

Abstract
Objective
Small fiber neuropathy (SFN) is clinically and etiologically heterogeneous. Although autoimmunity has been postulated to be pathophysiologically important in SFN, few autoantibodies have been described. We aimed to identify autoantibodies associated with idiopathic SFN (iSFN) by a novel high‐throughput protein microarray platform that captures autoantibodies expressed in the native conformational state.
Methods
Sera from 58 SFN patients and 20 age‐ and gender‐matched healthy controls (HCs) were screened against >1,600 immune‐related antigens. Fluorescent unit readout and postassay imaging were performed, followed by composite data normalization and protein fold change (pFC) analysis. Analysis of an independent validation cohort of 33 SFN patients against the same 20 HCs was conducted to identify reproducible proteins in both cohorts.
Results
Nine autoantibodies were screened with statistical significance and pFC criteria in both cohorts, with at least 50% change in serum levels. Three proteins showed consistently high fold changes in main and validation cohorts: MX1 (FC = 2.99 and 3.07, respectively, p = 0.003, q = 0.076), DBNL (FC = 2.11 and 2.16, respectively, p = 0.009, q < 0.003), and KRT8 (FC = 1.65 and 1.70, respectively, p = 0.043, q < 0.003). Further subgroup analysis into iSFN and SFN by secondary causes (secondary SFN) in the main cohort showed that MX1 is higher in iSFN compared to secondary SFN (FC = 1.61 vs 0.106, p = 0.009).
Interpretation
Novel autoantibodies MX1, DBNL, and KRT8 are found in iSFN. MX1 may allow diagnostic subtyping of iSFN patients. ANN NEUROL 2022;91:66–77
Small fiber neuropathy (SFN) is a heterogeneous disease characterized by pain, dysesthesia, and autonomic dysfunction, exclusively involves small nerve fibers, and spares the large nerve fibers. The minimum prevalence of SFN in Switzerland is calculated to be 131.5 per 100,000 population, 1 although the exact prevalence worldwide is unknown. Although diabetes and vitamin B12 deficiency are common causes of SFN, no cause is found in around 50% of the cases. 2 In a proportion of these patients, treatment with immunomodulatory agents has been useful. 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10
Intravenous immunoglobulin (IVIg) has been trialed in 3 groups of SFN patients: idiopathic SFN (iSFN), 3 , 4 SFN associated with systemic autoimmune diseases5, 6, 7, 8 and SFN associated with sarcoidosis. 9 , 10 The similarities shared among these 3 groups suggest an autoimmune and inflammatory mechanism of action. However, prior studies were retrospective and descriptive small case series or reports, and quality evidence is lacking. To date, the only double‐blinded, randomized controlled trial performed on iSFN patients contradicts this, showing a statistically nonsignificant improvement of neuropathic pain in 40% of iSFN patients treated with IVIg, compared to 30% with placebo. 11 Improvement of neuropathic pain was defined as a 1‐point improvement on the Pain Intensity Numerical Rating Scale score. 11 This underlines the pitfalls of case reports or open case studies, and the importance of double‐blind randomized trials. Nevertheless, the highly complex nature of autoimmunity implies that the lack of statistically significant effect on neuropathic pain with IVIg monotherapy in patients with iSFN cannot totally discount the possibility of an autoimmune cause in a proportion of SFN patients. SFN is a diagnostically and therapeutically challenging disease, and further understanding of the pathophysiology is required to determine the most targeted treatments for SFN patients.
Antibodies have long been implicated in various types of large fiber neuropathies and assist in classifying the disease phenotype. 12 Despite this, the pathophysiological mechanisms of SFN remain enigmatic. SFN may involve multiple immunological processes that can be further understood with proteomics. Proteins fold into spatial conformations depending on the molecular and ionic interactions in their primary structure. Traditional protein analysis techniques require isolation of the constituent proteins, proteolysis, purification, and identification. 13 Conventional techniques of protein identification involve methods such as enzyme‐linked immunosorbent assay and Western blotting, whereas newer techniques utilize mass spectrometry methods to analyze the proteins. 14 High‐throughput techniques have been made possible with the development of protein microarrays and chips, 14 and may prove useful in heterogeneous diseases where multiple mechanisms may be at work. Despite these new advances, common protein purification methods denature the native conformation of the protein, 15 affecting interaction between antigen and antibody, and subsequently the sensitivity and specificity of antibody detection. Accordingly, previous studies that have used conventional assay platforms, proteomics, or Western blotting failed to identify antibody targets for SFN. 16 , 17 , 18 , 19
Using a novel autoantibody technology (Sengenics Immunome Protein Array 20 ), we endeavored to identify putative autoantibodies in SFN. This is a validated high‐throughput technology that utilizes an array of correctly folded and functional full‐length human proteins for the detection of autoantibodies. 20 We propose that iSFN has a strong immunological component. SFN autoantibodies may now be detected in their original, physiological, and functional conformation with this new protein array platform that consists of >1,600 proteins selected on the basis of their involvement in the immune system. This study identifies novel autoantibodies associated with iSFN that can guide future cohort studies to validate their clinical and pathological significance and subtyping of SFN.
Subjects and Methods
Diagnosis and Subject Recruitment
Two cohorts of patients were recruited. For the main cohort, 59 adult patients 21 years of age and older with neuropathic pain and/or autonomic symptoms, and a diagnosis of SFN based on the NEURODIAB criteria, 21 with no apparent cause for SFN during index visit, were recruited after written informed consent from the Neurology Clinic at the National University Hospital in Singapore from January 1, 2015 to June 30, 2020. Clinical information including the symptom duration of SFN, medication use, past medical history of conditions associated with SFN, and family history of neuropathy was obtained. Healthy subjects matched for age by 5‐year intervals from 21 to 75 years and for gender were included as controls. The study was approved by the National Healthcare Group Domain Specific Review Board (Domain A, DSRB number 2015/00238).
For the validation cohort, 36 SFN patients with the same above‐mentioned criteria were recruited from the National University Hospital, Singapore, and Lucerne Cantonal Hospital, Lucerne, Switzerland from 1 July 2020–30 June 2021. The latter was an extension of the Swiss Cohort Study that was approved by the respective ethics committee (Project ID 2018–00762) and is also based on individual consent.
Classification of SFN Patients
Patients were further grouped by (1) those with positive symptoms or negative signs, (2) topography of length‐dependent or non–length‐dependent symptoms, (3) the presence or absence of autonomic symptoms, (4) the presence or absence of positive autoimmune blood tests, and (5) the most likely etiology of SFN.
Positive symptoms were defined as pain, burning, electric‐shock–like sensation, hypersensitivity, changes in thermal sensation, itchiness, hyperalgesia, allodynia, and heat sensation. Negative signs were defined as a reduction in any of the sensory modalities of light touch, pain, or temperature. Length‐dependent symptoms were defined as starting in the distal extremities of the legs and the hands symmetrically and ascending upward, whereas non–length‐dependent symptoms were defined as symptoms that were asymmetric, patchy, or involving the face and trunk prior to involvement of the extremities. The SFN diagnosis was based on the NEURODIAB criteria. 21 Accordingly, “possible SFN” was diagnosed when symptoms and/or signs of small fiber damage were present; “probable SFN” was diagnosed if clinical signs of small fiber damage were present, with a normal sural nerve conduction studies (NCS); and “definite SFN” was diagnosed if there were clinical signs of small fiber damage, with normal sural NCS and abnormal quantitative sensory test (QST) thresholds at the foot and/or reduced intraepidermal nerve fiber density (IENFD) at the ankle. Autonomic symptoms were defined as the absence or presence of chronic constipation or diarrhea, postural dizziness, palpitations with tachycardia, urinary symptoms, dry eyes, dry mouth, erectile dysfunction, and hyper‐ or hypohidrosis. The presence of positive autoimmune blood tests was defined as any of the following: antinuclear antibodies (ANA) detectable at ≥1:80 dilution, positivity of anti–extractable nuclear antigen antibodies (anti‐ENA), antineutrophil cytoplasmic antibodies (ANCA), or high erythrocyte sedimentation rate (ESR) after correction for age and gender.
The most likely underlying etiology of SFN was further reviewed by three neurologists independently (A.C.Y.C., Y.F.C., E.W.‐S.), and grouped into iSFN or SFN by secondary causes (secondary SFN), including systemic rheumatological disease, diabetes mellitus, impaired glucose tolerance, nutritional deficiency, hormonal and metabolic disease, alcohol‐ or drug‐related, underlying malignancy, and genetic disorders, if further testing or serial follow‐up returned positive.
Laboratory, Electrodiagnostic, and Histological Tests
Laboratory parameters including serum creatinine, vitamins B1 and B12, fasting lipid and glucose, glycosylated hemoglobin (HbA1C), myeloma screening panel with protein electrophoresis and immunofixation, thyroid function tests, hepatitis B, hepatitis C, human immunodeficiency virus, dry blood spot testing for Fabry disease, and inflammatory markers (ESR, ANA, anti‐ENA, ANCA) were tested and compared. 22 ESR was tested by centrifugation and photometric detection, ANA were tested by immunofluorescence, and both ANCA and anti‐ENA were tested by enzyme immunoassay at the Clinical Chemistry Laboratory of the National University Hospital, Singapore. NCS were performed for all patients. Electrophysiological tests, including needle electromyography, QST, quantitative sudomotor axonal reflex testing, and tilt table testing, were obtained if clinically indicated. Most skin biopsies were taken at 10cm above the lateral malleolus and at the proximal thigh, stained with protein‐gene product 9.5 for the IENFD, and viewed by bright‐field immunohistochemistry according to the immunoperoxidase method protocol. 23 Reduced IENFD was taken as <5th percentile corrected for age and gender. 23 A small number of patients who had skin biopsies taken at the hypothenar eminence were compared to our own laboratory's normal values. 24
Proteomics and Bioinformatics
Peripheral blood samples (3ml) were collected from each subject via venipuncture, centrifuged at 2,800rpm for 15 minutes at 10°C, and stored at −80°C for subsequent analyses. Fifty‐nine SFN patient samples, 20 healthy control (HC) samples, and 5 pooled normal sera were screened for >1,600 proteins on the Sengenics KREX platform in the main analysis. Thirty‐six SFN patient samples, 20 HC samples, and 5 pooled normal sera were screened independently by the same platform in the validation cohort. Preproteomic testing of the validation cohort of 36 SFN and 20 HCs excluded 1 SFN sample that did not pass quality control, and 2 were removed during bioinformatic analysis because they were outliers or failed normalization.
A biotin carboxyl carrier protein (BCCP) folding marker is cloned in‐frame with the gene encoding the protein of interest. BCCP acts as a protein folding marker and a protein solubility enhancer that fuses to either the N‐ or C‐terminal of the protein of interest. Full length proteins are expressed as fusions to the BCCP folding marker, which is biotinylated in vivo only when the protein is correctly folded. Misfolded proteins no longer have a way of attaching to streptavidin‐coated surface (Fig 1). This is followed by image analyses, data extraction and preprocessing, quality control, and composite global normalization.
FIGURE 1.
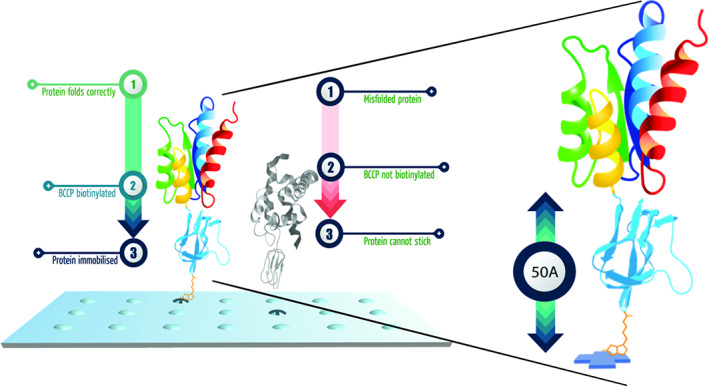
Sengenics Immunome KREX Protein Array platform. The biotin carboxyl carrier protein (BCCP) folding marker acts as a marker for correctly folded proteins. The unique BCCP folding marker conserves the native protein conformation. Proteins are immobilized on the array only when they are properly folded and biotinylated on the BCCP folding marker. [Color figure can be viewed at www.annalsofneurology.org]
The data were first examined by a projection‐based dimension reduction analysis (partial least squares discriminant analysis [PLS‐DA]) 25 and heatmap visualization. Differential protein abundance analysis was carried out with 2‐sample t tests, followed by multiple testing correction by q value wherever it is applicable. 26 Multiple testing correction (q < 0.1) was applied to the validation cohort analysis only, because the total number of significantly differential proteins was small in the main cohort (using p < 0.05), in which case the overall number of type I errors is already low. The minimum fold change (FC) required for statistical significance was set at 50%. Part of the statistical analysis was carried out using R (v4.0.5).
Subgroup Statistical Analysis
Subgroup analysis of protein FCs between iSFN and secondary SFN was also performed with Mann–Whitney U test. A two‐tailed p value of <0.05 was considered statistically significant. Statistical analyses were performed with IBM SPSS Statistics for Windows, version 28.0.0.0 (190) (IBM Corp., Armonk, N.Y., USA).
Results
In the main cohort of the 59 recruited patients, 1 was excluded because the diagnostic criteria for SFN were not met due to abnormal NCS. Final analyses included 58 patients (Fig 2). Most patients were male, with a mean age of 50.0 ± 13.5 years. The most common preceding medical conditions of the patients included a history of autoimmunity (20.7%), myeloradiculopathy (17.2%), and hyperlipidemia (17.2%; Table S1).
FIGURE 2.
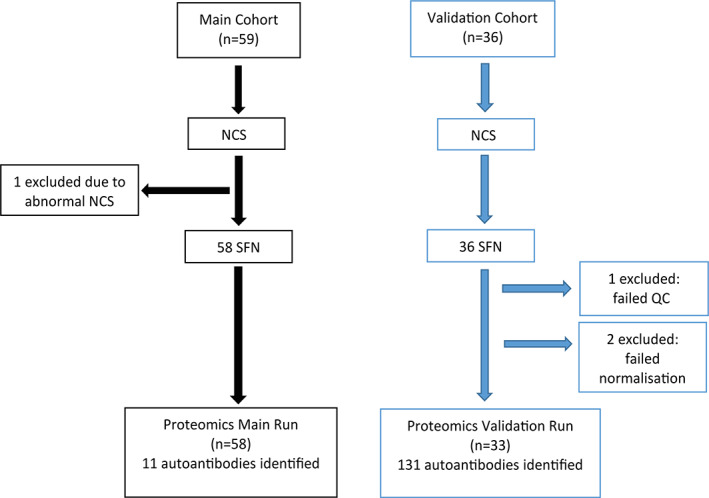
Flowchart of recruitment, proteomics testing, and data analysis of small fiber neuropathy (SFN) patients. Fifty‐nine patients were recruited to the main cohort. One patient was excluded due to failure in meeting diagnostic criteria, and the remaining samples (n = 58) were analyzed and compared with 20 healthy controls who were age‐ and gender‐matched. Thirty‐six patients were recruited to the validation cohort and were run independently and compared with 20 healthy controls. Three samples were excluded due to failure in quality control (QC) or normalization (n = 33). Bioinformatic analysis was performed to identify reproducible autoantigens. NCS = nerve conduction studies. [Color figure can be viewed at www.annalsofneurology.org]
Most patients presented with positive symptoms (51.7%) and in a length‐dependent pattern (51.7%). Autonomic symptoms were prevalent in the majority of patients (55.2%). All except 9 (15.5%) patients underwent skin biopsy, of whom 62.1% were confirmed to have SFN with IENFD lower than the 5th percentile for age and gender. For patients who did not have a positive skin biopsy, the definite diagnosis of SFN was made with abnormal QST in 2 or more limbs in 12% of patients. Definite SFN was thus confirmed in 43 (74.1%) SFN patients based on the NEURODIAB criteria. 21 Positive autoimmunity, as defined by ANA of 1:80 or greater, high ESR for age and gender, and presence of anti‐ENA or ANCA, were found in 29.3% in SFN. iSFN was diagnosed in 34 (58.6%) of patients with further testing and follow‐up, whereas 41.4% were diagnosed with secondary SFN. Among the secondary SFN patients, most were diagnosed with a systemic rheumatological disease (25.9%), followed by diabetes mellitus or impaired glucose tolerance (13.8%; see Table S1). Several patients had missing data for topography, autonomic symptoms, and autoimmunity.
The same recruitment strategy was employed for the validation cohort. The most likely etiology was evaluated by 2 neurologists (A.C.Y.C. and E.W.S.) independently and stratified into idiopathic versus secondary. The same criteria were employed at both hospitals' pathology centers. Comparison between the main cohort and the validation cohort showed no differences in the age, symptom description, topography, autonomic symptoms, prevalence of iSFN, and diagnostic certainty of SFN. However, there were more females in the validation cohort compared to the main cohort (78.8% vs 46.6%, p = 0.016; Table 1).
TABLE 1.
Baseline Demographic Comparisons between SFN Main and Validation Cohorts
| Characteristic | Main Cohort | Validation Cohort | p |
|---|---|---|---|
| Mean (± SD) or n (% of total) | |||
| Total | 58 | 33 | |
| Gender | |||
| M | 31 (53.4%) | 7 (21.2%) | 0.016 a |
| F | 27 (46.6%) | 26 (78.8%) | |
| Age, yr | 50.0 (±13.6) | 46.3 (±12.5) | 0.209 |
| Symptom description and signs | |||
| Positive | 30 (51.7%) | 25 (75.8%) | 0.051 |
| Negative | 13 (22.4%) | 2 (6.1%) | |
| Both positive and negative | 15 (25.9%) | 6 (18.2%) | |
| Topography based on symptoms | |||
| Length dependent | 26 (44.8%) | 21 (85.0%) | 0.116 |
| Non–length dependent | 30 (51.7%) | 12 (15.0%) | |
| Not available | 2 (3.45%) | 0 (0%) | |
| Autonomic symptoms b | |||
| No | 14 (24.1%) | 16 (48.5%) | 0.103 |
| Yes | 32 (55.2%) | 17 (51.5%) | |
| Not available | 12 (20.7%) | ||
| SFN diagnostic category | |||
| Possible | 6 (10.3%) | 1 (3.0%) | 0.065 |
| Probable | 9 (15.5%) | 2 (3.0%) | |
| Definite | 43 (74%) | 31 (93.9%) | |
| Etiology | |||
| Idiopathic | 34 (58.6%) | 18 (54.5%) | 0.826 |
| Secondary causes | 24 (41.4%) | 15 (45.5%) | |
Statistically significant.
Presence or absence of chronic diarrhea, constipation, urinary hesitancy or incontinence in the absence of other urological disorders, hyper‐/hypohidrosis, dry eyes/mouth/skin, chronic postural intolerance or orthostatic blood pressure drop, and chronic palpitations in the absence of cardiological disease.
F= female; M= male; SD= standard deviation; SFN= small fiber neuropathy.
Proteomic data showed 11 autoantibodies that were statistically significant (p < 0.05) when comparing 58 SFN and 20 HC sera samples in the main cohort, whereas the validation cohort identified 131 autoantibodies that were statistically significant (see Fig 2). This is consistent with the finding that, in the supervised PLS‐DA projection analysis, secondary SFN cases showed more pronounced differences from the HCs in the validation cohort (Fig 3). Among the 11 proteins, 9 showed reproducibly significant differences between SFN and HCs, including MX1, DBNL, LIN28A, KRT8, METTL3, NCOA5, PTPN1, ZNF276, and MIF. Another analysis was performed comparing the iSFN group to the HCs in both main and validation cohorts. MX1 and DBNL had the highest log2 FCs concurrently in the main and validation cohorts when comparing both SFN and iSFN to HCs, followed by KRT8 (Fig 4). In the SFN versus HCs analysis, MX1, DBNL, and KRT8 showed higher protein FC compared to HCs, which reached statistical significance (MX1: FC = 2.99 and 3.07, respectively, p = 0.003, q = 0.076; DBNL: FC = 2.11 and 2.16, respectively, p = 0.009, q = 0.003; KRT8: FC = 1.65 and 1.70, respectively, p = 0.043, q = 0.003). In the iSFN versus HCs analysis, MX1 showed the highest FC, but only met statistical significance in the main cohort (FC = 4.82 and 2.77, respectively, p = 0.001, q = 0.231). Scatterplot analysis further confirmed highest FC and correlation with MX1, followed by DBNL and KRT8 in the SFN versus HCs, and in iSFN versus HCs.
FIGURE 3.
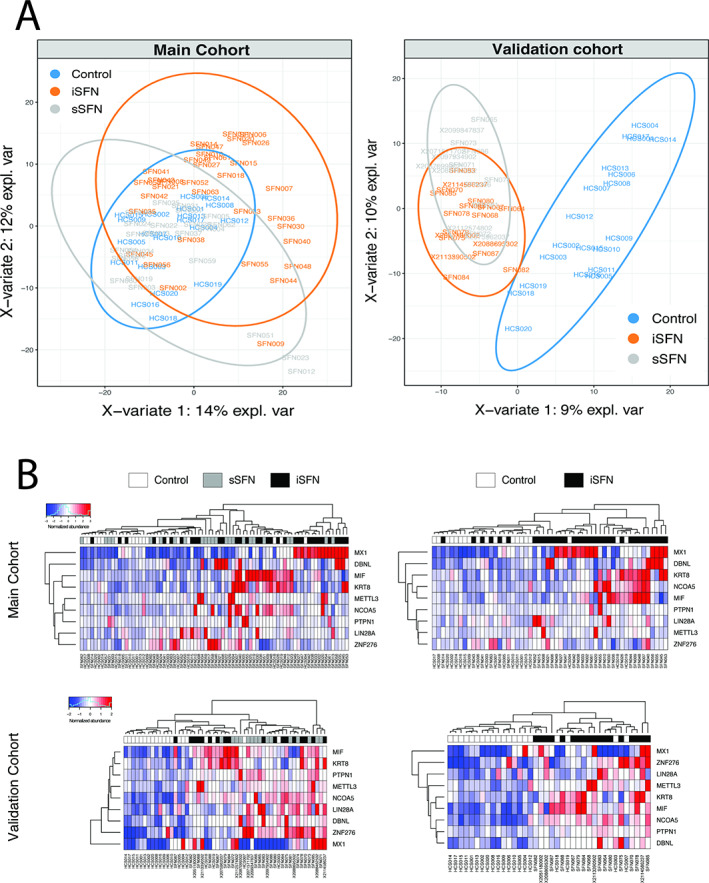
Global differences among the groups and autoantibodies with significantly altered serum levels between idiopathic small fiber neuropathy (iSFN; n = 34 for main cohort, n = 18 for validation cohort), secondary SFN (sSFN; n = 24 for main cohort, n = 15 for validation cohort), and healthy controls (HCs; n = 20). (A) Partial least squares discriminant analysis plots showing separability of subjects based on the overall molecular profiles between the iSFN, sSFN, and HC groups. Positive separation is seen between SFN and HCs in both main and validation cohorts. (B) Heatmap visualization of autoantibodies reproducibly altered between SFN, iSFN, and HCs of the main cohort and the validation cohort. [Color figure can be viewed at www.annalsofneurology.org]
FIGURE 4.
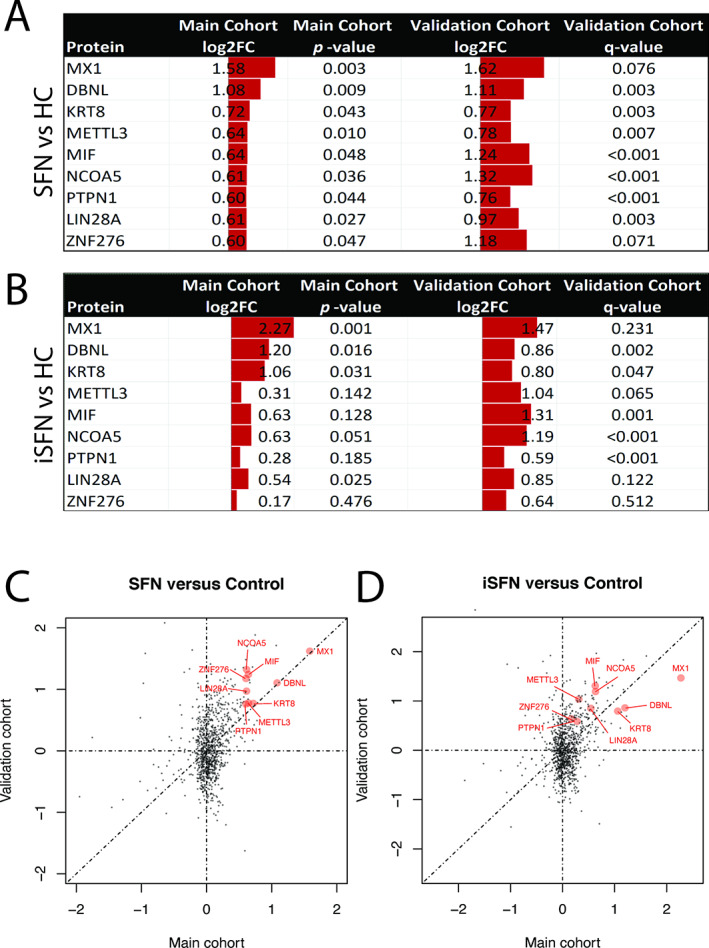
Nine proteins were significantly elevated in small fiber neuropathy (SFN) patients and idiopathic SFN (iSFN) compared to healthy controls (HC). (A, B) Common proteins between the main and validation cohorts with significant log2 fold change (FC) were identified. (C) Scatterplot analysis shows 9 proteins that were reproducibly altered in SFN in both cohorts with statistical significance. MX1, DBNL, and KRT8 showed the largest FCs in both cohorts. (D) In the iSFN analysis, MX1 showed the highest FC, but did not meet statistical significance in the validation cohort. DBNL and KRT8 showed the next highest FCs, which reached significance in both cohorts. p < 0.05 is considered to be significant, whereas q < 0.1 is considered to be significant in the validation cohort. [Color figure can be viewed at www.annalsofneurology.org]
Further subgroup analysis was conducted on the main cohort by comparing the FCs of the 9 reproducible proteins between patients of the iSFN and secondary SFN groups. Of the reproducible proteins in the main cohort, only MX1 had significantly higher FC in iSFN compared to secondary SFN (1.61 vs 0.106, p = 0.009; Table 2), confirming the heatmap visualization results of clustering at MX1. These 9 proteins are involved in metabolism and cellular processes, DNA and RNA functions and repair, antiviral activities, and inflammation. The functions of individual proteins and their disease associations are summarized in Table 3.
TABLE 2.
Protein Fold Changes of Shortlisted Proteins between Idiopathic SFN and Secondary SFN Patients in the Main Cohort
| Proteins | Idiopathic SFN | Secondary SFN | p |
|---|---|---|---|
| Median (5th to 95th percentile) | |||
| MX1 | 1.61 (−0.599 to 10.9) | 0.106 (−1.11 to 11.9) | 0.009 a |
| DBNL | 0.811 (−0.705 to 8.79) | 0.907 (−0.591 to 12.8) | 0.717 |
| KRT8 | 0.735 (−0.646 to 8.15) | 0.073 (−0.608 to 5.28) | 0.236 |
| LIN28A | −1.51 (−2.42 to 1.73) | −1.68 (−2.66 to 7.72) | 0.497 |
| METTL3 | 0.775 (0.226 to 5.10) | 1.18 (−0.029 to 9.07) | 0.072 |
| NCOA5 | 0.920 (−0.181 to 5.42) | 0.693 (−0.195 to 8.75) | 0.625 |
| PTPN1 | −0.260 (−0.780 to 2.81) | −0.158 (−0.764 to 11.9) | 0.195 |
| ZN276 | 2.23 (0.651 to 4.34) | 2.65 (0.930 to 11.4) | 0.160 |
| MIF | 0.300 (−0.904 to 5.98) | 0.533 (−0.254 to 6.22) | 0.449 |
Statistically significant.
SFN = small fiber neuropathy.
TABLE 3.
Functions of the Candidate Antigens and Their Physiological Function and/or Reported Disease Associations
| Associated Protein | Function 27 | Reported Disease Associations |
|---|---|---|
| MX1 (interferon‐induced GTP‐binding protein MX1) |
Interacts with the ankyrinlike repeat domain of the TRPC channels. 28 Antiviral activity against RNA and DNA viruses. 29 , 30 , 31 |
Neuropathic pain in mice 32 Intervertebral disc degeneration and consequent back pain in humans 33 |
| DBNL (drebrin‐like protein) | Involved in receptor‐mediated endocytosis, reorganizing the cytoskeleton to produce cell projections and synapse formations. It is an effector of antigen‐receptor signaling pathways in leukocytes, and it regulates T‐cell activation by bridging T‐cell receptors and the actin cytoskeleton to gene activation and endocytic processes. 34 | Alzheimer disease |
| KRT8 (keratin type II cytoskeletal 8) | Contractile apparatus to dystrophin at the costameres of striated muscle. |
Neuropathic pain Chronic inflammatory demyelinating polyneuropathy |
| LIN28A (protein lin‐28 homolog A) | RNA‐binding protein, inhibits processing of pre–let‐7 miRNAs and regulates translation of mRNAs that control developmental timing, pluripotency, and metabolism. |
Chronic neuropathic pain Primary tumors |
| METTL3 (N6‐adenosine‐methyltransferase catalytic subunit) |
Forms heterodimer with METTL14 that methylates adenosine residues of some RNAs and regulates processes such as circadian clock, differentiation of stem cells, cortical neurogenesis, response to DNA damage, differentiation of T cells, and primary mRNA processing. Involved in response to DNA damage. |
Inflammatory pain Neuropathic pain |
| NCOA5 (nuclear receptor coactivator 5) |
Has both coactivator and corepressor functions. Interactions with nuclear receptors. |
Lipoprotein disorders Psoriasis Behcet syndrome |
| PTPN1 (tyrosine‐protein phosphatase nonreceptor type 1) |
Tyrosine‐protein phosphatase that regulates endoplasmic reticulum unfolded protein response. May play important role in signal transduction cascades. May regulate signaling pathway, which modulates cell reorganization and cell–cell repulsion. |
Type 2 diabetes |
| ZNF276 (zinc finger protein 276) | May be involved in transcriptional regulation. | Nil |
| MIF (macrophage migration inhibitory factor) |
Proinflammatory cytokine. Mediates and regulates the function of macrophages in host defense, and counteracts the anti‐inflammatory activity of glucocorticoids. |
Inflammatory and neuropathic pain Severe sepsis and septic shock |
Extracted from the UniProt Consortium 27 unless otherwise specified.
TRPC = transient receptor potential canonical.
Discussion
Our study identifies novel autoantibodies involved in SFN by a validated high‐throughput protein microarray technology. First, bioinformatic analysis of all SFN patients was performed with main and validation cohorts to identify 9 reproducible autoantibodies. Second, our focused analysis identified 3 proteins with the highest FC and correlation, namely MX1, DBNL, and KRT8. Third, as clustering was seen in the heatmap analysis, a subgroup analysis was performed comparing iSFN and secondary SFN, which identified MX1 as a potential marker that may help to differentiate idiopathic from secondary forms of SFN.
A secondary analysis comparing the iSFN patients with HCs also showed the highest FCs in MX1, DBNL, and KRT8 in both cohorts. Although the p value of the main cohort was statistically significant, the q value of the validation cohort did not meet statistical significance. This may be due to the significantly smaller sample of iSFN in the validation cohort; further studies are required to confirm this.
The advantage of the high‐throughput protein microarray technology compared to conventional immunoassays is the detection and quantification of a large variety of proteins in their original conformation. In this study, autoantibodies against 9 proteins were found to occur at statistically higher levels in SFN patients than in HCs. MX1, LIN28A, KRT8, METTL3, and MIF have been shown to be directly associated with inflammatory neuropathy and neuropathic pain, whereas others are involved in cellular processes of transcription, DNA and RNA functions, and repair. 27
The diagnosis of SFN is increasing with improved awareness of the disease, but the underlying pathophysiology is still unknown in many patients despite thorough investigations. The response of iSFN patients toward immunotherapy is still controversial, and these patients are proposed to have an autoimmune etiology, 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 although the mechanism is not known. Dabby et al 16 described the presence of antisulfatide antibodies in SFN, which was perhaps one of the first studies suggesting that SFN may be an antibody‐mediated process. 16 A retrospective study by Levine et al 18 evaluated the rate of serum antibodies in 155 patients with confirmed SFN and found that 37% of patients had immunoglobulin M (IgM) against trisulfated heparan disaccharide (TS‐HDS), which is a cell‐surface protein, whereas 15% had IgG against fibroblast growth factor receptor 3 (FGFR3), an intracellular protein. TS‐HDS antibodies were statistically more frequent in patients with SFN compared to controls. Tholance et al's findings supported Levine et al's study on the presence of anti‐FGFR3 antibodies in SFN and found that FGFR3‐positive patients suffered significantly more from non–length‐dependent symptoms, suggesting dorsal root ganglia involvement. 17 In 2018, Fujii et al discovered anti–plexin D1 antibodies that cause neuropathic pain, 35 and confirmed their prevalence and pathogenicity in SFN in 2021. 36 Yuki et al 19 described a subpopulation of acute SFN patients' IgG that colocalized to voltage‐gated sodium channels in the small nerve fibers, suggesting a possible role of antibodies against surface antigens expressed on the neuronal cell body and axon but not on myelinated nerve fibers. However, further proteomic approaches failed to identify the target antigens. 19
In our study, scatterplot and heatmap analyses identified clustering of iSFN patients in autoantibodies against MX1, DBNL, and KRT8. MX1 is known as the interferon‐induced GTP‐binding protein MX1. Interferons induce cell‐autonomous defenses in any cell type involved in the adaptive and innate branches of the immune system. Anti‐MX1 antibodies are significant in both main and validation cohorts with the highest FC, as can be seen in Figure 4. Patients with iSFN also show a higher level of anti‐MX1 compared to controls or secondary SFN, suggesting a role of interferons in the pathogenesis of iSFN. MX1 has been found to interact with the ankyrinlike repeat domain of the transient receptor potential canonical (TRPC) channels, specifically TRPC6, and the subtypes TRPC3, TRPC4, and TRPC5 are downregulated after nerve injury. 28 MX1 was found to have a functional effect on TRPC6 activity by potentiating the intracellular influx of calcium through the channels. 28 TRPC channels are mammalian calcium‐permeable transmembrane cation channels that are present in the brain, cerebral arteries, astrocytes, neurons, and pyramidal cells. They serve critical physiological functions such as neuronal differentiation and mechanosensation, and have been associated with various neurological disorders. 37 More importantly, TRPC6 channels are found in mouse dorsal root ganglia 32 and play a pivotal role in neuropathic pain. One recent human study showed enhanced protein and mRNA expression of TRPC6 in patients with intervertebral disc degeneration and consequent back pain, 33 inferring that the high level of anti‐MX1 in iSFN patients may cause downstream effects of neuropathic pain via its interaction with TRPC6, although further research is required to elucidate this. Whether this protein is pathogenic or merely an epiphenomenon needs to be further studied.
DBNL (drebrin‐like protein), one of the 9 proteins that were found to be reproducible, had the second highest FC in both main and validation cohorts and may prove to be important in the pathophysiology of SFN, although it failed to show significance in differentiating between iSFN and secondary SFN. DBNL is an adapter protein that plays a role in receptor‐mediated endocytosis, and reorganizing the cytoskeleton to produce cell projections and synapse formations, 34 suggesting a disruption in the core cellular processes of the neurons.
KRT8 (keratin type II cytoskeletal 8), which ranks third in the list of 9 reproducible proteins, is a contractile apparatus to dystrophin that occurs in the costameres of striated muscle. The KRT8 gene has been found to be downregulated in chronic inflammatory demyelinating neuropathy (CIDP) patients compared to control and Charcot–Marie–Tooth disease groups, and is presumed to be involved in the development of the disease. 38 CIDP is an inflammatory neuropathy for which many antibodies have diagnostic and therapeutic implications. 39 The identification of KRT8 in iSFN and CIDP may suggest a similar pathophysiological mechanism.
During subgroup analysis comparing iSFN and secondary SFN, the only protein showing significantly higher FCs in the iSFN group was anti‐MX1. Clustering of anti‐MX1 was seen mainly in the iSFN patients in the heatmaps, further supporting anti‐MX1 as a potentially important biomarker of iSFN. What role anti‐MX1 plays in SFN requires further validation studies.
The strength of this study is its novelty, as few proteomic analyses on SFN patients have been performed to date. Certainly, no prior proteomic studies have been performed employing the native conformational structure of proteins and at such breadth. Our study identified a number of novel autoantibodies, shedding light onto their possible roles in SFN. Moreover, most iSFN patients are biopsy‐confirmed, and patients are categorized according to their level of diagnostic certainty based on the NEURODIAB criteria, 21 adding to the diagnostic certainty of this heterogeneous disease.
Although one of the limitations include the small sample size of SFN patients and the retrospective analysis of patients' clinical parameters, an independent validation cohort from 2 tertiary neurology centers was performed independently to ensure reproducibility of the results. Bioinformatic analysis with strict normalization and partial least squares discriminant analysis ensured separability between the SFN and control subjects.
A further limitation of the study includes variability of the investigations performed and clinical parameters for each patient, which may lead to missing data. Although a standardized approach to SFN patients was utilized, 22 parts of the clinical evaluation may be customized by the managing neurologist depending on the patient's presentation, leading to the inevitable possibility of missing data. Missing data would be excluded in the subgroup analysis, which would lead to a smaller sample size, hence a higher rate of type II error. 40 Notably, the prevalence and proportion of iSFN may not be fully reflective of the general SFN population, as attempts were made to filter iSFN patients since the index visit. Moreover, during the subgrouping of patients for the likely etiology of SFN, patients may have had more than one etiology, but the decision was made to categorize them into one predominant etiology. As a result, the effect of the secondary etiologies may be minimized or eliminated during analysis. However, few patients had more than one possible etiology.
The diagnostic criteria of SFN are constantly evolving, 41 , 42 , 43 which may affect the results of the study, but the most widely accepted NEURODIAB 21 criteria have been incorporated for the purposes of this study. The most recently proposed criteria by Devigili et al suggest that a combination of clinical, functional, and structural approaches to the diagnosis of SFN may be most reliable. 43 Although the new criteria are stricter and may reduce the rate of false positive iSFN, there are still limitations to skin biopsy and QST, and narrowing the diagnosis to this extent in this study may contrarily result in false negative results. A fine balance needs to be met, and although we have chosen to use the NEURODIAB criteria, most of the iSFN patients in our cohort had met all 3—clinical, functional, and structural approaches—proposed by the new criteria. Due to constantly evolving diagnostic criteria, immunological and biochemical tests may be of utmost value in the diagnostic armamentarium of iSFN, and should perhaps be added to the diagnostic criteria.
In conclusion, this pilot study reveals novel autoantibodies in iSFN, supporting the hypothesis that an immune‐mediated process may be involved. This paves the way for validation studies and in‐depth analysis of these autoantibodies. Further elucidation of the pathophysiology of SFN enables clinical subtyping, and the development of targeted treatments for iSFN patients who are currently treated merely symptomatically and inadequately.
Author Contributions
A.C.Y.C., H.Y.W., A.M., E.W.‐S., H.C., and V.K.S. contributed to the conception and design of the study. All authors contributed to the acquisition and analysis of data. A.C.Y.C., H.Y.W., and H.C. contributed to drafting the text or preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
TABLE S1. Supporting information.
Acknowledgments
This research is supported by the Singapore Ministry of Health's National Medical Research Council under its Clinician–Scientist Individual Research Grant New Investigator Grant (CNIG18nov‐0009).
We thank all the study participants, Drs K.B. Tan and J. Wong for histological analysis of the skin biopsy samples, and L. Wong for assisting with the administration involved in the study.
[Correction added on December 3, 2021, after first online publication: The affiliation for Einar Wilder‐Smith has been updated.]
References
- 1. Bitzi LM, Lehnick D, Wilder‐Smith EP. Small fibre neuropathy: Swiss cohort characterization. Muscle Nerve 2021;64:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Basantsova NY, Starshinova AA, Dori A, et al. Small‐fiber neuropathy definition, diagnosis, and treatment. Neurol Sci 2019;40:1343–1350. [DOI] [PubMed] [Google Scholar]
- 3. Liu X, Treister R, Lang M, Oaklander AL. IVIg for apparently autoimmune small‐fiber polyneuropathy: first analysis of efficacy and safety. Ther Adv Neurol Disord 2018;11:1756285617744484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Greef BT, Geerts M, Hoeijmakers JG, et al. Intravenous immunoglobulin therapy for small fiber neuropathy: study protocol for a randomized controlled trial. Trials 2016;17:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sala TP, Villedieu M, Damian L, et al. Long‐term efficacy of immunoglobulins in small fiber neuropathy related to Sjögren's syndrome. J Neurol 2020;267:3499–3507. [DOI] [PubMed] [Google Scholar]
- 6. Wakasugi D, Kato T, Gono T, et al. Extreme efficacy of intravenous immunoglobulin therapy for severe burning pain in a patient with small fiber neuropathy associated with primary Sjögren's syndrome. Mod Rheumatol 2009;19:437–440. [DOI] [PubMed] [Google Scholar]
- 7. Morozumi S, Kawagashira Y, Iijima M, et al. Intravenous immunoglobulin treatment for painful sensory neuropathy associated with Sjögren's syndrome. J Neurol Sci 2009;279:57–61. [DOI] [PubMed] [Google Scholar]
- 8. Kizawa M, Mori K, Iijima M, et al. Intravenous immunoglobulin treatment in painful sensory neuropathy without sensory ataxia associated with Sjögren's syndrome. J Neurol Neurosurg Psychiatry 2006;77:967–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Parambil JG, Tavee JO, Zhou L, et al. Efficacy of intravenous immunoglobulin for small fiber neuropathy associated with sarcoidosis. Respir Med 2011;105:101–105. [DOI] [PubMed] [Google Scholar]
- 10. Tavee JO, Karwa K, Ahmed Z, et al. Sarcoidosis‐associated small fiber neuropathy in a large cohort: clinical aspects and response to IVIG and anti‐TNF alpha treatment. Respir Med 2017;126:135–138. [DOI] [PubMed] [Google Scholar]
- 11. Geerts M, de Greef BT, Sopacua M, et al. Intravenous immunoglobulin therapy in patients with painful idiopathic small fiber neuropathy. Neurology 2021;96:e2534–e2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kamm C, Zettl UK. Autoimmune disorders affecting both the central and peripheral nervous system. Autoimmun Rev 2012;11:196–202. [DOI] [PubMed] [Google Scholar]
- 13. Patterson SD, Aebersold RH. Proteomics: the first decade and beyond. Nat Genet 2003;33:311–323. [DOI] [PubMed] [Google Scholar]
- 14. Aslam B, Basit M, Nisar MA, et al. Proteomics: technologies and their applications. J Chromatogr Sci 2017;55:182–196. [DOI] [PubMed] [Google Scholar]
- 15. Mak A, Kow NY, Ismail NH, et al. Detection of putative autoantibodies in systemic lupus erythematous using a novel native‐conformation protein microarray platform. Lupus 2020;29:1948–1954. [DOI] [PubMed] [Google Scholar]
- 16. Dabby R, Weimer LH, Hays AP, et al. Antisulfatide antibodies in neuropathy: clinical and electrophysiologic correlates. Neurology 2000;54:1448–1452. [DOI] [PubMed] [Google Scholar]
- 17. Tholance Y, Moritz CP, Rosier C, et al. Clinical characterisation of sensory neuropathy with anti‐FGFR3 autoantibodies. J Neurol Neurosurg Psychiatry 2020;91:49–57. [DOI] [PubMed] [Google Scholar]
- 18. Levine TD, Kafaie J, Zeidman LA, et al. Cryptogenic small‐fiber neuropathies: serum autoantibody binding to trisulfated heparan disaccharide and fibroblast growth factor receptor‐3. Muscle Nerve 2020;61:512–515. [DOI] [PubMed] [Google Scholar]
- 19. Yuki N, Chan AC, Wong AHY, et al. Acute painful autoimmune neuropathy: a variant of Guillain‐Barré syndrome. Muscle Nerve 2018;57:320–324. [DOI] [PubMed] [Google Scholar]
- 20. Duarte JG, Blackburn JM. Advances in the development of human protein microarrays. Expert Rev Proteomics 2017;14:627–641. [DOI] [PubMed] [Google Scholar]
- 21. Tesfaye S, Boulton AJ, Dyck PJ, et al. Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care 2010;33:2285–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chan AC, Wilder‐Smith E. Clinical reasoning: burning hands and feet. Neurology 2015;84:e146–e152. [DOI] [PubMed] [Google Scholar]
- 23. Lauria G, Bakkers M, Schmitz C, et al. Intraepidermal nerve fiber density at the distal leg: a worldwide normative reference study. J Peripher Nerv Syst 2010;15:202–207. [DOI] [PubMed] [Google Scholar]
- 24. Wilder‐Smith EP, Chow A. Comparison of a simple method for quantitation of intraepidermal nerve fibres with a standard image analysis method using hypothenar skin. J Neurol 2006;253:1011–1015. [DOI] [PubMed] [Google Scholar]
- 25. Barker M, Rayens W. Partial least squares for discrimination. J Chemomet 2003;17:166–173. [Google Scholar]
- 26. Storey JD. The positive false discovery rate: a Bayesian interpretation and the q‐value. Ann Stat 2003;31:2013–2035. [Google Scholar]
- 27. Consortium TU . UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 2020;49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lussier MP, Cayouette S, Lepage PK, et al. MxA, a member of the dynamin superfamily, interacts with the ankyrin‐like repeat domain of TRPC. J Biol Chem 2005;280:19393–19400. [DOI] [PubMed] [Google Scholar]
- 29. Kochs G, Janzen C, Hohenberg H, Haller O. Antivirally active MxA protein sequesters La Crosse virus nucleocapsid protein into perinuclear complexes. Proc Natl Acad Sci U S A 2002;99:3153–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Andersson I, Bladh L, Mousavi‐Jazi M, et al. Human MxA protein inhibits the replication of Crimean‐Congo hemorrhagic fever virus. J Virol 2004;78:4323–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Turan K, Mibayashi M, Sugiyama K, et al. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res 2004;32:643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Elg S, Marmigere F, Mattsson JP, Ernfors P. Cellular subtype distribution and developmental regulation of TRPC channel members in the mouse dorsal root ganglion. J Comp Neurol 2007;503:35–46. [DOI] [PubMed] [Google Scholar]
- 33. Sadowska A, Hitzl W, Karol A, et al. Differential regulation of TRP channel gene and protein expression by intervertebral disc degeneration and back pain. Sci Rep 2019;9:18889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Le Bras S, Foucault I, Foussat A, et al. Recruitment of the actin‐binding protein HIP‐55 to the immunological synapse regulates T cell receptor signaling and endocytosis. J Biol Chem 2004;279:15550–15560. [DOI] [PubMed] [Google Scholar]
- 35. Fujii T, Yamasaki R, Iinuma K, et al. A novel autoantibody against plexin D1 in patients with neuropathic pain. Ann Neurol 2018;84:208–224. [DOI] [PubMed] [Google Scholar]
- 36. Fujii T, Lee E‐J, Miyachi Y, et al. Antiplexin D1 antibodies relate to small fiber neuropathy and induce neuropathic pain in animals. Neurol Neuroimmunol Neuroinflam 2021;8:e1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang R, Tu S, Zhang J, Shao A. Roles of TRP channels in neurological diseases. Oxid Med Cell Longev 2020;2020:7289194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee G, Xiang Z, Brannagan TH III, et al. Differential gene expression in chronic inflammatory demyelinating polyneuropathy (CIDP) skin biopsies. J Neurol Sci 2010;290:115–122. [DOI] [PubMed] [Google Scholar]
- 39. Querol L, Devaux J, Rojas‐Garcia R, Illa I. Autoantibodies in chronic inflammatory neuropathies: diagnostic and therapeutic implications. Nat Rev Neurol 2017;13:533–547. [DOI] [PubMed] [Google Scholar]
- 40. Columb M, Atkinson M. Statistical analysis: sample size and power estimations. BJA Educat 2015;16:159–161. [Google Scholar]
- 41. Blackmore D, Siddiqi ZA. Diagnostic criteria for small fiber neuropathy. J Clin Neuromuscul Dis 2017;18:125–131. [DOI] [PubMed] [Google Scholar]
- 42. Thaisetthawatkul P, Fernandes Filho JAM, Herrmann DN. Contribution of QSART to the diagnosis of small fiber neuropathy. Muscle Nerve 2013;48:883–888. [DOI] [PubMed] [Google Scholar]
- 43. Devigili G, Rinaldo S, Lombardi R, et al. Diagnostic criteria for small fibre neuropathy in clinical practice and research. Brain 2019;142:3728–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1. Supporting information.