

Abstract
Inflammatory conditions represent the largest class of chronic skin disease, but the molecular dysregulation underlying many individual cases remains unclear. scRNA-seq has increased precision in dissecting the complex mixture of immune and stromal cell perturbations in inflammatory skin disease states. We single-cell profiled CD45+ immune cell transcriptomes from 31 patient skin samples (7 atopic dermatitis, 8 psoriasis vulgaris, 2 lichen planus, 1 bullous pemphigoid, 6 clinical/histopathologically indeterminate rashes, and 7 healthy controls). Our data revealed active proliferative expansion of the Treg and Trm components and universal T cell exhaustion in human rashes, with a relative attenuation of antigen-presenting cells. Skin-resident memory T cells showed the greatest transcriptional dysregulation in both atopic dermatitis and psoriasis, while atopic dermatitis also demonstrated recurrent abnormalities in ILC and CD8+ cytotoxic lymphocytes. Transcript signatures differentiating these rash types included genes previously implicated in Th2/Th17 diatheses, segregated in unbiased functional networks, and accurately identified disease class in untrained validation data sets. Notably, these gene signatures were able to classify clinicopathologically ambiguous rashes with diagnoses consistent with therapeutic response. Thus, we have defined major classes of human inflammatory skin disease at the molecular level and described a quantitative method to classify indeterminate instances of pathologic inflammation. To make this approach accessible to the scientific community, we created a proof-of-principle web interface (RashX) where scientists and clinicians can visualize their patient-level rash scRNA-seq-derived data in the context of our Th2/Th17 transcriptional framework.
One Sentence Summary:
scRNA-seq of immune cells can distinguish different inflammatory skin diseases and classify indeterminate rashes.
INTRODUCTION
Atopic dermatitis (AD) and psoriasis vulgaris (PV), prototypical inflammatory skin diseases, collectively affect about 10% of adults in the United States (1–3). AD is classically viewed as a Th2 skewed inflammatory disease, with psoriasis displaying a Th1/Th17 predominance. Biologic therapies now successfully target specific, dysregulated immune pathways in each disease - IL4Rα or JAK1 inhibitors in atopic dermatitis and IL-17 or IL-23 antagonists in psoriasis. While these drugs represent remarkable advances in treatment, ~20–50% of patients still do not achieve significant improvement on a given drug (4–6). One major challenge is the clinical heterogeneity of inflammatory skin disease (7), which can preclude a definitive diagnosis and complicates optimal treatment choice. A molecular endotyping approach could offer clinicians more objective criteria to classify inflammatory disease and provide a rational basis for therapeutic selection.
Identification of molecular abnormalities defining inflammatory skin disease is impeded by bulk cell-based profiling methods, which likely obscure cell type-specific transcriptional dysregulation. Studies have attempted to overcome disease heterogeneity by comparing internally controlled patient populations (8). Single-cell RNA sequencing (scRNA-seq) has t identified new disease-specific genes across diverse inflammatory pathologies (9) (10–13). Such high-resolution methods capturing simultaneous molecular portraits of different cell types are particularly appealing in human skin, where the complex composition of tissue limits mechanistic biology. Single-cell RNA-seq studies of AD-affected skin reveal increased type 2/type 22 T cells, inflammatory dendritic cells, and tissue resident memory T cells (12, 14, 15). Dupilumab-treated AD is characterized by persistence of transcriptionally defined mature dendritic cell subsets and tissue resident memory subpopulations (i.e. type 2/type 22 T cells, and TH2A)(16). In psoriasis, single cell transcriptomic studies show increased Th17/Tc17 cells in active lesions, where two non-exhausted, CXCL13-expressing Tc17 subpopulations correlate with disease severity (9, 12). However, these advances do not clarify whether inflammatory skin diseases as a whole can be reliably molecularly classified on the patient level.
In this study, we hypothesized that by focusing exclusively on cutaneous immune cells and applying increased analytic resolution (i.e. 41 cutaneous immune cell type classes), scRNA-seq approaches could improve identification and understanding of molecular abnormalities discriminating atopic dermatitis and psoriasis vulgaris. T cells and antigen-presenting cells (APCs) from rashes were profiled using unbiased single-cell droplet microfluidics RNA (10X Genomics) (17) and protein epitope (cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq)) (18) approaches. Our study targeted a series of 8 PV, 7 AD, and also 6 clinical/histopathologically indeterminate rashes (CIRs) which presented in adulthood and harbored clinical and histopathologic features of both psoriasis and atopic dermatitis. To more precisely defined the cell type-specific transcriptional alterations distinguishing these diseases, we used molecular signatures to investigate and classify individual, indeterminate cases of cutaneous inflammation displaying clinical or histopathologic features of both atopic dermatitis and psoriasis. With this approach, we created a webtool to help clinicians and scientists identify their clinically indeterminate skin rash cases.
RESULTS
scRNA-seq detected conserved skin CD45+ immune cell classes
Our skin samples included 8 samples of psoriasis vulgaris, 7 samples of atopic dermatitis, 1 case of bullous pemphigoid, 2 cases of lichen planus, 6 CIRs harboring clinical and histopathologic features of both psoriasis and atopic dermatitis, and 7 healthy controls. Additional clinical characteristics for each sample are described in Table S1. After enzymatic digestion of donor skin biopsies, we flow sorted live CD45+ cells and performed Chromium 3’ single cell RNA-seq and CITE-seq protein epitope sequencing. We obtained transcriptomic data from 158,037 single cells after quality control filtering (removal of doublets and poor-quality cells). Using Seurat, clustering was performed using Louvain community detection-based modularity optimization (19). We first utilized a resolution parameter of 0.4 based on clustree optimization approaches (20), delineating 16 immune cell clusters, including six lymphocyte clusters (CD3+ or KLRB1+), nine antigen presenting cell clusters (HLA-DRA+), and a mast cell cluster highly expressing TPSAB1 (tryptase) (Fig. S1A, S1B and Table S2).
We speculated that more finely demarcating immune cell populations might reveal new cell type-specific expression differences, either between healthy and inflamed skin or between disease classes. We separately subclustered the CD3+/KLRB1+ lymphocyte populations and HLA-DRA+ APC populations into 23 and 24 higher resolution classes, respectively, again based on clustree-based optimization. After removal of clusters containing mostly non-immune or low-quality cells, 21 CD3+/KLRB1+ clusters and 19 HLA-DR+ clusters were retained (Fig. S1C). Including the previously described mast cell cluster, this classification generated 41 final clusters (displayed as a uniform manifold approximation and projection (UMAP) representation in Fig. 1A, Table S3, S4). Larger immune clusters were well represented across normal and major disease classes, suggesting limited distortion from sample-specific batch effects (Fig. 1B and S1D).
Fig. 1. Immune single cell landscape of rash-affected and normal human skin (31 samples).
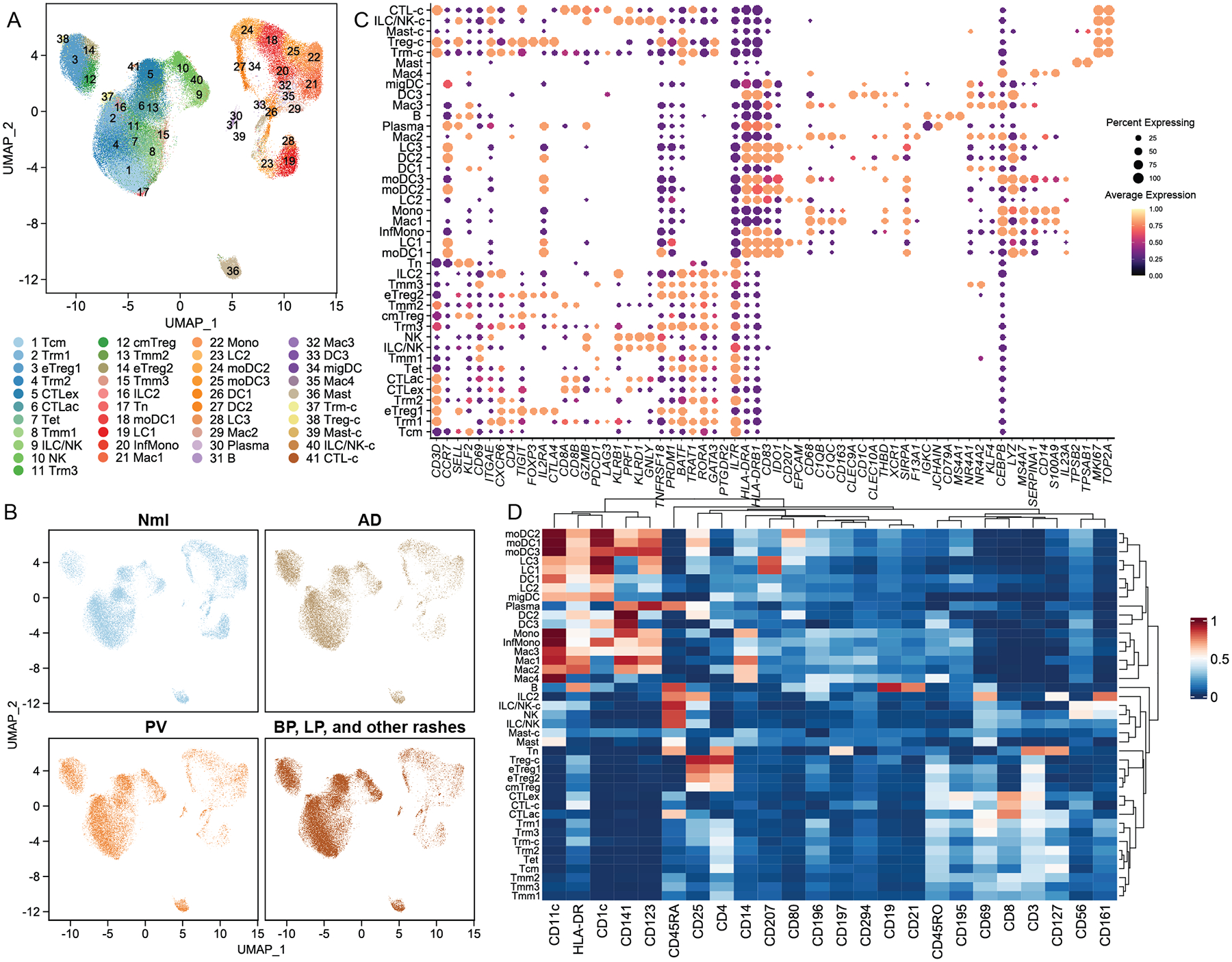
A) UMAP representation of 41 single cell RNA-sequencing (scRNA-seq) defined immune cell classes. B) UMAP representation of cell distribution across immune cell classes for disease classes and healthy control skin. C) Expression of critical marker transcripts (columns) distinguishing immune cell classes (rows). Size of dots represents the fraction of cells expressing a particular marker, and color intensity indicates mean normalized scaled expression levels. D) Expression of protein epitope (CITE-seq) markers for the 41 transcript-based immune cell clusters. Color intensity represents fraction of cells expressing a given marker.
We next assigned identities to the 41 immune cell populations by comparing marker genes for each cluster (i.e. the most differentially expressed transcripts or epitope markers between cells in that cluster versus all other cells; Table S5) against canonical markers for established cell types. Among T cells, we identified two CCR7+, SELL+, KLF2+ (21), CD3D+ populations, one corresponding to central memory cells (Tcm) with elevated CD69 and CD45RO protein epitope expression (CITE-seq data) and one smaller cluster representing a naïve T cell population with high CD45RA protein expression (Fig. 1C, 1D). We also identified three CCR7+/SELL− migratory memory classes (Tmm1, Tmm2, and Tmm3) (22). Three CD3D+ T cell clusters expressed tissue-resident memory T cell marker transcripts ITGAE (CD103) and CXCR6 and the CD69 protein epitope (CITE-seq data) (23), which we termed Trm1, Trm2, and Trm3, contained both CD4+ and CD8A+ cells (Fig. 1C, 1D). Three other CD4+ populations were identified as regulatory T cells based on the expression of FOXP3, TIGIT, CTLA4, IL2RA (CD25), and IKZF2 (Helios) (24, 25) (Fig. 1C). Two of these regulatory T cell populations displayed effector markers (TNFRSF18, PRDM1) (26) and were termed eTreg1 and eTreg2, while a third CD45RO+ class harboring SELL and CCR7 transcript was classified as central memory Tregs (cmTreg, Fig. 1C, 1D) (27). A small CD3D+ T cell cluster bearing elevated levels of numerous epigenetic regulators, transcription factors, and lncRNAs such as BATF, SNHG12, and ZFAS1, was named Tet.
Two CD8A+CD8B+ clusters were defined as cytotoxic T cells based on expression of GZMB, NKG7 and CCL5, including an activated cluster (CTLac) expressing TNFRSF4, TNFRSF18, and CD96 (28, 29), and a closely related class enriched in canonical exhaustion markers such as PDCD1 and LAG3 (CTLex) (30) (Fig. 1C). We also defined 3 innate lymphoid/natural killer cell populations as KLRB1 (CD161)+ with absent or relatively low CD3D, CD19/MS4A1, CD14, HLA-DRA expression (31). Of these, one small cluster expressed the type 2 transcriptional factors GATA3 and PTGDR2, and was identified as an innate lymphoid cell population (ILC2)(12). Two populations of KLRD1+, GNLY+, PRF1+, GZMB+ cells were assigned as either NK or ILC/NK cells, with the former expressing high levels of the CD56 epitope by CITE-seq (Fig. 1C, 1D).
We also identified a group of myeloid lineage subpopulations (32) (Fig. 1C, 1D). Four macrophage populations were enriched for CD68, CEBPB, and FCER1G (33). Two were distinguished by complement transcripts C1QB and C1QC as well as the scavenger receptor CD163 (Mac1 and Mac3), another by alternative activation and suppression markers NR4A1, NR4A2, KLF4 (Mac2) (12), and the fourth by monocyte markers CD14 and S100A9 (Mac4). Three Langerhans cells populations were identified based on CD207 transcript and protein and CD1c and EPCAM expression (LC1, LC2, and LC3).
Five monocyte or monocyte-derived cell populations shared expression of monocyte-associated genes MS4A7, LYZ, and SERPINA1 (34) (Fig. 1C). One cluster representing classical monocytes (Mono) expressed higher levels of CD14 transcript and protein and S100A9 (35) (Fig 1C, 1D), while we designated another inflammatory monocytes (InfMono), as it was enriched for inflammatory genes such as IL1B, IL23A, and CXCL3 (12). The three remaining clusters also expressed MHCII molecules (HLA-DRA, HLA-DRB1) and were labeled monocyte-derived DC (moDC1, moDC2, and moDC3). We identified four DC classes (HLA-DRA+), one enriched in CD1C and CLEC10A (DC1), with two others expressing CLEC9A, CLEC10A and XCR1 (DC2 and DC3) (12) (Fig. 1C). There was an additional migratory DC (migDC) population which expressed CD1C and migratory/mature markers, such as FSCN1, LAMP3, and CCR7 (36).
Two minor populations were enriched in immunoglobulin genes (IGHG, IGHA, IGKC, and JCHAIN), consistent with B cell lineage. One was identified as B cells (CD19+, MS4A2+, Fig. 1C), also enriched for CD19 and CD21 epitopes (Fig. 1D), and one as plasma cells (CD19−, MS4A2−, IGHG1/IGHG4+) (37). A large population of mast cells (Mast) was easily distinguished by expression of TPSAB1 (tryptase) and TPSB2 (38) (Fig. 1C).
Five clusters showed elevated cell division transcripts, such as MKI67, TOP2A, CENPF, and UBE2C (39) (Fig. 1C). Four of these cycling cell subpopulations mapped closely to existing T cell clusters and were named accordingly: Tregs (Treg-c), Trm (Trm-c), CD8+ cytotoxic T cells (CTL-c), and NK and ILC cells (NK/ILC-c). One mitotically active myeloid group expressed mast cell markers and was named Mast-c.
We clustered our CD45+ cells into a comparatively large number of classes to maximize discovery of molecular abnormalities that might be obscured in conflated populations. Our classification comported well with both other published scRNA-seq cutaneous immune cell landscapes (11, 12) (Fig. S1E, F) and canonical CD45+ classes. As the latter classifications were developed primarily from flow cytometry experiments in blood and other non-cutaneous tissues, we expected our approaches to yield some small differences in subclass definition. Overall, our scRNA-seq derived analysis retrieved most well-established skin CD45+ immune cell populations, with robust representation in both rashes and healthy controls.
Inflamed skin is characterized by CD8+ T cell exhaustion and regulatory T cell expansion
To discern differences in immune cell composition between normal and rash-affected skin, we first calculated each cluster’s aggregate representation in each of these two states. This analysis showed inflammation was accompanied by relative increases in multiple lymphoid cell classes and proportionate decreases in myeloid populations (Fig. 2A). We next applied a weighted Gaussian linear model to compare normal and rash-derived CD45+ cell proportions (see Methods -Weighted Gaussian linear model for differential immune cell composition analysis), identifying 27 clusters with statistically significant alterations (Fig. 2B). Chronic inflammatory skin diseases showed an increase in exhausted CD8+ T cells (CTLex class up 80.4%, from 4.5% to 8.2%), highlighting marked exhaustion of CD8+ cells in virtually every rash sample assayed (Table S6). We also noted a substantial expansion of all three Treg classes, which were increased between 2.19 to 3.13-fold, generalizing a trend reported anecdotally in some rash types (40) (Table S6). Resident-memory T cell classes were also proportionately increased, with Trm1 up 71.4% from 6.3 to 10.7% and Trm2 up 108.7% from 4.3% to 9.0%, (Table S6). Mitotically active cell clusters were markedly expanded in rashes, revealing active proliferation of Trm, Treg, ILC/NK, and CD8+ T cell populations in lesional skin (Fig. 2B, Table S6).
Fig. 2. Enrichment of Treg, Trm, and exhausted CD8+T cell populations in rash-affected skin.
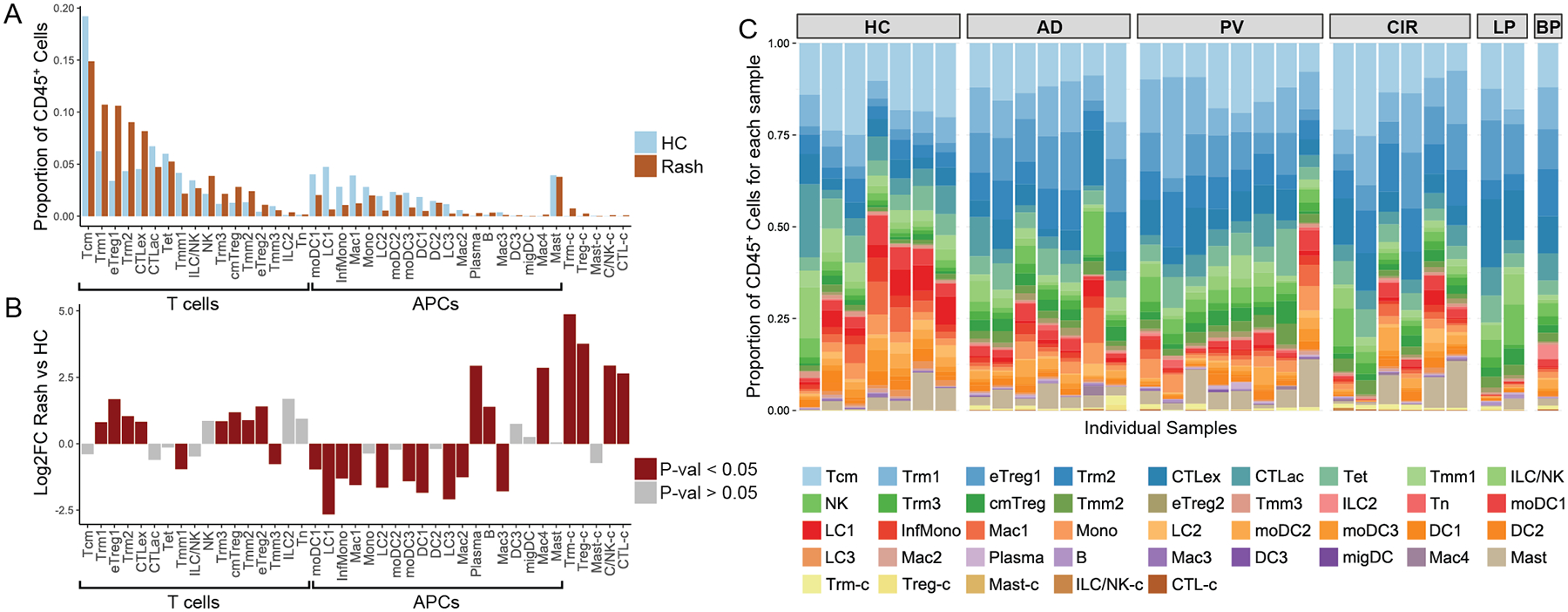
A) Distribution of immune cell populations for 7 healthy control (HC) and 24 rash-affected (Rash) skin samples. X-axis represents different immune cell populations. Y-axis represents proportion of CD45+ immune cells for each immune cell population in either rash-affected or normal skin. B) Quasi-binomial model Log2 fold change for rash-affected versus healthy control skin (Y-axis). X-axis represents different immune cell populations. Unpaired two sample t-test was used. Red colored bars indicate statistically significant changes (p-value < 0.05). C) Proportion of cells in each CD45+ immune cell population for each individual sample.
Analyses specific to atopic dermatitis and psoriasis were also performed. Cell population frequency assessment for PV and AD showed similar patterns for CTLac, Trm1, and Trm2 cell populations (in AD down 34.9%, up 70.2%, and up 109.6%, respectively; in PV down 29.2%, up 84.5%, and up 89.1%, respectively, Fig S3 and Table S6). However, CTLex and NK cells were more elevated in PV than in AD (in PV versus healthy control samples up 74.6% and 99.4%, respectively; in AD up 23.0 and 20.4%, respectively; Fig. S3 and Table S6). While absolute cell numbers were low, plasma and B cells also both showed significant increases in rash samples in aggregate and for plasma cells in both AD and PV (relative to healthy control skin, Table S6). Both disease-level analyses again showed expanded Treg and Trm cells as well as exhausted CD8+ T cells, suggesting critical roles for these subpopulations in initiating or maintaining cutaneous inflammation.
Distinct, cell-type specific patterning differentiates forms of pathologic skin inflammation
We applied a hurdle model-based approach (MAST) (41) across all 41 CD45+ cell populations to detect differentially expressed genes (DEGs) between 1) lesional psoriasis and healthy control skin (PV versus HC), 2) lesional atopic and healthy control skin (AD versus HC), and 3) lesional atopic dermatitis versus psoriatic skin (AD versus PV; Table S7). In each comparison, we required an absolute log2 fold-change difference of 0.425 and p value < 0.001 of genes for further consideration.
The Trm1, CTLac, ILC/NK, Trm3, moDC1, InfMono, Mono, and LC2 populations showed abundant DEGs differentiating inflamed versus control skin (Fig. 3A, Table S11). For example, the number of DEGs for PV versus HC and AD versus HC comparisons in Trm1 were 531 and 327, respectively; in CTLac 306 and 574; and in InfMono, 522 and 451 (Fig. 3A, Table S11). In distinguishing atopic dermatitis from psoriasis, these 8 T cell and APC populations also displayed the most statistically significant DEGs for the AD versus PV comparison, although fewer than disease-normal comparisons, likely attributable to shared inflammatory gene regulation in the two conditions (Fig. 3A). For example, DEGs that differentiated AD and PV numbered 95 in Trm1, 137 in CTLac, and 242 in InfMono (Fig. 3A, Table S11).
Fig. 3. Conservatively selected transcriptional abnormalities discriminating psoriasis and atopic dermatitis demonstrate cell-type specific patterning.
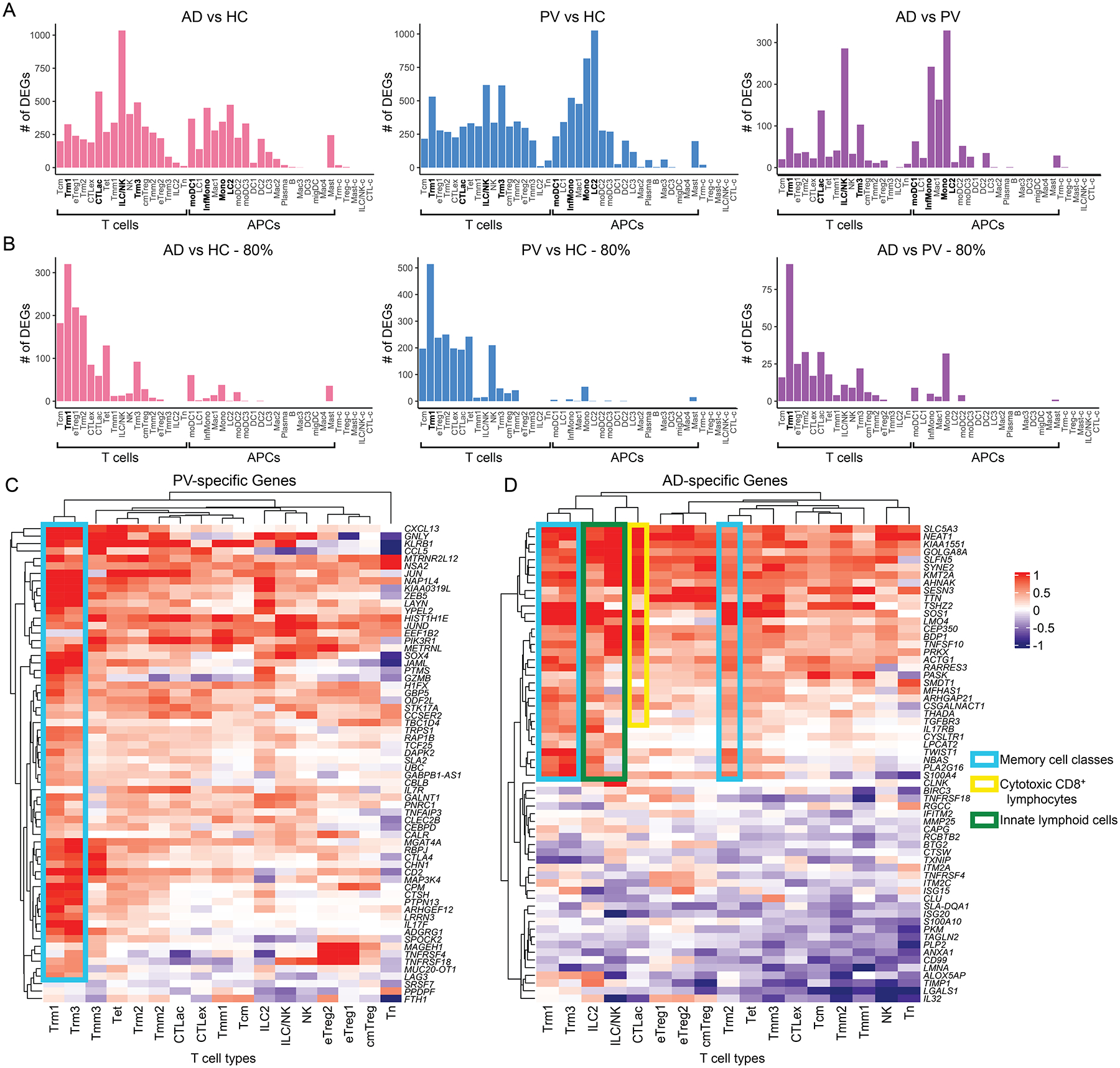
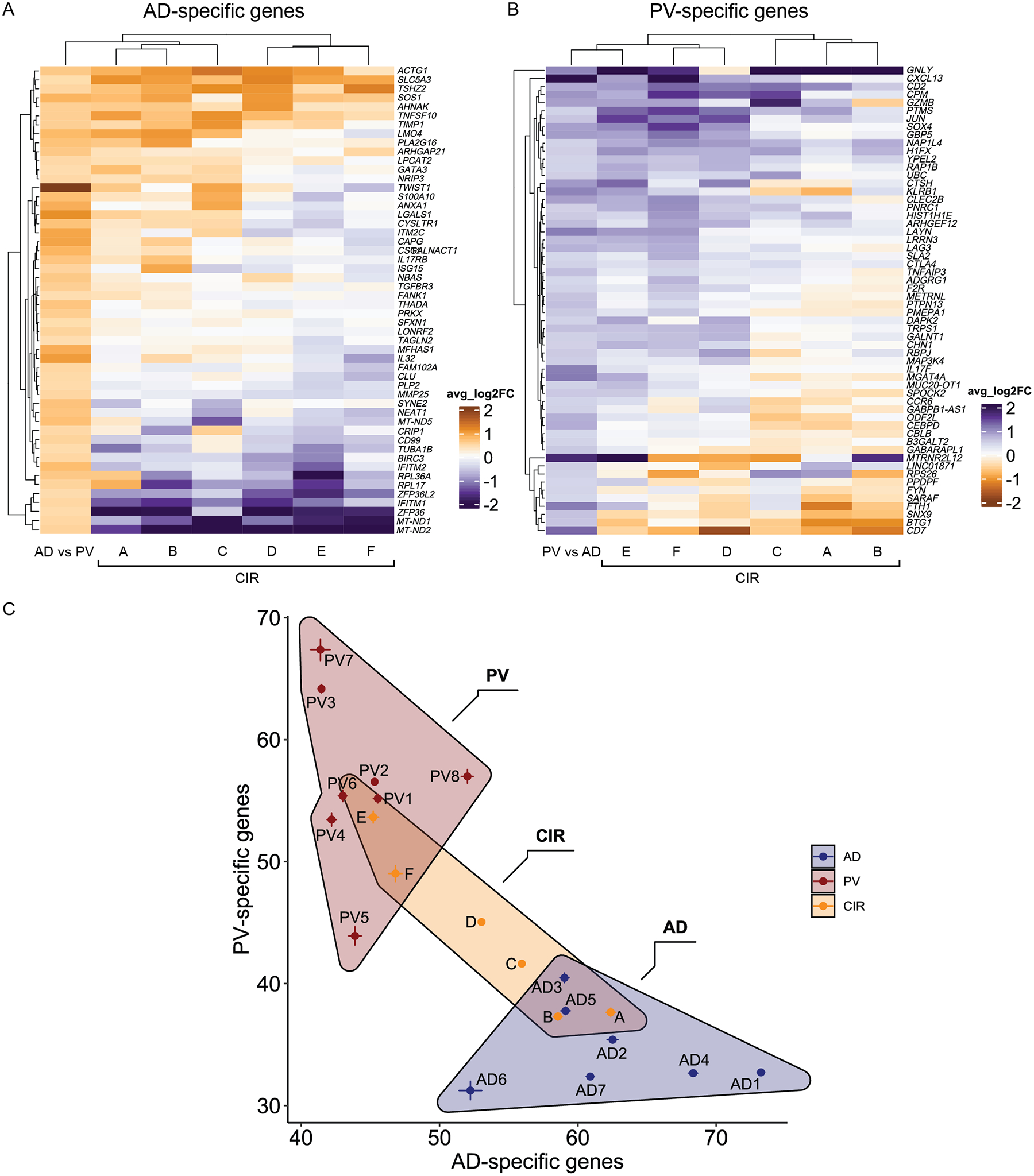
A) Number of DEGs per immune cell population for atopic dermatitis (AD) and psoriasis (PV) based on MAST statistical framework. Number of DEGs (adjust-p-val < 0.001, absolute log2FC > 0.425; on y-axis) for each immune cell population (x-axis) for 7 AD versus 7 healthy control (HC) sample comparisons (left), 8 PV versus 7 HC samples (middle), and 7 AD versus 8 PV samples (right). B) Number of DEGs per immune cell population for AD and PV comparisons as in A but for DEGs present in 80% of samples from a disease class. C and D) Heatmap showing immune cell population-specific transcriptional patterns for PV-specific genes (C) and AD-specific genes (D) (Table S7) across lymphocyte subtypes (columns). Color key reflects the avg_log2FC for 8 PV versus 7 HC samples (C) or 7 AD versus 7 HC samples (D).
The treatment of single cells as distinct data sources in DEG approaches such as MAST generates very low p values for even relatively small fold-change differences. We reasoned that such small differences were more likely to be biologically meaningful if observed in multiple samples. We therefore applied a stringent filter in which at least 80% of individual atopic dermatitis or psoriasis samples were required to display statistically significant differential expression for a given DEG, in comparison to all healthy control samples. The largest number of statistically significant DEGs surviving this heterogeneity filter generally resided in the clusters containing the most cells, likely because of the increased effective sample size in these comparisons. However, Trm1 cells possessed a disproportionately large number of DEGs in the 3 comparisons (e.g. in the PV versus Nml comparison, 514 DEGs for Trm1 cells compared to 197 for Tcm and 238 for eTreg1 cells; Fig. 3B and Table S11).
We next examined psoriasis versus atopic dermatitis transcriptional abnormalities that met the aforementioned criteria for at least one lymphocyte population. We reasoned that DEGs identified in this way would be dysregulated in most of our patient samples, but exclude non-specific inflammatory genes present in both diseases. We observed such psoriasis-specific upregulated genes were heavily concentrated in skin resident-memory classes Trm1 and Trm3, although there was also marked involvement of all migratory memory classes (Fig. 3C). An abundance of Th17-linked transcripts was immediately apparent. In addition to the known psoriasis markers IL17F and CXCL13 (9), these psoriasis-specific upregulated DEGs include granulysin GNLY and CTLA4, whose loci have been linked genetically to psoriasis (42, 43), KLRB1, which is downregulated by the TNFα-blocker alefacept in psoriatic lesions (44), MGAT4, which is downregulated during ustekinumab treatment of psoriatic arthritis (45), and PIK3R1, whose germline loss of function impairs cutaneous immunity (46). We also detected recurrent overexpression of signaling components like MAP3K4 and PTPN13 restricted to the Trm1 and Trm3 classes, possibly helping explain why such disease-specific transcripts have not been reported in prior bulk analysis studies (Table S7).
Skin- resident memory classes also prominently expressed atopic dermatitis-specific upregulated DEGs (Fig. 3D), including the known Th1-inhibiting transcriptional regulator TWIST1(47), IL17RB, which has been implicated in the related Th2 diathesis of asthma(48), and the candidate atopic dermatitis susceptibility loci NBAS and CYSLTR1 (49, 50). A similar number of atopic dermatitis-specific transcripts were also highly elevated in ILC classes and effector memory CTLs, including MLL1 (KMT2A), an epigenetic regulator required to maintain Th2 memory cell responses (51), the long non-coding RNA NEAT1 which upregulates Th2 cytokines in CD4+ T cells, and the experimentally Th2-inducible transcript AHNAK (52). This pattern reinforced existing models positing a central role for ILC (53) and CD8+ T cells (54, 55) in the pathogenesis of atopic diatheses (Table S7).
For both psoriasis and atopic dermatitis, fewer DEGs were detected in regulatory T and exhausted CD8+ T cells (Figure 3A, 3B, Table S11). Differentiating transcripts were also sparse in APC classes, which frequently contained less than 100 cells per sample, underpowering them for DEG discovery (Fig. 3A and 3B, Table S11). However, we did detect elevated expression of the antimicrobial gene epiregulin in atopic patients, consistent with its previous discovery in non-lesional skin from atopics (56), in both classical monocytes (Mono) and the macrophage class Mac1. Atopic monocytes also harbored elevated transcripts of the inflammatory protein S100A4, previously identified as a pro-Th2 mediator (57), and AREG was highly expressed in mast cells from psoriatic skin, potentially contributing to psoriasiform epidermal hyperplasia (53) (Table S7). Similar patterns of DEGs were observed when differential expression analyses were re-performed utilizing an alternative, non-parametric (Mann-Whitney) approach (Fig. S4).
Skin resident-memory T cell DEGs distinguish psoriasis versus atopic dermatitis samples in a validation cohort
To further assess disease specificity of these transcripts, we focused on lymphocyte subpopulations, in which the vast majority of DEGs were discovered. We generated gene set average expression scores for these DEGs that met the stringent criteria as an AD or PV-specific gene in at least 80% of samples for at least one lympphoycte subpopulation (Table S7, Methods - Differential Expression analysis between rash-affected and normal skin). As in the Fig. 3C and 3D heatmaps, we saw that AD-specific gene expression was accentuated in skin-resident memory T cells, ILC classes, and effector memory CTLs of AD samples, whereas PV-specific genes were most prominently represented in skin-resident memory T cells of PV samples (Fig. 4A and 4B).
Fig. 4. Atopic dermatitis and Psoriasis vulgaris -specific gene module scores are elevated for their respective disease classes and classify samples from an external dataset.
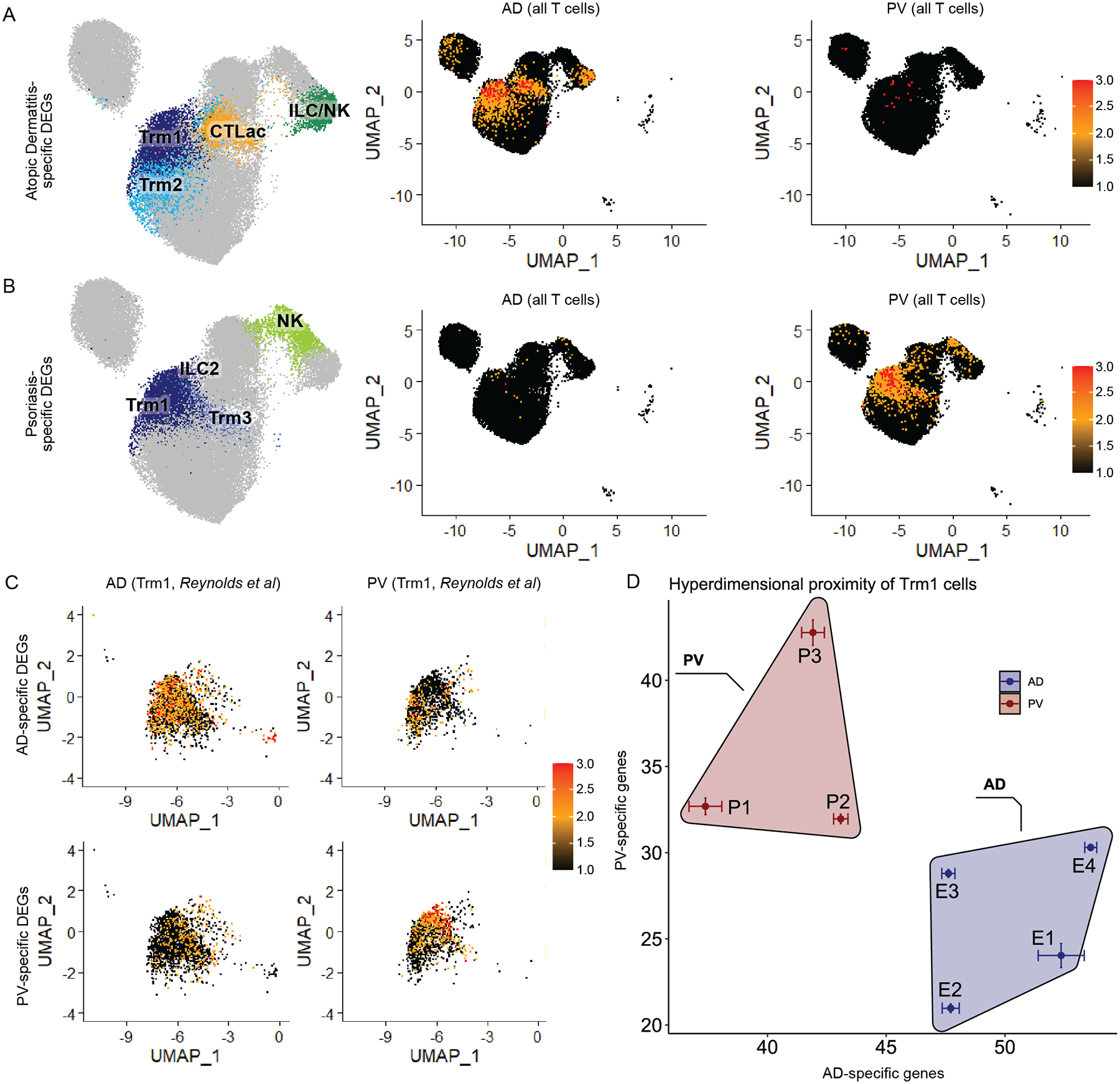
A) AD- and B) PV-specific lymphocyte DEG gene set (Table S7) expression scores (calculated by Seurat AddModuleScore) displayed for lymphocytes on a single cell level from 7 AD samples (middle panels) and 8 PV samples (right panels) in pseudocolored feature plots. Left panels label cell populations that corresponded to high scoring AD and PV gene set scores. C) AD- and PV- specific Trm1 DEG gene set (Table S7) scores displayed on a single cell level for Trm1 cells in pseudocolored feature plots for four atopic dermatitis (left) or three psoriasis (right) samples from the Reynolds et al dataset. D) Hyperdimensionality plot classification of Reynolds et al. AD and PV validation cohort using AD- and PV- specific Trm1 DEG gene set modules (Table S7).
We examined the Trm1 cell class, as they harbored the largest number of conserved psoriasis and atopic dermatitis-specific DEGs (Fig. 3) and are functionally implicated in both diseases (59, 60). As the Trm1 population likely contained closely interrelated cell types (e.g. Tc17 cells express Trm protein epitopes (61)), we manually gated for Tc2/Th2 cells, Tc17, and Th17 cells to better understand cell subtypes within this cluster. In PV samples, there was a large proportional increase of Tc17 and Th17 cells, whereas in AD samples there were more Tc2/Th2 cells, consistent with previous studies (12) (Fig. S5). For Trm1 cells, the CD4:CD8 cell ratios in AD and PV samples were roughly comparable (Fig. S6).
While the relative proportions of these Trm1 cell subtypes alone could not discriminate AD from PV samples, we reasoned that DEGs from this population might. We thus tested the discriminative power of Trm1 AD- and PV-specific genes in an unrelated, external dataset (three psoriasis and four atopic dermatitis samples from Reynolds et al. (12)). First, we calculated gene set expression scores for our Trm1 AD- or PV-specific genes in a transcriptionally analogous Trm population from the Reynolds et al samples (Figure 4C, Table S7, Methods – differential expression analysis between rash-affected and normal skin). We also utilized these AD- and PV- specific Trm1 DEGs to visually map relatedness of these external dataset samples (Figure 4D, Methods – hyperdimensionality proximity analysis). By both measures, our disease specific DEGs accurately identified the two rash types in the Reynolds et al dataset (Fig. 4C and D). Thus, we demonstrated the potential to identify rash type, on a patient level, based entirely on molecular data in Trm1 cells.
Transcriptional abnormalities differentiating psoriasis and atopic dermatitis in skin resident-memory cells segregate within unbiased functional networks
We sought to assess the functional significance of the hundreds of DEGs identified by our scRNA-seq approach. We again focused on the Trm1 cluster, which contained the established psoriasis genes IL17F, IFNG, and CXCL13 (9). We mapped psoriasis and atopic dermatitis-specific Trm1 DEGs (Table S7) onto a co-expression correlation network built on our scRNA-seq data. Gene-gene transcriptional correlation values were calculated and the resulting network visualized by qgraph (62). The AD and PV-specific genes segregated sharply in this network (Fig. 5A), which was expected, because such differences in single-cell expression aided in their discovery. We next evaluated the functional interrelatedness of these psoriasis and atopic dermatitis-specific genes. We looked for interactions amongst these DEGs utilizing an external database of known and predicted protein-protein interactions (STRING), and identified 98 protein nodes (genes), which formed 394 interactions (Fig. 5B) (63). As visually apparent, the psoriasis vulgaris and atopic dermatitis-associated genes occupied distinct sectors of the protein-protein functional interaction network, notably recapitulating their segregation in the transcriptional expression network (Fig. 5A). We quantified this functional segregation by calculating the normalized cut score (64) between AD- and PV- specific genes, which compares the weighted number of edges connecting two groups relative to the weighted number of edges within each group. These two groups showed significantly smaller linkages between the two groups than in multiple permutation tests that randomly assigned these genes to the AD and PV specific categories (p=0.001, Fig. 5B). The segregation of PV and AD-specific Trm1 genes in both transcriptional and protein interaction networks further supports a model in which distinct functional networks drive these two disease classes.
Fig. 5. Atopic dermatitis and Psoriasis vulgaris -specific DEGs segregate discretely based on unbiased pathway and network analysis.
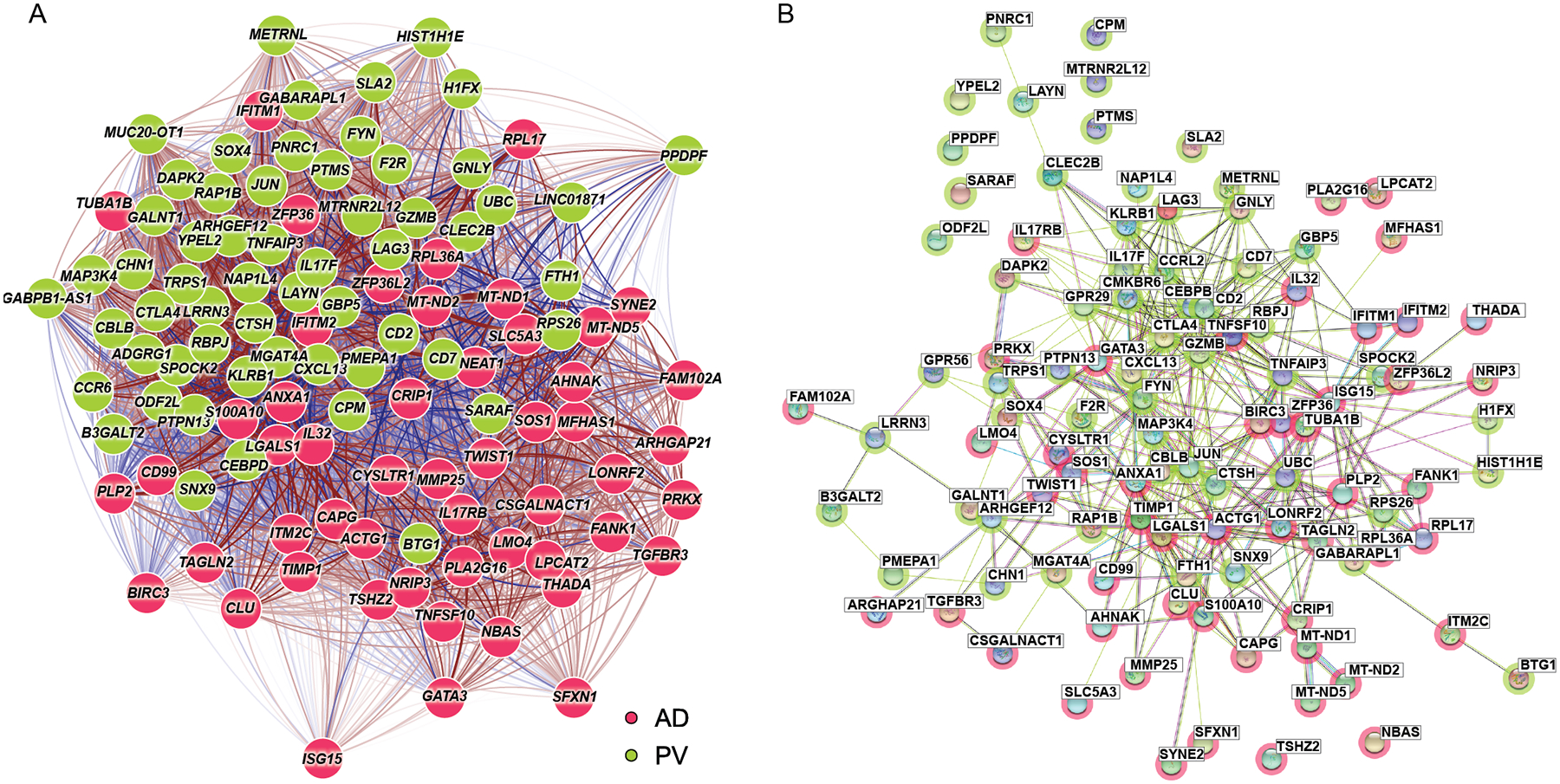
A) Transcriptional network (qgraph) for Trm1 AD- and PV- specific genes (Table S7). The Fruchterman-Reingold algorithm was used to determine the network layout. Color shading of the gene nodes denotes AD (green) or psoriasis (red)–upregulated genes. Red lines represent positive correlation while blue lines represent negative correlation. Edges and links are shown for the correlation values from scRNA-seq data, with stronger edge intensity or thicker connecting links signifying higher correlation. B) STRING protein-protein network analysis for the same Trm1 AD- and PV- specific genes. Color shading of the nodes denotes AD (green) or psoriasis (red)–specific genes.
Clinically and histopathologically indeterminate rashes (CIRs), bullous pemphigoid, and lichen planus share molecular features with atopic dermatitis or psoriasis
Many adult-onset rashes do not completely match clinical and histopathologic criteria for classic atopic dermatitis or psoriasis, leading to diagnostic and therapeutic ambiguity. Instead, these clinically indeterminate cases harbor overlapping eczematous/spongiotic features (as typically seen in atopic dermatitis) and psoriasiform patterns, either by histopathology or clinical presentation (65). These patients also lacked a clinical history consistent with canonical atopic dermatitis, i.e. eczematous rashes or lichenification in flexural areas beginning in early childhood. We termed such cases Clinical/histopathologically Indeterminate Rashes (CIRs). Six CIRs were profiled by scRNA-seq in this study, whose clinical features were summarized in Table S1. All CIR patients were offered a trial on the IL4Rα-blocking biologic dupilumab, three of whom accepted. Two of the patients (CIR-A and CIR-B) showed near-resolution of itch and clinically appreciable lesions within two months of treatment initiation. The third patient, CIR-E, suffered from a dermatitis with some eczematous features, including a spongiotic histopathology, in a background of known psoriasis. This patient’s rash showed no improvement after three months of dupilumab treatment.
We utilized our psoriasis-distinguishing and atopic-distinguishing DEGs from the Trm1 population from Fig. 5 to group these samples (Table S7). CIR-A and CIR-B, the dupilumab (atopic dermatitis therapy)-responsive cases, segregated more closely with atopic dermatitis (Fig. 6A and B). The lone dupilumab failure case, CIR-E, segregated with psoriasis. To develop a test of significance, we calculated all-versus-all distances in the hyperdimensional plane for AD- and PV-specific genes in Trm1 cells and then asked if the proximity of any CIR profile was closer to the PV centroid or the AD centroid than expected by random chance. The statistically derived distances show samples CIR-A, -B, and -C are significantly more similar to atopic dermatitis, while CIR-E and -F are more similar to psoriasis; (Fig. 6C, Table S8). The histopathology of the CIRs that molecularly stratified with AD did not more closely resemble AD, nor did the histology of the CIRs that molecularly stratified with PV more closely resemble PV. As we were unable to classify CIRs merely by the presence of the previously described Tc2/Th2 and Tc17 subpopulations within the Trm1 cluster, these observations reinforced the potential value in molecularly classification using disease-specific gene sets. We were interested in how our psoriasis- and atopic dermatitis-distinguishing DEGs would classify our two lichen planus and one bullous pemphigoid samples. In both the heatmap representations and hyper dimensionality map (Fig. S7, Table S8), we saw the bullous pemphigoid sample segregate more closely with the AD samples and the lichen planus samples with the psoriasis samples.
Fig. 6. Clinical/histopathologically Indeterminate Rashes (CIRs) show molecular stratification with atopic dermatitis or psoriasis-specific DEGs.
A) Heatmap showing relative expression levels (avg_log2FC) for each CIR Trm1 cell population relative to Trm 1 cells from all 7 healthy controls. Column 1 shows avg_log2FC values for Trm1 cells from 7 AD versus 8 PV samples. Genes depicted are Trm1 AD-specific genes (Table S7). B) Same as A except genes depicted are Trm1 PV-specific genes (Table S7). Column 1 shows avg_log2FC values for Trm1 cells from all PV versus all AD samples. C) Hyperdimensionality plot showing stratification of CIR samples relative to AD and PV samples. Each AD, PV, and CIR sample is mapped based on aggregate gene score of Trm1 population AD-specific genes (x-axis) and PV-specific genes (y-axis). One-sided Mann-Whitney tests used to calculate significance and p-values shown in Table S8.
A web interface (RashX) to visualize patient-level rash scRNA-seq-derived data in context of this Th2/Th17 sample framework
Atypical rashes that are not easily classified into canonical clinical categories, such as the CIRs, occur frequently in dermatology clinics. We constructed a proof-of-principle interface, RashX (hosted at https://rashX.ucsf.edu), in which transcriptional abnormalities from any individual rash’s Trm1 population can be placed in context of the Th2/Th17 atopic dermatitis-psoriasis stratification shown in Fig. 6. To support a universal input format, our portal accepts standard 10X Genomics scRNA-seq immune cell dataset matrices (in a RDS file format). The web interface then identifies cells in the external dataset that are most similar to our Trm1 population. Differential gene expression analysis for the Trm1 disease specific genes (Table S7) is then performed on the external dataset’s Trm1 cells in comparison to those from our healthy controls. Heatmaps for these AD- and PV-specific genes and a hyperdimensionality plot as in Fig. 6 are then generated for the external sample. To illustrate use of RashX, we included dataset matrices from an external atopic dermatitis sample and a psoriasis sample hosted at the website portal. Sample web portal outputs showed that these samples segregated closely to their parent class (Fig. S8). Thus, we have created a globally accessible, convenient resource allowing any researcher to place their disease sample scRNA-seq data in context of our Trm1 Th2/Th17 AD- and PV-framework.
DISCUSSION
Here we present single cell transcriptomic profiles of chronic inflammatory skin disease, encompassing 24 rash and 7 healthy control samples. We identified 41 T cell and antigen presenting cell subpopulations within these samples and produced a detailed global portrait of molecular derangements unique to atopic dermatitis and psoriasis vulgaris. Globally, we found proliferation-driven expansion of the Treg and Trm components and universal cytotoxic T cell exhaustion in diverse forms of chronic skin inflammation. Our findings indicated that Tregs usually proliferate in rashes in an attempt to control pathogenic skin inflammation, but that one or more qualitative factors prevented successful regulation. Cytotoxic lymphocyte exhaustion was evident in every case; however, these cells harbored relatively few distinguishing abnormalities between disease classes, suggestive of a shared end state rather than a causative force.
While patient-level studies have previously attempted to identify psoriasis- and atopic dermatitis-specific genetic changes, a limited number of genes have shown reproducibility between reports (66–70). We suspect a major limitation in these prior bulk profiling experiments is conflation of transcriptional differences arising in distinct cell populations. For example, both our AD and PV disease-specific transcriptional signatures were only discovered in CD69+ CD103+ resident memory T cells, which are play central roles in chronic inflammatory disease. In skin disease, there is significant Trm cell infiltration of active psoriatic lesions. (61). Underscoring Trm functional importance, grafting of prepsoriatic human skin (and associated Trm cells) onto mice leads to development of psoriatic lesions, while blockade of cutaneous T cell migration by E-selectin inhibition does not improve psoriasis (71, 72). Trm cells are also enriched in joints and mediate arthritis flares (73) and expansion and/or rejunevation of CD8+ pulmonary Trm cell populations post-influenza infection are associated with aberrant fibrosis (74). The discovery of our atopic dermatitis and psoriasis-specific transcriptional signatures in Trm cells highlights the importance of this cell population in chronic tissue inflammation and identifies a large set of candidate effectors potentially driving each disease, which can now be functionally investigated.
These transcripts distinguishing atopic dermatitis and psoriasis in Trm cells included previously described Th2 and Th17-specific genetic abnormalities in each class (e.g IL17F, CXCL13 GNLY and NBAS (9, 38, 49, 50)). However, our data tied both known and unknown dysregulated genes into coherent signatures demonstrating recurrent patient-level segregation between the two disease classes. Each gene set also showed significantly enhanced interconnection in unbiased functional networks, indicative of biological cooperativity. The PV- and AD-specific Trm1 signatures sets performed robustly, not only in classifying psoriasis and atopic samples from an unrelated, external dataset, but also in discerning the Th2/Th17 bias of bullous pemphigoid and lichen planus samples. These commonalities between the bullous pemphigoid sample and atopic dermatitis were consistent with the prominent type 2 inflammation of bullous pemphigoid and its reported response to dupilumab (75). Similarly, lichen planus exhibits Th1/Th17 activity, with reports of clinical improvement after administration of IL17 antagonists used for psoriasis (76, 77).
Our set of AD- and PV-specific Trm1 DEGs were able to match a subset of clinicopathologically ambiguous cases of skin inflammation into AD- or PV-like classes, consistent with therapeutic response to IL4Rα blockade. Although our CIRs represented only a small initial test set, these results suggest that clinical response in some fraction of such rashes may be predicted based on their molecular similarity to existing diseases. Our findings thus make the case for larger, unbiased therapeutic trials based on the precision medicine approach described here.
A limitation of our study is that while comparable in size to other studies (9, 11, 12), co-variates (e.g. age and anatomic location) may contribute some variability given the relatively small sample size of our dataset. Furthermore, our unbiased CD45+ immune cell profiling strategy inevitably leads to certain populations, in particular APCs, harboring significantly fewer profiled single cells hindering discovery of their unique molecular abnormalities. Expansion of our dataset with a larger cohort of samples would both enable detection of complementary signatures in other subpopulations, as well as further validate/generalize our T cell population discoveries. In addition, we anticipate future samples utilizing scRNA-seq approaches integrated with clonality assessment, will allow more complex and informative analyses of inflammatory dysregulation.
Our data in combination with published rash samples (1,2), represent the beginnings of an inflammatory skin disease database where scRNA-seq profiles of any rash can be compared. Our web interface at https://rashX.ucsf.edu provides an immediate means to avail our data and analytic methods to the translational community at large. By placing transcriptomic features of individual rashes in the context of a large, existing dataset, we seek to eventually generate a standardized framework to link molecular features to disease prognosis and drug response, based on contributions from clinical centers worldwide.
MATERIALS AND METHODS
Study Design
We used CD45+ immune cell single cell RNA-seq to 1) more precisely define immune cell type-specific molecular abnormalities distinguishing canonical inflammatory skin diseases (i.e. psoriasis vulgaris and classic atopic dermatitis) and 2) to utilize these patterns to better understand individual, indeterminate cases of cutaneous inflammation displaying overlapping clinical or histopathologic features of both diseases. We assessed CD45+ immune cells (~6,000 cells per sample) using 10X Genomics droplet-based 3’ scRNA-seq from 31 enzymatically dissociated skin samples (7 atopic dermatitis, 8 psoriasis vulgaris, 2 lichen planus, 1 bullous pemphigoid, 6 clinicopathologically indeterminate rash lesional samples, and 7 normal controls). Detailed information regarding clinical characteristics of patient samples are in Table S1.
Sample collection
Written informed consent was obtained from normal and rash-affected skin donors under protocols approved by the University of California, San Francisco Institutional Review Board. Six millimeter full thickness punch biopsies were obtained from lesional rash-affected skin while mammoplasty and abdominoplasty surgical skin tissue discards were utilized for normal samples. Diagnoses were made by a board-certified dermatologist based on clinical evaluation, as well as histopathology of a representative hematoxylin and eosin-stained section of each skin sample reviewed by a board-certified dermatopathologist. Psoriatic and atopic dermatitis patients were judged to have moderate to severe disease based on Psoriasis and Eczema Area and Severity Index scores, respectively. The clinical characteristics and histopathologic diagnosis of patient samples were detailed in Supplementary Table S1. Samples were obtained from patients off of systemic immunosuppressives for at least 4 weeks (except for one sample) and topical steroids to the sampled area for at least two weeks prior to biopsy.
Immune cell isolation
Skin samples were initially placed in ice cold PBS immediately after the biopsy for up to 8 hours prior to processing. Samples were then finely minced and transferred to 3 mL of enzymatic digestion buffer (RPMI 1640 medium, 10% heat-inactivated fetal bovine serum, 200 U/ml collagenase type IV (Worthington, LS004188), and 200ug/ml DNase (Sigma, DN25–100MG)) at 37 °C for 16–18 hours. After overnight digestion, samples were transferred to 50 ml conical tubes, shaken vigorously for 30 seconds, and then filtered through a 100-μm cell strainer. Samples where CITE-seq antibodies were added prior to flow sorting (designated as “pre” for CD45 and Totalseq A order in Table S10), were then pelleted by centrifugation at 400g for 5 minutes. Cell surface receptors were blocked by adding 5 μL Human TruStain FcX (BioLegend) in 100uL of cell staining buffer (Biolegend) at 4°C for 10 minutes. Following blocking, TotalSeq-A antibodies (Biolegend, see Table S10) at a concentration of 1 ug of antibody per 1 million cells and a 1:20 dilution of an anti-CD45 antibody conjugated to APC fluorophore (ThermoFisher) were then added for 30 minutes at 4°C. Cells were washed by PBS with 2% FBS and samples were resuspended in 400 μL cell staining buffer and filtered through a 40-μm filter before adding DAPI to a final concentration of 1ug/mL and flow sorting on a SH800 cell sorter (Sony Biotechnology). The samples were then washed one time and resuspended in cell staining buffer prior to library preparation. Samples where CITE-seq antibodies were added subsequent to flow sorting (designated as “post” for CD45 and Totalseq A order in Table S10) were pelleted by centrifugation at 400g for 5 minutes and then incubated with a 1:20 dilution of an anti-CD45 antibody conjugated to APC fluorophore (ThermoFisher) in cell staining buffer (Biolegend) for 30 min at 4°C. Cells were washed by PBS with 2% FBS and samples were resuspended in 400 μL cell staining buffer and filtered through a 40-μm filter before adding DAPI to a final concentration of 1 ug/mL and flow sorting on a SH800 cell sorter (Sony Biotechnology). After gating out cell debris and doublets, DAPI negative and CD45 positive live immune cells were collected into 3mL cell staining buffer. Immune cells were then spiked with 5% murine splenocytes to serve as a non-specific background staining control for CITE-seq antibodies (see Table S1 for spike-in information). Cell surface receptors were blocked by adding 5 μL Human TruStain FcX (BioLegend) in a 100uL reaction volume at 4°C for 10 minutes. Following blocking, TotalSeq-A antibodies (Biolegend, see Table S11) were added at a concentration of 0.5 ug/1 million cells per antibody for 30 minutes at 4°C. The sample was then washed three times and resuspended in cell staining buffer prior to library preparation.
scRNA-seq and CITE-seq library preparation and sequencing
scRNA-seq libraries profiling ~6000 cutaneous immune cells/sample were prepared by the Genomics Core Facility, UCSF Institute for Human Genetics using the Chromium Single cell 3’ Solution V2 or V3 kit (10x Genomics, Pleasanton, CA) per manufacturer’s protocol. For CITE-seq samples, 0.2 pmol of ADT (antibody derived tag) additive primer was added at the RNA library cDNA amplification step. CITE-seq libraries were then prepared according to TotalSeq-A antibody manufacturer’s protocol (BioLegend). In brief, 70ul of ADT-containing cDNA amplification supernatant was purified with two rounds of 2X SPRI beads (Beckman-Coulter) then amplified for 14–20 cycles using HiFi HotStart ReadyMix (2X, KAPA, Roche Sequencing & Life Science, Wilmington, MA) and 2.5 uM of oligos corresponding to SI PCR primer and Truseq Small RNA RPI1–6 primers. After amplification, the resulting application products were purified by a 1.2X SPRI bead cleanup, then quantified with Qubit dsDNA HS Assay Kit. Quality for scRNA and ADT libraries was assessed by a TapeStation D1000 ScreenTape (Agilent Technologies) and quantitated using the Kapa library quantitation kit prior to sequencing. mRNA and ADT libraries were sequenced using an Illumina HiSeq 4000 with paired end 150 base pair sequencing parameters Detailed information on sequencing results, including total read count and sequencing saturations are shown in Table S9.
Single cell RNA and CITE-seq data processing
Single-cell RNA-seq and antibody-derived-tag (ADT) FASTQ files were aligned and quantified using Cell Ranger Software (version 3.0.2, 10x Genomics) against human GRCh38 transcriptome (v 3.0.0). For samples with mouse splenocytes spike-in, FASTQ files were aligned to a combined human and mouse genome reference GRCh38+mm10 (v3.1.0). Empty droplets were removed as part of internal Cell Ranger quality control algorithms. Quality of cells was assessed based on the total number of detected genes per cell and the percentage of mitochondrial gene counts, with removal of cells containing less than 100 or over 6000 unique genes. Cells with percentage of reads mapped to mitochondrial genes exceeding 20% were also removed.
Doublets were detected and removed by the function scDblFinder (R package, v1.5.7) (78). Similar results were obtained using the DoubletFinder package (79). Counts were normalized using the NormalizeData function in Seurat using the scale factor (1e4) with natural-log transformation. 2000 highly variable genes (HVGs) were then selected using FindVariableFeatures() in Seurat using the “versust” method. These HVGs were then scaled and centered based on individual expression values
ADT samples with fraction antibody reads usable > 30%, antibody reads in cells > 30%, fraction unrecognized antibody <30%, and median umis per cell >100 were kept for downstream analysis (Table S11). For ADT data, we used Seurat CLR normalization to calculate the mean and standard deviation for a given antibody in mouse cells (to estimate non-specific background staining) and then used a cutoff of 1.5*SD + mean where the ADT density distribution for mouse and human cells were clearly separated (18)(Fig. S9 and Table S10).
Dimension reduction and unsupervised clustering
Principal components were computed using the RunPCA() function in Seurat based on HVGs. We then provided this PCA matrix to the Harmony algorithm (80) within the Seurat workflow using sample, flow order, and mouse spike-in as technical covariates for batch correction. The batch-corrected coordinate space was then used for dimensional reduction and embedding as a Uniform Manifold Approximation and Projection (UMAP) representation under the RunUMAP() function in Seurat. The batch-corrected coordinate space was also used to compute the nearest neighbor graph by FindNeighbors() function (Seurat). The nearest neighbor graph was applied to FindClusters() with the Louvain algorithm. Fourteen clusters with a resolution of 0.4 were retained based on clustree results.
Cluster annotation
Cluster-specific differential expressed genes were detected using the FindMarkers function in Seurat running MAST, and was limited to genes with expression greater than 25% in either of the two populations tested, combined with a fold change cutoff of 0.25 (log2 scale). The p-values were adjusted using the Bonferroni correction for multiple testing. Two major groups (HLA-DRA+ myeloid cells and CD3+ or KLRB1+ lymphoid cells) were subclustered for additional rounds of feature detection, embedding, dimensionality reduction, visualization and clustering under the above described clustering workflow, with further subclustering of an indeterminate CD3+/KLRB1+ cluster enriched in cycling transcripts. (Fig. S1C, Table S3, S4). After removal of clusters containing a sizable proportion of non-immune or low-quality cells, 21 CD3+/KLRB1+ clusters and 19 HLA−DR+ clusters were retained. The final 41 cluster immune cell object was comprised of these subclustered populations and the original mast cell cluster and underwent supervised clustering based on 1328 features derived from the significant HVFs-derived cluster markers.
Weighted Gaussian linear model for differential immune cell composition analysis
A weighted Gaussian linear model was utilized to analyze differential immune cell composition between rash-affected and normal samples. We denote Nik as the number of cells in a given sample (i) and a given cluster (k) and Ni=∑k Nik as the total number of cells in sample i. For each cluster k and sample i, let pik = Nik/Ni. We applied weighted linear models on the log cluster compositions, log(pik), with sample disease status as the predictor and the standard deviation of log proportion, obtained using the delta method, as weights. Differential clusters were then selected with adjusted p value <0.05 for the two sample t test for the coefficient for disease status, which characterizes the average changes of the log proportion between rash-affected and normal samples.
Differential Expression analysis between rash-affected and normal skin.
Differential expression analysis was performed between disease groups on a per cluster basis using the FindMarkers function from the Seurat R package using MAST (41) or Wilcox (Wilcoxon rank sum test) differential expression tests. For AD (or PV) versus Nml DEGs, we performed a DE analysis with all AD (or PV) samples combined against all normal combined on a per cluster basis, then retained DEGs with adj_p_val < 0.001 & abs avg_log2FC >0.425. For AD versus PV DEGs, we merged all statistically significant (p-val < 0.001, |avg_log2FC|>0.425) DEGs from the above AD versus Nml and for PV versus Nml DEG comparisons. Using this gene list, we performed differential expression analysis between AD versus PV samples. We then retained DEGs with |avg_log2FC|>0.425 & adj_p_val < 0.001. For the 80% heterogeneity filter DEGs, we created a gene set containing significant AD (or PV) versus Nml from every cluster to perform DE analysis for each individual AD (or PV) sample versus all Nmls, then retained DEGs with adj_p_val < 0.001 & abs avg_log2FC >0.425. The DEGs from the 7 or 8 individual AD (or PV) comparisons were then combined and genes that were significantly expressed in at least 80% of AD individual samples comparisons were retained. For the 80% heterogeneity filter AD versus PV comparison, we merged all statistically significant (p-val < 0.001, |avg_log2FC|>0.425) DEGs for the AD versus Nml (80% filter) and PV versus Nml (80% filter) DEG comparisons. Using this gene set, we performed differential expression analysis between all AD versus PV samples. We then retained DEGs with |avg_log2FC|>0.425 & adj_p_val < 0.001. DEG gene set scores calculating average expression levels on a single cell minus aggregated expression of a control gene set were generated using AddModuleScore() in the Seurat package(81).
Transcriptional network visualization
To map the pairwise (bivariate) single cell transcriptional correlations between variables and the structures that emerge from these pairwise correlations, we used a method analogous to Correlation Network Analysis (82). We generated a subsetted data matrix for Trm1 cluster cells (each listed in a separate row) with reads counts for AD-specific and PV-specific genes from Trm1 (|avg_log2FC| > 0.425, adj_ p_val < 0.001; each in a separate column). Then the cor() function in the R stats packages (83) was used to obtain Spearman coefficients of gene-gene correlation. We then plotted an overall transcriptional network graph in the R package qgraph (version 1.3.2) (62) using the correlation matrix. The coordinates of each node were computed using the Fruchterman-Reingold algorithm, where the attraction between nodes is proportional to the strength of the correlation between two genes (nodes).
STRING protein interaction network visualization
AD versus PV-specific transcripts (|avg_log2FC|>0.425, adj_ p_val < 0.001) from Trm1 were mapped onto the STRING protein interaction network. A combined score greater than 0.4 was used as a cutoff. The retained interactions were visualized by igraph. Each node corresponds to a gene and each edge represents existing experimental and database evidence for protein interactions. We quantified the segregation and tested the significance between the AD-specific and PV-specific nodes on this graph by performing the permutation test conditional on the observed network. We used the STRINGdb package in R to link the genes to proteins in the STRING database and subset the network to only genes in the list of 110 DE genes using the string_db$map object in STRINGdb. This resulted in 108 proteins (2 genes did not map to a protein). The resulting subnetwork constrained to these 108 proteins consisted of 98 nodes (proteins) with connections to other nodes. We randomly permute the AD- or PV- specific labels for the 98 nodes (genes) and calculate the normalized cut score; we repeated this 100,000 times to construct a null distribution for the normalized cut score, conditional on this gene network. A p-value was calculated as the proportion of the resampled normalized cut scores smaller than the observed normalized cut score, resulting in a p-value of 0.001.
Hyperdimensionality proximity analysis
Having identified two sets of DEGs in AD and PV from Trm1 cells (Table S7), we wished to visualize how samples occurred in this hyperdimensional space, summarized by these two axes. We took the aggregate gene approach described in Cao et al (84), in which genes along one axis for any particular cell are summed over normalized gene counts with pseudocount values of 1. Since there were 2 gene sets of interest, we calculated these 2 AD and PV signatures for each cell. Cells were then grouped within sample by geometric mean. We then considered these samples bound to arbitrary regions within this hyperdimensional space and visualized these regions with hull plots using the R/ggforce(85) with concavity=5. We tested whether indeterminate (CIR), BP, LP, or external dataset AD/PV samples were more proximate to the PV or AD region by Canberra distance matrix obtained from the ‘vegan’ R package (86) among indeterminate samples and PV/AD samples across the hull plane and then testing for the lesser distance using one-sided Mann-Whitney tests.
Statistical Analyses
Statistical analyses were performed with Rstudio v.1.4.1717 and GraphPad Prism (version 8.0; GraphPad Software, La Jolla, California). Differential immune cell composition between two groups was analyzed with a weighted Gaussian linear model. For single-cell analysis, utilized Seurat utilized a GLM-framework that treats cellular detection rate as a covariate or a nonparametric Wilcoxon rank sum test with Bonferroni correction. The length of a segment connecting two gene signatures was measured using Euclidean distance or Canberra distance. Pearson’s r was calculated with the R function cor(). Adjusted p < 0.05 is considered significant for Seurat-based analyses, while p <0.05 was used for other analyses.
Supplementary Material
Acknowledgments:
We wish to thank Rachel Sevey for assistance with figure illustration
Funding:
This project was supported in part by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health K08AR067243 to JBC, the LEO foundation to RJC, NIH NIAMS R01AR061106 to TM (administered by the Northern California Institute for Research and Education), NIH NIAMS K08AR075880 to RRG, and NIH/NCRR UCSF-CTSI Grant Number UL1TR001872. JBC is a recipient of research awards from the National Eczema Association and National Psoriasis Foundation. This study was supported in part by funds from Sun Pharmaceutical Industries. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or the Department of Veterans Affairs.
Competing interests:
JBC and RJC are investigators for Sun Pharmaceutical Industries, LEO Pharmaceuticals, and Sanofi (grants to their institution)
Data and materials availability:
Sequencing BAM files are deposited at the European Genome-Phenome Archive (EGA), which is hosted by the EBI and the CRG, under accession number EGAS00001005271. The processed Seurat objects with cell identities are available at Zenodo (https://zenodo.org/deposit/5228495). All statistical analysis and plotting of scRNA-seq and cell surface protein data were performed using Rstudio software (Version 1.4.1717). All analysis scripts are available at the online repository https://github.com/Yale73/scRNA-seq-for-diverse-human-rashes. All data are available in the main text or the supplementary materials. The online rash mapping webtool is available at https://rashX.ucsf.edu. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Jeffrey Cheng (Jeffrey.Cheng@ucsf.edu) or Raymond Cho (Raymond.cho@ucsf.edu).
References and Notes
- 1.Rachakonda TD, Schupp CW, Armstrong AW, Psoriasis prevalence among adults in the United States. J Am Acad Dermatol 70, 512–516 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Shaw TE, Currie GP, Koudelka CW, Simpson EL, Eczema prevalence in the United States: data from the 2003 National Survey of Children’s Health. J Invest Dermatol 131, 67–73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiesa Fuxench ZC, Block JK, Boguniewicz M, Boyle J, Fonacier L, Gelfand JM, Grayson MH, Margolis DJ, Mitchell L, Silverberg JI, Schwartz L, Simpson EL, Ong PY, Atopic Dermatitis in America Study: A Cross-Sectional Study Examining the Prevalence and Disease Burden of Atopic Dermatitis in the US Adult Population. J Invest Dermatol 139, 583–590 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, Silverberg JI, Deleuran M, Kataoka Y, Lacour J-P, Kingo K, Worm M, Poulin Y, Wollenberg A, Soo Y, Graham NMH, Pirozzi G, Akinlade B, Staudinger H, Mastey V, Eckert L, Gadkari A, Stahl N, Yancopoulos GD, Ardeleanu M, Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N Engl J Med 375, 2335–2348 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Ferreira S, Guttman-Yassky E, Torres T, Selective JAK1 Inhibitors for the Treatment of Atopic Dermatitis: Focus on Upadacitinib and Abrocitinib. Am J Clin Dermatol 21, 783–798 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Menter A, Strober BE, Kaplan DH, Kivelevitch D, Prater EF, Stoff B, Armstrong AW, Connor C, Cordoro KM, Davis DMR, Elewski BE, Gelfand JM, Gordon KB, Gottlieb AB, Kavanaugh A, Kiselica M, Korman NJ, Kroshinsky D, Lebwohl M, Leonardi CL, Lichten J, Lim HW, Mehta NN, Paller AS, Parra SL, Pathy AL, Rupani RN, Siegel M, Wong EB, Wu JJ, Hariharan V, Elmets CA, Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol 80, 1029–1072 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Tsoi LC, Rodriguez E, Degenhardt F, Baurecht H, Wehkamp U, Volks N, Szymczak S, Swindell WR, Sarkar MK, Raja K, Shao S, Patrick M, Gao Y, Uppala R, Perez White BE, Getsios S, Harms PW, Maverakis E, Elder JT, Franke A, Gudjonsson JE, Weidinger S, Atopic Dermatitis Is an IL-13-Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. J. Invest. Dermatol 139, 1480–1489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quaranta M, Knapp B, Garzorz N, Mattii M, Pullabhatla V, Pennino D, Andres C, Traidl-Hoffmann C, Cavani A, Theis FJ, Ring J, Schmidt-Weber CB, Eyerich S, Eyerich K, Intraindividual genome expression analysis reveals a specific molecular signature of psoriasis and eczema. Science Translational Medicine 6, 244ra90–244ra90 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Liu J, Chang H-W, Huang Z-M, Nakamura M, Sekhon S, Ahn R, Munoz-Sandoval P, Bhattarai S, Beck KM, Sanchez IM, Yang E, Pauli M, Arron ST, Fung-Leung W-P, Munoz E, Liu X, Bhutani T, North J, Fourie AM, Rosenblum MD, Liao W, Single-cell RNA sequencing of psoriatic skin identifies pathogenic Tc17 cell subsets and reveals distinctions between CD8+ T cells in autoimmunity and cancer. Journal of Allergy and Clinical Immunology 0 (2020), doi: 10.1016/j.jaci.2020.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng JB, Sedgewick AJ, Finnegan AI, Harirchian P, Lee J, Kwon S, Fassett MS, Golovato J, Gray M, Ghadially R, Liao W, Perez White BE, Mauro TM, Mully T, Kim EA, Sbitany H, Neuhaus IM, Grekin RC, Yu SS, Gray JW, Purdom E, Paus R, Vaske CJ, Benz SC, Song JS, Cho RJ, Transcriptional Programming of Normal and Inflamed Human Epidermis at Single-Cell Resolution. Cell Reports 25, 871–883 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bangert C, Rindler K, Krausgruber T, Alkon N, Thaler FM, Kurz H, Ayub T, Demirtas D, Fortelny N, Vorstandlechner V, Bauer WM, Quint T, Mildner M, Jonak C, Elbe-Bürger A, Griss J, Bock C, Brunner PM, Persistence of mature dendritic cells, TH2A, and Tc2 cells characterize clinically resolved atopic dermatitis under IL-4Rα blockade. Science Immunology 6 (2021), doi: 10.1126/sciimmunol.abe2749. [DOI] [PubMed] [Google Scholar]
- 12.Reynolds G, Vegh P, Fletcher J, Poyner EFM, Stephenson E, Goh I, Botting RA, Huang N, Olabi B, Dubois A, Dixon D, Green K, Maunder D, Engelbert J, Efremova M, Polański K, Jardine L, Jones C, Ness T, Horsfall D, McGrath J, Carey C, Popescu D-M, Webb S, Wang X, Sayer B, Park J-E, Negri VA, Belokhvostova D, Lynch MD, McDonald D, Filby A, Hagai T, Meyer KB, Husain A, Coxhead J, Vento-Tormo R, Behjati S, Lisgo S, Villani A-C, Bacardit J, Jones PH, O’Toole EA, Ogg GS, Rajan N, Reynolds NJ, Teichmann SA, Watt FM, Haniffa M, Developmental cell programs are co-opted in inflammatory skin disease. Science 371 (2021), doi: 10.1126/science.aba6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Cook C, Sedgewick AJ, Zhang S, Fassett MS, Ricardo-Gonzalez RR, Harirchian P, Kashem SW, Hanakawa S, Leistico JR, North JP, Taylor MA, Zhang W, Man M-Q, Charruyer A, Beliakova-Bethell N, Benz SC, Ghadially R, Mauro TM, Kaplan DH, Kabashima K, Choi J, Song JS, Cho RJ, Cheng JB, Single-Cell Profiling Reveals Divergent, Globally Patterned Immune Responses in Murine Skin Inflammation. iScience 23, 101582 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He H, Suryawanshi H, Morozov P, Gay-Mimbrera J, Del Duca E, Kim HJ, Kameyama N, Estrada Y, Der E, Krueger JG, Ruano J, Tuschl T, Guttman-Yassky E, Single-cell transcriptome analysis of human skin identifies novel fibroblast subpopulation and enrichment of immune subsets in atopic dermatitis. J Allergy Clin Immunol 145, 1615–1628 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Rojahn TB, Vorstandlechner V, Krausgruber T, Bauer WM, Alkon N, Bangert C, Thaler FM, Sadeghyar F, Fortelny N, Gernedl V, Rindler K, Elbe-Bürger A, Bock C, Mildner M, Brunner PM, Single-cell transcriptomics combined with interstitial fluid proteomics defines cell type-specific immune regulation in atopic dermatitis. J Allergy Clin Immunol 146, 1056–1069 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Bangert C, Rindler K, Krausgruber T, Alkon N, Thaler FM, Kurz H, Ayub T, Demirtas D, Fortelny N, Vorstandlechner V, Bauer WM, Quint T, Mildner M, Jonak C, Elbe-Bürger A, Griss J, Bock C, Brunner PM, Persistence of mature dendritic cells, TH2A, and Tc2 cells characterize clinically resolved atopic dermatitis under IL-4Rα blockade. Science Immunology 6 (2021), doi: 10.1126/sciimmunol.abe2749. [DOI] [PubMed] [Google Scholar]
- 17.Salomon R, Kaczorowski D, Valdes-Mora F, Nordon RE, Neild A, Farbehi N, Bartonicek N, Gallego-Ortega D, Droplet-based single cell RNAseq tools: a practical guide. Lab Chip 19, 1706–1727 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, Smibert P, Simultaneous epitope and transcriptome measurement in single cells. Nature Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zappia L, Oshlack A, Clustering trees: a visualization for evaluating clusterings at multiple resolutions. Gigascience 7 (2018), doi: 10.1093/gigascience/giy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi H, Song H, Jung YW, The Roles of CCR7 for the Homing of Memory CD8+ T Cells into Their Survival Niches. Immune Netw 20 (2020), doi: 10.4110/in.2020.20.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, Elco CP, Huang V, Matos TR, Kupper TS, Clark RA, Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med 7, 279ra39 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willemsen M, Linkutė R, Luiten RM, Matos TR, Skin-resident memory T cells as a potential new therapeutic target in vitiligo and melanoma. Pigment Cell & Melanoma Research 32, 612–622 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szabo PA, Levitin HM, Miron M, Snyder ME, Senda T, Yuan J, Cheng YL, Bush EC, Dogra P, Thapa P, Farber DL, Sims PA, Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat Commun 10, 4706 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mijnheer G, Lutter L, Mokry M, van der Wal M, Scholman R, Fleskens V, Pandit A, Tao W, Wekking M, Vervoort S, Roberts C, Petrelli A, Peeters JGC, Knijff M, de Roock S, Vastert S, Taams LS, van Loosdregt J, van Wijk F, Conserved human effector Treg cell transcriptomic and epigenetic signature in arthritic joint inflammation. Nat Commun 12, 2710 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaeth M, Wang Y-H, Eckstein M, Yang J, Silverman GJ, Lacruz RS, Kannan K, Feske S, Tissue resident and follicular Treg cell differentiation is regulated by CRAC channels. Nat Commun 10, 1183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boix-Giner F, Millan O, San Segundo D, Muñoz-Cacho P, Mancebo E, Llorente S, Rafael-Valdivia L, Rimola A, Fábrega E, Mrowiec A, Allende L, Minguela A, Bolarín JM, Paz-Artal E, López-Hoyos M, Brunet M, Muro M, High frequency of central memory regulatory T cells allows detection of liver recipients at risk of early acute rejection within the first month after transplantation. International Immunology 28, 55–64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ward-Kavanagh LK, Lin WW, Šedý JR, Ware CF, The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity 44, 1005–1019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiang EY, de Almeida PE, de Almeida Nagata DE, Bowles KH, Du X, Chitre AS, Banta KL, Kwon Y, McKenzie B, Mittman S, Cubas R, Anderson KR, Warming S, Grogan JL, CD96 functions as a co-stimulatory receptor to enhance CD8+ T cell activation and effector responses. Eur J Immunol 50, 891–902 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Nguyen LT, Ohashi PS, Clinical blockade of PD1 and LAG3 — potential mechanisms of action. Nat Rev Immunol 15, 45–56 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Li S, Morita H, Sokolowska M, Tan G, Boonpiyathad T, Opitz L, Orimo K, Archer SK, Jansen K, Tang MLK, Purcell D, Plebanski M, Akdis CA, Gene expression signatures of circulating human type 1, 2, and 3 innate lymphoid cells. J. Allergy Clin. Immunol 143, 2321–2325 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Rizzo G, Vafadarnejad E, Arampatzi P, Silvestre J-S, Zernecke A, Saliba A-E, Cochain C, Single-cell transcriptomic profiling maps monocyte/macrophage transitions after myocardial infarction in mice. bioRxiv, 2020.04.14.040451 (2020). [Google Scholar]
- 33.Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba A-E, Zernecke A, Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ Res 122, 1661–1674 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, He Y, Wang L, Zhang Q, Kim A, Gao R, Orf J, Wang T, Sawant D, Kang J, Bhatt D, Lu D, Li C-M, Rapaport AS, Perez K, Ye Y, Wang S, Hu X, Ren X, Ouyang W, Shen Z, Egen JG, Zhang Z, Yu X, Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 181, 442–459.e29 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, Smibert P, Large-scale simultaneous measurement of epitopes and transcriptomes in single cells. Nat Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, Modak M, Carotta S, Haslinger C, Kind D, Peet GW, Zhong G, Lu S, Zhu W, Mao Y, Xiao M, Bergmann M, Hu X, Kerkar SP, Vogt AB, Pflanz S, Liu K, Peng J, Ren X, Zhang Z, Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 179, 829–845.e20 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Tan Y, Sun F, Hou L, Zhang C, Ge T, Yu H, Wu C, Zhu Y, Duan L, Wu L, Song N, Zhang L, Zhang W, Wang D, Chen C, Wu C, Jiang G, Zhang P, Single-cell transcriptome and antigen-immunoglobin analysis reveals the diversity of B cells in non-small cell lung cancer. Genome Biology 21, 152 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang J, Faiz A, Berg M, Carpaij OA, Vermeulen CJ, Brouwer S, Hesse L, Teichmann SA, ten Hacken NHT, Timens W, van den Berge M, Nawijn MC, Gene signatures from scRNA-seq accurately quantify mast cells in biopsies in asthma. Clinical & Experimental Allergy 50, 1428–1431 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scott R, Ghule P, Stein J, Stein G, Cell cycle gene expression networks discovered using systems biology: Significance in carcinogenesis. J Cell Physiol 230, 2533–2542 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi R, Kano Y, Yamazaki Y, Kimishima M, Mizukawa Y, Shiohara T, Defective Regulatory T Cells In Patients with Severe Drug Eruptions: Timing of the Dysfunction Is Associated with the Pathological Phenotype and Outcome. The Journal of Immunology 182, 8071–8079 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, Linsley PS, Gottardo R, MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ermis E, Celik SK, Solak N, Genc GC, Dursun A, The role of GNLY gene polymorphisms in psoriasis pathogenesis. An Bras Dermatol 94, 198–203 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dursun HG, Yılmaz HO, Dursun R, Kulaksızoğlu S, Association of Cytotoxic T Lymphocyte Antigen-4 Gene Polymorphisms with Psoriasis Vulgaris: A Case-Control Study in Turkish Population. Journal of Immunology Research 2018, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haider AS, Lowes MA, Gardner H, Bandaru R, Darabi K, Chamian F, Kikuchi T, Gilleaudeau P, Whalen MS, Cardinale I, Novitskaya I, Krueger JG, Novel insight into the agonistic mechanism of alefacept in vivo: differentially expressed genes may serve as biomarkers of response in psoriasis patients. J Immunol 178, 7442–7449 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Fiechter RH, de Jong HM, van Mens LJJ, Fluri IA, Tas SW, Baeten DLP, Yeremenko NG, van de Sande MGH, IL-12p40/IL-23p40 Blockade With Ustekinumab Decreases the Synovial Inflammatory Infiltrate Through Modulation of Multiple Signaling Pathways Including MAPK-ERK and Wnt. Front Immunol 12, 611656 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nunes-Santos CJ, Uzel G, Rosenzweig SD, PI3K pathway defects leading to immunodeficiency and immune dysregulation. J Allergy Clin Immunol 143, 1676–1687 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Niesner U, Albrecht I, Janke M, Doebis C, Loddenkemper C, Lexberg MH, Eulenburg K, Kreher S, Koeck J, Baumgrass R, Bonhagen K, Kamradt T, Enghard P, Humrich JY, Rutz S, Schulze-Topphoff U, Aktas O, Bartfeld S, Radbruch H, Hegazy AN, Löhning M, Baumgart DC, Duchmann R, Rudwaleit M, Häupl T, Gitelman I, Krenn V, Gruen J, Sieper J, Zeitz M, Wiedenmann B, Zipp F, Hamann A, Janitz M, Scheffold A, Burmester GR, Chang HD, Radbruch A, Autoregulation of Th1-mediated inflammation by twist1. J Exp Med 205, 1889–1901 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seumois G, Zapardiel-Gonzalo J, White B, Singh D, Schulten V, Dillon M, Hinz D, Broide DH, Sette A, Peters B, Vijayanand P, Transcriptional Profiling of Th2 Cells Identifies Pathogenic Features Associated with Asthma. J Immunol 197, 655–664 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim KW, Myers RA, Lee JH, Igartua C, Lee KE, Kim YH, Kim E-J, Yoon D, Lee J-S, Hirota T, Tamari M, Takahashi A, Kubo M, Choi J-M, Kim K-E, Nicolae DL, Ober C, Sohn MH, Genome-wide association study of recalcitrant atopic dermatitis in Korean children. J Allergy Clin Immunol 136, 678–684.e4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arriba-Mendez S, Sanz C, Isidoro-Garcia M, Davild I, Laffond E, Horeno E, Avila C, Lorente F, 927T>C polymorphism of the cysteinyl-leukotriene type-1 receptor (CYSLTR1) gene in children with asthma and atopic dermatitis. Pediatr Allergy Immunol 17, 323–328 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Yamashita M, Hirahara K, Shinnakasu R, Hosokawa H, Norikane S, Kimura MY, Hasegawa A, Nakayama T, Crucial role of MLL for the maintenance of memory T helper type 2 cell responses. Immunity 24, 611–622 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Tibbitt CA, Stark JM, Martens L, Ma J, Mold JE, Deswarte K, Oliynyk G, Feng X, Lambrecht BN, De Bleser P, Nylén S, Hammad H, Arsenian Henriksson M, Saeys Y, Coquet JM, Single-Cell RNA Sequencing of the T Helper Cell Response to House Dust Mites Defines a Distinct Gene Expression Signature in Airway Th2 Cells. Immunity 51, 169–184.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Rafei-Shamsabadi DA, Klose CSN, Halim TYF, Tanriver Y, Jakob T, Context Dependent Role of Type 2 Innate Lymphoid Cells in Allergic Skin Inflammation. Frontiers in Immunology 10, 2591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vocanson M, Hennino A, Rozières A, Poyet G, Nicolas J-F, Effector and regulatory mechanisms in allergic contact dermatitis. Allergy 64, 1699–1714 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Hijnen D, Knol EF, Gent YY, Giovannone B, Beijn S, Kupper TS, Bruijnzeel-Koomen CAFM, Clark RA, CD8+ T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-γ, IL-13, IL-17 and IL-22. J Invest Dermatol 133, 10.1038/jid.2012.456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suárez-Fariñas M, Tintle S, Shemer A, Chiricozzi A, Nograles K, Cardinale I, Duan S, Bowcock A, Krueger JG, Guttman-Yassky E, Non-lesional atopic dermatitis (AD) skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J Allergy Clin Immunol 127, 954–64.e1–4 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bruhn S, Fang Y, Barrenäs F, Gustafsson M, Zhang H, Konstantinell A, Krönke A, Sönnichsen B, Bresnick A, Dulyaninova N, Wang H, Zhao Y, Klingelhöfer J, Ambartsumian N, Beck MK, Nestor C, Bona E, Xiang Z, Benson M, A Generally Applicable Translational Strategy Identifies S100A4 as a Candidate Gene in Allergy. Sci Transl Med 6, 218ra4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhagavathula N, Nerusu KC, Fisher GJ, Liu G, Thakur AB, Gemmell L, Kumar S, Xu ZH, Hinton P, Tsurushita N, Landolfi NF, Voorhees JJ, Varani J, Amphiregulin and epidermal hyperplasia: amphiregulin is required to maintain the psoriatic phenotype of human skin grafts on severe combined immunodeficient mice. Am J Pathol 166, 1009–1016 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pan Y, Kupper TS, Metabolic Reprogramming and Longevity of Tissue-Resident Memory T Cells. Front Immunol 9, 1347 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clark RA, Skin-Resident T Cells: The Ups and Downs of On Site Immunity. Journal of Investigative Dermatology 130, 362–370 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheuk S, Wikén M, Blomqvist L, Nylén S, Talme T, Ståhle M, Eidsmo L, Epidermal Th22 and Tc17 Cells Form a Localized Disease Memory in Clinically Healed Psoriasis. J.I 192, 3111–3120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Epskamp S, Cramer AOJ, Waldorp LJ, Schmittmann VD, Borsboom D, qgraph: Network Visualizations of Relationships in Psychometric Data. Journal of Statistical Software 48, 1–18 (2012). [Google Scholar]
- 63.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, von Mering C, STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research 47, D607–D613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shi J, Malik J, Normalized cuts and image segmentation. IEEE Transactions on Pattern Analysis and Machine Intelligence 22, 888–905 (2000). [Google Scholar]
- 65.Cohen JN, Bowman S, Laszik ZG, North JP, Clinicopathologic overlap of psoriasis, eczema, and psoriasiform dermatoses: A retrospective study of T helper type 2 and 17 subsets, interleukin 36, and β-defensin 2 in spongiotic psoriasiform dermatitis, sebopsoriasis, and tumor necrosis factor α inhibitor-associated dermatitis. J. Am. Acad. Dermatol 82, 430–439 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Rawat A, Rinchai D, Toufiq M, Marr AK, Kino T, Garand M, Tatari-Calderone Z, Kabeer BSA, Krishnamoorthy N, Bedognetti D, Karim MY, Sastry KS, Chaussabel D, A Neutrophil-Driven Inflammatory Signature Characterizes the Blood Transcriptome Fingerprint of Psoriasis. Front Immunol 11, 587946 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsoi LC, Rodriguez E, Degenhardt F, Baurecht H, Wehkamp U, Volks N, Szymczak S, Swindell WR, Sarkar MK, Raja K, Shao S, Patrick M, Gao Y, Uppala R, Perez White BE, Getsios S, Harms PW, Maverakis E, Elder JT, Franke A, Gudjonsson JE, Weidinger S, Atopic Dermatitis Is an IL-13-Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. J Invest Dermatol 139, 1480–1489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keermann M, Kõks S, Reimann E, Prans E, Abram K, Kingo K, Transcriptional landscape of psoriasis identifies the involvement of IL36 and IL36RN. BMC Genomics 16, 322 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quaranta M, Knapp B, Garzorz N, Mattii M, Pullabhatla V, Pennino D, Andres C, Traidl-Hoffmann C, Cavani A, Theis FJ, Ring J, Schmidt-Weber CB, Eyerich S, Eyerich K, Intraindividual genome expression analysis reveals a specific molecular signature of psoriasis and eczema. Science Translational Medicine (2014), doi: 10.1126/scitranslmed.3008946. [DOI] [PubMed] [Google Scholar]
- 70.Li B, Tsoi LC, Swindell WR, Gudjonsson JE, Tejasvi T, Johnston A, Ding J, Stuart PE, Xing X, Kochkodan JJ, Voorhees JJ, Kang HM, Nair RP, Abecasis GR, Elder JT, Transcriptome analysis of psoriasis in a large case-control sample: RNA-seq provides insights into disease mechanisms. J Invest Dermatol 134, 1828–1838 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bhushan M, Bleiker TO, Ballsdon AE, Allen MH, Sopwith M, Robinson MK, Clarke C, Weller RPJB, Graham-Brown R. a. C., Keefe M, Barker JNWN, Griffiths CEM, Anti-E-selectin is ineffective in the treatment of psoriasis: a randomized trial. Br J Dermatol 146, 824–831 (2002). [DOI] [PubMed] [Google Scholar]
- 72.Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, Nestle FO, Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med 199, 731–736 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chang MH, Levescot A, Nelson-Maney N, Blaustein RB, Winden KD, Morris A, Wactor A, Balu S, Grieshaber-Bouyer R, Wei K, Henderson LA, Iwakura Y, Clark RA, Rao DA, Fuhlbrigge RC, Nigrovic PA, Arthritis flares mediated by tissue-resident memory T cells in the joint. Cell Rep 37, 109902 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Z, Wang S, Goplen NP, Li C, Cheon IS, Dai Q, Huang S, Shan J, Ma C, Ye Z, Xiang M, Limper AH, Porquera E-C, Kohlmeier JE, Kaplan MH, Zhang N, Johnson AJ, Vassallo R, Sun J, PD-1hi CD8+ resident memory T cells balance immunity and fibrotic sequelae. Sci Immunol 4, eaaw1217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abdat R, Waldman RA, de Bedout V, Czernik A, Mcleod M, King B, Gordon S, Ahmed R, Nichols A, Rothe M, Rosmarin D, Dupilumab as a novel therapy for bullous pemphigoid: A multicenter case series. J Am Acad Dermatol 83, 46–52 (2020). [DOI] [PubMed] [Google Scholar]
- 76.Schmidt T, Solimani F, Pollmann R, Stein R, Schmidt A, Stulberg I, Kühn K, Eming R, Eubel V, Kind P, Arweiler N, Sitaru C, Hertl M, TH1/TH17 cell recognition of desmoglein 3 and bullous pemphigoid antigen 180 in patients with lichen planus. Journal of Allergy and Clinical Immunology 142, 669–672.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 77.Solimani F, Pollmann R, Schmidt T, Schmidt A, Zheng X, Savai R, Mühlenbein S, Pickert J, Eubel V, Möbs C, Eming R, Hertl M, Therapeutic Targeting of Th17/Tc17 Cells Leads to Clinical Improvement of Lichen Planus. Frontiers in Immunology 10, 1808 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Germain P-L, Lun A, scDblFinder: scDblFinder (Bioconductor version: Release (3.13), 2021; https://bioconductor.org/packages/scDblFinder/).
- 79.McGinnis CS, Murrow LM, Gartner ZJ, DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Systems 8, 329–337.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P-R, Raychaudhuri S, Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, Fallahi-Sichani M, Dutton-Regester K, Lin J-R, Cohen O, Shah P, Lu D, Genshaft AS, Hughes TK, Ziegler CGK, Kazer SW, Gaillard A, Kolb KE, Villani A-C, Johannessen CM, Andreev AY, Van Allen EM, Bertagnolli M, Sorger PK, Sullivan RJ, Flaherty KT, Frederick DT, Jané-Valbuena J, Yoon CH, Rozenblatt-Rosen O, Shalek AK, Regev A, Garraway LA, Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Langfelder P, Horvath S, Fast R Functions for Robust Correlations and Hierarchical Clustering. J Stat Softw 46, i11 (2012). [PMC free article] [PubMed] [Google Scholar]
- 83.R: The R Project for Statistical Computing (available at https://www.r-project.org/).
- 84.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, Trapnell C, Shendure J, The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Accelerating ggplot2 (available at https://ggforce.data-imaginist.com/).
- 86.Dixon P, VEGAN, a package of R functions for community ecology. Journal of Vegetation Science 14, 927–930 (2003). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing BAM files are deposited at the European Genome-Phenome Archive (EGA), which is hosted by the EBI and the CRG, under accession number EGAS00001005271. The processed Seurat objects with cell identities are available at Zenodo (https://zenodo.org/deposit/5228495). All statistical analysis and plotting of scRNA-seq and cell surface protein data were performed using Rstudio software (Version 1.4.1717). All analysis scripts are available at the online repository https://github.com/Yale73/scRNA-seq-for-diverse-human-rashes. All data are available in the main text or the supplementary materials. The online rash mapping webtool is available at https://rashX.ucsf.edu. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Jeffrey Cheng (Jeffrey.Cheng@ucsf.edu) or Raymond Cho (Raymond.cho@ucsf.edu).