

Abstract
Background/Aim: Hydration and hydroxyurea (HU) can modify sickle cell disease (SCD) severity. Optimal nutrition and L-glutamine (Gln) may provide further amelioration.
Patients and Methods: Reviews of medical records and nutrition surveys were used to investigate severity of pediatric patients with SCD in relation to nutrition, growth, hematologic parameters, and disease-modifying agents.
Results: Among 25 females and 25 males (9.1±7 years), beta-globin genotypes were: HbSS/Sβ0, 60%; HbSC, 32%; HbSβ+, 8%. The mean number of annual pain crises (APC) was 0.97±1.1. APCs increased ≥2-fold as HbF dropped to <10% with age. Proper hydration and nutrition correlated with younger ages and fewer APCs. Height and weight Z-scores were ≤–1SD in 20% of 35 surveyed patients (12±7.8 years), who had more APCs (2.5±2.5 vs. 1±1.3, p=0.03). Prealbumin levels were overall low. Twenty-two of 28 patients on HU reported ≥90% adherence – with higher mean corpuscular volume (92±9.6 vs. 74±10 f/l, p<0.01). Seventy percent of Gln prescriptions were filled. Compliance over 23 months was ≥70% in 12 patients, including 2 on chronic transfusion. Of 10 evaluable patients, 6 (8.8±2.2 years) had fewer APCs with Gln (mean 0.2 vs. 0.9, p=0.016), with increasing prealbumin levels (14.1 to 15.8 mg/dl, p=0.1).
Conclusion: Younger, and well-nourished, well-hydrated patients have a milder clinic course. Disease severity was the worse in undernourished teenagers with suboptimal compliance. L-Glutamine with prealbumin monitoring should be considered for further evaluation in pediatric SCD.
Keywords: Sickle cell disease, L-glutamine, nutrition
Sickle cell disease (SCD) affects millions of people worldwide, including more than 100,000 Americans (1). SCD is caused by occurrence of two mutated β-globin genes (a sickle allele combined with either a sickle, hemoglobin C, or β-thalassemia allele), which disrupt the architecture of red blood cells (RBCs) due to hemoglobin S (2). Sickle-shaped RBCs cause adhesive vaso-occlusion coupled with endothelial damage and hemolysis (3), which produce acute and chronic complications (4), including painful crises (5).
Clinical control aimed at amelioration of SCD severity is challenging but achievable in many cases. Adequate hydration and nutrition, and medications - both established and novel agents - can contribute to disease management (2,6-8).
One of the undesirable outcomes in SCD is poor growth (9), which can be worsened by nutritional deficiencies (10). The role of optimized nutrition in SCD must be emphasized as a way to meet the increased energy expenditures and higher metabolic demands associated with chronic diseases (11). Moreover, certain nutritional compounds may have therapeutic effects. For example, the anti-oxidative property of L-glutamine (Gln) is believed to be the basis for its action against RBC sickling (2).
In 2018, after a Phase 3 study, a purified form of Gln called Endari® was added to the armamentarium of FDA-approved, disease-modifying agents for SCD (2,12) - in addition to hydroxyurea (HU) (13,14). Both agents are given orally, HU daily and Gln twice a day, and patient compliance can significantly affect outcomes. Due to the increased metabolic demands of chronic illness and oxidative stress, optimal nutrition and use of Gln may contribute to disease control. Thus, we hypothesized that better nutrition and Gln adherence may contribute to reduction of pain crises in SCD.
Pediatric patients with SCD typically are treated at comprehensive sub-specialty centers, for which our center includes pediatric hematologists, nurses, a dietician, and a clinical social worker. Clinic patients are seen for the full spectrum of preventive and health maintenance services, including well child care, prophylactic penicillin until 5 years of age, immunizations, and annual transcranial doppler examinations. Growth and development are assessed carefully, and patients are referred to specialists for further evaluation and management as needed. Evidence-based guidelines are being implemented for management of SCD (3), including guidance aimed at cardiopulmonary and kidney complications (15). Patients in pain crisis are transferred to the emergency department for acute management and/or inpatient admission.
HU therapy is considered and/or offered to nearly all infants (9 months of age and older), children, and adolescents. Gln is offered to patients 5 years of age and older. The importance of compliance is emphasized for HU, Gln, and other medications, along with thorough reconciliation and refills as needed.
We retrospectively reviewed hydration and nutritional status, clinical disease severity, and laboratory parameters of pediatric patients with SCD at our center. We assessed for potential associations among nutritional status, clinical measures, and laboratory results, including hematologic values [fetal hemoglobin (HbF) percentage, mean corpuscular volume (MCV), and platelet counts]. Medication compliance was included in the assessments. Finally, we describe our early experience with L-glutamine in pediatric patients.
Patients and Methods
An IRB-approved retrospective chart review and data extraction were conducted, along with voluntary nutritional surveys in pediatric patients with SCD. All patients with SCD aged 1 year or older who were actively and continuously treated at our institution within the 3 years prior to study were included (our clinic typically treats patients until 21 years old – few patients were 21 years old 3 years prior, who turned up to 23.9 at the time of data analysis). Patient demographics, physical growth parameters (with percentiles and Z scores), and clinical and laboratory data were obtained from electronic medical records (EMR) of regularly scheduled clinic visits, emergency department visits, and inpatient hospitalizations. Clinical and laboratory data were limited to those in our current EMR (from October 2014). Data extraction was through 2020, then statistical analyses were performed.
Earlier in study, patients were voluntarily surveyed with questionnaires to assess hydration, nutrition, and diet histories. Only 35 of 50 patients/families volunteered for this survey. Data collected included: amounts of water and liquid intake, number of meals per day and meal recalls. Caloric intake was estimated from meal recalls. The number of calories consumed per day was divided by the number of calories required for the patient, according to height, weight, and age (16). These calorie measurements were used to group patients based upon their percent of caloric consumption relative to their caloric need. A 100% cutoff was used to dichotomize patients into properly nourished versus poorly nourished groups. Similarly, 8-ounce cup estimates were used to assess proper hydration (100% of required daily maintenance fluids). In summary, patients were considered to have appropriate overall hydration and nutrition, if at least 100% of the recommended daily fluid and caloric intakes were reported, respectively. (8) Height and weight Z-scores were used to evaluate growth.
Emergency department visits and hospitalizations were used to determine the annual rate of pain crises (APC=number of pain crises requiring emergency department visits or hospitalizations divided by duration of follow-up in years). The APC value was used to dichotomize the clinical severities of the disease courses: <1 for mild and ³1 for moderate/severe.
Further evaluations were carried out for associations between disease severity and clinical or laboratory variables representing nutrition, growth, hematologic parameters [such as fetal hemoglobin (HbF) and mean MCV], and compliance with treatment (HU and/or Gln).
Laboratory values. For fetal hemoglobin, the latest available values were used, typically within the last year of the study. Low fetal hemoglobin referred to levels less than 10% in this study (17). Complete blood count (CBC) parameters for each patient were extracted from within the last year before data analyses and were used to calculate group averages – including MCV. All available prealbumin data collected during clinic visits within the last 3 years were used for analyzing averages per patient groups and trends. Age-specific prealbumin reference values were used in analyses of prealbumin data. Utilized laboratory values were random as a result of the retrospective character of this study and were not separated to baseline versus acute crises; however many were collected during routine clinical visits.
HU and Gln data were extracted from electronic prescriptions. Amounts of Gln dispensed were confirmed with the pharmacy. Reported compliance was quantified based on patient or parent estimations of percent doses missed versus taken, for both HU and Gln.
Statistical analysis. For categorical variables, Fisher exact tests were used to compare outcome variables including patient characteristics among groups. For continuous variables, a two sample Student t-test was used. Age was used as a continuous variable for examination of associations among fetal hemoglobin, disease severity, and prealbumin. Descriptive statistics, standard deviations and standard errors of the mean (SEM, in Figure 1) were also computed for the parameters of interest. A flowchart for patients on Gln was generated based on the descriptive statistics. Statistical tests were performed using Excel and p-values <0.05 were considered statistically significant. Graph Pad Prism (GraphPad Software, San Diego, CA, USA) was used to generate the majority of the figures.
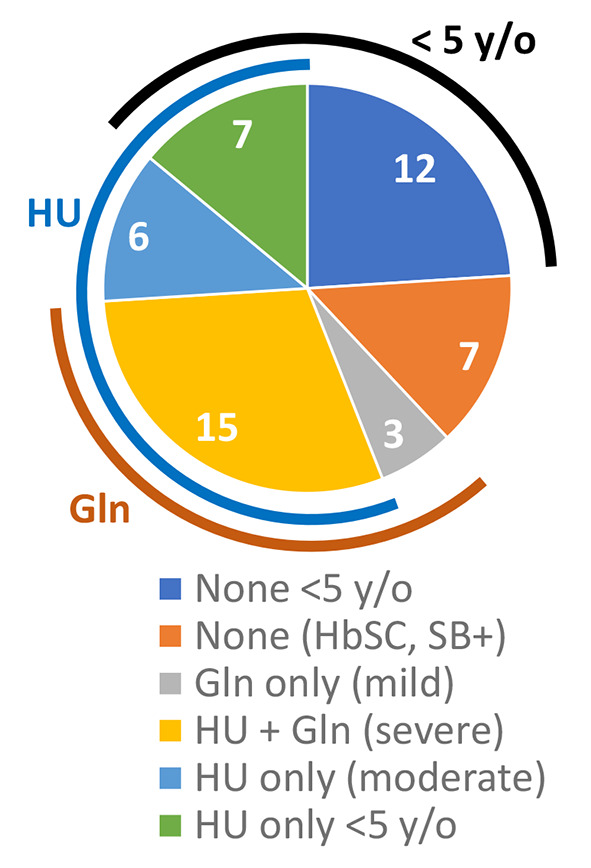
Figure 1. Hydroxyurea and L-glutamine in different age groups. Patients were grouped based on age (<5 years; 5 or older) and whether they were on disease-modifying agent(s). Nineteen patients with mild disease were not on any of the agents: 7 had HbSC or Sβ+, and 12 were under 5 years of age. Most of the patients on treatment with both agents had a severe phenotype. The remaining patients had a mild to moderate phenotype and were on only one agent.
Results
Patient characteristics. Our cohort included 50 patients with SCD (25 females and 25 males), with an average age of 9.1±7 years (range=1-23.9 years). The cohort was 88% African American, 10% Hispanic, and 2% mixed. There were 60% HbSS or Sβ0 (n=30), 32% HbSC (n=16), and 8% HbSβ+ (n=4) individuals in the cohort. As shown in Figure 1, 56% of our patients were on HU (n=28), the majority being HbSS or Sβ0, while others had HbSC disease. Patient demographics, clinical severity, laboratory values, medication use, and nutritional status are summarized in Table I. Statistical comparisons with the Student’s t-test between groups with mild versus severe disease showed significantly younger ages associated with APC <1. As expected, higher proportions of patients with severe disease had HbSS or Sβ0 and were more likely to be on HU (by Fisher exact test, Table I). Two patients were on chronic transfusions and were excluded from these analyses.
Table I. Patient characteristics.

APC: Annual pain crises; MCV: mean corpuscular volume; SD: standard deviation.
Nutritional status and hydration. The average BMI percentile was 56.4±32 (mean±SDEV). Based on the Z-scores, 28% of the heights were at least one standard deviation below the average, while 24% of the patients had weights at least one standard deviation below the average, indicating stunted growth. Only 16% of patients had a BMI one standard deviation below the average.
The percentage of patients with appropriate caloric intakes was also estimated. More than 70% of 35 surveyed patients (n=25) consumed 100% or more of their required fluid intake (i.e., were appropriately or well hydrated), whereas only 43% had 100% or more of their required caloric intake (i.e., were appropriately or well nourished). We then determined average (±SDEV) APC values for 3 groups: Group 1: 0.87±1.1 for well hydrated/well nourished (n=14, average age 3.1±2.3 years). Group 2: 1.3±2 for well hydrated/poorly nourished (n=11, average age 11.6±6.3 years). Group 3: 2.03±1.9 for poorly hydrated/poorly nourished (n=9, average age 13.4±5.2 years). One 3.7-year-old patient with an APC of 0.9 had proper nutrition but poor hydration.
There were no correlations between caloric intake and BMI/Z-scores versus disease severity for the cohort of 50 patients. However, 20% of surveyed patients (average age 12±7.8 years) had Z-scores less than -1 SD for both height and weight during the survey. Their mean APC value was significantly higher, compared to APC values for the remaining surveyed patients (2.5±2.5 vs. 1±1.3, p=0.03 by t-test).
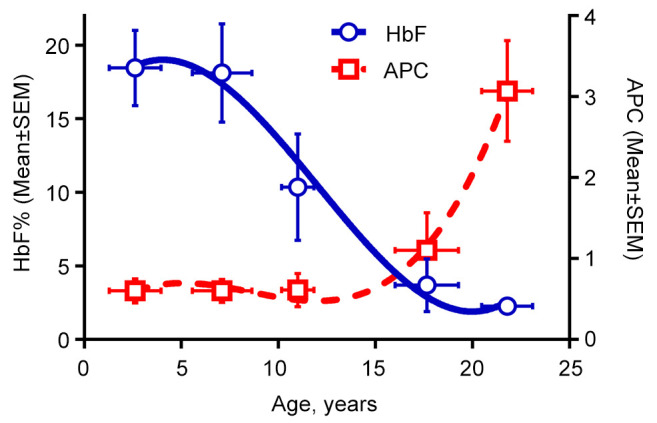
Associations between age, Hb F, disease severity, and prealbumin. The average number of annual pain crises requiring hospital visits or admissions was 0.97±1.14 in the cohort. HbF expectedly dropped with age. As shown in Figure 2, HbF levels <10% in teenage and older patients were associated with APC values up to ≥2-fold higher, compared to younger patients (despite similar proportions of HbSS/Sβ0).
Figure 2. Age, fetal hemoglobin, and pain crises. Relationships between the age groups, mean HbF, and APC rate: at about 10-12 years of age and older, HbF generally drops to under 10%, which coincides with significantly increased APC. Numbers of patients from left to right in different age groups were: n=19 (1-4.9 years); n=12 (5-9.9 years); n=6 (10-14.9 years), n=7 (15-19.9 years), and n=6 (≥20 years). SEM: Standard error of mean; APC: annual pain crisis; HbF: fetal hemoglobin.
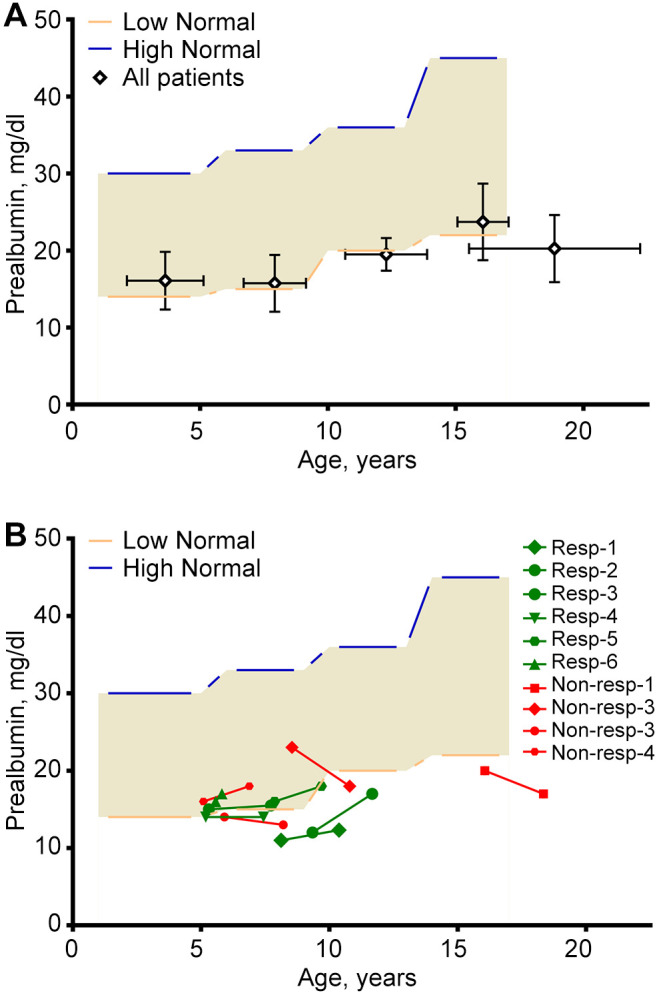
Prealbumin levels trended to be low, especially in older patients (Figure 3A). However more than half of patients on Gln showed an increase in Prealbumin levels and also corresponded to a decrease in APC (Figure 3B).
Figure 3. Prealbumin versus age. Light brown areas represent reference ranges for different age groups: 1-5, 6-9, 10-13, and 14-17 years. (A) Mean (±SD) values of prealbumin in corresponding age groups. Numbers of prealbumin samples in different age groups, from left to right: n=53, n=38, n=2, n=36, and n=28 (for >17 years). (B) Paired prealbumin levels (0-6 mo) before and (in average 23 months) after Gln in 10 patients. Up-sloping green lines represent patients with increased prealbumin and decreased APC values (i.e., L-glutamine responders). Red lines represent prealbumin levels for non-responding patients (i.e., their APCs did not decrease after Gln). APC: Annual pain crisis.
Hydroxyurea and L-glutamine adherence. Twenty-eight patients (56%) were on HU (22/28 with HbSS/Sβ0; age 10.8±7.4 years). Most of the remaining patients with milder disease, who were not on HU, had HbSC or Sβ+ (Figure 1). Twenty-two patients reported good hydroxyurea adherence, typically ≥90%, which was associated with higher MCVs (92±9.6 vs. 74±10 f/l; p<0.01), a trend toward higher HbF levels, and younger ages (15 vs. 9% and 9.7±7 vs. 14±7.7 years, respectively; p=NS; Figure 4). Hydroxyurea adherence data was obtained during routine visits per patient reporting.
Figure 4. HU adherence versus MCV, HbF, age, and APC. Corresponding panels A-D show the difference in MCV, HbF, age and APC based on HU adherence (50-80% adherence vs. 90-100% adherence). Statistically significant difference (based on t-test) between the HU adherence groups exists only for MCV, which is higher with better hydroxyurea adherence. Box plots with error bars represent means and standard deviations. APC: Annual pain crisis; HbF: fetal hemoglobin; MCV: mean corpuscular volume; HU: hydroxyurea.

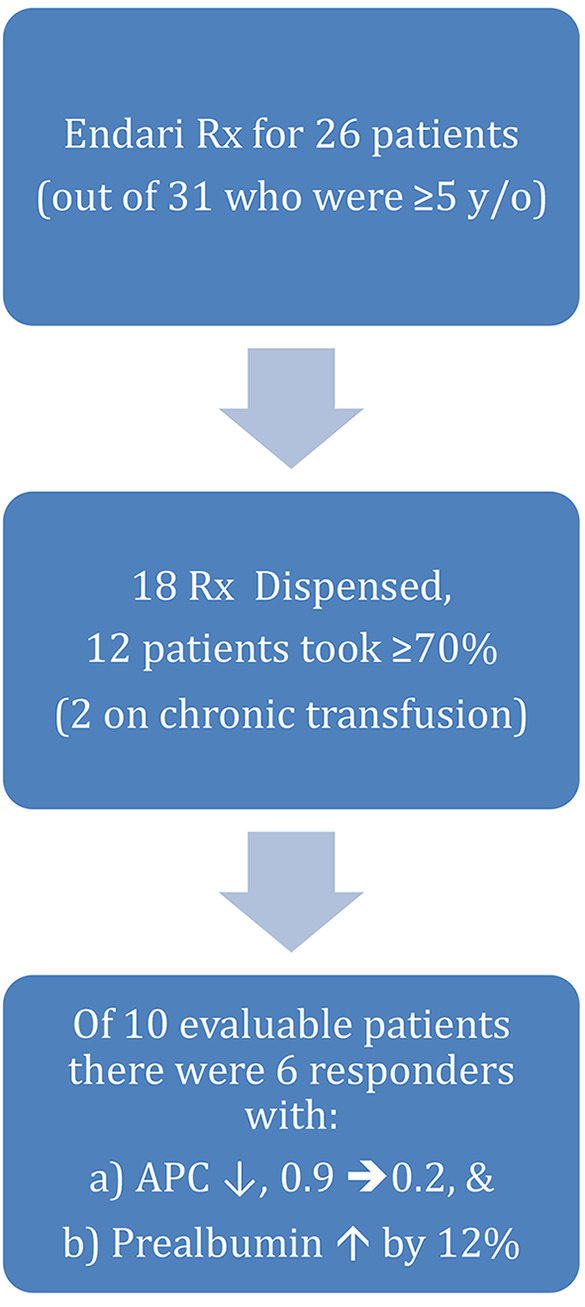
The majority (83%) of our patients 5 years of age or older were prescribed Gln, and 70% of the prescriptions were dispensed (Figure 5). Data regarding dispensing rate was obtained by the specialty pharmacy. Some refused Gln, due to abdominal pain or taste, and a quarter of the patients reported Gln intakes of <50%, while on both HU and Gln. Overall Gln compliance in 23 months was ≥70% in 12 patients. Gln adherence data was also obtained during routine visits per patient or parental reporting. Patients on chronic transfusion were excluded from further analyses. Of the 10 evaluable patients, 6 (8.8±2.2 years) had a significantly lower APC with Gln (mean APC 0.9, compared to 0.2; p=0.016 by paired t-test), coupled with a 12% rise in prealbumin (14.1 to 15.8 mg/dl; p=0.1; Figure 4B) within in average 23-month use (Figure 3B). The majority of the patients who were prescribed Gln were already on HU.
Figure 5. Flowchart showing L-Glutamine prescriptions, dispensing, intake, and effect.
Discussion
Complex pathophysiology makes it challenging to control SCD severity, which can be influenced by hydration and disease-modifying agents, such as HU (18). Proper hydration, nutrition, and medication compliance all contribute to amelioration of SCD morbidity. The small size of this study and patients’ subgroups do not allow separation of confounding variables and multivariate evaluation. For example, factors associated with increasing age (worse nutrition, less sleep, worse medication adherence, etc.) will also be associated with increased acute care utilization and worse disease control. In contrary, well-hydrated patients may have a milder clinical course because they are also younger. Analyzing the impact of HU within the groups with good nutrition vs. poor nutrition would also be more feasible for larger cohorts. Nevertheless, increasing age and decreasing HbF over time remain the main factors driving the severity of SCD in this study. Proper hydration alone without proper nutrition did not ameliorate SCD severity. Most of our patients met their hydration requirement. However, only 43% consumed 100% of their caloric requirement. Moreover, about a quarter of our patients had stunted growth, as demonstrated by heights and weights.
Twenty percent of the surveyed patients in this study had lower weights and heights than the average values for their age groups, as shown by Z-scores, despite an adequate caloric intake. In addition, patients with both poor hydration and nutrition tended to be older. We believe these results show the interplay between disease management and proper nutritional assessment when evaluating patients with SCD. Inadequate nutritional intake, micronutrient deficiency, endocrine dysfunction, and hyper-metabolism have been suggested as possible factors that may contribute to growth failure in children with SCD (19). Patients with SCD have an increase in energy expenditure and a decrease in protein synthesis, due to the chronic disease state, which further impedes growth (6). Our data supports emphasizing the importance of nutritional assessment for patients with SCD. This will help ensure dietary adequacy, which often worsens with age, especially during adolescence (20).
Our analyses of nutritional and hydration status were limited to only two-thirds of our pediatric SCD population. Specifically, we were able to interview 35 out of 50 patients, mostly due to time and patient availability to complete the questionnaire. Limitations also include meal recall bias and use of a sedentary parameter when calculating caloric requirement, thus underestimating the caloric needs of active patients. Due to our small patient cohort we included all genotypes of sickle cell disease in our analysis of nutrition and hydration, though the metabolic needs and lab values of patients with different genotypes may slightly differ (21). For instance, MCV can be lower for HbSC disease and patients with thalassemia co-inheritance may not achieve an MCV of >90 despite excellent adherence.
Endari (L-glutamine) received FDA approval in 2017 as a disease modifier for SCD after a phase 3 trial showed a reduction in vaso-occlusive crises in patients taking the medication over a 48-week period (2). It is thought that Gln raises the proportion of the reduced form of nicotinamide adenine dinucleotides (NAD) in RBCs, thus reducing oxidative stress (22). In our study we assessed effects of Gln in relation to disease severity and overall nutrition. Though the analyses of disease severity, pain crises, hospitalizations, and lab values in the Gln group were limited to small numbers of patients and short follow-up durations, there was a small group within the cohort of patients taking Gln that had a significant reduction in annual pain crises, with prealbumin levels trending up. The reduction of pain crises supports the findings of the initial phase 3 trial of Gln. Gln has also been shown to decrease resting energy expenditure in children and adolescents with SCD, perhaps also contributing to its overall effect on nutritional status (23). Another limitation was our medication adherence data was obtained by patient reporting and thus was subject to recall bias. We did have dispensing data from our local specialty pharmacy to help support patients’ claims of adherence, however medication possession ration (MPR) can be considered in larger prospective studies as suggested previously (24). Absolute neutrophil count in addition to MCV can also be used for drug titration.
A recent publication showed a correlation between SCD severity and albumin levels and suggested the use of albumin as a biomarker for disease severity (25). Similarly, we saw a trend of improved prealbumin levels in patients compliant with Gln, suggesting the possible use of prealbumin as a biomarker for disease severity. Prealbumin has been used as a marker for nutritional evaluation in hospitalized patients (26). Our data for prealbumin as a biomarker for SCD was not statistically significant possibly due to low numbers; however, it supports the hypothesis that prealbumin may have a role in evaluation of SCD modifying therapy. This potential role is not necessarily diminished by debatable cause and effect relationship: rapid RBC turnover with more severe disease results in higher metabolism therefore lower prealbumin can be reflective of increased metabolic demands rather than differences in diet and disease management. Nevertheless dietary management and disease control with modifying therapies may minimize this metabolic gap and at the same time improve prealbumin which can be measured. Albumin investigations can also be considered for pediatric SCD studies (25).
In conclusion, well-nourished and well-hydrated patients were younger and had a milder disease course, whereas more severe disease was observed in poorly nourished older patients, often with suboptimal compliance. Gln can be a clinically meaningful addition to HU and should be further studied in pediatric patients with SCD. Prealbumin may be investigated along with potential biomarkers for SCD control using Gln.
Funding
Moran Gotesman received a consulting fee for an Emmaus advisory board.
Conflicts of Interest
The Authors have no conflicts of interest to declare in relation to this study.
Authors’ Contributions
All Authors contributed to writing and editing the manuscript. Guy Elgar, Laura Hernandez Santiago and Abigail Alvarez helped with data collection. Moran Gotesman, Eduard Panosyan and Youngju Pak did the data anlaysis and statistics.
References
- 1.Telen MJ. Curative vs. targeted therapy for SCD: does it make more sense to address the root cause than target downstream events. Blood Adv. 2020;4(14):3457–3465. doi: 10.1182/bloodadvances.2020001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niihara Y, Miller ST, Kanter J, Lanzkron S, Smith WR, Hsu LL, Gordeuk VR, Viswanathan K, Sarnaik S, Osunkwo I, Guillaume E, Sadanandan S, Sieger L, Lasky JL, Panosyan EH, Blake OA, New TN, Bellevue R, Tran LT, Razon RL, Stark CW, Neumayr LD, Vichinsky EP, Investigators of the phase 3 trial of l-Glutamine in sickle cell disease A phase 3 trial of l-Glutamine in sickle cell disease. N Engl J Med. 2018;379(3):226–235. doi: 10.1056/NEJMoa1715971. [DOI] [PubMed] [Google Scholar]
- 3.Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, Jordan L, Lanzkron SM, Lottenberg R, Savage WJ, Tanabe PJ, Ware RE, Murad MH, Goldsmith JC, Ortiz E, Fulwood R, Horton A, John-Sowah J. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033–1048. doi: 10.1001/jama.2014.10517. [DOI] [PubMed] [Google Scholar]
- 4.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031. doi: 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- 5.Aich A, Jones MK, Gupta K. Pain and sickle cell disease. Curr Opin Hematol. 2019;26(3):131–138. doi: 10.1097/MOH.0000000000000491. [DOI] [PubMed] [Google Scholar]
- 6.Mandese V, Marotti F, Bedetti L, Bigi E, Palazzi G, Iughetti L. Effects of nutritional intake on disease severity in children with sickle cell disease. Nutr J. 2016;15(1):46. doi: 10.1186/s12937-016-0159-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smaldone A, Manwani D, Green NS. Greater number of perceived barriers to hydroxyurea associated with poorer health-related quality of life in youth with sickle cell disease. Pediatr Blood Cancer. 2019;66(7):e27740. doi: 10.1002/pbc.27740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hyacinth HI, Gee BE, Hibbert JM. The role of nutrition in sickle cell disease. Nutr Metab Insights. 2010;3:57–67. doi: 10.4137/NMI.S5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henderson RA, Saavedra JM, Dover GJ. Prevalence of impaired growth in children with homozygous sickle cell anemia. Am J Med Sci. 1994;307(6):405–407. doi: 10.1097/00000441-199406000-00004. [DOI] [PubMed] [Google Scholar]
- 10.Hyacinth HI, Adekeye OA, Yilgwan CS. Malnutrition in sickle cell anemia: implications for infection, growth, and maturation. J Soc Behav Health Sci. 2013;7(1):10.5590/JSBHS.2013.07.1.02. doi: 10.5590/JSBHS.2013.07.1.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singhal A, Davies P, Sahota A, Thomas PW, Serjeant GR. Resting metabolic rate in homozygous sickle cell disease. Am J Clin Nutr. 1993;57(1):32–34. doi: 10.1093/ajcn/57.1.32. [DOI] [PubMed] [Google Scholar]
- 12.Aschenbrenner DS. New drug for sickle cell disease. Am J Nurs. 2017;117(11):20–21. doi: 10.1097/01.NAJ.0000526743.46055.ef. [DOI] [PubMed] [Google Scholar]
- 13.Schuchard SB, Lissick JR, Nickel A, Watson D, Moquist KL, Blaylark RM, Nelson SC. Hydroxyurea use in young infants with sickle cell disease. Pediatr Blood Cancer. 2019;66(7):e27650. doi: 10.1002/pbc.27650. [DOI] [PubMed] [Google Scholar]
- 14.Park H, Bhatti S, Chakravorty S. Effectiveness of hydroxycarbamide in children with sickle cell disease - Analysis of dose-response metrics in a large birth cohort in a tertiary sickle cell centre. Pediatr Blood Cancer. 2019;66(7):e27615. doi: 10.1002/pbc.27615. [DOI] [PubMed] [Google Scholar]
- 15.Liem RI, Lanzkron S, D Coates T, DeCastro L, Desai AA, Ataga KI, Cohen RT, Haynes J, Osunkwo I, Lebensburger JD, Lash JP, Wun T, Verhovsek M, Ontala E, Blaylark R, Alahdab F, Katabi A, Mustafa RA. American Society of Hematology 2019 guidelines for sickle cell disease: cardiopulmonary and kidney disease. Blood Adv. 2019;3(23):3867–3897. doi: 10.1182/bloodadvances.2019000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otten JJ, Hellwig JP, Meyers LD. Washington, DC, USA, National Academies Press. 2006. DRI, Dietary Reference Intakes: the essential guide to nutrient requirements. [Google Scholar]
- 17.Antwi-Boasiako C, Frimpong E, Ababio GK, Dzudzor B, Ekem I, Gyan B, Sodzi-Tettey NA, Antwi DA. Sickle cell disease: Reappraisal of the role of foetal haemoglobin levels in the frequency of vaso-occlusive crisis. Ghana Med J. 2015;49(2):102–106. doi: 10.4314/gmj.v49i2.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinberg MH. Fetal hemoglobin in sickle cell anemia. Blood. 2020;136(21):2392–2400. doi: 10.1182/blood.2020007645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett EL. Understanding growth failure in children with homozygous sickle-cell disease. J Pediatr Oncol Nurs. 2011;28(2):67–74. doi: 10.1177/1043454210382421. [DOI] [PubMed] [Google Scholar]
- 20.Kawchak DA, Schall JI, Zemel BS, Ohene-Frempong K, Stallings VA. Adequacy of dietary intake declines with age in children with sickle cell disease. J Am Diet Assoc. 2007;107(5):843–848. doi: 10.1016/j.jada.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 21.Reid M. Nutrition and sickle cell disease. C R Biol. 2013;336(3):159–163. doi: 10.1016/j.crvi.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 22.Quinn CT. l-Glutamine for sickle cell anemia: more questions than answers. Blood. 2018;132(7):689–693. doi: 10.1182/blood-2018-03-834440. [DOI] [PubMed] [Google Scholar]
- 23.Williams R, Olivi S, Li CS, Storm M, Cremer L, Mackert P, Wang W. Oral glutamine supplementation decreases resting energy expenditure in children and adolescents with sickle cell anemia. J Pediatr Hematol Oncol. 2004;26(10):619–625. doi: 10.1097/01.mph.0000140651.65591.b8. [DOI] [PubMed] [Google Scholar]
- 24.Ogu UO, Thomas M, Chan F, Vattappally L, Sebastian G, Crouch A, You S, Minniti CP. L-glutamine use in adults with sickle cell disease: Clinical trials where success meets reality. Am J Hematol. 2021;96(1):E38–E40. doi: 10.1002/ajh.26021. [DOI] [PubMed] [Google Scholar]
- 25.Nouraie M, Ashley-Koch AE, Garrett ME, Sritharan N, Zhang Y, Little J, Gordeuk VR, Gladwin MT, Telen MJ, Kato GJ. Serum albumin is independently associated with higher mortality in adult sickle cell patients: Results of three independent cohorts. PLoS One. 2020;15(8):e0237543. doi: 10.1371/journal.pone.0237543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beck FK, Rosenthal TC. Prealbumin: a marker for nutritional evaluation. Am Fam Physician. 2002;65(8):1575–1578. [PubMed] [Google Scholar]