

Abstract
Methanol, a simple polar solvent, has been widely identified as an attractive carbon source to produce chemicals and fuels in bioprocesses. Specifically, to achieve recombinant protein production from methylotrophic yeasts, such as Pichia pastoris, this organic solvent can be used as a sole carbon source for growth and maintenance as well as an inducer for protein expression. However, if methanol feeding is not controlled well in such a fermentation process, accumulation of the solvent in the growth media will have a detrimental effect on the cells. Hence, monitoring the levels of methanol in these fermentation processes is a crucial step to ensure a healthy culture and maximum protein production. There are various techniques elaborated in the literature for monitoring methanol in cell cultures, but often, they appear to be expensive methods that are less affordable for many laboratories. This is because, in addition to the sophisticated equipment that is required for the analysis, the complexity of the samples retrieved from the bioprocesses necessitates laborious processing steps often involving expensive tools. In this study, a fast, simple, and sensitive method is developed to process biological samples by using the salting-out-assisted liquid–liquid extraction technique to quantify the concentration of methanol and ethanol using gas chromatography. On comparing the combinations of widely available salts and solvents, it was noticed that salting out using potassium carbonate followed by the liquid–liquid extraction of the analyte using ethyl acetate showed the best recovery. Followed by this, a validation test for the developed method was performed, which resulted in good peak resolution, linearity, and limit of detection for the quantitation of methanol and ethanol. By further assessing the tested combination, it was confirmed that its application could be extended to other matrices. Such an approach facilitates the possibility to monitor and control the methanol levels in fermentation and aids in bioprocess optimization.
Introduction
Nowadays, yeasts are utilized as a popular host to produce heterologous proteins instead of using conventional extraction methods from their natural source. Methanol is a commonly used organic compound in fermentation processes involving the methylotrophic yeast Pichia pastoris, an important host for recombinant protein production. This species has a tightly regulated alcohol oxidase 1 promoter (pAOX1), which induces the gene expression at suitable methanol concentrations. There have been studies showcasing the relevance of methanol feeding in the bioproduction process as well as identifying this feeding as an important parameter to enhance the production of recombinant proteins.1,2 The utilization of methanol as an inducer in P. pastoris fermentation can be challenging for large-scale cultivations, which include fed-batch and continuous processes. This is because, in addition to serving as an induction source for protein expression, it also functions as an energy source for the host cells.3 Monitoring the methanol levels in P. pastoris bioprocesses is very crucial during the fermentation since it can significantly affect the viability of the cells as well as the production yield of the protein of interest.4 Therefore, monitoring and controlling the concentration of methanol throughout the bioproduction is essential.
At present, different analytical methods have been adopted by researchers to quantify the methanol concentration present in the fermentation media. These techniques often involve expensive analytical instruments, assay kits, and chemicals. Several published papers are elaborating on the use of chemical and enzymatic methods for quantifying the volatile components present in biological samples. Using conventional techniques such as calorimetric methods for the analysis of methanol is often time-consuming and considered a tedious task.5 Enzymatic assays, for instance, facilitate the quantitation of analytes in the absence of expensive instrumentation and excessive sample preparation. However, the sensitivity, speed, and reproducibility of the method are inferior compared to chromatographic techniques. Difficulties caused in assay standardization, storage requirements to avoid enzyme instabilities, and ambiguous results are some major concerns arising from enzymatic methods. The flow injection analysis (FIA) is a suitable alternative to overcome these downsides of low reliability and reproducibility of conventional enzymatic analyses. However, this technique poses the need for an automated sample dilution system to work in the low linear range.6 Sequential injection analysis (SIA), an alternative to FIA, facilitates the measurement of methanol through automatic sampling from the bioreactor for enzymatic analysis.7 A major advantage of using SIA over FIA is that the hindrances noticed from the latter can be reduced in SIA. Another possible approach toward determining the concentration of methanol in the medium is by using monitoring techniques such as sensors that can be used directly in contact with the process fluids (on-line sensors) or used outside the bioreactor (off-line sensors). Often, sensors are complex, expensive, labor-intensive, and not compatible with standard sterilization procedures. Hence, only a few are available for bioprocess monitoring.8 Some of the major drawbacks of using online sensors for monitoring the concentrations are the response time and low precision of the measurement.6,9
High-performance liquid chromatography (HPLC) and gas chromatography (GC) are considered as some of the most accurate and widely used techniques to identify and quantify methanol in the research labs. Nunes et al.10 summarize in their work the commonly used techniques for methanol analysis and emphasizes the usage of GC and HPLC for the simultaneous determination of methanol and acetic acid. GC, specifically, is considered a precise and reliable method to determine low molecular weight volatile components11,12 and has been extensively used in forensic and research labs.13 The highlight of this technique is its ability for efficient separation and quantification of analytes present in complex matrices.14 However, they come with the disadvantage of requiring extensive and time-consuming processing of the samples retrieved from bioprocesses. These samples often tend to be too complex or contain components that are harmful to be injected into the analytical instrument. Most of the P. pastoris fermentation is performed in either the basal salt media (BSM) or the buffered minimal glycerol/methanol (BMG/BMM) media. The growth medium is often supplemented with methanol through exponential feeding to ensure sufficient availability of the substrate for the induction of the protein. The samples retrieved from such a process include several metabolites that are secreted into the media, which may interfere with the chromatographic analysis. Therefore, the processing step is crucial to optimize the quantification step as well as for the elongation of the column life. For specific compounds, an additional step such as derivatization helps in increasing the volatility and chromatographic performance. This technique can be used for the analyses of compounds such as alcohols, acids, and amines that are difficult to analyze due to the presence of reactive hydrogen. A major drawback of using this technique is the potential errors arising from the incomplete chemical conversion of the analyte. Additionally, a chemical conversion often involves reagents that are expensive.15
A technique that is available today and worth investigating in the bioproduction field is headspace analysis (HSA). Muna et al.16 elaborate in their paper on a highly sensitive, rapid, and reliable procedure for the quantification of methanol present in working environments through HSA. In this method, the quantification is performed by coupling the headspace injection with the GC flame ionization detection (GC–FID) method using a capillary column. However, the efficiency of these techniques may vary depending on the matrix of the biological samples; furthermore, it necessitates a headspace sampler to perform the analyses. Headspace sampling can be an effective technique for the separation of volatile components from complex samples. It is one of the most promising techniques for analyses of volatile components due to its ability to facilitate the detection of trace quantities with limited sample preparation. In this technique, the sample is placed in a closed thermostated glass vial where the volatile analyte attains an equilibrium distribution between the gas and solid/liquid phase. The gas phase is then injected into the GC for analysis. The non-volatile part of the sample remains in the vial and therefore prevents accumulation in the inlet and column. The extraction efficiency (EE) of the HSA can be improved by choosing the means of separating the analyte from the complex mixture. It can be achieved either by the static mode, where the volatile analyte enters the gaseous state by thermal or chemical means or, dynamic mode, where the analytes are attained by passing large volumes of inert gas over the sample. Such an emerging technique can facilitate the analyses of dozens of samples. However, such a set of serial analyses is preferably performed using automatic and semiautomatic attachments to gas chromatographs.15 Additionally, compared to a conventional GC setup, the HSA requires thermostatic conditions ranging from 30 to 250 °C. Moreover, compared to a conventionally used GC–FID design, the commonly used static headspace design requires a long time for reaching the interfacial equilibrium and gives unreliable storage stability of samples.
The addition of internal standards can compensate for fluctuations noticed from the sample analyses or changes in extraction efficiencies. When choosing an internal standard, a compound similar to the analyte of interest is selected and added to the blank, standards, and samples. Wang et al.17 mentioned the usage of internal standards to overcome the difficulty in the simultaneous determination of methanol and ethanol present in large quantities in alcoholic beverages.
Sample processing is a vital step to facilitate the analytical quantification of components present in a complex biological mixture. Samples for GC should be prepared in a manner such that the high molecular weight components in the sample are removed. However, the steps necessary to remove interferences from such samples can be a tedious task depending on the complexity of the matrix. Several techniques have been recommended in the literature to quantify methanol in complex sample matrices. Some of these are (i) derivatization of sample matrices such as water–ethanol,18 aqueous,19 and alcohol;20 (ii) direct injection of samples such as plasma,21 whole blood,22 alcohol, food products,23 and wine;24 (iii) HSA of wine,25 washing filtrate,26 and air;16 (iv) solid-phase extraction of air;27 and (v) dilution of cell cultures.28 One of the most important and widely used techniques for the separation of a volatile component from biological samples is extraction. Liquid–liquid extraction is a commonly used technique to attain a compound out of a mixture by using a solvent. For this, an organic solvent is selected that facilitates the separation of hydrophobic compounds from the hydrophilic compounds, which prefer the polar aqueous phase. In this type of treatment, extraction can be useful to separate the sample into two immiscible phases, and the analyte of interest can be recovered from the organic phase. The compounds attained via this method are separated based on their relative solubilities.29,41 However, some analytes do not exhibit a complete separation using the organic solvents alone. In such cases, incorporating an additional technique such as salting out can enhance the EE of these compounds from aqueous solutions. The EE can be calculated from the amount of solute present in the extract relative to the total amount of solute present in the initial sample. Salting-out-assisted liquid–liquid extraction (SALLE) can, therefore, serve as a simple and cost-efficient method to separate the water-miscible organic solvent from aqueous biological samples within a short analysis time.29 It is a well-known technique traditionally used for sample clean-up processes and has been favored for several LC/MS/MS bioanalyses.30 In this technique, a high concentration of salts is used for the separation of water-miscible organic solvents from aqueous solutions. To achieve this, the easiest and most economical technique is to use inexpensive salts that are commonly available in the research labs. The biological samples contain particulates from the growth medium as well as the metabolites produced during the fermentation that can interfere with the analytical method by forming precipitates in the analytical instrument. Moreover, the high salt media, which is typically used for P. pastoris fermentation, and the sugars used for the growth of the organism need to be removed prior to injection of the sample as they can cause blockage in the chromatography column. Such a technique commonly includes salts such as sodium chloride and ammonium sulfate in combination with an extraction solvent such as acetonitrile.31 Liquid–liquid extraction is performed by mixing two immiscible phases so that the compounds in the aqueous phase will diffuse into the organic phase. Some compounds require rigorous mixing of the mixtures with organic solvents for a prolonged period to facilitate sufficient separation. The separation of the phases is achieved by incorporating centrifugation or using semi-permeable membranes.32 Selecting the appropriate solvent for the extraction of the compound of interest is an important step. While selecting a suitable solvent, characteristics such as solubility and polarity need to be considered. The SALLE technique has not been reported in the literature for the quantification of methanol present in the samples retrieved from yeast bioprocesses.
Therefore, this study aims to develop and validate a sample processing method for GC capable of separating, identifying, and quantifying the methanol present in the biological samples derived from P. pastoris fermentation. To achieve this, the conditions for methanol analysis and the identification of the best combination of a salt and a solvent for effective separation of methanol from biological systems have been assessed. To demonstrate the reliability of the technique, phase separation has been assessed in simple and complex matrices. To illustrate the accuracy of the method, the most commonly used P. pastoris fermentation media are also investigated in this study. Additionally, the efficiency of the selected method to separate and identify methanol in the presence of ethanol, which is a common metabolite in yeast fermentation, is quantified. This simple and accurate method is thereby expected to provide a hassle-free and economical processing step to separate methanol from a complex matrix through phase separation and quantify using a widely available GC–FID. To the author’s knowledge, this study is the first to achieve optimization of the SALLE approach for the identification and quantification of methanol concentrations present in a complex biological matrix with high salt concentrations and cellular metabolites. Compared to the existing labor, material, and equipment-intensive methods for methanol detection, this study presents a simpler alternative.
Materials and Methods
Chemicals and Media Compositions
For the method development and standard preparation, GC ultra-grade ethyl acetate (≥99.9%, Carl Roth, Belgium), ethanol absolute 100% HPLC grade (Chem-Lab, Belgium), and methanol (≥99.5%, VWR, France) were used. The peaks attained from the standard and sample were compared based on their retention times for the identification of methanol and ethanol. For the salting-out technique, sodium sulfate (Honeywell, Austria), sodium chloride (Sigma-Aldrich, Germany), potassium carbonate (Vel, Belgium), and ammonium sulfate (Chem-Lab, Belgium) were used. The extraction of methanol and ethanol was studied using GC ultra-grade ethyl acetate (≥99.9%, Carl Roth, Germany), diethyl ether (99+%, Acros Organics, USA), methyl-tert-butylether (MTBE ≥99.5%, Carl Roth, Germany), and ≥99.8% chloroform (Sigma-Aldrich, Germany). The calibration standards were made using Milli-Q water (18.2 MΩ cm at 25 °C, Millipore). A detailed list of the different salts and solvents used in the SALLE technique is mentioned in Table 1. The media compositions used in this study were as described in Tables S1–S3 and the supplier information is listed in Table S4.
Table 1. Salts and Solvents Used for the SALLE Technique.
| salts |
solvents |
||
|---|---|---|---|
| IUPAC name | chemical formula | IUPAC name | chemical formula |
| sodium chloride | NaCl | ethyl acetate | C4H8O2 |
| sodium sulfate | Na2SO4 | diethyl ether | C5H12O |
| ammonium sulfate | (NH4)2SO4 | methyl-tert-butyl ether | CHCl3 |
| potassium carbonate | K2CO3 | chloroform | (C2H5)2O |
Instrumentation
For the quantification of methanol and ethanol present in the samples, an Agilent 8860 GC system (Agilent Technologies, California, USA) equipped with a flame ionization detector (FID, Agilent technologies) was used. The chromatographic separation was performed on an HP-5 capillary column of 30 m × 0.320 mm i.d. and × 0.25 μm film thickness (Agilent Technologies, USA). A sample volume of 1 μL was injected using an ALS syringe (10 μL, fixed needle, 23–26s/42/cone, Agilent technologies, USA) in the split/splitless mode depending on the concentration range of the analyte present in the sample. The conditions selected for the analysis of methanol and ethanol are listed out in Table 2. The detector and injector temperatures were 300 and 225 °C, respectively. The oven temperature program was 40 °C (held for 3 min), increased at 60 °C/min to reach 225 °C. The carrier gas used was helium at 1.5 mL/min. The air and hydrogen flow rates were 300 and 40 mL/min, respectively. The data from GC runs were analyzed using OpenLab CDS software (version 2.4). The resulting peaks were identified by comparing the retention times and quantified against the prepared standards. Each sample was prepared in triplicates and injected five times to determine the mean and standard deviation.
Table 2. Conditions Used in GC for Methanol and Ethanol Detection.
| parameter | Agilent 8860 GC system |
|---|---|
| oven/column temperature | 40–225 °C |
| running time | 8.08 min |
| carrier gas flow rate | 1.5 mL/min |
| detector temperature | 300 °C |
| H2 flow | 40 mL/min |
| Airflow | 300 mL/min |
| makeup flow | 25 mL/min |
Selection of Salts and Extraction Solvents
The initial parameter investigated was the choice of salt and solvent for extraction and recovery of the solute of interest. For the salting-out technique, a total of four salts were considered, which were sodium chloride (NaCl), sodium sulfate (Na2SO4), ammonium sulfate ((NH4)2SO4), and potassium carbonate (K2CO3). For the liquid–liquid extraction, a total of four solvents were considered, which were ethyl acetate (C4H8O2), diethyl ether ((C2H5)2O), methyl-tert-butyl ether (MTBE, (C5H12O)), and chloroform (CHCl3). A total of 16 combinations of salts and solvents were tested to understand their effect on the EE. The best combination of salt and solvent was selected based on the EE of methanol and ethanol.
A 0.1% (w/v) solution of methanol and ethanol in Milli-Q water was prepared in a 100 mL volumetric flask. The prepared solution (700 μL) was transferred into a sterile 2.0 mL Eppendorf tube (Fischer Scientific, USA). To this solution, the selected salt was added according to the quantity listed out in Table 3 based on the solubility of each salt. The solution with the salt was then vortexed for 2 min on a shaker (VELP Scientifica Srl, Italy) at 1200 rpm and 20 °C. After shaking, 700 μL of solvent was added into the Eppendorf using a 1 mL glass pipette. The mixture was vortexed for 15 min at 1200 rpm and 20 °C. The solution was then centrifuged (centrifuge 5810 R, Eppendorf) at 4000 rpm for 2 min and 20 °C to facilitate the phase separation. Following this step, the top phase from the supernatant was carefully pipetted out into a GC sample vial (Fischer Scientific, Poland) and injected immediately.
Table 3. Properties of Different Salts Tested in the Extraction Study.
| salt | temperature (°C) | solubility in water (g/100 mL) | quantity tested (g/0.7 mL) |
|---|---|---|---|
| NaCl | 20 | 36.0 | 0.252 |
| Na2SO4 | 25 | 28.1 | 0.194 |
| K2CO3 | 20 | 110.3 | 0.772 |
| (NH4)2SO4 | 20 | 74.4 | 0.520 |
Standard Solutions
For the construction of a standard curve, the standard solutions with known concentrations of the analyte were prepared. A 10% (w/v) standard stock solution of methanol and ethanol was prepared in a 100 mL volumetric flask by dissolving in Milli-Q water. From this, standard working solutions at 12 concentration levels (0.049, 0.098, 0.195, 0.390, 0.781, 1.562, 3.125, 6.25, 12.5, 25, 50, and 100 g/L) were attained by diluting the above stock solution with Milli-Q water in a 1:2 ratio. All solutions were processed just before injection. This processing consisted of salting-out of the analyte using K2CO3, followed by liquid–liquid extraction with ethyl acetate for all concentrations.
Method Validation
To verify the analytical performance of the developed methodology, validation parameters were assessed. These parameters include resolution, recovery, linearity, limit of detection (LOD), and limit of quantification (LOQ).
Peak Resolution
The peak resolution between methanol and ethanol was estimated by injecting a processed sample containing 0.1% (w/v) of both analytes in Milli-Q water. To attain an acceptable peak separation between the two volatile components, the chromatographic method was initially optimized by altering the split ratios. For this, a series of split ratios were tested comprising 10/1, 15/1, 20/1, and 30/1. Once an adequate separation was attained between the two analytes, the peak resolution was calculated using the half-width method.
Extraction Recovery
A four-by-four factorial design was used for testing the combination of four salts and solvents as indicated in Table 1 to achieve maximum extraction of the analyte, resulting in a total of 16 combinations. All the 16 combinations were prepared in triplicates and injected five times, resulting in a total of 240 injections. The best condition of the factorial design was selected by evaluating the chromatographic responses, and the recovery values were calculated from all 16 combinations. The extraction recovery was calculated from the ratio between the area attained from an extracted sample and the area attained from a sample without extraction expressed as a percentage.
Linearity and Range
Calibration curves for methanol and ethanol were prepared by injecting standard solutions ranging from 0.195 to 10 g/L. The peak area of the standards was plotted against the analyte concentrations. Standard calibration curves of the compounds were achieved by calculation of the regression line using the least-squares method. The range of the analytical method was determined by checking the resolution and linearity of the upper and lower concentration of the analytes.
Residual Effect and Matrix Effect
The specificity of the developed method was corroborated by injecting solutions containing pure methanol and ethanol. To check this, solutions containing the two analytes were prepared using deionized water and Milli-Q water. Furthermore, additional samples were prepared by spiking the most relevant P. pastoris fermentation media (Tables S1–S3) used in recombinant protein production. The concentration of the analytes studied to evaluate the effect of the sample matrix was set at 0.1% (w/v). The sample processing was done in triplicates, and each sample was injected five times to evaluate the accuracy of the method.
Limit of Detection and Limit of Quantification
The LOD was calculated based on the ratio between the standard deviation of the response and the slope from the calibration curve of the standards multiplied by a value of 3. The LOQ was taken as the lowest concentration of methanol and ethanol in the calibration curve that can be reproducibly quantified.
Repeatability
For checking the repeatability of the method, intra-day and inter-day measurements were performed. For this, a sample containing a concentration of 0.1% (w/v) of methanol and ethanol was processed and injected three times within the same day to check intra-day precision. For inter-day, new samples were processed in triplicates, and each sample was injected three times over three consecutive days.
Analysis of Bioreactor Samples
To confirm the applicability of the selected method, samples from an actual bioreactor experiment were processed and injected into the gas chromatograph. For this, a bioreactor experiment involving a concentration of 0.1% (w/v) methanol in BSM was conducted in the batch mode. The sample from the bioprocess is filtered using a 0.2 μm filter (Sarstedt, Germany) attached to a 10 mL syringe (BD Discardit II, USA) into a 15 mL sterile falcon tube. These samples were stored at −80 °C until further analysis. For the preparation of the sample solutions, the samples were thawed, and approximately 700 μL of 0.2 μm filtered sample was transferred into a sterile 2.0 mL Eppendorf tube (Fischer Scientific, USA). To this, 0.772 g of K2CO3 was added and vortexed for 2 min on a shaker (VELP Scientifica Srl, Italy) at 1200 rpm and 20 °C. After shaking, 700 μL of ethyl acetate was added into the Eppendorf using a 1 mL glass pipette. The mixture was vortexed for 15 min at 1200 rpm and 20 °C. The solution was then centrifuged (centrifuge 5810 R, Eppendorf) at 4000 rpm for 2 min and 20 °C to facilitate the phase separation. Following this step, the top phase from the supernatant was carefully pipetted out into a GC sample vial (Fischer Scientific, Poland) and injected immediately. The concentration of methanol present in the growth media during the fermentation was measured as a function of time using GC.
Culture Conditions for Fermentation Analysis
The strain used for the bioprocess was P. pastoris GS115 (Mut+) that was genetically engineered to produce a recombinant sweet protein, thaumatin. The genetic modification of the strain was performed at the VIB protein core (Zwijnaarde, Belgium) by cloning the pre-thaumatin II-pro gene into the expression vector pAOXZ. The construction of the expression vector enables the organism to secrete the sweet protein directly into the growth media. To produce the protein of interest, the strain was grown in 1.5 L of BSM within a computer-controlled bioreactor having a total capacity of 6 L (BioStat B, Sartorius, Germany).
Stock cultures were stored at −80 °C in a 5% (w/v) yeast peptone dextrose (YPD, Carl Roth, Germany) broth and 25% (w/v) glycerol (99+% p, Chem Lab, Germany). A purity plate was made by spreading a loopful of the thawed stock culture on a Petri plate containing YPD media and bacteriological agar (VWR, Belgium). After incubating the plates for 72 h at 30 °C, three colonies from the YPDA plate were transferred into an Erlenmeyer flask with 20 mL (5% w/v) YPD broth. The Erlenmeyer flask was incubated at 30 °C on an orbital shaker (Grant Instruments Ltd, England) at 220 rpm for 24 h, and 20 μL of this preculture was transferred into a 250 mL Schott bottle containing 80 mL (5% w/v) YPD broth. The Schott bottle was incubated for 72 h at 20 °C on orbital shakers at 160 rpm. From this second preculture, 0.5 mL of the preculture was mixed with 4.5 mL of 0.85% (w/v) sodium chloride (Sigma-Aldrich, Germany) diluent solution. After vortexing the solution, a total of 5 mL of diluted preculture was injected into the bioreactor using a sterile syringe (BD Discardit II, USA) such that the initial concentration in the media would be approximately 10 ln (cfu/mL). Samples were taken every 1 hour for methanol analysis. Prior to the analysis, the samples were processed as described in Figure 1.
Figure 1.

Different steps involved in sample processing for methanol analysis: (a) thawing of the sample in a 15 mL falcon tube, (b) transferring 700 μL of sample to a 1 mL Eppendorf tube, (c) addition of K2CO3 to 700 μL of sample, (d) vortexing the sample for 2 min, (e) addition of 700 μL of organic solvent, (f) vortexing the sample for 15 min and centrifugation for 2 min, (g) analyte separated to the top organic layer, and (h) transfer of top organic layer to a GC vial for analysis.
To check the accuracy of the considered method, the samples from the fermentation were additionally analyzed using an Agilent 1260 Infinity II LC System equipped with a refractive index detector (Agilent Technologies, USA). A fermentation monitoring column Aminex HP-X 87H (300 × 7.8 mm, Bio-Rad Laboratories, USA) maintained at 60 °C was utilized for the estimation of methanol. The mobile phase used was 0.01 M H2SO4 at a flow rate of 0.6 mL/min by isocratic elution. Prior to analyses, the samples were filtered using a 0.2 μm filter (Sarstedt, Germany). The samples were analyzed in triplicates, and their mean values were considered for data plotting.
Data Handling
All sample processing was performed in triplicates, and each sample was injected five times. The raw data attained from the analysis was examined using the OpenLab CDS software (Agilent Technologies). All the data processing was performed using Microsoft Excel, and the results estimated were expressed as their mean and standard deviation. MATLAB (R2020b) was utilized to generate codes for plotting the figures.
Results and Discussion
In this study, a fast and reliable sample processing method was developed and validated to accurately identify and quantify the amount of methanol and ethanol present in biological matrices retrieved from a P. pastoris fermentation. The detection and quantification were performed using a GC–FID that is commonly available in research facilities and the Agilent OpenLab CDS software. The objective of the sample preparation step is to remove the unwanted compounds/impurities present in the complex matrices retrieved from bioprocesses. This step enhances the analytical method by achieving a desirable sensitivity/selectivity and reduces carryover issues. The interferences that need to be eliminated from the sample for GC–FID detection include salts/ions, proteins, phospholipids, and other contaminants that can compromise the data quality as well as cause deterioration of the equipment in the long term. The detection and quantification of methanol and ethanol present in the complex mixtures were achieved by incorporating a salting-out step, followed by liquid–liquid extraction before injecting into the GC. The following sections summarize the results attained from the various experiments conducted in this study. First, the effect of the combinations between salt and solvents was studied to attain the maximum EE for methanol and ethanol without compromising the peak resolution. Followed by the selection of the best combination of salt and solvent, the validation characteristics such as linearity, range, detection limit, quantitation limit specificity, repeatability, and precision are evaluated.
Optimization of the SALLE Method
In the SALLE technique, the selection of the extraction salt and solvent is an important aspect. For instance, the type and the amount of salt that is used can affect the degree of phase separation.31 Similarly, when choosing a suitable organic liquid, a major consideration should be given for its solvent power to attain the desired compound into the organic phase as well as the compatibility of the solvent with the separation and detection of the desired compound during chromatography. Additionally, while selecting a suitable solvent for liquid–liquid extraction, care must be taken to ensure that the selected solvent is miscible in water, is polar, and has the ability to induce phase separation when the selected salt is added into the sample.30 There have been several studies indicating how the EE is influenced by the type of the solvent used33,34 and also how the combination of extraction parameters affects the efficiency. The combination of salt and solvent works better because the salt addition augments the ionic strength of the aqueous sample. This reduces the solubility of the organic analyte present in the solution and facilitates efficient extraction of this analyte in the sequential sample processing step.
There is an explicit need to optimize the addition of salt and solvent to maximize the EE. In this study, four different inorganic salts in combination with four different organic solvents were tested to check the efficiency of separation. The four salts that were selected are commonly used for SALLE, that is, sodium sulfate (Na2SO4), ammonium sulfate ((NH4)2SO4), sodium chloride (NaCl), and potassium carbonate (K2CO3). The solvents used were ethyl acetate (C4H8O2), diethyl ether ((C2H5)2O), methyl-tert-butyl ether (MTBE, (C5H12O)), and chloroform (CHCl3). A total of 16 combinations were tested, out of which the best combination that exhibited the highest peak intensity and a good resolution was selected.
Generally, neutral salts such as NaCl are favored in the SALLE technique. This is because neutral salts aid in controlling the pH of the sample during salt addition and therefore maintain the extraction conditions at the optimal level. However, as can be seen in Table 4, K2CO3 and (NH4)2SO4 gave better results in combination with the solvents used in comparison to NaCl and Na2SO4. The latter two salts, being pH neutral in nature, showed a lower efficiency in extracting methanol from the mixture. On the other hand, the acidic salt (NH4)2SO4 and the basic salt K2CO3 prevailed as more effective, irrespective of the solvent that was used solutions even. Among these two salts, the basic salt, K2CO3, showed a better performance. In the study conducted by Sazali et al.,31 water-miscible acetonitrile was used as an extractant in combination with high salt conditions. The salts examined in this particular study were (NH4)2SO4 and NaCl. The results attained from their study demonstrated that (NH4)2SO4 showed a better separation and peak area compared to NaCl, demonstrating that the former is an effective salting-out agent. However, when a comparison between (NH4)2SO4 and K2CO3 in combination with different solvents was made in this study, the latter showed a greater yield. This result is analogous to the findings of Xie et al.,35 where K2CO3 was identified as the best salt for extracting acetone, butanol, and ethanol present in samples attained from a prefractionator when compared to acidic and neutral salts. In their study, a series of acidic, basic, and neutral salts were used to test the best salting-out effect. Out of the selected salts, even though NaCl and (NH4)2SO4 were identified as the best neutral and acidic salts, considering the constituents present in the sample, K2CO3 was chosen as the optimal salt. Xie et al.35 substantiate the finding based on the relationship between dehydration and solubility of the selected salts, where K2CO3 exhibited the least organic content in the water phase and the least water content in the organic phase, resulting in the best salting-out effect. This is due to the hydrophilic nature of K2CO3 and the hydroxyl ions aiding in the breakage of the hydrogen bonds resulting in greater separation of organic residues from the aqueous phase. Although neutral and acidic salts exhibited a general salting-out effect, they did not show adequate separation due to the absence of ionized cation and hydroxyl ions.35
Table 4. Combinations of Salts and Solvents Tested for the Recovery of Methanol and Ethanol and Their Estimated EEs (%).
| salts |
|||||
|---|---|---|---|---|---|
| analyte | solvents | NaCl | Na2SO4 | NH4SO4 | K2CO3 |
| methanol | C4H8O2 | 22 | 24 | 33 | 58 |
| C5H12O | 16 | 17 | 24 | 27 | |
| CHCl3 | 8 | 7 | 12 | 13 | |
| (C2H5)2O | 15 | 16 | 23 | 28 | |
| ethanol | C4H8O2 | 50 | 48 | 68 | 92 |
| C5H12O | 41 | 38 | 48 | 65 | |
| CHCl3 | 31 | 27 | 47 | 53 | |
| (C2H5)2O | |||||
A liquid–liquid extraction involves two immiscible solvents with different polarities. The location of the separated non-polar extraction solvents depends on their density. In this study, the solvents used for separating methanol and ethanol from the aqueous mixture include ethyl acetate, MTBE, chloroform, and ether. The results attained from the extraction study as shown in Table 4 indicate that the EE was highest for ethyl acetate and the lowest for chloroform in all the combinations of salts tested. The combination of inorganic salt with diethyl ether failed to provide a peak for ethanol with sufficient resolution. Due to the overlapping of the ethanol peak with an impurity peak arising from the solvent, the results from this combination are not included in Table 4. Ethyl acetate is a preferable choice in liquid–liquid extraction due to its biphasic behavior, which enables it to extract both polar and non-polar compounds. Furthermore, it has been reported that ethyl acetate is expected to generate higher yields of extracts in combination with water.36
Therefore, from the percentage recoveries reported in Table 4, it can be concluded that the combination of the inorganic salt K2CO3 and organic solvent ethyl acetate showed the best performance. The estimated percentage extraction recoveries for methanol and ethanol using this combination were 58 and 92%, respectively. Moreover, ethyl acetate is a widely used solvent in liquid–liquid extraction due to its low toxicity and cost. Hence, this combination was selected for the subsequent study. The remaining combinations resulted in comparatively lower recovery values where the lowest was noticed for the combination of Na2SO4 and chloroform.
Peak Resolution
In this study, the chromatograms attained from the sample injections displayed two well-distinguishable peaks of methanol and ethanol, indicating a good separation of the compounds of interest. To ensure adequate separation between the peaks and to avoid overestimation of the area under the peaks, the method was modified concerning the split ratios used. The optimized split ratio was assessed by testing the following values: 10/1, 15/1, 20/1, and 30/1. It was seen that the ratio of 30/1 resulted in the best resolution. Therefore, this value was selected for the remaining study. From Figure 2a, the peaks attained for methanol and ethanol can be observed at 2.058 and 2.155 min, respectively. As revealed by the chromatogram attained for methanol and ethanol, additional components are eluted at later retention times, indicating the presence of impurities in the solvent. The additional peaks arising in the chromatogram of the analysis of ethyl acetate used in the study, as shown in Figure 2b, indicate the presence of minor impurities. These impurities are inevitable given that GC grade ethyl acetate was used, and they generally do not interfere with the analysis of methanol and ethanol. Moreover, the developed method ensures that the peaks of target analytes are well separated, and hence, the possible interferences from the impurities can be eliminated. To substantiate the findings, the peak resolution (RS) of methanol and ethanol was calculated by using the half-width method with eq 1, where R1 and R2 are the retention times of ethanol and methanol, and W1 and W2 are the corresponding peak widths measured at half the peak height. The resulting value from the above equation was 1.87, which indicates a good peak resolution between the two components. Hence, the value confirms a good chromatographic separation of methanol and ethanol after extraction with the selected method.
| 1 |
Figure 2.
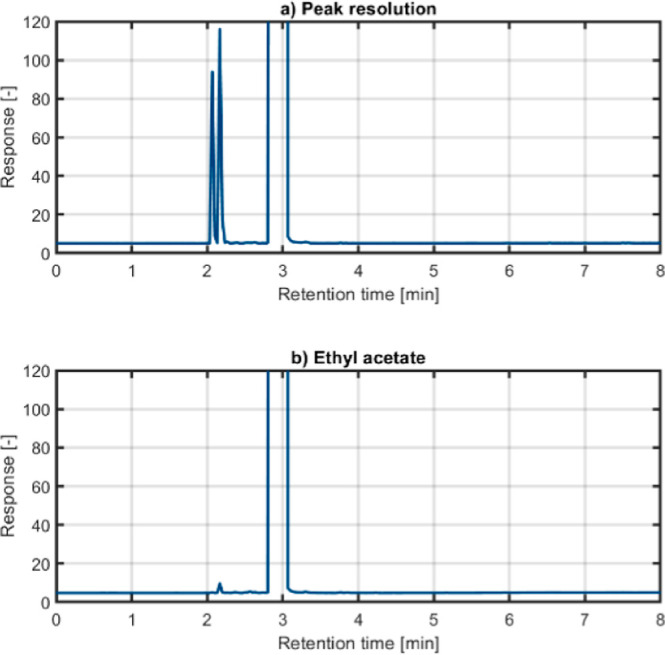
(a) Chromatogram showing the peak resolution of two analytes of interest, methanol and ethanol extracted from aqueous mixtures using the SALLE technique. (b) Chromatogram of pure ethyl acetate, indicating the presence of impurities in the solvent used for sample treatment.
Extraction Efficiency
The EE % of the tested combination was calculated to assess the recovery of the analytes. To achieve this, a neat blank, that is, a pure component (methanol/ethanol) in ethyl acetate without extraction, is injected into the GC. The ratio between the area attained from an extracted sample and the area attained from a sample without extraction that has the same analyte concentration expressed as a percentage gives the EE of the considered process. A constant EE is required to obtain a linear calibration curve. To check this, the EE was tested for concentrations ranging from 0.049 to 100 g/L of methanol and ethanol. The recoveries calculated for a wide range of standard concentrations are plotted against the known concentration of the analyte before sample treatment. It was observed that the EE for methanol is virtually constant over a wide range of concentrations tested except for the two lowest concentrations: 0.098 and 0.049 g/L. Hence, these values were eliminated from the calibration curve and are assumed to fall below the LOQ. In the case of ethanol, some discrepancies are noticed at extremely low concentrations. The recoveries attained for ethanol over a wide range of known concentrations are not constant. An increasing trend for extraction recoveries is noticed at lower concentrations (data not shown). This trend is assumed to originate from the presence of ethanol in the ethyl acetate that was used for liquid–liquid extraction. Given that ethyl acetate is an acetate ester formed between acetic acid and ethanol, it seems evident that some traces of ethanol appear in this solvent. These traces of ethanol in the solvent affect the extraction recoveries that are attained because the measured concentrations of ethanol are an overestimation of the real amount present. The total quantity of ethanol in the final extract is a combination of the extracted quantity from the sample and the ethanol present in the solvent. Hence, the distribution between the extracted ethanol and the ethanol present in the solvent should be estimated. This can be achieved by finding a correlation between the concentration of ethanol present in the final extract (Cextract), EE, extracted concentration from the sample (Csample), and concentration in the ethyl acetate (CEtAc), assuming a constant EE from the original sample irrespective of the sample concentration. Hence, a general equation denoting the relationship between the above can be given as
| 2 |
This equation includes the assumptions that the EE from the original sample is a constant and that the concentration of ethanol present in the final extract is the sum of the extracted concentration (EE*Csample) and the concentration in the ethyl acetate CEtAc. From this correlation, the values of EE and CEtAc, which are independent of the concentration in the sample, can be estimated. Therefore, the EE and the concentration of ethanol in ethyl acetate CEtAc were estimated to be 94.24% and 0.134 g/L, respectively. The comparison of the ethanol concentration in the sample and the extract is made in Figure 3. The relationship of eq 2 with the estimated parameters is depicted in the same figure. Equation 2 and its estimated parameters were used to calculate the extraction yield in an equivalent manner to the calculation using experimental results. This demonstrated a good agreement with the experimental extraction efficiencies and therefore confirmed the determination of the real extraction yield of 94.24% based on eq 2.
Figure 3.

Ethanol concentration in the extract as a function of the ethanol concentration in the sample. The line represents the relationship between both concentrations that is established in eq 2.
Linearity and Range
Linearity was analyzed from the regression of the calibration curves acquired from the ratio between the peak area and the concentration of the analyte. The criteria for acceptance of linearity are marked for a coefficient of determination (R2) of at least 0.99. In the concentration range that was studied, which ranged from 0.049 to 100 g/L, the analytical response was linear with an R2 value of 0.994 for methanol and 0.928 for ethanol, indicating a proportional increase of peak area to the analyte concentration. The discrepancies noted from the EE calculation of methanol and ethanol specifically for extremely low concentrations indicate the need for its elimination from the calibration curve. The linearity curve resulting from the elimination of these values (0.049 and 0.098 g/L) gives an R2 value of 1.000 for methanol and 0.997 for ethanol samples. However, it should be considered that the linear parameters from the ethanol calibration curve are estimated without considering the ethanol present in ethyl acetate. Figure 4a,b shows the linearity achieved for methanol and ethanol in the concentration range 0.195–100 g/L. Table 5 shows the parameters obtained for the calibration curves of the two analytes extracted using K2CO3 and ethyl acetate.
Figure 4.

Linearity curves attained for samples in the concentration range of 0.195–100 g/L for (a) methanol and (b) ethanol.
Table 5. Calibration Curve Parameters Attained for Methanol and Ethanol Concentration Ranging from 0.195 to 100 g/L.
| component | a | b | R2 | linear range |
|---|---|---|---|---|
| methanol | 0.994 | 4.433 | 1.000 | 0.195–100 g/L |
| ethanol | 0.928 | 5.472 | 0.997 | 0.195–100 g/L |
The constant EE attained for methanol extraction supports the linearity of the calibration curve attained for this method. Furthermore, it is worthy to know the influence of CEtAc on the calibration curve of ethanol. A linear correlation can be made between the concentration of ethanol in the extract estimated previously (Cextract) and peak area. As can be seen in Figure 5, the response is linear with an R2 value of 0.998, indicating a good correlation. Table 5 shows the data indicating the linearity parameters attained for methanol and ethanol concentration studied.
Figure 5.

Linearity curve attained for the concentration of ethanol in the extract estimated (g/L) and peak area (A).
Limit of Quantification and Limit of Detection
According to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines,37 the LOQ is defined as the lowest concentration of the analyte present in the sample that can be detected and measured with an acceptable level of precision and accuracy. Therefore, the LOQ value is taken as the lowest value on the linearity curve that gives an R2 value higher than 0.99. As a result, the LOQ estimated from the calibration curve is 0.195 g/L for both methanol and ethanol. To confirm the reproducibility and accuracy of the measurement, the selected concentration (0.195 g/L) of methanol and ethanol was extracted 10 times, and each processed sample was injected three times using the selected GC–FID method. The coefficient of variation attained for methanol and ethanol is 1.98 and 2.34%, respectively, showing an acceptable reproducibility.
The ICH guidelines define the LOD as the lowest concentration of the analyte that can be detected but not necessarily quantitated. Following the assessment of linearity, the LOD value was calculated based on the root-mean-square error approach (RMSE). A numerical factor of 3 is multiplied with the RMSE value as recommended by the International Union of Pure and Applied Chemistry (IUPAC)38 to estimate the LOQ value. The LOD is considered as a signal other than the noise that is detected by the instrument, which is not necessarily quantifiable. To express such a signal in terms of concentration, a calibration procedure is to be considered and can be calculated from the following equation
| 3 |
where C is the concentration of the analyte of interest present in the sample, A is the peak area, b is the intercept of the calibration curve on the vertical axis, and a is the slope of the calibration curve. Therefore, the LOD value estimated for methanol was 0.012 g/L, and the LOQ value was taken as 0.195 g/L since it guarantees the linearity of the calibration curve. However, in the case of ethanol, the effect of ethyl acetate has to be taken into consideration due to the overestimation of the ethanol concentration in the extract. From the calibration curve constructed between the concentration of ethanol in the extract (Cextract) and peak area (A), the following equation can be considered
| 4 |
Combining eqs 2 and 4, the actual concentration of the sample can be estimated according to the equation
| 5 |
Therefore, from the above correlation, the LOD for ethanol quantification was estimated to be 0.004 g/L. The LOQ and LOD values estimated for methanol and ethanol quantification are listed out in Table 6 along with the calibration equation.
Table 6. Limit of Detection and Limit of Quantification Estimated for Methanol and Ethanol Quantification.
| component | calibration equation | LOD (g/L) | LOQ (g/L) |
|---|---|---|---|
| methanol | ln(A) = 0.994·ln(C) + 4.433 | 0.012 | 0.195 |
| ethanol | ln(A) = 0.928·ln(C) + 5.472 | 0.004 | 0.195 |
Residual Effect and Matrix Effect
The large amount of salts added to the samples facilitates the migration of volatile components toward the organic phase. The partitioning of the volatile components into the organic phase can differ depending on the composition of the biological sample.39 To check the effect of the matrix used in actual bioprocessing, quantification of known quantities of ethanol and methanol was performed in distilled water and three different growth media that are commonly used in P. pastoris fermentation.
Extraction of Methanol and Ethanol from Distilled Water
To indicate the EE of the analytes of interest, experiments were conducted by dissolving methanol and ethanol in distilled water at a concentration of 1 g/L. These samples were processed by first salting out using K2CO3, followed by liquid–liquid extraction using ethyl acetate. The results indicate that the EE attained using distilled water as a matrix does not interfere with the analysis. The EE attained from distilled water is shown in Table 7. It can be seen that the EE value attained for distilled water is in line with the recoveries calculated over the entire range of the calibration curve of methanol and ethanol.
Table 7. Extraction Recovery of Methanol and Ethanol from Distilled Water and Commonly Used P. pastoris Fermentation Media.
| EE (%) |
||
|---|---|---|
| matrix | methanol | ethanol |
| distilled water | 67 | 105 |
| BMGY media | 73 | 115 |
| FM22 media | 61 | 92 |
| BSM | 69 | 110 |
Extraction of Methanol and Ethanol from Commonly Used P. pastoris Fermentation Media
To study the effect of media components used in the bioprocess on the EE of the targeted analytes, three commonly used fermentation media were selected to perform the SALLE technique using K2CO3 and ethyl acetate. These media were FM22, BSM, and BMGY. The results indicated in Table 7 show that the selected pretreatment works very well irrespective of the sample matrices that have been selected. The data attained also suggests that there is a difference in the extraction efficiencies between the matrices that have been involved. Pintać et al.36 detail in their study that the EE of a solvent can be greatly influenced by the matrix involved. In this study, the first two media are minimal media containing several salts that lead to the presence of Na+, K+, Mg2+, Ca2+, SO42–, Cl–, and PO43–, whereas the latter one is a complex media comprising mainly peptone, yeast extract, and yeast nitrogen base. The extraction efficiencies attained for these matrices are seen to be increasing in the order BMGY > BSM > distilled water > FM22. BSM and FM22 media are observed to have a lower EE, which indicates that the extraction is impacted by the presence of various salts present in the media. One of the most important parameters that can affect the extraction is the pH of the sample.40 Hence, the pH variations between the different media are expected to influence the final extraction yield. Overall, the extraction efficiencies were found to be in the same order of magnitude, irrespective of the matrix, and indicate proximity to the values estimated from the calibration curve. However, to obtain a high precision on the quantification, it is advised to use a similar matrix as the samples for measuring the calibration standards to achieve a similar EE.
Repeatability and Precision
The precision of the selected method was determined by checking the intraday and interday measurements (RSD %). The intraday precision of the method was evaluated by analyzing three different concentrations of methanol and ethanol (0.195, 0.781, and 3.125 g/L) within the calibration curve. To determine the variability in the pretreatment if reproduced on the same day, three replicates of each concentration were extracted using the selected SALLE technique and injected in triplicates. In the case of interday precision, the three concentrations of methanol and ethanol samples (0.195, 0.781, and 3.125 g/L) were extracted in three replicates on consecutive days, and the samples were injected in triplicate. The coefficient of variation determined for all the samples of methanol and ethanol for intraday and interday precision tests are shown in Table 8. The coefficient of variation attained for both measurements was between 0.5 and 1.6%, indicating very good repeatability of the sample pretreatment technique.
Table 8. Intraday and Interday Precision for Methanol and Ethanol Estimated at Three Different Concentrations of 0.195, 0.781, and 3.125 g/L.
| intraday | interday | ||
|---|---|---|---|
| component | concentration (g/L) | precision (% RSD) | precision (% RSD) |
| methanol | 0.195 | 1.55 | 1.55 |
| 0.781 | 0.79 | 0.99 | |
| 3.125 | 0.66 | 0.77 | |
| ethanol | 0.195 | 1.17 | 1.15 |
| 0.781 | 0.58 | 0.98 | |
| 3.125 | 0.64 | 0.70 |
Analysis of Methanol in the Fermentation Sample
To check the applicability of the developed method, the bioreactor samples retrieved from a P. pastoris fermentation were processed using K2CO3 and ethyl acetate. A batch bioreactor experiment was conducted with an initial methanol concentration of 10 g/L. The entire bioprocess lasted for 96 h, and a sample was withdrawn every hour during the daytime throughout the experiment. Each of these samples was treated using K2CO3 and ethyl acetate to quantify the methanol content at each time point. The organic phase containing the analyte of interest (methanol) was injected into the GC. The results attained from these injections are shown in Figure 6. To illustrate the change in methanol concentration throughout a P. pastoris fermentation process, a graph is plotted to indicate the consumption of methanol in a batch mode fermentation process together with the increase in cell biomass. This graph demonstrates that a smooth evolution of the methanol concentration as a function of time can be determined to monitor the bioprocesses.
Figure 6.

Growth and substrate consumption of P. pastoris utilizing methanol in the batch mode at a concentration of 10 g/L.
Comparison against an HPLC Method
The accuracy of the developed method was verified by comparing with the accuracy of an HPLC method to assess the concentration of methanol present in the fermentation samples. This was performed in accordance with the published work of Parpinello and Versari.41 According to this method, the samples were injected into the system without any preprocessing step. First, a calibration curve was constructed and plotted for methanol samples ranging from 0.195 to 100 g/L as shown in Figure 7a. It was observed that the linearity attained using the HPLC method showed a lower coefficient of determination (R2), which was approximately 0.941 in comparison with the GC method. Therefore, the concentration range in the calibration curve was adjusted to attain a higher R2 value. The adjusted calibration curve is indicated in Figure 7b, where a methanol concentration range of 1.563–100 g/L was used, resulting in an R2 value of 0.991. In comparison, the SALLE method in combination with GC showed an R2 value of 1.000, indicating a good linearity of the method within the range of 0.195–100 g/L. Therefore, it was concluded from the comparison that the GC method shows a better linearity over a large range of concentrations compared to the HPLC method.
Figure 7.

Linearity curve attained for methanol samples using HPLC in the concentration range of (a) 0.195–100 g/L with an R2 value of 0.9419 and (b) 1.563–100 g/L with an R2 value of 0.991.
Based on the calibration curve attained from the HPLC method, the concentration of methanol in the bioreactor samples was estimated. It was observed from these results, as indicated in Figure 8, that the estimated values resulted in an initial concentration of 7.410 g/L, which was much lower than the actual initial concentration of 10 g/L of methanol in the sample. The differences observed in the estimated values of methanol from the HPLC analysis can be arising from interferences present in the methanol samples, which are not pretreated before injection. The SALLE technique, on the other hand, ensures the removal of these interferences from the samples, thereby ensuring the accurate estimation of methanol concentration.
Figure 8.

Growth and substrate consumption of P. pastoris utilizing methanol in the batch mode at a concentration of 10 g/L analyzed using HPLC.
Hence, comparing the results attained from HPLC and GC, it was observed that the SALLE technique utilizing GC provided a more accurate quantification of the methanol concentration present in the fermentation samples over a wider range of concentrations.
Conclusions
In this study, the SALLE technique is adopted for the determination of methanol and ethanol present in P. pastoris fermentation samples using GC–FID. The optimized and validated SALLE method found in this study using K2CO3 and ethyl acetate is simple, inexpensive, efficient, and reproducible to use for the detection and quantification of methanol and ethanol in biological mixtures. Moreover, the recovery percentages estimated from this method are high, which is widely preferred in extraction techniques and strengthens its usage in various applications. The technique established through this study can be easily applied to biological samples with minimal volumes of the sample and solvent. Moreover, this process will allow a fast analysis of the volatile components considered, which makes it convenient to use for routine analysis. Furthermore, it was demonstrated that it can be applied to matrices that are used in P. pastoris fermentation processes. The recovery of the analyte after such a processing step was good and showed good reproducibility and linearity for actual samples retrieved from bioprocesses. To the author’s knowledge, such a technique for analyses and quantification of methanol and ethanol found in yeast fermentation has not yet been investigated and is therefore envisaged to be an alternative for laborious or expensive techniques required for routine analysis.
Acknowledgments
This work was funded by the KU Leuven Research Fund through project C24/18/046 by the Research Foundation Flanders (FWO) through project G0B4121N and the European Commission within the framework of the Erasmus+ FOOD4S Programme (Erasmus Mundus Joint Master Degree in Food Systems Engineering, Technology and Business 619864-EPP-1-2020-1-BE-EPPKA1-JMD-MOB). Author S.A. was funded by the Research Foundation Flanders (FWO), grant number 1224620N.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00055.
Media formulations used for P. pastoris fermentation involving BSM and FM22 media, media formulation used for the preparation of BMGY media, supplementary salt formulations PTM1 and PTM4 used for BSM and FM22, respectively, and list of chemicals and their suppliers used in the media formulation for P. pastoris fermentation (PDF)
Author Contributions
# J.A. and S.A. contributed equally. J.A., S.A., and J.F.M.V.I. contributed to conceptualization; S.A. contributed to methodology; J.A. contributed to investigation; J.A. and S.A. contributed to validation; J.A. and S.A. contributed to formal analysis; S.A. contributed to software; J.F.M.V.I. contributed to resources; J.A. and S.A. contributed to data curation; J.A. contributed to writing—original draft preparation; S.A. and J.F.M.V.I. contributed to writing—review and editing; J.A and S.A. contributed to visualization; J.F.M.V.I. contributed to supervision; and S.A. and J.F.M.V.I. contributed to funding acquisition. All authors have read and agreed to the published version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Garrigós-Martínez J.; Vuoristo K.; Nieto-Taype M. A.; Tähtiharju J.; Uusitalo J.; Tukiainen P.; Schmid C.; Tolstorukov I.; Madden K.; Penttilä M.; Montesinos-Seguí J. L. Bioprocess performance analysis of novel methanol-independent promoters for recombinant protein production with Pichia pastoris. Microb. Cell Fact. 2021, 20, 1–12. 10.1186/s12934-021-01564-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Anumanthan A.; Gao X.-G.; Ilangovan K.; Suzara V. V.; Düzgüneş N.; Renugopalakrishnan V. Expression of recombinant proteins in Pichia pastoris. Appl. Biochem. Biotechnol. 2007, 142, 105–124. 10.1007/s12010-007-0003-x. [DOI] [PubMed] [Google Scholar]
- Katakura Y.; Zhang W.; Zhuang G.; Omasa T.; Kishimoto M.; Goto Y.; Suga K.-I. Effect of methanol concentration on the production of human β2-glycoprotein I domain V by a recombinant Pichia pastoris: a simple system for the control of methanol concentration using a semiconductor gas sensor. J. Ferment. Bioeng. 1998, 86, 482–487. 10.1016/s0922-338x(98)80156-6. [DOI] [Google Scholar]
- Sreekrishna K.; Brankamp R. G.; Kropp K. E.; Blankenship D. T.; Tsay J.-T.; Smith P. L.; Wierschke J. D.; Subramaniam A.; Birkenberger L. A. Strategies for optimal synthesis and secretion of heterologous proteins in the methylotrophic yeast Pichia pastoris. Gene 1997, 190, 55–62. 10.1016/s0378-1119(96)00672-5. [DOI] [PubMed] [Google Scholar]
- Lee C. Y.; Acree T. E.; Butts R. M. Determination of methyl alcohol in wine by gas chromatography. Anal. Chem. 1975, 47, 747–748. 10.1021/ac60354a048. [DOI] [Google Scholar]
- Almuzara C.; Cos O.; Baeza M.; Gabriel D.; Valero F. Methanol determination in Pichia pastoris cultures by flow injection analysis. Biotechnol. Lett. 2002, 24, 413–417. 10.1023/a:1014554324117. [DOI] [Google Scholar]
- Macauley-Patrick S.; Fazenda M. L.; McNeil B.; Harvey L. M. Heterologous protein production using the Pichia pastoris expression system. Yeast 2005, 22, 249–270. 10.1002/yea.1208. [DOI] [PubMed] [Google Scholar]
- Biechele P.; Busse C.; Solle D.; Scheper T.; Reardon K. Sensor systems for bioprocess monitoring. Eng. Life Sci. 2015, 15, 469–488. 10.1002/elsc.201500014. [DOI] [Google Scholar]
- Guarna M. M.; Lesnicki G. J.; Tam B. M.; Robinson J.; Radziminski C. Z.; Hasenwinkle D.; Boraston A.; Jervis E.; MacGillivray R. T. A.; Turner R. F. B.; Kilburn D. G. On-line monitoring and control of methanol concentration in shake-flask cultures of Pichia pastoris. Biotechnol. Bioeng. 1997, 56, 279–286. 10.1002/(sici)1097-0290(19971105)56:3<279::aid-bit5>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Nunes C.; Rocha S. M.; Saraiva J.; Coimbra M. A. Simple and solvent-free methodology for simultaneous quantification of methanol and acetic acid content of plant polysaccharides based on headspace solid phase microextraction-gas chromatography (HS-SPME-GC-FID). Carbohydr. Polym. 2006, 64, 306–311. 10.1016/j.carbpol.2005.11.039. [DOI] [Google Scholar]
- Kwiecien N. W.; Bailey D. J.; Rush M. J. P.; Cole J. S.; Ulbrich A.; Hebert A. S.; Westphall M. S.; Coon J. J. High-resolution filtering for improved small molecule identification via GC/MS. Anal. Chem. 2015, 87, 8328–8335. 10.1021/acs.analchem.5b01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins M. J.; Langford V. S. Standard Validation Protocol for Selected Ion Flow Tube Mass Spectrometry Methods Applied to Direct Headspace Analysis of Aqueous Volatile Organic Compounds. Anal. Chem. 2021, 93, 8386–8392. 10.1021/acs.analchem.1c01310. [DOI] [PubMed] [Google Scholar]
- Bursová M.; Hložek T.; Čabala R. Simultaneous determination of methanol, ethanol and formic acid in serum and urine by headspace GC-FID. J. Anal. Toxicol. 2015, 39, 741–745. 10.1093/jat/bkv075. [DOI] [PubMed] [Google Scholar]
- Xavier-Junior F. H.; Maciuk A.; Rochelle do Vale Morais A.; Alencar E. d. N.; Garcia V. L.; Tabosa do Egito E. S.; Vauthier C. Development of a gas chromatography method for the analysis of copaiba oil. J. Chromatogr. Sci. 2017, 55, 969–978. 10.1093/chromsci/bmx065. [DOI] [PubMed] [Google Scholar]
- Rodinkov O. V.; Bugaichenko A. S.; Moskvin L. N. Static Headspace Analysis and Its Current Status. J. Anal. Chem. 2020, 75, 1–17. 10.1134/s106193482001013x. [DOI] [Google Scholar]
- Muna E. D. M.; Bizarri C. H. B.; Maciel J. R. M.; Da Rocha G. P.; de Araújo I. O. Method validation for methanol quantification present in working places. J. Phys.: Conf. Ser. 2015, 575, 012031. 10.1088/1742-6596/575/1/012031. [DOI] [Google Scholar]
- Wang M.-L.; Wang J.-T.; Choong Y.-M. Simultaneous quantification of methanol and ethanol in alcoholic beverage using a rapid gas chromatographic method coupling with dual internal standards. Food Chem. 2004, 86, 609–615. 10.1016/j.foodchem.2003.10.029. [DOI] [Google Scholar]
- Chen S. H.; Wu H. L.; Yen C. H.; Wu S. M.; Lin S. J.; Kou H. S. Trace determination of methanol in water–ethanol solution by derivatization and high-performance liquid chromatography. J. Chromatogr. A 1998, 799, 93–99. 10.1016/s0021-9673(97)01055-8. [DOI] [PubMed] [Google Scholar]
- Valdez D.; Reier J. C. A simplified procedure for the derivatization of alcohols at dilute levels in aqueous solutions with 3, 5-dinitrobenzoyl chloride. J. Chromatogr. Sci. 1986, 24, 356–360. 10.1093/chromsci/24.8.356. [DOI] [Google Scholar]
- Haj-Yehia A. I.; Benet L. Z. Determination of alcohols by high-performance liquid chromatography with fluorimetric detection after precolumn derivatization with 2-(4-carboxyphenyl)-6-methoxybenzofuran. J. Chromatogr. A 1996, 724, 107–115. 10.1016/0021-9673(95)00986-8. [DOI] [Google Scholar]
- Livesey J. F.; Perkins S. L.; Tokessy N. E.; Maddock M. J. Simultaneous determination of alcohols and ethylene glycol in serum by packed-or capillary-column gas chromatography. Clin. Chem. 1995, 41, 300–305. 10.1093/clinchem/41.2.300. [DOI] [PubMed] [Google Scholar]
- Pollack G. M.; Kawagoe J. L. Determination of methanol in whole blood by capillary gas chromatography with direct on-column injection. J. Chromatogr. B: Biomed. Sci. Appl. 1991, 570, 406–411. 10.1016/0378-4347(91)80546-o. [DOI] [PubMed] [Google Scholar]
- Tulashie S. K.; Appiah A. P.; Torku G. D.; Darko A. Y.; Wiredu A. Determination of methanol and ethanol concentrations in local and foreign alcoholic drinks and food products (Banku, Ga kenkey, Fante kenkey and Hausa koko) in Ghana. Int. J. Food Contam. 2017, 4, 1–5. 10.1186/s40550-017-0059-5. [DOI] [Google Scholar]
- Caruso R.; Gambino G. L.; Scordino M.; Sabatino L.; Traulo P.; Gagliano G. Gas chromatographic quantitative analysis of methanol in wine: Operative conditions, optimization and calibration model choice. Nat. Prod. Commun. 2011, 6, 1939. 10.1177/1934578x1100601237. [DOI] [PubMed] [Google Scholar]
- Zhang C.-Y.; Lin N.-B.; Chai X.-S.; Zhong-Li D. G.; Barnes D. G. A rapid method for simultaneously determining ethanol and methanol content in wines by full evaporation headspace gas chromatography. Food Chem. 2015, 183, 169–172. 10.1016/j.foodchem.2015.03.048. [DOI] [PubMed] [Google Scholar]
- Hu H.-C.; Chai X.-S. Determination of methanol in pulp washing filtrates by desiccated full evaporation headspace gas chromatography. J. Chromatogr. A 2012, 1222, 1–4. 10.1016/j.chroma.2011.11.045. [DOI] [PubMed] [Google Scholar]
- de Paula Pereira P.; Sousa S. E.; de Freitas F. T.; De Andrade J. B. Determination of methanol and ethanol by gas chromatography following air sampling onto florisil cartridges and their concentrations at urban sites in the three largest cities in Brazil. Talanta 1999, 49, 245–252. 10.1016/s0039-9140(98)00376-2. [DOI] [PubMed] [Google Scholar]
- Pontes H.; Guedes de Pinho P.; Casal S.; Carmo H.; Santos A.; Magalhaes T.; Remiao F.; Carvalho F.; Bastos M. L. GC determination of acetone, acetaldehyde, ethanol, and methanol in biological matrices and cell culture. J. Chromatogr. Sci. 2009, 47, 272–278. 10.1093/chromsci/47.4.272. [DOI] [PubMed] [Google Scholar]
- Gezahegn T.; Tegegne B.; Zewge F.; Chandravanshi B. S. Salting-out assisted liquid–liquid extraction for the determination of ciprofloxacin residues in water samples by high performance liquid chromatography–diode array detector. BMC Chem. 2019, 13, 1–10. 10.1186/s13065-019-0543-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y. Q.; Weng N. Salting-out assisted liquid–liquid extraction for bioanalysis. Bioanalysis 2013, 5, 1583–1598. 10.4155/bio.13.117. [DOI] [PubMed] [Google Scholar]
- Sazali N. H.; Alshishani A.; Saad B.; Chew K. Y.; Chong M. M.; Miskam M. Salting-out assisted liquid–liquid extraction coupled with high-performance liquid chromatography for the determination of vitamin D3 in milk samples. R. Soc. Open Sci. 2019, 6, 190952. 10.1098/rsos.190952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M. S.; Ji Q.; Zhang J.; El-Shourbagy T. A. Historical review of sample preparation for chromatographic bioanalysis: pros and cons. Drug Dev. Res. 2007, 68, 107–133. 10.1002/ddr.20173. [DOI] [Google Scholar]
- Filip S.; Pavlić B.; Vidović S.; Vladić J.; Zeković Z. Optimization of microwave-assisted extraction of polyphenolic compounds from Ocimum basilicum by response surface methodology. Food Anal. Methods 2017, 10, 2270–2280. 10.1007/s12161-017-0792-7. [DOI] [Google Scholar]
- Złotek U.; Mikulska S.; Nagajek M.; Świeca M. The effect of different solvents and number of extraction steps on the polyphenol content and antioxidant capacity of basil leaves (Ocimum basilicum L.) extracts. Saudi J. Biol. Sci. 2016, 23, 628–633. 10.1016/j.sjbs.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S.; Yi C.; Qiu X. Energy-saving recovery of acetone, butanol, and ethanol from a prefractionator by the salting-out method. J. Chem. Eng. Data 2013, 58, 3297–3303. 10.1021/je400740z. [DOI] [Google Scholar]
- Pintać D.; Majkić T.; Torović L.; Orčić D.; Beara I.; Simin N.; Mimica-Dukić N.; Lesjak M. Solvent selection for efficient extraction of bioactive compounds from grape pomace. Ind. Crops Prod. 2018, 111, 379–390. 10.1016/j.indcrop.2017.10.038. [DOI] [Google Scholar]
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use . ICH Harmonised Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology Q2 (R1) (consulted 22.7.2021), 2021. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf.
- Currie L. A. IUPAC Nomenclature in evaluation of analytical methods including detection and quantification capabilities. Pure Appl. Chem. 1995, 67, 1699–1723. 10.1351/pac199567101699. [DOI] [Google Scholar]
- Watts M. T.; Mcdonald O. L. The effect of sodium chloride concentration, water content, and protein on the gas chromatographic headspace analysis of ethanol in plasma. Am. J. Clin. Pathol. 1990, 93, 357–362. 10.1093/ajcp/93.3.357. [DOI] [PubMed] [Google Scholar]
- Orooji N.; Takdastan A.; Jalilzadeh Yengejeh R.; Jorfi S.; Davami A. H. A quick and inexpensive method to determine 2, 4-dichlorophenoxyacetic acid residues in water samples by HPLC. Desalin. Water Treat. 2021, 217, 329–338. 10.5004/dwt.2021.26905. [DOI] [Google Scholar]
- Parpinello G. P.; Versari A. A simple high-performance liquid chromatography method for the analysis of glucose, glycerol, and methanol in a bioprocess. J. Chromatogr. Sci. 2000, 38, 259–261. 10.1093/chromsci/38.6.259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.