

Abstract
Background and Aims
Hepatic ischemia–reperfusion injury (IRI) is a common complication of hepatectomy and liver transplantation. However, the mechanisms underlying hepatic IRI have not been fully elucidated. Regulator of G‐protein signaling 14 (RGS14) is a multifunctional scaffolding protein that integrates the G‐protein and mitogen‐activated protein kinase (MAPK) signaling pathways. However, the role of RGS14 in hepatic IRI remains unclear.
Approach and Results
We found that RGS14 expression increased in mice subjected to hepatic ischemia–reperfusion (IR) surgery and during hypoxia reoxygenation in hepatocytes. We constructed global RGS14 knockout (RGS14‐KO) and hepatocyte‐specific RGS14 transgenic (RGS14‐TG) mice to establish 70% hepatic IRI models. Histological hematoxylin and eosin staining, levels of alanine aminotransferase and aspartate aminotransferase, expression of inflammatory factors, and apoptosis were used to assess liver damage and function in these models. We found that RGS14 deficiency significantly aggravated IR‐induced liver injury and activated hepatic inflammatory responses and apoptosis in vivo and in vitro. Conversely, RGS14 overexpression exerted the opposite effect of the RGS14‐deficient models. Phosphorylation of TGF‐β‐activated kinase 1 (TAK1) and its downstream effectors c‐Jun N‐terminal kinase (JNK) and p38 increased in the liver tissues of RGS14‐KO mice but was repressed in those of RGS14‐TG mice. Furthermore, inhibition of TAK1 phosphorylation rescued the effect of RGS14 deficiency on JNK and p38 activation, thus blocking the inflammatory responses and apoptosis.
Conclusions
RGS14 plays a protective role in hepatic IR by inhibiting activation of the TAK1–JNK/p38 signaling pathway. This may be a potential therapeutic strategy for reducing incidences of hepatic IRI in the future.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- Bad
Bcl2‐associated agonist of cell death
- Bax
Bcl2‐associated x
- Bcl2
B‐cell lymphoma 2
- Ccl2
C‐C motif chemokine ligand 2
- Cxcl10
chemokine (C‐X‐C motif) ligand 10
- DEG
differentially expressed gene
- ERK
extracellular signal–regulated protein kinase
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- GSEA
gene set enrichment analysis
- GST
glutathione S‐transferase
- HR
hypoxia reoxygenation
- IR
ischemia–reperfusion
- IRI
ischemia–reperfusion injury
- JNK
c‐Jun N‐terminal kinase
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KO
knockout
- MAPK
mitogen‐activated protein kinase
- NTG
nontransgenic
- p‐
phosphorylated
- RGS14
regulator of G protein signaling 14
- RNA‐seq
RNA sequencing
- sh‐
short hairpin
- TAK1
TGF‐β‐activated kinase 1
- TG
transgenic
- TUNEL
terminal deoxynucleotidyl transferase uridine triphosphate nick end labeling
- WT
wild‐type
- 5Z‐7‐Ox
5Z‐7‐oxozeaenol
INTRODUCTION
Hepatic ischemia–reperfusion injury (IRI) is a common complication reported following hepatectomy and liver transplantation. In addition, abdominal trauma,[ 1 ] hemorrhagic shock,[ 2 ] myocardial ischemia, and stroke may result in low blood flow that results in the infliction of postreperfusion injury. Hepatic IRI affects the recovery and prognosis of patients as it can rapidly induce acute liver inflammation and lead to elevated levels of serum transaminase levels, which may result in the occurrence of severe liver cell damage and organ dysfunction.[ 3 , 4 ] Despite its remarkable clinical importance, the adverse consequences of hepatic IRI remain elusive in clinical practice. Thus, it is necessary to study the mechanism of hepatic IRI in order to provide solutions to these clinical problems.
Since the 1990s, research has been conducted on the mechanism of hepatic IRI, which involves the roles played by cellular complement, factors, and mediators, such as those in reactive oxygen species (ROS) generation, neutrophil infiltration, excessive endoplasmic reticulum stress activation, and microcirculation dysfunction.[ 5 , 6 , 7 ] Studies have also shown that inflammation and apoptosis play important roles in IRI.[ 8 ] As an aerobic organ, the liver is extremely susceptible to hypoxic stress; furthermore, under the conditions of ischemia and hypoxia, the morphology and function of the liver undergo changes, which can lead to liver cell death through apoptosis and necrosis.[ 9 ] The occurrence of reperfusion triggers an inflammatory response that leads to the production of proinflammatory cytokines, such as Il1b and Tnfα. These cytokines can induce neutrophil activation and migration from the endothelial lumen to the liver parenchyma[ 10 ] and can activate the mitogen‐activated protein kinase (MAPK) and NF‐κB signaling pathways, further enhancing the inflammatory response. In addition to the production of inflammatory factors, reperfusion leads to the production of considerable amounts of chemokines, accelerates the recruitment and activation of neutrophils, and promotes the process of IRI. Kupffer cells, neutrophils, CD4+ T cells, and natural killer cells constitute the main cell players in hepatic IRI, while complement factors, cytokines, and chemokines constitute the main humoral factors. Apoptosis also plays an important role in hepatic IRI. Apoptosis is the process of programmed cell death that helps to maintain tissue homeostasis by removing redundant or damaged cells; excessive inflammatory reactions eventually lead to the occurrence of hepatic necrosis and apoptosis following IRI.[ 11 ]
Regulator of G‐protein signaling 14 (RGS14) is a complex scaffolding protein in the RGS family that contains a conserved RGS domain. Almost all RGS proteins are guanosine triphosphatase–activating proteins and negatively regulate signal transduction.[ 12 , 13 ] RGS14 possesses multiple signaling regulatory elements. In addition to the canonical RGS domain and a GoLoco/G‐protein regulatory motif, RGS14 contains two tandem Ras/Rap‐binding domains that preferentially establish interactions with activated H‐Ras‐guanosine triphosphate (GTP) and Raf kinases.[ 14 ] RGS14 plays essential roles in the promotion of the birth process,[ 15 ] cellular mitosis,[ 16 , 17 ] and phagocytosis by activating αMβ2 integrin expression.[ 18 ] RGS14 is highly expressed in lymphocytes, monocytes, and dendritic cells.[ 19 ] Moreover, RGS14 can affect the recruitment of chemokines to lymphocytes by regulating the expression of chemokine receptors, thereby affecting the adhesion and migration of lymphocytes.[ 20 ] Previous reports of RGS14 focused on its effects in the CA2 region of the hippocampal area.[ 14 , 21 , 22 ] However, the exact role of RGS14 in hepatocytes, particularly in response to hepatic IRI, has not been characterized. In the present study, we explored whether RGS14 expression was altered in hepatic IRI and further investigated the involvement of RGS14 in hepatic IRI and its underlying mechanisms.
MATERIALS AND METHODS
Animals
Male mice were housed in polycarbonate cages within a specific pathogen‐free, temperature‐controlled facility, with a 12‐h light/dark cycle. Food and water were provided to the animals ad libitum. Animal protocols were approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University and conducted in accordance with the guidelines outlined by the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. RGS14 knockout (RGS14‐KO) and hepatocyte‐specific RGS14 transgene (RGS14‐TG) strains were generated as described in detail in the Supporting Information.
Hepatic IRI model
We used an established partial (70%) liver warm ischemia model, as described.[ 23 ] After 1 h of ischemia, the clamp was removed for reperfusion. Sham‐operated mice were subjected to the same procedure, without vascular occlusion.
Liver damage assessment
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured using the ADVIA 2400 Chemistry System (Siemens, Tarrytown, NY) according to the manufacturer's instructions. Histopathological analysis was performed per described methods.[ 23 ]
Immunofluorescence staining and immunohistochemistry
Immunofluorescence staining and immunohistochemistry procedures are described in detail in the Supporting Information.
Cell culture and hypoxia–reoxygenation experiment
Cell culture and hypoxia–reoxygenation (HR) procedures were performed per methods described.[ 24 , 25 ]
Real‐time quantitative PCR
Quantitative PCR analyses were performed using the ChamQ SYBR qPCR Master Mix (catalog no. Q311‐02; Vazyme, Nanjing, China) to quantify mRNA expression. The primer sequences used in this study are shown in Table S1.
Western blotting
The ChemiDoc MP imaging system (Bio‐Rad, Hercules, CA) was used to detect protein signals. ImageJ software was used to quantify protein levels. All antibodies used for western blotting are shown in Table S2.
Plasmid construction
Plasmid construction procedures are detailed in the Supporting Information.
Construction of stable cell lines
The primers used for RGS14 knockdown and overexpression plasmid construction are detailed in Table S3. L02 stable cell lines were established per described methods using plasmid and lentiviral vectors.[ 26 ] The stable cell lines were identified by western blotting and PCR analysis.
Immunoprecipitation assays
Coimmunoprecipitation assays were performed as described[ 26 ] to identify the interactions of RGS14 with other factors. Glutathione S‐transferase (GST) precipitation assays were performed to examine the direct interaction established between RGS14 and TGF‐β‐activated kinase 1 (TAK1).
RNA‐sequencing and data analysis
RNA‐sequencing (RNA‐seq) and data analyses are detailed in the Supporting Information.
Statistical analysis
All statistical analyses were conducted using SPSS. All data are expressed as mean ± SD. For comparison between the two groups, a two‐tailed Student t test (normal distribution data) was used. One‐way ANOVA was used for comparisons between multiple groups, Bonferroni analyses were used for data of homogeneity of variance, and Tamhane's T2 analysis was used for data of heteroscedasticity. Kruskal‐Wallis nonparametric analysis was performed for nonnormal distribution data among multiple groups. Statistical significance was set at p < 0.05.
RESULTS
RGS14 expression is up‐regulated in both in vivo and in vitro hepatic IRI
To analyze whether RGS14 expression was associated with hepatic IRI, we first analyzed the expression of RGS14 in a public database (PRJNA407106), which involved different time points of hepatic IRI. The results showed that RGS14 gene expression increased gradually after ischemia with prolonged reperfusion times (3, 6, 12, and 24 h) (Figure 1A). Further, we validated this result by establishing a hepatic IRI model using C57 wild‐type (WT) mice. RGS14 mRNA levels and protein expression gradually increased with reperfusion (3, 6, and 24 h) (Figure 1B,C). Moreover, immunohistochemical staining results further confirmed that RGS14 expression was significantly up‐regulated in the liver tissues, especially in hepatocytes of mice subjected to IRI (Figure 1D) when compared with the sham group. To further study the expression changes of RGS14 in hepatocytes, L02 hepatocytes were exposed to HR stimulation. Consistent with these in vivo results, RGS14 mRNA levels (Figure 1E) and RGS14 protein expression (Figure 1F) in cultured hepatocytes also significantly increased at 6 h after HR stimulus. These results suggested that RGS14‐up‐regulated expression was associated with hepatic IRI and that RGS14 might play an important role in this process.
FIGURE 1.
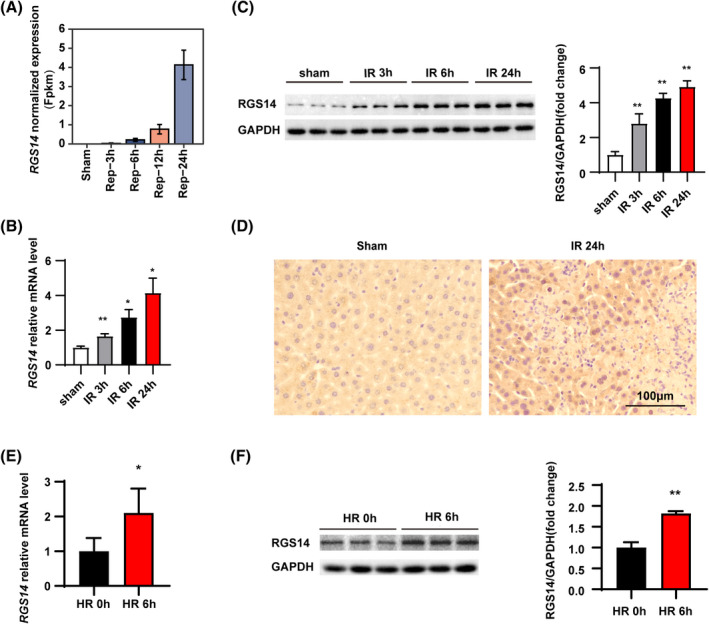
RGS14 expression is up‐regulated in both in vivo and in vitro hepatic IRI. (A) Public database showing changes in the expression of the RGS14 gene after sham treatment or ischemia for 1 h, followed by reperfusion for 3, 6, 12, and 24 h. (B) Real‐time quantitative PCR (RT‐qPCR) analysis of RGS14 mRNA levels in livers from WT mice subjected to sham treatment or ischemia for 1 h, followed by reperfusion for 3, 6, and 24 h (n = 4/group). (C) Western blot analysis and quantification of RGS14 protein expression in livers from WT mice subjected to sham treatment or ischemia for 1 h, followed by reperfusion for 3, 6, and 24 h (n = 3/group). (D) Immunohistochemical staining of RGS14 expression in livers of mice subjected to sham treatment or at 24 h after hepatic IR (n = 4/group). Scale bar, 100 μm. (E) RT‐qPCR analysis of RGS14 mRNA levels in cultured L02 hepatocytes after HR stimulation (n = 4 independent experiments). (F) Western blot analysis and quantification of RGS14 expression in cultured L02 hepatocytes after HR stimulation (n = 3 independent experiments). GAPDH served as a loading control. All data are shown as the mean ± SD. *p < 0.05, **p < 0.01. FPKM, fragments per kilobase of exon model per million mapped fragments
RGS14 alleviates liver damage during hepatic IRI
To investigate the effect of RGS14 on hepatic IRI, RGS14‐KO mice were constructed and subjected to hepatic IRI operation. Western blots results confirmed RGS14 deficiency in the livers of RGS14‐KO mice (Figure 2A). Serum AST and ALT levels and necrotic area were assessed to evaluate the extent of liver damage. In response to hepatic IRI, serum AST and ALT levels were significantly increased in WT mice, and RGS14 deficiency markedly increased serum AST and ALT levels compared to those of the WT group (Figure 2B). The necrotic area in the liver gradually increased at 3 and 6 h after reperfusion. Compared with WT controls, RGS14 deficiency markedly increased the necrotic area (Figure 2C). We further established hepatocyte‐specific RGS14‐TG mice to evaluate the effect of RGS14 overexpression on hepatic IRI. Western blot results showed overexpression of RGS14 in the livers of RGS14‐TG mice (Figure 2D). In contrast to the aggravation of liver damage owing to RGS14 deficiency, RGS14 overexpression resulted in decreased ALT and AST serum levels (Figure 2E) and reduced the necrotic area (Figure 2F) compared with that observed in the nontransgenic (NTG) controls at 3 and 6 h after hepatic IRI. These findings demonstrate that RGS14 reduces liver damage during hepatic IRI.
FIGURE 2.
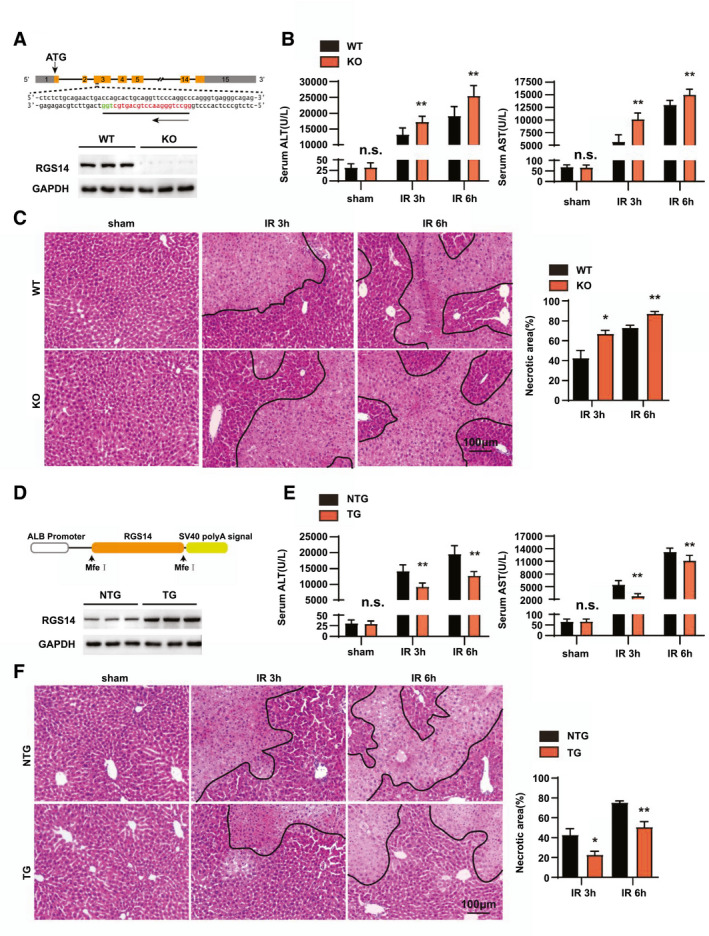
RGS14 alleviates liver damage during hepatic IRI. (A) Diagram of the RGS14‐KO strategy and RGS14 protein expression in the livers of WT and RGS14‐KO mice (n = 3/group). (B) Serum ALT and AST levels in WT and RGS14‐KO mice in the sham group and at 3 and 6 h after hepatic IR (n = 8/group). (C) Representative hematoxylin and eosin staining and statistics showing necrotic areas of liver tissue from WT and RGS14‐KO mice at 3 and 6 h after hepatic IR (n = 5–6/group). Scale bar, 100 μm. (D) Diagram of RGS14‐TG strategy and RGS14 protein expression in the livers of NTG and RGS14‐TG mice (n = 3/group). (E) Serum ALT and AST levels in NTG and RGS14‐TG mice in the sham group and at 3 and 6 h after hepatic IR (n = 8/group). (F) Representative hematoxylin and eosin staining and statistics showing necrotic areas of liver tissue from NTG and RGS14‐TG mice at 3 and 6 h after hepatic IR (n = 5–6/group). GAPDH served as a loading control. Scale bar, 100 μm. All data are shown as the mean ± SD. *p < 0.05, **p < 0.01. ALB, albumin; Mfe I, restriction enzyme; n.s., not significant
RGS14 alleviates the inflammation response in hepatic IRI
Activated inflammation plays an important role in the hepatic IRI process, and suppression of the augmented inflammatory response is a promising strategy to treat IRI.[ 27 ] We further investigated whether RGS14 could affect inflammatory responses induced by hepatic IRI. Immunofluorescence staining results showed that in response to hepatic IRI, the number of CD11b‐positive inflammatory cells was significantly increased in the liver of WT mice, while RGS14‐KO mice exhibited markedly higher CD11b‐positive cell infiltration in the liver tissues than that observed in WT mice (Figure 3A). In addition, we detected expression of inflammatory factors; and the results indicated that the mRNA levels of chemokine (C‐X‐C motif) ligand 10 (Cxcl10), C‐C motif chemokine ligand 2 (Ccl2), Il6, Il1b, and Tnf were significantly increased in the liver of RGS14‐KO mice compared to WT mice at 6 h after IRI (Figure 3B). Additionally, western blotting revealed enhanced activation of NF‐κB signaling in the liver tissues of RGS14‐KO mice compared to WT controls (Figure 3C). In contrast to RGS14 deficiency, RGS14‐TG mice exhibited reduced inflammatory cell infiltration, lower inflammatory cytokine, and lower chemokine expression, as well as reduced NF‐κB activation compared to NTG mice after hepatic IRI (Figure 3D–F). These findings suggest that RGS14 alleviates inflammatory responses during hepatic IRI.
FIGURE 3.
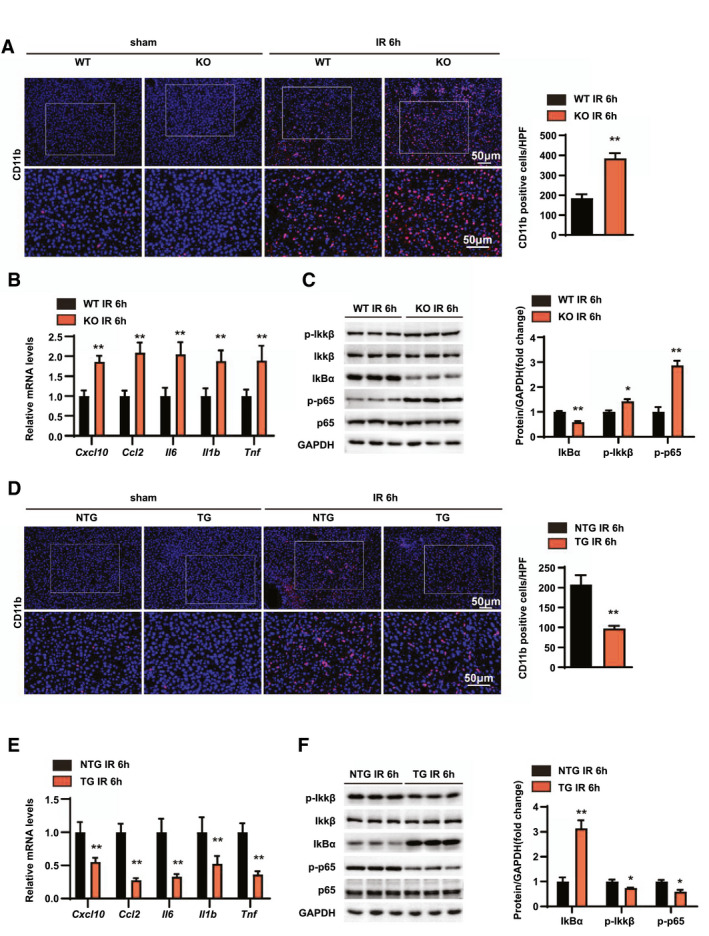
RGS14 alleviates inflammation in hepatic IRI. (A) Immunofluorescence staining of CD11b‐positive inflammatory cells (red) and statistics in ischemic liver sections from mice in the indicated groups (n = 4/group). (B) Relative mRNA expression levels of Cxcl10, Ccl2, Il6, Il1b, and Tnf in liver tissues of WT and RGS14‐KO mice at 6 h after IR (n = 4/group). (C) Western blot analysis of NF‐κB signaling in livers from WT and RGS14‐KO mice at 6 h after IR (n = 3/group). (D) Immunofluorescence staining of CD11b‐positive inflammatory cells (red) and statistics in ischemic liver sections from mice in the indicated groups (n = 4/group). (E) Relative mRNA expression levels of Cxcl10, Ccl2, Il6, Il1b, and Tnf in liver tissues of NTG and RGS14‐TG mice at 6 h after IR (n = 4/group). (F) Western blot analysis of NF‐κB signaling in livers from NTG and RGS14‐TG mice at 6 h after IR (n = 3/group). GAPDH served as a loading control. All data are shown as the mean ± SD. *p < 0.05, **p < 0.01. HPF, high‐power field; IκBα, inhibitory κBα; IKKβ, IκB kinase β
RGS14 alleviates apoptosis in hepatic IRI
Apoptosis is another essential step in the pathogenesis of hepatic IRI.[ 24 ] Therefore, terminal deoxynucleotidyl transferase uridine triphosphate nick end labeling (TUNEL) staining was performed to detect the effect of RGS14 on hepatocyte apoptosis after hepatic IRI. In response to IRI, apoptotic cell numbers increased compared to the sham group, and RGS14‐KO mice exhibited more apoptotic cells (Figure 4A). Also, immunohistochemical staining showed that cleaved caspase‐3 expression was significantly higher in liver tissues of RGS14‐deficient mice compared to the WT group (Figure 4A). Additionally, mRNA levels of the antiapoptotic gene B‐cell lymphoma 2 (Bcl2) were significantly decreased and levels of the proapoptotic genes Bcl2‐associated agonist of death (Bad) and Bcl2‐associated x (Bax) were increased in the liver of the RGS14‐KO mice at 6 h after IRI compared with those observed in WT mice (Figure 4B). Consistent with these results, the protein expression levels of proapoptotic factors (Bax, cleaved‐caspase 3) were increased and the expression level of the antiapoptotic factor Bcl2 were significantly decreased in the RGS14‐KO group in comparison with the control group (Figure 4C). In contrast, RGS14‐TG mice exhibited fewer apoptotic cells, higher expression of antiapoptotic genes and proteins, and lower expression of proapoptotic genes and proteins than NTG mice after hepatic IRI (Figure 4D–F). Together, these results suggest that RGS14 suppresses apoptosis during hepatic IRI.
FIGURE 4.
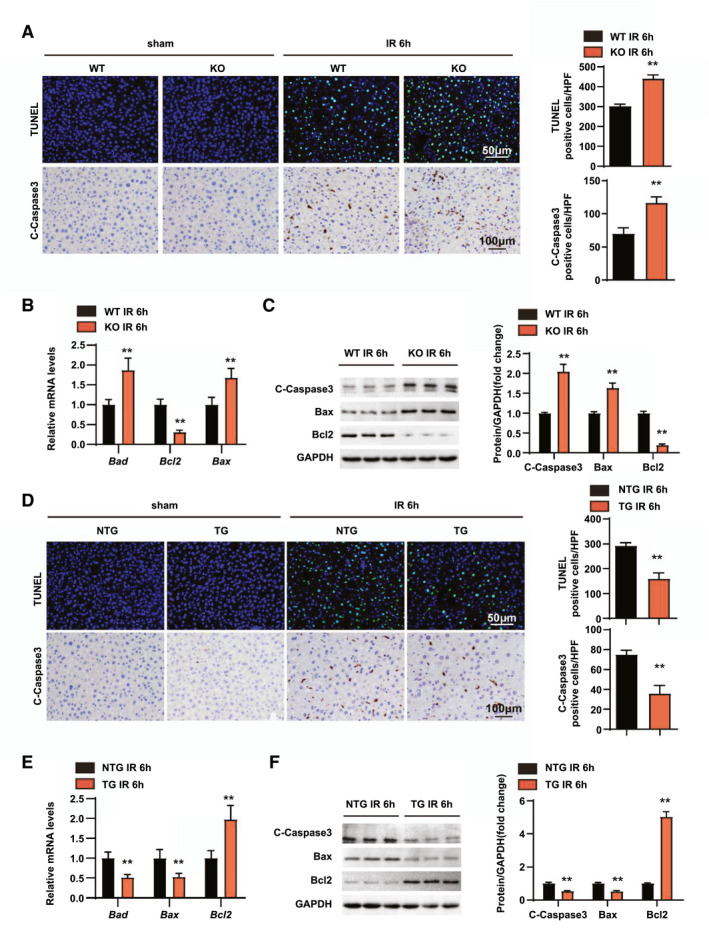
RGS14 alleviates apoptosis in hepatic IRI. (A) TUNEL staining and statistics in ischemic liver sections from mice in the indicated groups (n = 4/group). Immunohistochemical staining of cleaved‐caspase‐3 expression and statistics in ischemic liver sections from mice in the indicated groups (n = 6/group). (B) Relative mRNA expression levels of Bad, Bcl2, and Bax in liver tissues of WT and RGS14‐KO mice at 6 h after IR (n = 4/group). (C) Western blot analysis of cleaved‐caspase‐3, Bax, and Bcl2 expression in livers from WT and RGS14‐KO mice at 6 h after IR (n = 3/group). (D) TUNEL staining and statistics in ischemic liver sections from mice in the indicated groups (n = 4/group). Immunohistochemical staining of cleaved‐caspase‐3 expression and statistics in ischemic liver sections from mice in the indicated groups (n = 6/group). (E) Relative mRNA expression levels of Bad, Bcl2, and Bax in liver tissues of NTG and RGS14‐TG mice at 6 h after IR (n = 4/group). (F) Western blot analysis of cleaved‐caspase‐3, Bax, and Bcl2 expression in livers from NTG and RGS14‐TG mice at 6 h after IR (n = 3/group). GAPDH served as a loading control. All data are shown as the mean ± SD. *p < 0.05, **p < 0.01. C‐Caspase3, cleaved caspase‐3; HPF, high‐power field
RGS14 alleviates inflammation and apoptosis in hepatocyte HR stimulation
Based on our in vivo studies, we have demonstrated that RGS14 alleviates hepatic injury, inflammation, and apoptosis in hepatic IRI. To further explore the regulation of RGS14 on hepatocytes, RGS14‐knockdown hepatocytes were generated and subjected to HR stimulation (Figure 5A). Similar to the in vivo results, the mRNA levels of inflammatory chemokines (Ccl2) and cytokines (Tnf and Il1b) were significantly up‐regulated in RGS14 knockdown hepatocytes after an HR challenge (Figure 5B). Moreover, enhanced activation of NF‐κB signaling was observed in RGS14 knockdown hepatocytes (Figure 5C). Additionally, the mRNA and protein expression levels of proapoptotic molecules (Bax, cleaved caspase‐3) were increased and the mRNA and protein expression of antiapoptotic Bcl2 were decreased in RGS14 knockdown hepatocytes compared with the controls in response to HR (Figure 5D,E). On the other hand, RGS14‐overexpressing hepatocyte cell lines were constructed to evaluate the effects of RGS14 overexpression on inflammation and apoptosis of HR‐stimulated hepatocytes (Figure 5F). In contrast to RGS14 knockdown, expression levels of the inflammatory cytokines, chemokines (Figure 5G,H), and proapoptosis‐related factors (Figure 5I,J) were significantly reduced and those of antiapoptosis‐related molecules were increased compared to the control group. Taken together, these results suggest that RGS14 alleviated inflammation and apoptosis in hepatocytes after HR stimulation.
FIGURE 5.
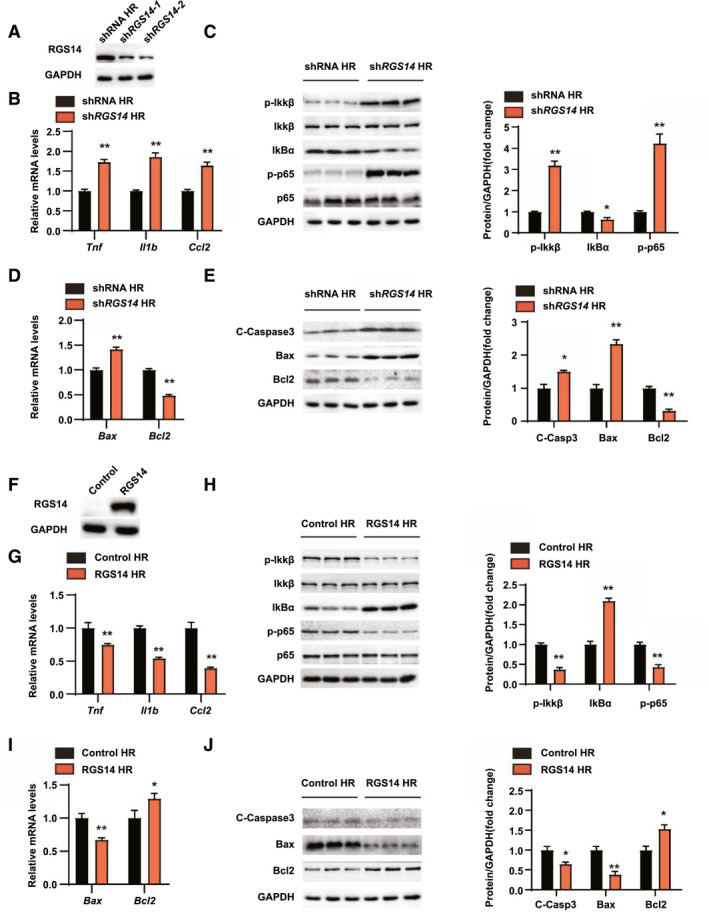
RGS14 alleviates inflammation and apoptosis during hepatic HR. (A) RGS14 protein expression in hepatocytes infected with shRNA or shRGS14 lentivirus. (B) mRNA levels of proinflammatory factors (Tnf, Il1b, and Ccl2) in hepatocytes from the indicated groups after HR challenge. (C) Expressions of NF‐κB signaling pathway proteins in hepatocytes from the indicated groups after HR challenge. (D) mRNA levels of Bax and Bcl2 in hepatocytes from the indicated groups after HR challenge. (E) Expression of apoptosis signaling pathway proteins in hepatocytes from the indicated groups after HR challenge. (F) RGS14 protein expression in hepatocytes infected with Control or RGS14 overexpressing lentivirus. (G) mRNA levels of proinflammatory factors (Tnf, Il1b, and Ccl2) in the indicated groups after HR challenge. (H) Expression of NF‐κB signaling pathway proteins in the indicated groups after HR challenge. (I) mRNA levels of Bax and Bcl2 in the indicated groups after HR challenge. (J) Expression of apoptosis signaling pathway proteins in the indicated groups after HR challenge. GAPDH served as a loading control. n = 3 independent experiments. All data are shown as the mean ± SD. *p < 0.05, **p < 0.01. C‐Caspase3, cleaved caspase‐3; IκBα, inhibitory κBα; IKKβ, IκB kinase β
RGS14 suppresses TAK1–c‐Jun N‐terminal kinase/p38 signaling during hepatic IRI
To investigate the underlying mechanism of hepatic IRI driven by RGS14, we performed an RNA‐seq analysis in liver tissue from RGS14‐KO mice and WT mice following hepatic IRI (Figure 6A). A hierarchical clustering dendrogram analysis showed the distribution profiles of RNA‐seq (Figure 6B). Gene set enrichment analysis (GSEA) showed that inflammation and apoptotic process–related pathways and involved genes were all up‐regulated by RGS14 deficiency (Figure 6C,D). Volcano mapping further confirmed that the majority of differential expression genes (DEGs; fold change >2, p < 0.05) were up‐regulated (Figure 6E). Metascape enrichment analysis showed that these genes influenced each other to form a complex regulatory network; among these, genes related to inflammation were at the key node of regulation (Figure 6F). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis showed that the MAPK pathway was the most significantly enriched signaling pathway associated with the DEGs during IRI (Figure 6G). To further verify the regulatory effects of RGS14 on the MAPK signaling pathway, western blot was used to detect the activation of MAPK signaling in vivo and in vitro. RGS14 deficiency elevated c‐Jun N‐terminal kinase (JNK), p38, and TAK1 phosphorylation levels, while RGS14 overexpression suppressed JNK, p38, and TAK1 phosphorylation levels at 6 h after reperfusion. However, neither RGS14 deficiency nor overexpression affected extracellular signal–regulated kinase (ERK) phosphorylation levels (Figure 6H,I). In addition, the effect of knockdown or overexpression of RGS14 in HR‐stimulated hepatocytes on this signaling pathway is consistent with what is seen in vivo (Figure 6J,K). Taken together, these results showed that RGS14 suppresses TAK1–JNK/p38 signaling during hepatic IRI.
FIGURE 6.
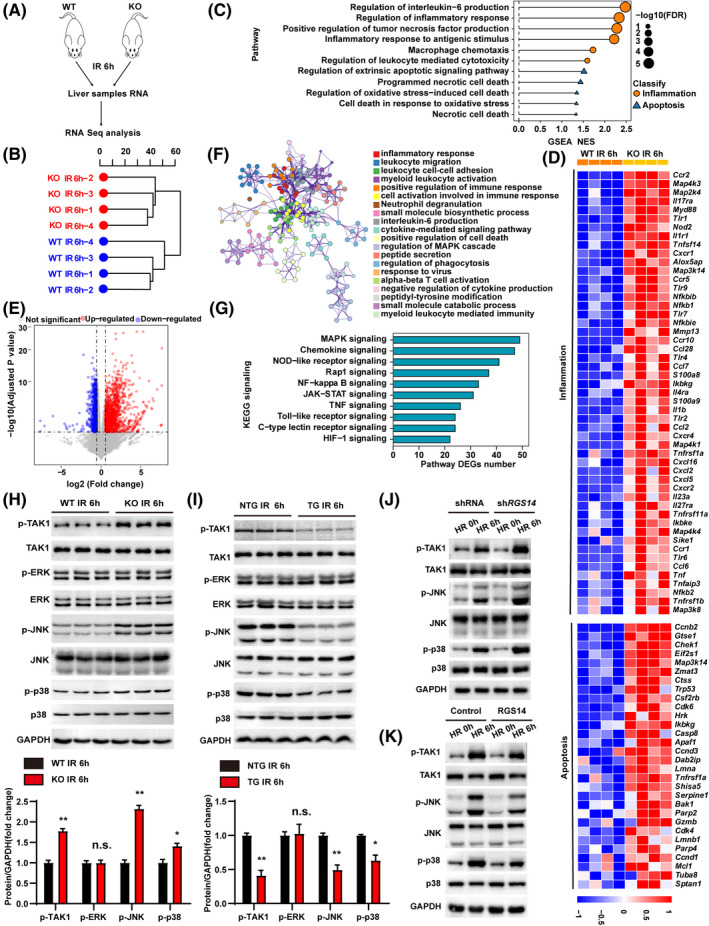
RGS14 inactivates TAK1–JNK/p38 signaling during hepatic IRI. (A) RNA‐seq construction plan for liver tissues from RGS14‐KO and WT mice at 6 h after hepatic IRI. (B) Hierarchical clustering dendrogram showing the distribution profiles of RNA‐seq. (C) GSEA of RNA‐seq data showing the enrichment of inflammation and apoptosis‐related pathways in the livers of RGS14‐KO and WT mice after hepatic IRI. The 11 most significantly enriched pathways are shown. Black dots represent −log10 p values for the enriched gene ontology pathways. Orange dots represent inflammation‐related pathways. Blue triangles represent apoptosis‐related pathways. (D) Heatmap showing the expression of inflammation‐related and apoptosis‐related genes of the leading‐edge subset in the livers of RGS14‐KO and WT mice detected by RNA‐seq analyses. (E) Volcano map showing DEGs in livers of RGS14‐KO and WT mice after hepatic IRI. The red dots represent up‐regulated DEGs, and the blue dots represent down‐regulated DEGs. The gray dots represent genes without significant differences in expression. (F) Metascape enrichment analysis showing groups of several categories based on gene functional relevance, and construction of a network was based on relevance and similarity. In the figure, different colors are used to represent different categories. (G) The top 10 most significantly enriched pathways contributing to RGS14 function were determined by KEGG enrichment analysis of the livers of RGS14‐KO and WT mice after hepatic IRI. (H) Western blot analysis of the levels of total and phosphorylated TAK1, JNK, ERK, and p38 in RGS14‐KO and WT mice at 6 h after IR (n = 3/group). (I) Western blot analysis of the levels of total and phosphorylated TAK1, JNK, ERK, and p38 in NTG and RGS14‐TG mice at 6 h after IR (n = 3/group). (J) Western blot analysis of the levels of total and phosphorylated TAK1, JNK, and p38 in shRNA control or RGS14‐konckdown hepatocytes subjected to HR challenge. (K) Western blot analysis of the levels of total and phosphorylated TAK1, JNK, and p38 in Flag control or RGS14‐overexpressing hepatocytes subjected to HR challenge. GAPDH served as a loading control. (A–G) n = 4/group. (J–K) n = 3 independent experiments. All data are shown as the mean ± SD. *p < 0.05, **p < 0.01. Alox5ap, arachidonate 5‐lipoxygenase‐activating protein; Apaf1, apoptotic peptidase activating factor 1; Bak1, BCL2‐antagonist/killer 1; Casp8, caspase 8; Ccl2, C‐C motif chemokine 2; Ccl28, C‐C motif chemokine 28; Ccl6, C‐C motif chemokine 6; Ccl7, C‐C motif chemokine 7; Ccnb2, cyclin B2; Ccnd1, cyclin D1; Ccnd3, cyclin D3; Ccr1, C‐C chemokine receptor type 1; Ccr10, C‐C chemokine receptor type 10; Ccr2, C‐C chemokine receptor type 2; Ccr5, C‐C chemokine receptor type 5; Cdk4, cyclin‐dependent kinase 4; Cdk6, cyclin dependent kinase 6; Chek1, checkpoint kinase 1; Csf2rb, colony stimulating factor 2 receptor subunit beta; Ctss, cathepsin S; Cxcl16, C‐X‐C motif chemokine 16; Cxcl2, C‐X‐C motif chemokine 2; Cxcl5, C‐X‐C motif chemokine 5; Cxcr1, C‐X‐C chemokine receptor type 1; Cxcr2, C‐X‐C chemokine receptor type 2; Cxcr4, C‐X‐C chemokine receptor type 4; Dab2ip, Disabled 2 interacting protein; Eif2s1, eukaryotic translation initiation factor 2 subunit 1 alpha; FDR, false discovery rate; Gtse1, G two S phase expressed protein 1; Gzmb, granzyme B; HIF‐1, hypoxia‐inducible factor 1; Hrk, harakiri, BCL2 interacting protein; Ikbke, inhibitor of kappaB kinase epsilon; Ikbkg, inhibitor of kappaB kinase gamma; Il17ra, interleukin‐17 receptor A; Il1b, interleukin‐1 beta; Il1r1 , interleukin‐1 receptor type 1; Il23a, interleukin‐23 subunit alpha; Il27ra, interleukin‐27 receptor subunit alpha; Il4ra, interleukin‐4 receptor subunit alpha; JAK‐STAT, Janus kinase–signal transducer and activator of transcription; Lmna, lamin‐A; Lmnb1, lamin B1; Map2k4, mitogen‐activated protein kinase kinase 4; Map3k14, mitogen‐activated protein kinase kinase kinase 14; Map3k8, mitogen‐activated protein kinase kinase kinase 8; Map4k1, mitogen‐activated protein kinase kinase kinase kinase 1; Map4k3, mitogen‐activated protein kinase kinase kinase kinase 3; Map4k4, mitogen‐activated protein kinase kinase kinase kinase 4; Mcl1, myeloid cell leukemia sequence 1; Mmp13, matrix metallopeptidase 13; Myd88, myeloid differentiation primary response gene 88; n.s., not significant; NES, normalized enrichment score; Nfkb1, nuclear factor NF‐kappa‐B p105 subunit; Nfkb2, nuclear factor NF‐kappa‐B p100 subunit; Nfkbib, NF‐kappa‐B inhibitor beta; Nfkbie, NF‐kappa‐B inhibitor epsilon; NOD, nonobese diabetic; Nod2, nucleotide‐binding oligomerization domain containing 2; Parp2, poly (ADP‐ribose) polymerase family, member 2; Parp4, poly (ADP‐ribose) polymerase family, member 4; S100a8, S100 calcium binding protein A8; S100a9, S100 calcium binding protein A9; Serpine1, serine (or cysteine) peptidase inhibitor, clade E, member 1; Shisa5, shisa family member 5; Sike1, suppressor of IKBKE 1; Sptan1, spectrin alpha, non‐erythrocytic 1; Tlr1, toll‐like receptor 1; Tlr2, toll‐like receptor 2; Tlr4, toll‐like receptor 4; Tlr6, toll‐like receptor 6; Tlr7, toll‐like receptor 7; Tlr9, toll‐like receptor 9; Tnf, tumor necrosis factor; Tnfaip3, tumor necrosis factor alpha‐induced protein 3; Tnfrsf11a, tumor necrosis factor receptor superfamily member 11A; Tnfrsf1a, tumor necrosis factor receptor superfamily member 1A; Tnfrsf1b, tumor necrosis factor receptor superfamily member 1B; Tnfsf14, tumor necrosis factor ligand superfamily member 14; Trp53, transformation related protein 53; Tuba8, tubulin alpha 8; Zmat3, zinc finger matrin type 3
RGS14 interacts with TAK1
To further explore the potential molecular mechanism by which RGS14 regulated TAK1 activation, we first detected the localization of RGS14 and TAK1 and found that RGS14 colocalized with TAK1 in the cytoplasm by immunofluorescence (Figure 7A). Next, coimmunoprecipitation experiments were conducted to investigate the interaction of RGS14 and TAK1. The results showed that RGS14 established an interaction with TAK1 in HEK293T cells (Figure 7B). Moreover, we performed GST precipitation assays to verify the direct interaction established between RGS14 and TAK1 (Figure 7C). The subsequent molecular mapping assay results revealed that the N‐terminal segment (1–300 amino acids) of TAK1 was responsible for its interaction with RGS14 (Figure 7D), while the 374–566‐amino acid domain of RGS14 enabled binding with TAK1 (Figure 7E).
FIGURE 7.
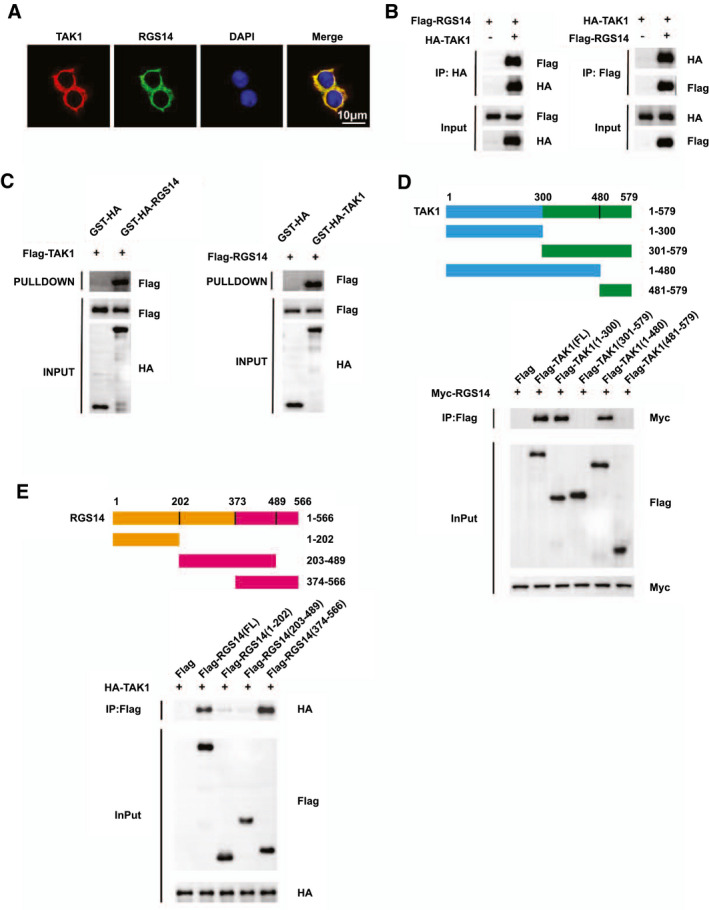
RGS14 establishes interaction with TAK1. (A) Representative confocal images showing colocalization between RGS14 (green) and TAK1 (red) in L02 cells transfected with HA‐tagged RGS14 and Flag‐tagged TAK1 plasmids. Nuclei were stained using DAPI (blue). Scale bar, 10 μm. (B) Flag‐tagged RGS14 and HA‐tagged TAK1 plasmids were cotransfected into HEK‐293T cells. Anti‐HA antibody (left panel) and anti‐Flag antibody (right panel) were used for immunoprecipitation. (C) The interaction of RGS14 with TAK1 was assayed by GST precipitation, and the purified GST‐HA was used as a control. (D) Full‐length MYC‐RGS14 and various truncated forms of Flag‐TAK1 were cotransfected into HEK‐293T cells. An anti‐Flag antibody was used for immunoprecipitation. (E) Full‐length HA‐TAK1 and various truncated forms of Flag‐RGS14 were cotransfected into HEK‐293T cells. An anti‐Flag antibody was used for immunoprecipitation. Results are representative of three independent experiments. HA, hemagglutinin; IP, immunoprecipitation
TAK1 mediates the effect of RGS14 in hepatic IRI
To further evaluate whether TAK1 mediated the effect of RGS14 on hepatic IRI, TAK1 activity was blocked in L02 hepatocytes using the specific TAK1 inhibitor 5Z‐7‐oxozeaenol (5Z‐7‐Ox). Compared with the DMSO control group, the inhibitor group showed a notable reduction in the activation of TAK1 and downstream JNK/p38 in RGS14 knockdown hepatocytes subjected to HR challenge (Figure 8A). Moreover, the promotional effects of RGS14 deficiency on the inflammatory responses and apoptosis were all blocked by 5Z‐7‐Ox (Figure 8B–E). Taken together, these results demonstrated that TAK1 mediated the effect of RGS14 in hepatic IRI.
FIGURE 8.
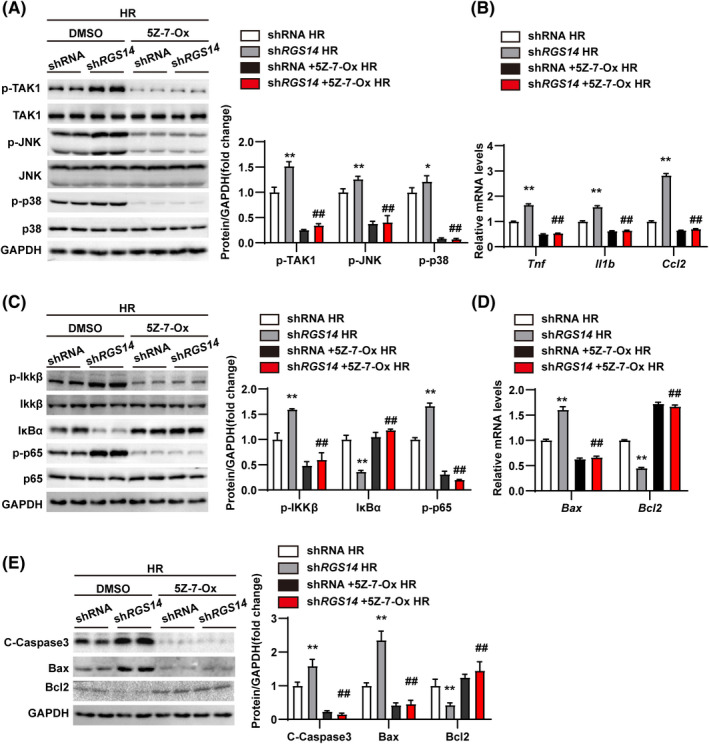
TAK1 mediates the effect of RGS14 on liver IRI. (A) Western blot analysis of the levels of total and phosphorylated TAK1, JNK, and p38 in shRNA and shRGS14 hepatocytes treated with DMSO or TAK1 inhibitor (5Z‐7‐Ox) and subjected to HR challenge. (B) mRNA levels of proinflammatory factors (Tnf, Il1b, and Ccl2) in shRNA and shRGS14 hepatocytes treated with DMSO or 5Z‐7‐Ox and subjected to HR challenge. (C) Western blot analysis of the activation of NF‐κB signaling in shRNA and shRGS14 hepatocytes treated with DMSO or 5Z‐7‐Ox and subjected to HR challenge. (D) mRNA levels of Bax and Bcl2 in shRNA and shRGS14 hepatocytes treated with DMSO or 5Z‐7‐Ox and subjected to HR challenge. (E) Western blot analysis of Bax, Bcl2, and cleaved caspase‐3 expression in shRNA and shRGS14 hepatocytes treated with DMSO or 5Z‐7‐Ox and subjected to HR challenge. GAPDH served as a loading control. Results are representative of three independent experiments. All data are shown as the mean ± SD. **p < 0.01, shRNA group compared with shRGS14 group; ## p < 0.01, shRGS14 + 5Z‐7‐Ox group compared with shRGS14 group. C‐Caspase3, cleaved caspase‐3; IκBα, inhibitory κBα; IKKβ, IκB kinase β
DISCUSSION
Hepatic IRI continues to be responsible for postoperative morbidity and mortality in patients undergoing liver surgery. The activation of inflammation and cell apoptosis are key events in the process of hepatic IRI. Several members of the RGS protein family play important regulatory roles in the occurrence and development of inflammatory responses,[ 28 ] apoptosis,[ 29 , 30 ] and IRI.[ 31 ] As a member of the RGS family, RGS14 harbors multiple protein binding domains and is an integrator of the G‐protein and MAPK signaling pathways.[ 32 ] However, whether RGS14 functions in hepatic IRI has not been reported. In this study, we found that expression of RGS14 increased during hepatic IRI in in vitro and in vivo models. Furthermore, RGS14 deficiency aggravated hepatic IRI, while RGS14 overexpression alleviated hepatic IRI. Mechanistically, we found that RGS14 deficiency activated the phosphorylation of TAK1–JNK/p38, while the effects of RGS14 deficiency could be blocked by TAK1 inhibitor. These findings suggest that RGS14 protects against hepatic IRI by inhibiting activation of TAK1 and downstream P38/JNK signaling.
The inflammatory response plays an important role in hepatic IRI, and inhibition of inflammation can effectively ameliorate the expression of factors associated with this type of injury.[ 33 , 34 ] Kupffer cell activation is a central event in the early stage of reperfusion because these cells release ROS and proinflammatory molecules, such as Il1b and Tnfα.[ 35 ] The proinflammatory cytokines Il1b and Tnfα can induce the activation of neutrophils and their migration from the endothelial lumen to the liver parenchyma, leading to neutrophil aggregation and adhesion.[ 36 ] In our study, the RGS14‐KO mice exhibited more inflammatory reactions and CD11b‐positive inflammatory cell infiltration in the liver tissues after IRI surgery. TNFα can also activate the MAPK and NF‐κB signaling pathways to further enhance the inflammatory response.[ 37 ] In addition to the production of inflammatory factors, a considerable number of chemokines further accelerate the recruitment and activation of neutrophils in the reperfusion stage[ 38 ] and promote the process of IRI. In our study, RGS14 deficiency resulted in an increased inflammatory response by promoting the NF‐κB signaling pathway, while RGS14 overexpression inhibited NF‐κB activation and down‐regulated the expression of Tnfα and Il1b at both the mRNA and protein levels. These results show that RGS14 can alleviate hepatic IRI by regulating inflammation.
An excessive inflammatory response ultimately induces apoptosis and necrosis.[ 27 ] Apoptosis occurs through extrinsic or intrinsic pathways that contribute to liver IRI.[ 39 ] Previous studies have shown that inhibition of apoptosis is a protective mechanism against hepatic IRI or other types of liver injury.[ 40 , 41 ] We found that RGS14 deficiency aggravated the apoptosis of liver IRI, causing an increase in the number of apoptotic and cleaved caspase‐3–positive cells. Concordantly, Bax, which promotes intrinsic apoptosis, showed increased expression, and Bcl2, which inhibits mitochondrial apoptosis, showed significantly decreased expression in RGS14‐KO liver tissues; additionally, RGS14 overexpression exerted the opposite effect and reduced the expression of apoptosis signaling factors.
RNA‐seq analysis identified that the MAPK pathway was the most significantly enriched pathway affected by RGS14 deficiency. There are three well‐characterized MAPK subgroups: ERKs, JNKs, and p38 MAPK. However, we found that RGS14 suppressed the activation of p38 and JNK, with no effect on ERK, indicating that RGS14 protected against hepatic ischemia–reperfusion (IR) progress thorough its regulation of p38/JNK signaling. Previous studies have demonstrated that activation of p38 and JNK can promote liver damage in IRI.[ 42 , 43 ] Once activated, JNK/p38 are translocated to the nucleus, where they phosphorylate and activate different transcription factors and transactivate target genes, resulting in the increase of inflammatory factors TNFα, IL1β, and Ccl2.[ 11 ] In addition, activation of p38 phosphorylation reportedly causes mitochondrial translocation of Bax, whereas activation of JNK phosphorylation can cause the phosphorylation of Bcl2.[ 44 ]
TAK1 (also known as MAP3K7) is a serine/threonine kinase located upstream of the MAPK pathway. When TAK1 is activated, it can be phosphorylated, ubiquitinated, acetylated, or glycosylated, resulting in the occurrence of corresponding reactions through these different signaling pathways.[ 45 ] Other studies have reported that several molecules or proteins can suppress TAK1 activation to alleviate hepatic IRI.[ 25 , 45 ] Moreover, it was reported that RGS5, which is also a member of the G‐protein family, can directly interact with TAK1 and regulate its activation in hepatocytes.[ 26 ] In the present study, we revealed that RGS14 interacted with TAK1 through the 374–566‐amino acid domain, and hepatic IRI caused by RGS14 deficiency was blocked by the TAK1 inhibitor 5Z‐7‐Ox. These results further demonstrate that RGS14 plays a protective role in hepatic IRI by suppressing the activation of TAK1. However, how RGS14 regulates TAK1 still needs to be further studied by searching for its binding protein through a protein spectrum.
In conclusion, we studied the role of RGS14 in hepatic IRI and found that RGS14 alleviated hepatic IRI through its interaction with TAK1 and the JNK/p38 signaling axis. Our findings provide insights into hepatic IRI and suggest that targeting RGS14 may be a potential strategy to confer protection against hepatic IRI.
CONFLICT OF INTEREST
None of the authors have any potential conflict of interests to disclose.
AUTHOR CONTRIBUTIONS
Jia‐Kai Zhang, Ming‐Jie Ding, Hui Liu, Ji‐Hua Shi, and Shui‐Jun Zhang participated in research design. Jia‐Kai Zhang, Ming‐Jie Ding, Hui Liu, Ji‐Hua Shi, Z‐H.W., Pei‐Hao Wen, Yi Zhang, Bing Yan, Dan‐Feng Guo, Xiao‐Dan Zhang, Ruo‐Lin Tao, Zhi‐Ping Yan, Yan Zhang, and Zhen Liu, conducted experiments. Jia‐Kai Zhang, Ming‐Jie Ding, Hui Liu, and Ji‐Hua Shi drafted the paper. Shui‐Jun Zhang supervised the study. All authors performed data analysis and interpretation and read and approved the final manuscript.
Supporting information
Supplementary Material
Zhang J‐K, Ding M‐J, Liu H, Shi J‐H, Wang Z‐H, Wen P‐H, et al. Regulator of G‐protein signaling 14 protects the liver from ischemia–reperfusion injury by suppressing TGF‐β‐activated kinase 1 activation. Hepatology. 2022;75:338–352. doi: 10.1002/hep.32133
Jia‐Kai Zhang, Ming‐Jie Ding, Hui Liu, and Ji‐Hua Shi contributed equally to this work.
Funding information
Supported by grants from the National Natural Science Foundation of China (81971881), Central‐China Science and Technology Innovative Talent Program in Henan Province (214200510027), The Medical Science and Technology Program of Henan Province China (SBGJ2018002 and SBGJ2018023, SB201901045). Gandanxiangzhao (GDXZ2019005)
Contributor Information
Wen‐Zhi Guo, Email: fccguowz@zzu.edu.cn.
Shui‐Jun Zhang, Email: zhangshuijun@zzu.edu.cn.
REFERENCES
- 1. Velmahos GC, Toutouzas KG, Radin R, Chan L, Rhee PM, Tillou A, et al. High success with nonoperative management of blunt hepatic trauma: the liver is a sturdy organ. Arch Surg. 2003;138:475–80. [DOI] [PubMed] [Google Scholar]
- 2. Sun P, Zhang P, Wang P‐X, Zhu L‐H, Du Y, Tian S, et al. Mindin deficiency protects the liver against ischemia/reperfusion injury. J Hepatol. 2015;63:1198–211. [DOI] [PubMed] [Google Scholar]
- 3. de Rougemont O, Lehmann K, Clavien PA. Preconditioning, organ preservation, and postconditioning to prevent ischemia–reperfusion injury to the liver. Liver Transpl. 2009;15:1172–82. [DOI] [PubMed] [Google Scholar]
- 4. Cursio R, Colosetti P, Gugenheim J. Autophagy and liver ischemia–reperfusion injury. Biomed Res Int. 2015;2015:417590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Y‐Q, Ding N, Zeng Y‐F, Xiang Y‐Y, Yang M‐W, Hong F‐F, et al. New progress in roles of nitric oxide during hepatic ischemia reperfusion injury. World J Gastroenterol. 2017;23:2505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jaeschke H. Molecular mechanisms of hepatic ischemia–reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284:G15–26. [DOI] [PubMed] [Google Scholar]
- 7. Gracia‐Sancho J, Villarreal G, Zhang Y, Yu JX, Liu Y, Tullius SG, et al. Flow cessation triggers endothelial dysfunction during organ cold storage conditions: strategies for pharmacologic intervention. Transplantation. 2010;90:142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu B, Qian J. Cytoprotective role of heme oxygenase‐1 in liver ischemia reperfusion injury. Int J Clin Exp Med. 2015;8:19867–73. [PMC free article] [PubMed] [Google Scholar]
- 9. Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125:1246–57. [DOI] [PubMed] [Google Scholar]
- 10. Llacuna L, Marí M, Lluis JM, García‐Ruiz C, Fernández‐Checa JC, Morales A. Reactive oxygen species mediate liver injury through parenchymal nuclear factor‐kappaB inactivation in prolonged ischemia/reperfusion. Am J Pathol. 2009;174:1776–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. King LA, Toledo AH, Rivera‐Chavez FA, Toledo‐Pereyra LH. Role of p38 and JNK in liver ischemia and reperfusion. J Hepatobiliary Pancreat Surg. 2009;16:763–70. [DOI] [PubMed] [Google Scholar]
- 12. Koelle MR. A new family of G‐protein regulators—the RGS proteins. Curr Opin Cell Biol. 1997;9:143–7. [DOI] [PubMed] [Google Scholar]
- 13. Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. RGS family members: GTPase‐activating proteins for heterotrimeric G‐protein alpha‐subunits. Nature. 1996;383:172–5. [DOI] [PubMed] [Google Scholar]
- 14. Evans PR, Lee SE, Smith Y, Hepler JR. Postnatal developmental expression of regulator of G protein signaling 14 (RGS14) in the mouse brain. J Comp Neurol. 2014;522:186–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ladds G, Zervou S, Vatish M, Thornton S, Davey J. Regulators of G protein signalling proteins in the human myometrium. Eur J Pharmacol. 2009;610:23–8. [DOI] [PubMed] [Google Scholar]
- 16. Martin‐McCaffrey L, Willard FS, Oliveira‐dos‐Santos AJ, Natale DRC, Snow BE, Kimple RJ, et al. RGS14 is a mitotic spindle protein essential from the first division of the mammalian zygote. Dev Cell. 2004;7:763–9. [DOI] [PubMed] [Google Scholar]
- 17. Martin‐McCaffrey L, Willard FS, Pajak A, Dagnino L, Siderovski DP, D'Souza SJ. RGS14 is a microtubule‐associated protein. Cell Cycle. 2005;4:953–60. [DOI] [PubMed] [Google Scholar]
- 18. Lim J, Thompson J, May RC, Hotchin NA, Caron E. Regulator of G‐protein signalling‐14 (RGS14) regulates the activation of αMβ2 integrin during phagocytosis. PLoS ONE. 2013;8:e69163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cho H, Kozasa T, Takekoshi K, De Gunzburg J, Kehrl J. RGS14, a GTPase‐activating protein for Gialpha, attenuates Gialpha‐ and G13alpha‐mediated signaling pathways. Mol Pharmacol. 2000;58:569–76. [DOI] [PubMed] [Google Scholar]
- 20. Moratz C, Harrison K, Kehrl JH. Regulation of chemokine‐induced lymphocyte migration by RGS proteins. Methods Enzymol. 2004;389:15–32. [DOI] [PubMed] [Google Scholar]
- 21. Lee SE, Simons SB, Heldt SA, Zhao M, Schroeder JP, Vellano CP, et al. RGS14 is a natural suppressor of both synaptic plasticity in CA2 neurons and hippocampal‐based learning and memory. Proc Natl Acad Sci U S A. 2010;107:16994–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vellano CP, Brown NE, Blumer JB, Hepler JR. Assembly and function of the regulator of G protein signaling 14 (RGS14)·H‐Ras signaling complex in live cells are regulated by Gαi1 and Gαi‐linked G protein‐coupled receptors. J Biol Chem. 2013;288:3620–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang P‐X, Zhang R, Huang L, Zhu L‐H, Jiang D‐S, Chen H‐Z, et al. Interferon regulatory factor 9 is a key mediator of hepatic ischemia/reperfusion injury. J Hepatol. 2015;62:111–20. [DOI] [PubMed] [Google Scholar]
- 24. Chen S‐Y, Zhang H‐P, Li J, Shi J‐H, Tang H‐W, Zhang YI, et al. Tripartite motif‐containing 27 attenuates liver ischemia/reperfusion injury by suppressing transforming growth factor β–activated kinase 1 (TAK1) by TAK1 binding protein 2/3 degradation. Hepatology. 2021;73:738–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yan Z‐Z, Huang Y‐P, Wang X, Wang H‐P, Ren F, Tian R‐F, et al. Integrated omics reveals tollip as a regulator and therapeutic target for hepatic ischemia–reperfusion injury in mice. Hepatology. 2019;70:1750–69. [DOI] [PubMed] [Google Scholar]
- 26. Wang J, Ma J, Nie H, Zhang X‐J, Zhang P, She Z‐G, et al. Hepatic regulator of G protein signaling 5 ameliorates nonalcoholic fatty liver disease by suppressing transforming growth factor beta–activated kinase 1‐c‐Jun‐N‐terminal kinase/p38 signaling. Hepatology. 2021;73:104–25. [DOI] [PubMed] [Google Scholar]
- 27. Kan C, Ungelenk L, Lupp A, Dirsch O, Dahmen U. Ischemia–reperfusion injury in aged livers—the energy metabolism, inflammatory response, and autophagy. Transplantation. 2018;102:368–77. [DOI] [PubMed] [Google Scholar]
- 28. Fang XI, Chung J, Olsen E, Snider I, Earls RH, Jeon J, et al. Depletion of regulator‐of‐G‐protein signaling‐10 in mice exaggerates high‐fat diet–induced insulin resistance and inflammation, and this effect is mitigated by dietary green tea extract. Nutr Res. 2019;70:50–9. [DOI] [PubMed] [Google Scholar]
- 29. Wang Z, Huang HE, He W, Kong B, Hu HE, Fan Y, et al. Regulator of G‐protein signaling 5 protects cardiomyocytes against apoptosis during in vitro cardiac ischemia–reperfusion in mice by inhibiting both JNK1/2 and P38 signaling pathways. Biochem Biophys Res Commun. 2016;473:551–7. [DOI] [PubMed] [Google Scholar]
- 30. Huang J, Stewart A, Maity B, Hagen J, Fagan RL, Yang J, et al. RGS6 suppresses Ras‐induced cellular transformation by facilitating Tip60‐mediated Dnmt1 degradation and promoting apoptosis. Oncogene. 2014;33:3604–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Siedlecki AM, Jin X, Thomas W, Hruska KA, Muslin AJ. RGS4, a GTPase activator, improves renal function in ischemia–reperfusion injury. Kidney Int. 2011;80:263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shu F, Ramineni S, Hepler J. RGS14 is a multifunctional scaffold that integrates G protein and Ras/Raf MAPkinase signalling pathways. Cell Signal. 2010;22:366–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu L, Zhou H, Ni M, Wang X, Busuttil R, Kupiec‐Weglinski J, et al. Innate immune regulations and liver ischemia–reperfusion injury. Transplantation. 2016;100:2601–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nastos C, Kalimeris K, Papoutsidakis N, Tasoulis M‐K, Lykoudis PM, Theodoraki K, et al. Global consequences of liver ischemia/reperfusion injury. Oxid Med Cell Longev. 2014;2014:906965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ni D, Wei H, Chen W, Bao Q, Rosenkrans ZT, Barnhart TE, et al. Ceria nanoparticles meet hepatic ischemia–reperfusion injury: the perfect imperfection. Adv Mater. 2019;31:e1902956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Welborn MB 3rd, Moldawer LL, Seeger JM, Minter RM, Huber TS. Role of endogenous interleukin‐10 in local and distant organ injury after visceral ischemia–reperfusion. Shock. 2003;20:35–40. [DOI] [PubMed] [Google Scholar]
- 37. Colletti LM, Cortis A, Lukacs N, Kunkel SL, Green M, Strieter RM. Tumor necrosis factor up‐regulates intercellular adhesion molecule 1, which is important in the neutrophil‐dependent lung and liver injury associated with hepatic ischemia and reperfusion in the rat. Shock. 1998;10:182–91. [DOI] [PubMed] [Google Scholar]
- 38. Djeu JY, Matsushima K, Oppenheim JJ, Shiotsuki K, Blanchard DK. Functional activation of human neutrophils by recombinant monocyte‐derived neutrophil chemotactic factor/IL‐8. J Immunol. 1990;144:2205–10. [PubMed] [Google Scholar]
- 39. Deng J, Feng J, Liu T, Lu X, Wang W, Liu N, et al. Beraprost sodium preconditioning prevents inflammation, apoptosis, and autophagy during hepatic ischemia–reperfusion injury in mice via the P38 and JNK pathways. Drug Des Devel Ther. 2018;12:4067–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Feng J, Zhang Q, Mo W, Wu L, Li S, Li J, et al. Salidroside pretreatment attenuates apoptosis and autophagy during hepatic ischemia–reperfusion injury by inhibiting the mitogen‐activated protein kinase pathway in mice. Drug Des Devel Ther. 2017;11:1989–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu L, Wang C, Li J, Li S, Feng J, Liu T, et al. Hepatoprotective effect of quercetin via TRAF6/JNK pathway in acute hepatitis. Biomed Pharmacother. 2017;96:1137–46. [DOI] [PubMed] [Google Scholar]
- 42. Liu T, Xia Y, Li J, Li S, Feng J, Wu L, et al. Shikonin attenuates concanavalin A–induced acute liver injury in mice via inhibition of the JNK pathway. Mediators Inflamm. 2016;2016:2748367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qin J‐J, Mao W, Wang X, Sun P, Cheng D, Tian S, et al. Caspase recruitment domain 6 protects against hepatic ischemia/reperfusion injury by suppressing ASK1. J Hepatol. 2018;69:1110–22. [DOI] [PubMed] [Google Scholar]
- 44. Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1‐mediated phosphorylation of Bcl‐2 regulates starvation‐induced autophagy. Mol Cell. 2008;30:678–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hirata Y, Takahashi M, Morishita T, Noguchi T, Matsuzawa A. Post‐translational modifications of the TAK1–TAB complex. Int J Mol Sci. 2017;18:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guo W‐Z, Fang H‐B, Cao S‐L, Chen S‐Y, Li J, Shi J‐H, et al. Six‐transmembrane epithelial antigen of the prostate 3 deficiency in hepatocytes protects the liver against ischemia–reperfusion injury by suppressing transforming growth factor‐β–activated kinase 1. Hepatology. 2020;71:1037–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material