

Abstract
A disintegrin and metalloproteases (ADAMs) are key mediators of cell signaling by ectodomain shedding of various growth factors, cytokines, receptors and adhesion molecules at the cellular membrane. ADAMs regulate cell proliferation, cell growth, inflammation, and other regular cellular processes. ADAM17, the most extensively studied ADAM family member, is also known as tumor necrosis factor (TNF)-α converting enzyme (TACE). ADAMs-mediated shedding of cytokines such as TNF-α orchestrates immune system or inflammatory cascades and ADAMs-mediated shedding of growth factors causes cell growth or proliferation by transactivation of the growth factor receptors including epidermal growth factor receptor. Therefore, increased ADAMs-mediated shedding can induce inflammation, tissue remodeling and dysfunction associated with various cardiovascular diseases such as hypertension and atherosclerosis, and ADAMs can be a potential therapeutic target in these diseases. In this review, we focus on the role of ADAMs in cardiovascular pathophysiology and cardiovascular diseases. The main aim of this review is to stimulate new interest in this area by highlighting remarkable evidence.
Keywords: Signal transduction, Hypertension, Atherosclerosis, Heart disease, Vascular biology, Endothelium, Inflammation, Angiotensin
Introduction
A disintegrin and metalloprotease (ADAM) family proteins belong to a Zn2+-dependent protease superfamily that are expressed as type 1 transmembrane proteins. In human, 22 ADAM proteins have been identified: ADAM1, 2, 3B, 7, 8, 9, 10, 11, 12, 15, 17, 18, 19, 20, 21, 22, 23, 28, 29, 30, 32, and 33 in MEROPS database within the M12B Adamalysin subfamily of metallopeptidases; https://merops.sanger.ac.uk/. Almost half of ADAMs are exclusively or predominantly expressed in testis and epididymis, whereas 11 ADAMs (ADAM8, 9.10, 11, 12, 15, 17, 19, 22. 23 and 33) are ubiquitously expressed [1]. Interestingly, only 12 human ADAMs are proteolytically active (ADAM8, 9, 10, 12, 15, 17, 19, 20, 21, 28, 30 and 33). These ADAMs work as key mediators of cell signaling by ectodomain shedding of various growth factors, cytokines, receptors and adhesion molecules at the cellular membrane. The proteolytically inactive ADAMs are considered to participate in cellular communication through their adhesive properties. Accordingly, ADAMs regulate cell proliferation, cell growth, inflammation, and other cellular processes [2].
Among ADAM family members, ADAM17, also known as tumor necrosis factor (TNF)-α converting enzyme (TACE), is the most well-studied protein. ADAM17 was first purified and cloned in 1997 as a metalloproteinase that specifically cleaves precursor TNF-α [3, 4]. These findings completely changed the significance of ADAMs from mere adhesion molecules to important regulators of cell signaling.
ADAM17 consists of an N-terminal signal sequence, a prodomain, a catalytic domain with a typical HEXXHXXGXXH sequence, a disintegrin domain, a membrane proximal domain, a transmembrane domain and a cytoplasmic tail (Fig. 1) [5, 6]. ADAM17 exists as a multimer at the cell membrane, and this multimerization is mediated by an EGF-like domain [7]. The maturation of ADAM17 proenzyme requires furin-dependent processing at either a canonical proprotein convertase (PC) cleavage site at the boundary between the prodomain and catalytic domain [8] or an upstream PC cleavage site [9]. These cleavages are thought to be essential for adequate activation of ADAM17. ADAM17 is expressed very broadly in somatic tissues and a variety of growth factors, cytokines, receptors, adhesion molecules and other molecules have been revealed as substrates of ADAM17 by in vivo or in vitro studies (Table 1). After shedding, cleaved substrates can bind to the receptors on the same cell (autocrine), local cells (paracrine), or non-local cells by transport through blood (endocrine) [10, 11]. In this manner, ADAM17-mediated shedding of cytokines such as TNF-α precursor to produce soluble TNF-α orchestrates immune system or inflammatory cascades.
Fig. 1.

The general structure of ADAMs. A disintegrin and metalloproteases (ADAMs) consist of several domains. The prodomain keeps ADAMs inactive, and protein convertases such as furin cleave this prodomain in Golgi apparatus to activate ADAM17. The metalloprotease domain is a key domain that is involved in catalytic activity and ligand shedding. The disintegrin domain interacts with integrins and supports adhesion. This domain also serves to maintain the structure of extracellular region. The membrane proximal domain regulates substrate binding and shedding activity. ADAM10 and ADAM17 have membrane proximal domain and other ADAMs have EGF-like repeats, which regulate substrate binding and shedding activity. Cytoplasmic tail of ADAMs interacts with signaling molecules. Phosphorylation of cytoplasmic tail regulates the activation, trafficking and subcellular localization of ADAMs
Table 1.
Substrates of ADAM17
| Cytokines | Receptors | Adhesion molecules |
|---|---|---|
|
CD44 [117] FLT-3L [254] Jagged 1 [38] Kit-ligand 1 and 2 [255] LAG-3 [256] MICA [257] MICB [258] RANKL [259] TNF-α [20, 260, 190, 261, 4, 11] TNF beta [262] |
APOER [266] CD30 [267] CD40 [268] CD89 [269] EMMPRIN [135] EPCR [83] Ephrin B4 [135] GPIba [274] GPV [275] GPVI [276] IL-1R II [21] Integrin beta-1 [150] Leptin receptor [279] LOX-1 [280] M6P/IGF2R [281] NPR [284] P75 TNF receptor [113] Ptprz [286] syndecan 1 and 4 [287] Toll-like receptor 4 [288] TrkA [289] VEGFR [31] VPS10p [290] |
ALCAM [291] CD62L [292] collagen XVII [293] desmoglein-2 [291] EpCAM [294] ICAM-1 [111] JAM-A [123] L-selectin [113] L1-CAM [295] PTP-LAR [296] NCAM [297] nectin-4 [298] PECAM-1 [135] |
| Growth factors | Others | |
|
CSF-1 [138] Epigen [302] Epiregulin [190] IGFR1 [135] Neuregulin-1 [199] Tomoegulin-2 [307] |
CD163 [310] KIM-1 [311] Klotho [312] MerTK [313] PMEL17 [314] PrPc [315] Tim-3 [316] VASN [317] |
The shedding of epidermal growth factor receptor (EGFR) ligands is an important process since EGFR is an essential tyrosine kinase receptor in the development of various diseases. The role of EGFR in cancer is widely studied; however, recent evidence has demonstrated the importance of EGFR on cardiovascular physiology and pathophysiology. More specifically, G protein-coupled receptors (GPCRs)-mediated EGFR transactivation has been recognized as a key point of control governing cardiovascular outcomes [12]. GPCR activation causes initial heterotrimeric G protein dissociation. Subsequently, ligand-specific intermediates including intracellular Ca2+ and reactive oxygen species (ROS) are elevated and non-receptor tyrosine kinases are activated, followed by metalloprotease activation and shedding of EGFR ligands [13–15]. Among these metalloproteases, ADAM17 has been recognized as an essential mediator of EGFR ligand shedding and subsequent EGFR transactivation [13, 15, 16]. Upon activation, ADAM17 cleaves EGFR ligands such as heparin binding EGF-like growth factor precursor (HB-EGF) to produce mature soluble HB-EGF which then binds and activates EGFR. In addition, upon shedding, the cytoplasmic tail of EGFR ligands is recognized as a site of protein interactions which mediates several intra- and intercellular phenomena including ligand trafficking or migration to the cell surface, signal transduction, and gene expression via interaction with a transcriptional repressor [17–19]. Therefore, ectodomain shedding of EGFR ligands by ADAM17 can initiate a bidirectional signaling event with released growth factor and free shed remnant. Both TNF receptor 1 (TNFR1) and TNFR2 are also ADAM17 substrates [20, 21]; thus, creation of soluble TNFRs modulates soluble TNF-α availability as well as TNFR activities. In addition to TNF-α precursor and TNFR, interleukin-6 receptor (IL6R) is a critical substrate to mediate ADAM17 function. IL6 primarily binds to IL6R, which is specifically expressed in certain cell types such as leukocyte and hepatocyte. The IL6 IL6R complex then binds to ubiquitously expressed signaling receptor gp130 leading to activation of STAT1, STAT3 and the ERK cascade to mediate inflammatory responses. Cells only expressing gp130 cannot respond to IL6. However during trans-signaling, gp130 can be activated with IL6 complexed with soluble IL6R generated by ADAM17 shedding of the receptor [22]. Taken together, ADAM17-mediated shedding of growth factors and cytokines causes cell growth and inflammation, respectively (Fig. 2).
Fig. 2.
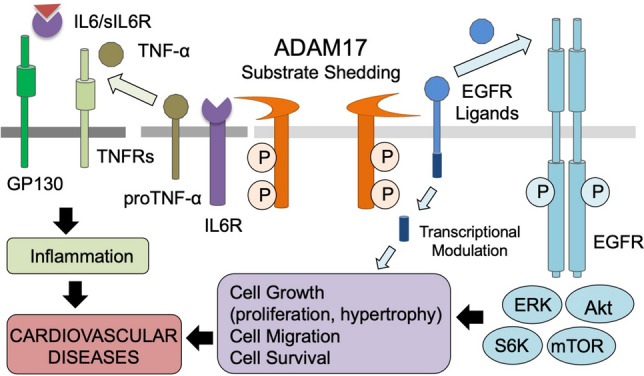
ADAM17 mediates cardiovascular diseases via ectodomain shedding. A variety of substrates including growth factors, cytokines, receptors, adhesion molecules are cleaved by ADAM17 and initiate or modulate intracellular signaling. The ectodomain sedding events can be occurred in cis (on the same cell) or trans (between two cells), and act in autocrine (on the same cell types), paracrine (on distinct resident cells) and/or endocrine (on distinct organs through circulation) manner. Therefore, these events involve single cell membrane (cis cleavage and autocrine signaling), two (cis and paracrine/endocrine or trans and paracrine) or three distinct cell-type membranes (trans and endocrine) expressing ADAM17, substrates and the receptors. Prototypical examples of ADAM17 substrate relationship are illustrated. Right: upon ADAM17 activation, cleaved EGFR ligands transactivate EGFR and initiate EGFR-mediated intracellular signaling including activation of ERK, Akt, mTOR and p70 S6K, resulting in cell proliferation or hypertrophy in an autocrine manner. In addition, the cytoplasmic tail of EGFR ligands is recognized as a site of protein interaction or translocate to nucleus which acts as a transcriptional modulator. Left: activated ADAM17 also regulate inflammation via the cleavages of inflammatory cytokines and their receptors. The examples shown are pro-NF-α shedding and TNFR activation as well as soluble IL6R (sIL6) generation to lead to the IL6-sIL6 complex, which can activate their receptor, GP130 in the absence of IL6R in a paracrine or endocrine manner
There are numerous regulators of ADAM17-dependent ectodomain EGFR ligand shedding including various extracellular stimuli, cellular protein modulators, phosphorylation in the cytosolic domain and its own disulfide switch [23, 24]. Certain regulators modulate ADAM17 activity via stabilization. Band 4.1 protein, ezrin, radixin, moesin (FERM) domain-containing protein 8 (FRMD8) stabilizes ADAM17 at the cell surface and supports ADAM17-mediated ligand shedding [25]. The sorting protein phosphofurin acidic cluster sorting protein 2 (PACS-2) co-localizes with ADAM17 on early endosomes, and loss of PACS-2 results in decreased ADAM17 recycling, stability upon internalization, cell surface expression, and EGFR ligand shedding [26].
Notably, TNF-α induces ADAM17 and Src-dependent EGFR activation, and initiates the extracellular signal-regulated kinase (ERK)-dependent guanine nucleotide exchange factors (GEF)-H1 and RhoA signaling pathway, suggesting a mechanistic link between inflammatory and proliferative pathophysiology [27]. Platelet-derived growth factor (PDGF) receptor β stimulation activates ADAM17 shedding of TNF-α or transforming growth factor (TGF)-α and subsequently initiates EGFR signaling pathways [28]. p38 mitogen-activated protein (MAP) kinase and Src are reported to activate ADAM17 via interaction with the cytoplasmic domain of ADAM17, increase ADAM17-mediated shedding of TGF-α family ligands and activate EGFR signaling [29–31].
A protein kinase C (PKC) activator, phorbol 12-myristate 13-acetate (PMA), induces ADAM17-mediated HB-EGF shedding and EGFR transactivation. PKC and ADAM17-dependent HB-EGF shedding is triggered by apically localized A1 adenosine receptor stimulation [32]. Notably, in EGFR ligands shedding, regulatory proteins such as PKCα, PKC-regulated protein phosphatase 1 inhibitor 14D (PPP1R14D), and PKCδ affect the shedding of some ADAM17 substrates without significant effect on protease activity [33]. In addition, PKCα and PPP1R14D act on ADAM17-mediated shedding of TGF-β, HB-EGF and amphiregulin, whereas PKCδ is required for ADAM17-mediated shedding of neuregulin [33], suggesting a complex regulation of EGFR ligand shedding.
Several phosphorylation sites such as Thr735 phosphorylation by ERK or p38 MAP kinase appear to be involved in ADAM17 activation [23, 34]. Polo-like kinase 2 (PLK2) interacts with and phosphorylates ADAM17 at Ser794 resulting in shedding of pro-TNF-α and TNF receptors, and PLK2 expression is up-regulated in inflammatory conditions [35]. ADAM17 Tyr702 is another important phosphorylation site for EGFR transactivation induced by G protein-coupled receptor (GPCR) agonist such as angiotensin II [13]. However, phosphorylation of the intracellular domain of ADAM17 may not always be essential for activation. Studies with ADAM17 chimeric construct showed that PMA-induced ADAM17 activation did not require the intracellular domain, but it required the transmembrane domain [36, 37]. While detailed mechanistic dissection of the mode of modulations is beyond the scope of this review article, these conflicting findings indicate that ADAM17 regulation by the modulations and the modulators are likely cell/tissue type and context-specific.
It is well established that mature ADAM17 is associated with lipid rafts, although some substrates such as Jagged-1 are cleaved by ADAM17 in lipid raft-independent pathways [38]. Many signaling proteins which involve ADAM17-mediated EGFR transactivation including EGFR, GPCRs such as AT1R, G proteins, Src family kinases and ADAM17 are localized to caveolae, a subset of lipid rafts [39–41]. Angiotensin II-induced transactivation of EGFR relies on ADAM17 compartmentalization in caveolae [41]. Caveolin-1, a major structural protein of caveolae, is required for TGF-β-mediated ADAM17 activation via phosphorylation of Src and NADPH Oxidase 1 (NOX1)-mediated ROS production [42]. Silencing of caveolin-1 in cultured VSMCs can prevent angiotensin II (AngII)-induced ADAM17 induction and activation [43]. However, inhibition of EGFR transactivation by over-expressing caveolin-1 was also observed in VSMCs stimulated by AngII [41]. Therefore, further investigation is needed to explore the contradictory data observed regarding the signaling relationship of ADAM17 and caveolin-1.
The conformational change in ADAM17 also affects ADAM17 activity. Changes in the redox environment like PMA-dependent induction of mitochondrial ROS enhance ADAM17 activity, and the inactivation of thiol isomerases, specifically protein disulfide isomerase (PDI), is reported as a key player. PDI regulates ADAM17 activity by conformational change in ADAM17 from an active “open form” to an inactive “closed form” [44]. In addition, thioredoxin-1 is reported to interact with the cytoplasmic domain of ADAM17 and negatively regulate ADAM17 activity [45, 46]. The noncatalytic domains of ADAM17 are also reported to regulate the ADAM17 activity via steric hindrance [47]. Conserved ADAM-seventeeN Dynamic Interaction Sequence (CANDIS) encoded by ADAM17 is a short juxtamembrane segment of 17 amino acid residues. CANDIS appears critical in substrate recognition, and also regulates the shedding activity of ADAM17 by interacting with lipid bilayers [48]. It has also been reported that the membrane proximal domain of ADAM17 provides a phosphatidylserine binding motif. ADAM17 is activated upon phosphatidylserine binding in several cell types including endothelial cells. Cells undergoing apoptosis will enhance phosphatidylserine in outer membrane. Thus, CANDIS and the membrane proximal domain likely provide a means to stimulate ADAM17 activity upon extracellular stress, such as those causing apoptosis [49]. In addition, site-specific O-glycosylation in juxtamembrane segment of several ADAM17 substrates mediated by distinct polypeptide N-acetylgalactosamine (GalNAc)-transferase (GalNAc-T) isoforms is also reported to widely modulate ADAM17-mediated shedding in a substrate-specific manner [50].
Catalytically inactive rhomboid protein (iRhom) 2 was identified as a key protein which controls the maturation and function of ADAM17 [51–53], regulating cytokine and growth factor signaling [54]. Due to its preferential expression in leukocytes, iRhom2 –/– mice are defective in myeloid-specific TNF-α shedding [53]. However, iRhom2 –/– mice showed decreased myeloid cell repopulation under stress due to defect in myeloid colony-stimulating factor receptor-1 (CSFR1) shedding. Moreover, in iRhom2 and related iRhom1 double knockout mouse tissues, there is a lack of ADAM17 maturation and reduced EGFR activation [55]. FERM domain-containing 8 (FRMD8) [25]/iTAP (iRhom Tail-Associated Protein[56] has been discovered which enhances stability of ADAM17 and iRhoms. It has also been shown that ERK-dependent phosphorylation of iRhom2 recruits 14-3-3 proteins which leads to iRhom2 dissociation from ADAM17 leading to ADAM17 activation [57]. It is interesting to note that iRom1 is preferentially and constitutively expressed in endothelial cells with transcriptional regulation by shear stress, whereas inflammatory cytokines can induce iRhom2 but not iRom1 [58]. Therefore, iRhoms appear to be the main focus of research into ADAM17 regulation.
Increased ADAM17-mediated shedding contributes to the progression of various cardiovascular diseases such as atherosclerosis or ischemia via both EGFR transactivation and inflammation. Thus, ADAM17 is a potential therapeutic target in these diseases. The role of ADAM17 in cancer and autoimmune diseases has been well documented [59–61]; here, we focus on the role of ADAM17 in cardiovascular pathophysiology and cardiovascular diseases. This review also includes a discussion of other ADAM family proteins which share cell-specific distribution, the HExGHxxGxxHD motif that is required for proteolytic activity, and, therefore, function with ADAM17. Notably, most substrates can be cleaved by a variety of ADAM family members, and this seemingly nonspecific relationship between substrates and ADAMs makes the physiology of ADAMs more complicated and interesting. The main aim of this review is to rejuvenate interest in ADAM research by highlighting remarkable evidence.
Adam17 and cardiovascular pathophysiology
ADAM17 is expressed in various cells including endothelial cells, vascular smooth muscle cells (VSMCs), fibroblasts, and monocytes. In cultured VSMCs, angiotensin II stimulation increases ADAM17 phosphorylation [13], protein expression, mRNA expression, and promoter activity [62]. Activation of ADAM17 via tyrosine phosphorylation contributes to HB-EGF shedding, EGFR transactivation [63], and subsequent growth promoting signals induced by angiotensin II [13].
Previous investigations of ADAM17 in cardiovascular pathophysiology have revealed ADAM17 to be a highly regulated controller of disease progression. The expression and activity of ADAM17 are regulated in a multi-layered and highly complicated manner as reviewed previously [64], and the regulation of ADAM17 in cardiovascular pathophysiology has also been investigated. Notably, the meaning of increased ADAM17 expression should be carefully interpreted. Systemically, ADAM17 overexpression mice show no enhancement in TNF-α shedding activity, suggesting that ADAM17 activity can be independent of transcriptional regulation and that excess ADAM17 does not necessarily result in enhanced shedding activity in vivo [65]. In this section, we highlight in vivo and in vitro findings regarding the role of ADAM17 and the regulation of ADAM17 in cardiovascular pathophysiology. Moreover, we review the clinical studies investigating the role of ADAM17 in human cardiovascular diseases.
ADAM17 and hypertension
In a mouse model of angiotensin II-induced hypertension with smooth muscle ADAM17 deletion or systemic pharmacological inhibition of ADAM17, vascular medial hypertrophy and perivascular fibrosis were attenuated [66]. This is because ADAM17 mediates angiotensin II-induced EGFR transactivation in vascular smooth muscle cells (VSMCs) causing growth promoting signal transduction [12]. Thus, inhibition of EGFR also mitigated hypertensive vascular remodeling in mice infused with angiotensin II [67]. Interestingly, blood pressure remains high in these models with ADAM17/EGFR inhibition at 2-week time point; whereas, less hypertension was reported at 1-week point [68], suggesting unique roles of ADAM17 in hypertensive vascular pathology. How does the acute signaling events via the ADAM17/EGFR system mediate chronic vascular pathology? This seems to involve feed-forward induction of ADAM17 transcript via ER stress and subsequent unfolded protein response (UPR). Upon AngII stimulation, chronic UPR markers were induced in vitro in VSMCs and in vivo in the vasculature. Suppression of ER stress and UPR via chemical chaperoning, thus, attenuated vascular ADAM17 induction and associated vascular pathology [67]. At the cellular level, AngII-induced UPR seems insufficient to attenuate protein misfolding leading to protein aggregate formation in VSMCs. The sustained proteotoxicity prolongs UPR, enhances inflammatory response and senescence [69, 70]. Accordingly, chronic activation of the vascular ADAM17/EGFR system seems to contribute to premature inflamm-aging via protein aggregation [71, 72]. Interestingly, vascular ADAM17 promoter can also be activated via hypoxia inducible factor 1α upon AngII stimulation in VSMCs. Thus, vascular ADAM17 activation may be exaggerated under ischemic conditions [62]. This novel concept of ADAM17 in mediating hypertension and chronic vascular pathology is illustrated in Fig. 3.
Fig. 3.
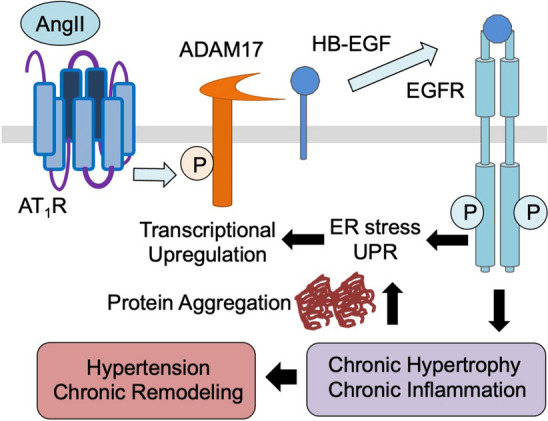
The potential molecular mechanism by which Angiotensin II signaling via ADAM17 mediates chronic vascular pathology in hypertension. Angiotensin II rapidly activates ADAM17 via its Tyr702 phosphorylation through the GPCR, AT1 receptor (AT1R) in VSMCs. This leads to proHB-EGF shedding and subsequent EGFR transactivation. Enhanced protein synthesis results in protein misfolding causing protein aggregate formation. Protein aggregates enhance ER stress and UPR which transcriptionally upregulate ADAM17 thus create the feed-forward loop of sustained signaling leading to hypertensive vascular remodeling
ADAM17 also influences blood pressure via a brain-dependent mechanism. Deoxycorticosterone acetate (DOCA)-salt treatment enhanced ADAM17 expression and activity in the hypothalamus, significantly reduced an ADAM17 substrate, angiotensin-converting enzyme 2 (ACE2) expression and activity in brain, resulting in increased blood pressure, inflammation, hypothalamic angiotensin II levels, and causing autonomic dysfunction. Accordingly, knockdown of ADAM17 in the brain can blunt the development of hypertension and restore ACE2 activity and baroreflex function, indicating that ADAM17-mediated shedding of ACE2 contributes to the development of neurogenic hypertension [73]. With neuron selective ADAM17 knockout mice, the mechanism appears to involve ADAM17-dependent ACE2 inactivation in pre-sympathetic neurons within the paraventricular nucleus [74]. Moreover, in the brain of hypertensive patients ADAM17-mediated ACE2 shedding seems to be promoted by angiotensin II suggesting the involvement of ADAM17 in neurogenic hypertension in human [75].
ADAM17, atherosclerosis and neointima formation
ADAM17 is highly expressed in aortic lesions in atherosclerosis-prone sites in high-fat diet-fed apolipoprotein E knockout mice, and ADAM17 may contribute to the elevated levels of circulating soluble TNF-α receptors [76]. In addition, ADAM17 is recognized as a candidate gene of atherosclerosis susceptibility since ADAM17 mRNA expression and activity is increased in association with atherosclerosis resistance in low density lipoprotein (LDL) receptor deficient mice [77]. ADAM17 gene silencing by injecting shRNA into the abdominal aortic plaque enhances plaque stability and improves vascular positive remodeling via attenuation of local inflammation, neovascularization and matrix metalloproteinase (MMP) activation, and enhancement of collagen production [78]. In addition, genetical or pharmacological inhibition of ADAM17 prevents neointimal hyperplasia after vascular injury [79]. Since ADAM17–/– mice are not viable, ADAM17 hypomorphic mice have been generated, which have barely detectable levels of ADAM17 in all tissues [80]. Contrary to results with ADAM17 inhibition, a study using ADAM17 hypomorphic mice revealed that ADAM17 deficiency enhances atherosclerosis via TNF receptor 2 (TNFR2) signaling [81], suggesting that moderate activation of ADAM17 had atheroprotective effects by preventing the endogenous TNFR2 overactivation. The cell-type-specific difference in the role of ADAM17 could be one reason for these controversial findings. Indeed, it has been reported that myeloid ADAM17 deletion is detrimental; whereas, endothelial ADAM17 deletion appears protective against atherosclerosis development [82].
ADAM17 expression is reported in human atherosclerotic plaques [76]. Microparticles isolated from human atherosclerotic plaques are shown to carry active ADAM17 on their surface. These microparticles enhance the shedding of TNF-α, TNF receptor 1 (TNFR1), and endothelial protein C receptor (EPCR) at endothelial cells, indicating ADAM17-positive microparticles could regulate the inflammatory balance in culprit lesions [83]. Moreover, the ADAM17 at advanced human atherosclerotic lesions is in its catalytically active form and ADAM17-expressing cells are co-localized with CD68-positive cells of monocytic origin [84]. These results suggest the contribution of ADAM17 in monocyte homing, migration, and proliferation in human atherosclerotic lesions.
ADAM17 and aortic aneurysms
ADAM17 is identified as a central gene associated with angiotensin II-induced abdominal aortic aneurysm (AAA) in genome-wide transcriptional profiling [85]. ADAM17 expression is enhanced in experimental models of AAA, and temporal and systemic deletion of ADAM17 prevents AAA development in association with attenuating inflammation elicited by TNF-α [86]. AAA as well as enhanced ADAM17 expression and EGFR phosphorylation in experimental AAA are markedly attenuated in caveolin-1 knockout mice, supporting ADAM17 compartmentalization in caveolae in VSMCs [43]. Consistent with these findings, VSMC ADAM17 silencing or systemic pharmacological ADAM17 inhibition attenuated AAA in mice with angiotensin II infusion [87]. Cleavage of an EGFR ligand appears critical since inhibition of EGFR is sufficient to prevent angiotensin II-dependent AAA in mice [88]. How does ADAM17-dependent EGFR transactivation lead to chronic vascular cell dysfunction to contribute to AAA? As recognized in hypertension, ER stress and UPR seem key drivers for the VSMC phenotype involved in AAA [89].
It is also interesting to note the role mitochondrial morphology plays in AAA. Mitochondrial fission fusion events are critical for mitochondrial homeostasis. However, under several stressed conditions such as those with cardiovascular diseases, the balance shifts toward more fission leading to mitochondrial dysfunction and mitochondrial oxidative stress. Thus, mitochondrial fission sustains inflammatory responses [71]. A GTPase Drp1 is a master regulator of mitochondrial fission. In human AAA as well as AngII-dependent model of AAA, Drp1 expression appears enhanced. Moreover, in abdominal aortic VSMC, the critical ADAM17/EGFR downstream effector ERK phosphorylates and activates Drp1 leading to mitochondrial fission. AngII stimulated mitochondrial oxygen consumption which was attenuated with a Drp1 inhibitor. In addition, inhibition of mitochondrial fission attenuated AngII-dependent AAA, which was associated with prevention of aortic ER stress/UPR and senescence. Inflammatory leukocyte infiltration was also attenuated [90]. There is additional evidence linking ADAM17 and mitochondrial fission in the cardiovascular system. In cultured aortic endothelial cells, TNF-α stimulated Drp1-dependent mitochondrial fission and nuclear factor-κB-dependent inflammatory responses. Genetic inhibition of Drp1 attenuated nuclear factor-κB activation and subsequent inflammatory responses in endothelial cells with TNF-α exposure. Thus, in endothelial Drp1-deleted mice, leukocyte adhesion to endothelium in response to TNF-α injection was attenuated [91]. In addition, Drp1 appears to be indispensable for AngII-induced senescence in endothelial cells [92]. Potential overall contributions of ADAM17 in AAA and associated inflammation via Drp1-dependent mitochondrial fission are illustrated in Fig. 4.
Fig. 4.
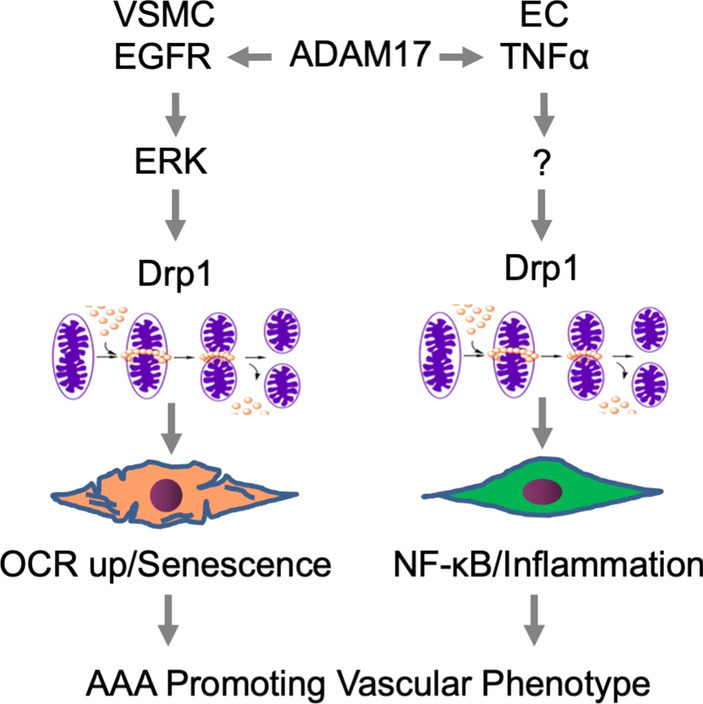
Vascular ADAM17 activation mediates smooth muscle cell senescence and endothelial inflammation thus changes vascular cell phenotypes leading to AAA. In VSMCs, ADAM17-dependent EGFR activation causes mitochondrial fission via Drp1 which leads to enhanced oxygen consumption and senescence. In EC, ADAM17-dependent TNF-α production stimulates Drp1-dependent mitochondrial fission and subsequent mitochondrial ROS production and NF-κB activation thus sustains EC inflammation. The vascular phenotype changes caused by ADAM17 activation, thus, contribute to AAA development
Similar to AAA, experimental thoracic aortic aneurysm (TAA) model showed significant elevation of expression of ADAM17 in the vasculature [93]. Interestingly, VSMC-specific ADAM17 deletion attenuates TAA formation via fibrosis, inflammation, and adverse aortic remodeling, whereas EC-specific ADAM17 deletion also attenuates TAA progression by protecting the integrity of adherens junction and tight junctions in an adventitial elastase exposure model [94].
In human AAA sample obtained during surgical operation, ADAM17 is overexpressed in the aortic wall [86] compared to normal aortae. Enzymatically active ADAM10 and ADAM17 are carried on membrane microvesicles in the intraluminal thrombus of human AAA [95]. ADAM17 expression is higher in the transition zone than in the mid-portion of aneurysm, and ADAM17 is expressed in CD68-positive macrophages in the media and adventitia obtained from the transition zone in AAA [96]. In addition, ADAM17 promoter polymorphism rs12692386 is reported to associate with AAA, enhanced ADAM17 expression and circulated TNF-α [97]. Taken together, these data indicate ADAM17 is important in the pathogenesis of AAA in humans.
ADAM17 in mediating vascular inflammation
Atherosclerosis is accelerated by chronic inflammation. Macrophages and monocytes are recognized as contributors to the inflammatory component of atherogenesis [98]. TIMP-3 overexpression in macrophage attenuates atherosclerosis in LDL receptor knockout mice [99]. ADAM17-mediated shedding of colony-stimulating factor 1 (CSF-1) on the cell surface of neutrophils and macrophages enhances macrophage proliferation in state of acute and chronic inflammation [100]. Accordingly, monocyte ADAM17, not endothelial ADAM17, facilitates the completion of trans-endothelial migration by accelerating the rate of diapedesis [101]. ADAM17 induced shedding of CSF [102] and functional suppression of macrophage via CSFR1 has been reported [103]. However, participation of CSFR1 in atherosclerosis has also been reported [104]. Thus, further clarification seems needed regarding the overall contribution of the ligand as well as the receptor shedding by ADAM17 in atherosclerosis and associated inflammation.
Adhesion molecules such as vascular cell adhesion protein 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), and L-selectin expressed in endothelium are other key players in atherosclerosis development by regulating leukocyte recruitment [105–108]. ADAM17 cleaves these molecules and regulates inflammation. ADAM17-mediated shedding of VCAM-1 produces soluble form of VCAM-1 [109]. This process is supported by the findings suggesting that circulating VCAM-1 can be a marker of atherosclerotic lesions in diabetes patients with atherosclerosis [110]. Ectodomain shedding of ICAM-1 is also ADAM17 dependent, and pharmacological or genetic inhibition of ADAM17 can block the ICAM-1 shedding, resulting in up-regulation of cell adhesive function [111]. Similar to soluble VCAM-1, circulating ICAM-1 is also reported to serve as a molecular marker for atherosclerosis [112]. L-selectin constitutively expressed by most circulating leucocytes including neutrophils is critical in directing these cells to the sites of inflammation. ADAM17-deficient cells are impaired in L-selectin shedding, showing that L-selectin is cleaved by ADAM17 [113], even though there is ADAM17-independent shedding of L-selectin [114]. Rapid ADAM17-mediated L-selectin shedding increases rolling velocity and enhances leukocyte accumulation on the vascular wall [115]. Constitutive shedding by ADAM17 also regulates soluble L-selectin that affects interactions between leukocyte and endothelium [116]. CD44, a glycoprotein which promotes cell adhesion and migration, recruits inflammatory cells to vessel wall and activates vascular cells in atherogenic conditions, and CD44 undergoes ADAM17-dependent cleavage [117].
Stimulation of the thromboxane A2 receptor induces rapid ADAM17-mediated shedding of cell surface CX3CL1, a key factor in recruiting monocytes. Shedding of CX3CL1 results in recruitment of leukocytes to vascular inflammatory sites and enhanced adhesion once recruited [118]. ADAM17 also affects vascular permeability by shedding of adhesion molecules in cell junction. JAM-A is another molecule known to facilitate vascular inflammation by promoting the migration of endothelial cells [119, 120] and monocytes [121, 122], as well as angiogenesis. ADAM17-mediated shedding of JAM-A is caused at vascular inflammation sites, and this shedding of JAM-A down-regulates transmigration of monocytes and increases endothelial permeability within the endothelial junctions [123]. There seem to be certain players that affect vascular inflammation via ADAM17 activity modulation. Neutrophil activation upon E-selectin binding or endothelial adhesion promotes redistribution and co-clustering of ADAM17 and L-selectin, modulating the process of rolling, activation, arrest, and transmigration of neutrophils [124]. Oxidative conditions such as H2O2 treatment induce L-selectin shedding and thiol-disulfide conversion occurring in extracellular region of ADAM17 are involved in this reaction [125]. Taken together, ADAM17 regulates vascular inflammation in various manner including inflammatory activation, regulation of vascular permeability, rolling, adhesion, and transmigration of leukocytes.
ADAM17 mechanism of angiogenesis and neovascularization
Accumulating evidence suggests that ADAM17 promotes angiogenesis through stimulation of endothelial cell proliferation, invasion, network formation, and MMP-2 activation [126, 127]. Vascular endothelial growth factor (VEGF)-A and the receptor vascular endothelial growth factor receptor 2 (VEGFR2) are essential for angiogenesis, and VEGFR2 is known to coordinate endothelial cell migration, capillary formation, and vascular permeability [128]. VEGF-A activates ADAM17 via ERK pathway, resulting in shedding of VEGFR2 and other substrates, and ADAM17 selective inhibition attenuates VEGFR2-induced ERK phosphorylation [31]. VEGF-A/VEGFR2 causes migration of human umbilical vein endothelial cells (HUVECs), and fibroblast growth factor 7 (FGF7)/FGF receptor 2-IIIb (FGFR2b) causes cell migration in epithelial cells. These migrations depend on EGFR/ERK signaling and ADAM17-mediated HB-EGF shedding [37]. In addition, a study using HUVECs showed that IL6 and interferon-γ caused ADAM17-dependent shedding of neuregulin. Based on several cytokine measurements, this neuregulin shedding is speculated to contribute to inflammation-associated angiogenesis [129]. ADAM17-mediated HB-EGF shedding and subsequent EGFR transactivation in retinal endothelial cells are also reported as key components in ocular neovascularization [130]. Genetic variation at Tgfbm3 or pharmacological inhibition of ADAM17 modulates postnatal circulating endothelial progenitor cell (CEPC) numbers through TGF-β receptor 1 activity, suggesting that variant ADAM17 is an innate modifier of adult angiogenesis since CEPC numbers correlate with angiogenic potential [131]. Finally, ADAM17 inhibition enhances the expression of thrombospondin -1 (TSP1), an anti-angiogenic factor, and overexpression of ADAM17 down-regulates TSP1 in endothelial cells, suggesting that ADAM17 positively regulates angiogenesis by its negative feedback of TSP1 [132].
Cdc42, a Ras-related GTPase, has an important role in cell migration, proliferation, and survival. Contrary to the positive regulatory roles of ADAM17 as described, the deletion of Cdc42 increases ADAM17-dependent VEGFR2 shedding, thus impairing angiogenesis in mice [133]. Flt, one of the VEGF receptors at the cell surface, consists of a homodimer or heterodimer with VEGFR2. ADAM17-mediated ectodomain shedding of Flt antagonizes VEGF when Flt is co-expressed with VEGFR [134]. The regulatory roles of ADAM17 in angiogenesis could be varied by the given pathology and require further investigation.
Mice with conditionally inactivated ADAM17 in smooth muscle cells (Adam17/flox/flox/sm22α-Cre mice) show no clear effects on angiogenesis [135]. On the other hand, mice with conditionally inactivated ADAM17 in endothelial cells (Adam17/flox/flox/Tie2-Cre mice) show significantly reduced pathological neovascularization, although they have no obvious defects in developmental angiogenesis [135]. Similarly, endothelial ADAM17 knockdown with both constitutive and inducible VE-cadherin Cre mice is reported to reduce collateral circulation formation [136]. These results indicate the essential role of endothelial ADAM17 in neovascularization. Study showing that retinal neovascularization is attenuated by ADAM17 inhibition with in vivo angiogenesis model supports this finding [137]. The distinct finding regarding the developmental angiogenesis likely involves different endothelial Cre driver-expression in distinct stage of the development. It is also important to determine the detailed substrate(s) and the activation mechanisms by which ADAM17 mediates angiogenesis under physiological (developmental), pathophysiological (retinal angiogenesis) or anti-pathological (collateral angiogenesis upon hypoxia) conditions.
ADAM17, cardiac development and diseases
ADAM17−/− mice die shortly after birth, with defects in the aortic, pulmonic, and tricuspid valves of their heart [138]. Similarly, mice lacking the Zn2+ binding domain of ADAM17 (ADAM17 Δzn/Δzn), which inactivates metalloproteinase activity, die shortly after birth [113]. ADAM17 Δzn/Δzn embryos present defective cardiac valvulogenesis [139], abnormal vascular beds and internal hemorrhages [140]. The waved with open eyes (woe) mouse is a model of syntenic human ocular disorders. Woe is a hypomorphic mutation in ADAM17 where a small amount of functional ADAM17 is produced in woe animals, and they show enlarged heart and defects in the semilunar cardiac valves [141]. In addition, endothelial cell-specific ADAM17-deleted mice show cardiac valve enlargement during embryogenesis and progressive cardiomegaly and pronounced systolic dysfunction as adults, showing that endothelial ADAM17 may be necessary in normal cardiac development and homeostasis [142]. These results demonstrate the role of ADAM17 in the development of cardiac system and valves.
Expression of ADAM17 in the left ventricle is up-regulated in an abdominal artery coarctation-induced model of myocardial hypertrophy with increased expression of a NADPH oxidase, Nox4, showing that ADAM17 activation is required in pathological cardiac hypertrophy [143]. And cardiac protective effects of some drugs such as peroxisome proliferator-activated receptors (PPAR)-α agonists or Nox1/4 inhibitor are involved in a reduction of ADAM17 expression [144, 145]. Furthermore, treatment with ADAM17 small-interfering RNA can prevent angiotensin II-induced cardiac hypertrophy and fibrosis, with inhibition of angiotensin II-induced overexpression of markers of myocardial hypertrophy and fibrosis such as brain natriuretic peptide (BNP), α-skeletal actin, β myosin heavy chain (β-MHC), type I collagen, type II collagen, and fibronectin [146] or MMP-2 [147]. Interestingly, angiotensin II-induced cardiac hypertrophy is attenuated by VSMC-specific ADAM17 silencing [66, 68], showing that angiotensin II-induced vascular EGFR activation may be a specific requirement for the cardiac phenotype.
In addition to these findings, cardiomyocyte-specific ADAM17 knockdown mice showed lower mortality rate and less cardiac dysfunction caused by myocardial infarction with reduced activation and expression of VEGFR2 in infarcted myocardium, highlighting the detrimental role of cardiomyocyte ADAM17 in recovery after myocardial infarction via suppression of angiogenesis [148]. The myocardial infarction experimental model also showed that enhanced ADAM17 expression, along with decreased TIMP-3 and increased TNF-α expression within one week after acute myocardial infarction, is associated with cardiac remodeling [149]. These data indicate the potential benefit of ADAM17 inhibition in cardiac diseases.
On the contrary, myocardial hypertrophy and dysfunction induced by transverse aortic constriction are enhanced in cardiomyocyte-specific ADAM17 knockdown mice, and upregulation of integrin β1 induced by this pressure overload is also enhanced in ADAM17 knockdown animal. However, hypertrophy induced by a sub-pressor dose of angiotensin II is not affected by cardiac ADAM17 knockdown, suggesting that ADAM17 has a protective function in pressure-overload cardiomyopathy [150]. In addition, iRhom2–/– mice showed defective inflammatory responses at both acute M1 and chronic M2 phases resulting in impaired cardiac repair upon myocardial infarction. This phenotype is explained by defective control of myeloid TNF-α/TNFR signaling.
Regarding the role of ADAM17 in human heart, the coronary arteries obtained from aged or obese patients showed increased vascular endothelial ADAM17 activity suggesting the development of remote coronary microvascular dysfunction [151]. Systemic levels of ADAM17 and TNF-α are higher in acute myocardial infarction (AMI) patients compared to patients with stable angina. ADAM17 is highly expressed at the site of ruptured plaques in AMI patients, and this local ADAM17 expression level is independently and significantly correlated with adverse cardiac events during follow-up period [152]. Both spontaneous and ADAM17 activator-stimulated levels of ADAM17 and TNF-α are higher in peripheral blood mononuclear cells obtained from AMI patients compared to normal subjects, and these levels are correlated with in-hospital complications [153]. Moreover, a score evaluated from ADAM17 circulating substrates (soluble ICAM-1, soluble VCAM-1, soluble IL6 receptor, and soluble TNFR1) is reported to be able to predict recurring cardiovascular events [154]. Collectively, clinical studies further support the detrimental roles of ADAM17 in human myocardial diseases.
ADAM17 and kidney diseases
Mice infused with angiotensin II for 2 months suffer from ADAM17-mediated shedding of TGF-α and subsequent EGFR transactivation-dependent renal lesions such as glomerulosclerosis, tubular atrophy, and interstitial fibrosis [155]. In addition, ADAM17 is induced and redistributed in angiotensin II-damaged kidneys and inhibition of ADAM17 can blunt angiotensin II-induced renal lesions [155]. Similarly, fibrosis after ischemia–reperfusion injury or unilateral ureteral obstruction is attenuated in ADAM17 hypomorphic mice or mice with inducible silencing of ADAM17 in proximal tubule [156]. The non-receptor tyrosine kinase, focal adhesion kinase (FAK), is suggested as a key regulator of Src-mediated ADAM17 Tyr702 phosphorylation and subsequent profibrotic responses in mesangial cells under high glucose condition [157]. In streptozotocin-induced diabetic mice, Src inhibitors also attenuate ADAM17 activation in the kidney cortex, albuminuria, glomerular collagen accumulation, that are associated with attenuation of ERK and EGFR phosphorylation [158]. These data suggest the critical role of ADAM17 in renal fibrosis.
In lupus nephritis, iRom2/ADAM17-mediated TNF-α and EGFR signaling pathways also cause renal damage [159]. Polycystic kidney disease (PKD) is a genetic disorder leading to the formation of multiple cysts in kidneys. The study of animal models of autosomal recessive PKD has revealed that ADAM17 expression is increased in the collecting duct epithelial cells in the cystic kidneys. Activation of ADAM17 induces constitutive shedding of HB-EGF, amphiregulin and TGF-α, resulting in EGFR/ERK pathway activation and maintains higher cell proliferation in PKD cells [160].
Clinical studies further suggest that ADAM17 plays important roles in human renal diseases. Patients with acute kidney injury or chronic kidney disease (CKD) have high soluble amphiregulin in their urine and both ADAM17 and amphiregulin expression are strongly correlated with markers of fibrosis in kidney biopsies [156]. In various human renal diseases, ADAM17 is strongly induced in podocytes, proximal tubules, and peritubular capillaries, and renal ADAM17 expression is significantly associated with glomerular and interstitial injury or renal function [161]. Urinary ADAM17 is increased in type 2 diabetes patients and could be used as an early biomarker to detect CKD [162]. Moreover, large clinical studies showed that high ADAMs activity level is independently correlated with CKD progression and onset of cardiovascular events in CKD patients [163, 164].
ADAM17 and metabolic disorders
ADAM17 activation is considered as one of the major drivers causing insulin resistance associated with metabolic disorders. In insulin receptor haplo-insufficient (Insr ±) diabetic mice, pharmacological inhibition of ADAM17 by TAPI-1 can reduce blood glucose level and vascular inflammation [165]. In addition, knock down of tissue inhibitor of metalloproteinases-3 (TIMP-3), an inhibitor for ADAM17 and MMPs, in Insr ± mice aggravates blood glucose level and vascular inflammation [165]. On the contrary, TIMP-3 overexpression in macrophage can protect mice from increasing insulin resistance, adipose tissue inflammation, and non-alcoholic fatty liver [166].
There is additional evidence to support these findings. High-fat diet causes increased body weight, liver weight, epididymal adipose tissue weight, systolic blood pressure, fasting blood glucose and lipid levels, and decreased adiponectin level, and these changes are attenuated in temporal systemic ADAM17 deletion (TaceMx1) mice. In addition, increased macrophage infiltration and the expression of TNF-α and monocyte chemoattractant protein-1 (MCP-1) in epididymal adipose tissue induced by high-fat diet are also attenuated in TaceMx1, suggesting that ADAM17 is an important mediator in the development of obesity-induced metabolic disorders [167]. ADAM17 ± mice are partially protected from obesity and insulin resistance compared with wild type mice [168], and ADAM17 inhibitor can improve insulin sensitivity in fructose-fed rats [169] or high-fat diet-fed mice [170]. Deletion of iRom2 also protects against diet-induced obesity [171]. In addition, macrophage metabolic reprogramming has been suggested to enhance aortic dissection via hypoxia-inducible factor 1α (HIF-1α)-dependent ADAM17 induction [172]. In line with the requirement of iRhom2 in myeloid TNF-α production, iRhom2–/– mice are protected against high-fat diet-induced adipose tissue inflammation, weight gain and insulin resistance [173]. Taken together, ADAM17 and iRhom2 should be recognized to play an important role in metabolic disorders and diabetes.
ADAM17 SNPs and loss-of-function mutations
ADAM17 SNPs (rs10495565, rs12474540, and rs17524594) associate with the presence of pulmonary arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 (HHT1), indicating genetic variation in ADAM17 can promote a TGF-β-regulated vascular diseases [131]. In addition, ADAM17 SNPs (rs6705408, rs10495563, and rs6432017) are associated with incidence of Kawasaki disease and interaction with TGF-β signaling is suggested [174]. Two ADAM17 SNPs (m1254A > G and i33708A > G) also contribute to obesity risk [175]. However. the relation of these SNPs and ADAM17 expression or activity remains unstudied. ADAM17 SNP Ser746Leu and -154A allele have been reported to increase soluble TNF-α plasma levels and the risk of cardiovascular death [176]. In addition, further studies may enable us to use a tailor-made approach for cardiovascular diseases based on information from ADAM17 SNPs.
Regarding the loss-of-function mutation, a late-onset familial Alzheimer disease was identified to co-segregate with rare heterozygous ADAM17 single nucleotide variant rs142946965 [177]. This causes ADAM17 mutation R215I directly adjacent to pro-protein convertase cleavage motif 210–214 and severely impairs ADAM17 maturation leading to amyloid β formation. In addition, heterozygous mutation of ADAM17 Y42D and L659P are associated with incidence of Fallot tetralogy and loss of HB-EGF shedding [178]. Finally, two distinct homozygous loss-of-function mutations of human ADAM17 have been reported (c.603-606delCAGA and c.308dupA). The siblings with 603-606delCAGA demonstrated skin lesions and diarrhea. While one of the siblings (a girl) died at age of 12, the affected boy has survived with loss of ADAM17 expression, diminished TNF-α production and left ventricular dilatation [179]. The c.308dupA patient demonstrated skin lesions, diarrhea and severely diminished levels of plasma TNF-α and IL2. Interestingly, this patient developed unexpected hypertension. Recurrent sepsis was the cause of death at 10 months [180].
Other ADAMs in cardiovascular pathophysiology
In addition to ADAM17, ADAM8, 9, 10, 12, 15, 19, 28 and 33 are expressed on various cells including endothelial cells, smooth muscle cells, and leukocytes, and they also have proteolytic activity. Accumulating data suggest that other ADAMs play notable roles in cardiovascular pathophysiology by mediating inflammation, angiogenesis, cell proliferation, and cell migration (Fig. 5 and Table 2). Among these ADAM families, ADAM10 is most broadly expressed and is closely related to ADAM17 in its structure and function. Therefore, ADAM10 is recognized as another important shedding proteinase which mediates various signal transduction. Important for cardiovascular pathophysiology, ADAM10 can affect inflammation by cleaving CD44 [117], CX3CL1 [181], C-X-C motif chemokine ligand 16 (CXCL16) [182], IL6 receptor [183], receptor for advanced glycation end products (RAGE) [184], and TNF-α [185]. It also affects angiogenesis by cleaving JAM-A [123], Notch [186], neuropilin 1 (NRP-1) [31, 187], VEGFR2 [188] and vascular endothelial (VE)-cadherin [189]. It affects cell proliferation or migration by cleaving betacellulin [190, 191] and HB-EGF [192], affects collagen turnover by cleaving discordin domain receptor family, member 1 (DDR1) [193], affects apoptosis by cleaving receptor activator of nuclear factor κ-B ligand (RANKL) [194], and affects blood pressure by cleaving corin [195]. Furthermore, ADAM10 can affect acute kidney injury by cleaving meprin A, a membrane-associated metalloproteinase in proximal tubules, since meprin A is one of the key players in acute kidney injury [196]. However, as mentioned previously, most substrates can be cleaved by multiple ADAMs. The interaction between each ADAM and its substrates depends on pathophysiologic condition. Furthermore, some substrates are cleaved by their respective ADAMs in different manner and in different cell component. ADAM10-mediated Notch shedding is ligand dependent; whereas, ADAM17-mediated Notch shedding is ligand-independent [197]. Neural cell adhesion molecule L1/CD171 and CD44 are cleaved by ADAM17 at cell surface and soluble forms are released into the extracellular space; whereas, they are cleaved by ADAM10 in endosomes and soluble forms are released from cell as exosomes [198]. Neuregulin, cleaved by ADAM17 at cell surface, is cleaved in the Golgi apparatus by ADAM19 [199]. There are highly complicated relations between ADAMs and substrates that should be elucidated further. Because of this complexity, the approach to consider ADAMs as therapeutic targets is challenging.
Fig. 5.
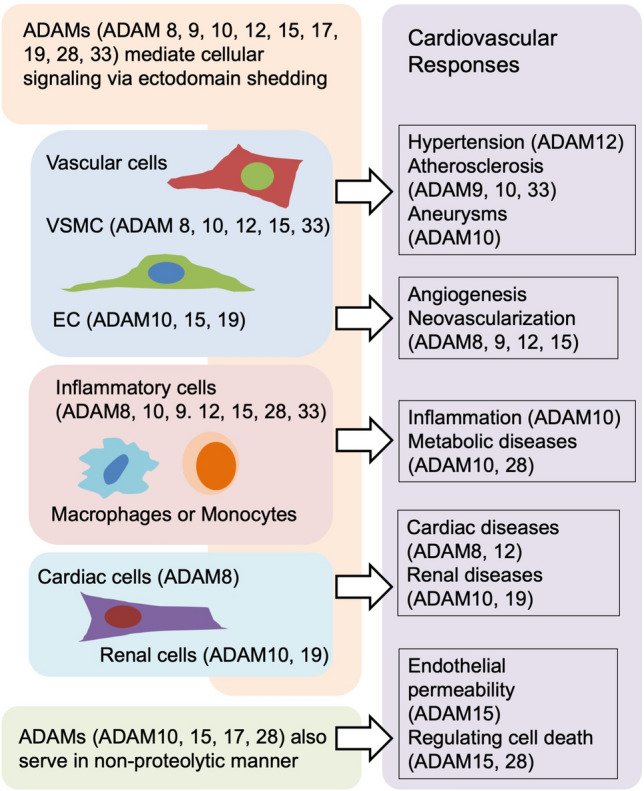
Cell-type-specific role of non-ADAM17 ADAMs in cardiovascular diseases. Non-ADAM17 ADAMs are expressed in various cell types and regulate cellular signaling within and between these cells. Non-ADAM17 ADAMs thereby mediate cardiovascular pathophysiology including hypertension, atherosclerosis and cardiovascular inflammation. The figures are created based on the references used in the other ADAMs section
Table 2.
Cardiovascular-related substrates of ADAMs
| Inflammation | Angiogenesis | Proliferation/migration | Others | |
|---|---|---|---|---|
| ADAM8 |
CD16 [318] CD23 [319] CX3CL1 [318] L-selectin [320] TNF alpha [318] TNFR1 [321] VCAM-1 [322] |
TGF alpha [318] | ||
| ADAM9 |
CD40 [210] VCAM-1 [210] |
Notch [282] Tie-2 [210] VE-cadherin [210] VEGFRII [210] |
HB-EGF [323] |
EphB4 [210] |
| ADAM10 |
CD44 [117] CX3CL1 [181] CXCL16 [182] IL6R [183] RAGE [184] TNF alpha [185] |
E-cadherin [326] JAM-A [123] N-cadoherin [221] Notch2 [327] NRP-1 [31] VEGFRII [188] VE-cadherin [189] |
HB-EGF [63] |
CD84 [328] Corin [195] DDR1 [193] Klotho [109] Meprin A [196] Neuregulin [329] RANKL [194] |
| ADAM12 |
E-cadherin [330] IFGBP3 [331] IFGBP5 [331] Notch1 [282] |
Betacellulin [332] HB-EGF [333] |
||
| ADAM15 |
E-caderin [334] VEGFR [335] |
FGFR2iiib [331] | ADAM10 [325] | |
| ADAM17 |
CD40 [268] CD44 [117] CD163 [310] ICAM-1 [111] IL-1R II [21] L-selectin [113] L1-CAM [295] PECAM-1 [135] |
JAM-A [123] VEGFR [31] |
IGFR1 [135] SEMA4D [305] syndecan 1 and 4 [287] |
EPCR [83] Ephrin B4 [135] Jagged 1 [38] Klotho [312] RANKL [259] |
| ADAM19 |
Alpha 2 macroglobulin [336] |
Neuregulin [231, 338] |
(pro)renin receptor [339] RANKL [337] |
|
| ADAM28 | TNF alpha [234] | IGFBP3 [340] | CTGF [341] | vWF [342] |
| ADAM33 | RANKL [343] |
ADAMs can also serve in non-proteolytic manner. ADAM15 regulates endothelial permeability and neutrophil migration by promoting Src/ERK signaling in a protease activity-independent manner [200], and subsequently contributes to atherosclerosis [201]. ADAM28 is reported to bind to C1q and attenuates C1q-induced cell death [202]. It binds to P-selectin glycoprotein ligand-1 (PSGL-1) to promote leukocyte rolling, adhesion to endothelial cells and subsequent inflammation [203]. It also binds to integrin α4 β1 and enhances cell adhesion to VCAM-1 and regulates spatial and temporal trans-endothelial migration of lymphocytes [204]. In the following section, we highlight the role of these ADAMs in cardiovascular pathophysiology.
Lessons from genetically modified animal models
ADAM8 –/– mice [205], ADAM9 –/– mice [206], ADAM15 –/– mice [207], and ADAM33 –/– mice [208] are viable and do not show an obvious phenotype under normal conditions. However, there are reduced retinal neovascularization in an experimental retinopathy model in ADAM8 –/– mice [209], ADAM9 –/– mice [210] and ADAM15 –/– mice [207]. ADAM10 –/– mice die before birth with defects in cardiovascular system [211]. ADAM19 –/– mice also die perinatally, likely as a result of cardiac valve and vasculature defects [212, 213]. In addition, knockdown of MMP-7 attenuates angiotensin II-induced myocardial ADAM12 overexpression, hypertension and cardiac hypertrophy, showing the importance of MMP-7/ADAM12 signaling axis in hypertensive cardiac disorders [214].
Mice lacking the ADAM10 gene primarily in endothelial cells show multiple cardiac and vascular defects similar to Notch1 mutants [186, 215], suggesting that Notch signaling pathway is a key player in ADAM10-mediated cardiovascular development. ADAM10-mediated Notch signaling also promotes the development and maturation of the glomerular vasculature [216]. Using a similar model, Notch1 and Notch4 were shown to control the development of several organ-specific vascular beds in an ADAM10-dependent manner [217]. Moreover, collecting duct-specific ADAM10 knockout mice show defects in urine concentration, polyuria, and hydronephrosis, along with reduction of Notch activity in the collecting duct epithelium [218]. Transplantation of bone marrow from myeloid-specific ADAM10 knockout mice to atherogenic model mice does not affect the plaque size, but increases plaque collagen content, indicating that myeloid ADAM10 modulates atherosclerotic plaque stability. ADAM10-deficient macrophages further showed anti-inflammatory phenotype with increased IL-10 and decreased pro-inflammatory factors such as IL-12 [219]. In addition, AngII-induced AAA in mice is exaggerated in ADAM15–/– mice. This is due to defect in thrombospondin-1 processing by ADAM15 causing thrombospondin-1-dependent apoptosis of VSMCs [220].
Other ADAMs in cardiovascular disease, human findings
In human atherosclerotic lesions, ADAM10 is expressed and its expression is associated with plaque progression and neovascularization [188]. Increased ADAM10 expression in human atherosclerotic lesions is associated with decreased N-cadherin when apoptosis increases [221]. This association between ADAM10 and vascular remodeling is further supported by some animal models. A study with CaCl2-induced TAA model showed that ADAM10 expression was significantly increased in intima and media of TAA [93]. A study using diabetic minipigs showed that ADAM10 expression was increased in vascular segments obtained from coronary artery restenosis, implicating the role of ADAM10 in neointimal formation [222]. ADAM9 and ADAM15 also express in human atherosclerotic lesions [223], co-localized with CD68-positive cells of monocytic origin in the plaques [84]. ADAM8 and ADAM15 are highly expressed in the media layer in patients with ascending aortic dissection compared to that in patients with dilatation of the ascending aorta [224]. Although ADAM8 is up-regulated in atherosclerotic lesions and expressed in circulating neutrophils and macrophages in humans, whole body and hematopoietic ADAM8 deletion did not alter atherosclerotic plaque development [225]. In high-graded carotid artery lesions, macrophages and smooth muscle cells are positive for ADAM8, ADAM10, ADAM12, ADAM15, and ADAM17. The luminal surface of endothelial cells is positive for ADAM15, and neo-vessels are positive for ADAM12 [226]. ADAM33 is expressed in smooth muscle cells and inflammatory cells within human atherosclerotic lesions [227]. Moreover, ADAM33 SNPs are reported to correlate with the extent of atherosclerosis in coronary artery disease patients [227] and cardiovascular mortality [228]. The risk alleles of ADAM8 SNPs are associated with elevated serum soluble ADAM8 and the risk of myocardial infarction in two independent cohorts [229]. This result is supported by animal study showing that myocardial infarction increased remote ADAM8 expression in rat heart [230]. In line with ADAM15–/– mice enhancing AAA, ADAM15 expression appears decreased in human AAA samples as well as AngII model of mouse AAA [220]. Collectively, several ADAMs seem to contribute to arterial physiology, progression of atherosclerosis and ischemic heart diseases in humans.
In human kidneys, mesangial ADAM19 expression is associated with glomerular damage, and ADAM19 in proximal tubules and in peritubular capillaries is associated with interstitial fibrosis, and tubular ADAM19 is associated with declining renal function [231]. These data indicate the role of ADAM19 in renal profibrotic and proinflammatory processes [231]. In addition, study with renal transplant patients showed that ADAM19 mRNA was significantly higher in chronic allograft nephropathy, and ADAM19 expression in renal endothelium was significantly higher in acute rejection [232].
RAGE is widely recognized to have an important role in the pathogenesis of diabetic complications, and Type 1 diabetes patients have significantly higher serum soluble RAGE, along with an increase in serum ADAM10 [233]. Finally, ADAM28 expression in blood mononuclear cells significantly correlates with parameters of metabolic syndrome including body mass index and relative fat, suggesting the role of ADAM28 in human metabolic conditions [234].
Therapeutic potential
As reviewed elsewhere, various ADAM17 inhibitors have been synthesized which selectively inhibit ADAM17 and do not inhibit other metalloproteinases [235, 236]. Using animal models, the efficacy of ADAM17 inhibition is reported in not only inflammatory diseases such as rheumatoid arthritis [237] but also cardiovascular disorders such as renal fibrosis [155, 238], intestinal reperfusion injury [239], or polycystic kidney disease [240, 241]. Similarly, mice with genetically modulated ADAM17 indicate the positive potential of ADAM17 inhibition in inflammation such as septic shock [80, 138, 242]. A9B8 is a human/mouse cross-reactive inhibitory antibody against ADAM17. A9B8 treatment attenuated EGFR transactivation in cultured VSMCs. Moreover, it attenuated cardiovascular pathology in mice infused with angiotensin II [66]. A9B8 also effectively prevented AAA development and rupture in a mouse model [87]. In addition, the auto-inhibitory ADAM17 prodomain which inhibits ADAM17, but not ADAM10, can attenuate TNF-α secretion. This peptide inhibitor appears effective in ADAM17-dependent models of inflammatory diseases including rheumatoid arthritis [243]. In spite of these promising in vivo results, pre-clinical trials and clinical trials using ADAM17 inhibitors had to be discontinued due to hepatotoxicity [237] or lack of efficacy [244]. One of the reasons can be that ADAM17 inhibition affects normal physiological conditions. ADAM17 –/– mice die shortly after birth because of a variety of defects [138] but mice with reduced ADAM17 level in all tissues (ADAM17 ex/ex) show substantially increased susceptibility to inflammation [80], indicating that adequate therapy window should be set for ADAM17 inhibitors. Since ADAM17 inhibitors have demonstrated adverse side effects clinically, certain regulators of ADAM17 can also be considered as therapeutic targets. One such regulator, iRhom2, is an essential determinant of ADAM17-dependent shedding in leukocytes by mediating ADAM17 maturation, and iRhom2 is potential target for selective inactivation of the pro-inflammatory roles of ADAM17 activation [245].
Another approach is to analyze and utilize ADAMs-modulating aspects of existing drugs. Aspirin is widely used for the prevention of thrombosis of coronary artery and cerebral artery. Aspirin at high concentrations is reported to induce ADAM17-mediated shedding of glycoprotein (GP)Ib α and GPV [246]. Non-steroidal anti-inflammatory drugs (NSAIDs) with diphenylamine structure causes a reduction in the neutrophil intracellular ATP concentration, and this reduction is related with ADAM17-dependent L-selectin shedding at leukocyte surface [247]. 1,25-dihydroxyvitamin D, the hormonal form of vitamin D, has a potential anti-inflammatory and anti-atherosclerotic effect, and is widely used for chronic kidney disease patients, because 1,25-dihydroxyvitamin D is proven to significantly improve not only secondary hyperparathyroidism but patients’ survival via renal and cardiovascular protective effects [248]. 1,25-dihydroxyvitamin D inhibits ADAM17 expression through the induction of C/EBP beta [249], and prevents ADAM17/TNF-α-mediated secondary hyperparathyroidism, fibrotic and inflammatory lesions to the renal parenchyma, and systemic inflammation [250]. 1,25-dihydroxyvitamin D also causes ADAM10-dependent TNFR1 shedding, thus blocking TNF-α function in VSMC [251]. These agents regulating ADAMs activity could be considered as a novel therapeutic approach if the mechanisms are clarified further.
Concluding remarks
Since ADAMs are ubiquitously expressed in somatic cells and they cleave various substrates, ADAMs, especially ADAM17, have important and highly intricate roles in cell signaling. The accumulation of research in this area steadily shed light on the role of ADAM17 and other ADAMs in cardiovascular diseases. Although ADAM17 and some of the other ADAMs are essential for normal development or cardiovascular homeostasis, excess of these ADAMs activation aggravates inflammatory response and cardiovascular pathophysiology, and ADAM17 inhibition is thought to be promising therapeutic target for cardiovascular and renal diseases. We hope further research based on existing evidence highlighted in this review will elucidate ADAMs-mediated signal transduction and pathophysiology of cardiovascular diseases, and embody the therapeutic potential with pharmacological targeting.
Author contributions
TK and SE proposed the idea and writing. KE and RS proof-read the article and performed critical revision in organization and discussion.
Funding
This work was supported by National Institute of Health grants, HL128324 (S.E.), HL133248 (S.E.), DK111042 (R.S. and S.E.), and NS109382 (S.E.).
Compliance with ethical standards
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cho C. Testicular and epididymal ADAMs: expression and function during fertilization. Nat Rev Urol. 2012;9(10):550–560. doi: 10.1038/nrurol.2012.167. [DOI] [PubMed] [Google Scholar]
- 2.Brocker CN, Vasiliou V, Nebert DW. Evolutionary divergence and functions of the ADAM and ADAMTS gene families. Hum Genomics. 2009;4(1):43–55. doi: 10.1186/1479-7364-4-1-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moss M, Jin S, Milla M, Bickett D, Burkhart W, Carter H, Chen W, Clay W, Didsbury J, Hassler D, Hoffman C, Kost T, Lambert M, Leesnitzer M, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton L, Schoenen F, Seaton T, Su J, Becherer J. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385(6618):733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 4.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385(6618):729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 5.Black RA, White JM. ADAMs: focus on the protease domain. Curr Opin Cell Biol. 1998;10(5):654–659. doi: 10.1016/S0955-0674(98)80042-2. [DOI] [PubMed] [Google Scholar]
- 6.Blobel CP. Metalloprotease-disintegrins: links to cell adhesion and cleavage of TNF alpha and Notch. Cell. 1997;90(4):589–592. doi: 10.1016/S0092-8674(00)80519-X. [DOI] [PubMed] [Google Scholar]
- 7.Lorenzen I, Trad A, Grotzinger J. Multimerisation of A disintegrin and metalloprotease protein-17 (ADAM17) is mediated by its EGF-like domain. Biochem Biophys Res Commun. 2011;415(2):330–336. doi: 10.1016/j.bbrc.2011.10.056. [DOI] [PubMed] [Google Scholar]
- 8.Schlondorff J, Becherer JD, Blobel CP. Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE) Biochem J. 2000;347(Pt 1):131–138. doi: 10.1042/bj3470131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong E, Maretzky T, Peleg Y, Blobel CP, Sagi I. The functional maturation of A disintegrin and metalloproteinase (ADAM) 9, 10, and 17 requires processing at a newly identified proprotein convertase (PC) cleavage site. J Biol Chem. 2015;290(19):12135–12146. doi: 10.1074/jbc.M114.624072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiley H, Woolf M, Opresko L, Burke P, Will B, Morgan J, Lauffenburger D. Removal of the membrane-anchoring domain of epidermal growth factor leads to intracrine signaling and disruption of mammary epithelial cell organization. J Cell Biol. 1998;143(5):1317–1328. doi: 10.1083/jcb.143.5.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borrell-Pages M, Rojo F, Albanell J, Baselga J, Arribas J. TACE is required for the activation of the EGFR by TGF-alpha in tumors. EMBO J. 2003;22(5):1114–1124. doi: 10.1093/emboj/cdg111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forrester SJ, Kawai T, O'Brien S, Thomas W, Harris RC, Eguchi S. Epidermal growth factor receptor transactivation: mechanisms, pathophysiology, and potential therapies in the cardiovascular system. Annu Rev Pharmacol Toxicol. 2016;56:627–653. doi: 10.1146/annurev-pharmtox-070115-095427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elliott KJ, Bourne AM, Takayanagi T, Takaguri A, Kobayashi T, Eguchi K, Eguchi S. ADAM17 silencing by adenovirus encoding miRNA-embedded siRNA revealed essential signal transduction by angiotensin II in vascular smooth muscle cells. J Mol Cell Cardiol. 2013;62:1–7. doi: 10.1016/j.yjmcc.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.George AJ, Hannan RD, Thomas WG. Unravelling the molecular complexity of GPCR-mediated EGFR transactivation using functional genomics approaches. FEBS J. 2013;280(21):5258–5268. doi: 10.1111/febs.12509. [DOI] [PubMed] [Google Scholar]
- 15.Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol. 2006;291(1):C1–10. doi: 10.1152/ajpcell.00620.2005. [DOI] [PubMed] [Google Scholar]
- 16.Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6(1):32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- 17.Kinugasa Y, Hieda M, Hori M, Higashiyama S. The carboxyl-terminal fragment of pro-HB-EGF reverses Bcl6-mediated gene repression. J Biol Chem. 2007;282(20):14797–14806. doi: 10.1074/jbc.M611036200. [DOI] [PubMed] [Google Scholar]
- 18.Nanba D, Mammoto A, Hashimoto K, Higashiyama S. Proteolytic release of the carboxy-terminal fragment of proHB-EGF causes nuclear export of PLZF. J Cell Biol. 2003;163(3):489–502. doi: 10.1083/jcb.200303017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hieda M, Isokane M, Koizumi M, Higashi C, Tachibana T, Shudou M, Taguchi T, Hieda Y, Higashiyama S. Membrane-anchored growth factor, HB-EGF, on the cell surface targeted to the inner nuclear membrane. J Cell Biol. 2008;180(4):763–769. doi: 10.1083/jcb.200710022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bell JH, Herrera AH, Li Y, Walcheck B. Role of ADAM17 in the ectodomain shedding of TNF-alpha and its receptors by neutrophils and macrophages. J Leukoc Biol. 2007;82(1):173–176. doi: 10.1189/jlb.0307193. [DOI] [PubMed] [Google Scholar]
- 21.Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Shows D, Peschon JJ, Black RA. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J Biol Chem. 2000;275(19):14608–14614. doi: 10.1074/jbc.275.19.14608. [DOI] [PubMed] [Google Scholar]
- 22.Scheller J, Ohnesorge N, Rose-John S. Interleukin-6 trans-signalling in chronic inflammation and cancer. Scand J Immunol. 2006;63(5):321–329. doi: 10.1111/j.1365-3083.2006.01750.x. [DOI] [PubMed] [Google Scholar]
- 23.Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45(2):146–169. doi: 10.3109/10409231003628015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krossa S, Scheidig AJ, Grotzinger J, Lorenzen I. Redundancy of protein disulfide isomerases in the catalysis of the inactivating disulfide switch in A disintegrin and metalloprotease 17. Sci Rep. 2018;8(1):1103. doi: 10.1038/s41598-018-19429-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kunzel U, Grieve AG, Meng Y, Sieber B, Cowley SA, Freeman M. FRMD8 promotes inflammatory and growth factor signalling by stabilising the iRhom/ADAM17 sheddase complex. Elife. 2018 doi: 10.7554/eLife.35012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dombernowsky SL, Samsoe-Petersen J, Petersen CH, Instrell R, Hedegaard AM, Thomas L, Atkins KM, Auclair S, Albrechtsen R, Mygind KJ, Frohlich C, Howell M, Parker P, Thomas G, Kveiborg M. The sorting protein PACS-2 promotes ErbB signalling by regulating recycling of the metalloproteinase ADAM17. Nat Commun. 2015;6:7518. doi: 10.1038/ncomms8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kakiashvili E, Dan Q, Vandermeer M, Zhang Y, Waheed F, Pham M, Szaszi K. The epidermal growth factor receptor mediates tumor necrosis factor-alpha-induced activation of the ERK/GEF-H1/RhoA pathway in tubular epithelium. J Biol Chem. 2011;286(11):9268–9279. doi: 10.1074/jbc.M110.179903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mendelson K, Swendeman S, Saftig P, Blobel CP. Stimulation of platelet-derived growth factor receptor beta (PDGFRbeta) activates ADAM17 and promotes metalloproteinase-dependent cross-talk between the PDGFRbeta and epidermal growth factor receptor (EGFR) signaling pathways. J Biol Chem. 2010;285(32):25024–25032. doi: 10.1074/jbc.M110.102566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu P, Derynck R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol Cell. 2010;37(4):551–566. doi: 10.1016/j.molcel.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gooz M, Gooz P, Luttrell LM, Raymond JR. 5-HT2A receptor induces ERK phosphorylation and proliferation through ADAM-17 tumor necrosis factor-alpha-converting enzyme (TACE) activation and heparin-bound epidermal growth factor-like growth factor (HB-EGF) shedding in mesangial cells. J Biol Chem. 2006;281(30):21004–21012. doi: 10.1074/jbc.M512096200. [DOI] [PubMed] [Google Scholar]
- 31.Swendeman S, Mendelson K, Weskamp G, Horiuchi K, Deutsch U, Scherle P, Hooper A, Rafii S, Blobel CP. VEGF-A stimulates ADAM17-dependent shedding of VEGFR2 and crosstalk between VEGFR2 and ERK signaling. Circ Res. 2008;103(9):916–918. doi: 10.1161/CIRCRESAHA.108.184416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prakasam HS, Gallo LI, Li H, Ruiz WG, Hallows KR, Apodaca G. A1 adenosine receptor-stimulated exocytosis in bladder umbrella cells requires phosphorylation of ADAM17 Ser-811 and EGF receptor transactivation. Mol Biol Cell. 2014;25(23):3798–3812. doi: 10.1091/mbc.E14-03-0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dang M, Armbruster N, Miller MA, Cermeno E, Hartmann M, Bell GW, Root DE, Lauffenburger DA, Lodish HF, Herrlich A. Regulated ADAM17-dependent EGF family ligand release by substrate-selecting signaling pathways. Proc Natl Acad Sci U S A. 2013;110(24):9776–9781. doi: 10.1073/pnas.1307478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kommaddi RP, Thomas R, Ceni C, Daigneault K, Barker PA. Trk-dependent ADAM17 activation facilitates neurotrophin survival signaling. FASEB J. 2011;25(6):2061–2070. doi: 10.1096/fj.10-173740. [DOI] [PubMed] [Google Scholar]
- 35.Schwarz J, Schmidt S, Will O, Koudelka T, Kohler K, Boss M, Rabe B, Tholey A, Scheller J, Schmidt-Arras D, Schwake M, Rose-John S, Chalaris A. Polo-like kinase 2, a novel ADAM17 signaling component, regulates tumor necrosis factor alpha ectodomain shedding. J Biol Chem. 2014;289(5):3080–3093. doi: 10.1074/jbc.M113.536847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Gall SM, Maretzky T, Issuree PDA, Niu XD, Reiss K, Saftig P, Khokha R, Lundell D, Blobel CP. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J Cell Sci. 2010;123(22):3913–3922. doi: 10.1242/jcs.069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maretzky T, Evers A, Zhou W, Swendeman SL, Wong PM, Rafii S, Reiss K, Blobel CP. Migration of growth factor-stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nat Commun. 2011;2:229. doi: 10.1038/ncomms1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parr-Sturgess CA, Rushton DJ, Parkin ET. Ectodomain shedding of the Notch ligand Jagged1 is mediated by ADAM17, but is not a lipid-raft-associated event. Biochem J. 2010;432(2):283–294. doi: 10.1042/BJ20100321. [DOI] [PubMed] [Google Scholar]
- 39.Ushio-Fukai M, Alexander RW. Caveolin-dependent angiotensin II type 1 receptor signaling in vascular smooth muscle. Hypertension. 2006;48(5):797–803. doi: 10.1161/01.HYP.0000242907.70697.5d. [DOI] [PubMed] [Google Scholar]
- 40.Gratton JP, Bernatchez P, Sessa WC. Caveolae and caveolins in the cardiovascular system. Circ Res. 2004;94(11):1408–1417. doi: 10.1161/01.RES.0000129178.56294.17. [DOI] [PubMed] [Google Scholar]
- 41.Takaguri A, Shirai H, Kimura K, Hinoki A, Eguchi K, Carlile-Klusacek M, Yang B, Rizzo V, Eguchi S. Caveolin-1 negatively regulates a metalloprotease-dependent epidermal growth factor receptor transactivation by angiotensin II. J Mol Cell Cardiol. 2011;50(3):545–551. doi: 10.1016/j.yjmcc.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moreno-Caceres J, Mainez J, Mayoral R, Martin-Sanz P, Egea G, Fabregat I. Caveolin-1-dependent activation of the metalloprotease TACE/ADAM17 by TGF-beta in hepatocytes requires activation of Src and the NADPH oxidase NOX1. FEBS J. 2016 doi: 10.1111/febs.13669. [DOI] [PubMed] [Google Scholar]
- 43.Takayanagi T, Crawford KJ, Kobayashi T, Obama T, Tsuji T, Elliott KJ, Hashimoto T, Rizzo V, Eguchi S. Caveolin 1 is critical for abdominal aortic aneurysm formation induced by angiotensin II and inhibition of lysyl oxidase. Clin Sci (Lond) 2014;126(11):785–794. doi: 10.1042/CS20130660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willems SH, Tape CJ, Stanley PL, Taylor NA, Mills IG, Neal DE, McCafferty J, Murphy G. Thiol isomerases negatively regulate the cellular shedding activity of ADAM17. Biochem J. 2010;428(3):439–450. doi: 10.1042/BJ20100179. [DOI] [PubMed] [Google Scholar]
- 45.Aragao AZ, Nogueira ML, Granato DC, Simabuco FM, Honorato RV, Hoffman Z, Yokoo S, Laurindo FR, Squina FM, Zeri AC, Oliveira PS, Sherman NE, Paes Leme AF. Identification of novel interaction between ADAM17 (a disintegrin and metalloprotease 17) and thioredoxin-1. J Biol Chem. 2012;287(51):43071–43082. doi: 10.1074/jbc.M112.364513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Granato DC, e Costa RAP, Kawahara R, Yokoo S, Aragao AZ, Domingues RR, Pauletti BA, Honorato RV, Fattori J, Figueira ACM, Oliveira PSL, Consonni SR, Fernandes D, Laurindo F, Hansen HP, Paes Leme AF. Thioredoxin-1 negatively modulates ADAM17 activity through direct binding and indirect reductive activity. Antioxid Redox Signal. 2018;29(8):717–734. doi: 10.1089/ars.2017.7297. [DOI] [PubMed] [Google Scholar]
- 47.Stawikowska R, Cudic M, Giulianotti M, Houghten RA, Fields GB, Minond D. Activity of ADAM17 (a disintegrin and metalloprotease 17) is regulated by its noncatalytic domains and secondary structure of its substrates. J Biol Chem. 2013;288(31):22871–22879. doi: 10.1074/jbc.M113.462267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dusterhoft S, Michalek M, Kordowski F, Oldefest M, Sommer A, Roseler J, Reiss K, Grotzinger J, Lorenzen I. Extracellular juxtamembrane segment of ADAM17 interacts with membranes and is essential for its shedding activity. Biochemistry. 2015;54(38):5791–5801. doi: 10.1021/acs.biochem.5b00497. [DOI] [PubMed] [Google Scholar]
- 49.Sommer A, Kordowski F, Büch J, Maretzky T, Evers A, Andrä J, Düsterhöft S, Michalek M, Lorenzen I, Somasundaram P, Tholey A, Sönnichsen FD, Kunzelmann K, Heinbockel L, Nehls C, Gutsmann T, Grötzinger J, Bhakdi S, Reiss K. Phosphatidylserine exposure is required for ADAM17 sheddase function. Nat Commun. 2016;7:11523. doi: 10.1038/ncomms11523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goth CK, Halim A, Khetarpal SA, Rader DJ, Clausen H, Schjoldager KT. A systematic study of modulation of ADAM-mediated ectodomain shedding by site-specific O-glycosylation. Proc Natl Acad Sci U S A. 2015;112(47):14623–14628. doi: 10.1073/pnas.1511175112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McIlwain D, Lang P, Maretzky T, Hamada K, Ohishi K, Maney S, Berger T, Murthy A, Duncan G, Xu H, Lang K, Häussinger D, Wakeham A, Itie-Youten A, Khokha R, Ohashi P, Blobel C, Mak T. iRhom2 regulation of TACE controls TNF-mediated protection against listeria and Responses to LPS. Science. 2012;335(6065):229–232. doi: 10.1126/science.1214448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maretzky T, McIlwain D, Issuree P, Li X, Malapeira J, Amin S, Lang P, Mak T, Blobel C. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc Natl Acad Sci U S A. 2013;110(28):11433–11438. doi: 10.1073/pnas.1302553110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adrain C, Zettl M, Christova Y, Taylor N, Freeman M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science. 2012;335(6065):225–228. doi: 10.1126/science.1214400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grieve AG, Xu H, Kunzel U, Bambrough P, Sieber B, Freeman M. Phosphorylation of iRhom2 at the plasma membrane controls mammalian TACE-dependent inflammatory and growth factor signalling. Elife. 2017 doi: 10.7554/eLife.23968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li X, Maretzky T, Weskamp G, Monette S, Qing X, Issuree PD, Crawford HC, McIlwain DR, Mak TW, Salmon JE, Blobel CP. iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc Natl Acad Sci U S A. 2015;112(19):6080–6085. doi: 10.1073/pnas.1505649112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oikonomidi I, Burbridge E, Cavadas M, Sullivan G, Collis B, Naegele H, Clancy D, Brezinova J, Hu T, Bileck A, Gerner C, Bolado A, von Kriegsheim A, Martin SJ, Steinberg F, Strisovsky K, Adrain C. iTAP, a novel iRhom interactor, controls TNF secretion by policing the stability of iRhom/TACE. Elife. 2018 doi: 10.7554/eLife.35032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cavadas M, Oikonomidi I, Gaspar CJ, Burbridge E, Badenes M, Felix I, Bolado A, Hu T, Bileck A, Gerner C, Domingos PM, von Kriegsheim A, Adrain C. Phosphorylation of iRhom2 controls stimulated proteolytic shedding by the metalloprotease ADAM17/TACE. Cell Rep. 2017;21(3):745–757. doi: 10.1016/j.celrep.2017.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Babendreyer A, Rojas-González DM, Giese AA, Fellendorf S, Düsterhöft S, Mela P, Ludwig A. Differential induction of the ADAM17 regulators iRhom1 and 2 in endothelial cells. Front Cardiovasc Med. 2020 doi: 10.3389/fcvm.2020.610344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scheller J, Chalaris A, Garbers C, Rose-John S. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol. 2011;32(8):380–387. doi: 10.1016/j.it.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 60.Pruessmeyer J, Ludwig A. The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin Cell Dev Biol. 2009;20(2):164–174. doi: 10.1016/j.semcdb.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 61.Lisi S, D’Amore M, Sisto M. ADAM17 at the interface between inflammation and autoimmunity. Immunol Lett. 2014;162(1 Pt A):159–169. doi: 10.1016/j.imlet.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 62.Obama T, Takayanagi T, Kobayashi T, Bourne AM, Elliott KJ, Charbonneau M, Dubois CM, Eguchi S. Vascular induction of a disintegrin and metalloprotease 17 by angiotensin II through hypoxia inducible factor 1alpha. Am J Hypertens. 2015;28(1):10–14. doi: 10.1093/ajh/hpu094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ohtsu H, Dempsey PJ, Frank GD, Brailoiu E, Higuchi S, Suzuki H, Nakashima H, Eguchi K, Eguchi S. ADAM17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arterioscler Thromb Vasc Biol. 2006;26(9):e133–137. doi: 10.1161/01.ATV.0000236203.90331.d0. [DOI] [PubMed] [Google Scholar]
- 64.Murphy G. Regulation of the proteolytic disintegrin metalloproteinases, the ‘Sheddases’. Semin Cell Dev Biol. 2009;20(2):138–145. doi: 10.1016/j.semcdb.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 65.Yoda M, Kimura T, Tohmonda T, Morioka H, Matsumoto M, Okada Y, Toyama Y, Horiuchi K. Systemic overexpression of TNFalpha-converting enzyme does not lead to enhanced shedding activity in vivo. PLoS ONE. 2013;8(1):e54412. doi: 10.1371/journal.pone.0054412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takayanagi T, Forrester SJ, Kawai T, Obama T, Tsuji T, Elliott KJ, Nuti E, Rossello A, Kwok HF, Scalia R, Rizzo V, Eguchi S. Vascular ADAM17 as a novel therapeutic target in mediating cardiovascular hypertrophy and perivascular fibrosis induced by angiotensin II. Hypertension. 2016;68(4):949–955. doi: 10.1161/HYPERTENSIONAHA.116.07620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takayanagi T, Kawai T, Forrester SJ, Obama T, Tsuji T, Fukuda Y, Elliott KJ, Tilley DG, Davisson RL, Park JY, Eguchi S. Role of epidermal growth factor receptor and endoplasmic reticulum stress in vascular remodeling induced by angiotensin II. Hypertension. 2015;65(6):1349–1355. doi: 10.1161/HYPERTENSIONAHA.115.05344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen M, Morton J, Davidge ST, Kassiri Z. Loss of smooth muscle cell disintegrin and metalloproteinase 17 transiently suppresses angiotensin II-induced hypertension and end-organ damage. J Mol Cell Cardiol. 2017;103:11–21. doi: 10.1016/j.yjmcc.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 69.Cicalese S, Okuno K, Eguchi S. Novel methods article detection of protein aggregation and proteotoxicity induced by angiotensin II in vascular smooth muscle cells. J Cardiovasc Pharmacol. 2020 doi: 10.1097/fjc.0000000000000934. [DOI] [PubMed] [Google Scholar]
- 70.Cicalese S, Okuno K, Elliott KJ, Kawai T, Scalia R, Rizzo V, Eguchi S. 78 kDa glucose-regulated protein attenuates protein aggregation and monocyte adhesion induced by angiotensin II in vascular cells. Int J Mol Sci. 2020 doi: 10.3390/ijms21144980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cooper HA, Scalia R, Rizzo V, Eguchi S. Angiotensin II- and Alzheimer-type cardiovascular aging. Circ Res. 2018;123(6):651–653. doi: 10.1161/circresaha.118.313477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Okuno K, Cicalese S, Elliott KJ, Kawai T, Hashimoto T, Eguchi S. Targeting molecular mechanism of vascular smooth muscle senescence induced by angiotensin II, a potential therapy via senolytics and senomorphics. Int J Mol Sci. 2020 doi: 10.3390/ijms21186579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xia H, Sriramula S, Chhabra KH, Lazartigues E. Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension. Circ Res. 2013;113(9):1087–1096. doi: 10.1161/CIRCRESAHA.113.301811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mukerjee S, Gao H, Xu J, Sato R, Zsombok A, Lazartigues E. ACE2 and ADAM17 interaction regulates the activity of presympathetic neurons. Hypertension. 2019;74(5):1181–1191. doi: 10.1161/hypertensionaha.119.13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu J, Sriramula S, Xia H, Moreno-Walton L, Culicchia F, Domenig O, Poglitsch M, Lazartigues E. Clinical relevance and role of neuronal AT1 receptors in ADAM17-mediated ACE2 shedding in neurogenic hypertension. Circ Res. 2017;121(1):43–55. doi: 10.1161/CIRCRESAHA.116.310509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Canault M, Peiretti F, Kopp F, Bonardo B, Bonzi MF, Coudeyre JC, Alessi MC, Juhan-Vague I, Nalbone G. The TNF alpha converting enzyme (TACE/ADAM17) is expressed in the atherosclerotic lesions of apolipoprotein E-deficient mice: possible contribution to elevated plasma levels of soluble TNF alpha receptors. Atherosclerosis. 2006;187(1):82–91. doi: 10.1016/j.atherosclerosis.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 77.Holdt LM, Thiery J, Breslow JL, Teupser D. Increased ADAM17 mRNA expression and activity is associated with atherosclerosis resistance in LDL-receptor deficient mice. Arterioscler Thromb Vasc Biol. 2008;28(6):1097–1103. doi: 10.1161/ATVBAHA.108.165654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao X, Kong J, Zhao Y, Wang X, Bu P, Zhang C, Zhang Y. Gene silencing of TACE enhances plaque stability and improves vascular remodeling in a rabbit model of atherosclerosis. Sci Rep. 2015;5:17939. doi: 10.1038/srep17939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takaguri A, Kimura K, Hinoki A, Bourne AM, Autieri MV, Eguchi S. A disintegrin and metalloprotease 17 mediates neointimal hyperplasia in vasculature. Hypertension. 2011;57(4):841–845. doi: 10.1161/HYPERTENSIONAHA.110.166892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chalaris A, Adam N, Sina C, Rosenstiel P, Lehmann-Koch J, Schirmacher P, Hartmann D, Cichy J, Gavrilova O, Schreiber S, Jostock T, Matthews V, Hasler R, Becker C, Neurath MF, Reiss K, Saftig P, Scheller J, Rose-John S. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J Exp Med. 2010;207(8):1617–1624. doi: 10.1084/jem.20092366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nicolaou A, Zhao Z, Northoff BH, Sass K, Herbst A, Kohlmaier A, Chalaris A, Wolfrum C, Weber C, Steffens S, Rose-John S, Teupser D, Holdt LM. Adam17 deficiency promotes atherosclerosis by enhanced TNFR2 signaling in mice. Arterioscler Thromb Vasc Biol. 2017;37(2):247–257. doi: 10.1161/ATVBAHA.116.308682. [DOI] [PubMed] [Google Scholar]
- 82.van der Vorst EP, Zhao Z, Rami M, Holdt LM, Teupser D, Steffens S, Weber C. Contrasting effects of myeloid and endothelial ADAM17 on atherosclerosis development. Thromb Haemost. 2017;117(3):644–646. doi: 10.1160/TH16-09-0674. [DOI] [PubMed] [Google Scholar]
- 83.Canault M, Leroyer AS, Peiretti F, Leseche G, Tedgui A, Bonardo B, Alessi MC, Boulanger CM, Nalbone G. Microparticles of human atherosclerotic plaques enhance the shedding of the tumor necrosis factor-alpha converting enzyme/ADAM17 substrates, tumor necrosis factor and tumor necrosis factor receptor-1. Am J Pathol. 2007;171(5):1713–1723. doi: 10.2353/ajpath.2007.070021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Oksala N, Levula M, Airla N, Pelto-Huikko M, Ortiz RM, Jarvinen O, Salenius JP, Ozsait B, Komurcu-Bayrak E, Erginel-Unaltuna N, Huovila AP, Kytomaki L, Soini JT, Kahonen M, Karhunen PJ, Laaksonen R, Lehtimaki T. ADAM-9, ADAM-15, and ADAM-17 are upregulated in macrophages in advanced human atherosclerotic plaques in aorta and carotid and femoral arteries–Tampere vascular study. Ann Med. 2009;41(4):279–290. doi: 10.1080/07853890802649738. [DOI] [PubMed] [Google Scholar]
- 85.Spin JM, Hsu M, Azuma J, Tedesco MM, Deng A, Dyer JS, Maegdefessel L, Dalman RL, Tsao PS. Transcriptional profiling and network analysis of the murine angiotensin II-induced abdominal aortic aneurysm. Physiol Genomics. 2011;43(17):993–1003. doi: 10.1152/physiolgenomics.00044.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kaneko H, Anzai T, Horiuchi K, Kohno T, Nagai T, Anzai A, Takahashi T, Sasaki A, Shimoda M, Maekawa Y, Shimizu H, Yoshikawa T, Okada Y, Yozu R, Fukuda K. Tumor necrosis factor-alpha converting enzyme is a key mediator of abdominal aortic aneurysm development. Atherosclerosis. 2011;218(2):470–478. doi: 10.1016/j.atherosclerosis.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 87.Kawai T, Takayanagi T, Forrester SJ, Preston KJ, Obama T, Tsuji T, Kobayashi T, Boyer MJ, Cooper HA, Kwok HF, Hashimoto T, Scalia R, Rizzo V, Eguchi S. Vascular ADAM17 (a disintegrin and metalloproteinase domain 17) is required for angiotensin II/beta-aminopropionitrile-induced abdominal aortic aneurysm. Hypertension. 2017;70(5):959–963. doi: 10.1161/HYPERTENSIONAHA.117.09822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Obama T, Tsuji T, Kobayashi T, Fukuda Y, Takayanagi T, Taro Y, Kawai T, Forrester SJ, Elliott KJ, Choi E, Daugherty A, Rizzo V, Eguchi S. Epidermal growth factor receptor inhibitor protects against abdominal aortic aneurysm in a mouse model. Clin Sci (Lond) 2015;128(9):559–565. doi: 10.1042/cs20140696. [DOI] [PubMed] [Google Scholar]
- 89.Miyao M, Cicalese S, Cooper HA, Eguchi S. Endoplasmic reticulum stress and mitochondrial biogenesis are potential therapeutic targets for abdominal aortic aneurysm. Clin Sci (Lond) 2019;133(19):2023–2028. doi: 10.1042/cs20190648. [DOI] [PubMed] [Google Scholar]
- 90.Cooper HA, Cicalese S, Preston KJ, Kawai T, Okuno K, Choi ET, Kasahara S, Uchida HA, Otaka N, Scalia R, Rizzo V, Eguchi S. Targeting mitochondrial fission as a potential therapeutic for abdominal aortic aneurysm. Cardiovasc Res. 2020 doi: 10.1093/cvr/cvaa133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Forrester SJ, Preston KJ, Cooper HA, Boyer MJ, Escoto KM, Poltronetti AJ, Elliott KJ, Kuroda R, Miyao M, Sesaki H, Akiyama T, Kimura Y, Rizzo V, Scalia R, Eguchi S. Mitochondrial fission mediates endothelial inflammation. Hypertension. 2020;76(1):267–276. doi: 10.1161/hypertensionaha.120.14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miyao M, Cicalese S, Kawai T, Cooper HA, Boyer MJ, Elliott KJ, Forrester SJ, Kuroda R, Rizzo V, Hashimoto T, Scalia R, Eguchi S. Involvement of senescence and mitochondrial fission in endothelial cell pro-inflammatory phenotype induced by angiotensin II. Int J Mol Sci. 2020 doi: 10.3390/ijms21093112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Geng L, Wang W, Chen Y, Cao J, Lu L, Chen Q, He R, Shen W. Elevation of ADAM10, ADAM17, MMP-2 and MMP-9 expression with media degeneration features CaCl2-induced thoracic aortic aneurysm in a rat model. Exp Mol Pathol. 2010;89(1):72–81. doi: 10.1016/j.yexmp.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 94.Shen M, Hu M, Fedak PWM, Oudit GY, Kassiri Z. Cell-specific functions of adam17 regulate the progression of thoracic aortic aneurysm. Circ Res. 2018;123(3):372–388. doi: 10.1161/CIRCRESAHA.118.313181. [DOI] [PubMed] [Google Scholar]
- 95.Folkesson M, Li C, Frebelius S, Swedenborg J, Wagsater D, Williams KJ, Eriksson P, Roy J, Liu ML. Proteolytically active ADAM10 and ADAM17 carried on membrane microvesicles in human abdominal aortic aneurysms. Thromb Haemost. 2015;114(6):1165–1174. doi: 10.1160/TH14-10-0899. [DOI] [PubMed] [Google Scholar]
- 96.Satoh H, Nakamura M, Satoh M, Nakajima T, Izumoto H, Maesawa C, Kawazoe K, Masuda T, Hiramori K. Expression and localization of tumour necrosis factor-alpha and its converting enzyme in human abdominal aortic aneurysm. Clin Sci (Lond) 2004;106(3):301–306. doi: 10.1042/CS20030189. [DOI] [PubMed] [Google Scholar]
- 97.Li Y, Yang C, Ma G, Cui L, Gu X, Chen Y, Zhao B, Wang H, Li K. Analysis of ADAM17 polymorphisms and susceptibility to sporadic abdominal aortic aneurysm. Cell Physiol Biochem. 2014;33(5):1426–1438. doi: 10.1159/000358708. [DOI] [PubMed] [Google Scholar]
- 98.Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res. 2015;107(3):321–330. doi: 10.1093/cvr/cvv147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Casagrande V, Menghini R, Menini S, Marino A, Marchetti V, Cavalera M, Fabrizi M, Hribal ML, Pugliese G, Gentileschi P, Schillaci O, Porzio O, Lauro D, Sbraccia P, Lauro R, Federici M. Overexpression of tissue inhibitor of metalloproteinase 3 in macrophages reduces atherosclerosis in low-density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol. 2012;32(1):74–81. doi: 10.1161/ATVBAHA.111.238402. [DOI] [PubMed] [Google Scholar]
- 100.Tang J, Frey JM, Wilson CL, Moncada-Pazos A, Levet C, Freeman M, Rosenfeld ME, Stanley ER, Raines EW, Bornfeldt KE. Neutrophil and macrophage cell surface CSF-1 shed by ADAM17 drives mouse macrophage proliferation in acute and chronic inflammation. Mol Cell Biol. 2018 doi: 10.1128/MCB.00103-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tsubota Y, Frey JM, Tai PW, Welikson RE, Raines EW. Monocyte ADAM17 promotes diapedesis during transendothelial migration: identification of steps and substrates targeted by metalloproteinases. J Immunol. 2013;190(8):4236–4244. doi: 10.4049/jimmunol.1300046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rovida E, Paccagnini A, Del Rosso M, Peschon J, Dello Sbarba P. TNF-alpha-converting enzyme cleaves the macrophage colony-stimulating factor receptor in macrophages undergoing activation. J Immunol. 2001;166(3):1583–1589. doi: 10.4049/jimmunol.166.3.1583. [DOI] [PubMed] [Google Scholar]
- 103.Willman CL, Stewart CC, Miller V, Yi TL, Tomasi TB. Regulation of MHC class II gene expression in macrophages by hematopoietic colony-stimulating factors (CSF). Induction by granulocyte/macrophage CSF and inhibition by CSF-1. J Exp Med. 1989;170(5):1559–1567. doi: 10.1084/jem.170.5.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Metharom P, Martin K, Kumar AH, Sawhney N, Cronin MF, McCarthy DG, Maguire AR, Caplice NM. Pleiotropic role for monocyte C-fms protein in response to vascular injury: potential therapeutic target. Atherosclerosis. 2011;216(1):74–82. doi: 10.1016/j.atherosclerosis.2011.01.037. [DOI] [PubMed] [Google Scholar]
- 105.Davies MJ, Gordon JL, Gearing AJ, Pigott R, Woolf N, Katz D, Kyriakopoulos A. The expression of the adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human atherosclerosis. J Pathol. 1993;171(3):223–229. doi: 10.1002/path.1711710311. [DOI] [PubMed] [Google Scholar]
- 106.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107(10):1255–1262. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Eriksson EE, Xie X, Werr J, Thoren P, Lindbom L. Importance of primary capture and L-selectin-dependent secondary capture in leukocyte accumulation in inflammation and atherosclerosis in vivo. J Exp Med. 2001;194(2):205–218. doi: 10.1084/jem.194.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med. 2006;203(5):1273–1282. doi: 10.1084/jem.20052205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Garton KJ, Gough PJ, Philalay J, Wille PT, Blobel CP, Whitehead RH, Dempsey PJ, Raines EW. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17) J Biol Chem. 2003;278(39):37459–37464. doi: 10.1074/jbc.M305877200. [DOI] [PubMed] [Google Scholar]
- 110.Otsuki M, Hashimoto K, Morimoto Y, Kishimoto T, Kasayama S. Circulating vascular cell adhesion molecule-1 (VCAM-1) in atherosclerotic NIDDM patients. Diabetes. 1997;46(12):2096–2101. doi: 10.2337/diab.46.12.2096. [DOI] [PubMed] [Google Scholar]
- 111.Tsakadze NL, Sithu SD, Sen U, English WR, Murphy G, D'Souza SE. Tumor necrosis factor-alpha-converting enzyme (TACE/ADAM-17) mediates the ectodomain cleavage of intercellular adhesion molecule-1 (ICAM-1) J Biol Chem. 2006;281(6):3157–3164. doi: 10.1074/jbc.M510797200. [DOI] [PubMed] [Google Scholar]
- 112.Hwang SJ, Ballantyne CM, Sharrett AR, Smith LC, Davis CE, Gotto AM, Jr, Boerwinkle E. Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: the atherosclerosis risk in communities (ARIC) study. Circulation. 1997;96(12):4219–4225. doi: 10.1161/01.CIR.96.12.4219. [DOI] [PubMed] [Google Scholar]
- 113.Peschon J, Slack J, Reddy P, Stocking K, Sunnarborg S, Lee D, Russell W, Castner B, Johnson R, Fitzner J, Boyce R, Nelson N, Kozlosky C, Wolfson M, Rauch C, Cerretti D, Paxton R, March C, Black R. An essential role for ectodomain shedding in mammalian development. Science. 1998;282(5392):1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 114.Walcheck B. ADAM-17-independent shedding of L-selectin. J Leukoc Biol. 2003;74(3):389–394. doi: 10.1189/jlb.0403141. [DOI] [PubMed] [Google Scholar]
- 115.Walcheck B, Kahn J, Fisher JM, Wang BB, Fisk RS, Payan DG, Feehan C, Betageri R, Darlak K, Spatola AF, Kishimoto TK. Neutrophil rolling altered by inhibition of L-selectin shedding in vitro. Nature. 1996;380(6576):720–723. doi: 10.1038/380720a0. [DOI] [PubMed] [Google Scholar]
- 116.Schleiffenbaum B, Spertini O, Tedder TF. Soluble L-selectin is present in human plasma at high levels and retains functional activity. J Cell Biol. 1992;119(1):229–238. doi: 10.1083/jcb.119.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nagano O, Murakami D, Hartmann D, De Strooper B, Saftig P, Iwatsubo T, Nakajima M, Shinohara M, Saya H. Cell-matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca(2+) influx and PKC activation. J Cell Biol. 2004;165(6):893–902. doi: 10.1083/jcb.200310024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tole S, Durkan AM, Huang YW, Liu GY, Leung A, Jones LL, Taylor JA, Robinson LA. Thromboxane prostanoid receptor stimulation induces shedding of the transmembrane chemokine CX3CL1 yet enhances CX3CL1-dependent leukocyte adhesion. Am J Physiol Cell Physiol. 2010;298(6):C1469–1480. doi: 10.1152/ajpcell.00380.2009. [DOI] [PubMed] [Google Scholar]
- 119.Cooke VG, Naik MU, Naik UP. Fibroblast growth factor-2 failed to induce angiogenesis in junctional adhesion molecule-A-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26(9):2005–2011. doi: 10.1161/01.ATV.0000234923.79173.99. [DOI] [PubMed] [Google Scholar]
- 120.Naik MU, Naik UP. Junctional adhesion molecule-A-induced endothelial cell migration on vitronectin is integrin alpha v beta 3 specific. J Cell Sci. 2006;119(Pt 3):490–499. doi: 10.1242/jcs.02771. [DOI] [PubMed] [Google Scholar]
- 121.Woodfin A, Reichel CA, Khandoga A, Corada M, Voisin MB, Scheiermann C, Haskard DO, Dejana E, Krombach F, Nourshargh S. JAM-A mediates neutrophil transmigration in a stimulus-specific manner in vivo: evidence for sequential roles for JAM-A and PECAM-1 in neutrophil transmigration. Blood. 2007;110(6):1848–1856. doi: 10.1182/blood-2006-09-047431. [DOI] [PubMed] [Google Scholar]
- 122.Khandoga A, Kessler JS, Meissner H, Hanschen M, Corada M, Motoike T, Enders G, Dejana E, Krombach F. Junctional adhesion molecule-A deficiency increases hepatic ischemia-reperfusion injury despite reduction of neutrophil transendothelial migration. Blood. 2005;106(2):725–733. doi: 10.1182/blood-2004-11-4416. [DOI] [PubMed] [Google Scholar]
- 123.Koenen RR, Pruessmeyer J, Soehnlein O, Fraemohs L, Zernecke A, Schwarz N, Reiss K, Sarabi A, Lindbom L, Hackeng TM, Weber C, Ludwig A. Regulated release and functional modulation of junctional adhesion molecule A by disintegrin metalloproteinases. Blood. 2009;113(19):4799–4809. doi: 10.1182/blood-2008-04-152330. [DOI] [PubMed] [Google Scholar]
- 124.Schaff U, Mattila PE, Simon SI, Walcheck B. Neutrophil adhesion to E-selectin under shear promotes the redistribution and co-clustering of ADAM17 and its proteolytic substrate L-selectin. J Leukoc Biol. 2008;83(1):99–105. doi: 10.1189/jlb.0507304. [DOI] [PubMed] [Google Scholar]
- 125.Wang Y, Herrera AH, Li Y, Belani KK, Walcheck B. Regulation of mature ADAM17 by redox agents for L-selectin shedding. J Immunol. 2009;182(4):2449–2457. doi: 10.4049/jimmunol.0802770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gooz P, Gooz M, Baldys A, Hoffman S. ADAM-17 regulates endothelial cell morphology, proliferation, and in vitro angiogenesis. Biochem Biophys Res Commun. 2009;380(1):33–38. doi: 10.1016/j.bbrc.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kwak HI, Mendoza EA, Bayless KJ. ADAM17 co-purifies with TIMP-3 and modulates endothelial invasion responses in three-dimensional collagen matrices. Matrix Biol. 2009;28(8):470–479. doi: 10.1016/j.matbio.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 128.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7(5):359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 129.Kalinowski A, Plowes NJ, Huang Q, Berdejo-Izquierdo C, Russell RR, Russell KS. Metalloproteinase-dependent cleavage of neuregulin and autocrine stimulation of vascular endothelial cells. FASEB J. 2010;24(7):2567–2575. doi: 10.1096/fj.08-129072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Inoue Y, Shimazawa M, Nakamura S, Takata S, Hashimoto Y, Izawa H, Masuda T, Tsuruma K, Sakaue T, Nakayama H, Higashiyama S, Hara H. Both autocrine signaling and paracrine signaling of HB-EGF enhance ocular neovascularization. Arterioscler Thromb Vasc Biol. 2018;38(1):174–185. doi: 10.1161/ATVBAHA.117.310337. [DOI] [PubMed] [Google Scholar]
- 131.Kawasaki K, Freimuth J, Meyer DS, Lee MM, Tochimoto-Okamoto A, Benzinou M, Clermont FF, Wu G, Roy R, Letteboer TG, Ploos van Amstel JK, Giraud S, Dupuis-Girod S, Lesca G, Westermann CJ, Coffey RJ, Jr, Akhurst RJ. Genetic variants of Adam17 differentially regulate TGFbeta signaling to modify vascular pathology in mice and humans. Proc Natl Acad Sci U S A. 2014;111(21):7723–7728. doi: 10.1073/pnas.1318761111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Caolo V, Swennen G, Chalaris A, Wagenaar A, Verbruggen S, Rose-John S, Molin DG, Vooijs M, Post MJ. ADAM10 and ADAM17 have opposite roles during sprouting angiogenesis. Angiogenesis. 2015;18(1):13–22. doi: 10.1007/s10456-014-9443-4. [DOI] [PubMed] [Google Scholar]
- 133.Jin Y, Liu Y, Lin Q, Li J, Druso JE, Antonyak MA, Meininger CJ, Zhang SL, Dostal DE, Guan JL, Cerione RA, Peng X. Deletion of Cdc42 enhances ADAM17-mediated vascular endothelial growth factor receptor 2 shedding and impairs vascular endothelial cell survival and vasculogenesis. Mol Cell Biol. 2013;33(21):4181–4197. doi: 10.1128/MCB.00650-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Raikwar NS, Liu KZ, Thomas CP. N-terminal cleavage and release of the ectodomain of Flt1 is mediated via ADAM10 and ADAM 17 and regulated by VEGFR2 and the Flt1 intracellular domain. PLoS ONE. 2014;9(11):e112794. doi: 10.1371/journal.pone.0112794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Weskamp G, Mendelson K, Swendeman S, Le Gall S, Ma Y, Lyman S, Hinoki A, Eguchi S, Guaiquil V, Horiuchi K, Blobel CP. Pathological neovascularization is reduced by inactivation of ADAM17 in endothelial cells but not in pericytes. Circ Res. 2010;106(5):932–940. doi: 10.1161/CIRCRESAHA.109.207415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lucitti JL, Mackey JK, Morrison JC, Haigh JJ, Adams RH, Faber JE. Formation of the collateral circulation is regulated by vascular endothelial growth factor-A and a disintegrin and metalloprotease family members 10 and 17. Circ Res. 2012;111(12):1539–1550. doi: 10.1161/CIRCRESAHA.112.279109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chikaraishi Y, Shimazawa M, Yokota K, Yoshino K, Hara H. CB-12181, a new azasugar-based matrix metalloproteinase/tumor necrosis factor-alpha converting enzyme inhibitor, inhibits vascular endothelial growth factor-induced angiogenesis in vitro and retinal neovascularization in vivo. Curr Neurovasc Res. 2009;6(3):140–147. doi: 10.2174/156720209788970072. [DOI] [PubMed] [Google Scholar]
- 138.Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, Blobel C. Cutting edge: TNF-α-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol. 2007;179(5):2686–2689. doi: 10.4049/jimmunol.179.5.2686. [DOI] [PubMed] [Google Scholar]
- 139.Jackson LF, Qiu TH, Sunnarborg SW, Chang A, Zhang C, Patterson C, Lee DC. Defective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant BMP signaling. EMBO J. 2003;22(11):2704–2716. doi: 10.1093/emboj/cdg264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Canault M, Certel K, Schatzberg D, Wagner DD, Hynes RO. The lack of ADAM17 activity during embryonic development causes hemorrhage and impairs vessel formation. PLoS ONE. 2010;5(10):e13433. doi: 10.1371/journal.pone.0013433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hassemer EL, Le Gall SM, Liegel R, McNally M, Chang B, Zeiss CJ, Dubielzig RD, Horiuchi K, Kimura T, Okada Y, Blobel CP, Sidjanin DJ. The waved with open eyelids (woe) locus is a hypomorphic mouse mutation in Adam17. Genetics. 2010;185(1):245–255. doi: 10.1534/genetics.109.113167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wilson CL, Gough PJ, Chang CA, Chan CK, Frey JM, Liu Y, Braun KR, Chin MT, Wight TN, Raines EW. Endothelial deletion of ADAM17 in mice results in defective remodeling of the semilunar valves and cardiac dysfunction in adults. Mech Dev. 2013;130(4–5):272–289. doi: 10.1016/j.mod.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zeng SY, Chen X, Chen SR, Li Q, Wang YH, Zou J, Cao WW, Luo JN, Gao H, Liu PQ. Upregulation of Nox4 promotes angiotensin II-induced epidermal growth factor receptor activation and subsequent cardiac hypertrophy by increasing ADAM17 expression. Can J Cardiol. 2013;29(10):1310–1319. doi: 10.1016/j.cjca.2013.04.026. [DOI] [PubMed] [Google Scholar]
- 144.Zeng SY, Lu HQ, Yan QJ, Zou J. A reduction in ADAM17 expression is involved in the protective effect of the PPAR-alpha activator fenofibrate on pressure overload-induced cardiac hypertrophy. PPAR Res. 2018;2018:7916953. doi: 10.1155/2018/7916953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zeng SY, Yang L, Yan QJ, Gao L, Lu HQ, Yan PK. Nox1/4 dual inhibitor GKT137831 attenuates hypertensive cardiac remodelling associating with the inhibition of ADAM17-dependent proinflammatory cytokines-induced signalling pathways in the rats with abdominal artery constriction. Biomed Pharmacother. 2019;109:1907–1914. doi: 10.1016/j.biopha.2018.11.077. [DOI] [PubMed] [Google Scholar]
- 146.Wang X, Oka T, Chow FL, Cooper SB, Odenbach J, Lopaschuk GD, Kassiri Z, Fernandez-Patron C. Tumor necrosis factor-alpha-converting enzyme is a key regulator of agonist-induced cardiac hypertrophy and fibrosis. Hypertension. 2009;54(3):575–582. doi: 10.1161/HYPERTENSIONAHA.108.127670. [DOI] [PubMed] [Google Scholar]
- 147.Odenbach J, Wang X, Cooper S, Chow FL, Oka T, Lopaschuk G, Kassiri Z, Fernandez-Patron C. MMP-2 mediates angiotensin II-induced hypertension under the transcriptional control of MMP-7 and TACE. Hypertension. 2011;57(1):123–130. doi: 10.1161/HYPERTENSIONAHA.110.159525. [DOI] [PubMed] [Google Scholar]
- 148.Fan D, Takawale A, Shen M, Wang W, Wang X, Basu R, Oudit GY, Kassiri Z. Cardiomyocyte a disintegrin and metalloproteinase 17 (ADAM17) is essential in post-myocardial infarction repair by regulating angiogenesis. Circ Heart Fail. 2015;8(5):970–979. doi: 10.1161/CIRCHEARTFAILURE.114.002029. [DOI] [PubMed] [Google Scholar]
- 149.Zheng DY, Zhao J, Yang JM, Wang M, Zhang XT. Enhanced ADAM17 expression is associated with cardiac remodeling in rats with acute myocardial infarction. Life Sci. 2016 doi: 10.1016/j.lfs.2016.02.097. [DOI] [PubMed] [Google Scholar]
- 150.Fan D, Takawale A, Shen M, Samokhvalov V, Basu R, Patel V, Wang X, Fernandez-Patron C, Seubert JM, Oudit GY, Kassiri Z. A Disintegrin and metalloprotease-17 regulates pressure overload-induced myocardial hypertrophy and dysfunction through proteolytic processing of integrin beta1. Hypertension. 2016;68(4):937–948. doi: 10.1161/HYPERTENSIONAHA.116.07566. [DOI] [PubMed] [Google Scholar]
- 151.Dou H, Feher A, Davila AC, Romero MJ, Patel VS, Kamath VM, Gooz MB, Rudic RD, Lucas R, Fulton DJ, Weintraub NL, Bagi Z. Role of adipose tissue endothelial ADAM17 in age-related coronary microvascular dysfunction. Arterioscler Thromb Vasc Biol. 2017;37(6):1180–1193. doi: 10.1161/ATVBAHA.117.309430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Satoh M, Ishikawa Y, Itoh T, Minami Y, Takahashi Y, Nakamura M. The expression of TNF-alpha converting enzyme at the site of ruptured plaques in patients with acute myocardial infarction. Eur J Clin Invest. 2008;38(2):97–105. doi: 10.1111/j.1365-2362.2007.01912.x. [DOI] [PubMed] [Google Scholar]
- 153.Shimoda Y, Satoh M, Nakamura M, Akatsu T, Hiramori K. Activated tumour necrosis factor-alpha shedding process is associated with in-hospital complication in patients with acute myocardial infarction. Clin Sci (Lond) 2005;108(4):339–347. doi: 10.1042/CS20040229. [DOI] [PubMed] [Google Scholar]
- 154.Rizza S, Copetti M, Cardellini M, Menghini R, Pecchioli C, Luzi A, Di Cola G, Porzio O, Ippoliti A, Romeo F, Pellegrini F, Federici M. A score including ADAM17 substrates correlates to recurring cardiovascular event in subjects with atherosclerosis. Atherosclerosis. 2015;239(2):459–464. doi: 10.1016/j.atherosclerosis.2015.01.029. [DOI] [PubMed] [Google Scholar]
- 155.Lautrette A, Li S, Alili R, Sunnarborg SW, Burtin M, Lee DC, Friedlander G, Terzi F. Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: a new therapeutic approach. Nat Med. 2005;11(8):867–874. doi: 10.1038/nm1275. [DOI] [PubMed] [Google Scholar]
- 156.Kefaloyianni E, Muthu ML, Kaeppler J, Sun X, Sabbisetti V, Chalaris A, Rose-John S, Wong E, Sagi I, Waikar SS, Rennke H, Humphreys BD, Bonventre JV, Herrlich A. ADAM17 substrate release in proximal tubule drives kidney fibrosis. JCI Insight. 2016 doi: 10.1172/jci.insight.87023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li R, Wang T, Walia K, Gao B, Krepinsky JC. Regulation of profibrotic responses by ADAM17 activation in high glucose requires its C-terminus and FAK. J Cell Sci. 2018 doi: 10.1242/jcs.208629. [DOI] [PubMed] [Google Scholar]
- 158.Taniguchi K, Xia L, Goldberg HJ, Lee KW, Shah A, Stavar L, Masson EA, Momen A, Shikatani EA, John R, Husain M, Fantus IG. Inhibition of Src kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes. 2013;62(11):3874–3886. doi: 10.2337/db12-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Qing X, Chinenov Y, Redecha P, Madaio M, Roelofs JJ, Farber G, Issuree PD, Donlin L, McLlwain DR, Mak TW, Blobel CP, Salmon JE. iRhom2 promotes lupus nephritis through TNF-alpha and EGFR signaling. J Clin Invest. 2018;128(4):1397–1412. doi: 10.1172/JCI97650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Beck Gooz M, Maldonado EN, Dang Y, Amria MY, Higashiyama S, Abboud HE, Lemasters JJ, Bell PD. ADAM17 promotes proliferation of collecting duct kidney epithelial cells through ERK activation and increased glycolysis in polycystic kidney disease. Am J Physiol Renal Physiol. 2014;307(5):F551–559. doi: 10.1152/ajprenal.00218.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Melenhorst WB, Visser L, Timmer A, van den Heuvel MC, Stegeman CA, van Goor H. ADAM17 upregulation in human renal disease: a role in modulating TGF-alpha availability? Am J Physiol Renal Physiol. 2009;297(3):F781–790. doi: 10.1152/ajprenal.90610.2008. [DOI] [PubMed] [Google Scholar]
- 162.Gutta S, Grobe N, Kumbaji M, Osman H, Saklayen M, Li G, Elased KM. Increased urinary angiotensin converting enzyme 2 and neprilysin in patients with type 2 diabetes. Am J Physiol Renal Physiol. 2018;315(2):F263–F274. doi: 10.1152/ajprenal.00565.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Palau V, Riera M, Duran X, Valdivielso JM, Betriu A, Fernandez E, Pascual J, Soler MJ. Circulating ADAMs are associated with renal and cardiovascular outcomes in chronic kidney disease patients. Nephrol Dial Transplant. 2018 doi: 10.1093/ndt/gfy240. [DOI] [PubMed] [Google Scholar]
- 164.Palau V, Riera M, Duran X, Valdivielso JM, Betriu A, Fernandez E, Pascual J, Soler MJ. Circulating ADAMs are associated with renal and cardiovascular outcomes in chronic kidney disease patients. Nephrol Dial Transplant. 2020;35(1):130–138. doi: 10.1093/ndt/gfy240. [DOI] [PubMed] [Google Scholar]
- 165.Federici M, Hribal ML, Menghini R, Kanno H, Marchetti V, Porzio O, Sunnarborg SW, Rizza S, Serino M, Cunsolo V, Lauro D, Mauriello A, Smookler DS, Sbraccia P, Sesti G, Lee DC, Khokha R, Accili D, Lauro R. Timp3 deficiency in insulin receptor-haploinsufficient mice promotes diabetes and vascular inflammation via increased TNF-alpha. J Clin Invest. 2005;115(12):3494–3505. doi: 10.1172/JCI26052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Menghini R, Casagrande V, Menini S, Marino A, Marzano V, Hribal ML, Gentileschi P, Lauro D, Schillaci O, Pugliese G, Sbraccia P, Urbani A, Lauro R, Federici M. TIMP3 overexpression in macrophages protects from insulin resistance, adipose inflammation, and nonalcoholic fatty liver disease in mice. Diabetes. 2012;61(2):454–462. doi: 10.2337/db11-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kaneko H, Anzai T, Horiuchi K, Morimoto K, Anzai A, Nagai T, Sugano Y, Maekawa Y, Itoh H, Yoshikawa T, Okada Y, Ogawa S, Fukuda K. Tumor necrosis factor-α converting enzyme inactivation ameliorates high-fat diet-induced insulin resistance and altered energy homeostasis. Circ J. 2011;75(10):2482–2490. doi: 10.1253/circj.CJ-11-0182. [DOI] [PubMed] [Google Scholar]
- 168.Serino M, Menghini R, Fiorentino L, Amoruso R, Mauriello A, Lauro D, Sbraccia P, Hribal ML, Lauro R, Federici M. Mice heterozygous for tumor necrosis factor-alpha converting enzyme are protected from obesity-induced insulin resistance and diabetes. Diabetes. 2007;56(10):2541–2546. doi: 10.2337/db07-0360. [DOI] [PubMed] [Google Scholar]
- 169.Togashi N, Ura N, Higashiura K, Murakami H, Shimamoto K. Effect of TNF-alpha–converting enzyme inhibitor on insulin resistance in fructose-fed rats. Hypertension. 2002;39(2 Pt 2):578–580. doi: 10.1161/hy0202.103290. [DOI] [PubMed] [Google Scholar]
- 170.de Meijer VE, Le HD, Meisel JA, Sharma AK, Popov Y, Puder M. Tumor necrosis factor alpha-converting enzyme inhibition reverses hepatic steatosis and improves insulin sensitivity markers and surgical outcome in mice. PLoS ONE. 2011;6(9):e25587. doi: 10.1371/journal.pone.0025587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Badenes M, Amin A, Gonzalez-Garcia I, Felix I, Burbridge E, Cavadas M, Ortega FJ, de Carvalho E, Faisca P, Carobbio S, Seixas E, Pedroso D, Neves-Costa A, Moita LF, Fernandez-Real JM, Vidal-Puig A, Domingos A, Lopez M, Adrain C. Deletion of iRhom2 protects against diet-induced obesity by increasing thermogenesis. Mol Metab. 2020;31:67–84. doi: 10.1016/j.molmet.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lian G, Li X, Zhang L, Zhang Y, Sun L, Zhang X, Liu H, Pang Y, Kong W, Zhang T, Wang X, Jiang C. Macrophage metabolic reprogramming aggravates aortic dissection through the HIF1alpha-ADAM17 pathway() EBioMedicine. 2019;49:291–304. doi: 10.1016/j.ebiom.2019.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Badenes M, Amin A, González-García I, Félix I, Burbridge E, Cavadas M, Ortega FJ, de Carvalho É, Faísca P, Carobbio S, Seixas E, Pedroso D, Neves-Costa A, Moita LF, Fernández-Real JM, Vidal-Puig A, Domingos A, López M, Adrain C. Deletion of iRhom2 protects against diet-induced obesity by increasing thermogenesis. Mol Metab. 2020;31:67–84. doi: 10.1016/j.molmet.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Peng Q, Deng Y, Yang X, Leng X, Yang Y, Liu H. Genetic variants of ADAM17 are implicated in the pathological process of Kawasaki disease and secondary coronary artery lesions via the TGF-beta/SMAD3 signaling pathway. Eur J Pediatr. 2016 doi: 10.1007/s00431-016-2696-8. [DOI] [PubMed] [Google Scholar]
- 175.Junyent M, Parnell LD, Lai CQ, Arnett DK, Tsai MY, Kabagambe EK, Straka RJ, Province M, An P, Smith CE, Lee YC, Borecki I, Ordovas JM. ADAM17_i33708A>G polymorphism interacts with dietary n-6 polyunsaturated fatty acids to modulate obesity risk in the genetics of lipid lowering drugs and diet network study. Nutr Metab Cardiovasc Dis. 2010;20(10):698–705. doi: 10.1016/j.numecd.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Morange PE, Tregouet DA, Godefroy T, Saut N, Bickel C, Rupprecht HJ, Lackner K, Barbaux S, Poirier O, Peiretti F, Nalbone G, Juhan-Vague I, Blankenberg S, Tiret L. Polymorphisms of the tumor necrosis factor-alpha (TNF) and the TNF-alpha converting enzyme (TACE/ADAM17) genes in relation to cardiovascular mortality: the AtheroGene study. J Mol Med (Berl) 2008;86(10):1153–1161. doi: 10.1007/s00109-008-0375-6. [DOI] [PubMed] [Google Scholar]
- 177.Hartl D, May P, Gu W, Mayhaus M, Pichler S, Spaniol C, Glaab E, Bobbili DR, Antony P, Koegelsberger S, Kurz A, Grimmer T, Morgan K, Vardarajan BN, Reitz C, Hardy J, Bras J, Guerreiro R, Balling R, Schneider JG, Riemenschneider M, Aesg A rare loss-of-function variant of ADAM17 is associated with late-onset familial Alzheimer disease. Mol Psychiatry. 2018 doi: 10.1038/s41380-018-0091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Xie Y, Ma A, Wang B, Peng R, Jing Y, Wang D, Finnell RH, Qiao B, Wang Y, Wang H, Zheng Y. Rare mutations of ADAM17 from TOFs induce hypertrophy in human embryonic stem cell-derived cardiomyocytes via HB-EGF signaling. Clin Sci (Lond) 2019;133(2):225–238. doi: 10.1042/cs20180842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Blaydon DC, Biancheri P, Di WL, Plagnol V, Cabral RM, Brooke MA, van Heel DA, Ruschendorf F, Toynbee M, Walne A, O’Toole EA, Martin JE, Lindley K, Vulliamy T, Abrams DJ, MacDonald TT, Harper JI, Kelsell DP. Inflammatory skin and bowel disease linked to ADAM17 deletion. N Engl J Med. 2011;365(16):1502–1508. doi: 10.1056/NEJMoa1100721. [DOI] [PubMed] [Google Scholar]
- 180.Bandsma RH, van Goor H, Yourshaw M, Horlings RK, Jonkman MF, Schölvinck EH, Karrenbeld A, Scheenstra R, Kömhoff M, Rump P, Koopman-Keemink Y, Nelson SF, Escher JC, Cutz E, Martín MG. Loss of ADAM17 is associated with severe multiorgan dysfunction. Hum Pathol. 2015;46(6):923–928. doi: 10.1016/j.humpath.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102(4):1186–1195. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- 182.Abel S, Hundhausen C, Mentlein R, Schulte A, Berkhout TA, Broadway N, Hartmann D, Sedlacek R, Dietrich S, Muetze B, Schuster B, Kallen KJ, Saftig P, Rose-John S, Ludwig A. The transmembrane CXC-chemokine ligand 16 is induced by IFN-gamma and TNF-alpha and shed by the activity of the disintegrin-like metalloproteinase ADAM10. J Immunol. 2004;172(10):6362–6372. doi: 10.4049/jimmunol.172.10.6362. [DOI] [PubMed] [Google Scholar]
- 183.Matthews V, Schuster B, Schutze S, Bussmeyer I, Ludwig A, Hundhausen C, Sadowski T, Saftig P, Hartmann D, Kallen KJ, Rose-John S. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE) J Biol Chem. 2003;278(40):38829–38839. doi: 10.1074/jbc.M210584200. [DOI] [PubMed] [Google Scholar]
- 184.Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, Reiss K, Saftig P, Bianchi ME. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10) FASEB J. 2008;22(10):3716–3727. doi: 10.1096/fj.08-109033. [DOI] [PubMed] [Google Scholar]
- 185.Hikita A, Tanaka N, Yamane S, Ikeda Y, Furukawa H, Tohma S, Suzuki R, Tanaka S, Mitomi H, Fukui N. Involvement of a disintegrin and metalloproteinase 10 and 17 in shedding of tumor necrosis factor-alpha. Biochem Cell Biol. 2009;87(4):581–593. doi: 10.1139/o09-015. [DOI] [PubMed] [Google Scholar]
- 186.Zhang C, Tian L, Chi C, Wu X, Yang X, Han M, Xu T, Zhuang Y, Deng K. Adam10 is essential for early embryonic cardiovascular development. Dev Dyn. 2010;239(10):2594–2602. doi: 10.1002/dvdy.22391. [DOI] [PubMed] [Google Scholar]
- 187.Mehta V, Fields L, Evans IM, Yamaji M, Pellet-Many C, Jones T, Mahmoud M, Zachary I. VEGF (vascular endothelial growth factor) induces NRP1 (neuropilin-1) cleavage via ADAMs (a disintegrin and metalloproteinase) 9 and 10 to generate novel carboxy-terminal NRP1 fragments that regulate angiogenic signaling. Arterioscler Thromb Vasc Biol. 2018 doi: 10.1161/ATVBAHA.118.311118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Donners MM, Wolfs IM, Olieslagers S, Mohammadi-Motahhari Z, Tchaikovski V, Heeneman S, van Buul JD, Caolo V, Molin DG, Post MJ, Waltenberger J. A disintegrin and metalloprotease 10 is a novel mediator of vascular endothelial growth factor-induced endothelial cell function in angiogenesis and is associated with atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30(11):2188–2195. doi: 10.1161/ATVBAHA.110.213124. [DOI] [PubMed] [Google Scholar]
- 189.Schulz B, Pruessmeyer J, Maretzky T, Ludwig A, Blobel CP, Saftig P, Reiss K. ADAM10 regulates endothelial permeability and T-Cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res. 2008;102(10):1192–1201. doi: 10.1161/CIRCRESAHA.107.169805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164(5):769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sanderson MP, Erickson SN, Gough PJ, Garton KJ, Wille PT, Raines EW, Dunbar AJ, Dempsey PJ. ADAM10 mediates ectodomain shedding of the betacellulin precursor activated by p-aminophenylmercuric acetate and extracellular calcium influx. J Biol Chem. 2005;280(3):1826–1837. doi: 10.1074/jbc.M408804200. [DOI] [PubMed] [Google Scholar]
- 192.Yan Y, Shirakabe K, Werb Z. The metalloprotease Kuzbanian (ADAM10) mediates the transactivation of EGF receptor by G protein-coupled receptors. J Cell Biol. 2002;158(2):221–226. doi: 10.1083/jcb.200112026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shitomi Y, Thogersen IB, Ito N, Leitinger B, Enghild JJ, Itoh Y. ADAM10 controls collagen signaling and cell migration on collagen by shedding the ectodomain of discoidin domain receptor 1 (DDR1) Mol Biol Cell. 2015;26(4):659–673. doi: 10.1091/mbc.E14-10-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hikita A, Yana I, Wakeyama H, Nakamura M, Kadono Y, Oshima Y, Nakamura K, Seiki M, Tanaka S. Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-kappaB ligand. J Biol Chem. 2006;281(48):36846–36855. doi: 10.1074/jbc.M606656200. [DOI] [PubMed] [Google Scholar]
- 195.Jiang J, Wu S, Wang W, Chen S, Peng J, Zhang X, Wu Q. Ectodomain shedding and autocleavage of the cardiac membrane protease corin. J Biol Chem. 2011;286(12):10066–10072. doi: 10.1074/jbc.M110.185082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Herzog C, Haun RS, Ludwig A, Shah SV, Kaushal GP. ADAM10 is the major sheddase responsible for the release of membrane-associated meprin A. J Biol Chem. 2014;289(19):13308–13322. doi: 10.1074/jbc.M114.559088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bozkulak EC, Weinmaster G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol. 2009;29(21):5679–5695. doi: 10.1128/MCB.00406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Stoeck A, Keller S, Riedle S, Sanderson MP, Runz S, Le Naour F, Gutwein P, Ludwig A, Rubinstein E, Altevogt P. A role for exosomes in the constitutive and stimulus-induced ectodomain cleavage of L1 and CD44. Biochem J. 2006;393(Pt 3):609–618. doi: 10.1042/BJ20051013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yokozeki T, Wakatsuki S, Hatsuzawa K, Black RA, Wada I, Sehara-Fujisawa A. Meltrin beta (ADAM19) mediates ectodomain shedding of Neuregulin beta1 in the Golgi apparatus: fluorescence correlation spectroscopic observation of the dynamics of ectodomain shedding in living cells. Genes Cells. 2007;12(3):329–343. doi: 10.1111/j.1365-2443.2007.01060.x. [DOI] [PubMed] [Google Scholar]
- 200.Sun C, Wu MH, Guo M, Day ML, Lee ES, Yuan SY. ADAM15 regulates endothelial permeability and neutrophil migration via Src/ERK1/2 signalling. Cardiovasc Res. 2010;87(2):348–355. doi: 10.1093/cvr/cvq060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sun C, Wu MH, Lee ES, Yuan SY. A disintegrin and metalloproteinase 15 contributes to atherosclerosis by mediating endothelial barrier dysfunction via Src family kinase activity. Arterioscler Thromb Vasc Biol. 2012;32(10):2444–2451. doi: 10.1161/ATVBAHA.112.252205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Miyamae Y, Mochizuki S, Shimoda M, Ohara K, Abe H, Yamashita S, Kazuno S, Ohtsuka T, Ochiai H, Kitagawa Y, Okada Y. ADAM28 is expressed by epithelial cells in human normal tissues And protects from C1q-induced cell death. FEBS J. 2016 doi: 10.1111/febs.13693. [DOI] [PubMed] [Google Scholar]
- 203.Shimoda M, Hashimoto G, Mochizuki S, Ikeda E, Nagai N, Ishida S, Okada Y. Binding of ADAM28 to P-selectin glycoprotein ligand-1 enhances P-selectin-mediated leukocyte adhesion to endothelial cells. J Biol Chem. 2007;282(35):25864–25874. doi: 10.1074/jbc.M702414200. [DOI] [PubMed] [Google Scholar]
- 204.McGinn OJ, English WR, Roberts S, Ager A, Newham P, Murphy G. Modulation of integrin alpha4beta1 by ADAM28 promotes lymphocyte adhesion and transendothelial migration. Cell Biol Int. 2011;35(10):1043–1053. doi: 10.1042/CBI20100885. [DOI] [PubMed] [Google Scholar]
- 205.Kelly K, Hutchinson G, Nebenius-Oosthuizen D, Smith AJ, Bartsch JW, Horiuchi K, Rittger A, Manova K, Docherty AJ, Blobel CP. Metalloprotease-disintegrin ADAM8: expression analysis and targeted deletion in mice. Dev Dyn. 2005;232(1):221–231. doi: 10.1002/dvdy.20221. [DOI] [PubMed] [Google Scholar]
- 206.Weskamp G, Cai H, Brodie TA, Higashyama S, Manova K, Ludwig T, Blobel CP. Mice Lacking the metalloprotease-disintegrin MDC9 (ADAM9) have no evident major abnormalities during development or adult life. Mol Cell Biol. 2002;22(5):1537–1544. doi: 10.1128/mcb.22.5.1537-1544.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Horiuchi K, Weskamp G, Lum L, Hammes HP, Cai H, Brodie TA, Ludwig T, Chiusaroli R, Baron R, Preissner KT, Manova K, Blobel CP. Potential role for ADAM15 in pathological neovascularization in mice. Mol Cell Biol. 2003;23(16):5614–5624. doi: 10.1128/MCB.23.16.5614-5624.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Chen C, Huang X, Sheppard D. ADAM33 is not essential for growth and development and does not modulate allergic asthma in mice. Mol Cell Biol. 2006;26(18):6950–6956. doi: 10.1128/MCB.00646-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Guaiquil VH, Swendeman S, Zhou W, Guaiquil P, Weskamp G, Bartsch JW, Blobel CP. ADAM8 is a negative regulator of retinal neovascularization and of the growth of heterotopically injected tumor cells in mice. J Mol Med (Berl) 2010;88(5):497–505. doi: 10.1007/s00109-010-0591-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Guaiquil V, Swendeman S, Yoshida T, Chavala S, Campochiaro PA, Blobel CP. ADAM9 is involved in pathological retinal neovascularization. Mol Cell Biol. 2009;29(10):2694–2703. doi: 10.1128/MCB.01460-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lubke T, Lena Illert A, von Figura K, Saftig P. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. 2002;11(21):2615–2624. doi: 10.1093/hmg/11.21.2615. [DOI] [PubMed] [Google Scholar]
- 212.Zhou HM, Weskamp G, Chesneau V, Sahin U, Vortkamp A, Horiuchi K, Chiusaroli R, Hahn R, Wilkes D, Fisher P, Baron R, Manova K, Basson CT, Hempstead B, Blobel CP. Essential role for ADAM19 in cardiovascular morphogenesis. Mol Cell Biol. 2004;24(1):96–104. doi: 10.1128/MCB.24.1.96-104.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kurohara K, Komatsu K, Kurisaki T, Masuda A, Irie N, Asano M, Sudo K, Nabeshima Y, Iwakura Y, Sehara-Fujisawa A. Essential roles of meltrin beta (ADAM19) in heart development. Dev Biol. 2004;267(1):14–28. doi: 10.1016/j.ydbio.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 214.Wang X, Chow FL, Oka T, Hao L, Lopez-Campistrous A, Kelly S, Cooper S, Odenbach J, Finegan BA, Schulz R, Kassiri Z, Lopaschuk GD, Fernandez-Patron C. Matrix metalloproteinase-7 and ADAM-12 (a disintegrin and metalloproteinase-12) define a signaling axis in agonist-induced hypertension and cardiac hypertrophy. Circulation. 2009;119(18):2480–2489. doi: 10.1161/CIRCULATIONAHA.108.835488. [DOI] [PubMed] [Google Scholar]
- 215.Glomski K, Monette S, Manova K, De Strooper B, Saftig P, Blobel CP. Deletion of Adam10 in endothelial cells leads to defects in organ-specific vascular structures. Blood. 2011;118(4):1163–1174. doi: 10.1182/blood-2011-04-348557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Farber G, Hurtado R, Loh S, Monette S, Mtui J, Kopan R, Quaggin S, Meyer-Schwesinger C, Herzlinger D, Scott RP, Blobel CP. Glomerular endothelial cell maturation depends on ADAM10, a key regulator of Notch signaling. Angiogenesis. 2018;21(2):335–347. doi: 10.1007/s10456-018-9599-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Alabi R, Glomski K, Haxaire C, Weskamp G, Monette S, Blobel CP. ADAM10-Dependent signaling through notch1 and notch4 controls development of organ-specific vascular beds. Circ Res. 2016 doi: 10.1161/CIRCRESAHA.115.307738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Guo Q, Wang Y, Tripathi P, Manda KR, Mukherjee M, Chaklader M, Austin PF, Surendran K, Chen F. Adam10 mediates the choice between principal cells and intercalated cells in the kidney. J Am Soc Nephrol. 2015;26(1):149–159. doi: 10.1681/ASN.2013070764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.van der Vorst EP, Jeurissen M, Wolfs IM, Keijbeck A, Theodorou K, Wijnands E, Schurgers L, Weber S, Gijbels MJ, Hamers AA, Dreymueller D, Rose-John S, de Winther MP, Ludwig A, Saftig P, Biessen EA, Donners MM. Myeloid A disintegrin and metalloproteinase domain 10 deficiency modulates atherosclerotic plaque composition by shifting the balance from inflammation toward fibrosis. Am J Pathol. 2015;185(4):1145–1155. doi: 10.1016/j.ajpath.2014.11.028. [DOI] [PubMed] [Google Scholar]
- 220.Jana S, Chute M, Hu M, Winkelaar G, Owen CA, Oudit GY, Kassiri Z. ADAM (a disintegrin and metalloproteinase) 15 deficiency exacerbates Ang II (Angiotensin II)-induced aortic remodeling leading to abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2020;40(8):1918–1934. doi: 10.1161/atvbaha.120.314600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Musumeci G, Coleman R, Imbesi R, Magro G, Parenti R, Szychlinska MA, Scuderi R, Cina CS, Castorina S, Castrogiovanni P. ADAM-10 could mediate cleavage of N-cadherin promoting apoptosis in human atherosclerotic lesions leading to vulnerable plaque: a morphological and immunohistochemical study. Acta Histochem. 2014;116(7):1148–1158. doi: 10.1016/j.acthis.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 222.Yang K, Lu L, Liu Y, Zhang Q, Pu LJ, Wang LJ, Zhu ZB, Wang YN, Meng H, Zhang XJ, Du R, Chen QJ, Shen WF. Increase of ADAM10 level in coronary artery in-stent restenosis segments in diabetic minipigs: high ADAM10 expression promoting growth and migration in human vascular smooth muscle cells via Notch 1 and 3. PLoS ONE. 2013;8(12):e83853. doi: 10.1371/journal.pone.0083853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Al-Fakhri N, Wilhelm J, Hahn M, Heidt M, Hehrlein FW, Endisch AM, Hupp T, Cherian SM, Bobryshev YV, Lord RS, Katz N. Increased expression of disintegrin-metalloproteinases ADAM-15 and ADAM-9 following upregulation of integrins alpha5beta1 and alphavbeta3 in atherosclerosis. J Cell Biochem. 2003;89(4):808–823. doi: 10.1002/jcb.10550. [DOI] [PubMed] [Google Scholar]
- 224.Levula M, Paavonen T, Valo T, Pelto-Huikko M, Laaksonen R, Kahonen M, Huovila A, Lehtimaki T, Tarkka M, Mennander AA. A disintegrin and metalloprotease -8 and -15 and susceptibility for ascending aortic dissection. Scand J Clin Lab Invest. 2011;71(6):515–522. doi: 10.3109/00365513.2011.591939. [DOI] [PubMed] [Google Scholar]
- 225.Theodorou K, van der Vorst EPC, Gijbels MJ, Wolfs IMJ, Jeurissen M, Theelen TL, Sluimer JC, Wijnands E, Cleutjens JP, Li Y, Jansen Y, Weber C, Ludwig A, Bentzon JF, Bartsch JW, Biessen EAL, Donners M. Whole body and hematopoietic ADAM8 deficiency does not influence advanced atherosclerotic lesion development, despite its association with human plaque progression. Sci Rep. 2017;7(1):11670. doi: 10.1038/s41598-017-10549-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Pelisek J, Pongratz J, Deutsch L, Reeps C, Stadlbauer T, Eckstein HH. Expression and cellular localization of metalloproteases ADAMs in high graded carotid artery lesions. Scand J Clin Lab Invest. 2012;72(8):648–656. doi: 10.3109/00365513.2012.734394. [DOI] [PubMed] [Google Scholar]
- 227.Holloway JW, Laxton RC, Rose-Zerilli MJ, Holloway JA, Andrews AL, Riaz Z, Wilson SJ, Simpson IA, Ye S. ADAM33 expression in atherosclerotic lesions and relationship of ADAM33 gene variation with atherosclerosis. Atherosclerosis. 2010;211(1):224–230. doi: 10.1016/j.atherosclerosis.2010.02.023. [DOI] [PubMed] [Google Scholar]
- 228.Figarska SM, Vonk JM, van Diemen CC, Postma DS, Boezen HM. ADAM33 gene polymorphisms and mortality. A Prospect Cohort Study PLoS One. 2013;8(7):e67768. doi: 10.1371/journal.pone.0067768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Raitoharju E, Seppala I, Levula M, Kuukasjarvi P, Laurikka J, Nikus K, Huovila AP, Oksala N, Klopp N, Illig T, Laaksonen R, Karhunen PJ, Viik J, Lehtinen R, Pelto-Huikko M, Tarkka M, Kahonen M, Lehtimaki T. Common variation in the ADAM8 gene affects serum sADAM8 concentrations and the risk of myocardial infarction in two independent cohorts. Atherosclerosis. 2011;218(1):127–133. doi: 10.1016/j.atherosclerosis.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 230.Vuohelainen V, Raitoharju E, Levula M, Lehtimaki T, Pelto-Huikko M, Honkanen T, Huovila A, Paavonen T, Tarkka M, Mennander A. Myocardial infarction induces early increased remote ADAM8 expression of rat hearts after cardiac arrest. Scand J Clin Lab Invest. 2011;71(7):553–562. doi: 10.3109/00365513.2011.591424. [DOI] [PubMed] [Google Scholar]
- 231.Melenhorst WB, van den Heuvel MC, Timmer A, Huitema S, Bulthuis M, Timens W, van Goor H. ADAM19 expression in human nephrogenesis and renal disease: associations with clinical and structural deterioration. Kidney Int. 2006;70(7):1269–1278. doi: 10.1038/sj.ki.5001753. [DOI] [PubMed] [Google Scholar]
- 232.Melenhorst WB, van den Heuvel MC, Stegeman CA, van der Leij J, Huitema S, van den Berg A, van Goor H. Upregulation of ADAM19 in chronic allograft nephropathy. Am J Transplant. 2006;6(7):1673–1681. doi: 10.1111/j.1600-6143.2006.01384.x. [DOI] [PubMed] [Google Scholar]
- 233.Lee AC, Lam JK, Shiu SW, Wong Y, Betteridge DJ, Tan KC. Serum level of soluble receptor for advanced glycation end products is associated with a disintegrin and metalloproteinase 10 in type 1 diabetes. PLoS ONE. 2015;10(9):e0137330. doi: 10.1371/journal.pone.0137330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Jowett JB, Okada Y, Leedman PJ, Curran JE, Johnson MP, Moses EK, Goring HH, Mochizuki S, Blangero J, Stone L, Allen H, Mitchell C, Matthews VB. ADAM28 is elevated in humans with the metabolic syndrome and is a novel sheddase of human tumour necrosis factor-alpha. Immunol Cell Biol. 2012;90(10):966–973. doi: 10.1038/icb.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bahia MS, Silakari O. Tumor necrosis factor alpha converting enzyme: an encouraging target for various inflammatory disorders. Chem Biol Drug Des. 2010;75(5):415–443. doi: 10.1111/j.1747-0285.2010.00950.x. [DOI] [PubMed] [Google Scholar]
- 236.DasGupta S, Murumkar PR, Giridhar R, Yadav MR. Current perspective of TACE inhibitors: a review. Bioorg Med Chem. 2009;17(2):444–459. doi: 10.1016/j.bmc.2008.11.067. [DOI] [PubMed] [Google Scholar]
- 237.Moss ML, Sklair-Tavron L, Nudelman R. Drug insight: tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat Clin Pract Rheumatol. 2008;4(6):300–309. doi: 10.1038/ncprheum0797. [DOI] [PubMed] [Google Scholar]
- 238.Mulder GM, Melenhorst WB, Celie JW, Kloosterhuis NJ, Hillebrands JL, Ploeg RJ, Seelen MA, Visser L, van Dijk MC, van Goor H. ADAM17 up-regulation in renal transplant dysfunction and non-transplant-related renal fibrosis. Nephrol Dial Transplant. 2012;27(5):2114–2122. doi: 10.1093/ndt/gfr583. [DOI] [PubMed] [Google Scholar]
- 239.Souza DG, Ferreira FL, Fagundes CT, Amaral FA, Vieira AT, Lisboa RA, Andrade MV, Trifilieff A, Teixeira MM. Effects of PKF242-484 and PKF241-466, novel dual inhibitors of TNF-alpha converting enzyme and matrix metalloproteinases, in a model of intestinal reperfusion injury in mice. Eur J Pharmacol. 2007;571(1):72–80. doi: 10.1016/j.ejphar.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 240.Dell KM, Nemo R, Sweeney WE, Jr, Levin JI, Frost P, Avner ED. A novel inhibitor of tumor necrosis factor-alpha converting enzyme ameliorates polycystic kidney disease. Kidney Int. 2001;60(4):1240–1248. doi: 10.1046/j.1523-1755.2001.00963.x. [DOI] [PubMed] [Google Scholar]
- 241.Sweeney WE, Jr, Hamahira K, Sweeney J, Garcia-Gatrell M, Frost P, Avner ED. Combination treatment of PKD utilizing dual inhibition of EGF-receptor activity and ligand bioavailability. Kidney Int. 2003;64(4):1310–1319. doi: 10.1046/j.1523-1755.2003.00232.x. [DOI] [PubMed] [Google Scholar]
- 242.Long C, Wang Y, Herrera AH, Horiuchi K, Walcheck B. In vivo role of leukocyte ADAM17 in the inflammatory and host responses during E. coli-mediated peritonitis. J Leukoc Biol. 2010;87(6):1097–1101. doi: 10.1189/jlb.1109763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Wong E, Cohen T, Romi E, Levin M, Peleg Y, Arad U, Yaron A, Milla ME, Sagi I. Harnessing the natural inhibitory domain to control TNFα Converting Enzyme (TACE) activity in vivo. Sci Rep. 2016;6:35598. doi: 10.1038/srep35598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Thabet MM, Huizinga TW. Drug evaluation: apratastat, a novel TACE/MMP inhibitor for rheumatoid arthritis. Curr Opin Investig Drugs. 2006;7(11):1014–1019. [PubMed] [Google Scholar]
- 245.Issuree PD, Maretzky T, McIlwain DR, Monette S, Qing X, Lang PA, Swendeman SL, Park-Min KH, Binder N, Kalliolias GD, Yarilina A, Horiuchi K, Ivashkiv LB, Mak TW, Salmon JE, Blobel CP. iRHOM2 is a critical pathogenic mediator of inflammatory arthritis. J Clin Invest. 2013;123(2):928–932. doi: 10.1172/JCI66168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Aktas B, Pozgajova M, Bergmeier W, Sunnarborg S, Offermanns S, Lee D, Wagner DD, Nieswandt B. Aspirin induces platelet receptor shedding via ADAM17 (TACE) J Biol Chem. 2005;280(48):39716–39722. doi: 10.1074/jbc.M507762200. [DOI] [PubMed] [Google Scholar]
- 247.Gomez-Gaviro MV, Gonzalez-Alvaro I, Dominguez-Jimenez C, Peschon J, Black RA, Sanchez-Madrid F, Diaz-Gonzalez F. Structure-function relationship and role of tumor necrosis factor-alpha-converting enzyme in the down-regulation of L-selectin by non-steroidal anti-inflammatory drugs. J Biol Chem. 2002;277(41):38212–38221. doi: 10.1074/jbc.M205142200. [DOI] [PubMed] [Google Scholar]
- 248.Teng M, Wolf M, Ofsthun MN, Lazarus JM, Hernan MA, Camargo CA, Jr, Thadhani R. Activated injectable vitamin D and hemodialysis survival: a historical cohort study. J Am Soc Nephrol. 2005;16(4):1115–1125. doi: 10.1681/ASN.2004070573. [DOI] [PubMed] [Google Scholar]
- 249.Arcidiacono MV, Yang J, Fernandez E, Dusso A. The induction of C/EBPbeta contributes to vitamin D inhibition of ADAM17 expression and parathyroid hyperplasia in kidney disease. Nephrol Dial Transplant. 2015;30(3):423–433. doi: 10.1093/ndt/gfu311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Dusso A, Arcidiacono MV, Yang J, Tokumoto M. Vitamin D inhibition of TACE and prevention of renal osteodystrophy and cardiovascular mortality. J Steroid Biochem Mol Biol. 2010;121(1–2):193–198. doi: 10.1016/j.jsbmb.2010.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Yang WS, Kim HW, Lee JM, Han NJ, Lee MJ, Park SK. 1,25-dihydroxyvitamin D3 causes ADAM10-dependent ectodomain shedding of tumor necrosis factor receptor 1 in vascular smooth muscle cells. Mol Pharmacol. 2015;87(3):533–542. doi: 10.1124/mol.114.097147. [DOI] [PubMed] [Google Scholar]
- 252.Garton K, Gough P, Blobel C, Murphy G, Greaves D, Dempsey P, Raines E. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1) J Biol Chem. 2001;276(41):37993–38001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
- 253.Schulte A, Schulz B, Andrzejewski MG, Hundhausen C, Mletzko S, Achilles J, Reiss K, Paliga K, Weber C, John SR, Ludwig A. Sequential processing of the transmembrane chemokines CX3CL1 and CXCL16 by alpha- and gamma-secretases. Biochem Biophys Res Commun. 2007;358(1):233–240. doi: 10.1016/j.bbrc.2007.04.100. [DOI] [PubMed] [Google Scholar]
- 254.Horiuchi K, Morioka H, Takaishi H, Akiyama H, Blobel CP, Toyama Y. Ectodomain shedding of FLT3 ligand is mediated by TNF-alpha converting enzyme. J Immunol. 2009;182(12):7408–7414. doi: 10.4049/jimmunol.0801931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Kawaguchi N, Horiuchi K, Becherer JD, Toyama Y, Besmer P, Blobel CP. Different ADAMs have distinct influences on Kit ligand processing: phorbol-ester-stimulated ectodomain shedding of Kitl1 by ADAM17 is reduced by ADAM19. J Cell Sci. 2007;120(Pt 6):943–952. doi: 10.1242/jcs.03403. [DOI] [PubMed] [Google Scholar]
- 256.Li N, Wang Y, Forbes K, Vignali KM, Heale BS, Saftig P, Hartmann D, Black RA, Rossi JJ, Blobel CP, Dempsey PJ, Workman CJ, Vignali DA. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007;26(2):494–504. doi: 10.1038/sj.emboj.7601520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, Stevanovic S, Rammensee HG, Steinle A. Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 2008;68(15):6368–6376. doi: 10.1158/0008-5472.CAN-07-6768. [DOI] [PubMed] [Google Scholar]
- 258.Boutet P, Aguera-Gonzalez S, Atkinson S, Pennington CJ, Edwards DR, Murphy G, Reyburn HT, Vales-Gomez M. Cutting edge: the metalloproteinase ADAM17/TNF-alpha-converting enzyme regulates proteolytic shedding of the MHC class I-related chain B protein. J Immunol. 2009;182(1):49–53. doi: 10.4049/jimmunol.182.1.49. [DOI] [PubMed] [Google Scholar]
- 259.Lum L, Wong B, Josien R, Becherer J, Erdjument-Bromage H, Schlöndorff J, Tempst P, Choi Y, Blobel C. Evidence for a role of a tumor necrosis factor-alpha (TNF-alpha)-converting enzyme-like protease in shedding of TRANCE, a TNF family member involved in osteoclastogenesis and dendritic cell survival. J Biol Chem. 1999;274(19):13613–13618. doi: 10.1074/jbc.274.19.13613. [DOI] [PubMed] [Google Scholar]
- 260.Kenny PA, Bissell MJ. Targeting TACE-dependent EGFR ligand shedding in breast cancer. J Clin Invest. 2007;117(2):337–345. doi: 10.1172/JCI29518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Althoff K, Mullberg J, Aasland D, Voltz N, Kallen K, Grotzinger J, Rose-John S. Recognition sequences and structural elements contribute to shedding susceptibility of membrane proteins. Biochem J. 2001;353(Pt 3):663–672. doi: 10.1042/bj3530663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Young J, Yu X, Wolslegel K, Nguyen A, Kung C, Chiang E, Kolumam G, Wei N, Wong WL, DeForge L, Townsend MJ, Grogan JL. Lymphotoxin-alphabeta heterotrimers are cleaved by metalloproteinases and contribute to synovitis in rheumatoid arthritis. Cytokine. 2010;51(1):78–86. doi: 10.1016/j.cyto.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 263.Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T, Yamamoto N, Sasazuki T, Ishizaka Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci U S A. 2008;105(22):7809–7814. doi: 10.1073/pnas.0711241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Lambert DW, Clarke NE, Hooper NM, Turner AJ. Calmodulin interacts with angiotensin-converting enzyme-2 (ACE2) and inhibits shedding of its ectodomain. FEBS Lett. 2008;582(2):385–390. doi: 10.1016/j.febslet.2007.11.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, Hooper NM, Turner AJ. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2) J Biol Chem. 2005;280(34):30113–30119. doi: 10.1074/jbc.M505111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Liu Q, Zhang J, Tran H, Verbeek MM, Reiss K, Estus S, Bu G. LRP1 shedding in human brain: roles of ADAM10 and ADAM17. Mol Neurodegener. 2009;4:17. doi: 10.1186/1750-1326-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Hansen H, Recke A, Reineke U, Von Tresckow B, Borchmann P, Von Strandmann E, Lange H, Lemke H, Engert A. The ectodomain shedding of CD30 is specifically regulated by peptide motifs in its cysteine-rich domains 2 and 5. FASEB J. 2004;18(7):893–895. doi: 10.1096/fj.03-0901fje. [DOI] [PubMed] [Google Scholar]
- 268.Contin C, Pitard V, Itai T, Nagata S, Moreau JF, Dechanet-Merville J. Membrane-anchored CD40 is processed by the tumor necrosis factor-alpha-converting enzyme. Implications for CD40 signaling. J Biol Chem. 2003;278(35):32801–32809. doi: 10.1074/jbc.M209993200. [DOI] [PubMed] [Google Scholar]
- 269.Peng M, Guo S, Yin N, Xue J, Shen L, Zhao Q, Zhang W. Ectodomain shedding of Fcalpha receptor is mediated by ADAM10 and ADAM17. Immunology. 2010;130(1):83–91. doi: 10.1111/j.1365-2567.2009.03215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Määttä J, Sundvall M, Junttila T, Peri L, Laine V, Isola J, Egeblad M, Elenius K. Proteolytic cleavage and phosphorylation of a tumor-associated ErbB4 isoform promote ligand-independent survival and cancer cell growth. Mol Biol Cell. 2006;17(1):67–79. doi: 10.1091/mbc.E05-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Rio C, Buxbaum J, Peschon J, Corfas G. Tumor necrosis factor-alpha-converting enzyme is required for cleavage of erbB4/HER4. J Biol Chem. 2000;275(14):10379–10387. doi: 10.1074/jbc.275.14.10379. [DOI] [PubMed] [Google Scholar]
- 272.Schantl J, Roza M, Van Kerkhof P, Strous G. The growth hormone receptor interacts with its sheddase, the tumour necrosis factor-alpha-converting enzyme (TACE) Biochem J. 2004;377(Pt 2):379–384. doi: 10.1042/bj20031321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Zhang Q, Thomas S, Xi S, Smithgall T, Siegfried J, Kamens J, Gooding W, Grandis J. SRC family kinases mediate epidermal growth factor receptor ligand cleavage, proliferation, and invasion of head and neck cancer cells. Cancer Res. 2004;64(17):6166–6173. doi: 10.1158/0008-5472.CAN-04-0504. [DOI] [PubMed] [Google Scholar]
- 274.Bergmeier W, Piffath CL, Cheng G, Dole VS, Zhang Y, von Andrian UH, Wagner DD. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates GPIbalpha shedding from platelets in vitro and in vivo. Circ Res. 2004;95(7):677–683. doi: 10.1161/01.RES.0000143899.73453.11. [DOI] [PubMed] [Google Scholar]
- 275.Rabie T, Strehl A, Ludwig A, Nieswandt B. Evidence for a role of ADAM17 (TACE) in the regulation of platelet glycoprotein V. J Biol Chem. 2005;280(15):14462–14468. doi: 10.1074/jbc.M500041200. [DOI] [PubMed] [Google Scholar]
- 276.Bender M, Hofmann S, Stegner D, Chalaris A, Bosl M, Braun A, Scheller J, Rose-John S, Nieswandt B. Differentially regulated GPVI ectodomain shedding by multiple platelet-expressed proteinases. Blood. 2010;116(17):3347–3355. doi: 10.1182/blood-2010-06-289108. [DOI] [PubMed] [Google Scholar]
- 277.Chalaris A, Rabe B, Paliga K, Lange H, Laskay T, Fielding CA, Jones SA, Rose-John S, Scheller J. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood. 2007;110(6):1748–1755. doi: 10.1182/blood-2007-01-067918. [DOI] [PubMed] [Google Scholar]
- 278.Althoff K, Reddy P, Voltz N, Rose-John S, Mullberg J. Shedding of interleukin-6 receptor and tumor necrosis factor alpha. Contribution of the stalk sequence to the cleavage pattern of transmembrane proteins. Eur J Biochem. 2000;267(9):2624–2631. doi: 10.1046/j.1432-1327.2000.01278.x. [DOI] [PubMed] [Google Scholar]
- 279.Sommer C, Lee S, Gulseth HL, Jensen J, Drevon CA, Birkeland KI. Soluble leptin receptor predicts insulin sensitivity and correlates with upregulation of metabolic pathways in men. J Clin Endocrinol Metab. 2018;103(3):1024–1032. doi: 10.1210/jc.2017-02126. [DOI] [PubMed] [Google Scholar]
- 280.Zhao XQ, Zhang MW, Wang F, Zhao YX, Li JJ, Wang XP, Bu PL, Yang JM, Liu XL, Zhang MX, Gao F, Zhang C, Zhang Y. CRP enhances soluble LOX-1 release from macrophages by activating TNF-alpha converting enzyme. J Lipid Res. 2011;52(5):923–933. doi: 10.1194/jlr.M015156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Leksa V, Loewe R, Binder B, Schiller HB, Eckerstorfer P, Forster F, Soler-Cardona A, Ondrovicova G, Kutejova E, Steinhuber E, Breuss J, Drach J, Petzelbauer P, Binder BR, Stockinger H. Soluble M6P/IGF2R released by TACE controls angiogenesis via blocking plasminogen activation. Circ Res. 2011;108(6):676–685. doi: 10.1161/CIRCRESAHA.110.234732. [DOI] [PubMed] [Google Scholar]
- 282.Dyczynska E, Sun D, Yi H, Sehara-Fujisawa A, Blobel CP, Zolkiewska A. Proteolytic processing of delta-like 1 by ADAM proteases. J Biol Chem. 2007;282(1):436–444. doi: 10.1074/jbc.M605451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israel A. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5(2):207–216. doi: 10.1016/S1097-2765(00)80417-7. [DOI] [PubMed] [Google Scholar]
- 284.Cho RW, Park JM, Wolff SB, Xu D, Hopf C, Kim JA, Reddy RC, Petralia RS, Perin MS, Linden DJ, Worley PF. mGluR1/5-dependent long-term depression requires the regulated ectodomain cleavage of neuronal pentraxin NPR by TACE. Neuron. 2008;57(6):858–871. doi: 10.1016/j.neuron.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Weskamp G, Schlondorff J, Lum L, Becherer JD, Kim TW, Saftig P, Hartmann D, Murphy G, Blobel CP. Evidence for a critical role of the tumor necrosis factor alpha convertase (TACE) in ectodomain shedding of the p75 neurotrophin receptor (p75NTR) J Biol Chem. 2004;279(6):4241–4249. doi: 10.1074/jbc.M307974200. [DOI] [PubMed] [Google Scholar]
- 286.Chow JP, Fujikawa A, Shimizu H, Suzuki R, Noda M. Metalloproteinase- and gamma-secretase-mediated cleavage of protein-tyrosine phosphatase receptor type Z. J Biol Chem. 2008;283(45):30879–30889. doi: 10.1074/jbc.M802976200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Pruessmeyer J, Martin C, Hess FM, Schwarz N, Schmidt S, Kogel T, Hoettecke N, Schmidt B, Sechi A, Uhlig S, Ludwig A. A disintegrin and metalloproteinase 17 (ADAM17) mediates inflammation-induced shedding of syndecan-1 and -4 by lung epithelial cells. J Biol Chem. 2010;285(1):555–564. doi: 10.1074/jbc.M109.059394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Yang WS, Kim JJ, Lee MJ, Lee EK, Park SK. ADAM17-Mediated ectodomain shedding of toll-like receptor 4 as a negative feedback regulation in lipopolysaccharide-activated aortic endothelial cells. Cell Physiol Biochem. 2018;45(5):1851–1862. doi: 10.1159/000487876. [DOI] [PubMed] [Google Scholar]
- 289.Diaz-Rodriguez E, Montero JC, Esparis-Ogando A, Yuste L, Pandiella A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor alpha-converting enzyme at threonine 735: a potential role in regulated shedding. Mol Biol Cell. 2002;13(6):2031–2044. doi: 10.1091/mbc.01-11-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Hermey G, Sjogaard SS, Petersen CM, Nykjaer A, Gliemann J. Tumour necrosis factor alpha-converting enzyme mediates ectodomain shedding of Vps10p-domain receptor family members. Biochem J. 2006;395(2):285–293. doi: 10.1042/BJ20051364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Bech-Serra JJ, Santiago-Josefat B, Esselens C, Saftig P, Baselga J, Arribas J, Canals F. Proteomic identification of desmoglein 2 and activated leukocyte cell adhesion molecule as substrates of ADAM17 and ADAM10 by difference gel electrophoresis. Mol Cell Biol. 2006;26(13):5086–5095. doi: 10.1128/MCB.02380-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Li Y, Brazzell J, Herrera A, Walcheck B. ADAM17 deficiency by mature neutrophils has differential effects on L-selectin shedding. Blood. 2006;108(7):2275–2279. doi: 10.1182/blood-2006-02-005827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Franzke CW, Tasanen K, Borradori L, Huotari V, Bruckner-Tuderman L. Shedding of collagen XVII/BP180: structural motifs influence cleavage from cell surface. J Biol Chem. 2004;279(23):24521–24529. doi: 10.1074/jbc.M308835200. [DOI] [PubMed] [Google Scholar]
- 294.Maetzel D, Denzel S, Mack B, Canis M, Went P, Benk M, Kieu C, Papior P, Baeuerle PA, Munz M, Gires O. Nuclear signalling by tumour-associated antigen EpCAM. Nat Cell Biol. 2009;11(2):162–171. doi: 10.1038/ncb1824. [DOI] [PubMed] [Google Scholar]
- 295.Maretzky T, Schulte M, Ludwig A, Rose-John S, Blobel C, Hartmann D, Altevogt P, Saftig P, Reiss K. L1 is sequentially processed by two differently activated metalloproteases and presenilin/gamma-secretase and regulates neural cell adhesion, cell migration, and neurite outgrowth. Mol Cell Biol. 2005;25(20):9040–9053. doi: 10.1128/MCB.25.20.9040-9053.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Ruhe JE, Streit S, Hart S, Ullrich A. EGFR signaling leads to downregulation of PTP-LAR via TACE-mediated proteolytic processing. Cell Signal. 2006;18(9):1515–1527. doi: 10.1016/j.cellsig.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 297.Kalus I, Bormann U, Mzoughi M, Schachner M, Kleene R. Proteolytic cleavage of the neural cell adhesion molecule by ADAM17/TACE is involved in neurite outgrowth. J Neurochem. 2006;98(1):78–88. doi: 10.1111/j.1471-4159.2006.03847.x. [DOI] [PubMed] [Google Scholar]
- 298.Fabre-Lafay S, Garrido-Urbani S, Reymond N, Goncalves A, Dubreuil P, Lopez M. Nectin-4, a new serological breast cancer marker, is a substrate for tumor necrosis factor-alpha-converting enzyme (TACE)/ADAM-17. J Biol Chem. 2005;280(20):19543–19550. doi: 10.1074/jbc.M410943200. [DOI] [PubMed] [Google Scholar]
- 299.Singh RJ, Mason JC, Lidington EA, Edwards DR, Nuttall RK, Khokha R, Knauper V, Murphy G, Gavrilovic J. Cytokine stimulated vascular cell adhesion molecule-1 (VCAM-1) ectodomain release is regulated by TIMP-3. Cardiovasc Res. 2005;67(1):39–49. doi: 10.1016/j.cardiores.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 300.Murthy A, Defamie V, Smookler DS, Di Grappa MA, Horiuchi K, Federici M, Sibilia M, Blobel CP, Khokha R. Ectodomain shedding of EGFR ligands and TNFR1 dictates hepatocyte apoptosis during fulminant hepatitis in mice. J Clin Invest. 2010;120(8):2731–2744. doi: 10.1172/JCI42686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Gschwind A, Hart S, Fischer O, Ullrich A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J. 2003;22(10):2411–2421. doi: 10.1093/emboj/cdg231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Sahin U, Blobel CP. Ectodomain shedding of the EGF-receptor ligand epigen is mediated by ADAM17. FEBS Lett. 2007;581(1):41–44. doi: 10.1016/j.febslet.2006.11.074. [DOI] [PubMed] [Google Scholar]
- 303.Schafer B, Gschwind A, Ullrich A. Multiple G-protein-coupled receptor signals converge on the epidermal growth factor receptor to promote migration and invasion. Oncogene. 2004;23(4):991–999. doi: 10.1038/sj.onc.1207278. [DOI] [PubMed] [Google Scholar]
- 304.Wang Y, Sul HS. Ectodomain shedding of preadipocyte factor 1 (Pref-1) by tumor necrosis factor alpha converting enzyme (TACE) and inhibition of adipocyte differentiation. Mol Cell Biol. 2006;26(14):5421–5435. doi: 10.1128/MCB.02437-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Zhu L, Bergmeier W, Wu J, Jiang H, Stalker T, Cieslak M, Fan R, Boumsell L, Kumanogoh A, Kikutani H, Tamagnone L, Wagner D, Milla M, Brass L. Regulated surface expression and shedding support a dual role for semaphorin 4D in platelet responses to vascular injury. Proc Natl Acad Sci U S A. 2007;104(5):1621–1626. doi: 10.1073/pnas.0606344104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Motani K, Kosako H. Activation of stimulator of interferon genes (STING) induces ADAM17-mediated shedding of the immune semaphorin SEMA4D. J Biol Chem. 2018;293(20):7717–7726. doi: 10.1074/jbc.RA118.002175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Ali N, Knauper V. Phorbol ester-induced shedding of the prostate cancer marker transmembrane protein with epidermal growth factor and two follistatin motifs 2 is mediated by the disintegrin and metalloproteinase-17. J Biol Chem. 2007;282(52):37378–37388. doi: 10.1074/jbc.M702170200. [DOI] [PubMed] [Google Scholar]
- 308.Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273(43):27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 309.Slack B, Ma L, Seah C. Constitutive shedding of the amyloid precursor protein ectodomain is up-regulated by tumour necrosis factor-alpha converting enzyme. Biochem J. 2001;357(Pt 3):787–794. doi: 10.1042/bj3570787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Etzerodt A, Maniecki MB, Moller K, Moller HJ, Moestrup SK. Tumor necrosis factor alpha-converting enzyme (TACE/ADAM17) mediates ectodomain shedding of the scavenger receptor CD163. J Leukoc Biol. 2010;88(6):1201–1205. doi: 10.1189/jlb.0410235. [DOI] [PubMed] [Google Scholar]
- 311.Gandhi R, Yi J, Ha J, Shi H, Ismail O, Nathoo S, Bonventre JV, Zhang X, Gunaratnam L. Accelerated receptor shedding inhibits kidney injury molecule-1 (KIM-1)-mediated efferocytosis. Am J Physiol Renal Physiol. 2014;307(2):F205–221. doi: 10.1152/ajprenal.00638.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci U S A. 2007;104(50):19796–19801. doi: 10.1073/pnas.0709805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Zhang Y, Wang Y, Zhou D, Zhang LS, Deng FX, Shu S, Wang LJ, Wu Y, Guo N, Zhou J, Yuan ZY. Angiotensin II deteriorates advanced atherosclerosis by promoting MerTK cleavage and impairing efferocytosis through the AT1R/ROS/p38 MAPK/ADAM17 pathway. Am J Physiol Cell Physiol. 2019;317(4):C776–C787. doi: 10.1152/ajpcell.00145.2019. [DOI] [PubMed] [Google Scholar]
- 314.Kummer MP, Maruyama H, Huelsmann C, Baches S, Weggen S, Koo EH. Formation of Pmel17 amyloid is regulated by juxtamembrane metalloproteinase cleavage, and the resulting C-terminal fragment is a substrate for gamma-secretase. J Biol Chem. 2009;284(4):2296–2306. doi: 10.1074/jbc.M808904200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Alfa Cisse M, Sunyach C, Slack BE, Fisher A, Vincent B, Checler F. M1 and M3 muscarinic receptors control physiological processing of cellular prion by modulating ADAM17 phosphorylation and activity. J Neurosci. 2007;27(15):4083–4092. doi: 10.1523/JNEUROSCI.5293-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Moller-Hackbarth K, Dewitz C, Schweigert O, Trad A, Garbers C, Rose-John S, Scheller J. A disintegrin and metalloprotease (ADAM) 10 and ADAM17 are major sheddases of T cell immunoglobulin and mucin domain 3 (Tim-3) J Biol Chem. 2013;288(48):34529–34544. doi: 10.1074/jbc.M113.488478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Malapeira J, Esselens C, Bech-Serra JJ, Canals F, Arribas J. ADAM17 (TACE) regulates TGFbeta signaling through the cleavage of vasorin. Oncogene. 2011;30(16):1912–1922. doi: 10.1038/onc.2010.565. [DOI] [PubMed] [Google Scholar]
- 318.Naus S, Reipschlager S, Wildeboer D, Lichtenthaler SF, Mitterreiter S, Guan Z, Moss ML, Bartsch JW. Identification of candidate substrates for ectodomain shedding by the metalloprotease-disintegrin ADAM8. Biol Chem. 2006;387(3):337–346. doi: 10.1515/BC.2006.045. [DOI] [PubMed] [Google Scholar]
- 319.Fourie AM, Coles F, Moreno V, Karlsson L. Catalytic activity of ADAM8, ADAM15, and MDC-L (ADAM28) on synthetic peptide substrates and in ectodomain cleavage of CD23. J Biol Chem. 2003;278(33):30469–30477. doi: 10.1074/jbc.M213157200. [DOI] [PubMed] [Google Scholar]
- 320.Gomez-Gaviro M, Dominguez-Luis M, Canchado J, Calafat J, Janssen H, Lara-Pezzi E, Fourie A, Tugores A, Valenzuela-Fernandez A, Mollinedo F, Sanchez-Madrid F, Diaz-Gonzalez F. Expression and regulation of the metalloproteinase ADAM-8 during human neutrophil pathophysiological activation and its catalytic activity on L-Selectin shedding. J Immunol. 2007;178(12):8053–8063. doi: 10.4049/jimmunol.178.12.8053. [DOI] [PubMed] [Google Scholar]
- 321.Bartsch JW, Wildeboer D, Koller G, Naus S, Rittger A, Moss ML, Minai Y, Jockusch H. Tumor necrosis factor-alpha (TNF-alpha) regulates shedding of TNF-alpha receptor 1 by the metalloprotease-disintegrin ADAM8: evidence for a protease-regulated feedback loop in neuroprotection. J Neurosci. 2010;30(36):12210–12218. doi: 10.1523/JNEUROSCI.1520-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Matsuno O, Miyazaki E, Nureki S, Ueno T, Kumamoto T, Higuchi Y. Role of ADAM8 in experimental asthma. Immunol Lett. 2006;102(1):67–73. doi: 10.1016/j.imlet.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 323.Izumi Y, Hirata M, Hasuwa H, Iwamoto R, Umata T, Miyado K, Tamai Y, Kurisaki T, Sehara-Fujisawa A, Ohno S, Mekada E. A metalloprotease-disintegrin, MDC9/meltrin-gamma/ADAM9 and PKCdelta are involved in TPA-induced ectodomain shedding of membrane-anchored heparin-binding EGF-like growth factor. EMBO J. 1998;17(24):7260–7272. doi: 10.1093/emboj/17.24.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Parkin E, Harris B. A disintegrin and metalloproteinase (ADAM)-mediated ectodomain shedding of ADAM10. J Neurochem. 2009;108(6):1464–1479. doi: 10.1111/j.1471-4159.2009.05907.x. [DOI] [PubMed] [Google Scholar]
- 325.Tousseyn T, Thathiah A, Jorissen E, Raemaekers T, Konietzko U, Reiss K, Maes E, Snellinx A, Serneels L, Nyabi O, Annaert W, Saftig P, Hartmann D, De Strooper B. ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the gamma-secretase. J Biol Chem. 2009;284(17):11738–11747. doi: 10.1074/jbc.M805894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Grabowska MM, Sandhu B, Day ML. EGF promotes the shedding of soluble E-cadherin in an ADAM10-dependent manner in prostate epithelial cells. Cell Signal. 2012;24(2):532–538. doi: 10.1016/j.cellsig.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.van Tetering G, van Diest P, Verlaan I, van der Wall E, Kopan R, Vooijs M. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J Biol Chem. 2009;284(45):31018–31027. doi: 10.1074/jbc.M109.006775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Hofmann S, Vogtle T, Bender M, Rose-John S, Nieswandt B. The SLAM family member CD84 is regulated by ADAM10 and calpain in platelets. J Thromb Haemost. 2012;10(12):2581–2592. doi: 10.1111/jth.12013. [DOI] [PubMed] [Google Scholar]
- 329.Fleck D, van Bebber F, Colombo A, Galante C, Schwenk BM, Rabe L, Hampel H, Novak B, Kremmer E, Tahirovic S, Edbauer D, Lichtenthaler SF, Schmid B, Willem M, Haass C. Dual cleavage of neuregulin 1 type III by BACE1 and ADAM17 liberates its EGF-like domain and allows paracrine signaling. J Neurosci. 2013;33(18):7856–7869. doi: 10.1523/JNEUROSCI.3372-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Aghababaei M, Hogg K, Perdu S, Robinson WP, Beristain AG. ADAM12-directed ectodomain shedding of E-cadherin potentiates trophoblast fusion. Cell Death Differ. 2015;22(12):1970–1984. doi: 10.1038/cdd.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Loechel F, Fox JW, Murphy G, Albrechtsen R, Wewer UM. ADAM 12-S cleaves IGFBP-3 and IGFBP-5 and is inhibited by TIMP-3. Biochem Biophys Res Commun. 2000;278(3):511–515. doi: 10.1006/bbrc.2000.3835. [DOI] [PubMed] [Google Scholar]
- 332.Horiuchi K, Le Gall S, Schulte M, Yamaguchi T, Reiss K, Murphy G, Toyama Y, Hartmann D, Saftig P, Blobel CP. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell. 2007;18(1):176–188. doi: 10.1091/mbc.E06-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Asakura M, Kitakaze M, Takashima S, Liao Y, Ishikura F, Yoshinaka T, Ohmoto H, Node K, Yoshino K, Ishiguro H, Asanuma H, Sanada S, Matsumura Y, Takeda H, Beppu S, Tada M, Hori M, Higashiyama S. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat Med. 2002;8(1):35–40. doi: 10.1038/nm0102-35. [DOI] [PubMed] [Google Scholar]
- 334.Najy AJ, Day KC, Day ML. The ectodomain shedding of E-cadherin by ADAM15 supports ErbB receptor activation. J Biol Chem. 2008;283(26):18393–18401. doi: 10.1074/jbc.M801329200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Xie B, Shen J, Dong A, Swaim M, Hackett SF, Wyder L, Worpenberg S, Barbieri S, Campochiaro PA. An Adam15 amplification loop promotes vascular endothelial growth factor-induced ocular neovascularization. FASEB J. 2008;22(8):2775–2783. doi: 10.1096/fj.07-099283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Wei P, Zhao YG, Zhuang L, Ruben S, Sang QX. Expression and enzymatic activity of human disintegrin and metalloproteinase ADAM19/meltrin beta. Biochem Biophys Res Commun. 2001;280(3):744–755. doi: 10.1006/bbrc.2000.4200. [DOI] [PubMed] [Google Scholar]
- 337.Chesneau V, Becherer JD, Zheng Y, Erdjument-Bromage H, Tempst P, Blobel CP. Catalytic properties of ADAM19. J Biol Chem. 2003;278(25):22331–22340. doi: 10.1074/jbc.M302781200. [DOI] [PubMed] [Google Scholar]
- 338.Shirakabe K, Wakatsuki S, Kurisaki T, Fujisawa-Sehara A. Roles of meltrin beta /ADAM19 in the processing of neuregulin. J Biol Chem. 2001;276(12):9352–9358. doi: 10.1074/jbc.M007913200. [DOI] [PubMed] [Google Scholar]
- 339.Yoshikawa A, Aizaki Y, Kusano K, Kishi F, Susumu T, Iida S, Ishiura S, Nishimura S, Shichiri M, Senbonmatsu T. The (pro)renin receptor is cleaved by ADAM19 in the Golgi leading to its secretion into extracellular space. Hypertens Res. 2011;34(5):599–605. doi: 10.1038/hr.2010.284. [DOI] [PubMed] [Google Scholar]
- 340.Mochizuki S, Shimoda M, Shiomi T, Fujii Y, Okada Y. ADAM28 is activated by MMP-7 (matrilysin-1) and cleaves insulin-like growth factor binding protein-3. Biochem Biophys Res Commun. 2004;315(1):79–84. doi: 10.1016/j.bbrc.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 341.Mochizuki S, Tanaka R, Shimoda M, Onuma J, Fujii Y, Jinno H, Okada Y. Connective tissue growth factor is a substrate of ADAM28. Biochem Biophys Res Commun. 2010;402(4):651–657. doi: 10.1016/j.bbrc.2010.10.077. [DOI] [PubMed] [Google Scholar]
- 342.Mochizuki S, Soejima K, Shimoda M, Abe H, Sasaki A, Okano HJ, Okano H, Okada Y. Effect of ADAM28 on carcinoma cell metastasis by cleavage of von Willebrand factor. J Natl Cancer Inst. 2012;104(12):906–922. doi: 10.1093/jnci/djs232. [DOI] [PubMed] [Google Scholar]
- 343.Zou J, Zhu F, Liu J, Wang W, Zhang R, Garlisi CG, Liu YH, Wang S, Shah H, Wan Y, Umland SP. Catalytic activity of human ADAM33. J Biol Chem. 2004;279(11):9818–9830. doi: 10.1074/jbc.M309696200. [DOI] [PubMed] [Google Scholar]