

Abstract
Aims
In patients with current or a history of hyperkalaemia, treatment with renin–angiotensin–aldosterone system inhibitors (RAASi) is often compromised. Patiromer, a novel potassium (K+) binder, may improve serum K+ levels and adherence to RAASi.
Methods
The DIAMOND trial will enroll ∼820 patients with heart failure with reduced ejection fraction (HFrEF; ejection fraction ≤40%). Patients meeting the screening criteria will enter a single‐blinded run‐in phase where they will be started or continued on a mineralocorticoid receptor antagonist (MRA) titrated to 50 mg/day and other RAASi therapy to ≥50% target dose, and patiromer. Patiromer will be titrated up to a maximum three packs/day (8.4 g/pack) to achieve optimal doses of RAASi without hyperkalaemia. The run‐in phase will last up to 12 weeks, following which patients will undergo double‐blind randomization in a 1:1 ratio to receive either continued patiromer or placebo (patiromer withdrawal). The primary endpoint is the mean difference in serum K+ from randomization between patiromer and placebo arms. Secondary endpoints will include hyperkalaemia events with K+ value >5.5 mEq/L, durable enablement of MRA at target dose, investigator‐reported adverse events of hyperkalaemia, hyperkalaemia‐related clinical endpoints and an overall RAASi use score (using a 0–8‐point scale) comprising all‐cause death, occurrence of cardiovascular hospitalization or usage of comprehensive heart failure medication.
Conclusion
The DIAMOND trial is designed to determine if patiromer can favourably impact K+ control in patients with HFrEF with hyperkalaemia or a history of hyperkalaemia leading to RAASi therapy compromise, and in turn improve RAASi use.
Keywords: Heart failure, Potassium, Hyperkalaemia, Patiromer, Renin–angiotensin–aldosterone system inhibitors, Mineralocorticoid receptor antagonists, Adherence, Trial design
Introduction
Renin–angiotensin–aldosterone system inhibitors (RAASi) are recommended for patients with heart failure (HF) with reduced ejection fraction (HFrEF). 1 , 2 These agents have multiple beneficial effects, but they increase the risk of hyperkalaemia, 3 which is more pronounced in patients with heart failure, who tend to be older and have chronic kidney disease (CKD) and diabetes mellitus. 4 , 5 , 6 This often contributes to suboptimal prescription of RAASi therapy. 3 , 7 , 8 , 9 Previous studies have demonstrated that after an episode of hyperkalaemia, RAASi doses are lowered in a large proportion of patients. 10 , 11 , 12 , 13 , 14 Many patients discontinue therapy permanently and suffer adverse outcomes. 12 Epstein et al. 10 observed that nearly 60% of patients with HF who discontinued RAASi experienced an adverse clinical event. Trevisan et al. 15 showed that stopping mineralocorticoid receptor antagonist (MRA) after a hyperkalaemia episode was associated with a lower risk of recurrent hyperkalaemia but a higher risk of death and cardiovascular (CV) events. Consequently, a gap remains between guideline recommendations and real‐world practice, where management of HF with optimal use and dose of RAASi is hindered by hyperkalaemia.
Patiromer is a sodium‐free, novel potassium (K+) binder that is well tolerated and efficacious in maintaining normokalaemia. The DIAMOND (Patiromer for the Management of Hyperkalemia in Subjects Receiving RAASi Medications for the Treatment of Heart Failure; NCT03888066) trial was initially designed to evaluate whether patiromer‐enabled RAASi therapy can improve clinical outcomes in patients with HFrEF with either hyperkalaemia or history of hyperkalaemia‐related compromise of RAASi therapy. However, related to the impact of ongoing coronavirus disease 2019 (COVID‐19) pandemic on clinical trials, the trial's primary endpoint was changed to serum K+ control prior to closeout and database lock or unblinding of treatment assignment.
Study design
Trial structure and oversight
The DIAMOND trial is a prospective phase 3b multinational, multicentre, double‐blind, randomized withdrawal, parallel‐group, placebo‐controlled trial that is designed to evaluate whether patiromer treatment in patients who developed hyperkalaemia while receiving RAASi medications will result in improvements in K+ concentration and in turn lead to optimization of RAASi use consistent with guidelines. The trial is conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice. An independent ethics committee approved the clinical protocol at every participating centre. All subjects provide written informed consent. The sponsor is Vifor Pharma, Inc.
The trial was designed by the Executive Committee, whose members included academic investigators and representatives of Vifor Pharma, Inc. The Executive Committee was responsible for developing the trial protocol, supervising the development of case report forms, and the statistical plan, overseeing the enrolment, and the quality of follow‐up. National leaders from different countries ensured that investigators remain committed to their responsibilities and encourage recruitment. A blinded Endpoint Adjudication Committee assessed all potential clinical events to evaluate whether they adhere to pre‐specified criteria for a clinical or a safety endpoint. An independent Data Monitoring Committee was responsible for assuring safety and making recommendations regarding the continuity or cessation of the trial. The members of these Committees are listed in online supplementary Appendix S1.
Study participants
Men and women aged ≥18 years who have chronic HFrEF, New York Heart Association functional class II–IV symptoms with a left ventricular ejection fraction of ≤40% as measured by any echocardiographic, radionuclide, magnetic resonance imaging, angiographic, or computed tomography within 12 months of randomization are eligible for enrolment (Table 1 ). Subjects are required to have hyperkalaemia at screening (defined by two K+ values of >5.0 mEq/L) while receiving an angiotensin‐converting enzyme inhibitor (ACEi)/angiotensin II receptor blocker (ARB)/angiotensin receptor–neprilysin inhibitor (ARNi), and/or an MRA. Alternatively, subjects are also eligible if they are normokalaemic at screening but have a prior history of RAASi discontinuation due to hyperkalaemia. Table 2 displays the B‐type natriuretic peptide and N‐terminal pro B‐type natriuretic peptide thresholds that had to be met for inclusion, according to history of atrial fibrillation and previous HF hospitalizations. Subjects with an estimated glomerular filtration rate (eGFR) <30 ml/min/1.73 m2 per the Chronic Kidney Disease Epidemiology Collaboration equation, acute decompensated HF within 4 weeks before screening, symptomatic hypotension or systolic blood pressure <90 mmHg, or any significant comorbidity which may impact the patient's clinical course, independent of HF, were excluded from the trial. A complete list of exclusion criteria is provided in Table 3 .
Table 1.
Inclusion criteria for the DIAMOND trial
|
| OR |
| Without hospitalization for HF or equivalent (e.g. emergency room or outpatient visit for worsening HF during which the subject received intravenous medications for the treatment of HF) within the last 12 months before screening |
|
ACEi, angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; ARNi, angiotensin receptor–neprilysin inhibitor; BNP, B‐type natriuretic peptide; CKD‐EPI, Chronic Kidney Disease Epidemiology Collaboration; HF, heart failure; K+, serum potassium; MRA, mineralocorticoid receptor antagonist; NT‐proBNP, N‐terminal pro B‐type natriuretic peptide; RAASi, renin–angiotensin–aldosterone system inhibitor.
For subjects treated with ARNi (sacubitril/valsartan) in the previous 4 weeks before screening, only NT‐proBNP values are to be considered.
Table 2.
B‐type natriuretic peptide and N‐terminal pro B‐type natriuretic peptide threshold levels (based on local laboratory), comorbidities, and previous hospitalizations
| Subjects with hospitalization for HF or equivalent a within last 12 months | Subjects with no hospitalization for HF or equivalent a within last 12 months | |
|---|---|---|
| Subjects presenting without atrial fibrillation when the blood sample was collected |
BNP b >150 pg/ml (18 pmol/L) or NT‐proBNP >600 pg/ml (71 pmol/L) |
BNP b >300 pg/ml (35 pmol/L) or NT‐proBNP >1200 pg/ml (142 pmol/L) |
| Subjects presenting with atrial fibrillation when the blood sample was collected |
BNP b >300 pg/ml (35 pmol/L) or NT‐proBNP >1200 pg/ml (142 pmol/L) |
BNP b >600 pg/ml (71 pmol/L) or NT‐proBNP >2400 pg/ml (284 pmol/L) |
BNP, B‐type natriuretic peptide; HF, heart failure; NT‐proBNP, N‐terminal pro B‐type natriuretic peptide.
For example urgent emergency room or outpatient visit for worsening HF during which the subject received intravenous medications for the treatment of HF.
For subjects treated with angiotensin receptor–neprilysin inhibitor (sacubitril/valsartan) in the previous 4 weeks before screening, only NT‐proBNP values are to be considered.
Table 3.
Exclusion criteria for the DIAMOND trial
|
Study visits and follow‐up
Following screening, eligible subjects are enrolled into a single‐blinded run‐in phase with weekly visits. The purpose of the run‐in phase is to control K+ levels with patiromer while simultaneously optimizing the use and doses of RAASi medications, including MRA (titrated to 50 mg/day). Patiromer is titrated up to a maximum of three packs/day (8.4 g/pack). After the run‐in phase that can last for up to 12 weeks, subjects undergo double‐blind randomization in a 1:1 ratio, to receive either continued patiromer or placebo (patiromer withdrawal) (Figure 1 ). Randomization is performed by using permuted block design and is stratified by geographic region. Randomized subjects continue the same number of packets of patiromer as established at the end of the run‐in phase and are instructed to continue the ACEi/ARB/ARNi and MRA regimen that was administered at the end of the run‐in phase.
Figure 1.
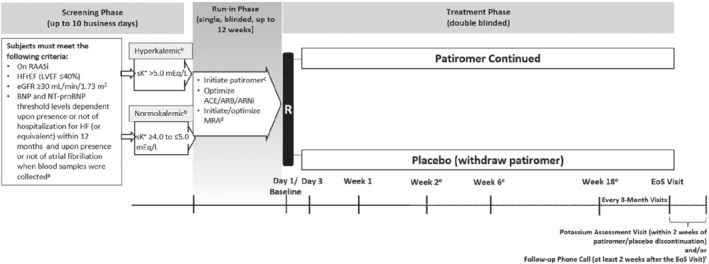
Design of the DIAMOND trial. ACEi, angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; ARNi, angiotensin receptor–neprilysin inhibitor; BNP, B‐type natriuretic peptide; eGFR, estimated glomerular filtration rate; EoS, end of study; HF, heart failure; HFrEF, heart failure with reduced ejection fraction; LVEF, left ventricular ejection fraction; MRA, mineralocorticoid receptor antagonist; NT‐proBNP, N‐terminal pro B‐type natriuretic peptide; R, randomization; RAASi, renin–angiotensin–aldosterone system inhibitor; sK+, serum potassium.
Prior to initiation of assigned patiromer/placebo, and as part of the randomization criteria, K+ concentration is measured at baseline. Thereafter, samples for the measurement are collected at every visit, starting from Day 3, and then at Weeks 1, 2, 6, 18 and every 3 months thereafter until the end of study (EoS). The original EoS date would have been set by the sponsor when sufficient number of subjects would have been enrolled to reach the expected number of events. However, the study endpoint was changed to control of K+ levels, and the total number of patients enrolled was adjusted. Patient‐reported outcomes are evaluated using the Kansas City Cardiomyopathy Questionnaire and EuroQol five‐dimension, five‐level (EQ‐5D‐5L) questionnaire. Subjects are monitored for changes in renal function, especially during medication adjustments and when there is a change in clinical status. All randomized subjects are followed for the occurrence of pre‐specified outcome events during the course of the trial. Subjects who prematurely discontinue patiromer/placebo should remain in the study for the collection of events data up to and including the common EoS and receive usual care during the study phase.
Primary and secondary endpoints
The original primary endpoint of the trial was to assess the efficacy of patiromer‐enabled optimization of RAASi therapy on time to CV death or first CV hospitalization. However, due to the impact of the COVID‐19 pandemic, slowing of the enrolment rate, and the shift in hospitalization patterns and the adaptations of the treatment practices for HF, there was a profoundly lower‐than‐expected enrolment rate as well as event rate. 16 Additionally, considering the risk to the patients as well as the logistics concern related to the impact on the COVID‐19 pandemic on the operations of the trial, the objective of the study was adjusted by the academic oversight team and the sponsor while the study remained blinded.
The primary endpoint was changed to mean change in K+ levels from baseline to all available post‐baseline K+ values (with sufficient numbers of patients at follow‐up visits) to be analysed by a mixed model for repeated measures approach. A Gaussian linear model for repeated measures will be used to evaluate the primary endpoint with treatment, geographic region, sex, baseline type 2 diabetes mellitus status and treatment by visit interaction serving as factors, and baseline K+ level and eGFR as covariates. All analyses will be restricted to serum K+ levels and not substituted by plasma values, as plasma values are systematically lower. 17 K+ values, either central or local laboratory measured, will be assessed at all visits, and if both values are present at a given visit, then central values will be used. Least squares mean changes from baseline will be reported for both treatment groups with 95% confidence intervals (CIs) as well as the difference between the least squares group means with 95% CI and p‐value testing the null hypothesis of no treatment effect.
Secondary efficacy endpoints will be analysed in a hierarchal manner and summarized descriptively through the calculation of point estimates by treatment group along with 95% CIs for the treatment differences. First, the time to the first event of hyperkalaemia (K+ > 5.5 mEq/L) will be analysed. Second, the time to event of reduction of the MRA dose below target (50 mg of spironolactone or eplerenone) will be assessed using a Cox proportional hazards regression model. Discontinuation of target dose would be required for at least 14 days, or less if at the EoS. Third, investigator‐reported adverse events of hyperkalaemia (time‐to‐first and recurrent) will be analysed using a negative binomial regression with the logarithm of the individual follow‐up time as offset. A joint frailty model of the total (first and recurrent) hyperkalaemia events and time to death as terminating event will be performed. Hyperkalaemia‐related specific outcomes will be analysed using the unmatched win‐ratio approach, with the following hierarchical components, all assessed during comparable follow‐up times: time to CV death; total number of CV hospitalizations; and total number of hyperkalaemia events with K+ > 6.5 mEq/L, >6.0–6.5 mEq/L and >5.0–6.0 mEq/L. In addition, the RAASi use score will be analysed using the win‐ratio approach for each pair of patients at the end of the comparable follow‐up period for that pair of patients. Hierarchical components of the RAASi use score are illustrated in Figure 2 . 18 This score, ranging from 0 to 8 points, will be analysed at the respective time points for each patient in each comparison, and consists of all‐cause death, total number of CV hospitalizations, and HF medication use. A list of other secondary endpoints, other endpoints and safety evaluations are provided in Table 4 .
Figure 2.
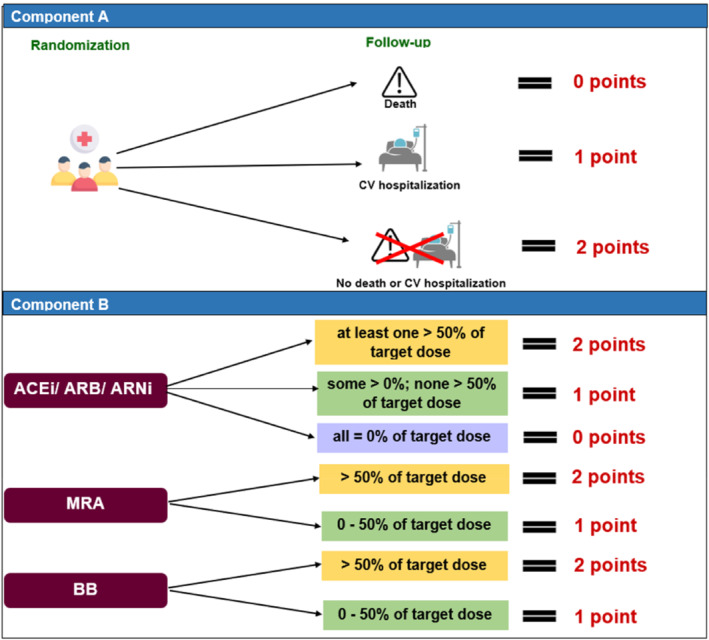
Hierarchical components of the renin–angiotensin–aldosterone system inhibitor use score. ACEi, angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; ARNi, angiotensin receptor–neprilysin inhibitor; BB, beta‐blocker; CV, cardiovascular; MRA, mineralocorticoid receptor antagonist.
Table 4.
List of secondary endpoints, other endpoints and safety evaluations
| Key secondary endpoints (hierarchically ordered) |
|
| Other secondary endpoints |
|
|
|
|
|
|
|
|
|
|
|
| Other endpoints |
|
|
|
|
|
|
|
|
|
|
|
|
|
| Safety evaluations |
|
|
|
|
|
ACEi, angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; ARNi, angiotensin receptor–neprilysin inhibitor; BNP, B‐type natriuretic peptide; CSS, clinical summary score; CV, cardiovascular; eGFR, estimated glomerular filtration rate; EoS, end of study; EQ‐5D‐5L, EuroQol five‐dimension, five‐level; HF, heart failure; K+, serum potassium; KCCQ, Kansas City Cardiomyopathy Questionnaire; MRA, mineralocorticoid receptor antagonist; NT‐proBNP, N‐terminal pro B‐type natriuretic peptide; NYHA, New York Heart Association; OSS, overall summary score; RAASi, renin–angiotensin–aldosterone system inhibitor; TSS, total symptom score.
Sample size and power calculation
The sample size of 410 subjects per treatment arm for a total of 820 subjects will provide 90% power to detect a difference between the placebo and the patiromer group on the mean change in K+ levels from baseline. This sample size calculation was based upon the following assumptions: 2‐sided alpha level of 5%, a difference between group means of 0.116, a standard deviation of 0.5, and a 5% loss to follow‐up. 19
Discussion
The DIAMOND trial will evaluate whether the use of patiromer, a novel K+ binder, allows better serum K+ control in patients with HFrEF who are hyperkalaemic or have a history of hyperkalaemia and are being optimized on RAASi therapy. Previous studies have consistently demonstrated an association between elevated K+ levels with lower use of RAASi, which has poor prognostic implications in HFrEF. 4 , 5 High K+ levels often lead to dose reduction or discontinuation of these proven therapies. 4 , 5 , 6 Patiromer is a non‐absorbable polymer that binds to free K+ in the gastrointestinal tract, thereby reducing the amount of K+ being absorbed in the blood. 20 This ensures maintenance of normokalaemia without incurring adverse events due to the suboptimal dosing of guideline‐recommended HF therapies.
The safety and efficacy of patiromer in patients with HF and/or CKD receiving RAASi therapy were initially established in several key clinical studies. In the 4‐week PEARL‐HF trial (Evaluation of Patiromer in Heart Failure Patients), patients treated with patiromer were significantly more likely to increase their dose of spironolactone from 25 to 50 mg/day compared with those receiving placebo without experiencing hyperkalaemia. 19 Similarly, in the 12‐week OPAL‐HK study (A Two‐Part, Single‐Blind, Phase 3 Study Evaluating the Efficacy and Safety of Patiromer for the Treatment of Hyperkalemia), 6% and 56% of patients in the patiromer and placebo groups, respectively, had to discontinue RAASi therapy due to recurrence of hyperkalaemia. 21 However, the reliability of these findings is limited by small sample size, short follow‐up period and restricted analysis of HF subgroups that were not well phenotyped. Similar to patiromer, sodium zirconium cyclosilicate (SZC) is also a Food and Drug Administration‐approved treatment for chronic hyperkalaemia. 22 , 23 The PRIORITIZE HF (Potassium Reduction Initiative to Optimize RAAS Inhibition Therapy With Sodium Zirconium Cyclosilicate in Heart Failure) trial was also evaluating the efficacy and safety of using SZC to initiate and intensify RAASi therapy in HFrEF. However, the trial was prematurely terminated due to the COVID‐19 pandemic, which resulted in a reduced sample size and inconclusive results. 24 The REALIZE‐K (Study to Assess Efficacy and Safety of SZC for the Management of High Potassium in Patients With Symptomatic HFrEF Receiving Spironolactone) trial is currently ongoing to determine if SZC can allow safe optimization of RAASi in patients with HFrEF while maintaining normokalaemia. 25
In circumstances where discontinuation of RAASi therapy is deemed necessary in HFrEF due to hyperkalaemia, guidelines suggest that the drug discontinuation period is kept to a minimum with the reintroduction of RAASi therapy when possible. 1 , 2 , 26 However, evidence suggests that a significant proportion of these patients are unable to benefit from these recommendations due to prescriber inertia or persistent risk or fear of hyperkalaemia. 5 , 6 Indeed, MRAs are often discontinued but rarely restarted after an episode of hyperkalaemia. 12 The DIAMOND study included high‐risk subjects (e.g. those with CKD, diabetes mellitus, older age, elevated natriuretic peptides) who pose the greatest therapeutic dilemma to clinicians, because they have the highest risk of developing hyperkalaemia but stand to benefit most from RAASi therapies. The DIAMOND trial will also assess clinical endpoints, such as time to CV death and CV hospitalization, but will not be powered for them. It will also evaluate quality‐of‐life measures. In addition, the trial will assess a novel composite endpoint including a RAASi use score using the win ratio. 18
The COVID‐19 pandemic continues to evolve and has affected all aspects of clinical research, including enrolment, conduct of study procedures, and the natural history trajectory of the disease process, which has had an impact on outcomes. The DIAMOND trial is the first, and so far only, trial that was designed to assess clinical outcomes with the use of an enablement strategy for RAASi therapy. Unfortunately, for the foreseeable future, this question will remain unanswered with the change in the plans for the DIAMOND trial described herein. These dynamics will continue to affect other trials and hinder development of novel therapies for various diseases, underscoring the need for adapting to alternate research strategies to achieve these goals.
The DIAMOND trial will be the largest trial ever performed to assess the impact of patiromer on K+ control in patients with HFrEF with hyperkalaemia or a history of hyperkalaemia on a background of guideline‐directed RAASi medication. Given the contraindication of hyperkalaemia with the usage of RAASi, this trial will add significantly to our knowledge about controlling K+ levels and the possible benefits of enabling RAASi in patients with HFrEF with the use of patiromer.
Funding
This study was sponsored and funded by Vifor Pharma.
Conflict of interest: J.B. reports consulting fees from Abbott, Adrenomed, Amgen, Applied Therapeutics, Array, AstraZeneca, Bayer, Boehringer Ingelheim, CVRx, G3 Pharma, Impulse Dynamics, Innolife, Janssen, LivaNova, Luitpold, Medtronic, Merck, Novartis, Novo Nordisk, Relypsa, Sequana Medical, and Vifor Pharma. S.D.A. reports consulting fees from Abbott, Bayer, Boehringer Ingelheim, Cardiac Dimension, Cordio, Impulse Dynamics, Novartis, Occlutech, Servier, and Vifor Pharma, and grant support from Abbott and Vifor Pharma. A.J.S.C. reports honoraria from Astra Zeneca, Bayer, Boehringer Ingelheim, Menarini, Novartis, Nutricia, Servier, Vifor, Abbott, Actimed, Arena, Cardiac Dimensions, Corvia, CVRx, Enopace, ESN Cleer, Faraday, Gore, Impulse Dynamics, Respicardia, and Viatris. F.D. reports support and stock options from Vifor Pharma as a Vifor employee. G.F. reports honoraria from Bayer and Boehringer Ingelheim, committee membership for Medtronic, Vifor Pharma, Amgen, Servier, and Novartis, and grants from the European Commission. T.F. reports consulting fees from Vifor Pharma, Bayer, Biosense Webster, CSL Behring, Galapagos, Minoryx, Novartis, LivaNova, Janssen, Roche, and honoraria from Fresenius Kabi. U.M.G. reports grants, support and stock options from Vifor Pharma. M.N.K. reports consulting fees from Amgen, Applied Therapeutics, AstraZeneca, Bayer, Boehringer Ingelheim, Eli Lilly, Esperion Therapeutics, Janssen, Merck, Novo Nordisk, Sanofi, and Vifor Pharma, grants from AstraZeneca and Boehringer Ingelheim, and grant support from Abbott and Vifor Pharma, and honoraria from AstraZeneca, Boehringer Ingelheim, and Novo Nordisk. L.H.L. reports grants from AstraZeneca, Vifor, Boston Scientific, Boehringer Ingelheim, and Novartis, consulting fees from Merck, Vifor Pharma, AstraZeneca, Bayer, Pharmacosmos, MedScape, Sanofi, Lexicon, Myokardia, Boehringer Ingelheim, and Servier, honoraria from Abbott, MedScape, Radcliffe, AstraZeneca, and Novartis, and stock options and patents with AnaCardio. M.M. reports consulting fees from Actelion, Amgen, AstraZeneca, Abbott Vascular, Bayer, Servier, Edwards Therapeutics, Livanova, Vifor Pharma, and WindTree Therapeutics. C.M.Q. reports support from Vifor Pharma as a Vifor employee. I.L.P. serves on an advisory board for Vifor Pharma Ltd and on the Steering Committee of the DIAMOND clinical trial. F.J.P. reports consulting fees from Vifor Pharma and NovoNordisk, honoraria from Servier, Pfizer, Novartis, and Boehringer Ingelheim, and presidency (unpaid) of the World Heart Federation. P.R. reports grants from AstraZeneca, Bayer, Fresenius, Novartis, Vifor Fresenius Medical Care Renal Pharma, Relypsa, and Vifor, consulting fees from Bayer, Idorsia, G3P, KBP, and Sanofi, honoraria from Sequana medical, AstraZeneca, Bayer, Fresenius, Novartis, Grunenthal, Stealth Peptides, Vifor Fresenius Medical Care Renal, NovoNordisk, Ablative Solutions, Corvidia Relypsa, and Vifor CardioRenal, and non‐financial support from Servier, Fresenius, and G3P. P.v.d.M. reports grants from Vifor Pharma, Pfizer, AstraZeneca, and Ionis, consulting fees from Vifor Pharma, Novartis and AstraZeneca, and honoraria from Vifor Pharma and Pharmacosmos. M.W. reports consulting fees from Vifor Pharma and AstraZeneca, and honoraria from Vifor Pharma. B.P. reports consulting fees from AstraZeneca, Boehringer Ingelheim/Lilly, Sanofi/Lexicon, SCPharmaceuticals, SQinnovations, G3 Pharmaceuticals, Sarfez, KBP biosciences, Cereno scientific, Phase bio, Proton Intel, and Vifor Pharma, stock options from SCPharmaceuticals, SQinnovations, G3 Pharmaceuticals, Sarfez, Cereno scientific, KBP biosciences, Proton Intel, and Vifor Pharma, US Patent 9931412–site specific delivery of eplerenone to the myocardium, and US Patent pending 63/045,783–Histone‐modulating agents for the treatment and prevention of organ damage. All other authors have nothing to disclose.
Supporting information
Appendix S1. Supporting Information.
References
- 1. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2016;18:891–975. [DOI] [PubMed] [Google Scholar]
- 2. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, et al. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2016;134:e282–93. [DOI] [PubMed] [Google Scholar]
- 3. Seliger SL. Hyperkalemia in patients with chronic renal failure. Nephrol Dial Transplant. 2019;34(Suppl 3):iii12–8. [DOI] [PubMed] [Google Scholar]
- 4. Bandak G, Sang Y, Gasparini A, Chang AR, Ballew SH, Evans M, et al. Hyperkalemia after initiating renin‐angiotensin system blockade: the Stockholm Creatinine Measurements (SCREAM) Project. J Am Heart Assoc. 2017;6:e005428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cooper LB, Benson L, Mentz RJ, Savarese G, DeVore AD, Carrero JJ, et al. Association between potassium level and outcomes in heart failure with reduced ejection fraction: a cohort study from the Swedish Heart Failure Registry. Eur J Heart Fail. 2020;22:1390–8. [DOI] [PubMed] [Google Scholar]
- 6. Beusekamp JC, Tromp J, van der Wal HH, Anker SD, Cleland JG, Dickstein K, et al. Potassium and the use of renin‐angiotensin‐aldosterone system inhibitors in heart failure with reduced ejection fraction: data from BIOSTAT‐CHF. Eur J Heart Fail. 2018;20:923–30. [DOI] [PubMed] [Google Scholar]
- 7. Maggioni AP, Anker SD, Dahlström U, Filippatos G, Ponikowski P, Zannad F, et al.; Heart Failure Association of the ESC . Are hospitalized or ambulatory patients with heart failure treated in accordance with European Society of Cardiology guidelines? Evidence from 12,440 patients of the ESC Heart Failure Long‐Term Registry. Eur J Heart Fail. 2013;15:1173–84. [DOI] [PubMed] [Google Scholar]
- 8. Savarese G, Carrero JJ, Pitt B, Anker SD, Rosano GMC, Dahlström U, et al. Factors associated with underuse of mineralocorticoid receptor antagonists in heart failure with reduced ejection fraction: an analysis of 11 215 patients from the Swedish Heart Failure Registry. Eur J Heart Fail. 2018;20:1326–34. [DOI] [PubMed] [Google Scholar]
- 9. Rossignol P, Lainscak M, Crespo‐Leiro MG, Laroche C, Piepoli MF, Filippatos G, et al.; Heart Failure Long‐Term Registry Investigators Group . Unravelling the interplay between hyperkalaemia, renin‐angiotensin‐aldosterone inhibitor use and clinical outcomes. Data from 9222 chronic heart failure patients of the ESC‐HFA‐EORP Heart Failure Long‐Term Registry. Eur J Heart Fail. 2020;22:1378–89. [DOI] [PubMed] [Google Scholar]
- 10. Epstein M, Reaven NL, Funk SE, McGaughey KJ, Oestreicher N, Knispel J. Evaluation of the treatment gap between clinical guidelines and the utilization of renin‐angiotensin‐aldosterone system inhibitors. Am J Manag Care. 2015;21(11 Suppl):S212–20. [PubMed] [Google Scholar]
- 11. Ouwerkerk W, Voors AA, Anker SD, Cleland JG, Dickstein K, Filippatos G, et al. Determinants and clinical outcome of uptitration of ACE‐inhibitors and beta‐blockers in patients with heart failure: a prospective European study. Eur Heart J. 2017;38:1883–90. [DOI] [PubMed] [Google Scholar]
- 12. Trevisan M, de Deco P, Xu H, Evans M, Lindholm B, Bellocco R, et al. Incidence, predictors and clinical management of hyperkalaemia in new users of mineralocorticoid receptor antagonists. Eur J Heart Fail. 2018;20:1217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ferreira JP, Butler J, Rossignol P, Pitt B, Anker SD, Kosiborod M, et al. Abnormalities of potassium in heart failure: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2020;75:2836–50. [DOI] [PubMed] [Google Scholar]
- 14. Beusekamp JC, Tromp J, Cleland JGF, Givertz MM, Metra M, O'Connor CM, et al. Hyperkalemia and treatment with RAAS inhibitors during acute heart failure hospitalizations and their association with mortality. JACC Heart Fail. 2019;7:970–9. [DOI] [PubMed] [Google Scholar]
- 15. Trevisan M, Fu EL, Xu Y, Savarese G, Dekker FW, Lund LH, et al. Stopping mineralocorticoid receptor antagonists after hyperkalaemia: trial emulation in data from routine care. Eur J Heart Fail. 2021;23:1698–707. [DOI] [PubMed] [Google Scholar]
- 16. DeFilippis EM, Reza N, Donald E, Givertz MM, Lindenfeld J, Jessup M. Considerations for heart failure care during the COVID‐19 pandemic. JACC Heart Fail. 2020;8:681–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cooper LB, Savarese G, Carrero JJ, Szabo B, Jernberg T, Jonsson Å, et al. Clinical and research implications of serum versus plasma potassium measurements. Eur J Heart Fail. 2019;21:536–7. [DOI] [PubMed] [Google Scholar]
- 18. Pocock SJ, Ariti CA, Collier TJ, Wang D. The win ratio: a new approach to the analysis of composite endpoints in clinical trials based on clinical priorities. Eur Heart J. 2012;33:176–82. [DOI] [PubMed] [Google Scholar]
- 19. Pitt B, Anker SD, Bushinsky DA, Kitzman DW, Zannad F, Huang IZ; PEARL‐HF Investigators . Evaluation of the efficacy and safety of RLY5016, a polymeric potassium binder, in a double‐blind, placebo‐controlled study in patients with chronic heart failure (the PEARL‐HF) trial. Eur Heart J. 2011;32:820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pitt B, Bushinsky DA, Kitzman DW, Ruschitzka F, Metra M, Filippatos G, et al.; Patiromer‐204 Investigators . Evaluation of an individualized dose titration regimen of patiromer to prevent hyperkalaemia in patients with heart failure and chronic kidney disease. ESC Heart Fail. 2018;5:257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pitt B, Bakris GL, Bushinsky DA, Garza D, Mayo MR, Stasiv Y, et al. Effect of patiromer on reducing serum potassium and preventing recurrent hyperkalaemia in patients with heart failure and chronic kidney disease on RAAS inhibitors. Eur J Heart Fail. 2015;17:1057–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. US Food and Drug Administration . FDA Drug Safety Communication: FDA recommends separating dosing of potassium‐lowering drug sodium polystyrene sulfonate (Kayexalate) from all other oral drugs. 2017 September 6 [cited 2021 Aug 19]. Available from: https://www.fda.gov/drugs/drug‐safety‐and‐availability/fda‐drug‐safety‐communication‐fda‐recommends‐separating‐dosing‐potassium‐lowering‐drug‐sodium.
- 23. US Food and Drug Administration . Lokelma prescribing information. 2018. [cited 2021 Aug 19]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/207078s000lbl.pdf.
- 24. ClinicalTrials.gov . Potassium Reduction Initiative to Optimize RAAS Inhibition Therapy With Sodium Zirconium Cyclosilicate in Heart Failure (PRIORITIZE HF). [cited 2021 Aug 19]. Available from: https://clinicaltrials.gov/ct2/show/results/NCT03532009.
- 25. ClinicalTrials.gov . Study to Assess Efficacy and Safety of SZC for the Management of High Potassium in Patients With Symptomatic HFrEF Receiving Spironolactone (REALIZE‐K). [cited 2021 Sep 19]. Available from: https://clinicaltrials.gov/ct2/show/NCT04676646.
- 26. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, et al.; ESC Scientific Document Group . 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021;42:3599–726. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.