

Summary
Little is known regarding the shared genetic architecture or causality underlying the phenotypic association observed for uterine leiomyoma (UL) and breast cancer (BC). Leveraging summary statistics from the hitherto largest genome-wide association study (GWAS) conducted in each trait, we investigated the genetic overlap and causal associations of UL with BC overall, as well as with its subtypes defined by the status of estrogen receptor (ER). We observed a positive genetic correlation between UL and BC overall (rg = 0.09, p = 6.00 × 10−3), which was consistent in ER+ subtype (rg = 0.06, p = 0.01) but not in ER− subtype (rg = 0.06, p = 0.08). Partitioning the whole genome into 1,703 independent regions, local genetic correlation was identified at 22q13.1 for UL with BC overall and with ER+ subtype. Significant genetic correlation was further discovered in 9 out of 14 functional categories, with the highest estimates observed in coding, H3K9ac, and repressed regions. Cross-trait meta-analysis identified 9 novel loci shared between UL and BC. Mendelian randomization demonstrated a significantly increased risk of BC overall (OR = 1.09, 95% CI = 1.01–1.18) and ER+ subtype (OR = 1.09, 95% CI = 1.01–1.17) for genetic liability to UL. No reverse causality was found. Our comprehensive genome-wide cross-trait analysis demonstrates a shared genetic basis, pleiotropic loci, as well as a putative causal relationship between UL and BC, highlighting an intrinsic link underlying these two complex female diseases.
Keywords: breast cancer, uterine leiomyoma, genetic correlation, pleiotropic loci, causal inference
Graphical abstract
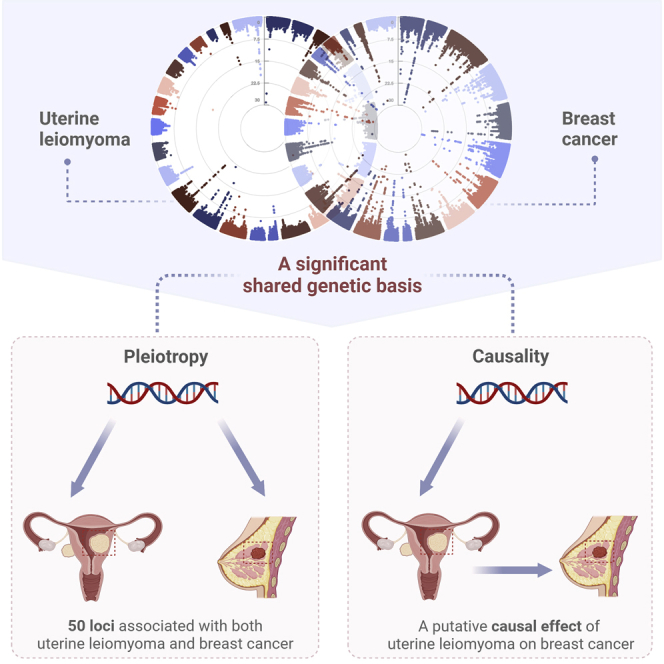
Wu et al. demonstrate a significant shared genetic basis, multiple pleiotropic loci, as well as a putative causal relationship between uterine leiomyoma and breast cancer, highlighting a biological link underlying these two complex female diseases. These findings provide important implications for future therapeutic strategy and risk prediction.
Introduction
Uterine leiomyoma (UL), also known as fibroids, are benign tumors affecting 5.4%–77.0% of women of reproductive age.1 Although the majority of UL are asymptomatic, nearly 25% of women with UL may experience heavy menstrual bleeding, abdominal pain, infertility, and increased risk of miscarriage.2 Breast cancer (BC), on the other hand, affects 1 in 10 women throughout their lifetimes and ranks first in both incidence and mortality of female cancers.3 A shared etiology underlying these two complex diseases has long been recognized. Several common risk factors including obesity, oral contraceptive use, hormone replacement therapy, and reproductive factors may lead to increased levels of estrogen and progesterone.4,5 Furthermore, epidemiological studies have evaluated and largely demonstrated a UL-BC phenotypic link. Leveraging data from 2,411 BC-affected individuals and the entire registered female population (aged 20 years or above, N = 162,449) of Gothenburg, Sweden, Lindegård et al. found a significantly increased risk of UL associated with non-fatal BC (observed/expected BC cases = 1.7, p < 0.01).6 Later on, a prospective cohort study of 57,747 African American women reported a non-significant but positive association for early-diagnosed UL with BC before age 40 (IRR = 1.39, 95% CI = 0.97–1.99).7 With a two-times augmented sample size and a median follow-up time of 6.5 years, Shen et al. found a 31% increased risk of BC in women diagnosed with UL compared to UL-free referents of East Asian ancestry (HR = 1.31, 95% CI = 1.13–1.52).8 Nevertheless, owing to the observational nature of conventional epidemiological studies, phenotypic correlations derived from these studies are likely subject to bias, confounding, and reverse causality.9
Linking traits through genetics overcomes at least one major challenge of observational studies—reverse causality—and with careful design can also address confounding. Indeed, current progress from genome-wide association studies (GWASs) have elucidated a considerable number of disease-associated variants (single nucleotide polymorphisms, SNPs) for both UL (n = 29) and BC (n > 150).10,11 The SNP heritability of UL and BC has further been quantified as 3%–13%10,12 and 13%–18%,11,13 respectively, indicating a non-trivial genetic component in disease susceptibility. Multiple loci (i.e., GREB1, NEK10, TERT, ESR1, TP53, and MCM8) influencing both traits have also been identified,10,12 suggesting the observed epidemiological association is at least in part attributable to a shared genetic architecture.
The accumulating amount of genome-wide genetic data enable the utilization of a recently developed statistical genetics tool, named genome-wide cross-trait analysis. This design offers an unprecedented opportunity to characterize comprehensively the shared genetic architecture and causal link across traits, driving forward epidemiologic associations with novel insights into the underlying biological mechanisms.9 Such analysis has several analytic aspects: a genetic correlation analysis to estimate global and local genetic correlation, a cross-trait meta-analysis to identify shared loci, and Mendelian randomization (MR) to make causal inference. Nevertheless, to the best of our knowledge, no genome-wide cross-trait analysis has been conducted to explore systematically the shared and distinct etiology underpinning UL and BC.
Therefore, in this study, we performed a comprehensive genome-wide cross-trait analysis to investigate the genetic overlap as well as the causal relationship underlying UL and BC. First, we quantified genetic correlation to understand shared genetic basis. Next, we applied cross-trait meta-analysis to identify pleiotropic loci, of which biological function was further annotated, leveraging information from high-quality functional resources. Finally, we performed a bidirectional two-sample MR analysis to infer putative causal relationships. The overall study design is shown in Figure 1.
Figure 1.

Overall study design of genome-wide cross-trait analysis
GWAS summary statistics for each trait of interest were retrieved from publicly available GWASs. A global genetic correlation analysis between uterine leiomyoma and breast cancer was conducted. The estimated global genetic correlation was further dissected at LD-defined regions and by functional categories. Cross-trait meta-analysis was applied to identify pleiotropic loci, and a bidirectional two-sample Mendelian randomization analysis was used to infer putative causal relationship. UL, uterine leiomyoma; BC, breast cancer; ER, estrogen receptor; GTEx, Genotype-Tissue Expression project; GWAS, genome-wide association study.
Material and methods
Data summary
This is a secondary analysis of existing GWASs. Summary statistics were retrieved from publicly available GWASs conducted for UL and BC. Details on the characteristics of each included dataset are presented in Table S1.
Uterine leiomyoma
The latest GWAS of UL was performed by Gallagher et al. in 2019,10 meta-analyzing data from five participating cohorts of the FibroGENE consortium (Women’s Genome Health Study [WGHS], Northern Finnish Birth Cohort [NFBC], QIMR Berghofer Medical Research Institute [QIMR], UK Biobank [UKB], and 23andMe). This GWAS combined 8.7 million variants in 35,474 UL-affected women and 267,505 female control subjects (all of European ancestry). UL was determined based on either self-report or clinical documentation. SNPs were imputed to the 1000 Genomes Project (1KGP) Phase 3 reference panel or the Haplo-type Reference Consortium (HRC) reference panel. A fixed-effect inverse-variance-weighted meta-analysis was conducted across all cohorts. Top-associated SNPs in the combined meta-analysis reaching a p threshold of 5 × 10−8 were reported.
We extracted relevant information of the 29 GWAS-identified UL-associated significant SNPs and used those SNPs as instrumental variables (IVs). Details on the characteristics of UL-associated IVs are shown in Table S2. We also retrieved full set summary statistics of UL.
Breast cancer
For BC overall, the most recent also the largest GWAS was performed by Zhang et al. in 2020,11 meta-analyzing data from 82 participating studies of the Breast Cancer Association Consortium (BCAC) and 11 other BC genetic studies. This GWAS combined 10.8 million variants in 133,384 BC-affected women and 113,789 female control subjects (all of European ancestry). SNPs were imputed to the 1KGP Phase 3 reference panel. A fixed-effect inverse-variance-weighted meta-analysis was conducted across all studies. Top-associated SNPs in the combined meta-analysis reaching a p threshold of 5 × 10−8 were reported. This GWAS confirmed 153 previous-reported BC SNPs and additionally identified 32 novel BC SNPs, for a total of 185 SNPs.
For subtype-specific BC, we retrieved summary statistics from the largest published GWAS on estrogen receptor (ER)+ and ER– BC performed by Michailidou et al. in 2017.13 This GWAS meta-analyzed data from BCAC and DRIVE (Discovery, Biology and Risk of Inherited Variants in Breast Cancer Consortium), combining 11.8 million variants in 122,977 BC-affected women (of which 69,501 were ER+ cases and 21,468 were ER–) and 105,974 female control subjects (all of European ancestry). Data imputed to the 1KGP reference panel were analyzed using a fixed-effects inverse-variance-weighted meta-analysis.
From both GWASs, we extracted effect sizes and relevant information for the 29 UL-associated IVs. We also extracted relevant information of the 185 GWAS-identified BC-associated significant SNPs (characteristics of which are shown in Table S3) and retrieved full set summary statistics of BC.
Statistical analysis
Global genetic correlation analysis
To evaluate a shared genetic basis between UL and BC, we performed a global genetic correlation analysis using linkage disequilibrium (LD) score regression (LDSC).14 LDSC estimates genetic correlation (rg) (ranging from −1 to 1) using only summary statistics, relying on the fact that GWAS effect size estimate for a given variant includes the effects of all variants in LD with that variant, which can be extended to the analysis of genetic correlation between traits if the χ2 statistics are replaced with the product of two z-scores from traits of interest. More precisely, by implementing the algorithms described below, LDSC uses the slope from the regression of z-scores on LD-score to estimate rg:
where z1j and z2j are the z-scores of SNP j from trait 1 and trait 2, N1 and N2 are the sample sizes for trait 1 and trait 2, is the genetic covariance, lj is the LD-score, M is number of SNPs, Ns is the number of overlapping samples, is the phenotypic correlation in overlapping samples, and h12 and h22 are the SNP heritability of trait 1 and trait 2.
LDSC analysis was performed using the known LD structure of European ancestry reference data from 1KGP and was restricted to only HapMap3 SNPs, recognized as well imputed in most studies to minimize bias due to low imputation quality. We conducted LDSC with and without a constrained intercept, as constraining the single-trait heritability intercept increases the accuracy of estimation when sample overlap and population stratification are minimal.14 A false discovery rate (FDR) corrected p value (Benjamin-Hochberg procedure) of 0.05 was used as significant threshold (FDR p < 0.05).
Local genetic correlation analysis
Global genetic correlations estimated by LDSC are based on aggregated information across all variants in the genome. It is possible that even though two traits are of negligible global genetic correlation, there are specific regions in the genome contributing to both traits. Therefore, we further calculated pairwise local genetic correlations for UL and BC using ρ-HESS.15 ρ-HESS provides a precise quantification of the similarity between pairs of traits driven by genetic variations at each specific region in the genome, using approximately 1,703 independent LD blocks with an average length of 1.6 Mb. Bonferroni correction was applied to adjust for multiple testing (two-tailed p < 0.05/1,703).15
Partitioned genetic correlation analysis
To investigate the contribution of genomic functional elements, we further partitioned the global UL-BC genetic correlation using stratified-LDSC.16 Fourteen functional categories were used: coding region, conserved region, DNase digital genomic foot-printing (DGF) region, DNase I hypersensitive sites (DHSs), fetal DHS, intronic region, promotor, repressed region, super-enhancer, transcribed region, and histone marks H3K27ac, H3K4me1, H3K4me3, and H3K9ac. Stratified-LDSC partitions SNPs into different functional categories and calculates LD-score for each given SNP to that category, which were used to estimate genetic correlation within that specific functional category.
Cross-trait meta-analysis
Shared genetic components suggest either genetic variants having independent effect on both traits (pleiotropy) or genetic variants influencing one trait via its effect on the other (causality). We next performed a cross-trait meta-analysis using Cross-Phenotype Association (CPASSOC)17 to identify pleiotropic loci. Using summary statistics of single SNP-trait associations from GWAS, CPASSOC provides two estimates, SHom and SHet. SHom is based on a fixed-effect meta-analysis approach and can be viewed as the maximum of weighted sum of trait-specific genetic effects. It is less powerful under the presence of between-study heterogeneity, which is common when meta-analyzing multiple traits. As an extension of SHom, SHet maintains statistical power even under the presence of heterogeneity by assigning more weights to the larger trait-specific effect sizes. This method (SHet) was thus adopted for the analysis herein.
Independent loci were obtained using the PLINK18 “clumping” function through applying the following parameters: --clump-p1 5e-8 --clump-p2 1e-5 --clump-r2 0.2 --clump-kb 500. Within each locus, the variant with the lowest p value was kept as the index SNP. Significant pleiotropic SNPs were defined as index variants satisfying pCPASSOC < 5 × 10−8 and psingle-trait < 1 × 10−3 (for both traits). SNPs that were not identified as significant by the original single-trait GWASs, meaning they themselves were independent (r2 < 0.20) of those previously reported genome-wide significant SNPs (of BC and UL), and none of their neighboring SNPs within 1.0 Mb region reached p < 5 × 10−8 in the single-trait GWAS (of BC and UL), were considered as novel pleiotropic SNPs and were prioritized in this study.
We used Ensemble Variant Effect Predictor (VEP)19 and 3DSNP20 for detailed functional annotation of the identified pleiotropic SNPs.
Fine mapping credible set analysis
The index SNP does not necessarily indicate the causal SNP.21 We further identified a credible set of variants that were 99% likely, based on posterior probability, to contain causal variants at each of the pleiotropic loci using FM-summary.22 Briefly, FM-summary is a Bayesian fine-mapping algorithm that maps only the primary signal and uses a flat prior with steepest descent approximation, assuming at least one causal variant exists within a given region.
Colocalization analysis
Another approach to understanding the shared genetic basis cross traits is to investigate whether the same variants are responsible for two GWAS signals or whether it is distinct genetic variants close to each other. We next performed a colocalization analysis through Coloc.23 Coloc uses a Bayesian algorithm to generate posterior probabilities for five mutually exclusive hypotheses regarding the sharing of causal variants in a genomic region, namely H0 (no association), H1 or H2 (association to one trait only), H3 (association to both traits, two distinct SNPs), and H4 (association to both traits, one shared SNP). We extracted summary statistics for variants within 1.0 Mb of the index SNP at each shared locus and calculated the posterior probability for H4 (PPH4) and H3 (PPH3). A locus was considered colocalized if PPH4 or PPH3 was greater than 0.5.
Pathway and GTEx tissue enrichment analysis
To gain biological insights for the novel pleiotropic SNPs identified from CPASSOC, we performed post-GWAS functional annotation by leveraging multiple resources. We applied the WebGestalt tool24 to assess the enrichment of novel shared genes in Gene Ontology (GO) biological processes and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. To identify tissues most relevant to the shared genes, we performed GTEx tissue enrichment analysis using functional mapping and annotation of genome-wide association studies (FUMA)25 GENE2FUNC process with 54 tissue types available from GTEx (v.8). The Benjamin-Hochberg procedure was used to correct for multiple testing.
Cell-type-specific enrichment of SNP heritability
To understand further the (dis)similarity across traits, we partitioned heritability using stratified-LDSC leveraging genome-wide genetic variants of UL and BC. We used 396 annotations constructed by the Roadmap project for six chromatin marks (DHS, H3K27ac, H3K36me3, H3K4me1, H3K4me3, and H3K9ac) in a set of 88 primary cell types or tissues. Each cell-type-specific annotation corresponded to a chromatin mark in a single cell type, and there were 396 such annotations in total. We further divided these 396 cell-type-specific annotations into 9 broad groups, namely adipose, central nervous system, digestive system, cardiovascular, musculoskeletal and connective tissue, immune and blood, liver, pancreas, and other.26 Annotation-specific enrichment values for each trait were calculated using LDSC and were transformed into color scale and visualized by hierarchical clustering. FDR-adjusted p value was applied based on the specific numbers of comparisons made in each analysis.
Mendelian randomization analysis
We finally performed a bidirectional two-sample MR analysis to detect a putative causal relationship. The inverse-variance weighted (IVW) approach was applied as our primary approach.27 This method pools the Wald ratio estimate of each SNP, obtained from dividing the SNP-outcome estimate by the SNP-exposure estimate, using a random-effects inverse-variance method that weights each ratio based on its standard error, and obtains the average casual effect estimates between two traits. Complementary to IVW, we also adopted MR-Egger regression and weighted median approach. MR-Egger regression can be used to detect and correct for bias due to directional pleiotropy.28 Weighted median approach provides a consistent estimate of the causal effect even when up to 50% of genetic variants are invalid.29 These two approaches were less powerful than IVW in detecting true causal effects and were therefore used to complement with findings from IVW. A causal estimate was considered significant if it was significant in IVW and showed directional consistency in MR-Egger regression and weighted median approach.
To validate MR model assumptions, that the IVs (1) are strongly associated with the exposure, (2) share no common cause with the outcome, and (3) affect the outcome only through the exposure,30 we conducted several important sensitivity analyses. First, we excluded palindromic IVs, SNPs whose alleles are represented by the same pair of nucleotides on the forward and the reverse strands, introducing ambiguity into the identification of effect allele. Second, we excluded pleiotropic SNPs which were associated with potential confounding phenotypes according to the GWAS catalog. Third, we performed leave-one-out analysis where one SNP was removed at a time and IVW was conducted based on the remaining SNPs. Finally, MR-Pleiotropy Residual Sum and Outlier (MR-PRESSO) method31 was applied to detect and correct for horizontal pleiotropy. To adjust for potential confounding effect acting in particular through body mass index (BMI), a risk factor known to influence both UL and BC,4,5 we also employed a multi-variable MR.32 To evaluate whether genetic liability to BC exerts a causal effect on UL, we conducted a reverse-direction MR where BC-associated independent SNPs were used as IVs.
Results
Global genetic correlation
Using unconstrained LDSC, a limited genetic correlation was found for UL with BC overall at marginal significance (rg = 0.07, p = 0.05). The estimate attenuated to null in BC subtypes (ER+: rg = 0.04, p = 0.35; ER−: rg = 0.05, p = 0.28). Considering the negligible sample overlap shared by UL and BC, we further constrained the intercepts of genetic covariance estimates to zero, through which LDSC could show greater power with slightly reduced standard errors. As a result, a significant positive genetic correlation was found for UL with BC overall (rg = 0.09, p = 6.00 × 10−3), which, when extended to BC subtypes, remained significant for ER+ subtype (rg = 0.06, p = 0.01, withstood multiple correction) but not for ER− subtype (rg = 0.06, p = 0.08) (Table 1).
Table 1.
Genome-wide genetic correlations between uterine leiomyoma and breast cancer using constrained and unconstrained LDSC
| Trait 1 | Trait 2 | Constrained LDSC |
Unconstrained LDSC |
||||
|---|---|---|---|---|---|---|---|
| rg | rg_SE | p | rg | rg_SE | p | ||
| Uterine leiomyoma | Breast cancer overall | 0.09 | 0.03 | 6.00 × 10−3 | 0.07 | 0.04 | 5.01 × 10−2 |
| Uterine leiomyoma | ER+ breast cancer | 0.06 | 0.03 | 1.00 × 10−2 | 0.04 | 0.04 | 0.35 |
| Uterine leiomyoma | ER– breast cancer | 0.06 | 0.04 | 0.08 | 0.05 | 0.05 | 0.28 |
rg, genetic correlation; SE, standard error; ER, estrogen receptor.
Local genetic correlation
Given the significant global genetic correlation, we further explored whether specific genomic regions conferred local genetic correlation (Figure 2, Tables S4–S6). After multiple correction, a strong local signal was found at 22q13.1 (chromosome 22: 39,307,894–40,545,797) for UL with both BC overall (p = 8.88 × 10−7) and ER+ subtype (p = 4.19 × 10−6). This genomic region harbors TNRC6B, a contributor to tumorigenesis of different cancers,33 and CBX7, associated with poor prognosis in ovarian clear cell adenocarcinoma.34 We did not observe any additional region that showed significant local genetic correlation.
Figure 2.

Local genetic correlation between uterine leiomyoma and breast cancer
(A) Manhattan plot showing the estimates of local genetic correlation, genetic covariance, and SNP heritability between uterine leiomyoma and breast cancer overall. Red bars represent loci showing significant local genetic correlation after multiple testing adjustment (p < 0.05/1,703).
(B) QQ-plot presenting region-specific p values from local genetic correlation between uterine leiomyoma and breast cancer overall.
(C) Manhattan plot showing the estimates of local genetic correlation, genetic covariance, and SNP heritability between uterine leiomyoma and ER+ breast cancer. Red bars represent loci showing significant local genetic correlation after multiple testing adjustment (p < 0.05/1,703).
(D) QQ-plot presenting region-specific p values from local genetic correlation between uterine leiomyoma and ER+ breast cancer. UL, uterine leiomyoma; BC, breast cancer; ER, estrogen receptor.
Partitioned genetic correlation
To characterize genetic overlap at the level of functional categories, we partitioned genetic correlation by 14 functional groups (Figure 3, Table S7). UL was significantly correlated with BC overall at 9 out of 14 functional categories, with rg ranging from 0.08 (H3K27ac) to 0.15 (coding). The repressed region (rg = 0.14), H3K9ac (rg = 0.12), and promotor region (rg = 0.11) also showed strong genetic correlation. Looking into BC subtypes, UL was significantly associated with ER+ BC at 6 functional categories. In addition to coding (rg = 0.16), H3K9ac showed the second strongest genetic correlation (rg = 0.13). For UL and ER– BC, no significant result was identified at any of these functional groups.
Figure 3.

Partitioned genetic correlation between uterine leiomyoma and breast cancer by genomic functional elements
Vertical axis represents genetic correlation. Horizontal axis represents 14 functional categories. Asterisks represent significance (p < 0.05), error bars represent the standard error of genetic correlation. UL, uterine leiomyoma; BC, breast cancer; ER, estrogen receptor; DGF, DNase digital genomic footprinting; DHS, DNase I hypersensitive sites.
Cross-trait meta-analysis and pleiotropic loci
Based on evidence of significant genetic overlap between UL and BC, we next interrogated at individual variant level to identify pleiotropic loci. A total of 8,170,973, 8,175,419, and 8,175,424 SNPs common between UL and BC overall, ER+ BC, and ER– BC were included in the cross-trait meta-analysis, respectively. As a result, 50 independent loci reached genome-wide significance in CPASSOC (fulfilling pCPASSOC < 5 × 10−8 and psingle-trait < 1 × 10−3), including 29 loci shared between UL and BC overall, 17 loci shared between UL and ER+ BC, and 14 loci shared between UL and ER– BC (Tables S8–S10). Among these shared loci, those closest to well-known oncogenes such as TERT, TNRC6B, and TP53 showed the strongest signals (i.e., index SNPs: rs10069690, rs2242652, rs4821942, and rs78378222).
After excluding SNPs that were in LD (r2 ≥ 0.20) with any of the previously reported single-trait-associated significant SNPs, we identified 8 novel pleiotropic SNPs for UL and BC overall of which 1 was also shared by UL and ER+ BC, and 1 novel pleiotropic SNP for UL and ER– BC, totaling 9 newly discovered SNPs (Table 2). Notably, rs3176337 (pCPASSOC = 7.87 × 10−11), the most significant novel SNP shared by UL and BC overall, was also identified as a novel pleiotropic SNP for UL and ER+ BC. This locus was near CDKN1A, a gene encoding a cyclin-dependent kinase inhibitor, which is a pivotal cell cycle regulator ensuring genomic stability and is often deregulated in human cancer.35 The second most significant novel SNP was mapped to PTPN11 (rs11066320, pCPASSOC = 1.31 × 10−09). PTPN11 is a member of the protein tyrosine phosphatase family, involved in a variety of cellular processes including cell growth, differentiation, mitotic cycle, and oncogenic transformation.36 Index SNP rs35840638 implicating ADAP2 was the third strongest shared signal (pCPASSOC = 2.17 × 10−09), specifically shared by UL and ER– BC. The fourth strongest signal was in close proximity to an intergenic region closest to GSTM1 (rs4147562, pCPASSOC = 2.83 × 10−09), which encodes a glutathione S-transferase that belongs to the mu class, and functions in the detoxification of electrophilic compounds such as products of oxidative stress and carcinogens.37 Other novel shared genetic variants were rs13001657 (intergenic region), rs62408878 (GNB4, MFN1), rs9316500 (DLEU1, RP11-175B12.2), rs3790110 (GNAO1), and rs2281925 (SLC2A4RG, ZBTB46), including regions previously implicated in energy metabolism and multiple cell cycle processes.38, 39, 40, 41 Detailed annotation for each SNP discovered by cross-trait meta-analysis is shown in Table S11.
Table 2.
Novel pleiotropic loci between uterine leiomyoma and breast cancer (pCPASSOC < 5 × 10−8, 5 × 10−8 < single trait p value < 1 × 10−3)
| SNP | Chr: Position | A1/A2 | UL |
BC |
pCPASSOC | Genes within clumping range | Linear closest genea | Interacting geneb | ||
|---|---|---|---|---|---|---|---|---|---|---|
| BETA | p | BETA | p | |||||||
| Uterine leiomyoma and breast cancer overall | ||||||||||
| rs4147562 | chr1: 110,230,073–110,230,099 | A/T | −0.46 | 2.9 10–05 | 0.04 | 3.5 10–05 | 2.8 10–09 | intergenic region | GSTM1 | GSTM4 |
| rs13001657 | chr2: 88,795,621–89,103,554 | A/G | 0.04 | 1.1 10–04 | 0.03 | 4.2 10–05 | 2.2 10–08 | ANKRD36BP2, EIF2AK3, LOC101928371, RPIA, TEX37 | – | EIF2AK3 |
| rs62408878 | chr3: 179,112,234–179,112,234 | T/C | 0.41 | 4.3 10–06 | −0.03 | 6.5 10–04 | 7.3 10–09 | intergenic region | GNB4, MFN1 | – |
| rs3176337 | chr6: 36,618,140–36,648,920 | A/C | 0.04 | 4.8 10–06 | −0.03 | 8.3 10–06 | 7.9 10–11 | CDKN1A, PANDAR | CDKN1A | MIR3925 |
| rs11066320 | chr12: 112,486,818–112,906,415 | A/G | −0.03 | 2.2 10–04 | −0.03 | 6.6 10–07 | 1.3 10–09 | HECTD4, MIR6861, NAA25, PTPN11, RPL6, TRAFD1 | PTPN11 | – |
| rs9316500 | chr13: 51,067,234–51,131,247 | T/G | 0.03 | 4.1 10–04 | 0.03 | 4.4 10–06 | 1.3 10–08 | DLEU1 | DLEU1, RP11-175B12.2 | – |
| rs3790110 | chr16: 56,372,907–56,547,254 | T/C | −0.03 | 5.0 10–04 | −0.03 | 6.2 10–07 | 4.1 10–09 | AMFR, BBS2, GNAO1, NUDT21, OGFOD1 | GNAO1 | CES5A |
| rs2281925 | chr20: 62,318,220–62,376,503 | A/G | 0.05 | 3.0 10–04 | 0.04 | 2.7 10–05 | 4.7 10–08 | ARFRP1, LIME1, RTEL1, RTEL1-TNFRSF6B, SLC2A4RG, TNFRSF6B, ZBTB46, ZGPAT | SLC2A4RG, ZBTB46 | ZGPAT |
| Uterine leiomyoma and ER+ breast cancer | ||||||||||
| rs3176337 | chr6: 36,618,140–36,648,920 | A/C | 0.04 | 4.8 10–06 | −0.03 | 1.7 10–04 | 1.5 10–08 | CDKN1A, PANDAR | CDKN1A | MIR3925 |
| Uterine leiomyoma and ER– breast cancer | ||||||||||
| rs35840638 | chr17: 29,166,302–29,318,794 | A/G | −0.05 | 2.3 10–07 | −0.05 | 1.9 × 10−04 | 2.2 10–09 | ADAP2, ATAD5, DPRXP4, RNF135, TEFM | ADAP2 | MIR4724 |
Position is under build 37 (hg19). SNP, single nucleotide polymorphism; Chr, chromosome; UL, uterine leiomyoma; BC, breast cancer; ER, estrogen receptor.
Linear closest genes of index SNPs were mapped by using VEP.
3D interacting genes of index SNPs were mapped by using 3DSNP.
Identification of causal variants and colocalization
Fine-mapping analysis assessed the 99% credible set of causal variants at each of the CPASSOC-identified pleiotropic loci, providing targets for downstream experimental analysis (results shown in Tables S12–S14). In general, we identified 138 candidate causal SNPs for novel shared loci between UL and BC overall, numbers specific to each index SNP were: rs4147562 (2), rs13001657 (23), rs62408878 (1), rs3176337 (5), rs11066320 (7), rs9316500 (11), rs3790110 (80), and rs2281925 (9). For novel loci shared between UL and BC subtypes, 17 and 13 candidate causal SNPs were discovered for rs3176337 and rs35840638, respectively.
Colocalization analysis was further performed to determine whether the genetic variants driving the association in two traits are the same or different. Most shared loci between UL and BC colocalized at the same candidate causal SNPs (PPH4 > 0.5) (22/50) or at different candidate causal SNPs (PPH3 > 0.5) (16/50), reinforcing shared causal associations (Table S15). Among the 9 novel pleiotropic loci, 2 loci (index SNP: rs11066320 and rs35840638) showed evidence of colocalization (PPH4 > 0.5).
Biological pathway, GTEx tissue, and SNP-heritability enrichment
After multiple correction, GO analysis across the 9 novel pleiotropic loci revealed enrichment in the benzene-containing compound metabolic process (GO:0042537, p = 9.87 × 10−06). KEGG analysis further identified significant enrichment in platinum drug resistance (hsa01524, p = 7.91 × 10−06). In GTEx tissue enrichment analysis, heart left ventricle was identified to be significantly enriched for the expression of novel shared genes underlying UL and BC (Figure S1). Results using all pleiotropic loci are shown in Tables S16 and S17 and Figure S2.
Partitioning SNP heritability by using 396 cell-type-specific annotations, we identified FDR-significant heritability enrichment for UL in smooth muscle and cardiovascular system (i.e., fetal heart, aorta, and right atrium). Although no significant enrichment was observed for BC overall or BC subtypes, they clustered closely with UL at each chromatin mark in musculoskeletal and connective, cardiovascular and digestive system, as well as other tissues or cell types such as ovary, obesity, mammary epithelial cells, fetal kidney, and primary B cells. Interestingly, different clustering patterns were observed comparing cell-type-specific enrichment for UL with ER+ versus ER– BC, where both UL and ER+ subtype were enriched for certain annotations while ER– subtype was not, for example, pancreas, fetal lung, and certain blood/immune system-related components (Figure S3).
Mendelian randomization
We finally conducted a two-sample MR using 28 GWAS-identified UL-associated SNPs as IVs (one SNP, rs2456181, was not available in the outcome GWASs). F-statistics for these IVs was 804.14, suggesting strong instruments (Tables S2 and S3). Using IVW, genetic liability to UL was significantly associated with an increased risk of BC overall (OR = 1.09, 95%CI = 1.01–1.18, p = 0.03). The estimates remained directionally consistent in MR-Egger regression and weighted median approach, despite larger statistical uncertainties. No sign of horizontal pleiotropy was detected (pMR-Egger intercept = 0.45). Subgroup analysis further identified such an association to be restricted to ER+ subtype (IVW OR = 1.09, 95%CI = 1.01–1.17, p = 0.03; pMR-Egger intercept = 0.94) but not to ER− subtype (IVW OR = 1.05, 95%CI = 0.88–1.25, p = 0.57; pMR-Egger intercept = 0.05) (Figure 4).
Figure 4.

Bidirectional causal relationship underlying uterine leiomyoma and breast cancer
(A) Estimates of causal effect for genetic liability to uterine leiomyoma with breast cancer overall.
(B) Estimates of causal effect for genetic liability to breast cancer overall with uterine leiomyoma.
(C) Estimates of causal effect for genetic liability to uterine leiomyoma with ER+ breast cancer.
(D) Estimates of causal effect for genetic liability to uterine leiomyoma with ER– breast cancer. Boxes represent the point estimates of causal effects, and error bars represent 95% confidence intervals. Inverse-variance weighted approach was adopted as the primary analysis. MR-Egger regression, weighted median, and MR-PRESSO approaches were adopted as sensitivity analysis. UL, uterine leiomyoma; BC, breast cancer; ER, estrogen receptor.
We performed important sensitivity analysis to verify MR model assumptions. The causal effect of UL in BC overall or ER+ subtype remained significant after excluding palindromic SNPs or pleiotropic SNPs. The leave-one-out analysis demonstrated that the observed causal relationship was not driven by any outlying variant (Figure S4). After removing outliers, MR-PRESSO yielded similar associations (BC overall OR = 1.05, p = 0.049; ER+ subtype OR = 1.09, p = 2.67 × 10−3; ER− subtype OR = 1.02, p = 0.72) (Figure 4). Multi-variable IVW taking into consideration BMI generated similar results (BC overall OR = 1.10, p = 0.02; ER+ subtype OR = 1.10, p = 0.03; ER− subtype OR = 1.06, p = 0.57), suggesting a causal association of UL with BC independent of obesity.
Using the 185 GWAS-identified BC-associated SNPs as IVs, we performed a reverse-direction MR. No evidence was found on the association between genetic liability to BC and UL risk (IVW OR = 1.00, p = 0.80; MR-Egger OR = 1.01, p = 0.68; weighted median OR = 1.00, p = 0.87) (Figure 4).
Discussion
To the best of our knowledge, this is the first large-scale genome-wide cross-trait analysis that systematically interrogates the shared genetic basis underlying UL and BC, two highly complicated and entangled disorders. We found evidence supporting a significant genetic correlation of UL with BC and with its ER+ subtype. When the whole genome was partitioned, significant correlations were further discovered within a specific genomic region (22q13.1) and functional categories (e.g., coding, H3K9ac, and repressed region). Using cross-trait meta-analysis, we identified multiple pleotropic loci with joint associations. Additionally, MR analysis highlighted a putative causal role of UL on BC risk, restricted to ER+ subtype.
Using LDSC with a constrained intercept, we detected a significant global genetic correlation of UL with BC overall, as well as with ER+ subtype. Constrained LDSC is known for improving the statistical power of rg estimated under the assumption of no sample overlap14 and was applied in our study for two reasons. First, the UL and BC GWASs shared no overlapping participating studies, and second, the intercepts of genetic covariance were estimated at around zero (∼0.001–0.007). Both reasons indicate an absence of bias from sample overlap or population stratification, justifying the utilization of the method. Partitioning the whole genome into 1,703 nearly independent regions, we found a strong local genetic correlation of UL at 22q13.1 with both overall and ER+ BC. This region harbors TNRC6B, a gene previously reported to be independently associated with UL and BC.10, 11, 12 In stratified-LDSC, significant genetic correlation was further observed in multiple annotated regions of the genome. The strongest partitioned rg was found, unsurprisingly, to be in the coding region, while partitioned rg was also high (or even higher than the global rg) in certain non-coding regions, including histone acetylation marks (i.e., H3K9ac) and histone modification marks (i.e., H3K4me1 and H3K27me3), highlighting their important roles in not only the progression of BC42 but also in the onset of disease. This observation is consistent with the idea that genetic variation within functional non-coding elements is also substantially involved in gene expression and regulation.43,44 Unfortunately, and consistently, no significant genetic correlation was observed for UL and ER– BC at any level from global rg to local rg.
While our findings demonstrate a putative shared etiology between UL and BC, it can be the result of pleiotropy (a situation in which a genetic variant or gene has effects on multiple traits) and/or causality (a situation in which a genetic variant has an effect on a trait via its genetic effect on an intermediate trait).9 In our downstream analysis performed to dissect such a complex genetic relationship, a total of 50 shared loci between UL and BC were identified of which 41 loci were previously reported to be significantly associated with UL and/or BC. These loci harbor genes previously implicated in risks of various carcinomas (i.e., CDKN1A, GSTM1, MFN1, TERT, TP53) or hormone-related traits (i.e., ESR1, GREB1, and MCM8). Multiple genes showed strong evidence of colocalization (PPH4 > 0.5), such as ATAD5, EXO1, HSPA4, MCM8, MLLT10, PTPN11, TERT, and TP53, demonstrating etiological connections. One advantage of meta-analyzing GWASs of different traits is that it improves the statistical power of detecting cross-trait genetic effects (especially for traits with smaller sample sizes) by combining association evidence from multiple GWASs, discovering signals which might not have reached genome-wide significance in a single-trait effort.45 Indeed, we found 9 novel loci to be jointly associated with both UL and BC, among which we highlight two interesting examples, SLC2A4RG/ZBTB46 and ADAP/MIR4724.
While SLC2A4RG and ZBTB46 were mapped by the same locus (index SNP: rs2281925), their involvement in breast tumorigenesis has rarely been studied. SLC2A4RG is a transcriptional activator of the glucose transporter SLC2A4.46 Overexpression of SLC2A4RG could lead to an inhibition of glioblastoma cell growth by downregulating the expression of cyclin-dependent kinases, suggesting the potential tumor-suppressor role of SLC2A4RG.47 By performing whole-exome sequencing on paired primary and metastatic tumors, a recent study found SLC2A4RG as a significantly mutated gene in metastatic BC, despite not reported to play a role in primary BC.48 ZBTB46 is another transcription factor specifically expressed by classical dendritic cells, functioning as a key immune regulator.49 Recent studies found ZBTB46 to be a novel tumor promoter for prostate cancer.50,51 Based on microarray expression profile analysis, a study of Chinese women found an upregulated long non-coding RNA, RP4-583P15.10, to be differentially expressed between BC tissues and paired adjacent tissues, located downstream of the natural antisense of ZBTB46.52
ADAP2 (index SNP: rs35840638) was identified as a significant pleiotropic locus affecting both UL and ER– BC. Furthermore, colocalization analysis showed that this locus had a great probability (98.2%) of containing a shared causal variant of both traits. Using DNA copy number data from 39 cancer types, Maroulio et al. identified ADAP2 as an essential gene limiting the extent of homozygous deletions in cancer genomes, suggesting its role in the survival of tumor cells.53 In addition, ADAP2 may be a potential indicator for the early diagnosis and prognosis of acute myeloid leukemia, suggested by Yu et al.54 However, there is no result with respect to ADAP2 function in BC. This locus also interacts with miRNA MIR4724 in a three-dimensional manner.20 miRNAs are a class of small non-coding RNAs regulating a wide range of physiological processes by repressing transcription or translation of their target genes, which may contribute to cancer etiology.55 MIR4724 has only been implied as a candidate miRNA biomarker for glaucoma.56 With an improved power from cross-trait GWASs, our study suggests MIR4724 as a potential risk gene underlying UL and BC.
By applying a comprehensive bidirectional MR design, we identified a potential causal relationship between genetic liability to UL and risk of BC, restricted to ER+ subtype. This was largely in line with our genetic correlation analysis on a negligible intrinsic relationship between UL and ER– BC. The strengths of our MR design include (1) genetic instruments derived from the hitherto largest GWAS based on hundreds of thousands of female participants for each trait, (2) reverse-direction MR clarifying the direction of the UL-BC relationship, (3) adjustment for obesity with multi-variable MR to understand the direct effect, and (4) the use of a variety of MR sensitivity analyses to guarantee model assumptions. Consequently, the estimated causal effects were directionally consistent across different statistical approaches and sensitivity analyses, supporting the validity of our findings. Our results are largely concordant with findings from previous case-control studies reporting positive associations of UL and BC6,57,58 and a finding from a prospective cohort study that women with a history of UL are at an elevated risk of reporting BC in the future.8 While biological mechanisms underlying the observed causal effect remain inconclusive, one might lie in the vertical pleiotropy (or “type II pleiotropy”),59 where genetic instruments for UL are associated with other traits (e.g., hormonal response), reflecting the downstream effects of UL that are potentially on the causal pathway linking UL to BC. Findings from our MR analysis have provided genetic evidence for the UL-BC association, highlighting a potential consideration of targeting women with UL for BC prevention (e.g., screening). Future efforts should be focused on the molecular characteristics of UL as well as its clinical factors (e.g., location, multiplicity, size, and recurrence) to target more precisely a population at risk for BC.60,61
Leveraging information on gene expression, we found largely consistent patterns of cell-type-specific heritability enrichment for UL and BC at multiple annotations including the cardiovascular system. Of note, GTEx tissue enrichment analysis also found a cardiovascular component, heart left ventricle, to be significantly enriched by UL-BC shared genes. UL was previously reported to be associated with hypertension and atherosclerosis.62,63 A recent prospective study found that, although the presence of UL was not associated with subsequent cardiovascular disease (CVD), risk factors for CVD (particularly BMI and hypertension) were substantially more prevalent in women with UL than in UL-free referents.64 It has also been recognized that BC and CVD share a number of common risk factors. Studies have recommended that early recognition and management of CVD risk factors are of utmost importance for BC treatment and prevention.65,66 Findings in our functional annotation analysis suggest possible common pathways leading to the comorbidity between UL and BC, which requires additional work to reveal the underlying pathophysiological mechanism.
As a subtype that possesses neither estrogen nor progesterone receptor (PR),67 ER– BC has been considered as less likely to be associated with UL (a hormone-responsive disorder) from a traditional perspective. Using information from genetic data, the current study identified multiple risk loci shared by UL and ER– BC, supporting a non-negligible pleiotropic effect. The similar enrichment patterns regarding the heritability for UL and ER– BC observed at various cell-type-specific annotations further corroborate potential common underlying etiology. Although no evidence on a significant genetic correlation or causal link was shown, it should be noted that the estimated global rg between UL and ER– BC is similar to that of the ER+ BC (rg = 0.06), with the number of ER– cases being less than a third of their ER+ counterparts. In fact, the significant genetic findings for ER– subtype are not entirely unexpected, as a substantial genetic correlation (rg = 0.60) between the two BC subtypes has already been uncovered by a recent large-scale GWAS.68
We acknowledge potential limitations of our study. First, to avoid bias from population stratification, all genetic data in this study were restricted to European ancestry, limiting the generalizability to other ethnic populations. Second, as most of the analytical software adopted by us do not currently support the management and analysis of sex chromosomes, we included only data from autosomes in our study (except in the MR analysis). This may lead to potentially undetected associations due to underrepresentation of X chromosome SNPs. Third, statistical power might be insufficient for the ER– subgroup, yielding null findings which might have been discovered with a larger sample size. Similarly, additional BC subtypes based on other hormonal receptor expression such as PR and human epidermal growth factor receptor 2 (HER2)69 were not investigated in this study due to limited sample size, for instance, the HER2 enriched-like BC cases analyzed by Zhang et al.11 (n = 2,884). Future studies with larger sample sizes of subtype-specific BC are warranted to extend our findings. Finally, we used limited numbers of UL-associated SNPs as IVs to detect the causal effect of UL on BC, making it difficult to rule out weak instrument bias. However, each SNP-exposure association had an F-statistic greater than 10, supporting the strength of the genetic variants. We emphasize that our inferred causal relationship is putative as it was generated based on GWAS summary statistics. Larger and more powerful GWASs for UL and BC are needed to establish definitively (or rule out) a potential causal link. Future longitudinal studies as well as experimental work are also warranted to investigate the biological mechanism underlying the observed genetic relationship.
To conclude, the current study furthers our understanding to the observational association between UL and BC by providing evidence of genetic correlation, revealing potential pleiotropic loci, and inferring a putative causal relationship. Our findings highlight an intrinsic link underlying these two complex female diseases and shed new light on the biological mechanisms; these findings might provide important directions for future therapeutic strategy as well as risk prediction.
Data and code availability
This study did not generate datasets or code.
Acknowledgments
Summary statistics for the genetic associations with uterine leiomyoma, breast cancer overall, and breast cancer subtypes were obtained from GWASs conducted by Gallagher et al., Zhang et al., and Michailidou et al. We are grateful to all investigators who shared genome-wide summary statistics. This work was supported by funds from the National Natural Science Foundation of China (No. 81874282), the National Key R&D Program of China (2020YFC2006505), the Health Commission of Sichuan Province (20PJ093), and the U.S. National Institutes of Health/Eunice Kennedy Shriver National Institute of Child Health and Human Development (HD060530). Graphical abstract was created with BioRender.com.
Declaration of interests
The authors declare no competing interests.
Published: July 7, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.05.015.
Contributor Information
Jiayuan Li, Email: lijiayuan73@163.com.
Xia Jiang, Email: xiajiang@scu.edu.cn.
Web resources
23andMe GWAS summary statistics, https://research.23andme.com/dataset-access/
3DSNP, http://cbportal.org/3dsnp/
Coloc, https://chr1swallace.github.io/coloc/
CPASSOC, http://hal.case.edu/∼xxz10/zhuweb/
FM-summary, https://github.com/hailianghuang/FM-summary
FUMA, https://fuma.ctglab.nl/
GWAS summary statistics for BC overall and subtype-specific BC, https://bcac.ccge.medschl.cam.ac.uk/bcacdata/oncoarray/oncoarray-and-combined-summary-result/
LDSC, https://github.com/bulik/ldsc
NHGRI-EBI GWAS Catalog: UL GWAS summary statistics (without 23andMe), https://www.ebi.ac.uk/gwas/downloads/summary-statistics
PLINK, https://www.cog-genomics.org/plink/1.9/
ρ-HESS, https://huwenboshi.github.io/hess/local_rhog/
TwoSampleMR, https://mrcieu.github.io/TwoSampleMR/
VEP, https://grch37.ensembl.org/info/docs/tools/vep/index.html
Webgestalt, http://webgestalt.org
Supplemental information
References
- 1.Stewart E.A., Cookson C.L., Gandolfo R.A., Schulze-Rath R. Epidemiology of uterine fibroids: a systematic review. BJOG. 2017;124:1501–1512. doi: 10.1111/1471-0528.14640. [DOI] [PubMed] [Google Scholar]
- 2.Marino J.L., Eskenazi B., Warner M., Samuels S., Vercellini P., Gavoni N., Olive D. Uterine leiomyoma and menstrual cycle characteristics in a population-based cohort study. Hum Reprod. 2004;19:2350–2355. doi: 10.1093/HUMREP/DEH407. [DOI] [PubMed] [Google Scholar]
- 3.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality Worldwide for 36 cancers in 185 Countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/CAAC.21660. [DOI] [PubMed] [Google Scholar]
- 4.Pavone D., Clemenza S., Sorbi F., Fambrini M., Petraglia F. Epidemiology and risk factors of uterine fibroids. Best Pract. Res. Clin. Obstet. Gynaecol. 2018;46:3–11. doi: 10.1016/J.BPOBGYN.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 5.Shah R., Rosso K., Nathanson S.D. Pathogenesis, prevention, diagnosis and treatment of breast cancer. World J. Clin. Oncol. 2014;5:283. doi: 10.5306/WJCO.V5.I3.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindegård B. Breast cancer among women from Gothenburg with regard to age, mortality and coexisting benign breast disease or leiomyoma uteri. Oncology. 1990;47:369–375. doi: 10.1159/000226850. [DOI] [PubMed] [Google Scholar]
- 7.Wise L.A., Radin R.G., Rosenberg L., Adams-Campbell L., Palmer J.R. History of uterine leiomyomata and incidence of breast cancer. Cancer Causes Control. 2015;26:1487–1493. doi: 10.1007/S10552-015-0647-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen T.-C., Hsia T.C., Hsiao C.L., Lin C.L., Yang C.Y., Soh K.S., Liu L.C., Chang W.S., Tsai C.W., Bau D.T. Patients with uterine leiomyoma exhibit a high incidence but low mortality rate for breast cancer. Oncotarget. 2017;8:33014–33023. doi: 10.18632/ONCOTARGET.16520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu Z., Hasegawa K., Camargo C.A., Liang L. Investigating asthma heterogeneity through shared and distinct genetics: insights from genome-wide cross-trait analysis. J. Allergy Clin. Immunol. 2021;147:796–807. doi: 10.1016/J.JACI.2020.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallagher C.S., Mäkinen N., Harris H.R., Rahmioglu N., Uimari O., Cook J.P., Shigesi N., Ferreira T., Velez-Edwards D.R., Edwards T.L., et al. Genome-wide association and epidemiological analyses reveal common genetic origins between uterine leiomyomata and endometriosis. Nat. Commun. 2019;10:4857. doi: 10.1038/s41467-019-12536-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H., Ahearn T.U., Lecarpentier J., Barnes D., Beesley J., Qi G., Jiang X., O’Mara T.A., Zhao N., Bolla M.K., et al. Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat. Genet. 2020;52:572–581. doi: 10.1038/s41588-020-0609-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rafnar T., Gunnarsson B., Stefansson O.A., Sulem P., Ingason A., Frigge M.L., Stefansdottir L., Sigurdsson J.K., Tragante V., Steinthorsdottir V., et al. Variants associating with uterine leiomyoma highlight genetic background shared by various cancers and hormone-related traits. Nat. Commun. 2018;9:3636. doi: 10.1038/S41467-018-05428-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michailidou K., Lindström S., Dennis J., Beesley J., Hui S., Kar S., Lemaçon A., Soucy P., Glubb D., Rostamianfar A., et al. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551:92–94. doi: 10.1038/nature24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bulik-Sullivan B., Finucane H.K., Anttila V., Gusev A., Day F.R., Loh P.R., Duncan L., Perry J.R.B., Patterson N., Robinson E.B., et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015;47:1236–1241. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi H., Mancuso N., Spendlove S., Pasaniuc B. Local genetic correlation gives insights into the shared genetic architecture of complex traits. Am. J. Hum. Genet. 2017;101:737–751. doi: 10.1016/J.AJHG.2017.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.R., Anttila V., Xu H., Zang C., Farh K., et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu X., Feng T., Tayo B., Liang J., Young J., Franceschini N., Smith J., Yanek L., Sun Y., Edwards T., et al. Meta-analysis of correlated traits via summary statistics from GWASs with an application in hypertension. Am. J. Hum. Genet. 2015;96:21–36. doi: 10.1016/J.AJHG.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zerbino D.R., Achuthan P., Akanni W., Amode M., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C.G., et al. Nucleic Acids Res. 2018;46:D754–D761. doi: 10.1093/NAR/GKX1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu Y., Quan C., Chen H., Bo X., Zhang C. 3DSNP: a database for linking human noncoding SNPs to their three-dimensional interacting genes. Nucleic Acids Res. 2017;45:D643–D649. doi: 10.1093/NAR/GKW1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaid D.J., Chen W., Larson N.B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 2018;19:491–504. doi: 10.1038/S41576-018-0016-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farh K.K.-H., Marson A., Zhu J., Kleinewietfeld M., Housley W.J., Beik S., Shoresh N., Whitton H., Ryan R.J.H., Shishkin A.A., et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2014;518:337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giambartolomei C., Vukcevic D., Schadt E.E., Franke L., Hingorani A.D., Wallace C., Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLOS Genet. 2014;10 doi: 10.1371/JOURNAL.PGEN.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang B., Kirov S., Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–W748. doi: 10.1093/NAR/GKI475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe K., Taskesen E., van Bochoven A., Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017;8:1826. doi: 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finucane H.K., Reshef Y.A., Anttila V., Slowikowski K., Gusev A., Byrnes A., Gazal S., Loh P.R., Lareau C., Shoresh N., et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 2018;50:621–629. doi: 10.1038/s41588-018-0081-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burgess S., Scott R.A., Timpson N.J., Davey Smith G., Thompson S.G. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur. J. Epidemiol. 2015;30:543–552. doi: 10.1007/S10654-015-0011-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowden J., Davey Smith G., Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015;44:512–525. doi: 10.1093/IJE/DYV080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bowden J., Davey Smith G., Haycock P.C., Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 2016;40:304–314. doi: 10.1002/GEPI.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davey Smith G., Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003;32:1–22. doi: 10.1093/IJE/DYG070. [DOI] [PubMed] [Google Scholar]
- 31.Verbanck M., Chen C.-Y., Neale B., Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018;50:693–698. doi: 10.1038/s41588-018-0099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burgess S., Thompson S.G. Multivariable mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am. J. Epidemiol. 2015;181:251–260. doi: 10.1093/AJE/KWU283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forouzanfar N., Baranova A., Milanizadeh S., Heravi-Moussavi A., Jebelli A., Abbaszadegan M.R. Novel candidate genes may be possible predisposing factors revealed by whole exome sequencing in familial esophageal squamous cell carcinoma. Tumor Biol. 2017;39 doi: 10.1177/1010428317699115. 101042831769911. [DOI] [PubMed] [Google Scholar]
- 34.Shinjo K., Yamashita Y., Yamamoto E., Akatsuka S., Uno N., Kamiya A., Niimi K., Sakaguchi Y., Nagasaka T., Takahashi T., et al. Expression of chromobox homolog 7 (CBX7) is associated with poor prognosis in ovarian clear cell adenocarcinoma via TRAIL-induced apoptotic pathway regulation. Int. J. Cancer. 2014;135:308–318. doi: 10.1002/IJC.28692. [DOI] [PubMed] [Google Scholar]
- 35.Kreis N.-N., Louwen F., Yuan J. The multifaceted p21 (Cip1/Waf1/CDKN1A) in cell differentiation, migration and cancer therapy. Cancers (Basel) 2019;11:1220. doi: 10.3390/CANCERS11091220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuban-Jankowska A., Gorska M., Knap N., Cappello F., Wozniak M. Protein tyrosine phosphatases in pathological process. Front. Biosci. 2015;20:4314–4388. doi: 10.2741/4314. [DOI] [PubMed] [Google Scholar]
- 37.Soto-Quintana O., Zúñiga-González G., Ramírez-Patiño R., Ramos-Silva A., Figuera L., Carrillo-Moreno D., Gutiérrez-Hurtado I., Puebla-Pérez A., Sánchez-Llamas B., Gallegos-Arreola M. Association of the GSTM1 null polymorphism with breast cancer in a Mexican population. Genet. Mol. Res. 2015;14:13066–13075. doi: 10.4238/2015.OCTOBER.26.2. [DOI] [PubMed] [Google Scholar]
- 38.Wang B., Li D., Rodriguez-Juarez R., Farfus A., Storozynsky Q., Malach M., Carpenter E., Filkowski J., Lykkesfeldt A.E., Kovalchuk O. A suppressive role of guanine nucleotide-binding protein subunit beta-4 inhibited by DNA methylation in the growth of anti-estrogen resistant breast cancer cells. BMC Cancer. 2018;18:817. doi: 10.1186/S12885-018-4711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang C., Xie X.X., Li W.J., Jiang D.Q. LncRNA DLEU1/microRNA-300/RAB22A axis regulates migration and invasion of breast cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2019;23:10410–10421. doi: 10.26355/EURREV_201912_19680. [DOI] [PubMed] [Google Scholar]
- 40.Song X., Zhang M., Chen L., Lin Q. Bioinformatic prediction of possible targets and mechanisms of action of the green tea compound epigallocatechin-3-gallate against breast cancer. Front. Mol. Biosci. 2017;4:43. doi: 10.3389/FMOLB.2017.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai W., Jiang L. Dysregulated mitochondrial dynamics and metabolism in obesity, diabetes, and cancer. Front. Endocrinol. (Lausanne) 2019;10:570. doi: 10.3389/FENDO.2019.00570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhuang J., Huo Q., Yang F., Xie N. Perspectives on the role of histone modification in breast cancer progression and the advanced technological tools to study epigenetic determinants of metastasis. Front. Genet. 2020;11:603552. doi: 10.3389/FGENE.2020.603552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elliott K., Larsson E. Non-coding driver mutations in human cancer. Nat. Rev. Cancer. 2021;21:500–509. doi: 10.1038/s41568-021-00371-z. [DOI] [PubMed] [Google Scholar]
- 44.Cuykendall T.N., Rubin M.A., Khurana E. Non-coding genetic variation in cancer. Curr. Opin. Syst. Biol. 2017;1:9–15. doi: 10.1016/J.COISB.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu Z., Anttila V., Smoller J.W., Lee P.H. Statistical power and utility of meta-analysis methods for cross-phenotype genome-wide association studies. PLoS One. 2018;13 doi: 10.1371/JOURNAL.PONE.0193256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sparling D.P., Griesel B.A., Weems J., Olson A.L. GLUT4 enhancer factor (GEF) interacts with MEF2A and HDAC5 to regulate the GLUT4 promoter in adipocytes. J. Biol. Chem. 2008;283:7429–7437. doi: 10.1074/JBC.M800481200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y., Yun D., Zou X., Jiang T., Li G., Hu L., Chen J., Xu J., Mao Y., Chen H., Lu D. Whole exome-wide association study identifies a missense variant in SLC2A4RG associated with glioblastoma risk. Am. J. Cancer Res. 2017;7:1937. [PMC free article] [PubMed] [Google Scholar]
- 48.Paul M.R., Pan T.C., Pant D.K., Shih N.N., Chen Y., Harvey K.L., Solomon A., Lieberman D., Morrissette J.J., Soucier-Ernst D., et al. Genomic landscape of metastatic breast cancer identifies preferentially dysregulated pathways and targets. J. Clin. Invest. 2020;130:4252–4265. doi: 10.1172/JCI129941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sallusto F. DCs: a dual bridge to protective immunity. Nat. Immunol. 2013;14:890–891. doi: 10.1038/ni.2693. [DOI] [PubMed] [Google Scholar]
- 50.Fararjeh A.S., Liu Y.N. ZBTB46, SPDEF, and ETV6: novel potential biomarkers and therapeutic targets in castration-resistant prostate cancer. Int. J. Mol. Sci. 2019;20:2802. doi: 10.3390/IJMS20112802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen W.Y., Tsai Y.C., Siu M.K., Yeh H.L., Chen C.L., Yin J.J., Huang J., Liu Y.N. Inhibition of the androgen receptor induces a novel tumor promoter, ZBTB46, for prostate cancer metastasis. Oncogene. 2017;36:6213–6224. doi: 10.1038/ONC.2017.226. [DOI] [PubMed] [Google Scholar]
- 52.Xu N., Wang F., Lv M., Cheng L. Microarray expression profile analysis of long non-coding RNAs in human breast cancer: a study of Chinese women. Biomed. Pharmacother. 2015;69:221–227. doi: 10.1016/J.BIOPHA.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 53.Pertesi M., Ekdahl L., Palm A., Johnsson E., Järvstråt L., Wihlborg A.K., Nilsson B. Essential genes shape cancer genomes through linear limitation of homozygous deletions. Commun. Biol. 2019;2:262. doi: 10.1038/s42003-019-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu Y.-H., Xin F., Dong L., Ge L., Zhai C.-Y., Shen X.-L. Weighted Gene Coexpression Network Analysis Identifies Critical Genes in Different Subtypes of Acute Myeloid Leukaemia. Biotechnol. Biotechnol. Equipment. 2020;34:925–936. doi: 10.1080/13102818.2020.1811767. [DOI] [Google Scholar]
- 55.Jeffrey S.S. Cancer biomarker profiling with microRNAs. Nat. Biotechnol. 2008;26:400–401. doi: 10.1038/nbt0408-400. [DOI] [PubMed] [Google Scholar]
- 56.Hindle A.G., Thoonen R., Jasien J.V., Grange R.M.H., Amin K., Wise J., Ozaki M., Ritch R., Malhotra R., Buys E.S. Identification of candidate miRNA biomarkers for Glaucoma. Invest Ophthalmol. Vis. Sci. 2019;60:134. doi: 10.1167/IOVS.18-24878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tseng J.J., Chen Y.H., Chiang H.Y., Lin C.H. Increased risk of breast cancer in women with uterine myoma: a nationwide, population-based, case-control study. J. Gynecol. Oncol. 2017;28:e35. doi: 10.3802/JGO.2017.28.E35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chuang S.-C., Wu G.-J., Lu Y.-S., Lin C.-H., Hsiung C.A. Associations between medical Conditions and breast cancer risk in Asians: a nationwide population-based study in Taiwan. PLoS One. 2015;10:e0143410. doi: 10.1371/JOURNAL.PONE.0143410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davey Smith G., Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 2014;23:R89–R98. doi: 10.1093/HMG/DDU328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sakai K., Tanikawa C., Hirasawa A., Chiyoda T., Yamagami W., Kataoka F., Susumu N., Terao C., Kamatani Y., Takahashi A., et al. Identification of a novel uterine leiomyoma GWAS locus in a Japanese population. Sci. Rep. 2020;10:1197. doi: 10.1038/s41598-020-58066-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dvorská D., Braný D., Danková Z., Danková Z., Halašová E., Halašová E., Višňovský J., Višňovský J. Molecular and clinical attributes of uterine leiomyomas. Tumor Biol. 2017;39 doi: 10.1177/1010428317710226. 101042831771022. [DOI] [PubMed] [Google Scholar]
- 62.Aksoy Y., Sivri N., Karaoz B., Sayin C., Yetkin E. Carotid intima-media thickness: a new marker of patients with uterine leiomyoma. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014;175:54–57. doi: 10.1016/J.EJOGRB.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 63.Boynton-Jarrett R., Rich-Edwards J., Malspeis S., Missmer S.A., Wright R. A prospective study of hypertension and risk of uterine leiomyomata. Am. J. Epidemiol. 2005;161:628–638. doi: 10.1093/AJE/KWI072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laughlin-Tommaso S.K., Fuchs E.L., Wellons M.F., Lewis C.E., Calderon-Margalit R., Stewart E.A., Schreiner P.J. Uterine fibroids and the risk of cardiovascular disease in the coronary artery risk development in young adult women’s study. J. Women’s Heal. 2019;28:46–52. doi: 10.1089/JWH.2018.7122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mehta L.S., Watson K.E., Barac A., Beckie T.M., Bittner V., Cruz-Flores S., Dent S., Kondapalli L., Ky B., Okwuosa T., et al. Cardiovascular disease and breast cancer: where these entities intersect: a scientific statement from the American heart association. Circulation. 2018;137:e30–e66. doi: 10.1161/CIR.0000000000000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Almuwaqqat Z., Meisel J.L., Barac A., Parashar S. Breast cancer and heart failure. Heart Fail Clin. 2019;15:65–75. doi: 10.1016/J.HFC.2018.08.007. [DOI] [PubMed] [Google Scholar]
- 67.Althuis M.D., Fergenbaum J.H., Garcia-Closas M., Brinton L.A., Madigan M.P., Sherman M.E. Etiology of hormone receptor-defined breast cancer: a systematic review of the literature. Cancer Epidemiol. Biomarkers Prev. 2004;13:1558–1568. https://pubmed.ncbi.nlm.nih.gov/15466970/ [PubMed] [Google Scholar]
- 68.Milne R.L., Kuchenbaecker K.B., Michailidou K., Beesley J., Kar S., Lindström S., Hui S., Lemaçon A., Soucy P., Dennis J., et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat. Genet. 2017;49:1767–1778. doi: 10.1038/NG.3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Onitilo A.A., Engel J.M., Greenlee R.T., Mukesh B.N. Breast cancer subtypes based on ER/PR and Her2 expression: comparison of clinicopathologic features and survival. Clin Med Res. 2009;7:4–13. doi: 10.3121/cmr.2008.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets or code.