebo, two GSK3358699) completed the study (ie, received all 14 doses) because cohorts 4 and 5 were stopped partway through the 14‐day treatment period (Figure 2). The four participants who completed part C were the sentinel participants from cohorts 4 and 5. The remaining participants in cohorts 4 and 5 of part C received between one and 13 doses before the cohorts were halted. The demographics in part C of the study were similar between the GSK3358699 and placebo groups (Table 1).
FIGURE 2.
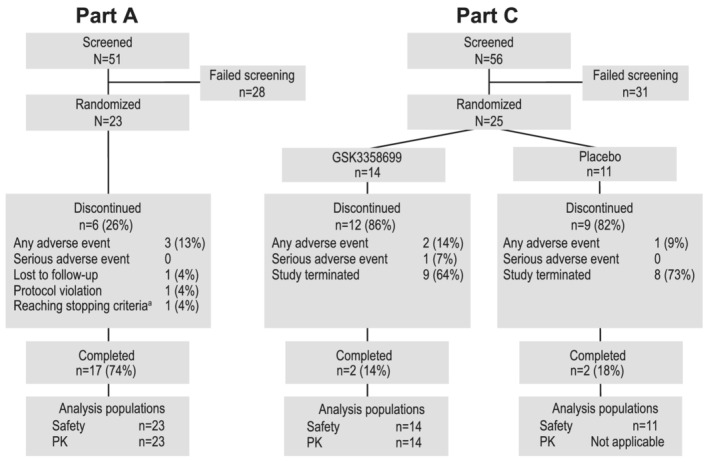
Participant disposition for parts A and C of the study. In part A, 23 participants were randomized and 17 completed the study. In part C, 25 patients were randomized and four completed the study (received all 14 doses). Reasons for discontinuation are shown in the diagram. PK, pharmacokinetics. aParticipant reached haematological stopping criteria‐low neutrophils
TABLE 1.
Demographics and baseline characteristics of study participants (safety population a )
| Demographic | Part A b total (N = 23) | Part C c | Part C total (N = 25) | |
|---|---|---|---|---|
| Placebo (N = 11) | GSK3358699 10 mg QD (N = 14) | |||
| Age, mean (range), y | 31 (18‐45) | 39 (29‐51) | 39 (20‐55) | 39 (20‐55) |
| Male, n (%) | 23 (100) | 11 (100) | 14 (100) | 25 (100) |
| BMI, mean (SD), kg/m2 | 25 (3) | 25 (3) d | 25 (3) | 25 (3) |
| Height, mean (SD), cm | 176 (6) | 181 (9) d | 175 (6) | 177 (8) |
| Weight, mean (SD), kg | 79 (12) | 81 (14) d | 77 (14) | 79 (14) |
| Ethnicity, n (%) | ||||
| Hispanic or Latino | 2 (9) | 3 (27) | 0 | 3 (12) |
| Not Hispanic or Latino | 21 (91) | 8 (73) | 14 (100) | 22 (88) |
| Race, n (%) | ||||
| Black or African American | 1 (4) | 1 (9) | 0 | 1 (4) |
| White | 21 (91) | 10 (91) | 14 (100) | 24 (96) |
| Multiple | 1 (4) | 0 | 0 | 0 |
Abbreviations: BMI, body mass index; QD, once daily; SD, standard deviation.
The safety population consisted of all randomized participants who received at least one dose of study treatment.
Part A was a single ascending‐dose crossover study in two interlocking cohorts. Each participant received a maximum of two single ascending oral doses of GSK3358699 (1, 3, 10, 20, 40 or 30 mg) and one dose of placebo.
In part C, participants received 10 mg of GSK3358699 or placebo daily for up to 14 days.
n = 10 for these parameters.
3.2. Safety and adverse events
In part A of the study, 21 participants experienced a total of 67 AEs (Table 2). All the AEs were considered mild or moderate in intensity and resolved by the end of the study. Headache was the most common treatment‐emergent AE after placebo administration (n = 2, 9%) or any dose of GSK3358699 (17‐33%). Treatment‐related AEs and AEs leading to withdrawal are summarized in Table 2. No deaths or serious AEs (SAEs) occurred in part A.
TABLE 2.
Adverse events reported in part A and part C of the study (safety population a )
| PART A b | ||||||||
|---|---|---|---|---|---|---|---|---|
| Adverse event, n (%) c | Placebo (N = 23) | GSK3358699 | ||||||
| 1 mg (n = 5) | 3 mg (n = 3) | 10 mg (n = 6) | 20 mg (n = 6) | 25 mg (n = 12) | 30 mg (n = 6) | 40 mg (n = 6) | ||
| Any AE | 4 (17) | 2 (40) | 3 (100) | 2 (33) | 3 (50) | 8 (67) | 4 (67) | 3 (50) |
| Any nonserious treatment‐related AE | 1 (4) | 0 | 1 (33) | 0 | 0 | 0 | 0 | 0 |
| Diarrhoea | 0 | 0 | 1 (33) | 0 | 0 | 0 | 0 | 0 |
| Nausea | 0 | 0 | 1 (33) | 0 | 0 | 0 | 0 | 0 |
| Transaminase increased | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| AEs leading to withdrawal | 0 | 1 (20) | 0 | 1 (17) | 0 | 0 | 0 | 0 |
| Neutropenia | 0 | 1 (20) | 0 | 0 | 0 | 0 | 0 | 0 |
| Ventricular tachycardia (nonsustained) | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 0 |
| PART C d | ||
|---|---|---|
| Adverse event, n (%) c | Placebo (N = 11) | GSK3358699 10 mg QD (N = 14) |
| Any AE | 7 (64) | 7 (50) |
| Any nonserious treatment‐related AE | 3 (27) | 3 (21) |
| Tachycardia | 1 (9) | 1 (7) |
| Atrial fibrillation | 1 (9) | 0 |
| Palpitations | 1 (9) | 0 |
| Ventricular extrasystoles | 0 | 1 (7) |
| Ventricular tachycardia (nonsustained) | 0 | 1 (7) |
| Serious AE | 0 | 1 (7) |
| Atrial fibrillation | 0 | 1 (7) |
| AEs leading to withdrawal | 1 (9) | 3 (21) |
| Atrial fibrillation | 0 | 1 (7) |
| Tachycardia | 0 | 1 (7) |
| Ventricular tachycardia (nonsustained) | 0 | 1 (7) |
| Keratitis | 1 (9) | 0 |
Abbreviation: AE, adverse event.
The safety population consisted of all randomized participants who received at least one dose of study treatment.
Part A was a single ascending‐dose crossover study in two interlocking cohorts. Each participant received a maximum of two single ascending oral doses of GSK3358699 (1, 3, 10, 20, 40 or 30 mg) and one dose of placebo.
The data presented represent the number of participants who experienced an adverse event.
In part C, participants received 10 mg of GSK3358699 or placebo daily for up to 14 days.
In part C of the study, 15 participants experienced a total of 23 AEs (Table 2). All the AEs were considered mild or moderate in intensity and resolved by the end of the study. Headache was the most common treatment‐emergent AE after placebo administration (n = 2, 18%); headache (n = 2, 14%) and NSVT (n = 2, 14%) were the most common AEs after GSK3358699 administration. Six participants had nonserious AEs that were considered treatment‐related (Table 2): one event each of tachycardia, atrial fibrillation (AF) and palpitations occurring in participants receiving placebo and one event each of tachycardia, ventricular extrasystoles and NSVT in participants receiving GSK3358699. One participant receiving GSK3358699 had experienced an SAE of AF (discussed further below), which was considered treatment‐related and led to study withdrawal (Table 2). Three other participants experienced AEs that led to study withdrawal: an AE of keratitis in a participant receiving placebo and one event each of tachycardia and NSVT in participants receiving GSK3358699. There were no deaths.
Three clinical chemistry values in part A, all following placebo administration, were of potential clinical importance. This included an increase in transaminases, which was also reported as an AE. Five haematology values in part A (two placebo, three GSK3358699) and seven in part C (six placebo, one GSK3358699) were also of potential clinical relevance. The SAE of AF in part C was the only clinically significant ECG abnormality reported in the study. Two clinically significant abnormalities in telemetry were reported in part C, one on day 1 and one on day 8, in participants receiving GSK3358699, which were also reported as AEs. No increases in QTcF above 450 ms or above 30 ms from baseline were observed in either part A or part C. There were no other clinically significant abnormalities in clinical chemistry evaluations, ECGs, telemetry or vital signs in the study.
As reported above, several cardiac AEs occurred during the study that were considered clinically significant (Table 3). Five cardiac AEs occurred in four out of 23 participants in part A of the study. All five events occurred after treatment with GSK3358699, but no clear dose relationship was apparent. Nine cardiac AEs occurred in nine participants out of 25 in part C of the study (6/11 in the GSK3358699 group and 3/14 in the placebo group). Two events of asymptomatic broad complex tachycardia (NSVT) occurred in cohort 4: one in a participant who had received one dose of 10 mg of GSK3358699 and one in a participant who had received eight doses of 10 mg of GSK3358699 (Supporting Information Table S1). These events, together with the two asymptomatic broad complex tachycardia (NSVT) events that occurred after GSK3358699 treatment in part A of the study, led to a temporary halt of cohort 4. All participants remained clinically well during these events. The majority of participants with cardiac events were referred to a local cardiologist for further investigation (laboratory tests, cardiograms, treadmill stress test and cardiac MRI) and discharged without any clinically significant findings of damage to the heart muscle. After review by an independent panel of internal and external cardiac specialists, the incidence of NSVT was determined to be in line with the expected background of NSVT observed in an otherwise healthy population and the four events were determined to be unrelated to treatment. The study was subsequently restarted with cohort 5 at the 10 mg of GSK3358699 dose.
TABLE 3.
Clinically significant adverse events reported in parts A and C of the study (safety population a )
| PART A b | ||||||||
|---|---|---|---|---|---|---|---|---|
| Adverse event, n (%) c | Placebo (N = 23) | GSK3358699 | ||||||
| 1 mg (n = 5) | 3 mg (n = 3) | 10 mg (n = 6) | 20 mg (n = 6) | 25 mg (n = 12) | 30 mg (n = 6) | 40 mg (n = 6) | ||
| Any cardiac event | 0 | 0 | 1 (33) | 1 (17) | 0 | 1 (8) | 1 (17) | 0 |
| Ventricular tachycardia (nonsustained) | 0 | 0 | 0 | 1 (17) | 0 | 0 | 1 (17) | 0 |
| Palpitations | 0 | 0 | 1 (33) | 0 | 0 | 0 | 0 | 0 |
| Sinus tachycardia | 0 | 0 | 0 | 0 | 0 | 1 (8) | 0 | 0 |
| PART C d | ||
|---|---|---|
| Adverse event, n (%) c | Placebo (N = 11) | GSK3358699 10 mg QD (N = 14) |
| Any cardiac event | 3 (27) | 6 (43) |
| Tachycardia | 1 (9) | 2 (14) |
| Atrial fibrillation | 1 (9) | 1 (7) |
| Atrial tachycardia | 0 | 1 (7) |
| Palpitations | 1 (9) | 0 |
| Ventricular extrasystoles | 0 | 1 (7) |
| Ventricular tachycardia (nonsustained) | 0 | 1 (7) |
Abbreviation: QD, once daily.
The safety population consisted of all randomized participants who received at least one dose of study treatment.
Part A was a single ascending‐dose crossover study in two interlocking cohorts. Each participant received a maximum of two single ascending oral doses of GSK3358699 (1, 3, 10, 20, 40 or 30 mg) and one dose of placebo.
The data presented represent the number of participants who experienced an adverse event.
In part C, participants received 10 mg of GSK3358699 or placebo daily for up to 14 days.
As illustrated in Tables 2 and 3, there was a total of two episodes of AF reported in part C of the study: one participant receiving placebo and one participant in the GSK3358699 group. The participant from the placebo group was a 48‐year‐old male with a body mass index (BMI) of 30.7 kg/m2. The episode of nonsustained AF, lasting for 8 beats, was observed during a period of extended telemetry following the AF observed in the participant discussed above. The AF occurred in the evening approximately 60 hours following the participant's last dose, lasting for approximately 5 seconds before spontaneously reverting to normal sinus rhythm. The participant was asymptomatic throughout and needed no intervention; blood tests, including troponin I, Ca2+, Mg2+, urea and electrolytes (U&Es), liver function test (LFT) and thyroid function test (TFT) were normal. The participant from the GSK3358699 group was a 56‐year‐old male with a BMI of 28.3 and had received a total of 60 mg of GSK3358699. They had no significant medical history and previously participated in five clinical trials at the same unit without event. The episode of sustained AF was observed pre‐dose, during the treatment period, in the morning on day 7 of treatment, 23 hours following the participant's preceding dose on day 6. The participant was asymptomatic throughout and, on examination, had an irregular pulse. The participant was transferred to the emergency department and received a single DC shock to restore sinus rhythm. Telemetry was extended for 48 hours with no further AF or significant cardiac arrythmia being detected; blood tests including troponin I, Ca2+, Mg2+, U&Es, LFT and TFT were normal. Following 24‐hour Holter monitoring, the participant was discharged and remained healthy on follow‐up.
Because the episode of AF in the participant treated with GSK3358699 required the participant to be hospitalized, it was considered an SAE and fulfilled the protocol‐stopping criteria. The study was therefore halted during cohort 5. After a review of the SAE and the other cardiac‐related AEs by the study sponsor's cardiology safety panel and safety governance boards, the decision was made to terminate the study.
3.3. Pharmacokinetics
All of the participants in part A received at least one single dose of GSK3358699. The median plasma concentration profiles over time for GSK3358699 and the metabolite GSK3206944 in part A are shown in Figure 3. After a single dose of up to 20 mg of GSK3358699, plasma concentrations of GSK3358699 were quantifiable up to 12 hours post‐dose. At doses of 25 mg and higher, plasma concentrations of GSK3358699 were quantifiable up to 24 hours post‐dose. Plasma concentrations of the metabolite GSK3206944 were quantifiable up to 12 hours post‐dose in all participants at all doses. At doses of 25 mg and higher, plasma concentrations of GSK3206944 were quantifiable in all participants up to 24 hours post‐dose and in the majority of participants at 48 hours post‐dose.
FIGURE 3.
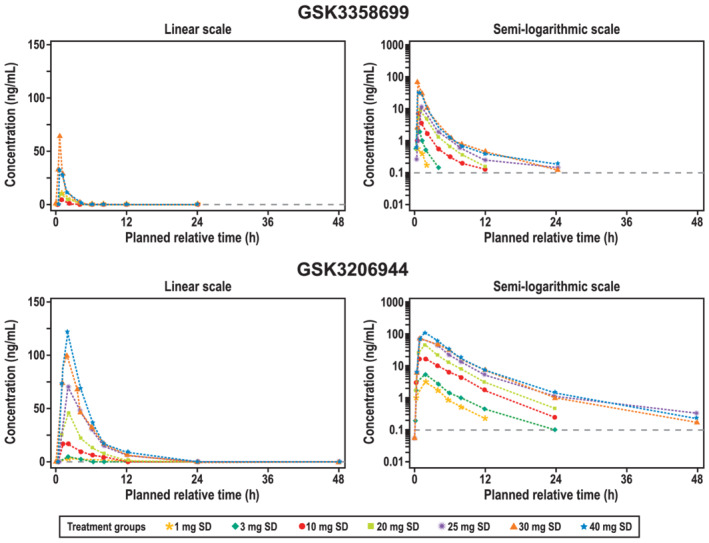
Median plasma concentration over time in part Aa of the study (pharmacokinetic populationb). Median plasma concentrations of GSK3358699 (top) and the metabolite GSK3206944 (bottom) over 48 hours following treatment with a single dose of GSK3358699 (1, 3, 10, 20, 30 or 40 mg) are shown on a linear (left) and semilogarithmic scale (right). SD, single dose. aPart A was a single ascending‐dose crossover study in two interlocking cohorts. Each participant received a maximum of two single ascending oral doses of GSK3358699 (1, 3, 10, 20, 40 or 30 mg) and one dose of placebo. bThe pharmacokinetic population included all participants who received an active dose of the study drug and for whom pharmacokinetic samples were obtained and analysed
In general, the exposure to GSK3358699 and GSK3206944 increased over the range of single GSK3358699 doses in part A (Table 4). Across the doses of 1 to 40 mg of GSK3358699, the AUC(0‐t) and AUC(0‐∞) increased in a greater than dose‐proportional manner, with the lower bound of the 90% CI for the slope estimate being greater than 1 (Table 5). The C max increased in a dose‐proportional manner and exceeded the pharmacokinetic‐stopping criteria at the 40 mg of GSK3358699 dose, which led to dose de‐escalation to 30 mg. The pharmacokinetic‐stopping criteria was also met at this dose level. The geometric mean of the exposures achieved following the 40 mg dose of GSK3358699 were lower than the geometric mean at the 30 mg dose (Table 4). The ranges of t max for GSK3358699 and GSK3206944 was 0.5‐1.0 and 1.0‐2.0 hours, respectively, after a single dose of 1‐40 mg of GSK3358699. The median t 1/2 (initial) for GSK3358699 and GSK3206944 ranged from 0.7 to 1.1 hours and from 2.1 to 2.9 hours, respectively, after a single dose of 1‐40 mg of GSK3358699. The median t 1/2 (terminal) increased with increasing dosage for both GSK3358699 and GSK3206944 (Table 4). At doses of 25 mg of GSK3358699 and higher, the t 1/2 (terminal) for GSK3358699 and GSK3206944 ranged from 4.8 to 6.9 hours and from 6.8 to 7.8 hours, respectively.
TABLE 4.
Summary of selected pharmacokinetic parameters in part A a of the study (pharmacokinetic population b )
| PK parameter | Treatment | n/N | GSK3358699 | GSK3206944 (metabolite) | ||
|---|---|---|---|---|---|---|
| Geometric mean (%CVb) | 95% CI | Geometric mean (%CVb) | 95% CI | |||
| AUC(0‐t), h*ng/mL | 1 mg SD | 5/5 | 0.8 (38.5) | (0.5, 1.3) | 15.1 (34.0) | (10.0, 22.8) |
| 3 mg SD | 3/3 | 2.2 (23.3) | (1.3, 4.0) | 28.4 (12.0) | (21.1, 38.3) | |
| 10 mg SD | 6/6 | 10.1 (44.0) | (6.5, 15.7) | 101.2 (27.4) | (76.3, 134.1) | |
| 20 mg SD | 6/6 | 23.0 (20.4) | (18.6, 28.4) | 221.9 (29.9) | (163.3, 301.6) | |
| 25 mg SD | 12/12 | 33.9 (22.5) | (29.4, 39.0) | 367.1 (13.9) | (336.2, 401.0) | |
| 30 mg SD | 6/6 | 76.7 (38.6) | (51.9, 113.4) | 494.7 (21.9) | (394.2, 620.9) | |
| 40 mg SD | 6/6 | 63.7 (30.8) | (46.5, 87.3) | 625.8 (32.7) | (448.0, 874.2) | |
| AUC(0‐∞), h*ng/mL | 1 mg SD | 0/5 | … | … | 17.1 (−) | … |
| 3 mg SD | 0/3 | … | … | 28.9 (−) | … | |
| 10 mg SD | 5/6 | 11.0 (45.7) | (6.4, 18.8) | 103.4 (25.8) | (79.3, 135.0) | |
| 20 mg SD | 6/6 | 23.5 (19.7) | (19.2, 28.9) | 192.6 (11.6) | (160.1, 231.6) | |
| 25 mg SD | 9/12 | 36.9 (23.2) | (31.0, 44.0) | 356.0 (13.7) | (317.8, 398.8) | |
| 30 mg SD | 4/6 | 76.9 (37.0) | (43.5, 135.9) | 511.8 (27.3) | (334.1, 784.1) | |
| 40 mg SD | 5/6 | 67.0 (33.1) | (44.9, 100.0) | 636.1 (31.9) | (458.9, 881.7) | |
| AUC(0‐24), h*ng/mL | 1 mg SD | 0/5 | … | … | 16.4 (−) | … |
| 3 mg SD | 0/3 | … | … | 28.1 (−) | … | |
| 10 mg SD | 5/6 | 11.0 (45.7) | (6.4, 18.8) | 98.8 (31.0) | (71.9, 135.9) | |
| 20 mg SD | 6/6 | 23.5 (19.7) | (19.1, 28.8) | 219.8 (28.8) | (163.4, 295.7) | |
| 25 mg SD | 10/12 | 34.5 (24.8) | (29.0, 41.0) | 348.7 (14.7) | (317.9, 382.6) | |
| 30 mg SD | 5/6 | 71.5 (34.8) | (47.0, 108.9) | 479.4 (22.8) | (378.5, 607.1) | |
| 40 mg SD | 6/6 | 64.1 (30.7) | (46.8, 87.8) | 599.5 (35.0) | (419.7, 856.5) | |
| C max, ng/mL | 1 mg SD | 5/5 | 0.9 (69.8) | (0.4, 1.9) | 3.1 (33.2) | (2.1, 4.7) |
| 3 mg SD | 3/3 | 1.9 (26.3) | (1.0, 3.6) | 6.0 (20.5) | (3.6, 9.9) | |
| 10 mg SD | 6/6 | 6.5 (48.2) | (4.0, 10.4) | 17.8 (49.9) | (10.8, 29.1) | |
| 20 mg SD | 6/6 | 15.0 (45.9) | (9.5, 23.7) | 48.5 (41.6) | (31.9, 73.7) | |
| 25 mg SD | 12/12 | 16.2 (39.6) | (12.7, 20.7) | 67.9 (31.3) | (55.9, 82.4) | |
| 30 mg SD | 6/6 | 49.8 (83.9) | (23.2, 107.2) | 106.7 (52.3) | (63.7, 178.7) | |
| 40 mg SD | 6/6 | 42.3 (98.5) | (17.8, 100.4) | 133.4 (42.6) | (86.9, 204.7) | |
| t max, h* | 1 mg SD | 5/5 | 0.5 (0.3, 1.0) | … | 1.0 (1.0, 2.0) | … |
| 3 mg SD | 3/3 | 0.5 (0.5, 1.0) | … | 2.0 (1.0, 2.0) | … | |
| 10 mg SD | 6/6 | 0.5 (0.5, 0.5) | … | 2.0 (1.0, 2.0) | … | |
| 20 mg SD | 6/6 | 1.0 (0.3, 1.0) | … | 2.0 (1.0, 2.1) | … | |
| 25 mg SD | 12/12 | 1.0 (0.2, 2.0) | … | 2.0 (2.0, 4.0) | … | |
| 30 mg SD | 6/6 | 0.8 (0.5, 1.0) | … | 2.0 (1.0, 2.0) | … | |
| 40 mg SD | 6/6 | 0.5 (0.5, 1.1) | … | 2.0 (1.0, 2.0) | … | |
| t 1/2 (terminal), h* | 1 mg SD | 0/5 | … | … | 5.0 (2.8, 7.1) | … |
| 3 mg SD | 0/3 | … | … | 4.9 (4.8, 5.0) | … | |
| 10 mg SD | 5/6 | 2.4 (2.0, 2.8) | … | 4.1 (3.0, 14.5) | … | |
| 20 mg SD | 6/6 | 2.8 (2.0, 3.4) | … | 3.8 (3.5, 4.3) | … | |
| 25 mg SD | 9/12 | 6.0 (2.1, 18.4) | … | 7.8 (3.8, 11.6) | … | |
| 30 mg SD | 4/6 | 4.8 (3.5, 11.8) | … | 7.0 (4.1, 8.8) | … | |
| 40 mg SD | 5/6 | 6.9 (2.1, 11.1) | … | 6.8 (3.0, 15.2) | … | |
| t 1/2 (initial), h* | 1 mg SD | 4/5 | 0.7 (0.6, 0.8) | … | 2.9 (2.7, 3.3) | … |
| 3 mg SD | 3/3 | 1.0 (0.5, 1.1) | … | 2.3 (2.2, 2.5) | … | |
| 10 mg SD | 6/6 | 0.7 (0.6, 1.6) | … | 2.5 (2.1, 3.2) | … | |
| 20 mg SD | 4/6 | 1.0 (0.7, 1.2) | … | 2.4 (1.8, 3.2) | … | |
| 25 mg SD | 11/12 | 1.1 (0.8, 2.8) | … | 2.2 (1.7, 3.9) | … | |
| 30 mg SD | 4/6 | 0.9 (0.8, 1.1) | … | 2.2 (1.9, 3.1) | … | |
| 40 mg SD | 5/6 | 1.0 (0.8, 1.2) | ‐ | 2.1 (1.9, 2.5) | ‐ | |
Abbreviations: AUC(0‐t), area under the concentration‐time curve from time zero to the time of the last quantifiable concentration; AUC(0‐∞), area under the concentration‐time curve extrapolated to infinity; AUC(0‐24), area under the concentration‐time curve from time zero to 24 hours post‐dose; C max, maximum observed concentration; CI, confidence interval; CVb, coefficient of variation; PK, pharmacokinetic; T max, time to reach C max; t 1/2 (terminal), apparent terminal half‐life; t 1/2 (initial), actual initial half‐life.
Part A was a single ascending‐dose crossover study in two interlocking cohorts. Each participant received a maximum of two single ascending oral doses of GSK3358699 (1, 3, 10, 20, 40 or 30 mg) and one dose of placebo.
The pharmacokinetic population included all participants who received an active dose of the study drug and for whom pharmacokinetic samples were obtained and analysed.
TABLE 5.
Summary of GSK3358699 dose proportionality model in part A a of the study (pharmacokinetic population b )
| PK parameter | Slope (SE) | 90% CI for slope | Fold increase for doubling dose | 90% CI for fold increase |
|---|---|---|---|---|
| AUC(0‐t), h*ng/mL | 1.2 (0.05) | (1.2, 1.3) | 2.4 | (2.2, 2.5) |
| AUC(0‐∞), h*ng/mL | 1.5 (0.1) | (1.2, 1.7) | 2.7 | (2.3, 3.3) |
| C max, ng/mL | 1.1 (0.1) | (0.9, 1.2) | 2.1 | (1.9, 2.3) |
AUC(0‐t), area under the concentration‐time curve from time zero to the time of the last quantifiable concentration; AUC(0‐∞), area under the concentration‐time curve extrapolated to infinity; CI, confidence interval; C max, maximum observed concentration; PK, pharmacokinetic; SE, standard error.
Part A was a single ascending‐dose crossover study in two interlocking cohorts. Each participant received a maximum of two single ascending oral doses of GSK3358699 (1, 3, 10, 20, 40 or 30 mg) and one dose of placebo.
The pharmacokinetic population included all participants who received an active dose of the study drug and for whom pharmacokinetic samples were obtained and analysed.
All 14 of the participants randomized to 10 mg of GSK3358699 in part C received at least one dose. Because cohorts 4 and 5 were halted, a full pharmacokinetic profile was only available for two of the 14 participants in part C. The median plasma concentration profiles over time for GSK3358699 and the metabolite GSK3206944 in part C are shown in Figure 4. The GSK3358699 C max, AUC(0‐tau) and AUC(0‐∞) after the first dose of 10 mg of GSK3358699 were comparable to the corresponding values in part A. The median t max values for GSK3358699 and GSK3206944 following the first dose were 0.5 and 2.0 hours, respectively (Table 6). The median t 1/2 (initial) values for GSK3358699 and GSK3206944 were 1.0 and 2.4 hours, respectively, and the median t 1/2 (terminal) values were 2.5 and 5.0 hours (Table 6).
FIGURE 4.
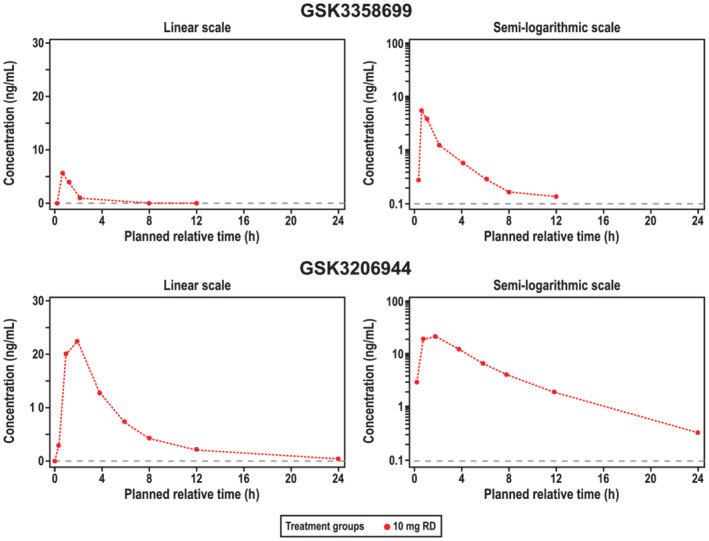
Median plasma concentration over time on day 1 of part C of the study (pharmacokinetic populationa). Median plasma concentrations of GSK3358699 (top) and the metabolite GSK3206944 (bottom) over 48 hours following treatment with 10 mg of GSK3358699 are shown on a linear (left) and semilogarithmic scale (right). As only two participants had a PK profile for day 14, only a median concentration‐time profile for day 1 is presented. RD, repeat dose. aThe pharmacokinetic population included all participants who received an active dose of the study drug and for whom pharmacokinetic samples were obtained and analysed
TABLE 6.
Summary of select pharmacokinetic parameters in part C a of the study (pharmacokinetic population b )
| PK parameter | Day | n/N | GSK3358699 | GSK3206944 (metabolite) | ||
|---|---|---|---|---|---|---|
| Geometric mean (%CVb) | 95% CI | Geometric mean (%CVb) | 95% CI | |||
| AUC(0‐t), h*ng/mL | 1 | 14/14 | 9.5 (43.4) | (7.4, 12.0) | 125.2 (42.3) | (99.0, 158.2) |
| 14 | 2/14 | 11.0 (−) | … | 108.7 (−) | … | |
| AUC(0‐∞), h*ng/mL | 1 | 7/14 | 9.8 (35.1) | (7.2, 13.4) | 128.4 (41.6) | (102.0, 161.8) |
| 14 | 0/14 | … | … | … | … | |
| AUC(0‐24), h*ng/mL | 1 | 7/14 | 9.8 (35.1) | (7.1, 13.4) | 125.4 (42.3) | (99.2, 158.4) |
| 14 | 1/14 | 8.0 (−) | … | 105.6 (−) | … | |
| AUC(0‐tau), h*ng/mL | 1 | 7/14 | 9.8 (35.1) | (7.1, 13.4) | 125.4 (42.3) | (99.2, 158.4) |
| 14 | 1/14 | 8.0 (−) | … | 105.6 (−) | … | |
| C max, ng/mL | 1 | 14/14 | 6.1 (54.0) | (4.6, 8.2) | 23.8 (44.2) | (18.7, 30.4) |
| 14 | 2/14 | 5.9 (−) | … | 15.3 (−) | … | |
| T max, h* | 1 | 14/14 | 0.5 (0.3, 2.0) | … | 2.0 (1.0, 2.0) | … |
| 14 | 2/14 | 0.5 (0.5, 0.6) | … | 1.0 (1.0, 1.0) | … | |
| t 1/2 (terminal), h* | 1 | 7/14 | 2.5 (1.4, 3.0) | … | 5.0 (3.3, 5.6) | … |
| 14 | 1/14 | 1.6 (−) | … | 6.2 (5.5, 6.8) | … | |
| t 1/2 (initial), h* | 1 | 13/14 | 1.0 (0.6, 1.5) | … | 2.4 (1.7, 4.0) | … |
| 14 | 2/14 | 0.8 (0.7, 0.8) | … | 3.7 (2.8, 4.5) | … | |
| R 0 | 14 | 1/14 | 0.6 (−) | … | 1.3 (−) | … |
| R S | 14 | 1/14 | 0.6 (−) | … | 1.2 (−) | … |
AUC(0‐t), area under the concentration‐time curve from time zero to the time of the last quantifiable concentration; AUC(0‐∞), area under the concentration‐time curve extrapolated to infinity; AUC(0‐24), area under the concentration‐time curve from time zero to 24 hours post‐dose; AUC(0‐tau), area under the concentration‐time curve from time zero to the end of the dosing period; CI, confidence interval; C max, maximum observed concentration; CVb, coefficient of variation; N, total number of participants; n, number of participants with nonmissing observations. PK, pharmacokinetic; R 0, accumulation between one single dose (day 1) and repeat dose (day 14); R S, steady state ratio; T max, time to reach C max; t 1/2 (terminal), apparent terminal half‐life; t 1/2 (initial), actual initial half‐life.
In part C, participants received 10 mg of GSK3358699 or placebo daily for up to 14 days.
The pharmacokinetic population included all participants who received an active dose of the study drug and for whom pharmacokinetic samples were obtained and analysed.
N, total number of participants; n, number of participants with nonmissing observations.
3.4. Intracellular GSK3206944 concentrations
The median monocytic intracellular GSK3206944 concentrations over time for part A are shown in Figure 5. For doses of GSK3358699 above 10 mg, intracellular GSK3206944 concentrations were quantifiable up to 4 hours post‐dose and for the 10 mg dose were quantifiable at 1 hour post‐dose only. Concentrations were not quantifiable for doses of 1 and 3 mg. None of the doses resulted in quantifiable concentrations at 8 hours or beyond. There was no evidence for enhanced intracellular GSK3206944 concentrations on repeat dosing at the 10 mg dose investigated (Figure 5).
FIGURE 5.
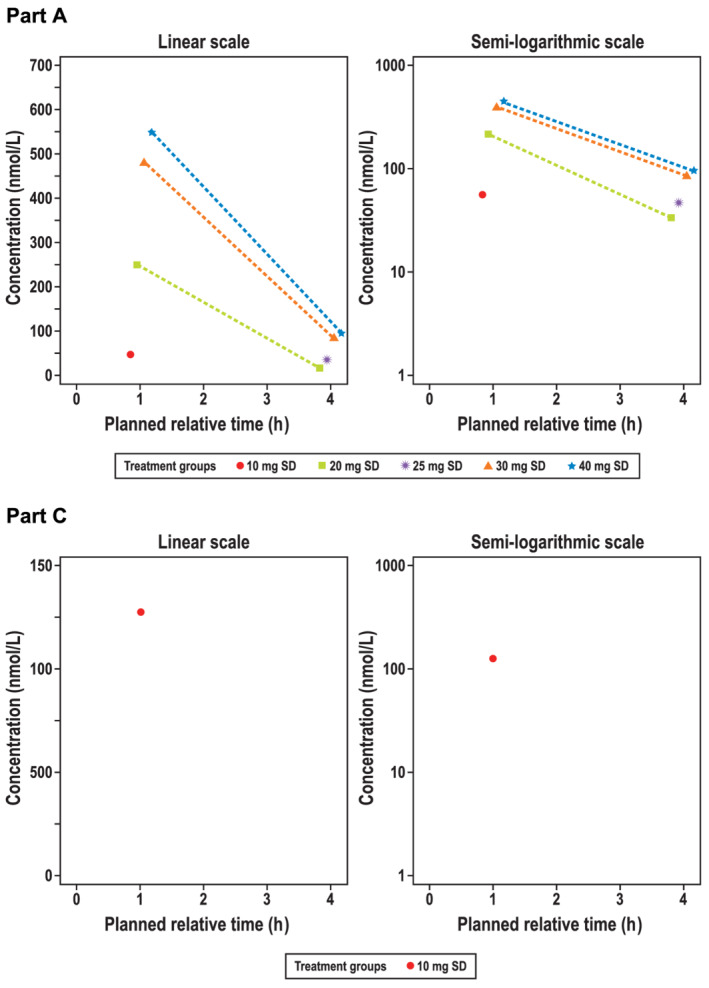
Median GSK3206944 intracellular molar concentration over time (linear and semilog) in (A) part Aa and (B) part Cb, day 1 (pharmacokinetic populationc). The median intracellular molar concentration of the metabolite GSK3206944 in monocytes over 4 hours in participants in part A treated with a single dose of GSK3358699 (10, 20, 35, 30 and 40 mg) are shown on a linear (top left) and semilogarithmic scale (top right). Concentrations were not quantifiable for single doses of 1 and 3 mg in part A. There was no evidence for enhanced intracellular GSK3206944 concentrations on repeat dosing at the 10 mg dose investigated in part C (bottom). SD, single dose. aPart A was a single ascending‐dose crossover study in two interlocking cohorts. Each participant received a maximum of two single ascending oral doses of GSK3358699 (1, 3, 10, 20, 40 or 30 mg) and one dose of placebo. bIn part C, participants received 10 mg of GSK3358699 or placebo daily for up to 14 days. cThe pharmacokinetic population included all participants who received an active dose of the study drug and for whom pharmacokinetic samples were obtained and analysed
3.5. Pharmacodynamics
The mean concentrations of MCP‐1 measured following ex vivo LPS stimulation of whole blood indicated dose‐dependent inhibition by GSK3358699 in part A (Table 7). The mean predicted percentage inhibition of MCP‐1 at approximately 1 hour post‐dose was 11% with placebo and 9%, 27%, 18%, 19%, 42% and 75% at GSK3358699 doses of 1, 3, 10, 20, 30 and 40 mg, respectively (Figure 6 and Table 7). The inhibition was no longer evident by 4 hours post‐dose. There was no clear impact of GSK3358699 on IL‐6 and TNF production at any dose in part A (Table 7). Large inter‐ and intracohort variability was observed for concentrations of MCP‐1, IL‐6 and TNF. Cytokine levels in the “null” samples were very low, indicating that there were no pre‐existing cytokines in the blood or tube contaminants that contributed to the large variability.
TABLE 7.
Mean predicted inhibition (%) of analytes 1 hour post‐dose in part A a of the study (safety population b )
| Analyte | Placebo (N = 23) mean % (SE) | 1 mg (N = 5) mean % (SE) | 3 mg (N = 3) mean % (SE) | 10 mg (N = 6) mean % (SE) | 20 mg (N = 6) mean % (SE) | 30 mg (N = 6) mean % (SE) | 40 mg (N = 6) mean % (SE) |
|---|---|---|---|---|---|---|---|
| MCP‐1 | 11 (±10) | 9 (±14) | 27 (−) | 18 (±9) | 19 (±10) | 42 (±12) | 75 (±13) |
| IL‐6 | 10 (±6) | 13 (±6) | 14 (±8) | −1 (±7) | 16 (±13) | 14 (±8) | 5 (±6) |
| TNF | 15 (±9) | 34 (±17) | 15 (±17) | −1 (±11) | 47 (±24) | 36 (±13) | 34 (±18) |
Abbreviations: IL‐6, interleukin‐6; MCP‐1, monocyte chemoattractant protein‐1; SE, standard error; TNF, tumour necrosis factor.
Part A was a single ascending‐dose crossover study in two interlocking cohorts. Each participant received a maximum of two single ascending oral doses of GSK3358699 (1, 3, 10, 20, 40 or 30 mg) and one dose of placebo.
The safety population consisted of all randomized participants who received at least one dose of study treatment.
FIGURE 6.
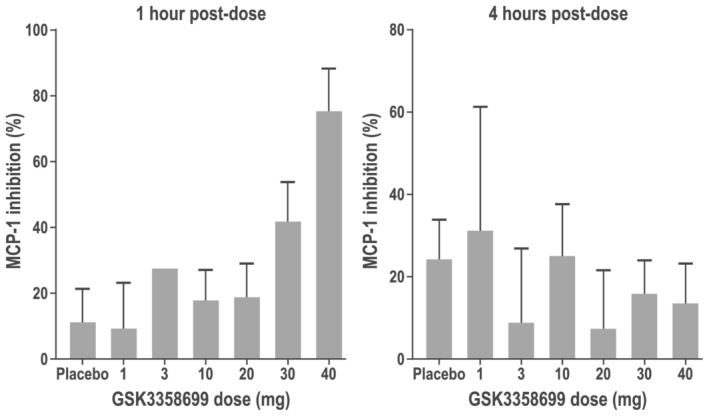
Mean predicted percentage inhibition of MCP‐1 in part Aa of the study (safety populationb). The mean predicted percentage inhibition of MCP‐1 at 1 hour post‐dose (left) and 4‐hours post dose (right) in participants in part A treated with single doses of GSK3358699 of 1, 3, 10, 20, 30 and 40 mg. aPart A was a single ascending‐dose crossover study in two interlocking cohorts. Each participant received a maximum of two single ascending oral doses of GSK3358699 (1, 3, 10, 20, 40 or 30 mg) and one dose of placebo. bThe safety population consisted of all randomized participants who received at least one dose of study treatment
In part C of the study, 10 mg of GSK3358699 did not appear to inhibit ex vivo LPS stimulated production of MCP‐1, IL‐6 or TNF (data not shown).
4. DISCUSSION
In this first‐in‐human study in healthy male volunteers, we examined the safety, tolerability, pharmacokinetic and pharmacodynamic profiles of GSK3358699, and the systemic and intracellular pharmacokinetic profiles of the metabolite GSK3206944. Single doses of GSK3358699 were generally well tolerated up to 40 mg and this targeted approach to BET inhibition largely mitigated the common dose‐limiting toxicities (ie, thrombocytopenia, gastrointestinal symptoms, QTc prolongation) observed preclinically and clinically with pan‐BET inhibitors. However, we observed a moderate increase in clinically significant cardiac AEs in some individuals, relative to placebo, with repeat dosing of GSK3358699.
GSK3358699 was systemically available following oral dosing, and the pharmacokinetic profile indicated that exposure to GSK3358699 and its metabolite GSK3206944 increases with increasing doses. GSK3358699 was designed to be a low exposure, short half‐life compound, but the AUC and half‐life of GSK3358699 were lower than expected based on preclinical data, indicating higher than predicted clearance. The increase in AUC for GSK3358699 was not dose‐proportional, possibly because of the lack of quantifiable plasma GSK3358699 at the lower doses at later time points. The GSK3358699 exposure at 30 mg was similar to that at 40 mg. However, it is difficult to conclude whether the 30 mg dose demonstrates unexpected behaviour or if the AUC was plateauing at 30 mg. The median initial GSK3358699 half‐life estimate was approximately 1 hour across all doses, resulting in limited GSK3358699 exposure.
Of note, intracellular concentrations of GSK3206944 were detectable in monocytes up to 4 hours post‐dose at doses of 20 mg of GSK3358699 and higher, despite GSK3358699 exposure levels that were lower than expected based on preclinical predictions. Our findings confirm that the esterase‐sensitive motif‐targeting approach generated the pharmacologically active acid metabolite in target cells, as intended. Based on the single dose data, it would have been more informative to study the degree of acid retention that could be achieved at doses greater than 20 mg, but this was precluded by early termination of part C of the study. Exposure to GSK3358699 and GSK3206944 after the first dose of 10 mg of GSK3358699 in part C of the study was comparable with the results from the corresponding dose in part A. Estimated accumulation of GSK3358699 and GSK3206944 at day 14 could not be accurately determined because data were limited to only two participants.
Previous in vitro experiments indicated that GSK3358699 potently inhibited MCP‐1, TNF and IL‐6 production in LPS‐stimulated human blood, and inhibition of LPS‐induced cytokine production in vitro has been demonstrated with other BET inhibitors. 3 , 9 , 10 , 11 Indeed, GSK3358699 demonstrated IC50 values of 7.4 nM, 9.3 nM and 18.6 nM for inhibition of MCP‐1, TNF and IL‐6, respectively, in standard preclinical human whole‐blood assays used routinely within our laboratories. Monocytes are one of the populations in whole blood that respond to LPS, and GSK3358699 is specifically hydrolysed to the acid metabolite GSK3206944 inside these cells, therefore we expected to see enhanced inhibition of cytokine production from monocytes. In addition, the systemic concentrations of GSK3358699 achieved throughout part A suggested significant inhibition of these pro‐inflammatory cytokines in participant blood stimulated ex vivo with LPS should be expected. To this end, participant blood was drawn directly into TruCulture tubes containing LPS at predetermined timepoints to establish a correlation between GSK3358699 blood concentration and the extent of MCP‐1, TNF and IL‐6 inhibition over time. For example, following a single 30‐mg dose of GSK3358699, a mean C max concentration of 49.8 ng/mL GSK3358699 was achieved (equivalent to a total blood concentration of 97.5 nM at C max). This concentration exceeds the human whole‐blood potencies measured in the preclinical whole‐blood assays discussed above. Unfortunately, as illustrated in Table 7, the measured percentage inhibition of TNF and IL‐6 at C max was highly variable and inconsistent with the observed preclinical data. This may in part be due to the variability in baseline cytokine values. Moreover, circadian effects on monocyte counts and activity are known to affect the absolute levels of LPS‐induced cytokine production in human whole blood. 12 Interestingly, as illustrated in Figure 6, MCP‐1 inhibition did show a level of dose dependency. However, based on the systemic concentrations achieved (Table 4), more significant inhibition of this sensitive cytokine might have been expected from the 20 mg and higher doses.
The pharmacodynamic profile of GSK3358699 following repeat dosing could not be adequately evaluated as part of this first‐in‐human study. The 10 mg dose explored during part C of the study was selected as an appropriate starting repeat dose specifically because it was not expected to drive significant levels of target engagement. Only two participants on active treatment (sentinels) were able to complete the full 14 days of dosing at this dose level prior to termination of the study. On this basis, there is insufficient data from which to conclude how GSK3358699 could have been dosed to drive pharmacodynamics and efficacy in a patient population.
The pharmacodynamic profile following single doses of GSK3358699 was difficult to interpret and is inconsistent with data from preclinical assays. The pharmacokinetic data shown in Table 4 and Figure 3 suggest that the desired profile was achieved for GSK3358699, and Figure 5 confirms exposure within target cells in this first‐in‐human study. However, the variability of the ex vivo whole‐blood LPS activation assay makes it difficult to form confident conclusions.
As mentioned previously, the study could not be completed as originally designed due in part to cardiac AEs that were observed during parts A and C of the study. Throughout this first‐in‐human study, participants were subject to extensive cardiovascular screening and monitoring, including 24‐hour Holter monitoring at screening and cardiac telemetry from 1 hour pre‐dose until 24 hours post‐dose. The observed cardiac AEs included an SAE of AF that required hospitalization, and four AEs of NSVT, which occurred at differing and irregular times of the day and night with no demonstrable relationship to dose, AUC or C max. No other clinically significant changes or trends in vital signs, ECGs, QTc intervals or safety laboratory tests were noted during the study.
The mechanism that may underpin these cardiac observations is, at the time of writing, unknown. However, a recent study showed cardiac muscle‐specific progressive destruction of mitochondria and a dose‐dependent decrease in respiration rate of heart mitochondria in mice treated with a BET inhibitor, highlighting a potential mechanism for cardiac toxicities. 13 Interestingly, BET inhibitors have also been proposed as a potential treatment for cardiovascular diseases, primarily driven through their potential to impact epigenetic reprogramming. 14 Further to this, selective BET inhibitors have been explored in large cardiovascular outcome trials. 15 Preclinically, GSK3358699 and GSK3206944 were extensively profiled, including across a suite of cardiac safety assays, and were shown to have low pro‐arrhythmic potential, consistent with our preclinical experience with other BET inhibitors.
NSVTs are a relatively common event in a healthy population and would be routinely identified in a study comprising extensive cardiac monitoring such as this one. New‐onset AF is not unusual in older age groups, and both cases reported as part of this study could be considered as arrhythmias commonly observed in clinical practice. However, the case of new‐onset sustained AF in the treatment group was unexpected and required hospitalization for a cardioversion procedure because a return to sinus rhythm did not occur spontaneously. There was also no clear precipitant of the AF and the participant had no risk factors, other than age. In addition, a series of 3‐month good laboratory practice (GLP) toxicology studies conducted in parallel to this first‐in‐human study in cynomolgus monkeys demonstrated troponin elevations associated with cardiac histopathology; similar findings were not observed in 1‐month toxicology studies. Taken together with the clinical event of sustained AF, a causal relationship could not be excluded.
The preclinical observations and episodes of NSVT and AF observed in part C led to redefined safety margins, limiting subsequent clinical evaluation to repeat doses of up to 5 mg, reducing confidence in the ability to fully interrogate the mechanism given the absence of a robust pharmacodynamic response from part A. Furthermore, patient populations for the planned clinical indications (rheumatoid arthritis and psoriasis) for GSK3358699 have increased cardiovascular risks, therefore alternative indications were considered based on a thorough benefit/risk assessment. An exhaustive review of the available clinical and preclinical data ultimately determined that GSK3358699 could not be developed within an acceptable therapeutic window.
In conclusion, GSK3358699 is systemically available following oral dosing, has a predictable pharmacokinetic profile and single doses are generally well tolerated up to 40 mg. Significant concentrations of the metabolite GSK3206944 can be detected in target cells up to 4 hours after dosing of GSK3358699, confirming the ESM‐targeting technology was effective. The totality of the data generated through part A and part C of this study, in combination with emerging preclinical data, led to a reassessment of the benefit and risk of GSK3358699 and termination of the study during part C.
CONFLICT OF INTEREST
J.A.B., J.B., M.S., P.K.M., P.E.S., W.A.F., G.K.W., J.C.B., D.F., H.C., S.H., B.D., J.J.O. and R.K.P. are employees of and hold stock in GlaxoSmithKline (GSK). E.J.B‐H. is an NHS employee and part‐time employee for GSK. P.W. is a complementary worker with GSK. S.D., L.E. and Y.C. are former employees of GSK, and Y.C. also holds stock in GSK.
CONTRIBUTORS
J.A.B., J.B., M.S., P.K.M., P.E.S., S.D., W.A.F., J.C.B., L.E., Y.C., D.F., E.J.B‐H., S.H., B.D. and R.K.P. made substantial contributions to conception and design. D.F., H.C., E.J.B‐H. and S.H. were involved in the acquisition of data. J.A.B., J.B., M.S., P.W., P.K.M., P.E.S., S.D., W.A.F., G.K.W., J.C.B., Y.C., D.F., H.C., B.D., J.J.O. and R.K.P. contributed to the analysis and interpretation of data. All authors reviewed and revised the manuscript critically for important intellectual content. All authors have given final approval of the version to be published and take public responsibility for the content. All agree to be accountable for all aspects of the work, ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Supporting information
Supporting Information Table S1 Summary of nonsustained ventricular tachycardia events that occurred during the study
ACKNOWLEDGMENTS
All listed authors meet the criteria for authorship set out by the International Committee for Medical Journal Editors. Editorial support (assembling tables and figures, collating author comments, copyediting, fact checking and referencing) and graphic services were provided by Erin P. Scott, PhD and Sarah Hummasti, PhD, AOIC, LLC and were funded by GSK. The authors thank Susanne Moore for her assistance with describing the preclinical data. Funding for this study (NCT03426995 available from www.clinicaltrials.gov) was provided by GlaxoSmithKline, Stevenage, UK.
Brown JA, Bal J, Simeoni M, et al. A randomized study of the safety and pharmacokinetics of GSK3358699, a mononuclear myeloid‐targeted bromodomain and extra‐terminal domain inhibitor. Br J Clin Pharmacol. 2022;88(5):2140-2155. doi: 10.1111/bcp.15137
The authors confirm that the PI for this paper is Dr Disala Fernando and that he had direct clinical responsibility for participants.
Funding information GlaxoSmithKline, Stevenage, UK
DATA AVAILABILITY STATEMENT
Within 6 months of this publication, anonymized individual participant data, the annotated case report form, protocol, reporting and analysis plan, data set specifications, raw dataset, analysis‐ready dataset and clinical study report will be available for research proposals approved by an independent review committee. Proposals should be submitted to www.clinicalstudydatarequest.com. A data access agreement will be required.
REFERENCES
- 1. Kulikowski E, Rakai BD, Wong NCW. Inhibitors of bromodomain and extra‐terminal proteins for treating multiple human diseases. Med Res Rev. 2021;41(1):223‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang N, Wu R, Tang D, Kang R. The BET family in immunity and disease. Signal Transduct Target Ther. 2021;6(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meng S, Zhang L, Tang Y, et al. BET inhibitor JQ1 blocks inflammation and bone destruction. J Dent Res. 2014;93(7):657‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nadeem A, Al‐Harbi NO, Al‐Harbi MM, et al. Imiquimod‐induced psoriasis‐like skin inflammation is suppressed by BET bromodomain inhibitor in mice through RORC/IL‐17A pathway modulation. Pharmacol Res. 2015;99:248‐257. [DOI] [PubMed] [Google Scholar]
- 5. Cheung K, Lu G, Sharma R, et al. BET N‐terminal bromodomain inhibition selectively blocks Th17 cell differentiation and ameliorates colitis in mice. Proc Natl Acad Sci USA. 2017;114(11):2952‐2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mele DA, Salmeron A, Ghosh S, Huang HR, Bryant BM, Lora JM. BET bromodomain inhibition suppresses TH17‐mediated pathology. J Exp Med. 2013;210(11):2181‐2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun Y, Han J, Wang Z, Li X, Sun Y, Hu Z. Safety and efficacy of Bromodomain and extra‐terminal inhibitors for the treatment of hematological malignancies and solid tumors: A systematic study of clinical trials. Front Pharmacol. 2020;11:621093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Needham LA, Davidson AH, Bawden LJ, et al. Drug targeting to monocytes and macrophages using esterase‐sensitive chemical motifs. J Pharmacol Exp Ther. 2011;339(1):132‐142. [DOI] [PubMed] [Google Scholar]
- 9. Chan CH, Fang C, Yarilina A, Prinjha RK, Qiao Y, Ivashkiv LB. BET bromodomain inhibition suppresses transcriptional responses to cytokine‐Jak‐STAT signaling in a gene‐specific manner in human monocytes. Eur J Immunol. 2015;45(1):287‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Belkina AC, Nikolajczyk BS, Denis GV. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol (Baltimore, MD: 1950). 2013;190:3670‐3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kokkola T, Suuronen T, Pesonen M, et al. BET inhibition upregulates SIRT1 and alleviates inflammatory responses. Chembiochem. 2015;16(14):1997‐2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Timmons GA, O'Siorain JR, Kennedy OD, Curtis AM, Early JO. Innate rhythms: Clocks at the center of monocyte and macrophage function. Front Immunol. 2020;11:1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Piquereau J, Boet A, Péchoux C, et al. The BET bromodomain inhibitor I‐BET‐151 induces structural and functional alterations of the heart mitochondria in healthy male mice and rats. Int J Mol Sci. 2019;20(7):1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Borck PC, Guo LW, Plutzky J. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ Res. 2020;126(9):1190‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ray KK, Nicholls SJ, Ginsberg HD, et al. Effect of selective BET protein inhibitor apabetalone on cardiovascular outcomes in patients with acute coronary syndrome and diabetes: Rationale, design, and baseline characteristics of the BETonMACE trial. Am Heart J. 2019;217:72‐83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Table S1 Summary of nonsustained ventricular tachycardia events that occurred during the study
Data Availability Statement
Within 6 months of this publication, anonymized individual participant data, the annotated case report form, protocol, reporting and analysis plan, data set specifications, raw dataset, analysis‐ready dataset and clinical study report will be available for research proposals approved by an independent review committee. Proposals should be submitted to www.clinicalstudydatarequest.com. A data access agreement will be required.