

Abstract
T-follicular helper cells (TFH) are a unique subset of T-cells with varied transcriptional profiles and functions. In the last 2016 WHO classification, lymphomas arising from TFH were included as a broad category and emphasis was given to separating them from other peripheral T cell lymphomas. The neoplasms derived from these mainly comprise angioimmunoblastic T-cell lymphoma, peripheral T-cell lymphoma with T-follicular helper cell phenotype, follicular T-cell lymphoma, and cutaneous CD4+ small-medium sized lymphoproliferative disorders. The TFH lymphomas comprise both indolent and aggressive forms. Additional immunohistochemistry to identify TFH cells like CD10, BCL6, ICOS, PD1, CXCL13 and mutations like RHOA, IDH2 is required for diagnosis and prognostication. The understanding of these has evolved over the years, and currently we review the updates and pathobiology of the above.
Keywords: Follicular helper T-cells, follicular T helper cell lymphoma, AITL, PTCL
Introduction
Peripheral T-cell lymphoma (PTCL) represents the neoplasm derived from post-thymic mature T-cells. With varying biological and clinical behavior, the heterogeneous group of Non-hodgkin lymphoma (NHL) encompasses 27 entities [1]. Defective maturation or differentiation of T-cells may be the causative factor for lymphomagenesis [2]. Their derivation from mature T-cells endows them with a unique morphological, immunophenotypic, and molecular phenotype. Since the T-cells themselves have many subtypes with varied cytokine milieus and functions, ascertaining the subtype of PTCL is of utmost importance. Many T-cell lymphomas were earlier classified as PTCL, NOS due to morphological overlap and gaps in knowledge about various genes and markers, as well as T-follicular helper T-cells (TFH). Now, the majority of PTCLs are defined entities such as angioimmunoblastic T cell lymphoma (AITL), anaplastic large cell lymphoma (ALCL) with or without ALK gene translocation, and PTCL, NOS, accounting for approximately 60% [3-5]. And hence, the main subtype resulting from the WHO 2016 reclassification are T-follicular helper cell derived neoplasms [6].
However, for better understanding of the demography and disease process and to aid patient management, reclassification of the diagnosed cases along with comprehensive diagnosis of the new cases is essential, for which the understanding of TFH cells is essential. Though rare, they usually have an aggressive course with a dismal outcome, and hence their timely and appropriate diagnosis may add value to the patient’s management.
Development of T-cells
T-cell lymphopoiesis is initiated in the bone marrow, with further maturation taking place in the thymus. Common lymphocyte progenitors enter the thymus after exiting the bone marrow environment to complete T-cell development [7]. Based on the molecular interactions, the maturation of T-cells occurs. A series of maturation steps take place in the thymic cortex and medulla, resulting in the formation of naive T-cells-single positive, either for CD4 or CD8. According to pathology, the different cytokine milieus lead to the development of various subtypes of T-cells. Adaptive immune responses mainly use CD4+ T-cells, which, via the various differentiated subtypes, participate in the immune response. The subtypes include TH1, TH2, TH17, T-regs, and TFH and each has a unique transcriptional and translational profile [8] (Figure 1; Table 1).
Figure 1.
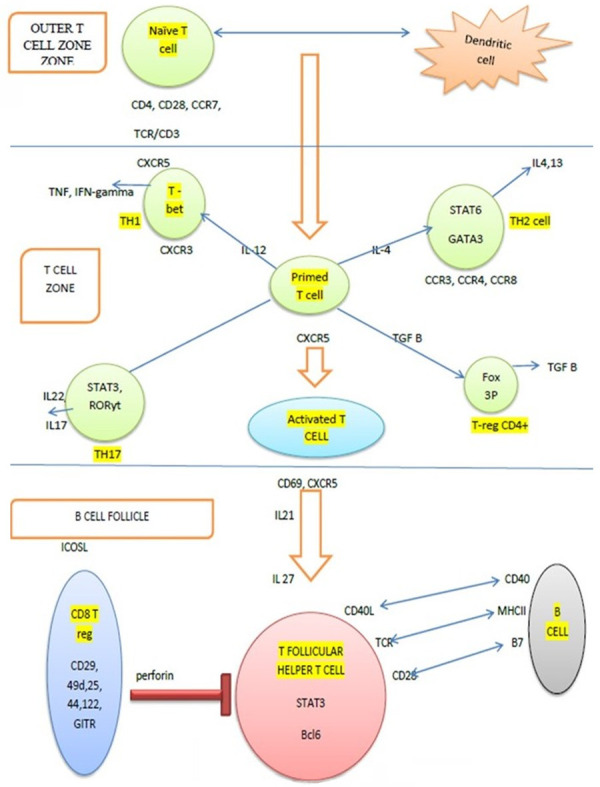
Development of T-follicular helper cells: Artwork shows TFH cell development in the lymphoid follicle. Interaction of naïve T-cell and dendritic cells in the outer T-cell zone prime the lymphocytes which in the respective cytokine milieu mature into the T-lymphocyte subsets. Activated T-cell with CXCR5 upon interaction with B-cell in the lymphoid follicle mature into the TFH cell and share a symbiotic relationship with B-cells.
Table 1.
Cytokine milieu for development and functions of TH cell subsets
| Cytokine milieu | Subset of T-cell | Markers for the cell | Cytokine secreted and function |
|---|---|---|---|
| IL12, IFN γ | TH1 | STAT4, TBX21, T-bet | IFN γ; Intracellular bacteria |
| IL4 | TH2 | STAT6, GATA3 | IL4, IL5, IL13 |
| IL6, TGFβ | TH17 | STAT3, ROR γ | IL17, IL21; Extracellular bacteria, fungi |
| IL6, IL21 | TFH | STAT3, BCL6, Ascl2 | IL21; B cell help |
| TGFβ | T reg | STAT5, FOXP3 | TGFβ, IL10; Regulatory function |
T-follicular helper cells (TFH)
Among all, TFH cell is a recently discovered entity in the last decade, having a unique biology. These T-cells are present in the B-cell zone of the lymph node and provide help to B-cells in antibody responses. Though they are home to the B-follicles like central memory T-cells, they also show loss of CCR7-like effector memory T-cells [9]. To induce the differentiation into the TFH lineage, interaction with an antigen of high affinity and co-stimulation is required [10]. BCL6 is essential for this differentiation. Unlike other T-cell subsets, TFH cell differentiation is a multifactorial process, with dendritic cell priming and expression of CXCR5 followed by migration to the border of the germinal follicle as the initial events. This process is regulated by IL6, ICOS, and TCR signals. The second stage is marked by the interaction of T-cells with the antigen-specific B-cells and they share a symbiotic relationship with them. The final stage of development occurs in the germinal center, where they acquire their unique phenotype [11] (Figure 1). These cells have a unique transcriptional profile and express markers e.g., PD-1, BCL6, CXCL13, CD10, ICOS, SAP, and CXCR5. Because PD1, BCL6, and TFH are all found on other T-cell subsets, no single marker should be used to define the TFH phenotype [10]. TFH cells are functionally plastic and hence, depending on the cytokine milieu, they can be reprogrammed into other lineages [12].
Autoimmune diseases have been found to be associated with dysregulation in TFH cells, which may cause uncontrolled stimulation of B-cells to produce antibodies and thereby cause damage [13,14]. This may further be implicated in lympho-proliferative neoplasms associated with chronic autoimmune diseases [10].
TFH cells are known to be the origin of both nodal and extranodal T-cell lymphomas like AITL, PTCL with TFH phenotype, follicular T-cell lymphoma and primary cutaneous CD4+ small to medium-sized lymphoproliferative lesions (Figure 2). Similar to non-neoplastic diseases, these lymphomas often show features of immune dysregulation. Apart from these, intratumoral CD4+ T-lymphocytes in follicular lymphomas have been shown to have a TFH phenotype and contribute to the lymphomagenesis by supporting B-cell survival [15]. CD4+ T-cells with a TFH phenotype have also been seen in the lymph node and peripheral blood in patients with chronic lymphocytic leukemia and are thought to support tumorigenesis by secreting IL21 [16].
Figure 2.
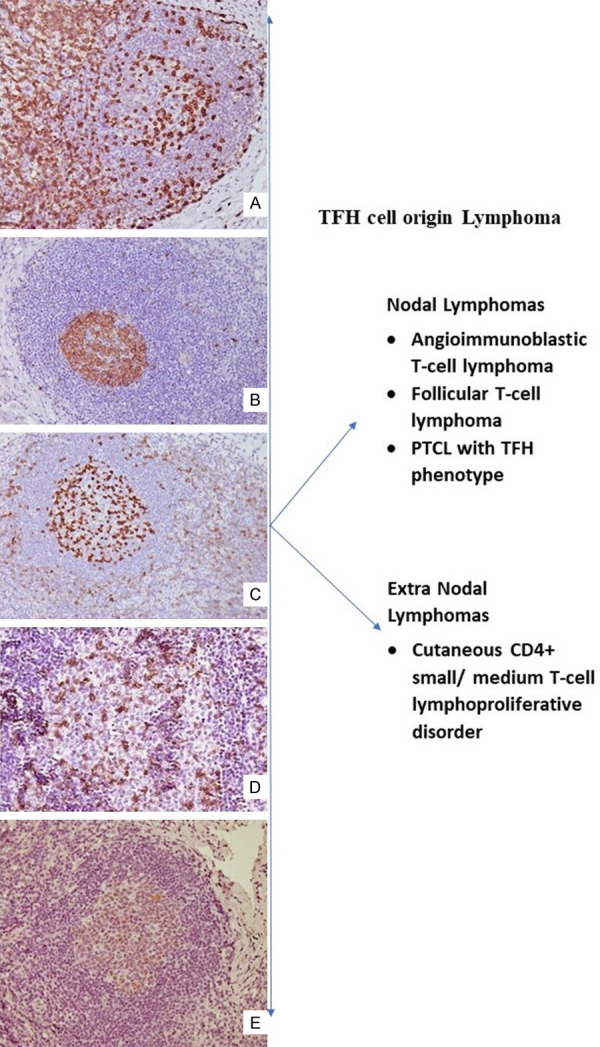
Characteristic immunophenotype of TFH: Microphotograph shows secondary follicle CD3 [A ×200] highlighting Follicular T helper (TFH) cell, CD10 [B ×200], PD1 [C ×200], ICOS [D ×200] and BCL6 [E ×200]. The right panel shows lymphoma arises from TFH cell.
Angioimmunoblastic T-cell lymphomas
Historic overview
With the initial recognition of the neoplasm as a non-neoplastic disease of dysregulation of the immune system, our conception of the neoplasm has come a long way. In the 1970s, it was considered to be angioimmunoblastic T-cell lymphadenopathy with dysproteinemia. With better understanding, it was added to the lymphoma group, and various clonal T-cell receptor (TCR) gene rearrangements were identified. With the awareness of the T-follicular helper cells, the plausible relationship with tumorigenesis was established in the years 2005-2007. However, the disease enjoys the status of a mystery, with never-ending puzzles.
Prevalence
AITL is the second most common PTCL subtype in the West and represents 1-3% of all NHLs and 20-30% of PTCL [1,17]. Many studies using new immunohistochemical and molecular markers have reclassified cases in the waste-basket of PTCL, NOS [18].
Etiology
The Epstein-Barr virus [EBV] and Human Herpesvirus 6 [HHV6] are thought to be associated with the pathogenesis and histological progression of AITL, and it has been observed that the onset of disease often mimics an infectious process [19]. Latency pattern II is usually manifested in T-cell lymphomas with viruses in the default program. The lytic cycle can stimulate the inflammatory microenvironment.
The relationship between EBV and AITL is debated; according to some authors, EBV acts as the neoplastic drive and causes rapid progression of the tumor, while others argue that the presence of EBV is due to a profound immunodeficiency due to AITL [19]. Though EBV is suspected to provide the neoplastic drive, the tumour cells are EBV negative. Also, a high viremia at the onset of the disease is proportionate to a poor response to therapy [20].
Various other molecular and genetic causes have been implicated, with the pathogenesis being multilineage and multistep. Clonal hematopoiesis caused by epigenetic modulators such as TET2 and DNMT3A results in CHIP lesion, and these mutations are found in all hematopoietic cells. IDH2 mutations can accumulate oncometabolites, causing a protumorigenic signal. RHOAG17V defines the lineage of the clonally expanded population of cells as the TFH phenotype. This is followed by multiple signaling pathways like MAPK, mTOR, and NF-kB along with the absence of inherent function of RHOA. All these accumulative effects lead to the clonal proliferation of the tumour cells, which is unregulated (Figure 3).
Figure 3.
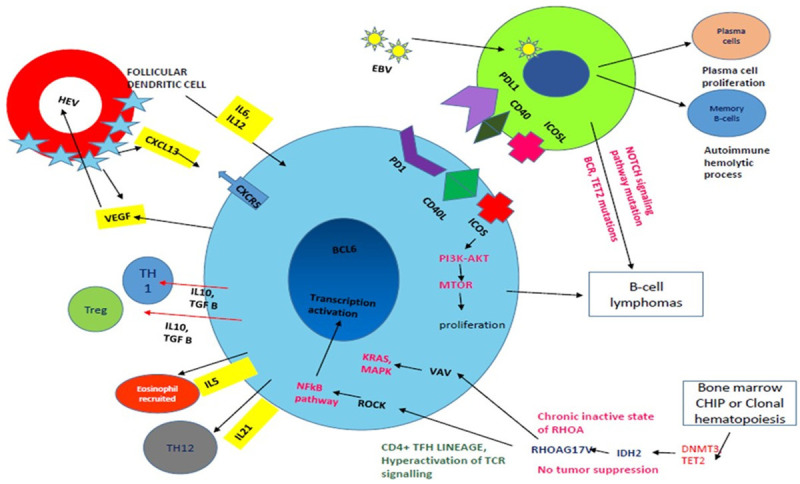
Pathogenesis of AITL: Art work shows TFH cells (Blue colour) interact with B-cells (Green colour) via PD1-PDL1, CD40L-CD40 and ICOSL-ICOS which leads to B-cell survival and proliferation. ICOS axis leads to TFH cell proliferation via p13AKT and mTOR pathway. Follicular dendritic cells secrete CXCL13, IL6, and IL2 creating conducive environment for proliferation of T-cells and in turn TFH cells produce VEGF which causes high endothelial venule proliferation (red colour circle). Eosinophils are recruited by IL5 secreted by TFH cells. EBV (small yellow circle with green spikes) infects the B-cells and further leads to T-cell proliferation and B-cell lymphomas rarely with gain of B-cell receptor, Tet2 mutations. Immunodeficiency is also caused by the virus. Increase in plasma cells and autoimmune haemolytic phenomenon can be associated with the B-cell proliferation associated. RHOA acts via the VAV-KRAS-MAPK and ROCK-NFkB pathway and affects the TFH cell differentiation and survival. Clonal haematopoiesis with various mutations can cause unregulated proliferation of the tumor cells.
Demographics
AITL affects middle-aged and elderly males, commonly with 50-65 years of age being the most common age of presentation. Though autoimmune conditions are seen in association with immune dysregulation, a male preponderance is seen worldwide [3,21].
Clinical presentation and serological profile
Primarily a nodal disease, extranodal sites like the spleen, liver, skin, and bone marrow can be involved. Patients usually present with lymphadenopathy, hepatosplenomegaly, and B-symptoms. Skin rash, pruritus, ascites, arthritis, pleural effusion can be seen with IgA and autoimmune mediated mechanisms. Though direct brain invasion is not seen, paraneoplastic neuropathy can be seen. With the disease process activating the B-cells, along with increased plasma cells, polyclonal hypergammaglobulinemia, and circulating immune complexes can be seen. Immunopositivity for rheumatoid factor and antismooth muscle antibodies can be found. An autoimmune phenomenon can also manifest as hemolytic anemia and cause peripheral blood spherocytosis and a positive direct Coomb’s test. Increased IgA can manifest as purpura, leukocytoclastic vasculitis and IgA-related renal disorders [nephropathy] [22,23]. Immunosuppression is also associated with disease, which may be due to dysfunctional T-cells along with the presence of tumour associated macrophages induced by IL10 and VEGF [18].
Staging
Ann Arbor staging was being used traditionally for the staging of lymphomas. Lugano staging, which indicates limited and advanced disease, modifies the criteria already being used for Ann Arbor. The Deauville five-point scale using fluorodeoxyglucose avidity is used for staging as well as for assessing treatment response. The baseline 18FDG SUVmax, along with extranodal site involvement, may serve as independent predictors of prognosis [24]. Bone marrow biopsies done for staging purposes showed nodular, interstitial, and diffuse infiltration of the tumour cells. Infiltration of AITL could be divided into three subgroups based on histomorphology: interstitial/micronodular with or without eosinophilia; diffuse involvement with eosinophilia; and patients with limited marrow involvement with eosinophilia [25].
Histomorphology
AITL is characterised by effacement of the lymph node architecture due to the presence of neoplastic T-cells. These are small to medium-sized atypical lymphoid cells with clear to pale cytoplasm. Increased vascularity is noted in the form of increased high endothelial venules [HEVs] and arborizing vessels in the paracortex. Normal venules should not be considered high endothelial venules and usually lack the specialised tall endothelial cells, thick basal lamina, and concentrically arranged reticular fibroblasts which are seen in HEVs [26]. With an inflammatory cytokine milieu in the tumour microenvironment, a polymorphous population of benign inflammatory cells like plasma cells, eosinophils, neutrophils, and histiocytes is noted.
On microscopy, three overlapping patterns are recognised [27]. In pattern 1, hyperplastic follicles are seen with the neoplastic cells surrounding the hyperplastic follicles with well-formed germinal centres. A well demarcated mantle cuff is often not seen [28]. In pattern 2, partial effacement of the architecture is seen, along with paracortical expansion and follicular remnants showing atretic and regressive changes. Pattern 3 is characterised by total effacement of the lymphoidal architecture. Histological evolution can be seen between the patterns, with a few cases showing a higher grade pattern on relapse or refractory disease [29] (Figure 4).
Figure 4.

Microphotograph shows: Diffuse effacement of architecture with paracortical expansion [A: H&E ×16] Presence of patent marginal sinus [B: H&E ×100] Intermediate sized atypical lymphoid cells having clear cytoplasm [C: H&E ×100] Polymorphous population with predominant eosinophil population [D: H&E x100] AITL with predominant lymphocyte population [E: H&E ×200] Polymorphous population with predominant plasma cells and Russel bodies [F: H&E ×100] Increased high endothelial venules [G: H&E ×100]. Eosinophilic hyaline material deposition [H: H&E ×100].
The patterns usually suggest a histological evolution and do not bear any consequences for the therapy. However, an increase in CD10 immunopositive tumour cells has been noted with an increasing pattern [19]. The patterns need to be assessed with caution since morphological mimics require differentiation, especially pattern 1. Pattern 1 AITL can closely resemble reactive hyperplasia, a progressive transformation of germinal centres. The presence of increased macrophages and decreased tangible body type in the germinal centre should alert the pathologist, requiring discrimination by an FDC marker. A characteristic irregular border with a sprouting pattern can be seen [30]. Pattern 2 can sometimes have a lollipop like pattern, resembling Castlemann disease [21]. The most common type pattern, 3, should be differentiated from Hodgkin’s lymphoma and T-cell rich large cell lymphoma. RS-like cells were seen in one case of PTCL, NOS, and their presence is said to have an association with PTCL with TFH phenotype [31].
Previous studies have indicated the relationship between AITL and DLBCL occurs simultaneously and either precedes the other. This could imply that bilineage tumorigenesis occurs as a result of transformation or by providing survival advantages to one another [32-34].
Immunophenotype
The neoplastic cells express most of the pan T-cell markers (CD3, CD2, and CD5) with less expression of CD7. Surface CD3 may be reduced by flow cytometry. Since the postulated normal counterpart and the cell of origin are said to be CD4-positive T-Follicular helper cells, the neoplastic cells express CD4 and the TFH markers. The TFH markers include PD-1, BCL6, CXCL13, CD10, ICOS, SAP, and CXCR5. Among these, PD1 and ICOS are more sensitive, while CXCL13 and CD10 are more specific. It is recommended to perform at least three of the markers, out of which two should be positive. Expression of one marker should never be considered diagnostic since TFH cells exhibit plasticity and TH1 and TH2 cells can show markers of TFH [35] (Figure 5).
Figure 5.

Immunophenotype of AITL: Microphotograph shows atypical lymphoid cells immunopositive for CD3 [A ×100] CD21 highlighting the expanded dendritic meshwork [B ×16] Loss of CD7 in the atypical lymphoid cells [C ×100] Characteristic immunophenotype of TFH atypical lymphoid cells PD1 [D ×100], Bcl6 [E ×100], ICOS [F ×100], CD10 [G ×100]. EBER ISH highlights the large atypical lymphoid cells [H ×100].
a. CXCL13 can be produced by TFH cells as well as follicular dendritic cells. It is known to be involved in B-cell maturation and plasma cell formation, and hence may be implicated in the autoimmune phenomenon. The marker can be positive in primary CNS lymphomas and gastric MALTomas. Immunopositivity can be seen as cytoplasmic with perinuclear dot.
Immunopositivity for CXCL13 was associated with mutant RHOA AITL in one of the study [36].
b. CD10 is a cell membrane metallopeptidase. Though a germinal center B-cell marker, its aberrant expression in the T-cells itself becomes a matter of concern.
c. ICOS [inducible co-stimulator, also known as CD278 is essential for T- and B-cell interaction for antigen processing and class switching. ICOS normally highlights the light zone, and increased expression can be seen upon activation. The tumor cells expressing ICOS usually co-express PD1, SAP and CD4 [37].
d. PD1 (programmed death 1, also known as CD279) controls the activation of T-lymphocytes. It can also be seen in ALCL, ATLL, and CLL etc. Usually PD1 serves as a better marker than CD10 or BCL6 which are also expressed on the B-cell and require double staining with CD4 for confirmation of neoplastic origin [38]. The expression of PD1, SAP and CXCL13 is considered malignant for the cells [39].
e. BCL6 (B-cell lymphoma 6 protein) is a zinc finger transcription factor and this also serves as a repressor for transcription. Expressed normally in the germinal centre, its expression in the interfollicular zone raises suspicion for an abnormal process. The interfollicular expansion of the BCL6 positive CD4+ T-cells can be due to the neoplastic process, which is aggravated by the outward migration of the BCL6+ CD4+ T-cells of the germinal centre [40].
f. SAP (signaling lymphocytic activation molecule-associated protein) is involved in humoral immune response.
g. CXCR5 It serves as the receptor for CXCL13. Their interaction can promote tumorigenesis via PI3K-AKT and MEK-ERK pathways [41].
h. The FDC meshwork show expansion and express CD21, CD23 and CD35. CD21 serves as a receptor for EBV or HHV8 entry and is believed to be more sensitive to detect the framework [42]. CD23 is a low affinity IgE receptor. CD21 and CD35 highlight both primary and secondary follicles; however, CD23 highlights the germinal centres only in the light zone [43]. The nodal germinal centre dark zone usually is highlighted less by these markers. Other lymphomas may also show their characteristic FDC pattern, and hence should be interpreted with caution [44]. FDC can also progress to follicular dendritic cell sarcoma [45].
i. The B immunoblasts can be EBV positive [80-95%] or negative, which can be demonstrated by in situ hybridization for EBV-encoded small RNA [EBER].
Controversial benign counterpart-AILD
Angioimmunoblastic lymphadenopathy with dysproteinemia (AILD), first described by Frizzera et al., is a rare entity with a debated existence due to the shared clinical, serological and histopathological features with AITL. Patients usually show syndromic presentation with fever, generalized lymphadenopathy, hepatosplenomegaly and rashes with increased susceptibility to infections [46]. Hemolytic anemia with a positive coombs test can be seen in AILD.
With a heterogeneous clinical course, mortality rates up to 60% have been reported. Both AILD and AITL have shown to have association with EBV whose role as a causative or an opportunistic infection is still pondered upon. These exhibit presence of EBV protein predominantly in the B-cells with a minor subset of T-cells expressing the same [47]. Due to the presence of clonal T-cell populations reported in studies [48] along with immunoglobulin chain rearrangements; AILD have been considered to be part of the spectrum of T-cell lymphomas, with possibly being the precursor to development of AITL.
Progression to DLBCL can also be seen [49,50]. Histomorphology matches an AITL with effacement of architecture with patent sinuses. Polymorphous cellular infiltrate is present in the node, consisting of mature lymphocytes, immunoblasts, macrophages, eosinophils, and plasma cells. Increased high endothelial venules are also noted, similar to AITL; however, cells with clear cytoplasm are not seen. The major population shows the immunophenotype of cytotoxic T-cells; however, the neoplastic cells are CD4 positive. Expansion of dendritic meshwork can be an accompanying feature. Diagnosis of AITL should be favoured when there is an abnormal T cell phenotype with loss of pan T-cell marker or expression of CD30 by numerous cells. Similarly, the presence of clonal populations or the presence of clonal cytogenetic abnormalities should raise the suspicion of lymphomas.
Genetic profile and mutational landscape
Clonal rearrangements in TR genes are seen in 75-90% of cases [51]. Trisomy of chromosomes 3, 5, and 21, gain of X, 22q, 19,11q13, and loss of 6q have also been reported. Next-generation sequencing has identified mutations in epigenetic modifiers such as IDH2 (20-30%), TET2 (50-80%), DNMT3A (20-30%), and small GTPase RHOA (60-70%) by next-generation sequencing [52]. IDH2 R172 mutations appear to be specific. Clonal rearrangements in TR genes are seen in 75-90% of cases [51]. Trisomy of chromosomes 3, 5 and 21, gain of X, 22q, 19,11q13, and loss of 6q have also been reported. Next-generation sequencing has identified mutations in epigenetic modifiers such as IDH2 (20-30%), TET2 (50-80%), DNMT3A (20-30%), and small GTPase RHOA (60-70%) by next-generation sequencing [52]. IDH2 R172 mutations appear to be specific for AITL. Other mutations such as FYN, PLG1, CD28, CTLA4-CD28 fusion and ITK-SYK fusion have been reported in some cases. A molecular classifier was developed, which was found to have a high rate of accuracy for AITL and ALK-ALCL [53]. Mutational heterogeneity leads to the understanding of two plausible pathways of lymphomagenesis, with classical being the most common [RHOA, DNMT3A, TET2, IDH2], the alternate involving VAV1 and an unknown pathway [54].
RHOA: It is the Ras Homolog Member A, located on chromosome 3p21.31. The gene has seven exons (GRCh38.p13 assembly) with exon 2 having the desired mutation in AITL. It covers 53854 bases. This gene has six transcripts [splice variants]: 2031 bp, 961 bp, 889 bp, 633 bp, 539 bp, and 388 bp. The RhoA protein can also have multiple isoforms. A member of the Rho family of small GTPases, this protein mainly works as a molecular switch in the signal transduction cascades. The active GTP bound state is important in cytoskeletal dynamics, mitogenesis, cell survival, arterial contractility, axogenesis, and phagocytosis [55]. This form is achieved by the action of guanine nucleotide exchange factors (GEFs) on the GDP bound inactive state, which through its action may act as a tumour suppressive protein. GTPase activating proteins (GAPs) increase the GTPase activity of Rho proteins. In benign conditions like spinal cord injury, vasospasm, hypertension, atherosclerosis, and myocardial hypertrophy, the Rho/Rho coiled-coil containing protein kinase (ROCK) signalling pathway has been found to play a substantial role [56]. The gene may also show overexpression, contributing to the neoplastic drive with tumour cell proliferation and metastatic potential.
With mutated RHOA, there is compromised binding and this mutant binding inhibits the binding of wild type as well. Because G1 to S phase cell cycle progression is reduced in the wild type, the mutated phenotype may increase tumour proliferation.
In AITL, the classical missense mutation of G17V (Glycine to valine) is noted in approximately 60% of cases worldwide. The mutation can be tested using molecular methods or commercially available antibodies. Low tumour burden due to polymorphism is associated with very low allele frequency [as low as 0.1 has been reported] and is difficult to detect using Sanger sequencing [57].
RHOA is over-expressed in a wide range of carcinomas of the breast, colon, lung, gastric, head and neck, bladder, testicular, and PTCLs. ATLL shows both activating (C16R, G14V) and inactivating mutations like G17E at different hotspots. The G17V mutant type serves as a dominant negative control for the enzyme.
Only a few studies have established the significance of mutant status on overall survival, with a survival advantage in one and no effect in another [58,59].
IDH2 (Isocitrate dehydrogenase 2): It is a cellular metabolism protein present in mitochondria that catalyzes the oxidative decarboxylation of isocitrate to 2-oxoglutarate. IDH2 mutations lead to the formation of an oncometabolite (2 hydroxy glutarate) that affects DNA and histone methylation. It has been implicated in several cancers, including sarcomas, hematolymphoid malignancies, colonic carcinomas, and glial tumors. IDH2 maps to chromosome 15q26.1 and covers 19433 bases. It is localised in the mitochondria. The transcript has 11 exons with three isoforms, with our locus of interest in exon 4. IDH2 mutations are highly specific for AITL, and arginine at position 172 can be missensed as threonine (R172T), lysine (R172K), glycine (R172G), or serine (R172S), with R172K being the most commonly observed [60]. IDH mutations improve prognosis in glial tumors; however, no significant difference has been observed in PTCL. The mutation can be tested using molecular methods or a commercially available antibody, which gives a granular cytoplasmic positivity suggestive of mitochondrial location. The combination of immunohistochemistry along with allele-specific PCR has a high sensitivity for detecting these mutations.
TET2: Ten eleven translocation, an iron-dependent oxygenase, is an epigenetic regulator and forms 5 hydroxymethylcytosine from 5 methycytosine. These mutations can be seen in T-cells, B-cells, and the other hematopoietic cells [61]. However, in T-cell lymphomas, these may point towards T-cell derivation. Though in AML, TET2 and IDH2 occur in a mutually exclusive form due to high levels of oncometabolites inhibiting the function of TET2, in AITL their co-occurrence has been seen. TET2 and RHOA are common associations in AITL. RHOA has been shown to occur prior to TET2 mutation and may play a role in lymphomagenesis [62]. The mutation can be a frameshift or nonsense type mutation. TET2 mutations can be seen in AITL (47%) and PTCL (38%) and are usually correlated with advanced disease stage with adverse prognostic factors like thrombocytopenia, higher IPI score and shorter PFS; however, OS is not affected [63].
DNMT3A: DNA methyltransferase is one of the commonly observed mutations and is usually seen with TET2. This also forms part of clonal hematopoiesis, and the hematopoietic cells usually show the presence of the mutation. In addition, this mutation can be acquired late during the disease process [64].
VAV1: G17V RHOA binds to VAV1 and phosphorylation of Tyr174 residue leads to activation of TCR signaling. VAV1 mutations along with VAV1-STAP2 fusion are seen which have not been observed in RHOA mutant cases [65].
ITK-SYK fusion: Initially thought to be a marker of PTCL with TFH phenotype; this fusion has also been described in AITL [66,67]. This fusion activates the T-cell signaling pathway after translocation between IL2 inducible T-cell kinase and spleen tyrosine kinase.
Prognostic scores
PTCL is an aggressive group of neoplasms; hence, prognostication can aid further patient management.
The IPI (International Prognostic Index) score is the most trusted and commonly used prognostic score for lymphomas. It is a scoring system based upon age, LDH levels, stage at presentation, ECOG performance status, and extranodal involvement with a maximum score of 5. Age-adjusted IPI is a modification of the above with patients considered only below 61 years of age and only considers three parameters: LDH levels, stage at presentation, and ECOG performance status. The PIT score, or Prognostic index for PTCL, considers LDH levels, age, ECOG performance status, and bone marrow involvement with a maximum score of 4. IgA levels, the presence of cytopenias like thrombocytopenia or anemia, and elevated white cell counts are also found to prognosticate patients with AITL [68].
Management
These are aggressive and require the administration of suitable chemotherapy. A CHOP regimen including cyclophosphamide, vincristine, prednisone, and doxorubicin is given. For patients >60 years of age, etoposide can be added. Due to immune dysregulation in AITL, immunosuppressants, cyclosporine, and thalidomide have also shown effects. Rituximab may also be added. An autologous hematopoietic stem cell transplant has proven to be useful.
The B-cells are also being studied to be targeted for therapeutic purposes. Various clinical trials are continuing, targeting different aspects of pathogenesis like VEGF, PD1 (immunotherapy), and ICOS (Figure 2).
Prognosis and overall survival
Male sex, mediastinal lymphadenopathy, and anaemia have adverse effects on the prognosis. A few studies have found that patients over the age of 60 with an increased total leukocyte count, elevated IgA levels, anemia, thrombocytopenia, and more than one extranodal site involvement fared worse than others [68]. The overall 5-year survival is less than compared to ALCL, which has a better response rate to therapy. According to one study, 32% each of AITL and PTCL had a 5-year overall survival rate, which was worse than compared to ALCL (70% for ALK+; 49% for ALK-) [3].
Peripheral T-cell lymphoma with TFH phenotype
These are a subset of PTCL having a TFH phenotype and some pathological features of AITL. However, they characteristically lack increased vascular pattern, follicular dendritic cell expansion, and a polymorphous infiltrate. This could imply a lack of microenvironment signature in the same [69]. Genetic alterations can be seen in TET2, RHOA, and DNMT3A. Due to similar phenotypic and genetic characteristics, these cases may represent a continuum with AITL, and some authors have suggested their constitution as a tumour cell-rich variant of AITL [70]. Factors like increased performance status, CRP levels, and BCL immunonegativity may be associated with a poor prognosis [71] (Figure 6).
Figure 6.

PTCL with TFH phenotype: Microphotograph shows effacement of lymph nodal architecture [H&E A ×100]. The cells are of intermediate size with mild nuclear irregularity [H&E B ×100]. The cells are immunopositive for CD3 [C ×100], BCL6 [D ×200] while negative for CD20 [E ×100]. CD23 shows loss of dendritic cell meshwork [F ×100].
Follicular T-cell lymphoma
It is a TFH phenotype neoplasm with a predominant follicular growth pattern and an absence of the characteristic features of AITL. A tumour of adults, it has predominantly nodal involvement with splenomegaly, skin rash, and B symptoms. The median survival is about 2 years. Unlike AITL, few cases have been shown to have limited involvement [69].
On microscopy, nodular or follicular proliferation of lymphoid cells is seen with effacement of the nodal architecture. The growth pattern can mimic follicular lymphoma or progressive transformation of germinal centres [72]. Reed Sternberg-like large cells can also be seen, which can be positive for CD30, CD15, and PAX5 but lack other B cell markers. On immunohistochemistry, it shows the TFH phenotype and EBV positivity can be seen in interfollicular B cells. These small B-cells show the presence of IgD (Figure 7).
Figure 7.

Follicular T-cell lymphoma: Microphotograph shows effacement of lymph nodal architecture by monomorphic nodules [H&E A ×100]. The nodule comprises small to intermediate size cells [H&E B ×100]. The cells are immunopositive for CD3 [C ×100], CD4 [E ×100], CD5 [G ×100], BCL6 [I ×100] while negative for CD20 [D ×100] and CD8 [F ×100]. CD23 shows mild expansion of dendritic cell meshwork [H ×100].
Translocation t [5,9] leading to ITK-SYK fusion can be seen, which was earlier supposed to be specific for follicular T-cell lymphoma; however, the fusion is seen to occur in AITL also [72]. Like other TFH cell-derived tumors, these tumours can also show TET2, DNMT3A, and RHOA mutations. However, with the limited data available, IDH2 mutations are not seen.
Primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder
These are clonal lymphoid proliferations of skin-homing CD4+ T-cells with TFH cell derivation and exhibit TR rearrangement. With a benign course and excellent prognosis, the term “lymphoma” is usually avoided. A disease of adults in their sixth decade, pediatric cases have also been reported. Patients commonly present with a slow-growing violaceous skin lesion on the head, neck, and extremities with an absence of patches like mycosis. A dense infiltration of the dermis by a small to intermediate sized T-cell lymphoid population along with histiocytes, plasma cells, and mature B-lymphocytes is identified. A recent study has highlighted two patterns of skin involvement, one of which is characterised by dense nodular or diffuse lymphoproliferation within the entire dermis and associated with the grenz zone, and the other is characterised by a subepidermal band like infiltrate in the superficial dermis or with peridnexal distribution and is associated with epidermotropism [73]. However, the prominence of epidermotropism should also raise the suspicion of mycosis. Angiocentricism and follicular dystrophy, along with angiocentrism and vascular changes, can also be seen. PD1, Bcl6, ICOS, and CXCL13 are commonly immunopositive in atypical cells, though they may show varied expression in different sized cells [73]. PD1-positive T-cells may show the formation of pseudo-rosettes. Differential staining for NFATc1 [Calcineurin/Nuclear Factor of activated T-cells-c1] can assist in distinguishing this LPD [nuclear] from mycosis [cytoplasmic] and pseudolymphomas [cytoplasmic] [74]. A low proliferation index of 20% is a must to rule out aggressive lymphomas [75]. Few authors have considered an aggressive form of the same as primary cutaneous helper T-cell lymphoma, which requires systemic therapy and shows systemic symptoms along with increased expression of CD10 [76].
Though the entity may share the morphological and immunophenotypic profile of AITL, the molecular profile of AITL is usually not manifested by this cutaneous disease.
Various morphological differentials like tumour stage of mycosis fungoides, primary cutaneous follicle centre lymphoma, cutaneous lymphoid hyperplasia, and primary cutaneous marginal zone lymphoma require careful evaluation of the biopsy before diagnosing this entity [77].
Numerous studies have indicated that with the new WHO classification, the increasing prevalence of TFH cell-derived neoplasms has been clarified. However, even with more defined entities, there still remains a grey area regarding the classification of PTCL. There is overlap in the morphology and immunophenotype, which increases the dependence on the newer markers and genetic profiles, which may not always be the solution to the problem. The use of all the TFH markers is not feasible, and even with the panel of sensitive and specific markers, some of the cases may be missed due to lack of confidence in a single marker’s immunopositivity and TFH-cell plasticity. Morphological overlap is a major hindrance. However, all these mimics have an aggressive course and are subjected to similar lines of therapy.
Molecular diagnostics, though essential, are limited. Development of less time-consuming and less cumbersome techniques for the detection of common mutations like RHOAG17V can improve diagnostics. However, all these tumors, especially AITL, are rich in background inflammatory cell infiltrate and hence require a sensitive test for detecting the low allele frequency.
Furthermore, apart from the cutaneous lymphoproliferative disorders, all the entities and PTCL, NOS have an aggressive course with a dismal outcome, which emphasizes the need for the look of targeted therapy.
The future is hopeful with better understanding, advancement of diagnostics, and the discovery of drugs targeting RHOA kinases.
Acknowledgements
SERB EEQ/2016/402, AIIMS-653 (Projects).
Disclosure of conflict of interest
None.
References
- 1.Bellei M, Chiattone CS, Luminari S, Pesce EA, Cabrera ME, de Souza CA, Gabús R, Zoppegno L, Zoppegno L, Milone J, Pavlovsky A, Connors JM, Foss FM, Horwitz SM, Liang R, Montoto S, Pileri SA, Polliack A, Vose JM, Zinzani PL, Zucca E, Federico M. T-cell lymphomas in South America and Europe. Rev Bras Hematol Hemoter. 2012;34:42–47. doi: 10.5581/1516-8484.20120013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pizzi M, Margolskee E, Inghirami G. Pathogenesis of peripheral T cell lymphoma. Annu Rev Pathol. 2018;13:293–320. doi: 10.1146/annurev-pathol-020117-043821. [DOI] [PubMed] [Google Scholar]
- 3.Vose J, Armitage J, Weisenburger D International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J. Clin. Oncol. 2008;26:4124–4130. doi: 10.1200/JCO.2008.16.4558. [DOI] [PubMed] [Google Scholar]
- 4.Savage KJ, Ferreri AJ, Zinzani PL, Pileri SA. Peripheral T-cell lymphoma--not otherwise specified. Crit Rev Oncol Hematol. 2011;79:321–329. doi: 10.1016/j.critrevonc.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 5.Rizvi MA, Evens AM, Tallman MS, Nelson BP, Rosen ST. T-cell non-Hodgkin lymphoma. Blood. 2006;107:1255–1264. doi: 10.1182/blood-2005-03-1306. [DOI] [PubMed] [Google Scholar]
- 6.Basha BM, Bryant SC, Rech KL, Feldman AL, Vrana JA, Shi M, Reed KA, King RL. Application of a 5 marker panel to the routine diagnosis of peripheral T-cell lymphoma with T-follicular helper phenotype. Am J Surg Pathol. 2019;43:1282–1290. doi: 10.1097/PAS.0000000000001315. [DOI] [PubMed] [Google Scholar]
- 7.Martin CH, Aifantis I, Scimone ML, von Andrian UH, Reizis B, von Boehmer H, Gounari F. Efficient thymic immigration of B220+ lymphoid-restricted bone marrow cells with T precursor potential. Nat Immunol. 2003;4:866–873. doi: 10.1038/ni965. [DOI] [PubMed] [Google Scholar]
- 8.Lone W, Alkhiniji A, Manikkam Umakanthan J, Iqbal J. Molecular insights into pathogenesis of peripheral T cell lymphoma: a review. Curr Hematol Malig Rep. 2018;13:318–328. doi: 10.1007/s11899-018-0460-z. [DOI] [PubMed] [Google Scholar]
- 9.Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, Förster R. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. 2000;192:1545–1552. doi: 10.1084/jem.192.11.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahearne MJ, Allchin RL, Fox CP, Wagner SD. Follicular helper T-cells: expanding roles in T-cell lymphoma and targets for treatment. Br J Haematol. 2014;166:326–335. doi: 10.1111/bjh.12941. [DOI] [PubMed] [Google Scholar]
- 11.Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41:529–542. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hegazy AN, Peine M, Helmstetter C, Panse I, Fröhlich A, Bergthaler A, Flatz L, Pinschewer DD, Radbruch A, Löhning M. Interferons direct Th2 cell reprogramming to generate a stable GATA-3(+)T-bet(+) cell subset with combined Th2 and Th1 cell functions. Immunity. 2010;32:116–128. doi: 10.1016/j.immuni.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Luo CM, Li Y, Liu WB, Feng HY, Wang HY, Huang X, Qiu L, Ouyang J. Expansion of circulating counterparts of follicular helper T cells in patients with myasthenia gravis. J Neuroimmunol. 2013;256:55–61. doi: 10.1016/j.jneuroim.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 14.Le Coz C, Joublin A, Pasquali JL, Korganow AS, Dumortier H, Monneaux F. Circulating TFH subset distribution is strongly affected in lupus patients with an active disease. PLoS One. 2013;8:e75319. doi: 10.1371/journal.pone.0075319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amé-Thomas P, Le Priol J, Yssel H, Caron G, Pangault C, Jean R, Martin N, Marafioti T, Gaulard P, Lamy T, Fest T, Semana G, Tarte K. Characterization of intratumoral follicular helper T cells in follicular lymphoma: role in the survival of malignant B cells. Leukemia. 2012;26:1053–1063. doi: 10.1038/leu.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahearne MJ, Willimott S, Piñon L, Kennedy DB, Miall F, Dyer MJ, Wagner SD. Enhancement of CD154/IL4 proliferation by the T follicular helper (Tfh) cytokine, IL21 and increased numbers of circulating cells resembling Tfh cells in chronic lymphocytic leukaemia. Br J Haematol. 2013;162:360–70. doi: 10.1111/bjh.12401. [DOI] [PubMed] [Google Scholar]
- 17.Rüdiger T, Weisenburger DD, Anderson JR, Armitage JO, Diebold J, MacLennan KA, Nathwani BN, Ullrich F, Müller-Hermelink HK Non-Hodgkin’s Lymphoma Classification Project. Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): results from the Non-Hodgkin’s Lymphoma Classification Project. Ann Oncol. 2002;13:140–149. doi: 10.1093/annonc/mdf033. [DOI] [PubMed] [Google Scholar]
- 18.Iqbal J, Wright G, Wang C, Rosenwald A, Gascoyne RD, Weisenburger DD, Greiner TC, Smith L, Guo S, Wilcox RA, Teh BT, Lim ST, Tan SY, Rimsza LM, Jaffe ES, Campo E, Martinez A, Delabie J, Braziel RM, Cook JR, Tubbs RR, Ott G, Geissinger E, Gaulard P, Piccaluga PP, Pileri SA, Au WY, Nakamura S, Seto M, Berger F, de Leval L, Connors JM, Armitage J, Vose J, Chan WC, Staudt LM Lymphoma Leukemia Molecular Profiling Project and the International Peripheral T-cell Lymphoma Project. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123:2915–2923. doi: 10.1182/blood-2013-11-536359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou YP, Attygalle AD, Chuang SS, Diss T, Ye HT, Liu HX, Hamoudi RA, Munson P, Bacon CM, Dogan A, Du MQ. Angioimmunoblastic T-cell lymphoma: histological progression associates with EBV and HHV6B viral load. Br J Haematol. 2007;138:44–53. doi: 10.1111/j.1365-2141.2007.06620.x. [DOI] [PubMed] [Google Scholar]
- 20.Delfau-Larue MH, de Leval L, Joly B, Plonquet A, Challine D, Parrens M, Delmer A, Salles G, Morschhauser F, Delarue R, Brice P, Bouabdallah R, Casasnovas O, Tilly H, Gaulard P, Haioun C. Targeting intratumoral B cells with rituximab in addition to CHOP in angioimmunoblastic T-cell lymphoma. A clinicobiological study of the GELA. Haematologica. 2012;97:1594–1602. doi: 10.3324/haematol.2011.061507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bal M, Gujral S, Gandhi J, Shet T, Epari S, Subramanian PG. Angioimmunoblastic T-Cell lymphoma: a critical analysis of clinical, morphologic and immunophenotypic features. Indian J Pathol Microbiol. 2010;53:640–645. doi: 10.4103/0377-4929.72010. [DOI] [PubMed] [Google Scholar]
- 22.Sugaya M, Nakamura K, Asahina A, Tamaki K. Leukocytoclastic vasculitis with IgA deposits in angioimmunoblastic T cell lymphoma. J Dermatol. 2001;28:32–37. doi: 10.1111/j.1346-8138.2001.tb00083.x. [DOI] [PubMed] [Google Scholar]
- 23.Togashi M, Wakui H, Kodama K, Kameoka Y, Komatsuda A, Nimura T, Ichinohasama R, Sawada K. Angioimmunoblastic T-cell lymphoma and membranous nephropathy: a still unreported association. Clin Exp Nephrol. 2010;14:288–293. doi: 10.1007/s10157-010-0266-3. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Yu WJ, Wu T, Xue YY, Zhang D, Xu HQ. The incremental prognostic value of baseline 18F-FDG PET/CT imaging in angioimmunoblastic T-cell lymphoma. Biomed Res Int. 2020;2020:4502489. doi: 10.1155/2020/4502489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerlach MM, Juskevicius D, Vela V, Dirnhofer S, Tzankov A. Bone marrow infiltration of angioimmunoblastic T-cell lymphoma: identification and prognostic impact of histologic patterns and diagnostic application of ancillary phenotypic and molecular analyses. Arch Pathol Lab Med. 2020;144:602–611. doi: 10.5858/arpa.2019-0007-OA. [DOI] [PubMed] [Google Scholar]
- 26.Hayasaka H, Taniguchi K, Fukai S, Miyasaka M. Neogenesis and development of the high endothelial venules that mediate lymphocyte trafficking. Cancer Sci. 2010;101:2302–2308. doi: 10.1111/j.1349-7006.2010.01687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attygalle A, Al-Jehani R, Diss TC, Munson P, Liu HX, Du MQ, Isaacson PG, Dogan A. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood. 2002;99:627–633. doi: 10.1182/blood.v99.2.627. [DOI] [PubMed] [Google Scholar]
- 28.Ree HJ, Kadin ME, Kikuchi M, Ko YH, Go JH, Suzumiya J, Kim DS. Angioimmunoblastic lymphoma (AILD-type T-cell lymphoma) with hyperplastic germinal centers. Am J Surg Pathol. 1998;22:643–655. doi: 10.1097/00000478-199806000-00001. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez-Justo M, Attygalle AD, Munson P, Roncador G, Marafioti T, Piris MA. Angioimmunoblastic T-cell lymphoma with hyperplastic germinal centres: a neoplasia with origin in the outer zone of the germinal centre? Clinicopathological and immunohistochemical study of 10 cases with follicular T-cell markers. Mod Pathol. 2009;22:753–761. doi: 10.1038/modpathol.2009.12. [DOI] [PubMed] [Google Scholar]
- 30.Warnke RA, Jones D, Hsi ED. Morphologic and immunophenotypic variants of nodal T-cell lymphomas and T-cell lymphoma mimics. Am J Clin Pathol. 2007;127:511–527. doi: 10.1309/QBLAMA321K9AD2XK. [DOI] [PubMed] [Google Scholar]
- 31.Eladl AE, Satou A, Elsayed AA, Suzuki Y, Kato S, Asano N, Nakamura S. Clinicopathological study of 30 cases of peripheral T-cell lymphoma with hodgkin and reed-sternberg-like B-cells from Japan. Am J Surg Pathol. 2017;41:506–516. doi: 10.1097/PAS.0000000000000813. [DOI] [PubMed] [Google Scholar]
- 32.Huang J, Zhang PH, Gao YH, Qiu LG. Sequential development of diffuse large B-cell lymphoma in a patient with angioimmunoblastic T-cell lymphoma. Diagn Cytopathol. 2012;40:346–351. doi: 10.1002/dc.21641. [DOI] [PubMed] [Google Scholar]
- 33.Hong RM, Sheng LX, Ouyang GF. Composite angioimmunoblastic T cell/diffuse large B-cell lymphoma treated with reduced-intensity conditioning HLA-haploidentical allo-HSCT: a case report and review of the literature. Int J Clin Exp Pathol. 2018;11:5473–5480. [PMC free article] [PubMed] [Google Scholar]
- 34.Wang YY, Xie BL, Chen Y, Huang ZQ, Tan H. Development of angioimmunoblastic T-cell lymphoma after treatment of diffuse large B-cell lymphoma: a case report and review of literature. Int J Clin Exp Pathol. 2014;7:3432–3438. [PMC free article] [PubMed] [Google Scholar]
- 35.Drieux F, Ruminy P, Abdel-Sater A, Lemonnier F, Viailly PJ, Fataccioli V, Marchand V, Bisig B, Letourneau A, Parrens M, Bossard C, Bruneau J, Dobay P, Veresezan L, Dupuy A, Pujals A, Robe C, Sako N, Copie-Bergman C, Delfau-Larue MH, Picquenot JM, Tilly H, Delarue R, Jardin F, de Leval L, Gaulard P. Defining signatures of peripheral T-cell lymphoma with a targeted 20-marker gene expression profiling assay. Haematologica. 2020;105:1582–1592. doi: 10.3324/haematol.2019.226647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manso R, González-Rincón J, Rodríguez-Justo M, Roncador G, Gómez S, Sánchez-Beato M, Piris MA, Rodríguez-Pinilla SM. Overlap at the molecular and immunohistochemical levels between angioimmunoblastic T-cell lymphoma and a subgroup of peripheral T-cell lymphomas without specific morphological features. Oncotarget. 2018;9:16124–16133. doi: 10.18632/oncotarget.24592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marafioti T, Paterson JC, Ballabio E, Chott A, Natkunam Y, Rodriguez-Justo M, Plonquet A, Rodriguez-Pinilla SM, Klapper W, Hansmann ML, Pileri SA, Isaacson PG, Stein H, Piris MA, Mason DY, Gaulard P. The inducible T-cell co-stimulator molecule is expressed on subsets of T cells and is a new marker of lymphomas of T follicular helper cell-derivation. Haematologica. 2010;95:432–439. doi: 10.3324/haematol.2009.010991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dorfman DM, Brown JA, Shahsafaei A, Freeman GJ. Programmed death-1 (PD-1) is a marker of germinal center-associated T cells and angioimmunoblastic T-cell lymphoma. Am J Surg Pathol. 2006;30:802–810. doi: 10.1097/01.pas.0000209855.28282.ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiba S, Sakata-Yanagimoto M. Advances in understanding of angioimmunoblastic T-cell lymphoma. Leukemia. 2020;34:2592–2606. doi: 10.1038/s41375-020-0990-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ree HJ, Kadin ME, Kikuchi M, Ko YH, Suzumiya J, Go JH. Bcl-6 expression in reactive follicular hyperplasia, follicular lymphoma, and angioimmunoblastic T-cell lymphoma with hyperplastic germinal centers: heterogeneity of intrafollicular T-cells and their altered distribution in the pathogenesis of angioimmunoblastic T-cell lymphoma. Hum Pathol. 1999;30:403–411. doi: 10.1016/s0046-8177(99)90115-6. [DOI] [PubMed] [Google Scholar]
- 41.Kazanietz MG, Durando M, Cooke M. CXCL13 and its receptor CXCR5 in cancer: inflammation, immune response, and beyond. Front Endocrinol (Lausanne) 2019;10:471. doi: 10.3389/fendo.2019.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Troxell ML, Schwartz EJ, van de Rijn M, Ross DT, Warnke RA, Higgins JP, Natkunam Y. Follicular dendritic cell immunohistochemical markers in angioimmunoblastic T-cell lymphoma. Appl Immunohistochem Mol Morphol. 2005;13:297–303. doi: 10.1097/01.pai.0000173053.45296.51. [DOI] [PubMed] [Google Scholar]
- 43.Kurshumliu F, Sadiku-Zehri F, Qerimi A, Vela Z, Jashari F, Bytyci S, Rashiti V, Sadiku S. Divergent immunohistochemical expression of CD21 and CD23 by follicular dendritic cells with increasing grade of follicular lymphoma. World J Surg Oncol. 2019;17:115. doi: 10.1186/s12957-019-1659-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carbone A, Gloghini A. Follicular dendritic cell pattern in early lymphomas involving follicles. Adv Anat Pathol. 2014;21:260–269. doi: 10.1097/PAP.0000000000000030. [DOI] [PubMed] [Google Scholar]
- 45.Benharroch D, Zekzer M, Nalbandyan K. Angioimmunoblastic T-cell lymphoma: a questionable association with follicular dendritic cell sarcoma. Case Rep Hematol. 2017;2017:9601094. doi: 10.1155/2017/9601094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frizzera G, Moran EM, Rappaport H. Angio-immunoblastic lymphadenopathy. Diagnosis and clinical course. Am J Med. 1975;59:803–818. doi: 10.1016/0002-9343(75)90466-0. [DOI] [PubMed] [Google Scholar]
- 47.Weiss LM, Jaffe ES, Liu XF, Chen YY, Shibata D, Medeiros LJ. Detection and localization of Epstein-Barr viral genomes in angioimmunoblastic lymphadenopathy and angioimmunoblastic lymphadenopathy-like lymphoma. Blood. 1992;79:1789–1795. [PubMed] [Google Scholar]
- 48.Jain S, Lone MR, Goswami A, Mandal T, Panda AK, Ramteke P, Srivastav T, Sharma MC, Gogia A, Sharma A, Bakhshi S, Mahapatra M, Kumar L, Mallick S. Lymphoma subtypes in India: a tertiary care center review. Clin Exp Med. 2021;21:315–321. doi: 10.1007/s10238-021-00683-2. [DOI] [PubMed] [Google Scholar]
- 49.O’Connor NT, Crick JA, Wainscoat JS, Gatter KC, Stein H, Falini B, Mason DY. Evidence for monoclonal T lymphocyte proliferation in angioimmunoblastic lymphadenopathy. J Clin Pathol. 1986;39:1229–1232. doi: 10.1136/jcp.39.11.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lipford EH, Smith HR, Pittaluga S, Jaffe ES, Steinberg AD, Cossman J. Clonality of angioimmunoblastic lymphadenopathy and implications for its evolution to malignant lymphoma. J Clin Invest. 1987;79:637–642. doi: 10.1172/JCI112860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Attygalle AD, Chuang SS, Diss TC, Du MQ, Isaacson PG, Dogan A. Distinguishing angioimmunoblastic T-cell lymphoma from peripheral T-cell lymphoma, unspecified, using morphology, immunophenotype and molecular genetics. Histopathology. 2007;50:498–508. doi: 10.1111/j.1365-2559.2007.02632.x. [DOI] [PubMed] [Google Scholar]
- 52.Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP, Parrens M, Martin A, Xerri L, Brousset P, Chan LC, Chan WC, Gaulard P, Mak TW. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119:1901–1903. doi: 10.1182/blood-2011-11-391748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Piccaluga PP, Agostinelli C, Califano A, Rossi M, Basso K, Zupo S, Went P, Klein U, Zinzani PL, Baccarani M, Dalla Favera R, Pileri SA. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. J Clin Invest. 2007;117:823–834. doi: 10.1172/JCI26833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Willemsen M, Abdul Hamid M, Winkens B, Zur Hausen A. Mutational heterogeneity of angioimmunoblastic T-cell lymphoma indicates distinct lymphomagenic pathways. Blood Cancer J. 2018;8:6. doi: 10.1038/s41408-017-0047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays. 2007;29:356–370. doi: 10.1002/bies.20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shimokawa H, Rashid M. Development of Rho-kinase inhibitors for cardiovascular medicine. Trends Pharmacol Sci. 2007;28:296–302. doi: 10.1016/j.tips.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 57.Nakamoto-Matsubara R, Sakata-Yanagimoto M, Enami T, Yoshida K, Yanagimoto S, Shiozawa Y, Nanmoku T, Satomi K, Muto H, Obara N, Kato T, Kurita N, Yokoyama Y, Izutsu K, Ota Y, Sanada M, Shimizu S, Komeno T, Sato Y, Ito T, Kitabayashi I, Takeuchi K, Nakamura N, Ogawa S, Chiba S. Detection of the G17V RHOA mutation in angioimmunoblastic T-cell lymphoma and related lymphomas using quantitative allele-specific PCR. PLoS One. 2014;9:e109714. doi: 10.1371/journal.pone.0109714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ondrejka SL, Grzywacz B, Bodo J, Makishima H, Polprasert C, Said JW, Przychodzen B, Maciejewski JP, Hsi ED. Angioimmunoblastic T-cell lymphomas with the RHOA p. Gly17Val mutation have classic clinical and pathologic features. Am J Surg Pathol. 2016;40:335–341. doi: 10.1097/PAS.0000000000000555. [DOI] [PubMed] [Google Scholar]
- 59.Butzmann A, Sridhar K, Jangam D, Kumar J, Sahoo MK, Shahmarvand N, Warnke R, Rangasamy E, Pinsky BA, Ohgami RS. A comprehensive analysis of RHOA mutation positive and negative angioimmunoblastic T-cell lymphomas by targeted deep sequencing, expression profiling and single cell digital image analysis. Int J Mol Med. 2020;46:1466–1476. doi: 10.3892/ijmm.2020.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dupuy A, Lemonnier F, Fataccioli V, Martin-Garcia N, Robe C, Pelletier R, Poullot E, Moktefi A, Mokhtari K, Rousselet MC, Traverse-Glehen A, Delarue R, Tournilhac O, Delfau-Larue MH, Haioun C, Ortonne N, Copie-Bergman C, de Leval L, Pujals A, Gaulard P. Multiple ways to detect IDH2 mutations in angioimmunoblastic T-cell lymphoma from immunohistochemistry to next-generation sequencing. J Mol Diagn. 2018;20:677–685. doi: 10.1016/j.jmoldx.2018.05.012. [DOI] [PubMed] [Google Scholar]
- 61.Schwartz FH, Cai Q, Fellmann E, Hartmann S, Mäyränpää MI, Karjalainen-Lindsberg ML, Sundström C, Scholtysik R, Hansmann ML, Küppers R. TET2 mutations in B cells of patients affected by angioimmunoblastic T-cell lymphoma. J Pathol. 2017;242:129–133. doi: 10.1002/path.4898. [DOI] [PubMed] [Google Scholar]
- 62.Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, Muto H, Tsuyama N, Sato-Otsubo A, Okuno Y, Sakata S, Kamada Y, Nakamoto-Matsubara R, Tran NB, Izutsu K, Sato Y, Ohta Y, Furuta J, Shimizu S, Komeno T, Sato Y, Ito T, Noguchi M, Noguchi E, Sanada M, Chiba K, Tanaka H, Suzukawa K, Nanmoku T, Hasegawa Y, Nureki O, Miyano S, Nakamura N, Takeuchi K, Ogawa S, Chiba S. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:171–175. doi: 10.1038/ng.2872. [DOI] [PubMed] [Google Scholar]
- 63.Lemonnier F, Couronné L, Parrens M, Jaïs JP, Travert M, Lamant L, Tournillac O, Rousset T, Fabiani B, Cairns RA, Mak T, Bastard C, Bernard OA, de Leval L, Gaulard P. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012;120:1466–1469. doi: 10.1182/blood-2012-02-408542. [DOI] [PubMed] [Google Scholar]
- 64.Odejide O, Weigert O, Lane AA, Toscano D, Lunning MA, Kopp N, Kim S, van Bodegom D, Bolla S, Schatz JH, Teruya-Feldstein J, Hochberg E, Louissaint A, Dorfman D, Stevenson K, Rodig SJ, Piccaluga PP, Jacobsen E, Pileri SA, Harris NL, Ferrero S, Inghirami G, Horwitz SM, Weinstock DM. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293–1296. doi: 10.1182/blood-2013-10-531509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fujisawa M, Sakata-Yanagimoto M, Nishizawa S, Komori D, Gershon P, Kiryu M, Tanzima S, Fukumoto K, Enami T, Muratani M, Yoshida K, Ogawa S, Matsue K, Nakamura N, Takeuchi K, Izutsu K, Fujimoto K, Teshima T, Miyoshi H, Gaulard P, Ohshima K, Chiba S. Activation of RHOA-VAV1 signaling in angioimmunoblastic T-cell lymphoma. Leukemia. 2018;32:694–702. doi: 10.1038/leu.2017.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Attygalle AD, Feldman AL, Dogan A. ITK/SYK translocation in angioimmunoblastic T-cell lymphoma. Am J Surg Pathol. 2013;37:1456–1457. doi: 10.1097/PAS.0b013e3182991415. [DOI] [PubMed] [Google Scholar]
- 67.Wang M, Zhang SW, Chuang SS, Ashton-Key M, Ochoa E, Bolli N, Vassiliou G, Gao ZF, Du MQ. Angioimmunoblastic T cell lymphoma: novel molecular insights by mutation profiling. Oncotarget. 2017;8:17763–17770. doi: 10.18632/oncotarget.14846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tokunaga T, Shimada K, Yamamoto K, Chihara D, Ichihashi T, Oshima R, Tanimoto M, Iwasaki T, Isoda A, Sakai A, Kobayashi H, Kitamura K, Matsue K, Taniwaki M, Tamashima S, Saburi Y, Masunari T, Naoe T, Nakamura S, Kinoshita T. Retrospective analysis of prognostic factors for angioimmunoblastic T-cell lymphoma: a multicenter cooperative study in Japan. Blood. 2012;119:2837–2843. doi: 10.1182/blood-2011-08-374371. [DOI] [PubMed] [Google Scholar]
- 69.Attygalle AD. Nodal T-cell lymphomas with a T-follicular helper cell phenotype. Diagnostic Histopathology. 2018;24:227–236. [Google Scholar]
- 70.Attygalle AD, Cabeçadas J, Gaulard P, Jaffe ES, de Jong D, Ko YH, Said J, Klapper W. Peripheral T-cell and NK-cell lymphomas and their mimics; taking a step forward - report on the lymphoma workshop of the XVIth meeting of the European Association for Haematopathology and the Society for Hematopathology. Histopathology. 2014;64:171–199. doi: 10.1111/his.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Suzuki T, Miyoshi H, Yanagida E, Kawamoto K, Yamada K, Takeuchi M, Ohshima K. Clinicopathological differences of nodal PTCL with TFH phenotype from AITL and PTCL, NOS, and detection of prognostic marker of nodal PTCL with TFH phenotype. 15th International conference on Malignant Lymphoma, Lugano. Hematol Oncol. 2019:276–277. [Google Scholar]
- 72.Huang YL, Moreau A, Dupuis J, Streubel B, Petit B, Le Gouill S, Martin-Garcia N, Copie-Bergman C, Gaillard F, Qubaja M, Fabiani B, Roncador G, Haioun C, Delfau-Larue MH, Marafioti T, Chott A, Gaulard P. Peripheral T-cell lymphomas with a follicular growth pattern are derived from follicular helper T cells (TFH) and may show overlapping features with angioimmunoblastic T-cell lymphomas. Am J Surg Pathol. 2009;33:682–690. doi: 10.1097/PAS.0b013e3181971591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beltzung F, Ortonne N, Pelletier L, Beylot-Barry M, Ingen-Housz-Oro S, Franck F, Pereira B, Godfraind C, Delfau MH, D’Incan M, Vergier B. Primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorders: a clinical, pathologic, and molecular study of 60 cases presenting with a single lesion: a multicenter study of the French Cutaneous Lymphoma Study Group. Am J Surg Pathol. 2020;44:862–872. doi: 10.1097/PAS.0000000000001470. [DOI] [PubMed] [Google Scholar]
- 74.Magro CM, Momtahen S. Differential NFATc1 expression in primary cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma and other forms of cutaneous T-cell Lymphoma and pseudolymphoma. Am J Dermatopathol. 2017;39:95–103. doi: 10.1097/DAD.0000000000000597. [DOI] [PubMed] [Google Scholar]
- 75.Surmanowicz P, Doherty S, Sivanand A, Parvinnejad N, Deschenes J, Schneider M, Hardin J, Gniadecki R. The clinical spectrum of primary cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoproliferative disorder: an updated systematic literature review and case series. Dermatology. 2021;237:618–628. doi: 10.1159/000511473. [DOI] [PubMed] [Google Scholar]
- 76.Gru AA, Wick MR, Eid M. Primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder-clinical and histopathologic features, differential diagnosis, and treatment. Semin Cutan Med Surg. 2018;37:39–48. doi: 10.12788/j.sder.2018.006. [DOI] [PubMed] [Google Scholar]
- 77.Salah E. Primary cutaneous CD4+ small/medium pleomorphic T-cell lymphoproliferative disorder: where do we stand? A systematic review. J Dtsch Dermatol Ges. 2019;17:123–136. doi: 10.1111/ddg.13691. [DOI] [PubMed] [Google Scholar]