

Abstract
Background
Mevidalen is a selective positive allosteric modulator (PAM) of the dopamine D1 receptor subtype.
Objective
To assess the safety and efficacy of mevidalen for treatment of cognition in patients with Lewy body dementia (LBD).
Methods
PRESENCE was a phase 2, 12‐week study in participants with LBD (N = 344) randomly assigned (1:1:1:1) to daily doses of mevidalen (10, 30, or 75 mg) or placebo. The primary outcome measure was change from baseline on Cognitive Drug Research Continuity of Attention (CoA) composite score. Secondary outcomes included Alzheimer's Disease Assessment Scale‐Cognitive Subscale 13 (ADAS‐cog13), Movement Disorder Society‐Unified Parkinson's Disease Rating Scale (MDS‐UPDRS), and Alzheimer's Disease Cooperative Study‐Clinical Global Impression of Change (ADCS‐CGIC). Numerous safety measures were collected.
Results
Mevidalen failed to meet primary or secondary cognition endpoints. Mevidalen resulted in significant, dose‐dependent improvements of MDS‐UPDRS total score (sum of Parts I−III, 10 mg P < 0.05, 30 mg P < 0.05, 75 mg P < 0.01, compared to placebo). The 30 mg and 75 mg mevidalen doses significantly improved ADCS‐CGIC scores compared to placebo (minimal or better improvement: 30 mg P < 0.01, 75 mg P < 0.01; moderate or better improvement: 30 mg P < 0.05, 75 mg P < 0.001). Increases in blood pressure, adverse events, and cardiovascular serious adverse events were most pronounced at the 75 mg dose.
Conclusions
Mevidalen harnesses a novel mechanism of action that improves motor symptoms associated with LBD on top of standard of care while improving or not worsening non‐motor symptoms associated with traditional dopaminergic therapy. © 2021 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society
Keywords: Lewy body dementia, selective positive allosteric modulator (PAM), dopamine D1 receptor, cognition, motor impairment
Lewy body dementia (LBD), including both Parkinson's disease dementia (PDD) and dementia with Lewy bodies (DLB), is the second most common neurodegenerative dementia after Alzheimer's disease (AD). 1 Intraneuronal inclusions of Lewy bodies throughout subcortical and cortical brain regions, containing primarily misfolded and aggregated α‐synuclein, are histopathological hallmarks of LBD. 2
Although once considered as two separate entities, the constellation of supportive pathological, clinical, imaging, and neurochemical data suggests PDD and DLB fall within a spectrum of the same disease. 3 , 4 , 5 , 6 , 7 They share neuroimaging characteristics with overlapping patterns of atrophy, glucose utilization, and neurotransmitter changes (cortical cholinergic deficits 8 and striatal/cortical dopaminergic deficits 9 ). Lewy body formation and propagation is accompanied by progressive neurodegeneration, particularly affecting the dopaminergic and cholinergic neurons 9 , 10 , 11 leading to overlap of the motor and cognitive impairments in PDD and DLB. Progressive executive dysfunction and visual–spatial abnormalities are also present in both PDD and DLB, but episodic memory remains relatively intact early in the disease process. 12 In addition, non‐motor features are similar for both PDD and LBD and include rapid eye movement (REM) sleep behavior disorder, hyposmia, prominent visual hallucinations, fluctuations in arousal and hypersomnolence, autonomic dysfunction, and depression/anxiety. 9
LBD is a multifaceted disease with core motor and non‐motor features often resulting in polypharmacy balancing symptomatic efficacy with medication side effects. The use of standard dopaminergic therapies is limited by non‐motor complications often resulting in under‐treatment of motor symptoms, 13 , 14 while rivastigmine remains the only medication approved in the United States (US) to treat cognitive impairment in PDD. 15 Additionally, use of medications to treat somnolence/fatigue, psychosis, cognition, and orthostatic hypotension may compound the side effect burden. 13 , 14
Mevidalen (LY3154207) represents a novel mechanism, a selective positive allosteric modulator (PAM) of the dopamine D1 receptor subtype (D1PAM). 16 It acts by increasing D1 receptor tone and its affinity for dopamine, thus amplifying the response to dopamine when and where it is released. 17 D1 agonists activate all D1 receptors they have access to, for the entire duration they are present, and often show bell‐shaped, dose−response relationships due to overstimulation at high doses and the tendency for tolerance development. 18 , 19 , 20 In contrast mevidalen, by potentiating the response to the remaining brain dopamine (or administered levodopa), would be subject to normal feedback control and have a lower propensity for overstimulation. Mevidalen has the potential to improve cognitive performance through enhancing frontal dopaminergic neurotransmission, activation of cortical neurons, enhancing synaptic plasticity, and D1‐mediated enhanced acetylcholine release. 17 Other potential effects observed in preclinical and phase 1 trials of mevidalen such as reduced daytime sleepiness, enhanced motor function, improved mood, and goal‐directed behaviors leading to reduced apathy (via activation of cortical and striatal D1 receptors) would also be beneficial in LBD. 17 , 21
The aim of this phase 2 proof‐of‐concept study (PRESENCE; NCT03305809) was to assess the safety and efficacy of mevidalen in patients with mild‐to‐moderate LBD (PDD or DLB) compared with placebo. The primary objective was to test the hypothesis that treatment with mevidalen administered for 12 weeks at 10, 30, or 75 mg daily would result in a significant improvement in cognition. Secondary objectives aimed to assess the impact of mevidalen treatment on the motor and non‐motor aspects of LBD.
1. Patients and Methods
1.1. Patient Population and Trial Design
PRESENCE (NCT03305809; ClinicalTrials.gov) was a multicenter, randomized, double‐blind, parallel‐group, placebo‐controlled, fixed‐dosage, phase 2 trial assessing the safety and efficacy of mevidalen (LY3154207) in participants with mild‐to‐moderate dementia associated with LBD (PDD and DLB). PRESENCE was initiated on November 9, 2017 and the last patient visit was on July 10, 2020. The study included a screening period of 7 to 14 days (visit 1–2), a pretreatment period of ≥11 days and ≤17 days (visit 2–3), a 12‐week treatment period (visits 3–11), and a 14‐day safety follow‐up period that included a 2‐week follow‐up visit (visit 12). An amendment (b) was introduced to the protocol after approximately half of the participants were enrolled to explicitly include individuals who met criteria for DLB, 22 to add a telephone visit (at visit 11 + 2 days), and to assess potential withdrawal symptoms.
Individuals aged 40–85 years with dementia (defined by a decline in cognition associated with functional impairment and with a Montreal Cognitive Assessment [MoCA] score of 10–23 [inclusive]) were enrolled. 23 Prospective participants were also required to meet the Movement Disorder Society‐Unified Parkinson's Disease Rating Scale (MDS‐UPDRS) diagnostic criteria for PD 24 or (after amendment b) McKeith criteria for DLB 22 with a modified Hoehn and Yahr (H&Y) stage of 0–4. 25 Participants being treated with anti‐parkinsonian agents, cognitive medications, antidepressant medications, or allowable antipsychotic medications were required to be on stable dosage for at least 3 weeks prior to screening, and to remain on stable dosage throughout the study. Finally, participants were required to have the support of a reliable study partner. Participants with a recent history of cardiac or cerebrovascular disease, on concomitant CYP3A4 inhibitors or inducers, or with elevated blood pressure (BP) at screening were excluded. Eligible participants were randomly assigned 1:1:1:1 to receive either orally administered mevidalen (10, 30, or 75 mg) or placebo once daily for a period of 12 weeks.
1.2. Oversight
The study was conducted at 77 sites in the US, Canada, and Puerto Rico. The trial received approval from the relevant ethics committees and participants provided written informed consent for study participation. The study was conducted in accordance with the Declaration of Helsinki and the Council for International Organizations of Medical Sciences International Ethical Guidelines. The sponsor held regularly scheduled trial level safety reviews on blinded data. An internal assessment committee (IAC) held unblinded safety data reviews to ensure the continuing safety of enrolled participants.
1.3. Clinical Outcome Assessments
1.3.1. Efficacy
1.3.1.1. Cognition
The primary objective was to assess the effect of mevidalen on cognition, as measured by the change in the Continuity of Attention (CoA) composite score of the Cognitive Drug Research‐Computerized Cognition Battery (CDR‐CCB) from baseline to week 12. 26 The CoA component of the CDR‐CCB was selected as the primary endpoint for this study as it measures attention and executive function, domains relevant to LBD and associated with frontostriatal D1 activity; is resistant to learning and confounding motor effects; and has demonstrated significant treatment effects in prior LBD trials. 27 , 28 A higher CoA score indicates better cognition on this scale. A secondary cognitive objective was to assess the effect of mevidalen on the 13‐item Alzheimer's Disease Assessment Scale‐Cognitive Subscale 13 (ADAS‐Cog13) score. The ADAS‐Cog13 is composed of the original 11‐item ADAS‐Cog, which measures memory, language, and praxis 29 but additionally includes Delayed Recall (episodic memory) and Number Cancellation (attention) items that have been shown as reliable and sensitive measures for assessing a wide range of dementia severity. 30 A higher ADAS‐Cog13 score indicates greater cognitive impairment. Cognitive tests were performed in the practically defined ON state. This was defined as best motor function or 1 hour after last dose of levodopa.
1.3.1.2. Other Secondary Objectives
Effect of mevidalen on PD severity was evaluated using the MDS‐UPDRS. 31 The scale is composed of four parts: MDS‐UPDRS Part I (non‐motor aspects of experiences of daily living) assesses a wide spectrum of non‐motor symptoms of PD, Part II (motor aspects of experience of daily living), Part III (motor examination), and Part IV (motor complications) used to assess the impact of treatment on motor complications (dyskinesias and motor fluctuations). The MDS‐UPDRS III was operationalized to be performed in the practically defined ON state (defined earlier). The MDS‐UPDRS total score is a sum of Parts I, II, and III. A higher score indicates greater severity or impairment.
Global efficacy was evaluated using the Alzheimer's Disease Cooperative Study‐Clinical Global Impression of Change (ADCS‐CGIC). This is a seven‐point, categorical scale that provides a single global rating of change from baseline 32 and includes a baseline interview to set subject‐specific probes for the follow‐up assessments. A score of 1 indicates marked improvement; 2, moderate improvement; 3, minimal improvement; 4, no change; 5, minimal worsening; 6, moderate worsening; and 7, marked worsening.
The effect of mevidalen on daytime sleepiness was evaluated using the Epworth Sleepiness Scale (ESS), a widely used scale in the field of sleep medicine as a subjective measure of a subject's sleepiness. It is an eight‐item questionnaire that measures one's chances of ‘dozing off’ in different daytime situations over the past week. Scores range from 0 to 24, with scores ≥10 indicating excessive daytime sleepiness. 33
1.3.2. Safety
Safety outcomes included assessments of reported adverse events (AEs), clinical laboratory tests, vital signs (including 6–8 hour in‐clinic BP and pulse rate [PR] monitoring), electrocardiograms, Columbia Suicide Severity Rating Scale, physical and neurological examinations, Questionnaire for Impulsive‐Compulsive Disorders in Parkinson's Disease (QUIP), and physical withdrawal symptoms.
1.3.3. Determination of Sample Size and Statistical Analysis
Positive outcome of the study was predefined as posterior probability of >0.67 that the effect size of the improvement of mevidalen over placebo on the CDR‐CoA scale was ≥0.2 points. If the true effect size for mevidalen relative to placebo on the CoA was 0.4, with the sample size of 344, there would be >80% probability of passing this criterion.
Participants were randomly assigned, in a blinded fashion, to one of the four treatments arms (placebo, LY3154207 10 mg, LY3154207 30 mg, LY3154207 75 mg) in a 1:1:1:1 ratio using a minimization procedure in order to balance for investigator, site, and current use of acetylcholinesterase inhibitors (AChEIs) (Yes, No) using an interactive web‐response system.
Unless otherwise specified, all efficacy analyses were conducted on the evaluable patient population (EPP). EPP included all data from all randomly assigned participants who received at least one dose of study drug and had the baseline efficacy assessment and at least one post‐dose efficacy assessment. Following reports of four vascular‐related serious adverse events (SAEs), occurring proximally to the initiation of the 75 mg mevidalen dose, and in conjunction with data from prior IAC reviews, the IAC recommended discontinuing the 75 mg dose group. The study was fully enrolled at the time of this decision, therefore all participants remaining in the study and randomly assigned to the 75 mg dose (14 participants at 13 sites) were prematurely discontinued from the study. No changes to the other doses, study conduct, or safety monitoring were made. The 14 participants who discontinued in the 75 mg mevidalen group were not included in the efficacy analyses but were included in the safety analyses. The primary and secondary efficacy analyses utilized a Bayesian mixed model repeated measures (MMRM) model. The MMRM model accounted for longitudinal data assessed throughout the study, after 1, 2, 4, 6, 8, 10, and 12 weeks of dosing. No adjustments were made for multiple comparisons.
Safety analyses were conducted on all participants who received at least one dose of mevidalen or placebo. Change in BP and PR from baseline up to week 12 were analyzed by an MMRM model with baseline value, treatment, and the relevant interaction terms.
2. Results
2.1. Trial Population and Baseline Characteristics
Of the 640 participants screened for inclusion in the trial, 344 participants were randomly assigned to receive mevidalen or placebo. The single most common reason for screen failure was out of range MoCA score, with most excluded patients recording scores that were above the allowable range. Participant allocation and flow through the trial are shown in Figure 1. The baseline demographic and clinical characteristics of these 344 participants are well‐balanced and shown in Table 1, although there was an imbalance of participants enrolled with a diagnosis for PDD or LBD, both overall and across treatment arms.
FIG. 1.
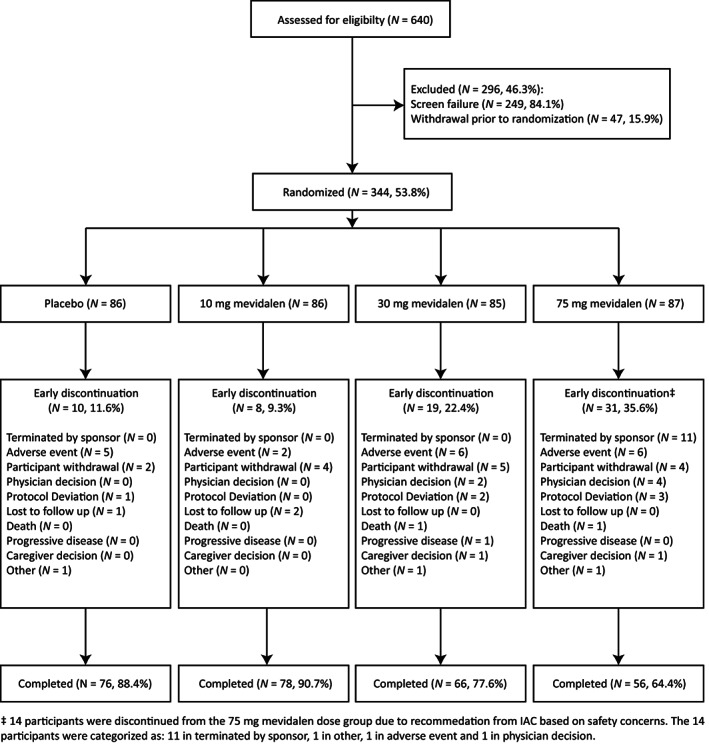
Enrollment, randomization, and treatment completion in PRESENCE. Abbreviation: N, number of participants.
TABLE 1.
Characteristics of participants at baseline
| Baseline demographics | Placebo (N = 86) | 10 mg LY3154207 (N = 86) | 30 mg LY3154207 (N = 85) | 75 mg LY3154207 (N = 87) | Overall (N = 344) |
|---|---|---|---|---|---|
| Male, % | 81.4 | 84.9 | 82.4 | 82.8 | 82.8 |
| Age, mean | 73.1 | 72.5 | 71.9 | 73.0 | 72.6 |
| White, % | 91.9 | 94.2 | 95.3 | 97.7 | 94.8 |
| AChEI use, % | 44.2 | 39.5 | 44.7 | 41.4 | 42.4 |
| DLB, % | 50.6 | 38.1 | 36.6 | 26.2 | 37.8* |
| PDD, % | 49.4 | 61.9 | 63.4 | 73.8 | 62.2* |
| Concomitant PD therapy use, % | 83.7 | 86.0 | 83.5 | 89.7 | 85.8 |
| LEDD (mg), mean | 621.2 | 610.1 | 606.9 | 677.8 | 629.5 |
|
MoCA Total score, % |
|||||
| Mild 17 , 18 , 19 , 20 , 21 , 22 , 23 | 68.6 | 69.8 | 80.0 | 69.0 | 71.8 |
| Moderate 10 , 11 , 12 , 13 , 14 , 15 , 16 | 31.4 | 30.2 | 20.0 | 31.0 | 28.2 |
| Clinical characteristics, mean (SD) | |||||
| CDR‐CoA | 28.15 (7.23) | 28.84 (7.50) | 28.81 (12.15) | 28.98 (6.19) | 28.70 (8.53) |
| ADAS‐cog13 | 27.52 (8.67) | 27.77 (11.22) | 25.07 (9.42) | 26.24 (10.32) | 26.65 (9.97) |
| MDS‐UPDRS | |||||
| Total | 61.77 (24.57) | 63.33 (25.19) | 64.10 (24.15) | 69.44 (23.47) | 64.70 (24.41) |
| Part I | 12.45 (6.04) | 13.03 (6.32) | 13.06 (6.34) | 13.00 (4.84) | 12.89 (5.89) |
| Part II | 15.92 (9.12) | 14.66 (8.39) | 15.26 (8.05) | 16.78 (7.96) | 15.66 (8.39) |
| Part III | 33.14 (14.68) | 35.85 (15.72) | 35.58 (14.71) | 39.35 (15.11) | 36.02 (15.16) |
| ESS | 10.88 (5.68) | 9.53 (4.82) | 9.32 (4.91) | 10.17 (5.83) | 9.98 (5.35) |
| MoCA total | 18.02 (3.54) | 18.19 (3.21) | 18.80 (3.00) | 17.89 (3.85) | 18.22 (3.42) |
| Modified H&Y | 2.53 (0.75) | 2.42 (0.67) | 2.51 (0.67) | 2.56 (0.80) | 2.51 (0.72) |
P < 0.05.
Abbreviations: N, number of participants; AChEI, acetylcholinesterase inhibitor; DLB, dementia with Lewy bodies; PDD, Parkinson's disease dementia; PD, Parkinson disease; LEDD, levodopa equivalent daily dose; MoCA, Montreal Cognitive Assessment; SD, standard deviation; CDR‐CoA, Cognitive Drug Research‐Continuity of Attention composite score; ADAS‐cog13, Alzheimer's Disease Assessment Scale‐Cognitive Subscale 13; MDS‐UPDRS, Movement Disorder Society‐Unified Parkinson Disease Rating Scale; ESS, Epworth Sleepiness Scale; H&Y, Hoehn and Yahr.
2.2. Cognitive Outcomes
The primary efficacy endpoint of change in the CoA composite score of the CDR‐CCB from baseline to week 12 was not met for any of the three doses of mevidalen (10, 30, and 75 mg) relative to placebo (Fig. 2A). The secondary cognitive outcome, ADAS‐cog13 scale change from baseline to week 12, did not show any statistically significant differences (P > 0.05) between mevidalen (10, 30, and 75 mg) and placebo (Fig. 2B). The difference from placebo in change from baseline for mevidalen in CDR‐CoA was +0.15 for 10 mg (95% CI −1.206, 1.496), +0.96 for 30 mg (95% CI −0.448, 2.362), and +0.26 for 75 mg (95% CI −1.310, 1.801). The difference from placebo in change from baseline for mevidalen for ADAS‐cog13 was −1.11 for 10 mg (95% CI −2.34, 1.54), −1.42 for 30 mg (95% CI −2.70, 1.28), and −1.59 for 75 mg (95% CI −2.98, 1.21). In addition, there was no statistically significant difference between the groups at any time point during mevidalen treatment in either CDR‐CoA or ADAS‐cog13.
FIG. 2.
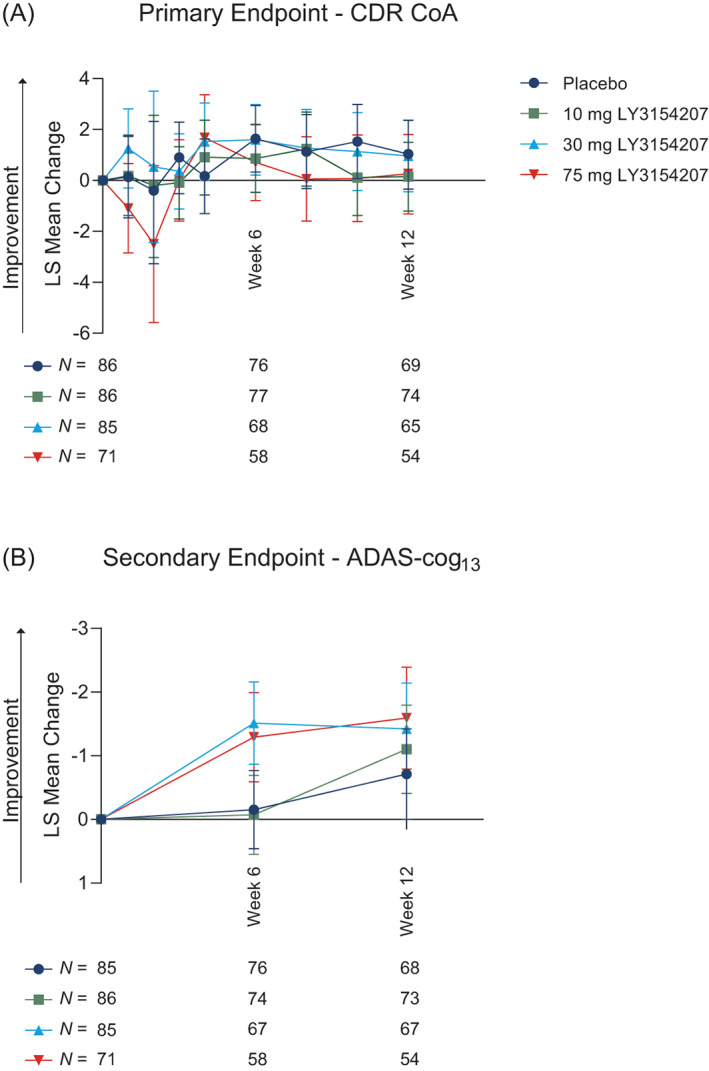
Cognitive outcomes using Cognitive Drug Research‐Continuity of Attention (CDR‐CoA) and Alzheimer's Disease Assessment Scale‐Cognitive Subscale 13 (ADAS‐cog13). CDR‐CoA (A) was the primary endpoint, while ADAS‐cog13 (B) was a key secondary endpoint in the PRESENCE trial. P values between treatment groups were non‐significant and are not shown. N numbers per group are shown below the associated time point. Abbreviations: LS, least squares; N, number of participants.
2.3. Other Secondary Efficacy Outcomes
In addition to cognition, PRESENCE was designed to evaluate the effect of mevidalen on other clinical features relevant to LBD, including motor function and non‐motor symptoms (wakefulness, mood, apathy, etc.). Mevidalen (10, 30, and 75 mg, compared to placebo) resulted in a significant improvement of MDS‐UPDRS total score (sum of Parts I−III) (Fig. 3A) and in MDS‐UPDRS Part I (Fig. 3B) from baseline to week 12. The 30 mg and 75 mg mevidalen doses also showed statistically significant improvements in MDS‐UPDRS Part II (Fig. 3C), while only 75 mg mevidalen showed a significant improvement in MDS‐UPDRS Part III (Fig. 3D). Detailed analyses of the MDS‐UPDRS subscales are shown in Figure S1. Of note, mevidalen treatment resulted in significant changes from baseline in items relevant for LBD including reductions in fatigue (P < 0.05 for 10 mg, P < 0.01 for 30 mg, and P < 0.05 for 75 mg); hallucinations and psychosis (P < 0.01 for 75 mg); daytime sleepiness (P < 0.05 for 10 mg, P < 0.01 for 30 mg, and P < 0.001 for 75 mg); and increase in time spent with dyskinesia (P < 0.01 for 75 mg).
FIG. 3.
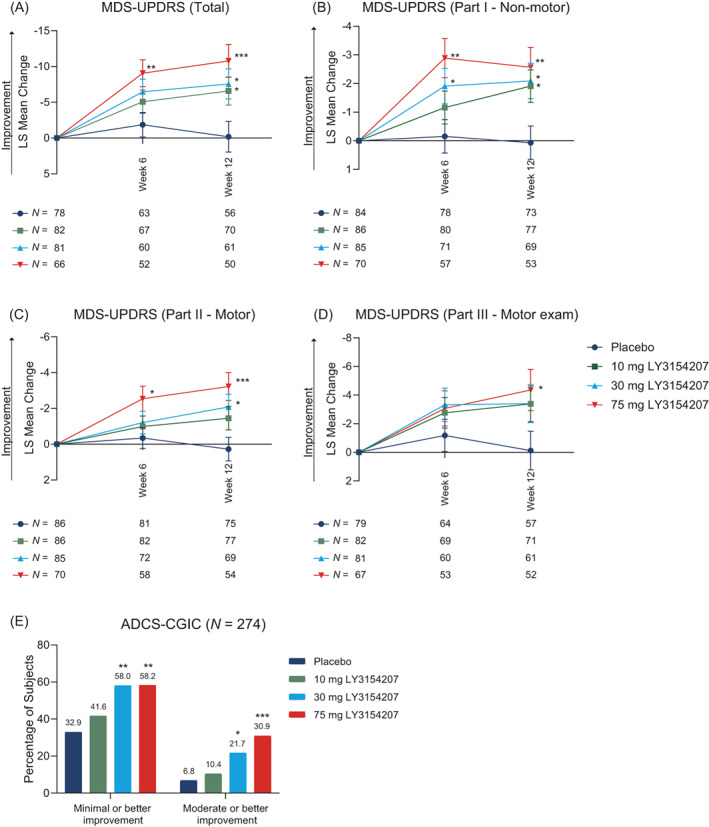
Motor and non‐motor secondary outcomes assessed by Movement Disorder Society‐Unified Parkinson Disease Rating Scale (MDS‐UPDRS) and global functioning assessed by Alzheimer's Disease Cooperative Study‐Clinical Global Impression of Change (ADCS‐CGIC). Total MDS‐UPDRS is the sum of Parts I−III (A), Part I (B), Part II (C), and Part III (D). Change in global functioning by ADCS‐CGIC are shown in (E). N numbers per group are shown below the associated time point. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: LS, least squares; N, number of participants.
When assessing improvement from baseline to week 12 in global functioning relative to placebo, the higher doses of mevidalen (30 mg and 75 mg) resulted in a statistically significant improvement in the ADCS‐CGIC score compared to placebo (minimal or better improvement: 30 mg P < 0.01, 75 mg P < 0.01; moderate or better improvement: 30 mg P < 0.05, 75 mg P < 0.001). Improvement in the ADCS‐CGIC score was not significant with 10 mg mevidalen treatment (Fig. 3E). Treatment with 75 mg mevidalen resulted in a significant improvement from baseline to week 12 compared to placebo (P < 0.05) in daytime sleepiness as measured by the ESS. Improvement in the ESS score was not significant with 10 mg or 30 mg mevidalen treatment (Fig. S2).
2.4. Safety and Summary of Adverse Events
Table 2 summarizes SAEs and TEAEs. Two deaths occurred during the study, one due to septic shock and considered by the investigator as unrelated to study treatment (30 mg mevidalen group) and the other cause was unknown but was considered by the investigator as related to study treatment (75 mg mevidalen group). A total of 21 (6.1%) participants in PRESENCE experienced at least one SAE with 47.6% of these occurring in the 75 mg group. Reports from four participants with vascular‐related SAEs, occurring in close temporal proximity to initiation of the 75 mg mevidalen dose, led the IAC to recommend discontinuing the 75 mg mevidalen group. The SAEs reported by these participants included congestive heart failure, hypertension, stroke, and hypertensive encephalopathy. A total of 214 participants (62.2%) in PRESENCE had at least one treatment‐emergent adverse event (TEAE) across all groups with a significant number of these occurring in the treatment groups (P < 0.05), and 74.7% of the 75 mg mevidalen group experiencing a TEAE (P < 0.01). The most common TEAE was falls, occurring in 10.2% of the total participants, with the highest proportion of these occurring in the 10 mg and 75 mg mevidalen groups (14.0% and 13.8%, respectively). There was an overall statistically significant incidence of fatigue (P < 0.05), headache (P < 0.05), and vomiting (P < 0.05) in treatment groups versus placebo, while incidence of headache also increased in the 30 mg mevidalen group (P < 0.05) in comparison to other treatment groups. Assessment for impulse control disorder symptoms was performed using QUIP for the duration of the study and no effect of mevidalen treatment was observed. Mevidalen treatment (75 mg) demonstrated persistent relative increases in systolic BP over 12 weeks (average increase of 5.67 ± 1.92 mmHg compared to baseline) (Fig. S3A) with minimal effects in diastolic BP (average increase of 1.24 ± 1.14 mmHg compared to baseline) (Fig. S3B). Additionally, an acute increase in PR was associated with the 75 mg mevidalen dose, which accommodated over the 12‐week treatment period (Fig. S3C).
TABLE 2.
Summary of safety outcomes/adverse events in PRESENCE
| Event, N (%) | Placebo (N = 86) | 10 mg LY3154207 (N = 86) | 30 mg LY3154207 (N = 85) | 75 mg LY3154207 (N = 87) | Overall (N = 344) |
|---|---|---|---|---|---|
| Deaths | 0 (0.0) | 0 (0.0) | 1 (1.2) | 1 (1.1) | 2 (0.6) |
| SAEs | 3 (3.5) | 5 (5.8) | 3 (3.5) | 10 (11.5) | 21 (6.1) |
| Participants with ≥1 TEAE | 44 (51.2) | 50 (58.1) | 55 (64.7) | 65 (74.7) ** | 214 (62.2) * |
| TEAEs occurring in at least 5% of participants, N (%) | |||||
| Falls | 4 (4.7) | 12 (14.0) | 7 (8.2) | 12 (13.8) | 35 (10.2) |
| Dizziness | 4 (4.7) | 4 (4.7) | 6 (7.1) | 10 (11.5) | 24 (7.0) |
| Nausea | 3 (3.5) | 2 (2.3) | 7 (8.2) | 8 (9.2) | 20 (5.8) |
| Hallucination | 4 (4.7) | 2 (2.3) | 5 (5.9) | 9 (10.3) | 20 (5.8) |
| Fatigue | 4 (4.7) | 2 (2.3) | 1 (1.2) | 9 (10.3) | 16 (4.7) * |
| Headache | 1 (1.2) | 2 (2.3) | 8 (9.4) * | 7 (8.0) | 18 (5.2) * |
| Dyskinesia | 2 (2.3) | 2 (2.3) | 3 (3.5) | 5 (5.7) | 12 (3.5) |
| Vomiting | 3 (3.5) | 0 (0.0) | 2 (2.4) | 7 (8.0) | 12 (3.5) * |
| Insomnia | 0 (0.0) | 4 (4.7) | 4 (4.7) | 5 (5.7) | 13 (3.8) |
| Diarrhea | 1 (1.2) | 2 (2.3) | 5 (5.9) | 2 (2.3) | 10 (2.9) |
Safety analysis included the 14 participants that were discontinued due to sponsor decision. Figures in bold type denote statistical significance.
P < 0.05.
P < 0.01.
Abbreviations: N, number of participants; SAE, serious adverse event; TEAE, treatment‐emergent adverse event.
3. Discussion
In this phase 2 trial, mevidalen failed to demonstrate cognitive efficacy in mild‐to‐moderate LBD. Despite this, mevidalen treatment resulted in robust global improvement as measured by the ADCS‐CGIC, a key secondary outcome that is often used as a registration co‐primary endpoint in therapeutic trials in dementia. This robust global improvement appears to be driven by improvements in secondary measures of motor and non‐motor function relevant to individuals with LBD.
Preclinical and phase 1 data predicted that mevidalen treatment could yield improvements in cognition; 17 , 21 however, we found that mevidalen failed to improve cognition in the CDR‐CoA and ADAS‐cog13 scales. There are several possible explanations for this lack of effect in these cognitive assessments. First, the treatment duration of 12 weeks may have been too short to fully capture any cognitive changes associated with treatment. The study aimed to enroll mild and moderate dementia but ended up highly weighted toward a mild population (although there was no imbalance among treatment arms) as indicated by baseline scores of MoCA, ADAS‐cog13, and CDR‐CoA. In particular, the average baseline CDR‐CoA score was near the scale's ceiling, reducing the ability to detect a treatment effect on this primary outcome measure. This ceiling effect was also perhaps in part due to the wide usage of AChEIs. Therefore, it is conceivable that a treated patient who is having benefit with an AChEI may not demonstrate improvement on the scale at the early 12‐week cognitive endpoint. Additionally, a placebo response at the 12‐week time point on the ADAS‐cog13 blunted the statistical effect of numerical improvement seen at the higher dose groups and may have shown greater separation if assessed after a longer treatment duration as one would anticipate worsening of cognition in the placebo group.
Despite the lack of cognitive benefit and consistent with the mechanism of action and preclinical and phase 1 data, mevidalen demonstrated significant and clinically meaningful benefits on parkinsonism as measured by the MDS‐UPDRS. Dose‐dependent and clinically meaningful improvements in the MDS‐UPDRS Part II (motor experiences of daily living) and Part III (motor examination) were seen, despite being in addition to stable doses of dopaminergic therapy to treat these symptoms. Consistent with other dopaminergic therapies, improvements were seen primarily in rigidity and bradykinesia domains with minimal benefit on tremor, suggesting that the motor benefit is being driven by dopaminergic stimulation and is not a non‐specific activating effect. In addition, dose‐dependent worsening of dyskinesias was anticipated and consistent with a dopaminergic mechanism. Finally, the benefit on motor experiences of daily living supports that this motoric benefit resulted in functional benefit in activities of daily living.
Unlike traditional dopaminergic therapies that often worsen non‐motor symptoms, mevidalen demonstrated statistically significant efficacy on MDS‐UPDRS Part I‐non‐motor experiences of daily living. Item analysis demonstrated that this benefit was largely driven by improvements in somnolence and fatigue, symptoms that are often difficult to treat and exacerbated by traditional dopaminergic therapies. These findings were consistent with its mechanism of action and both preclinical and phase 1 data. 17 , 21 , 34 Excessive daytime sleepiness affects up to 80% of LBD patients while fatigue affects 50% of patients with PD with increasing prevalence with cognitive impairment. Neither of these disabling non‐motor symptoms have effective treatments. 22 , 35 , 36 , 37 In addition, there was evidence of possible dose‐dependent improvements in hallucinations, despite the propensity of traditional dopaminergic therapy to exacerbate psychosis in LBD. 13 Mevidalen treatment therefore improves motor function in LBD on top of stable treatment while improving or not worsening commonly reported non‐motor complications of current therapies. 38
LBD is a multidimensional disease defined not only by the presence of dementia but by a range of core motor and non‐motor symptoms. These symptoms are difficult to address as treatment of one symptom often results in worsening of other symptoms (eg, improved motor symptoms with dopaminergic therapy worsens psychosis and/or somnolence). 13 The wide‐ranging symptoms, coupled with a high propensity for side effects, contributes to difficulty in disease management. 13 , 14 , 39 Mevidalen, as a D1PAM, harnesses a novel mechanism of action for the treatment of LBD that unlike existing dopaminergic therapies has the potential to address both motor and non‐motor symptoms relevant to LBD with a single therapy. Taken together, the motor and non‐motor benefits reflect a unique therapeutic profile for the symptomatic treatment of LBD. This multidimensional benefit appears to be clinically meaningful given the global improvement on the ADCS‐CGIC that while difficult to compare directly, somewhat exceeds the effects seen with a single‐dimensional treatment such as rivastigmine for cognition in PDD. 15
The PRESENCE trial confirmed the safety signal observed in preclinical and phase 1 studies with a dose‐dependent increase in BP and PR with initial dosing. 17 , 21 These BP and PR effects tended to accommodate over time; however, the accommodation was only partial at the 75 mg dose while lower doses did not differentiate from placebo with regard to BP and PR over 12 weeks. Even small persistent increases in BP over time have the potential to increase the risk of cardiovascular and cerebrovascular events which may limit the usefulness of the 75 mg dose in patients whose underlying medical condition might be compromised by increases in BP. 40 In addition, AE and SAE were more common at the 75 mg dose and tended to occur within 1 week of initiating therapy, suggesting that there may be an acute intolerance to high doses. This was consistent with the cardiovascular SAEs that occurred proximal to initiating dosing and led to the termination of the 75 mg dose. Future studies may need to explore dose titration as a strategy to mitigate the acute BP and PR effects and acute intolerability at higher doses, while evaluating doses between 30 mg and 75 mg may allow for identifying a dose that balances safety and efficacy.
3.1. Limitations
There were some limitations to the PRESENCE trial. First, the study was ongoing when the World Health Organization declared a pandemic secondary to COVID‐19, resulting in alternative methods of collecting key efficacy and safety data for a subset of participants enrolled at that time. Most participants were able to complete the study despite COVID‐19 restrictions. Sensitivity analyses with COVID‐impacted participant data censored did not demonstrate any impact on the overall study conclusions. Second, there was an imbalance of participants enrolled with a diagnosis for PDD or LBD, both overall and across treatment arms. While both are considered to fall into the over‐arching LBD population, 3 , 4 , 5 , 6 , 7 the imbalance across treatment arms was most notable in the placebo and 75 mg mevidalen treatment arms. Although the study was not powered to detect statistical differences in treatment effects in PDD versus DLB or within these subgroups, post hoc analyses did not suggest incongruence of response between PDD and DLB across primary and secondary outcome measures and no clear safety differences were identified. In addition, the population enrolled had significant motor impairment and most were treated with anti‐PD therapies. It is unclear if the efficacy seen in PRESENCE would translate to a population with less motor impairment or concomitant PD therapy use, as is more common in early DLB but not PDD. The 12‐week study duration was selected to align with available preclinical toxicology coverage at the time of study initiation and may have limited the ability to see differentiation from placebo on cognitive outcomes and to determine the persistence of benefit. Future studies will likely require longer duration treatment. Another limitation in terms of statistical analysis was that no adjustments were made for multiple comparisons which should be taken into account when interpreting the data and associated results. Finally, it appears that the primary cognitive outcome measure in the study lacked sufficient dynamic range to detect a treatment effect in the enrolled population; however, sensitivity analyses excluding those participants at the ceiling did not change the primary conclusion.
4. Conclusions
Treatment with mevidalen in patients with LBD did not improve cognition; however, future studies following participants for longer duration and using the ADAS‐cog13 may be useful in identifying a possible cognitive benefit. A significant safety signal at the 75 mg mevidalen dose led to the termination of this dose during the trial and may limit its utility. Future studies investigating mevidalen for the treatment of LBD should explore safety mitigation strategies such as dose titration and exploring doses between 30 mg and 75 mg to provide a more favorable benefit–risk ratio. The novel mechanism of mevidalen, and its potential to address motor and non‐motor symptoms relevant to LBD, differentiates it from traditional dopaminergic therapy and warrants further investigation of this unique therapeutic approach in an otherwise challenging disease.
Author Roles
(1) Research Project: A. Conception, B. Design, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique, C. Final Approval.
K.B.: 1A, 1B, 1C, 2B, 2C, 3B, 3C
L.M.: 1C, 2B, 2C, 3A, 3B, 3C
K.A.S.: 1A, 2C, 3B, 3C
P.A.: 1A, 1B, 1C, 2B, 2C, 3A, 3B, 3C
M.P.: 1C, 2B, 2C, 3B, 3C
J.S.: 1A, 2B, 2C, 3B, 3C
M.B.: 2C, 3B, 3C
Financial Disclosures
P.A., K.B., M.B., L.M., M.P., and J.S. are full‐time employees, salary recipients, and minor shareholders at Eli Lilly and Company. K.A.S. is a former full‐time employee (retired May 2021), former salary recipient, and minor shareholder at Eli Lilly and Company. J. Sims has received travel expenses from Eli Lilly and Company.
Supporting information
Fig. S1. (Related to Fig. 3). Movement Disorder Society‐Unified Parkinson Disease Rating Scale (MDS‐UPDRS) individual item analysis. Individual items of each part of the MDS‐UPDRS scale are shown, Part I in (A), Part II in (B), Part III in (C), and Part IV in (D). *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: LS, least squares
Fig. S2. Changes in daytime sleepiness as measured by the Epworth Sleepiness Scale. *P < 0.05. Abbreviations: ESS, Epworth Sleepiness Scale; LS, least squares
Fig. S3. Changes in cardiovascular outputs in PRESENCE. Baseline is defined as visit 2 time point matched of the mean of the three seated blood pressure measurements at each visit. *P < 0.05, **P < 0.01, ***P < 0.01. P values reflect significance of treatment difference relative to placebo for change from baseline. N numbers per group are shown below the associated time point. Abbreviations: LS, least squares; N, number of participants
Acknowledgments
Writing support and manuscript preparation was provided by Deirdre Hoban, PhD, an employee of Eli Lilly and Company. We thank all the patients and their families and caregivers who participated in this trial. We would like to acknowledge the site staff and site investigators, including but not limited to: Aaron Ellenbogen (Michigan Institute for Neurological Disorders (MIND), Farmington Hills, Michigan, USA), Adolfo Ramirez‐Zamora (Norman Fixel Institute For Neurological Diseases (FIND), Gainesville, Florida, USA), Alicia Cabrera (Clincloud, Llc., Maitland, Florida, USA), Amy Amara (University of Alabama Birmingham, Birmingham, Alabama, USA), Andrew Feigin (Nyu Langone, New York, New York, USA), Andrew Garner (Adirondack Medical Research, Glen Falls, New York, USA), Andrew Lerman (Visionary Investigators Network, Miami, Florida, USA), Anna Morenkova (University of California Irvine, Irvine, California, USA), Arifulla Khan (Northwest Clinical Research Center, Bellevue, Washington, USA), Arjun Tarakad (Baylor College of Medicine, Houston, Texas, USA), Ashok Patel (Bio Behavioral Health, Toms River, New Jersey, USA), Bruno Gallo (Suncoast Research Group, Miami, Florida, USA), Carmen Serrano (University of Puerto Rico, San Juan, Puerto Rico), Christian Schenk (Cortex, Psc., Las Piedras, Puerto Rico), Daniel Truong (Parkinson's & Movement Disorder Institute, Fountain Valley, California, USA), Danielle Feigenbaum (University of Southern California School of Medicine, Los Angeles, California, USA), David Grimes (Ottawa Hospital Research Institute, Parkinson's Disease and Movement Disorders Clinic, Ottawa, Ontario, Canada), David Shprecher (Banner Sun Health Research Institute, Sun City, Arizona, USA), David Weisman (Abington Neurological Associates, Ltd., Willow Grove, Pennsylvania, USA), Federico Rodriguez‐Porcel (Medical University of South Carolina, Charleston, South Carolina, USA), Fernando Pagan (Georgetown University Hospital, Washington, DC, USA), Horacio Capote (Dent Neurological Institute, Amherst, New York, USA), J. Antonelle de Marcaida (Hartford Healthcare Chase Movement Disorders Center, Vernon, Connecticut, USA), Jay Schneider (Thomas Jefferson University, Philadelphia, Philadelphia, USA), Julie Schwartzbard (Visionary Investigators Network, Aventura, Florida USA), Kara Wyant (University of Michigan, Ann Arbor, Michigan, USA), Kathleen Shannon (University of Wisconsin‐Madison Hospital and Health Clinic, Madison, Wisconsin, USA), Kinga Szigeti (Alzheimer's Disease and Memory Disorders Center, Buffalo, New York, USA), Kristi George (Josephson Wallack Munshower Neurology, Indianapolis, Indiana, USA), Laurice Yang (Stanford Neuroscience Health Center, Palo Alto, California, USA), Lin Zhang (University of California, Davis, Sacramento, California, USA), Margarita Almeida El‐Ramey (Visionary Investigators Network, Pembroke Pines, Florida, USA), Mark A. Goldstein (Jem Research Institute, Atlantis, Florida, USA), Michael Kleinman (Maine Medical Center‐Maine Medical Partners Neurology, Maine, USA), Nabila Dahodwala (Pennsylvania Hospital, Philadelphia, Pennsylvania, USA), Nirav Pavasia (Neurology Consultants of Dallas, Dallas, Texas, USA), Oleg Tcheremissine (Carolinas Healthcare System, North Charlotte, North Carolina, USA), Omid Omidvar (Collaborative Neuroscience Research, Llc., Long Beach, California, USA), Peter McAllister (New England Institute for Clinical Research, Stamford, Connecticut, USA), Pinky Agarwal (Evergreen Professional Plaza, Kirkland, Washington, USA), Rajeev Kumar (Rocky Mountain Movement Disorders Center, Englewood, Colorado, USA), Rajesh Pahwa (University of Kansas School of Medicine, Parkinson's Disease & Movement Disorders Center, Kansas City, Kansas, USA), Robert Riesenberg (Atlanta Center of Medical Research, Atlanta, Georgia, USA), Ryan Walsh (Barrow Neurological Institute, Barrow Neurology Clinic, Phoenix, Arizona, USA), Samantha Holden (University of Colorado Hospital, Aurora, Colorado, USA), Scott Sherman (University of Arizona Health Sciences, Department of Neurology, Tucson, Arizona, USA), Sharon Cohen (Toronto Memory Program, Toronto, Ontario, Canada), Shyamal Mehta (Mayo Clinic of Scottsdale, Scottsdale, Arizona, USA), Siddharth Kaul (Clinical Research Professionals, Chesterfield, Missouri, USA), Stephen Lee (Dartmouth Hitchcock Medical Center, Lebanon, New Hampshire, USA), Stewart Factor (Emory University, Atlanta, Georgia USA), Stuart Isaacson (Parkinson's Disease and Movement Disorders, Boca Raton, Florida, USA), Susan Steen (Axiom Clinical Research of Florida, Tampa, Florida, USA), Thyagarajan Subramanian (Penn State University Milton S. Hershey Medical Center, Pennsylvania, USA), Victor Faradji (Visionary Investigators Network, Miami, Florida, USA), Waleed El‐Feky (Texas Neurology, Dallas, Texas, USA), William Julio (Santa Cruz Behavioral Psc., Bayamón, Puerto Rico), Zoltan Mari (Cleveland Clinic of Las Vegas, Las Vegas, Nevada, USA), and Zoran Grujic (Central Dupage Hospital, Winfield, Illinois, USA).
Relevant conflicts of interest/financial disclosures: P.A., K.B., M.B., L.M., M.P., and J.S. are full‐time employees, salary recipients, and minor shareholders at Eli Lilly and Company. K.A.S. is a former full‐time employee (retired May 2021), former salary recipient, and minor shareholder at Eli Lilly and Company.
Funding agencies: This work is sponsored by Eli Lilly and Company.
Data Availability Statement
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, and blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
References
- 1. Hogan DB, Fiest KM, Roberts JI, Maxwell CJ, Dykeman J, Pringsheim T, et al. The prevalence and incidence of dementia with Lewy bodies: a systematic review. Can J Neurol Sci 2016;43(Suppl 1):S83–S95. [DOI] [PubMed] [Google Scholar]
- 2. Beyer K, Domingo‐Sabat M, Ariza A. Molecular pathology of Lewy body diseases. Int J Mol Sci 2009;10(3):724–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berg D, Postuma RB, Bloem B, Chan P, Dubois B, Gasser T, et al. Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson's disease. Mov Disord 2014;29(4):454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Friedman JH. Dementia with Lewy bodies and Parkinson disease dementia: it is the same disease! Parkinsonism Relat Disord 2018;46(Suppl 1):S6–S9. [DOI] [PubMed] [Google Scholar]
- 5. Gomperts SN. Lewy body dementias: dementia with Lewy bodies and Parkinson disease dementia. Continuum (Minneap Minn) 2016;22(2 Dementia):435–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jellinger KA. Dementia with Lewy bodies and Parkinson's disease‐dementia: current concepts and controversies. J Neural Transm (Vienna) 2018;125(4):615–650. [DOI] [PubMed] [Google Scholar]
- 7. Jellinger KA, Korczyn AD. Are dementia with Lewy bodies and Parkinson's disease dementia the same disease? BMC Med 2018;16(1):34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Colloby SJ, McKeith IG, Burn DJ, Wyper DJ, O'Brien JT, Taylor JP. Cholinergic and perfusion brain networks in Parkinson disease dementia. Neurology 2016;87(2):178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klein JC, Eggers C, Kalbe E, Weisenbach S, Hohmann C, Vollmar S, et al. Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology 2010;74(11):885–892. [DOI] [PubMed] [Google Scholar]
- 10. Colloby SJ, McParland S, O'Brien JT, Attems J. Neuropathological correlates of dopaminergic imaging in Alzheimer's disease and Lewy body dementias. Brain 2012;135(Pt 9):2798–2808. [DOI] [PubMed] [Google Scholar]
- 11. Harding AJ, Halliday GM. Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol 2001;102(4):355–363. [DOI] [PubMed] [Google Scholar]
- 12. Lippa CF, Duda JE, Grossman M, Hurtig HI, Aarsland D, Boeve BF, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 2007;68(11):812–819. [DOI] [PubMed] [Google Scholar]
- 13. Taylor JP, McKeith IG, Burn DJ, Boeve BF, Weintraub D, Bamford C, et al. New evidence on the management of Lewy body dementia. Lancet Neurol 2020;19(2):157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Walker Z, Possin KL, Boeve BF, Aarsland D. Lewy body dementias. Lancet 2015;386(10004):1683–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Emre M, Aarsland D, Albanese A, Byrne EJ, Deuschl G, De Deyn PP, et al. Rivastigmine for dementia associated with Parkinson's disease. N Engl J Med 2004;351(24):2509–2518. [DOI] [PubMed] [Google Scholar]
- 16. Hao J, Beck JP, Schaus JM, Krushinski JH, Chen Q, Beadle CD, et al. Synthesis and pharmacological characterization of 2‐(2,6‐dichlorophenyl)‐1‐((1S,3R)‐5‐(3‐hydroxy‐3‐methylbutyl)‐3‐(hydroxymethyl)‐1‐methyl‐3,4‐dihydroisoquinolin‐2(1H)‐yl)ethan‐1‐one (LY3154207), a potent, subtype selective, and orally available positive allosteric modulator of the human dopamine D1 receptor. J Med Chem 2019;62(19):8711–8732. [DOI] [PubMed] [Google Scholar]
- 17. Svensson KA, Hao J, Bruns RF. Positive allosteric modulators of the dopamine D1 receptor: a new mechanism for the treatment of neuropsychiatric disorders. Adv Pharmacol 2019;86:273–305. [DOI] [PubMed] [Google Scholar]
- 18. Arnsten AF, Girgis RR, Gray DL, Mailman RB. Novel dopamine therapeutics for cognitive deficits in schizophrenia. Biol Psychiatry 2017;81(1):67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gulwadi AG, Korpinen CD, Mailman RB, Nichols DE, Sit SY, Taber MT. Dinapsoline: characterization of a D1 dopamine receptor agonist in a rat model of Parkinson's disease. J Pharmacol Exp Ther 2001;296(2):338–344. [PubMed] [Google Scholar]
- 20. Svensson KA, Heinz BA, Schaus JM, Beck JP, Hao J, Krushinski JH, et al. An allosteric potentiator of the dopamine D1 receptor increases locomotor activity in human D1 knock‐in mice without causing stereotypy or tachyphylaxis. J Pharmacol Exp Ther 2017;360(1):117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilbraham D, Biglan KM, Svensson KA, Tsai M, Kielbasa W. Safety, tolerability, and pharmacokinetics of mevidalen (LY3154207), a centrally acting dopamine D1 receptor‐positive allosteric modulator (D1PAM), in healthy subjects. Clin Pharmacol Drug Dev 2020;10(4):393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology 2017;89(1):88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53(4):695–699. [DOI] [PubMed] [Google Scholar]
- 24. Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30(12):1591–1601. [DOI] [PubMed] [Google Scholar]
- 25. Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967;17(5):427–442. [DOI] [PubMed] [Google Scholar]
- 26. Simpson PM, Surmon DJ, Wesnes KA, Wilcock GK. The Cognitive Drug Research Computerized Assessment Systems for Elderly, Aami & Demented Patients: Wesnes KA, Simpson PM, White L & Pinker S. Cognitive Drug Research, 13 The Grove, Reading RG1 4RB. J Psychopharmacol 1992;6(1):108 [DOI] [PubMed] [Google Scholar]
- 27. Rowan E, McKeith IG, Saxby BK, O'Brien JT, Burn D, Mosimann U, et al. Effects of donepezil on central processing speed and attentional measures in Parkinson's disease with dementia and dementia with Lewy bodies. Dement Geriatr Cogn Disord 2007;23(3):161–167. [DOI] [PubMed] [Google Scholar]
- 28. Wesnes KA, McKeith I, Edgar C, Emre M, Lane R. Benefits of rivastigmine on attention in dementia associated with Parkinson disease. Neurology 2005;65(10):1654–1656. [DOI] [PubMed] [Google Scholar]
- 29. Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer's disease. Am J Psychiatry 1984;141(11):1356–1364. [DOI] [PubMed] [Google Scholar]
- 30. Mohs RC, Knopman D, Petersen RC, Ferris SH, Ernesto C, Grundman M, et al. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer's disease assessment scale that broaden its scope. The Alzheimer's Disease Cooperative Study. Alzheimer Dis Assoc Disord 1997;11(Suppl 2):S13–S21. [PubMed] [Google Scholar]
- 31. Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez‐Martin P, et al. Movement Disorder Society‐sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS‐UPDRS): scale presentation and clinimetric testing results. Mov Disord 2008;23(15):2129–2170. [DOI] [PubMed] [Google Scholar]
- 32. Schneider LS, Olin JT, Doody RS, Clark CM, Morris JC, Reisberg B, et al. Validity and reliability of the Alzheimer's Disease Cooperative Study‐Clinical Global Impression of Change. The Alzheimer's Disease Cooperative Study. Alzheimer Dis Assoc Disord 1997;11(Suppl 2):S22–S32. [DOI] [PubMed] [Google Scholar]
- 33. Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 1991;14(6):540–545. [DOI] [PubMed] [Google Scholar]
- 34. Schwartz MD, Kilduff TS. The neurobiology of sleep and wakefulness. Psychiatr Clin North Am 2015;38(4):615–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y, et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord 2007;22(12):1689–1707. quiz 837 [DOI] [PubMed] [Google Scholar]
- 36. Kluger BM, Herlofson K, Chou KL, Lou JS, Goetz CG, Lang AE, et al. Parkinson's disease‐related fatigue: a case definition and recommendations for clinical research. Mov Disord 2016;31(5):625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seppi K, Ray Chaudhuri K, Coelho M, Fox SH, Katzenschlager R, Perez Lloret S, et al. Update on treatments for nonmotor symptoms of Parkinson's disease‐an evidence‐based medicine review. Mov Disord 2019;34(2):180–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Borovac JA. Side effects of a dopamine agonist therapy for Parkinson's disease: a mini‐review of clinical pharmacology. Yale J Biol Med 2016;89(1):37–47. [PMC free article] [PubMed] [Google Scholar]
- 39. Verny M, Blanc F. Lewy body dementia: therapeutic propositions according to evidence based medicine and practice. Geriatr Psychol Neuropsychiatr Vieil 2019;17(2):189–197. [DOI] [PubMed] [Google Scholar]
- 40. Sager P, Heilbraun J, Turner JR, Gintant G, Geiger MJ, Kowey PR, et al. Assessment of drug‐induced increases in blood pressure during drug development: report from the Cardiac Safety Research Consortium. Am Heart J 2013;165(4):477–488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (Related to Fig. 3). Movement Disorder Society‐Unified Parkinson Disease Rating Scale (MDS‐UPDRS) individual item analysis. Individual items of each part of the MDS‐UPDRS scale are shown, Part I in (A), Part II in (B), Part III in (C), and Part IV in (D). *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: LS, least squares
Fig. S2. Changes in daytime sleepiness as measured by the Epworth Sleepiness Scale. *P < 0.05. Abbreviations: ESS, Epworth Sleepiness Scale; LS, least squares
Fig. S3. Changes in cardiovascular outputs in PRESENCE. Baseline is defined as visit 2 time point matched of the mean of the three seated blood pressure measurements at each visit. *P < 0.05, **P < 0.01, ***P < 0.01. P values reflect significance of treatment difference relative to placebo for change from baseline. N numbers per group are shown below the associated time point. Abbreviations: LS, least squares; N, number of participants
Data Availability Statement
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, and blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.