

Abstract
Background and Aims
Primary sclerosing cholangitis (PSC) is associated with increased risk of cholangiocarcinoma (CCA). Early and accurate CCA detection represents an unmet clinical need as the majority of patients with PSC are diagnosed at an advanced stage of malignancy. In the present study, we aimed at establishing robust DNA methylation biomarkers in bile for early and accurate diagnosis of CCA in PSC.
Approach and Results
Droplet digital PCR (ddPCR) was used to analyze 344 bile samples from 273 patients with sporadic and PSC‐associated CCA, PSC, and other nonmalignant liver diseases for promoter methylation of cysteine dioxygenase type 1, cannabinoid receptor interacting protein 1, septin 9, and vimentin. Receiver operating characteristic (ROC) curve analyses revealed high AUCs for all four markers (0.77–0.87) for CCA detection among patients with PSC. Including only samples from patients with PSC diagnosed with CCA ≤ 12 months following bile collection increased the accuracy for cancer detection, with a combined sensitivity of 100% (28/28) and a specificity of 90% (20/203). The specificity increased to 93% when only including patients with PSC with longtime follow‐up (> 36 months) as controls, and remained high (83%) when only including patients with PSC and dysplasia as controls (n = 23). Importantly, the bile samples from the CCA‐PSC ≤ 12 patients, all positive for the biomarkers, included both early‐stage and late‐stage CCA, different tumor growth patterns, anatomical locations, and carbohydrate antigen 19‐9 levels.
Conclusions
Using highly sensitive ddPCR to analyze robust epigenetic biomarkers, CCA in PSC was accurately detected in bile, irrespective of clinical and molecular features, up to 12 months before CCA diagnosis. The findings suggest a potential for these biomarkers to complement current detection and screening methods for CCA in patients with PSC.
Abbreviations
- CA 19‐9
carbohydrate antigen 19‐9
- CCA
cholangiocarcinoma
- CDO1
cysteine dioxygenase type 1
- CNRIP1
cannabinoid receptor interacting protein 1
- dCCA
extrahepatic CCA
- ddPCR
droplet digital PCR
- ERCP
endoscopic retrograde cholangiopancreatography
- FISH
fluorescence in situ hybridization
- iCCA
intrahepatic CCA
- NM
nonmalignant
- pCCA
perihilar CCA
- PSC
primary sclerosing cholangitis
- ROC
receiver operating characteristic
- SEPT9
septin 9
- VIM
vimentin
INTRODUCTION
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease, characterized by progressive inflammation and fibrosis of the biliary tree.[ 1 , 2 , 3 ] In the absence of any effective medical treatment, the majority of patients with PSC develop end‐stage liver disease, with liver transplantation representing the only curative option.[ 1 ] Although 5‐year survival after liver transplantation is good (exceeds 80%), development of cholangiocarcinoma (CCA) prior to transplantation represents a feared complication of PSC, with a major negative impact on life expectancy.[ 3 , 4 ]
Patients with PSC have a 160‐fold to 600‐fold increased lifetime risk of developing CCA compared to the general population, amounting to a 20% reported lifetime incidence of CCA.[ 4 , 5 , 6 , 7 ] CCA in PSC is commonly diagnosed at a young age (40–50 years), in contrast to sporadic CCA that often develops at a more advanced age (70–80 years).[ 5 ] Detecting CCA in PSC is challenging, partly due to nonspecific presenting features and overlapping findings in malignant and benign disease progression in PSC. Even with combined use of multiple detection modalities, including tumor serum marker carbohydrate antigen 19‐9 (CA 19‐9), imaging, biliary brushing for cytological and fluorescence in situ hybridization (FISH) evaluation, and intraductal biopsies, the diagnostic accuracy for CCA in PSC is low.[ 8 , 9 , 10 ] Consequently, CCA is commonly detected at an advanced stage. Owing to late diagnosis, CCA carries a dismal prognosis among patients with PSC. The median survival is < 12 months in patients with unresectable disease, and CCA currently represents the most frequent cause of PSC‐associated deaths.[ 5 ] If CCA is detected at an early stage, curative surgical resection or liver transplantation is possible, with reported 5‐year survival rates up to 35% and 74%, respectively.[ 11 , 12 ] As earlier tumor detection may significantly improve survival among patients with PSC and CCA, establishment of accurate detection methods for early‐stage CCA, which can be implemented in surveillance algorithms, represents a clear unmet need.
Aberrant DNA methylation alterations are valuable as cancer biomarkers, including early detection markers. They are highly prevalent in most cancer types, may be found in precancerous lesions or early‐stage cancers, and may further be detected in various body fluids that can be obtained by noninvasive or minimally invasive procedures.[ 13 ] In patients with CCA, aberrant DNA methylation has been observed in both blood and bile, emphasizing the potential of such markers for early detection of CCA.[ 13 , 14 ] We have previously identified a DNA methylation biomarker panel suitable for detecting CCA, with high sensitivity (85%) and specificity (98%), using biliary brushes.[ 15 ] In the current study we aimed at validating these markers in small volumes of bile for early and accurate detection of CCA in patients with PSC, using droplet digital PCR (ddPCR).
MATERIALS AND METHODS
Patient samples and clinical data
A total of 344 bile samples from 273 patients diagnosed with CCA (with or without underlying PSC), PSC with biliary dysplasia, nonmalignant (NM) PSC, and other benign liver diseases were included in the study. Patients were recruited at Oslo University Hospital, Rikshospitalet (Oslo, Norway; n = 219), Karolinska University Hospital (Stockholm, Sweden; n = 12), and Helsinki University Hospital (Helsinki, Finland; n = 42) between 2008 and 2019. Diagnosis of PSC was based on typical findings on magnetic resonance cholangiography and/or endoscopic retrograde cholangiopancreatography (ERCP) according to established criteria.[ 3 , 16 ] CCA diagnosis was established by histopathological analysis of tissue samples or through combined clinical, biochemical, radiological, and cytological assessments.[ 3 , 17 ] Diagnosis of low‐grade or high‐grade biliary dysplasia was based on evaluation of biliary brush specimens using standard cytological criteria or, in patients undergoing liver transplantation, by histopathological examination of the explanted liver.[ 18 , 19 ] In addition, all obtained brush cytologies were classified into the following categories: 0, acellular; 1, normal; 2, atypia; 3, suspicious of carcinoma; 4, diagnostic of carcinoma (Table S1). Patients with CCA receiving chemotherapy at the time of bile sampling were not included in the study.
Samples were split based on main disease category, including (1) patients with underlying PSC and (2) patients with non‐PSC‐related diseases (Figure 1). The PSC group was divided into two main groups: (1) CCA‐PSC (n = 38 [42 samples], including patients with CCA and concomitant PSC) and (2) PSC all (n = 205 [272 samples], including benign PSC without CCA). The CCA‐PSC group was further divided into subgroups based on time from bile sampling to a confirmed CCA diagnosis using standard diagnostic criteria, including (1) CCA‐PSC ≤ 12 (n = 28 [31 samples], ≤ 12 months from bile sampling to a confirmed CCA diagnosis) and (2) CCA‐PSC > 12 (n = 10 [11 samples], > 12 months from bile sampling to CCA diagnosis). We chose a threshold of 12 months from bile sampling to an established CCA diagnosis to increase the likelihood of CCA actually being present (CCA‐PSC ≤ 12) and simultaneously to see if the markers could detect CCA earlier (up to 12 months) than currently used detection methods. The benign PSC group was further divided into (1) PSC‐dysplasia (n = 23 [24 samples], patients with PSC and evidence of biliary dysplasia based on assessment of biliary brush cytology specimens or histological assessment of explant liver ±2 months from bile collection but with no evidence of CCA development), (2) PSC‐control > 36 (n = 170 [226 samples], patients with PSC with no evidence of CCA or biliary dysplasia based on histological assessment of explanted liver at or after bile collection or > 36 months of follow‐up [median 105 months] without established CCA or biliary dysplasia for nontransplanted patients), and (3) PSC‐control < 36 (n = 12 [22 samples], patients with PSC with no evidence of CCA or biliary dysplasia after bile collection but only including nontransplanted patients with < 36 months of follow‐up [median 26 months]). We chose a threshold of 36 months of follow‐up from bile sampling to divide the benign PSC group in two, to minimize the probability of presence of CCA in the strictest control group (PSC‐control > 36). We further included PSC‐dysplasia as a separate group to evaluate the methylation pattern in this potentially premalignant group. The non‐PSC group was divided into (1) CCA (n = 6 [6 samples]) and (2) other NM liver diseases (n = 24 [24 samples], including hereditary, idiopathic nonalcoholic fatty liver, biliary stone, and autoimmune liver disease other than PSC). The samples in the non‐PSC group were included to explore the methylation levels of the markers in non‐PSC‐related CCA and other NM liver diseases.
FIGURE 1.
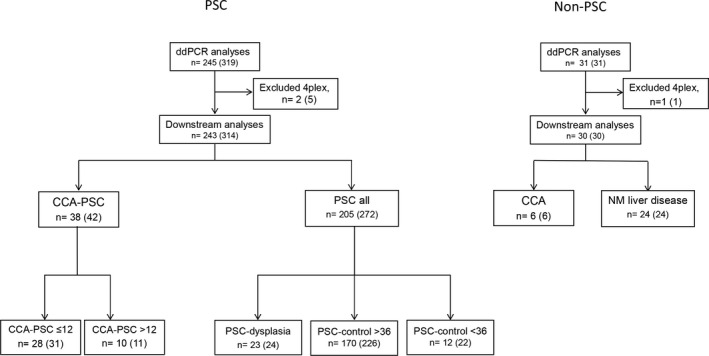
Flowchart of bile samples included in the study. Number of patients, followed by number of samples in parentheses. Samples are split based on main disease category, including (1) patients with underlying PSC (left panel) and (2) patients with non‐PSC‐related diseases (right panel). PSC samples (left panel) are divided into the following groups: (1) CCA‐PSC (n = 38, 42 samples), including CCA‐PSC ≤ 12 (n = 28, 31 samples; ≤ 12 months from bile sampling to CCA diagnosis) and CCA‐PSC > 12 (n = 10, 11 samples; > 12 months from bile sampling to CCA diagnosis), and (2) all PSC samples (without CCA, n = 205, 272 samples), including PSC‐dysplasia (n = 23, 24 samples; patients with PSC with evidence of biliary dysplasia based on assessment of biliary brush cytology specimens or histological assessment of explant liver ±2 months from bile collection), PSC‐control > 36 (n = 170, 226 samples; patients with PSC with no evidence of CCA or biliary dysplasia based on histological assessment of explanted liver at or after bile collection or > 36 months of follow‐up without established CCA or biliary dysplasia for nontransplanted patients), and PSC‐control < 36 (n = 12, 22 samples; patients with PSC with no evidence of CCA or biliary dysplasia after bile collection but only including nontransplanted patients with < 36 months of follow‐up). Non‐PSC samples (right panel) are divided into (1) CCA (n = 6, 6 samples, of which five were collected at the same time as a confirmed CCA diagnosis and one was collected 3 months prior to diagnosis) and (2) NM liver disease (n = 24, 24 samples; hereditary, idiopathic nonalcoholic fatty liver, biliary stone, and autoimmune liver disease other than PSC)
Bile was collected during ERCP, if possible before injecting contrast medium; at liver transplantation; or in one case at liver resection. All samples were stored at −80℃ or −20℃ (Karolinska University Hospital) prior to DNA extraction.
For summarized clinical and histopathological information of the included samples, see Tables 1 and 2; and for detailed information about each bile sample, see Table S1.
TABLE 1.
Clinical characteristics separated by groups
| CCA‐PSC (n = 38) | PSC (n = 205) | |
|---|---|---|
| Gender (male) | 76% | 76% |
| Median age CCA diagnosis, years (range) | 53.9 (46.0) | NA |
| Median age benign liver diagnosis, years (range) | 49.4 (51.1) | 31.0 (59.1) |
| IBD, number (%) | ||
| Crohn's | 5 (13) | 29 (14) |
| UC | 22 (58) | 115 (56) |
| Indet. colitis | 0 (0) | 3 (1) |
| No | 9 (24) | 50 (24) |
| NA | 2 (5) | 8 (4) |
| Cirrhosis, number (%) | ||
| Yes | 5 (13) | 39 (19) |
| No | 20 (53) | 152 (74) |
| NA | 13 (34) | 14 (7) |
| Acute cholangitis, number (%) | ||
| Yes | 2 (5) | 1 (0) |
| No | 36 (95) | 204 (100) |
| NA | 0 (0) | 0 (0) |
| Median CA19‐9 level, U/ml (range) | 47 (8,638) | 19 (384) |
| Missing | 3 | 10 |
| Dead | 74% | 3% |
Abbreviations: IBD, inflammatory bowel disease; Indet., indeterminate; NA, not available; UC, ulcerative colitis.
TABLE 2.
Histopathological data for patients with CCA‐PSC
| n = 38 | Number (%) |
|---|---|
| Tumor location | |
| iCCA | 14 (37) |
| pCCA | 15 (39) |
| dCCA | 7 (18) |
| xCCA | 2 (5) |
| AJCC (8th edition) | |
| AJCC 0 | 1 (3) |
| AJCC 1 | 1 (3) |
| AJCC 2 | 6 (16) |
| AJCC 3 | 15 (39) |
| AJCC 4 | 9 (24) |
| NA | 6 (16) |
| T | |
| Tis | 1 (3) |
| T1 | 5 (13) |
| T2 | 12 (32) |
| T3 | 5 (13) |
| T4 | 6 (16) |
| Tx | 9 (24) |
| N | |
| N0 | 5 (13) |
| N1 | 20 (53) |
| Nx | 13 (34) |
| M | |
| M0 | 29 (76) |
| M1 | 9 (24) |
| Resection margin | |
| R0 | 9 (24) |
| R1 | 6 (16) |
| NA | 23 (61) |
| Perineural growth | |
| Yes | 12 (32) |
| No | 2 (5) |
| NA | 24 (63) |
| Vascular encasement | |
| Yes | 6 (16) |
| No | 10 (26) |
| NA | 22 (58) |
| Differentiation | |
| Low (G3) | 4 (11) |
| Medium (G2) | 11 (29) |
| High (G1) | 1 (3) |
| NA | 22 (58) |
| Growth pattern | |
| Mass‐forming | 10 (26) |
| Periductal‐infiltrating | 6 (16) |
| Intraductal‐growing | 1 (3) |
| NA | 21 (55) |
The stage is from time of diagnosis.
Abbreviations: AJCC 0–4, American Joint Committee on Cancer classification stages 0–4; G, grade of differentiation; M, distant metastases; N, lymph node metastases; NA, not available; T, tumor stage evaluation; xCCA, unclassifiable CCA subtype.
DNA extraction
For DNA extraction, using aliquots of 100–200 µl of bile per sample, three methods were tested: High Pure Viral (Roche Diagnostics, Basel, Switzerland), PrepFiler (Thermo Fisher Scientific, MA), and a standard phenol chloroform procedure. The phenol chloroform procedure was performed as described,[ 15 ] while the manufacturers’ protocols were followed for the Pure Viral and the PrepFiler kits. Resulting DNA yields were measured using NanoDrop ND‐1000. The High Pure Viral kit provided the highest amount of DNA (mean output of four samples, 532 ng), followed by phenol chloroform (mean output of three samples, 72 ng) and PrepFiler (mean output of four samples, 22.5 ng) (Table S2).
To compare the amount of amplifiable DNA from the three extraction methods, samples were bisulfite‐treated (described in the following section), and 32 ng of bisulfite‐treated DNA (from each sample) was amplified by quantitative methylation specific PCR (qMSP) as described,[ 20 ] using the ALU primer and probe set. Following bisulfite treatment and qMSP amplification, phenol chloroform outperformed the two kits, with lower ALU cycle threshold values, indicating more amplifiable DNA (Table S2). For the 350 bile samples reported in the following, DNA was extracted from 100 to 200 µl bile using the phenol chloroform procedure.
Bisulfite treatment
Five‐hundred nanograms of DNA was bisulfite‐converted using the EpiTect bisulfite kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. Desulfonation of the samples was performed using a QIAcube (Qiagen) with elution in 40 µl buffer.
DNA methylation analyses
Primer and probe sequences for the target genes cysteine dioxygenase type 1 (CDO1), cannabinoid receptor interacting protein 1 (CNRIP1), septin 9 (SEPT9), and vimentin (VIM) and the 4Plex control comprising ephrin type A receptor 3, kelch repeat and BTB domain containing 4, pleckstrin homology and FYVE domain containing 1, and synaptotagmin 10 have been published.[ 15 , 21 ]
The QX200 Droplet Digital PCR System (BioRad) was used for DNA methylation analyses. The ddPCR consisted of 1 × ddPCR Supermix for Probes (BioRad), 818 nM of each primer, 182 nM of each probe, and 30 ng bisulfite‐converted DNA template, in a final volume of 22 µl. Droplet generation was performed in the QX200 Droplet Generator, using 20 µl of the ddPCR mixture and 70 µl droplet generation oil (BioRad). Samples were transferred to a 96‐well PCR plate (BioRad) and sealed in the PX1 PCR Plate Sealer (BioRad). The PCR was performed in a T100 Thermal Cycler (BioRad), using the manufacturer's recommended cycling conditions. Using a ramp rate of 2℃/s, amplification was performed at 95℃ for 10 min, before 40 cycles of 30 s at 94℃ and 1 min at 60℃, followed by enzyme deactivation at 98℃ for 10 min. The QX200 Droplet Reader (BioRad) was applied for detection of fluorescence signals.
Data analysis was performed using QuantaSoft, version 1.7.4.0917 (BioRad). An in‐house developed algorithm, PoDCall (https://bioconductor.org/packages/release/bioc/html/PoDCall.html), was applied for positive droplet calling as described.[ 21 ] Normalized DNA methylation levels were calculated by dividing the concentration (copies per microliter) of the target gene by the concentration (copies per microliter) of the 4Plex and multiplying by 400. For each target gene, samples with low DNA amount (4Plex concentration < 10 copies/µl) were excluded. Six samples had a 4Plex concentration < 10 for all the target genes (and were excluded altogether), and six samples had a 4Plex concentration < 10 for one to three genes and were excluded for the specific gene(s).
Statistical analysis
A Mann‐Whitney U test was used to test for differences in DNA methylation levels between the defined sample groups, and Fisher's exact test was used to examine the relation between methylation status (methylated or unmethylated for one or more of four markers) and sample group, as well as clinical and pathological data. Receiver operating characteristic (ROC) curve analysis was performed to evaluate the diagnostic biomarker potential of the individual markers. In addition, combined ROC curves were generated by summarizing the normalized methylation values for the four markers. The AUC, sensitivities, specificities, and 95% CIs were calculated. The cutoff providing the highest possible specificity (> 90%) without compromising the sensitivity was chosen for dichotomizing the samples. In cases where multiple samples from the same patient were analyzed, only one sample was selected for biomarker analysis. Samples in the group PSC‐dysplasia were selected over PSC‐control > 36, which were selected over PSC‐control < 36. For the remaining samples, random selection was performed, to select only one sample from each patient. Samples lacking methylation values (CDO1, n = 1 [0.3%]; CNRIP1, n = 1 [0.3%]; SEPT9, n = 1 [0.3%]; VIM, n = 4 [1.2%]) were excluded. In the analyses of the biomarker panel, only samples with information for all four markers were included. Four samples were excluded because of missing values. Overall survival was calculated from time of bile sampling until death from any cause, and cases were censored at last follow‐up. The effect of the four methylation markers was independently evaluated in a univariate approach, using the log rank method. The Kaplan‐Meier method was used to estimate the survival curve. The analyses were performed using GraphPad Prism 8.1.2 and IMB SPSS Statistics 25.
Written informed consent was obtained from all study subjects. For long‐archived samples, where this was not possible, an exception from informed consent was obtained by the local ethics committee to allow use of the samples. Study protocols were approved by the ethics committees of all the recruiting centers, as well as the Regional Committee for Medical and Health Research Ethics South Eastern Norway (6.2008.1723), the Swedish Ethical Review Authority (2013/2084‐31/2 with amendment 2016/1023, 2018/1786‐32), and Helsinki University Hospital (278/13/03/01/2009).
RESULTS
A total of 344 samples from 273 patients passed the inclusion criteria after ddPCR, including 44 patients (48 samples) with CCA, of whom 38 (42 samples) had concomitant PSC, and 229 (296 samples) patients with NM liver diseases, of whom 205 (272 samples) had PSC. All of these samples were subjected to downstream analyses, with the most detailed analyses performed among patients with CCA complicated with PSC and PSC controls (Figure 1; Table S1).
CCA‐PSC versus PSC‐controls
High accuracy for CCA detection in patients with underlying PSC up to 12 months prior to CCA diagnosis established by standard diagnostic modalities
To investigate the potential of the four DNA methylation markers to detect CCA in patients with underlying PSC, we included all CCA samples derived from patients with concomitant PSC (n = 38), and as controls we included all patients with PSC (n = 205), irrespective of the presence of dysplasia and length of follow‐up. First, we compared the normalized methylation levels of all four markers in the two groups, showing that samples from patients with CCA‐PSC displayed significantly higher methylation levels than samples from patients with PSC for all four markers (Mann‐Whitney U, p < 0.0001; Figure S1). Second, we did a ROC curve analysis to see how well the markers could differentiate between CCA‐PSC and PSC. Plotting the normalized DNA methylation values from the CCA‐PSCs against the normalized DNA methylation values from PSCs demonstrated that the individual biomarkers had high accuracy in distinguishing the two groups, with AUCs, sensitivities, and specificities ranging 0.77–0.87, 54%–76%, and 93%–98%, respectively (Figure 2A). Thirty of the 38 CCA samples had at least one methylated marker (79% sensitivity for the biomarker panel), while only 20 of the 203 samples (with valid DNA biomarker results) from patients with PSC were positive for the methylation panel (90% specificity). Combining the markers resulted in an AUC of 0.88 (Figure S2A).
FIGURE 2.
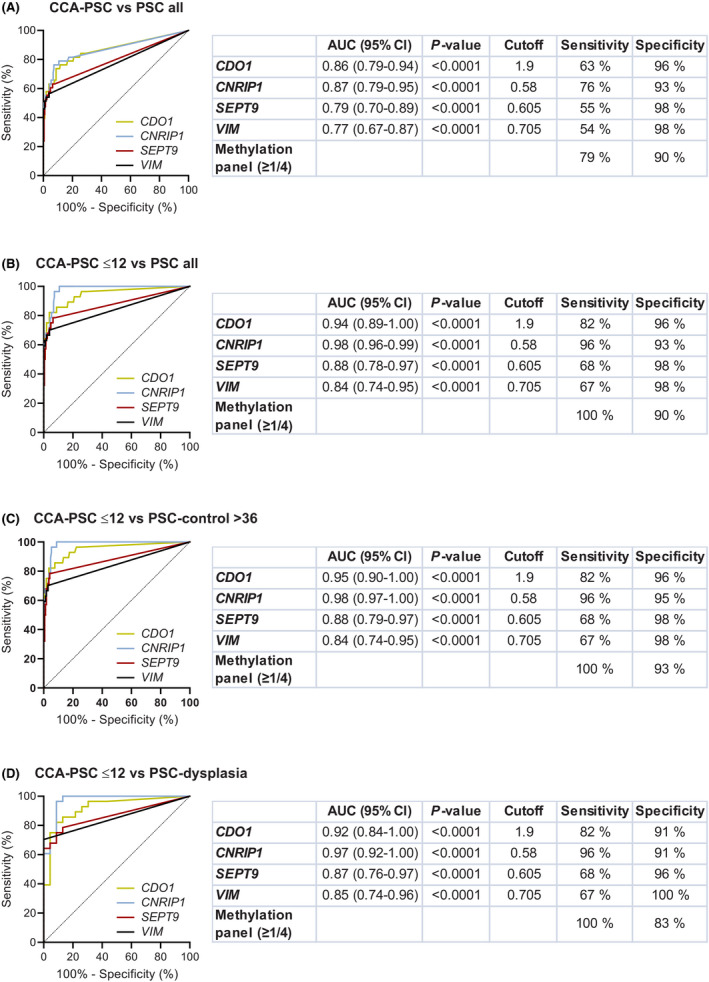
ROC curves, calculated AUCs, and sensitivity and specificity values for the four individual DNA methylation biomarkers in bile. (A) Samples from patients with CCA‐PSC (n = 38) versus all PSC (n = 205), (B) samples from patients with PSC diagnosed with CCA ≤ 12 months after bile sampling (CCA‐PSC ≤ 12, n = 28) versus PSC all (n = 205), (C) CCA‐PSC ≤ 12 (n = 28) versus PSC‐control > 36 (n = 170, including samples from patients with PSC showing no evidence of biliary dysplasia or CCA in explanted liver or with > 36 months of follow‐up), and (D) CCA‐PSC ≤ 12 (n = 28) versus PSC‐dysplasia (n = 23, including patients with PSC with evidence of biliary dysplasia based on assessment of biliary brush cytology specimens or histological assessment of explant liver ±2 months from bile collection)
Further, to see if time from bile sampling to a CCA diagnosis could affect the sensitivity of the DNA methylation markers, we included only CCA samples from patients diagnosed with CCA‐PSC ≤ 12 months from bile sampling (n = 28). As controls, we included (1) all PSC samples (n = 205), (2) samples from the PSC‐control > 36 group (n = 170; no evidence of CCA or biliary dysplasia based on histological assessment of explanted liver or > 36 months of follow‐up in nontransplanted patients), and (3) PSC‐dysplasia (n = 23; patients with PSC with evidence of biliary dysplasia based on assessment of biliary brush cytology specimens or histological assessment of explanted liver ±2 months from bile collection but with no evidence of CCA development). ROC curve analysis, plotting the normalized DNA methylation values for CCA‐PSC ≤ 12 against the normalized values for all PSC samples showed that the ability to separate the two groups was high, with AUCs of 0.98 (CNRIP1), 0.94 (CDO1), 0.88 (SEPT9), and 0.84 (VIM). The sensitivities and specificities of the individual markers ranged 67%–96% and 93%–98%, respectively (Figure 2B). Interestingly, the sensitivity for the biomarker panel (defined by one or more of four methylation positive markers) was 100% with a specificity of 90%. Including only the PSC‐control > 36 as controls increased the specificity for the biomarker panel to 93% (Figure 2C). Finally, we included only the PSC‐dysplasia samples as controls. The ability to distinguish the two groups remained high with AUCs ranging from 0.85 to 0.97 for the individual markers and a sensitivity and specificity for the biomarker panel of 100% and 83%, respectively (Figure 2D). AUCs for the combined biomarkers can be found in Figure S2B–D, including all PSC, PSC‐control > 36, and PSC‐dysplasia as controls, respectively.
Of further interest, the bile samples from the CCA‐PSC ≤ 12 patients, all positive for the methylation panel, were collected from patients with both early‐stage and late‐stage cancer at diagnosis (0–2 and 3–4), different tumor growth patterns (mass‐forming, periductal‐infiltrating, and intraductal‐growing), tumor size (< 3, 3–5, and > 5 cm), and anatomical location (intrahepatic CCA [iCCA], perihilar CCA [pCCA], and extrahepatic CCA [dCCA]) (Tables 2 and 3). Thus, the methylation panel achieved a sensitivity of 100% and a specificity of 90% for detecting CCA in patients with underlying PSC, including dysplasia, up to 12 months prior to a confirmed CCA diagnosis and irrespective of clinical and molecular features (Table 3). All CCA samples negative for methylation were analyzed > 12 months prior to CCA diagnosis (Table S1). Of note, no correlation was observed between age and methylation among CCA‐PSC for any of the four genes. For CNRIP1 and VIM a weak correlation was observed among patients with PSC (Table S3).
TABLE 3.
Associations between methylation of the biomarker panel and clinical and molecular features
| CCA‐PSC ≤ 12 | CCA‐PSC > 12 | |||||
|---|---|---|---|---|---|---|
| Total n | Pos. n (%) | Neg. n (%) | Total n | Pos. n (%) | Neg. n (%) | |
| No. of patients | 28 | 28 (100) | 0 (0) | 10 | 2 (20) | 8 (80) |
| Gender | ||||||
| Male | 20 | 20 (100) | 0 (0) | 9 | 1 (11) | 8 (89) |
| Female | 8 | 8 (100) | 0 (0) | 1 | 1 (100) | 0 (0) |
| AJCC (8th edition) | ||||||
| 0–2 | 7 | 7 (100) | 0 (0) | 1 | 0 (0) | 1 (100) |
| 3–4 | 19 | 19 (100) | 0 (0) | 5 | 1 (20) | 4 (80) |
| NA | 2 | 2 (100) | 0 (0) | 4 | 1 (25) | 3 (75) |
| Tumor location | ||||||
| iCCA | 8 | 8 (100) | 0 (0) | 6 | 1 (17) | 5 (83) |
| pCCA | 13 | 13 (100) | 0 (0) | 2 | 0 (0) | 2 (100) |
| dCCA | 5 | 5 (100) | 0 (0) | 2 | 1 (50) | 1 (50) |
| xCCA | 2 | 2 (100) | 0 (0) | – | – | – |
| Perineural growth | ||||||
| Yes | 11 | 11 (100) | 0 (0) | 1 | 0 (0) | 1 (100) |
| No | 2 | 2 (100) | 0 (0) | – | – | – |
| NA | 15 | 15 (100) | 0 (0) | 9 | 2 (22) | 7 (78) |
| Vascular encasement | ||||||
| Yes | 5 | 5 (100) | 0 (0) | 1 | 0 (0) | 1 (100) |
| No | 9 | 9 (100) | 0 (0) | 1 | 0 (0) | 1 (100) |
| NA | 14 | 14 (100) | 0 (0) | 8 | 2 (25) | 6 (75) |
| Differentiation | ||||||
| Low | 3 | 3 (100) | 0 (0) | 1 | 0 (0) | 1 (100) |
| Medium | 10 | 10 (100) | 0 (0) | 1 | 0 (0) | 1 (100) |
| High | 1 | 1 (100) | 0 (0) | – | – | – |
| NA | 14 | 24 (100) | 0 (0) | 8 | 2 (25) | 6 (75) |
| Growth pattern | ||||||
| Mass‐forming | 8 | 8 (100) | 0 (0) | 2 | 0 (0) | 2 (100) |
| Periductal‐infiltrating | 5 | 5 (100) | 0 (0) | 1 | 1 (100) | 0 (0) |
| Intraductal‐growing | 1 | 1 (100) | 0 (0) | – | – | – |
| NA | 14 | 14 (100) | 0 (0) | 7 | 1 (14) | 6 (86) |
| Tumor diameter | ||||||
| <3 cm | 7 | 7 (100) | 0 (0) | – | – | – |
| 3–5 cm | 8 | 8 (100) | 0 (0) | 1 | 0 (0) | 1 (100) |
| >5 cm | 6 | 6 (100) | 0 (0) | 4 | 1 (25) | 3 (75) |
| NA | 7 | 7 (100) | 0 (0) | 5 | 1 (20) | 4 (80) |
| Resection margins | ||||||
| R0 | 8 | 8 (100) | 0 (0) | 1 | 0 (0) | 1 (100) |
| R1 | 5 | 5 (100) | 0 (0) | 1 | 0 (0) | 1 (100) |
| NA | 15 | 15 (100) | 0 (0) | 8 | 2 (25) | 6 (75) |
| Age at bile sampling, years | ||||||
| 10–29 | 1 | 1 (100) | 0 (0) | – | – | – |
| 30–39 | 4 | 4 (100) | 0 (0) | 2 | 0 (0) | 2 (100) |
| 40–49 | 6 | 6 (100) | 0 (0) | 2 | 1 (50) | 1 (50) |
| 50–59 | 9 | 9 (100) | 0 (0) | 5 | 1 (20) | 4 (80) |
| 60–79 | 8 | 8 (100) | 0 (0) | 1 | 0 (0) | 1 (100) |
| IBD | ||||||
| UC | 19 | 19 (100) | 0 (0) | 3 | 0 | 3 (100) |
| Crohn's disease | 4 | 4 (100) | 0 (0) | 1 | 1 (100) | 0 (0) |
| None | 4 | 4 (100) | 0 (0) | 5 | 1 (20) | 4 (80) |
| NA | 1 | 1 (100) | 0 (0) | 1 | 0 (0) | 1 (0) |
| CA 19‐9 | ||||||
| >100 U/ml | 14 | 14 (100) | 0 (0) | 0 | – | – |
| <100 U/ml | 11 | 11 (100) | 0 (0) | 10 | 2 (20) | 8 (80) |
| NA | 3 | 3 (100) | 0 (0) | |||
| Bile origin | ||||||
| ERCP | 26 | 26 (100) | 0 (0) | 10 | 2 (20) | 8 (80) |
| Resection | 1 | 1 (100) | 0 (0) | – | – | – |
| Transplantation | 1 | 1 (100) | 0 (0) | – | – | – |
| Alive | ||||||
| Yes | 9 | 9 (100) | 0 (0) | 1 | 1 (100) | 0 (0) |
| No | 19 | 19 (100) | 0 (0) | 9 | 1 (11) | 8 (89) |
Abbreviations: AJCC 0–4, American Joint Committee on Cancer classification stages 0–4; IBD, inflammatory bowel disease; NA, not available; Neg., negative for the biomarker panel (none of the four markers methylated); Pos., positive for the biomarker panel (one or more of the four markers methylated); UC, ulcerative colitis.
DNA methylation in patients with PSC and underlying biliary dysplasia
To investigate if methylation levels of one or more markers in the four‐gene panel increased stepwise in bile derived from benign, dysplastic, and malignant stages of disease, we compared the methylation levels of the four genes in the PSC‐dysplasia group (n = 23) with the benign PSC‐control > 36 group (n = 170) and the CCA‐PSC ≤ 12 group (n = 28). The normalized methylation levels in the benign PSC and the PSC‐dysplasia groups compared with the CCA group were significantly different for all four markers (Figure S3). Although no significant difference in methylation levels for any of the four markers was observed between the PSC‐dysplasia group and the PSC‐control > 36 group, an increase in methylation status was observed from the PSC‐control > 36 group (7%) to the PSC‐dysplasia group (17%) and to CCA (100%; Fisher's exact, p < 0.001) (Table S4).
Comparison of the methylation panel with CA 19‐9 levels and brush cytology in indicating CCA in PSC
We also wanted to compare the results of the methylation panel in bile with the informativeness of CA 19‐9 levels and conventional brush cytology assessments in detecting CCA in PSC. Because FISH analyses were not available for the majority of the samples, no comparisons were made between FISH and the methylation panel.
Of the 28 CCA‐PSC ≤ 12 patients, CA 19‐9 levels were obtained from 25 patients, of whom 14 (56%) had a CA 19‐9 level > 100 U/ml. The biomarker panel was positive in all of these 14 patients (100% sensitivity). In addition, the biomarker panel was positive in all of the 11 samples with a CA 19‐9 level < 100 U/ml (Table 4 and Figure 3). Among the 203 patients with PSC with valid DNA biomarker results, CA 19‐9 measurements were available for 193 patients, including 13 (7%) with a CA 19‐9 level > 100 U/ml and 180 (93%) with a CA 19‐9 level < 100 U/ml. The biomarker panel was negative in 92% of the patients with CA 19‐9 > 100 U/ml (n = 13) and in 90% of the patients with CA 19‐9 < 100 U/ml (n = 180) (Table 4). Including only PSC‐control > 36, the biomarker panel was negative in 100% of the patients with CA 19‐9 > 100 U/ml. Thus, the high sensitivity (100%) of the biomarker panel for CCA diagnosis was independent of CA 19‐9 level, and the specificity remained high (> 90%) in patients with CA 19‐9 level above and below 100 U/ml.
TABLE 4.
Sensitivity and specificity of the biomarker panel in patients with CA 19‐9 level above and below 100 U/ml
| CCA‐PSC ≤ 12 | CCA‐PSC > 12 | PSC all | PSC‐control > 36 | PSC‐dysplasia | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total n | Pos. (%) | Neg. (%) | Total n | Pos. (%) | Neg. (%) | Total n | Pos. (%) | Neg. (%) | Total n | Pos. (%) | Neg. (%) | Total n | Pos. (%) | Neg. (%) | |
| No. of patients | 28 | 28 (100) | 0 (0) | 10 | 2 (20) | 8 (80) | 203 | 20 (10) | 183 (90) | 168 | 12 (7) | 156 (93) | 23 | 4 (17) | 19 (83) |
| CA 19‐9 | |||||||||||||||
| > 100 U/ml | 14 | 14 (100) | 0 (0) | 0 | 0 | 0 | 13 | 1 (8) | 12 (92) | 11 | 0 (0) | 11 (100) | 2 | 1 (50) | 1 (50) |
| < 100 U/ml | 11 | 11 (100) | 0 (0) | 10 | 2 (20) | 8 (80) | 180 | 18 (10) | 162 (90) | 150 | 12 (8) | 138 (92) | 21 | 3 (14) | 18 (86) |
| NA | 3 | 3 (100) | 0 (0) | – | – | – | 10 | 1 (10) | 9 (90) | 7 | 0 (0) | 7 (100) | – | – | – |
Group definitions: CCA‐PSC ≤ 12, patients with CCA and concomitant PSC and ≤ 12 months from bile sampling to a confirmed CCA diagnosis; CCA‐PSC > 12, patients with CCA and concomitant PSC and > 12 months from bile sampling to CCA diagnosis; PSC‐dysplasia, patients with PSC and evidence of biliary dysplasia based on assessment of biliary brush cytology specimens or histological assessment of explant liver ±2 months from bile collection; PSC‐control > 36, patients with PSC and no evidence of CCA or biliary dysplasia based on histological assessment of explanted liver at or after bile collection or > 36 months of follow‐up without established CCA or biliary dysplasia for nontransplanted patients; PSC all, benign PSC without CCA.
Abbreviations: NA, not available; Neg., negative for the biomarker panel (none of the four markers methylated); Pos., positive for the biomarker panel (one or more of the four markers methylated).
FIGURE 3.

Comparison of DNA methylation in bile with CA 19‐9 levels and biliary brush cytology. Red circle, sample scored as positive for methylation/cytology/CA 19‐9; white circle, sample scored as negative for methylation/cytology/CA 19‐9. The methylation panel was considered positive if one or more of the four biomarkers were methylated. Biliary brush cytology was scored positive if low/moderate‐grade or high‐grade dysplasia was identified and negative if only normal cells were identified. If more than one brush sample was taken, the case was scored based on the highest grade of dysplasia found. In cases where the sample was scored between two grades of dysplasia, the highest grade was used. For CA 19‐9 levels, 100 U/ml was used as the threshold: a CA 19‐9 level > 100 U/ml was considered positive, while a CA 19‐9 level < 100 U/ml was considered negative. The “b” samples are not included in the biomarker analysis. NA, not available; NC, acellular or nondiagnostic/scarce material not sufficient for further scoring
In the CCA‐PSC ≤ 12 group parallel cytology specimens from biliary brushings had been obtained for 22 patients (24 samples). Among these, brush cytology samples were not conclusive (acellular or nondiagnostic/scarce material) in seven samples (29%), of which all (100%) were positive for the methylation panel (Figure 3). Of the remaining 17 samples with conclusive results, 12 (71%) were scored as positive for low‐grade or high‐grade dysplasia, while the combined methylation panel was scored as positive in 100% of the samples (17/17) (Figure 3).
Among the PSC‐control > 36 group (n = 170), parallel bile (methylation) and brush cytology results were available for 127 patients. Among these patients brush cytology was not conclusive (acellular or nondiagnostic material) in 42 patients (33%). Of the remaining 85 patients, all scored as negative for dysplasia; 80 (94%) were also negative for the methylation panel.
High concordance in methylation status between multiple samples from the same patient
For 52 patients, including four with CCA‐PSC and 48 with PSC, multiple samples (two to four) were analyzed. For the majority of the patients where longitudinal sampling was present, disease category did not change during the sampling period (e.g., all samples from the same patient belonged to the CCA‐PSC ≤ 12 group or to the PSC‐control > 36 group). For the four patients with CCA (eight samples), a 100% concordance in methylation status for the biomarker panel was observed, while for the patients with PSC, 41 of 48 (85%) had concordant methylation results (Figure S4). None of the PSC samples with deviating results (one sample scored as negative for the biomarker panel and one sample scored as positive for the biomarker panel) were collected during ongoing acute cholangitis. However, the majority of the samples with deviating results had been collected > 2 years apart.
In addition, prior to this effort we analyzed the biomarkers in biliary brushes and tissue samples.[ 15 , 22 ] Figure S5 provides a direct comparison of methylation in bile, brush, and tissue of the four biomarkers in a subset of the samples (54 patients) included in previous efforts. In general, there was a high concordance in methylation status between the different materials for the same patients.
DNA methylation level and association with survival
To investigate if the level of methylation in bile could have an effect on survival, we did a log rank test. Dividing the CCA‐PSC ≤ 12 samples into high (normalized DNA methylation level > 10) and 0/low (normalized DNA methylation level < 10) level of methylation showed that patients with a high level of methylation had a tendency for shorter overall survival compared to patients with no or a low level of methylation (CDO1, p = 0.059; CNRIP1, p = 0.016; SEPT9, p = 0.078; and VIM p = 0.203) (Figure S6).
CCA without underlying PSC versus other NM liver disease
The methylation panel has potential to detect CCA in a non‐PSC setting
Finally, to investigate the potential of the biomarkers to also detect CCA in a non‐PSC setting, we compared the CCA samples derived from patients without underlying PSC (n = 6) with samples from patients with other NM liver diseases (n = 24). Results for the individual biomarkers are summarized in Figure S7, showing AUCs of 0.78 (CDO1), 1.00 (CNRIP), 0.75 (SEPT9), and 0.90 (VIM). Combined, the biomarker panel had a sensitivity of 100% and a specificity of 100% for distinguishing the two groups. Although we had few samples, these data show that the biomarkers have the potential to also differentiate CCA without underlying PSC from benign non‐PSC liver diseases. For clinical and histopathological data on CCA and other NM liver diseases, see Table S5A,B.
DISCUSSION
By analyzing an exceptionally large series of bile samples from well‐characterized patients, we demonstrate that four DNA methylation biomarkers accurately differentiate patients with CCA from patients with NM liver disease. The diagnostic accuracy for CCA was high both in sporadic and in PSC‐associated cancer, underscored by high specificity and the finding of positive methylation markers in all bile samples obtained up to 12 months prior to a confirmed CCA diagnosis. Furthermore, we have prior to this effort shown that the markers were highly methylated in CCA tumor tissue,[ 22 ] then in biliary brushes.[ 15 ] In the current study the utility of the methylation panel for early CCA diagnosis was validated using ddPCR in a limited amount of bile, emphasizing the robustness of the biomarkers. As such, this work represents a major advance in the field. The promising results suggest a potential for using these DNA methylation markers in bile (1) to complement current detection methods for CCA and (2) for CCA surveillance in patients with PSC. This also represents one of the largest liquid biopsy–derived biomarker studies in PSC‐CCA to date.[ 14 ]
Two previous studies have also suggested DNA methylation analyses of bile as a possible modality to detect CCA. Klump et al.[ 23 ] reported that cyclin‐dependent kinase inhibitor 2A (CDKN2A) was methylated in 52% (p16) and 48% (p14) of bile samples from patients with CCA and in only 6% of healthy individuals. Although promising, as a proof of concept for CCA detection using bile, CDKN2A methylation was also present in 46% of patients with PSC without an established CCA diagnosis, making this marker unsuitable for early detection of CCA in patients with PSC.[ 23 ] Shin et al.[ 24 ] analyzed 17 aberrantly methylated biomarkers in dCCA in a training and a validation set of bile specimens (53 CCA and 32 NM controls). The four best‐performing panels from these two sets were then analyzed in an independent test set (24 CCA and 16 NM controls). With 100% specificity and sensitivities of 70%, 74%, and 83% for the training, validation, and test sets, respectively, the best‐performing marker panel showed potential for accurate dCCA detection.[ 24 ] However, no PSC controls were included in the study. Because inflammation, as observed in PSC, is known to affect the methylation pattern,[ 25 ] the lack of PSC controls could affect the specificity of a potential detection test aimed at patients with PSC. In our study, the few false positives detected were all among the PSC controls; and if only including other NM liver diseases excluding PSC, we obtained a specificity of 100%. In general, we further observed higher methylation levels in patients with CCA and underlying PSC compared to those with CCA but without PSC (not shown). This has also been reported by others, using peptide markers in urine[ 26 ] and bile.[ 27 ]
Using blood to identify biomarkers is also attractive for cancer surveillance, considering both compliance and availability. Two genes, short stature homeobox 2 and SEPT9, have been reported to be methylated in 35% and 25%, respectively, of plasma samples from patients with CCA, with 99% specificity.[ 28 ] Although these findings are interesting and the specificity is good, the low sensitivity limits the usefulness of the markers as surveillance tools in a clinical setting. The performance in blood was furthermore poorer than that in tissue, leading one to question whether the amount of circulating cell free tumor DNA in blood is high enough for early and robust detection of CCA.[ 28 ] Because bile is drained directly from the biliary tract where the primary tumor resides, it is reasonable to believe that bile contains higher concentrations of tumor DNA from CCA compared to blood, and therefore may prove superior to blood for early and accurate CCA diagnosis.
So far, no evidence‐based surveillance strategy exists for CCA in patients with PSC. Current practice includes interval screening with serum‐based CA 19‐9 and ultrasound or MR imaging/cholangiopancreatography. If results are positive, ERCP with biliary brush cytology is typically performed,[ 8 , 29 , 30 , 31 , 32 ] often in combination with FISH.[ 31 ] Other surveillance strategies also exist, including using ERCP and brush cytology as primary surveillance tools.[ 33 , 34 ] Both CA 19‐9[ 31 ] and imaging[ 32 ] display suboptimal accuracy for CCA detection, although MRI was recently reported to be superior to ultrasound for detection of early‐stage CCA in asymptomatic patients with PSC.[ 35 ] The specificity of biliary brush for cytology and FISH is reported to be high (pooled specificity 97% and 96%, respectively), but the sensitivity is variable and often low (pooled sensitivity 47% and 50%, respectively).[ 36 , 37 ] False‐negative results may occur when the tumor resides in areas not accessible to or not targeted by brush cytology sampling. It can further be challenging to obtain adequate cytological material for evaluation due to the fibrotic and desmoplastic process in PSC and CCA.[ 19 ] Our findings demonstrate the challenge of obtaining appropriate biliary brush material for diagnostic purposes, with a large proportion of the brush cytologies in the CCA group and in the benign PSC group not being informative (~30%). The performance of biliary brush cytology is also largely dependent on the individual endoscopists and cytopathologists. This may lead to considerable variability in the diagnostic informativeness of biliary brush cytology between centers.[ 38 ] In contrast, it is easier to standardize methodologies for bile sampling and methylation profiling. Also, using bile as diagnostic material for CCA, the chance of obtaining representative material increases because cells and DNA are shed into bile independently of tumor localization. Indeed, using highly sensitive ddPCR methodology in limited amounts of bile, we successfully detected alterations in methylation levels that robustly differentiated patients with CCA from those without CCA, including in patients with underlying PSC. The high sensitivity for CCA detection was irrespective of clinical and molecular features, including cancer stage at diagnosis, tumor growth pattern, tumor size, anatomical location, and CA 19‐9 levels. The few samples from patients with PSC with a later confirmed CCA diagnosis not detected by the biomarkers included bile samples collected > 12 months prior to a clinical diagnosis. Hence, there is a high chance that these patients had not developed CCA at the time of bile sampling.
The current study included CCA‐PSC samples from three independent centers from three different countries. Unfortunately, due to the rarity of the disease, there were too few samples to include them as three independent series. We do, however, observe that the markers show 100% sensitivity among the CCA‐PSC ≤ 12 group, irrespective of center/country, with similar high specificities in the PSC‐control > 36 group (Sweden, 100%; Finland, 90%; Norway, 93%). Despite our promising findings, validation in larger multicenter sample series is warranted. Ideally, such analyses should be performed long‐term, longitudinally, in a large prospective cohort of bile samples from patients with PSC, to see whether patients with PSC who later develop CCA show early signs of methylation, and further to systematically compare the accuracy of the methylation panel with conventional diagnostic measures for CCA. The current study demonstrates that methylation changes may be detected up to 12 months prior to standard CCA detection methods. Of further interest, patients with biliary dysplasia, who are at higher risk of developing CCA,[ 39 ] displayed a higher methylation frequency (17%) compared to patients with PSC without findings of dysplasia (7%). Still, including longitudinal data and consecutive bile samples from patients with PSC could give a better indication of how long prior to malignancy these methylation changes are expected to occur. It might also give an indication of whether these changes are associated with biliary dysplasia or predominantly represent markers for CCA development that are independent of the dysplasia–carcinoma sequence. Four of 12 (33%) PSC samples from patients without an established CCA diagnosis but with < 36 months of follow‐up were methylated for the biomarkers. We cannot rule out that some of these might be false positives, caused by severe inflammation, cholestasis, or other benign factors in the bile ducts. Another possibility is that these patients represent a high‐risk group for CCA development. The PSC samples included in the study are derived from tertiary center referrals of patients undergoing ERCP, surgical resection, or liver transplantation. As such, the PSC samples included are enriched for advanced PSC cases, and it is not unlikely that a subgroup of these patients is at higher risk for CCA development. However, we observed no association between concurrent bacterial acute cholangitis or cirrhosis and DNA methylation level of any of the individual markers or with the methylation panel.
Interestingly, the individual DNA methylation markers analyzed in the current study for their diagnostic potential have also been evaluated by others for their prognostic value. Nakamoto et al. reported that the overall survival in patients with dCCA was poorer in patients with hypermethylated CDO1,[ 40 ] while a study by Chen et al. showed that low expression of CNRIP1, caused by DNA methylation, was associated with poor prognosis in patients with iCCA.[ 41 ] DNA methylation level of SEPT9 in tissue showed, on the other hand, no correlation with overall survival.[ 28 ] Although an in‐depth analysis of the prognostic value of the four biomarkers was outside the scope of the current study, we did a log rank test to investigate if the methylation level in bile could have an effect on survival, observing that our results were in agreement with what was reported by others.
In conclusion, by analyzing an unprecedented large series of bile samples predominantly derived from patients with PSC, we have shown that testing for DNA methylation biomarkers in bile detects CCAs in patients with PSC with high accuracy and at an earlier stage than conventional modalities. The findings suggest that these biomarkers have potential to complement standard modalities for improved CCA detection and potentially for CCA surveillance in patients with PSC. Prospective validation of the markers in a clinical setting is warranted.
CONFLICT OF INTEREST
Dr. Bergquist received grants from Gilead.
AUTHOR CONTRIBUTIONS
Hege Marie Vedeld: data acquisition and analysis, interpretation of data, manuscript drafting Marit Mæhle Grimsrud: data acquisition and analysis, interpretation of data, manuscript revision and approval. Kim Andresen: data acquisition and analysis, interpretation of data, manuscript revision and approval. Heidi D. Pharo: data acquisition and analysis, manuscript revision and approval. Erik von Seth: data acquisition, manuscript revision and approval. Tom H. Karlsen: data acquisition, manuscript revision and approval. Hilde Honne: data acquisition and analysis, manuscript revision and approval. Vemund Paulsen: data acquisition, manuscript revision and approval. Martti A Färkkilä: data acquisition, manuscript revision and approval. Annika Bergquist: data acquisition, manuscript revision and approval. Marine Jeanmougin: interpretation of data, manuscript revision and approval. Lars Aabakken: data acquisition, manuscript revision and approval. Kirsten M. Boberg: data acquisition, interpretation of data, manuscript revision and approval. Trine Folseraas: data acquisition and analysis, interpretation of data, manuscript revision and approval. Guro E. Lind: study concept and design, data acquisition and analysis, interpretation of data, manuscript revision and approval, study supervision.
Supporting information
Supplementary Material
Vedeld HM, Grimsrud MM, Andresen K, Pharo HD, von Seth E, Karlsen TH, et al. Early and accurate detection of cholangiocarcinoma in patients with primary sclerosing cholangitis by methylation markers in bile. Hepatology.2022;75:59–73. 10.1002/hep.32125
Hege Marie Vedeld and Marit Mæhle Grimsrud shared co‐first authorship.
Funding information
Supported by grants from the South‐Eastern Norway Regional Health Authority (project 40093, to G.E.L.; 2017016, to M.M.G.)
REFERENCES
- 1. Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. Primary sclerosing cholangitis. Lancet. 2013;382:1587–99. [DOI] [PubMed] [Google Scholar]
- 2. Schrumpf E, Boberg KM, Karlsen TH. Primary sclerosing cholangitis—the Norwegian experience. Scand J Gastroenterol. 2015;50:781–96. [DOI] [PubMed] [Google Scholar]
- 3. Karlsen TH, Folseraas T, Thorburn D, Vesterhus M. Primary sclerosing cholangitis—a comprehensive review. J Hepatol. 2017;67:1298–323. [DOI] [PubMed] [Google Scholar]
- 4. Bergquist A, Ekbom A, Olsson R, Kornfeldt D, Lööf L, Danielsson Å, et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol. 2002;36:321–7. [DOI] [PubMed] [Google Scholar]
- 5. Boonstra K, Weersma RK, van Erpecum KJ, Rauws EA, Spanier BWM, Poen AC, et al. Population‐based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology. 2013;58:2045–55. [DOI] [PubMed] [Google Scholar]
- 6. Tanaka A, Takamori Y, Toda G, Ohnishi S, Takikawa H. Outcome and prognostic factors of 391 Japanese patients with primary sclerosing cholangitis. Liver Int. 2008;28:983–9. [DOI] [PubMed] [Google Scholar]
- 7. Weismuller TJ, Trivedi PJ, Bergquist A, Imam M, Lenzen H, Ponsioen CY, et al. Patient age, sex, and inflammatory bowel disease phenotype associate with course of primary sclerosing cholangitis. Gastroenterology. 2017;152:1975–84.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Folseraas T, Boberg KM. Cancer risk and surveillance in primary sclerosing cholangitis. Clin Liver Dis. 2016;20:79–98. [DOI] [PubMed] [Google Scholar]
- 9. Razumilava N, Gores GJ. Surveillance for cholangiocarcinoma in patients with primary sclerosing cholangitis: effective and justified? Clin Liver Dis. 2016;8:43–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bowlus CL, Lim JK, Lindor KD. AGA clinical practice update on surveillance for hepatobiliary cancers in patients with primary sclerosing cholangitis: expert review. Clin Gastroenterol Hepatol. 2019;17:2416–22. [DOI] [PubMed] [Google Scholar]
- 11. Kang MJ, Jang J‐Y, Chang J, Shin YC, Lee D, Kim HB, et al. Actual long‐term survival outcome of 403 consecutive patients with hilar cholangiocarcinoma. World J Surg. 2016;40:2451–9. [DOI] [PubMed] [Google Scholar]
- 12. Azad AI, Rosen CB, Taner T, Heimbach JK, Gores GJ. Selected patients with unresectable perihilar cholangiocarcinoma (pCCA) derive long‐term benefit from liver transplantation. Cancers. 2020;12(11):3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vedeld HM, Goel A, Lind GE. Epigenetic biomarkers in gastrointestinal cancers: the current state and clinical perspectives. Semin Cancer Biol. 2018;51:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vedeld HM, Folseraas T, Lind GE. Detecting cholangiocarcinoma in patients with primary sclerosing cholangitis—the promise of DNA methylation and molecular biomarkers. JHEP Rep. 2020;2:100143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andresen K, Boberg KM, Vedeld HM, Honne H, Jebsen P, Hektoen M, et al. Four DNA methylation biomarkers in biliary brush samples accurately identify the presence of cholangiocarcinoma. Hepatology. 2015;61:1651–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lazaridis KN, LaRusso NF. Primary sclerosing cholangitis. N Engl J Med. 2016;375:1161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma—evolving concepts and therapeutic strategies. Nat Rev Clin Oncol. 2018;15:95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Majeed A, Castedal M, Arnelo U, Söderdahl G, Bergquist A, Said K. Optimizing the detection of biliary dysplasia in primary sclerosing cholangitis before liver transplantation. Scand J Gastroenterol. 2018;53:56–63. [DOI] [PubMed] [Google Scholar]
- 19. Boberg KM, Jebsen P, Clausen OP, Foss A, Aabakken L, Schrumpf E. Diagnostic benefit of biliary brush cytology in cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol. 2006;45:568–74. [DOI] [PubMed] [Google Scholar]
- 20. Lind GE, Danielsen SA, Ahlquist T, Merok MA, Andresen K, Skotheim RI, et al. Identification of an epigenetic biomarker panel with high sensitivity and specificity for colorectal cancer and adenomas. Mol Cancer. 2011;10:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pharo HD, Andresen K, Berg KCG, Lothe RA, Jeanmougin M, Lind GE. A robust internal control for high‐precision DNA methylation analyses by droplet digital PCR. Clin Epigenetics. 2018;10:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Andresen K, Boberg K, Vedeld H, Honne H, Hektoen M, Wadsworth C, et al. Novel target genes and a valid biomarker panel identified for cholangiocarcinoma. Epigenetics. 2012;7:1249–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klump B, Hsieh CJ, Dette S, Holzmann K, Kiebetalich R, Jung M, et al. Promoter methylation of INK4a/ARF as detected in bile—significance for the differential diagnosis in biliary disease. Clin Cancer Res. 2003;9:1773–8. [PubMed] [Google Scholar]
- 24. Shin S‐H, Lee K, Kim B‐H, Cho N‐Y, Jang J‐Y, Kim Y‐T, et al. Bile‐based detection of extrahepatic cholangiocarcinoma with quantitative DNA methylation markers and its high sensitivity. J Mol Diagn. 2012;14:256–63. [DOI] [PubMed] [Google Scholar]
- 25. Barnicle A, Seoighe C, Greally JM, Golden A, Egan LJ. Inflammation‐associated DNA methylation patterns in epithelium of ulcerative colitis. Epigenetics. 2017;12:591–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Metzger J, Negm AA, Plentz RR, Weismüller TJ, Wedemeyer J, Karlsen TH, et al. Urine proteomic analysis differentiates cholangiocarcinoma from primary sclerosing cholangitis and other benign biliary disorders. Gut. 2013;62:122–30. [DOI] [PubMed] [Google Scholar]
- 27. Lankisch TO, Metzger J, Negm AA, Voβkuhl K, Schiffer E, Siwy J, et al. Bile proteomic profiles differentiate cholangiocarcinoma from primary sclerosing cholangitis and choledocholithiasis. Hepatology. 2011;53:875–84. [DOI] [PubMed] [Google Scholar]
- 28. Branchi V, Schaefer P, Semaan A, Kania A, Lingohr P, Kalff JC, et al. Promoter hypermethylation of SHOX2 and SEPT9 is a potential biomarker for minimally invasive diagnosis in adenocarcinomas of the biliary tract. Clin Epigenetics. 2016;8:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Razumilava N, Gores GJ, Lindor KD. Cancer surveillance in patients with primary sclerosing cholangitis. Hepatology. 2011;54:1842–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eaton JE, Gossard AA, Talwalkar JA. Recall processes for biliary cytology in primary sclerosing cholangitis. Curr Opin Gastroenterol. 2014;30:287–94. [DOI] [PubMed] [Google Scholar]
- 31. Rizvi S, Eaton JE, Gores GJ. Primary sclerosing cholangitis as a premalignant biliary tract disease: surveillance and management. Clin Gastroenterol Hepatol. 2015;13:2152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Charatcharoenwitthaya P, Enders FB, Halling KC, Lindor KD. Utility of serum tumor markers, imaging, and biliary cytology for detecting cholangiocarcinoma in primary sclerosing cholangitis. Hepatology. 2008;48:1106–17. [DOI] [PubMed] [Google Scholar]
- 33. Boyd S, Mustonen H, Tenca A, Jokelainen K, Arola J, Färkkilä MA. Surveillance of primary sclerosing cholangitis with ERC and brush cytology: risk factors for cholangiocarcinoma. Scand J Gastroenterol. 2017;52:242–9. [DOI] [PubMed] [Google Scholar]
- 34. Boyd S, Tenca A, Jokelainen K, Mustonen H, Krogerus L, Arola J, et al. Screening primary sclerosing cholangitis and biliary dysplasia with endoscopic retrograde cholangiography and brush cytology: risk factors for biliary neoplasia. Endoscopy. 2016;48:432–9. [DOI] [PubMed] [Google Scholar]
- 35. Eaton JE, Welle CL, Bakhshi Z, Sheedy SP, Idilman IS, Gores GJ, et al. Early cholangiocarcinoma detection with magnetic resonance imaging versus ultrasound in primary sclerosing cholangitis. Hepatology. 2021;73:1868–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Njei B, McCarty TR, Varadarajulu S, Navaneethan U. Systematic review with meta‐analysis: endoscopic retrograde cholangiopancreatography‐based modalities for the diagnosis of cholangiocarcinoma in primary sclerosing cholangitis. Aliment Pharmacol Ther. 2016;44:1139–51. [DOI] [PubMed] [Google Scholar]
- 37. Trikudanathan G, Navaneethan U, Njei B, Vargo JJ, Parsi MA. Diagnostic yield of bile duct brushings for cholangiocarcinoma in primary sclerosing cholangitis: a systematic review and meta‐analysis. Gastrointest Endosc. 2014;79:783–9. [DOI] [PubMed] [Google Scholar]
- 38. Harewood GC, Baron TH, Stadheim LM, Kipp BR, Sebo TJ, Salomao DR. Prospective, blinded assessment of factors influencing the accuracy of biliary cytology interpretation. Am J Gastroenterol. 2004;99:1464–9. [DOI] [PubMed] [Google Scholar]
- 39. Lewis JT, Talwalkar JA, Rosen CB, Smyrk TC, Abraham SC. Precancerous bile duct pathology in end‐stage primary sclerosing cholangitis, with and without cholangiocarcinoma. Am J Surg Pathol. 2010;34:27–34. [DOI] [PubMed] [Google Scholar]
- 40. Nakamoto S, Kumamoto Y, Igarashi K, Fujiyama Y, Nishizawa N, Ei S, et al. Methylated promoter DNA of CDO1 gene and preoperative serum CA19‐9 are prognostic biomarkers in primary extrahepatic cholangiocarcinoma. PLoS ONE. 2018;13:e0205864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen D, Wu H, Feng X, Chen Y, Lv Z, Kota VG, et al. DNA methylation of cannabinoid receptor interacting protein 1 promotes pathogenesis of intrahepatic cholangiocarcinoma through suppressing Parkin‐dependent pyruvate kinase M2 ubiquitination. Hepatology. 2021;73(5):1816–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material