

Summary
Type I polyketide synthases (PKSs) are multidomain, multimodule enzymes capable of producing complex polyketide metabolites. These modules contain an acyltransferase (AT) domain, which selects acyl-CoA substrates to be incorporated into the metabolite scaffold. Herein, we reveal the sequences of three AT domains from a polyketide synthase (TgPKS2) from the apicomplexan parasite Toxoplasma gondii. Phylogenic analysis indicates these ATs (AT1, AT2, and AT3) are distinct from domains in well-characterized microbial biosynthetic gene clusters. Biochemical investigations revealed that AT1 and AT2 hydrolyze malonyl-CoA but the terminal AT3 domain is non-functional. We further identify an “on-off switch” residue that controls activity such that a single amino acid change in AT3 confers hydrolysis activity while the analogous mutation in AT2 eliminates activity. This biochemical analysis of AT domains from an apicomplexan PKS lays the foundation for further molecular and structural studies on PKSs from T. gondii and other protists.
Subject areas: Biological sciences, Biochemistry, Structural biology
Graphical abstract
Highlights
-
•
The apicomplexan parasite T. gondii encodes a type I polyketide synthase, TgPKS2
-
•
TgPKS2 has 3 acyltransferase (AT) domains with predicted selectivity for malonyl-CoA
-
•
AT1 and AT2 hydrolyze malonyl-CoA in vitro, while the AT3 domain is inactive
-
•
A single amino acid serves as an “on-off switch” residue that enables AT activity
Biological sciences; Biochemistry; Structural biology
Introduction
Polyketide synthases (PKSs) are a class of large multifunctional enzymes responsible for the biosynthesis of structurally and functionally diverse polyketide metabolites, ranging from simple fatty acids to complex macrolides (Fischbach and Walsh, 2006; Hertweck, 2009; Khosla et al., 2007; Walsh, 2008). Type I PKSs are complex biosynthetic proteins that contain one or more modules, each consisting of multiple domains that operate in an assembly-line fashion through the selection and incorporation of an acyl-CoA substrate (Keatinge-Clay, 2012). In a minimally complex system, modules contain an acyl carrier protein (ACP) that transports the covalently bound ketide chain between domains, an acyltransferase (AT) domain that recognizes an extending unit and loads it onto the upstream ACP, and a ketosynthase (KS) domain that catalyzes a Claisen-like condensation between the growing ketide chain and a specified extender unit selected by the AT domain (Figure 1) (Chen et al., 2007). Despite the numerous PKS gene clusters and their corresponding metabolites that have been characterized—chiefly from bacterial and fungal sources—genome sequencing suggests that a significant number of natural products have yet to be discovered (Scherlach and Hertweck, 2021).
Figure 1.

Schematic representation of the catalytic cycle of ketide chain elongation in a modular type I polyketide synthase
Chain extension is depicted through transfer between 3 sequential modules (1, 2, and 3) in an assembly-line type manner. This study focuses on the activity of the AT domain, and the role this domain plays in selection of CoA extender units for incorporation into the polyketide metabolite.
Advances in whole genome sequencing and bioinformatics have led to the discovery of previously unidentified biosynthetic gene clusters (BGCs), including those from organisms classically underrepresented in natural product discovery endeavors (Cooke et al., 2017; Ganley and Derbyshire, 2020; Ganley et al., 2017; Shou et al., 2016; Torres et al., 2020). A recent bioinformatic survey found that at least 95% of assembly-line PKSs identified are from bacteria, while only 1% are from eukaryotes (Nivina et al., 2019). Despite this small percentage, modular PKSs are found across eukaryotic lineages, including unicellular protists (Bushkin et al., 2013; Ganley et al., 2017; John et al., 2008; Sasso et al., 2012; Zhu et al., 2002), nematodes (Feng et al., 2021; Shou et al., 2016), arthropods (Brückner et al., 2020; Pankewitz and Hilker, 2008), and even chordates (Cooke et al., 2017), indicating that eukaryotic PKSs may be more phylogenetically widespread than previously assumed. One such example is PKSs from protistan lineages, including dinoflagellates, green algae, and apicomplexan parasites. The structures of many dinoflagellate polyketides have been known for decades, and include exquisitely complex, bioactive molecules (Chekan et al., 2020; Kimura et al., 2015; Nakanishi, 1985; Van Wagoner et al., 2014). While isotopic labeling studies have confirmed the acyl-CoA origin of these metabolites, a dinoflagellate polyketide has never been definitively linked to a specific PKS BGC (Van Wagoner et al., 2014). Even more underexplored among protists are the biosynthetic enzymes in the pathogenic phylum of apicomplexan parasites, including Toxoplasma gondii and Cryptosporidium spp. These disease-causing organisms have been known to harbor large modular PKSs within their genome for nearly two decades; however, there have been few studies to biochemically characterize these enzymes and no polyketide-like metabolites have been identified (Zhu et al., 2002). The evolutionary origin of PKS gene clusters in apicomplexan parasites remains poorly understood, and it remains unknown if these genes arose from horizontal gene transfer, convergent evolution, or another mechanism (John et al., 2008). Interestingly, the available sequences for BGCs from these organisms indicate that they are phylogenetically distinct from bacterial and fungal systems, suggesting the exciting possibility that they generate unique polyketide metabolites. Molecular insights into these PKSs would advance the current understanding of specialized apicomplexan metabolites, as well as the distinct molecular machines responsible for the biosynthesis of such molecules.
As a first step to understand PKSs from apicomplexan, we sought to evaluate the activity of AT domains within an assembly-line PKS from the model apicomplexan parasite T. gondii. Here, we uncover the presence of three AT domains from a T. gondii PKS (TgPKS2), named AT1, AT2, and AT3, and determine the substrate selectivity of these domains. Significantly, we discovered that the AT3 domain within TgPKS2 is inactive and identifies a single amino acid responsible for the varying activity between AT domains from this PKS. Thus, we reveal the presence of an “on-off switch: amino acid residue in AT domains from an apicomplexan parasite PKS that influences enzymatic activity. Broadly, this study lays the groundwork for further investigations into the biosynthetic potential of these pathogens to generate specialized metabolites.
Results and discussion
Identification and sequencing analysis of a type I PKS from T. gondii
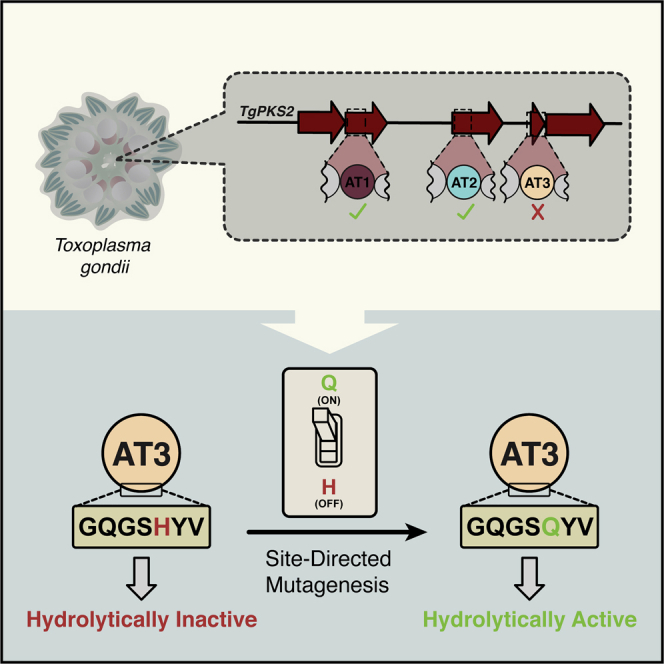
Preliminary analysis of the genome from the type I T. gondii strain GT1 using the biosynthetic gene cluster (BGC) prediction software fungiSMASH revealed the presence of a putative type I PKS, henceforth referred to as TgPKS2 (Figures 2A and S1A) (Medema et al., 2011). Further examination of this cluster revealed the presence of multiple assembly gaps, resulting in an inability to accurately predict protein domain architecture (Figure S1B). To resolve these gaps, we isolated parasite mRNA from cultures of the common laboratory strain type I T. gondii RH propagated in Vero cells for cDNA synthesis (Lau et al., 2016). Ambiguous regions of the cluster spanning the KS and AT domains were amplified with PCR and Sanger sequencing was used to deconvolute the coding sequences. Analysis of these sequences identified 3 putative AT domains, which we have named AT1, AT2, and AT3 (Figure 2B and Table S1). Our analysis additionally revealed the presence of a Type I fatty acid synthase (FAS) (Figure S1C). While Type I PKS and Type I FAS clusters share sequence similarities, the lack of predicted fully reducing domains in TgPKS2 (BlastP analysis) as well as comparison to similar putative PKS clusters from the apicomplexan parasites Eimeria tenella (VEuPathDB: ETH_00005790) and Besnoitia besnoiti (VEuPathDB: BESB_051670), neither of which have assembly gaps, supports the prediction that TgPKS2 is a Type I PKS and not an FAS. While our work clarified the sequence of each TgPKS2 AT domain, further sequencing efforts are necessary to parse out the full coding sequence and domain architecture of the estimated >18 kbp gene cluster.
Figure 2.

Identification of TgPKS2 biosynthetic gene cluster from T. gondii
(A) fungiSMASH analysis of T. gondii GT1 genome identified a Type I PKS, TgPKS2.
(B) Sequencing analysis of PKS2 revealed the presence of 3 AT domains (AT1, AT2, and AT3) each of which possess the GHS(L)G and HAFH fingerprint resides indicative of selectivity for malonyl-CoA.
(C) Phylogenetic comparison of AT domains from bacterial, fungal, and protistan organisms, suggests TgAT domains are distinct (purple). Proteins from previously studied PKS/FAS clusters are labeled with given protein names (i.e. DEBS AT3) or species and protein accession numbers (i.e. Vbra CEM29867.1). More information about the proteins used in tree construction can be found in Table S3.
Importantly, AT domain extender unit (i.e., substrate) selectivity can be predicted based on specificity-conferring sequences at the active site known as “fingerprint residues.” Specifically, the presence of an GHS(X)G motif and HAFH motif, where “X” corresponds to Ile, Val, or Leu, signifies selectivity for malonyl-CoA (Keatinge-Clay, 2017). All 3 TgPKS2 AT domains contain both the consensus sequences GHS(L)G and HAFH, indicating they likely incorporate malonyl-CoA extender units (Figure 2B) (Kalkreuter et al., 2021; Del Vecchio et al., 2003). Despite containing these known active site residues, AT domains from TgPKS2 appear to be distinct from bacterial and fungal AT domains based on phylogenetic analysis (Figure 2C). While the evolutionary origin of TgPKS2 remains an outstanding question, this sequence comparison hints that it may exhibit unexpected properties. To date, no PKS AT domains from any protist have been biochemically characterized, leaving a potentially diverse group of biosynthetic enzymes unexplored.
Evaluation of the hydrolytic activity of TgPKS2 AT domains
In bacterial PKS systems, differences in boundaries for AT domains have been shown to significantly influence catalytic activity. Specifically, previous work has demonstrated that the linker region (LR) between the KS and AT domains is important for proper activity of the AT domain (Liew et al., 2012; Tang et al., 2007; Wu et al., 2002; Yuzawa et al., 2012). Therefore, we carefully examined the KS-AT domain boundaries when rationally designing constructs for our biochemical studies. We found that the TgPKS2 KS-AT LRs were short compared to bacterial LRs and exhibited relatively low sequence similarity, confounding efforts to estimate the N-terminal boundary of the AT domain based on previously crystalized KS-AT didomains (Herbst et al., 2016; Khosla et al., 2007; Tang et al., 2006). We therefore aimed to evaluate both the standalone AT domains (lacking the LR) as well as the KS-AT didomain that contains the predicted LR (Figure S2). Toward this end, we first cloned the TgPKS2 KS-AT2 didomain construct with a C-terminal His-tag for expression in E. coli BL21(DE3), which contains the LR. SDS-PAGE and western blot evaluation of the KS-AT2 didomain (84.9 kDa) after induction revealed a C-terminal protein truncation (∼41 kDa) with no intact didomain in the soluble fraction even after adjusting the expression cells, IPTG concentration, induction time, and induction temperature to minimize protein cleavage. Despite this, we purified the truncated protein to homogeneity with affinity and size exclusion chromatography, and then used MALDI-TOF-MS to confirm the protein contained an intact AT and LR domain, but a truncated KS (Figure S2A and Table S2). We generated a homology model of the KS-AT2 didomain using AlphaFold2 to better understand this truncated protein (Jumper et al., 2021; Mirdita et al., 2021) (Figure S2B). Our model predicts that the C-terminus on the KS domain forms a small anti-parallel beta sheet (Figure 3A). We have termed these proteins that contain truncated KS domains ATX-extended (ATX-E) (Figure S2C). We predicted ATX-E proteins would be active and generated a plasmid to enable expression and purification of the AT2 domain with the extended N-terminus (AT2-E) as well as a shorter version containing the minimal AT2 domain without the LR (AT2). We further cloned, expressed, and purified analogous constructs of AT1 and AT3 (AT1, AT1-E, AT3, and AT3-E) (Figures 3B and S3A–S3C). As a supplement to our homology model, we used circular dichroism (CD) spectroscopy to evaluate the secondary structure of purified AT2-E (Figure S3D). We observed negative bands at 222 and 208 nm in the CD spectrum which is a consistent α-helical protein, in accordance with our model.
Figure 3.

Isolation of TgPKS2 AT and AT-E constructs
(A) Overlaid homology models of 3 TgAT domains from TgPKS2, highlighting the N-terminal extensions present in AT-E constructs.
(B) Depiction of each AT and AT-E domain construct and analysis of purified proteins by SDS-PAGE stained with Coomassie blue. Lane 1: BenchMark protein ladder, lane 2: AT1 (35.8 kDa), Lane 3: AT1-E (41.4 kDa), Lane 4: BenchMark protein ladder, lane 5: AT2 (34.8 kDa), Lane 6: AT2-E (40.5 kDa), Lane 7: BenchMark protein ladder, lane 8: AT3-E (41.5 kDa), Lane 9: AT3 (35.7 kDa).
We investigated the biochemical properties of our purified proteins to better understand the role of the LR in modulating TgPKS2 AT domain substrate specificity and activity. All six AT domain constructs were evaluated for self-acylation activity in the absence of an ACP domain—referred to as hydrolysis—toward malonyl-CoA and methylmalonyl-CoA utilizing an α-ketoglutarate dehydrogenase-coupled assay developed by Khosla and colleagues (Figure 4A) (Dunn et al., 2013). In this assay, the AT-mediated hydrolysis of the acyl-CoA substrate releases free coenzyme A (CoASH), which initiates the condensation of CoA and α-ketoglutarate (α-KG) to form succinyl-CoA. This requires the reduction of nicotinamide adenine dinucleotide (NAD+) to NADH (Figure S4A), which can be continuously monitored by fluorescence emission at 460 nm after excitation at 360 nM and fit to a first-order equation to obtain a reaction rate.
Figure 4.

Evaluation of AT domain hydrolytic activity
(A) Schematic of the α-ketoglutarate dehydrogenase (αKGDH)-coupled kinetic assay used to assess AT domain hydrolysis activity.
(B) Relative rates of hydrolysis between TgPKS2 AT and AT-E domains with malonyl-CoA (150 μM). Significance determined using an unpaired t-test; ns, not significant. Data shown as the average ±standard error of the mean of 3 (AT2/AT2-E and AT3/AT3-E) or 4 (AT1/AT1-E) replicate measurements.
(C) Michaelis-Menten saturation curves for TgPKS2 AT2 (green) and AT2-E (orange) in the presence of malonyl-CoA. Data shown as the average ± standard error of the mean of triplicate measurements. Plots displaying lower concentrations shown in Figures S4F and S4G.
(D) Table of kinetic values for the rates of hydrolysis from this study (TgAT2/TgAT2-E) compared to known kinetic values for other modular type I PKS AT domains as well as from bacterial iterative PKS and FAS systems.
The rates (V/[E]0) of our 6 isolated protein constructs were evaluated in the presence of malonyl-CoA or methylmalonyl-CoA (150 μM) (Figures 4B, S4B, and S4C). We observed that the rates of malonyl-CoA hydrolysis for AT1, AT1-E, AT2, and AT2-E ranged from approximately 3 to 11 min−1 (Figure 4B). The fluorescence signal changes in the presence of methylmalonyl-CoA for these constructs were not above the instrument’s signal to noise (Figure S4B), signifying methylmalonyl-CoA is not a substrate for these proteins. This experimental evidence for malonyl-CoA selectivity is consistent with our bioinformatic predictions. This data additionally revealed that the N-terminal extension did not significantly affect the hydrolysis rate for AT1/AT1-E and AT2/AT2-E (Figure 4B). The malonyl-CoA hydrolysis of these 4 constructs is within the same order of magnitude of other standalone AT domains from the microbial DEBS system, pikromycin synthase (PikA), and rapamycin synthase (RAPS) (Dunn et al., 2013; Stegemann and Grininger, 2021), suggesting the TgPKS2 AT1 and AT2 domains are relatively efficient. Interestingly, when our experimental parameters were applied to AT3 and AT3-E, a hydrolysis reaction was not observed with either malonyl-CoA or methylmalonyl-CoA (Figure S4C). This lack of activity observed for AT3 domain constructs was unexpected, as both proteins contain the malonyl-CoA consensus sequence, and the entire domain has an 89% and 93% amino acid identity to AT1 and AT2, respectively.
Because AT2 and AT2-E showed the highest expression levels (0.5 and 10 mg/L, respectively), these proteins were used to establish Michaelis-Menten kinetic values in the presence of malonyl-CoA (Figure 4C). Prior to these experiments, we confirmed that substrate turnover for both AT2 and AT2-E was dependent on enzyme concentration (0.12–0.48 μM) and linear over time (Figures S4D and S4E). Michaelis-Menten saturation curves for AT2 and AT2-E were generated by incubating each enzyme (100 nM) with varying concentrations of malonyl-CoA (0.5–100 μM). Reaction rates were monitored over time via the previously described αKGDH-coupled assay (Figure 4A). Both AT2 and AT2-E generated comparable kinetic values, with the observed catalytic efficiency (kcat/Km) of hydrolysis with malonyl-CoA being 9.4 μM−1 min−1 for AT2-E and 7.2 μM−1 min−1 for AT2 (Figures 4C, S4F, and S4G). Although slightly higher, these values are consistent with previously reported values from modular type I PKS assembly lines (Figure 4D). Based on these results, the presence of the LR in AT2-E did not significantly influence the Km or catalytic efficiency of the AT2 domain (Figure S4H). Despite predicted structural differences, comparison of the AT2-E and AT2 kinetic parameters to other PKS AT enzymes suggests that they are consistent with previous examples of type I modular PKSs, which often have catalytic efficiencies for hydrolysis between 1 and 10 μM−1 min−1 (Figure 4D).
TgPKS2 AT2 and AT3 hydrolytic activity is dependent on a single amino acid
The lack of observable hydrolytic activity for TgPKS2 AT3 and AT3-E (Figures 4B and S4C) prompted us to investigate a molecular explanation. Because all known residues critical for activity in bacterial AT domains are conserved in AT3, we needed to identify a unique feature outside of the established substrate binding residues (Park et al., 2014). To facilitate this, we investigated how the amino acid sequences of TgPKS2 AT1 and AT2 domains differed from AT3. AT3 shares a high amino acid sequence similarity to these proteins, thus comparison would implicate the fewest number of amino acids as important for activity. Fortuitously, a multiple sequence alignment of TgPKS2 AT1/2/3 displayed a single amino acid disparity within the N-terminal region where a His is observed at residue 72 in AT3 while AT1 and AT2 encode a Gln at this position (Figure 5A). We hypothesized this residue was responsible for conferring AT hydrolysis, which we tested by generating and isolating purified AT2-E-Q72H and AT3-E-H72Q mutant constructs (Figures S5A and S5B). To ensure that this single mutation did not dramatically disrupt protein folding, AT2-E-Q72H was evaluated with CD spectroscopy (Figure S5C). We observed that mutation of Q72H in AT2-E resulted in a complete loss of hydrolysis activity (Figures 5B and S5D), but no significant change in the protein secondary structure. These results are in agreement with our data on wild-type (WT) AT3-E which naturally contains the H72 residue. Furthermore, mutation of H72Q in AT3-E converted an inactive WT protein into one capable of hydrolysis of malonyl-CoA (150 μM), with a V/[E]0 similar to that of the active AT2-E (Figure 5B). Thus, mutation of this single residue allows for catalytic activity of the TgPKS2 AT3 domain. These results demonstrate that Q72 is necessary for hydrolysis activity in TgPKS2 AT domains, and that the presence of H72 renders these proteins non-functional.
Figure 5.

AT hydrolysis activity is controlled by a single amino acid residue
(A) Multiple sequence alignment of TgPKS2 AT domains, indicating the presence of a Q residue in AT1/AT2 as opposed to H in AT3. Homology models generated in AlphaFold2 were overlayed to visualize the 3-dimensional position of this residue in the three TgPKS2 AT domains.
(B) Relative rates of hydrolysis between AT2-E (WT), AT2-E Q72H, AT3-E (WT), and AT3-E H72Q domains with malonyl-CoA (150 μM). Data shown as the average ± the standard error of the mean of triplicate measurements. Significance was determined using an unpaired t-test; ∗∗p < 0.01.
(C) Depiction of the prevalence of histidine versus glutamine in 263 AT domains across bacterial, fungal, and protist species collected from fungiMASH and the MIBiG biosynthetic gene cluster repository (more information including species, protein accession numbers, and known metabolite product are available in Table S4).
The presence of inactive domains within type I PKS modules is common, but examples of inactive AT domains specifically, is rare. In previous reports, the absence of an AT domain catalytic serine residue (GHSX) has suggested the domain may be inactive. At least, one of the AT domains in the assembly lines for bengamide, desertomycin, conglobatin, and gephyronic acid polyketide biosynthesis is predicted to be inactive due to this missing catalytic residue. However, premature chain termination or module skipping has been observed in these systems for reasons that remain poorly understood (Hashimoto et al., 2020; Wenzel et al., 2015; Young et al., 2013; Zhou et al., 2015). Unlike these previous examples, TgAT3 inactivity does not stem from a missing catalytic residue but rather the presence of a His instead of Gln at position 72. It remains to be determined if AT3 prevents chain extension within the context of the full module, or if a yet unknown mechanism exists to aid in chain elongation.
Our findings illuminate an incidence of a naturally occurring “on-off switch” residue where toggling from His to Gln either completely abates or enables enzymatic activity. Such a rare occurrence is especially surprising for a residue outside of the enzyme’s known active site. While the biological reasoning behind the occurrence of H72 in the TgPKS2 AT3 domain remains unknown, an example of this type of variation within a single PKS has been described in KS and KSQ domains where the active site Cys found within a KS domain is replaced with a Gln (Bisang et al., 1999). Specifically, KS domains catalyze a Claisen-like condensation, while the KSQ domains function as a chain initiation factor and decarboxylate the corresponding acyl-CoA substrate (Robbins et al., 2016). However, this residue change is within the active site, not a proximal amino acid as seen in TgPKS2 AT domains. In fact, such an example of a single residue acting in this manner, in the active site or otherwise, has never been described in an AT domain from a PKS to the best of our knowledge. Taken together, this data demonstrate that Q72 is necessary for hydrolysis activity in TgPKS2 AT domains, and mutation of this residue in AT3 enables this activity.
With the significance of Q72 for hydrolytic activity of AT2 and AT3 in TgPKS2 established, we sought to investigate the prevalence of this mutation among PKS-AT domains. A BlastP analysis was performed on 263 AT domains from various species of bacteria, fungi, and protists (Figure 5C and Table S4A). We found that this Gln residue was highly conserved among AT domains in both prokaryotes and eukaryotes. All domains assessed contained either the Gln or His residue, with 253 AT domains having a Gln at this position, and only 10 containing a His (Figure 5C). T. gondii RH strain as well as the related apicomplexan species Cryptosporidium parvum and Cryptosporidium hominis each had one AT with a His, located in the 3rd module or later, but not in every AT domain within these organisms. The occurrence of AT domains with both Gln and His within PKSs suggests there may be strategic placement of the putatively inactive domain. Interestingly, of these 10 His-containing AT domains, the 7 non-apicomplexan examples are found in gene clusters that produce known polyketide metabolites (Table S4B). Based on the structures of these 7 molecules, and in some cases additional in vitro analysis of the modules, these AT domains appear to be active in the context of the full protein (Irschik et al., 2010; Lowell et al., 2017; Park et al., 2014; Romo et al., 2019; Zhou et al., 2019). Among these examples is the BGC for the production of tylactone macrolides, 16-membered ring macrolactones from Micromonospora chalcea, in which the AT in module 6 (JuvEIV, GenBank: ARW71486.1) contains a His at the position of interest. A similar BGC from Streptomyces fradiae produces natural products with the same core 16-membered ring macrolactone; however, the AT domain in module 6 (TylGIV, UniprotKB: O33957_STRFR) of this BGC contains a Gln instead of a His (Castonguay et al., 2008; Lowell et al., 2017).This indicates that this amino acid change does not affect AT catalytic activity in these systems and hints to a possible apicomplexan-specific role for Q/H72.
Upon additional investigation of the 7 His containing AT domains with known metabolites, we found that 3 of the bacterial domains have been shown to be active and exhibit selectivity toward ACP-linked substrates rather than CoA-linked substrates based on both in vitro biochemical characterization and final metabolite identity: zwittermicin, miharamycin A, and amipurimycin (Chan et al., 2006; Emmert et al., 2004; Goto et al., 1982; Park et al., 2014; Romo et al., 2019; Zhang et al., 2020). While at first specificity for ACP-linked substrate presents an intriguing possibility to explain this mutation in TgPKS2, AT3 lacks the sequence known to confer specificity toward ACP-linked extender units and possesses both malonyl-CoA consensus sequences (Figure S5E). Additionally, AT3 contains a conserved Arg residue that is implicated in CoA substrate specificity, whereas a Trp at this position has been shown to be requisite for ACP-bound substrate specificity (Zhang et al., 2020). While the function of TgAT domains that contain H72 is currently unknown, our data align with an “on-off switch” analogy where AT domains with this residue are unable to hydrolyze malonyl-CoA. Based on our data, it is possible this residue influences the structure of the synthesized metabolite in T. gondii. Future studies elucidating both microbial and apicomplexan polyketide structures with PKSs that contain Gln versus His in specific AT domains will be critical to inform a molecular role for this residue. Together, these results highlight the conservation of the Gln residue among both prokaryotic and eukaryotic organisms; however, its importance for enzyme activity and substrate selectivity in the context of the TgPKS2 gene cluster remains unknown.
H72Q mutation of TgPKS2 AT3 results in predicted conformational changes
Due to the lack of hydrolytic activity observed in the TgPKS2 AT3 domain, we questioned if His at position 72 results in a conformational change that could affect activity. Unfortunately, our attempts to crystalize the AT3 domain have been unsuccessful to date; therefore, we evaluated possible structural impacts of this residue with in silico modeling. First, we generated an AT3 H72Q homology models using AlphaFold2 to compare to our previously generated AT3 WT model (Figures 5A and 6). Next, we performed molecular dynamic (MD) simulations with wild-type AT3 and AT3 H72Q to examine relative conformational differences that could influence enzymatic activity. These models were subjected to 100 ns MD simulations and time averaged structures were generated. An overlay of the averaged structures revealed that the H/Q72 residue lies on a loop region directly adjacent to the active site pocket, and that the position of this loop region varies between the WT and mutant domains (Figure 6A). To quantify this observed conformational change within the backbone of the loop region, root mean square deviation (RMSD) values were calculated for the movement of the loop backbone (residues 67–78) of both proteins over 100 ns (Figures 6B, S6A, and S6B) (Darden et al., 1993; Götz et al., 2012; Maier et al., 2015). Based on this analysis, the loop region of the mutant (H72Q), which we have shown to be hydrolytically active (Figure 5B), displayed higher stability in our simulations when compared to the inactive wild-type domain (Figure 6B). The proposed increased stability of this region due to the single amino acid change from His to Gln could enable productive substrate interactions within the active site—something that may not occur in the wild-type protein—thus resulting in our observed lack of hydrolysis. To further investigate the AT domain active site, the distance between the catalytic dyad residues, Ser166 and His272, was determined throughout the MD simulations for wild-type AT3 and AT3 H72Q (Figure 6C). These calculations suggest that the distance between these residues is significantly greater in the wild-type protein (5.19 Å) when compared to AT3 H72Q (4.79 Å), where the active mutant also displayed greater flexibility throughout the simulation. Based on these findings, it is possible that a change in distance and/or orientation of the catalytic dyad residues in AT3 prevents proper catalytic activity.
Figure 6.

Molecular dynamic simulations of WT AT3 and AT3 H72Q
(A) Overlaid representative time-averaged structures for AT3 wild type (magenta) and AT3 H72Q (cyan) derived from the last 80 ns of MD simulations. H/Q72 is highlighted with a yellow box. Starting wild-type structure (transparent) shown. Homology models of proteins obtained from AlphaFold2.
(B) RMSD of the Cα from residues 67–78 (loop region) for AT3-E wild type (magenta and peach) and AT3-E H72Q (cyan and green) over 100 ns. Each simulation was performed in duplicate as displayed. Data suggest greater overall residue movements in this region for AT3-E wild type when compared to AT3-E H72Q.
(C) Boxplots showing the distance between residues of the catalytic dyad (Ser166 and His272) measured from the γ-oxygen of S166 to the Δ-nitrogen of His272 from duplicate simulations calculated for the final 80 ns of MD simulations, every 0.1 ns (n = 800). Significance was calculated using an unpaired t-test, ∗∗∗∗p < 0.0001.
Given the predicted change in the active site pocket based on our MD calculations, we turned to previous studies to evaluate known features of the AT active site. It has been proposed that the formation of an oxyanion hole within the AT active site is necessary for intermediate stabilization through hydrogen bonding of the deprotonated carbonyl oxygen of the malonate group (Ji et al., 2021; Keatinge-Clay et al., 2003; Liew et al., 2012). In the case of TgPKS2 AT3, the backbone amide groups of active site residues Q69 and L167 are predicted to form the core of the oxyanion hole, and both lie on the same loop region as H72 or are adjacent to it (Figure S6C). Our distance measurements indicate that H/Q72 is unlikely to interact directly with the catalytic residues (Figure S6D) and further, their presence (H or Q) does not greatly impact the electrostatic potential around the active site region or H/Q72 (Figure S6E). One possible explanation for the functional importance of position 72 is that the presence of His at this position results in a conformational change in the loop that disrupts the oxyanion hole. Consequently, this disruption could limit the ability of AT3 to hydrolyze substrate. Future structural and biochemical studies on substrate binding, including a crystal structure, will be critical to probing this hypothesis and understanding why position 72 is critical for activity.
Based on our study, the TgPKS2 AT3 domain may be inactive—or have an activity that is currently unforeseeable—however, it is still likely that the full-length enzyme is capable of producing its polyketide metabolite(s), the structures of which may be partially determined by AT3. As evidence toward this, we and others have shown that TgPKS2 is expressed in T. gondii cultures (Figure S6F) and homologs of TgPKS2 are differentially expressed in various life stages of Cryptosporidium spp. (Tandel et al., 2019; Waldman et al., 2020). Additionally, although inactive AT domains within enzymes are uncommon, as discussed above, there is precedence for other inactive domains within modular PKS assembly lines. Importantly, in examples known to date, the PKS still produces polyketide metabolites (Dutta et al., 2014; Garg et al., 2014; Gokhale et al., 1998; Moretto et al., 2019). As one possibility, an inactive AT3 domain could enable early chain elongation termination, effectively truncating the metabolite. Alternatively, His 72 may confer specificity for an undetermined substrate. For the prior, there are numerous examples of polyketide chain termination through noncanonical routes, including KS-mediated chain release via Dieckmann condensation (Little and Hertweck, 2021). We are currently investigating AT3 function within the context of its full module to shed light on these remaining questions. While this study will be critical to better understand TgPKS2, it is obstructed by technical challenges due to the large size of the modules, iterative DNA sequences within the cluster, and the atypical parasite model system. Additionally, structural studies on TgPKS2 may further illuminate the nuances of protein-protein interactions and substrate-induced conformational changes in that enable AT domain product formation, where our evidence to date suggests unique features exist when compared to microbial and fungal systems.
In summary, our investigations into the catalytic activity of AT domains from a PKS in the apicomplexan parasite T. gondii demonstrated the hydrolytic activity and acyl-CoA substrate specificity of three AT domains. Our studies revealed the importance of a non-active site residue at position 72 within TgPKS2 AT domains, which functioned as an “on-off switch” for malonyl-CoA hydrolysis such that mutagenesis of this single AT residue eliminated or imparted activity. While a molecular explanation for the role of this residue in hydrolysis remains elusive, our work highlights the distinct properties of TgPKS2 when compared to well-characterized model systems. Future efforts to reconstitute TgPKS2 or other protistan PKSs in vitro will be critical to uncover the polyketides generated by these organisms. Expanding our understanding of PKS properties that are unique to apicomplexans could advance the identification of novel specialized metabolites and enable small molecule structure predictions for these molecules based on BGC sequences.
Significance
Apicomplexan parasites, including Toxoplasma gondii, represent one of the most widespread pathogenic groups of organisms on Earth. Nearly two decades ago, the capacity of these organisms to produce polyketide natural products was discovered, yet there have been no secondary metabolites isolated or characterized from an apicomplexan parasite and their biosynthetic enzymes have not been biochemically studied. As whole genome sequencing continues to reveal the biosynthetic potential of previously underexplored organisms, biochemical studies into atypical BGCs are increasingly critical to facilitate metabolite identification. This study deconvolutes the coding region of three AT domains from the apicomplexan parasite T. gondii and evaluates their precursor selectivity and enzymatic hydrolysis. In so doing, one of the domains, AT3, was found to contain a single residue change in a highly conserved region that renders it inactive. Mutational studies confirmed that toggling this residue between a His and Gln enables or ablates activity—conferring a binary relationship between enzyme activity and the amino acid at position 72. In all, this biochemical investigation of PKS AT domains from an apicomplexan parasite establishes these organisms as a promising area to discover specialized metabolites.
Limitations of this study
In this study, we have utilized AlphaFold2 to generate homology models of TgPKS2 AT domains to aid in our molecular understanding of these proteins. Although AlphaFold2 allows for highly accurate structural predictions, crystal structures of TgPKS2 AT domains will be needed to validate hypothesis that stem from our models. Furthermore, AT3 could be studied in the context of the full-length TgPKS2 protein to understand its lack of hydrolysis activity. The technical challenges associated with obtaining material from cultures of an obligate intracellular parasite such as T. gondii make determining TgPKS2 activity in this organism arduous. As such, further genetic and biological studies will be necessary to determine the activity of TgAT domains within the context of the producing parasite.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HisProbe™-HRP Conjugate | ThermoFisher Scientific | Cat# 15165 |
| Bacterial and virus strains | ||
| BL21 (DE3) | New England Biolabs | Cat# C2527 |
| NEB5α | New England Biolabs | Cat# C2987 |
| Chemicals, peptides, and recombinant proteins | ||
| ProSignal® Femto ECL Reagent | Prometheus | Cat # 20-302 |
| GoScript Reverse Transcription System | Promega | Cat# A5003 |
| Ni-NTA agarose | Qiagen | Cat# 30210 |
| cOmplete Ultra EDTA-free | Sigma | Cat# 5892953001 |
| Q5® High-Fidelity DNA Polymerase | New England Biolabs | Cat# M0491 |
| Q5® Site-Directed Mutagenesis Kit | New England Biolabs | Cat# E05541 |
| NEBuilder® HiFi DNA Assembly Master Mix | New England Biolabs | Cat# E2621 |
| Malonyl coenzyme A lithium salt | Sigma-Aldrich | Cat# M4263 |
| Methylmalonyl coenzyme A tetralithium salt hydrate | Sigma-Aldrich | Cat# M1762 |
| α-Ketoglutarate Dehydrogenase from porcine heart | Sigma-Aldrich | Cat# K1502-20UN |
| T7 Tag-TgAT1-6×His | This Study | N/A |
| TgAT2-6×His | This Study | N/A |
| T7 Tag-TgAT3-6×His | This Study | N/A |
| T7 Tag-TgAT1-E-6×His | This Study | N/A |
| TgAT2-E-6×His | This Study | N/A |
| T7 Tag-TgAT3-E-6×His | This Study | N/A |
| TgAT2-E(Q72H)-6×His | This Study | N/A |
| T7 Tag-TgAT3-E(H72Q)-6×His | This Study | N/A |
| Critical commercial assays | ||
| Pierce™ Coomassie Plus Bradford Assay | ThermoFisher Scientific | Cat# 23236 |
| Experimental models: Cell lines | ||
| Vero | ATCC | Cat# CCL-81 |
| Experimental models: Organisms/strains | ||
| T. gondii RH MCherry | Laura Knoll (University of Wisconsin Madison) | N/A |
| Oligonucleotides | ||
| AT1 amplification/restriction digest primers AGCCGGATCCATGGTGTGGCTGTTTACTGGT, ATGAAGCTTGCAGCAATTCGGACCTAAGTG |
This paper | N/A |
| AT1-E amplification/restriction digest primers AGCCGGATCCATGGCCGGTGAAAAGACG, ATGAAGCTTACAGCAATTCGGCCCCAA |
This paper | N/A |
| AT2 amplification/restriction digest primers TATAGCCATATGATGGTGTGGTTATTCACGG, TATCGCCTCGAGATCTGCTCCCGAATCAC |
This paper | N/A |
| AT2-E amplification/restriction digest primers TATAGCCATATGATGCATCTTATGGCAGGTG AAAAAACA, TATCGCCTCGAGATCTGCTCCCGAATCAC |
This paper | N/A |
| AT3 amplification/restriction digest primers GCCGGATCCATGGTGTGGTTATTTACCGGGC, ATGAAGCTTCCCAATGTTGGCAAGTGTACGT |
This paper | N/A |
| AT3-E amplification/Gibson assembly primers AGCAAATGGGTCGCGGATCCCTAATGACCGGCGA GAAGAC, CTCGAGTGCGGCCGCAAGCTTAGCTCC ATTGACGTAGGCGG |
This paper | N/A |
| AT2-E(Q72H) Site-directed mutagenesis primers TATTAACGTAATGGGATCCTTGC, TGGCCAAAAGT CTGTACG |
This paper | N/A |
| AT3-E (H72Q) Site-directed mutagenesis primers TCAGGGATCCCAATACGTAAATATG, CCCGTGA ATAGCCAAACC |
This paper | N/A |
|
TgPKS2 Sequencing primers set 1 CCCTCGGGCGGACGATATAA, TGGAGCTGTCAGTTCATTCGGGT |
This paper | N/A |
|
TgPKS2 Sequencing primers set 2 GTCTGAATCTAAGGCCGCCT, CGAGATCGGCGCCCTT |
This paper | N/A |
|
TgPKS2 Sequencing primers set 3 GTTATCCGTTGCGGTAGTGGA, ACACCATC CTGTGGGTCCT |
This paper | N/A |
|
TgPKS2 Sequencing primers set 4 CCGTTGCGGTAGTGGAGA, TGCTACCGC TCCACCGAAC |
This paper | N/A |
|
TgPKS2 cDNA amplification primers AGACCTTGGTGCTTCAGACG, TGTCGGGC AATATCTACGG |
This paper | N/A |
| Recombinant DNA | ||
| pET-21a(+)_TgAT1_No_RBS | Twist Bioscience | N/A |
| pET-21a(+)_TgAT2_No_RBS | Twist Bioscience | N/A |
| pET-21a(+)_TgAT3_No_RBS | Twist Bioscience | N/A |
| pET-21a(+)_TgAT1 | This Study | N/A |
| pET-30b(+)_TgAT2 | This Study | N/A |
| pET-21a(+)_TgAT3 | This Study | N/A |
| pET-21a(+)_TgAT1-E | This study | N/A |
| pET-30b(+)_TgAT2-E | This study | N/A |
| pET-21a(+)_TgAT3-E | This study | N/A |
| pET-30b(+)_TgAT2-E(Q72H) | This Study | N/A |
| pET-21a(+)_TgAT3-E(H72Q) | This Study | N/A |
| pET-21a(+) | EDM Millipore | Cat# 69740 |
| pET-30b(+) | EDM Millipore | Cat# 69910 |
| Software and algorithms | ||
| ChemDraw Professional 18.0 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
| GraphPad Prism 9 | GraphPad software | graphpad.com |
| fungiSMASH 5.0 | (Blin et al., 2019) | https://fungismash.secondarymetabolites.org/#!/start |
| PyMOL version 2.5 | The PyMOL Molecular Graphics System, Schrödinger, LLC. | https://pymol.org/2/ |
| Clustal Omega | (Sievers et al., 2011) | http://www.clustal.org/omega/ |
| iTOL version 6.3.1 | (Letunic and Bork, 2021) | https://itol.embl.de/ |
| AlphaFold2 | (Jumper et al., 2021) | https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb#scrollTo = _sztQyz29DIC |
| MIBiG (Minimum Information about a Biosynthetic Gene cluster) | (Kautsar et al., 2020) | https://mibig.secondarymetabolites.org/ |
| IDT Codon Optimization Tool | Integrated DNA Technologies, Inc. | https://www.idtdna.com/CodonOpt |
| Other | ||
| HiLoad® 26/600 Superdex® 200 pg | GE Healthcare Life Sciences |
Cat#: 28-9893-36 |
| POROS™ HQ 10 μm Column, 4.6 × 100 mm, 1.7 mL | Applied Biosciences/ThermoFisher Scientific | Cat#: 1231226 |
| Black 96 well solid plates | Corning | Cat#: 3915 |
| LightCycler® 480 Multiwell Plate 96, white | Roche | Cat#: 04729692001 |
| Novex™ WedgeWell™ 4 to 20%, Tris-Glycine, 1.0 mm, Mini Protein Gel, 15 well | ThermoFisher Scientific | Cat#: XP04205BOX |
Resource availability
Lead contact
Emily R. Derbyshire (emily.derbyshire@duke.edu).
Materials availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Emily R. Derbyshire (emily.derbyshire@duke.edu).
Experimental model and subject details
Cell and parasite culture
Vero cells used for T. gondii infections were purchased from the American Type Culture Collection (ATCC) and MCherry expressing RH T. gondii parasites were a kind gift from Dr. Laura Knoll (University of Wisconsin Madison). T. gondii parasites were grown in Vero cells maintained in Dulbecco’s Modified Eagle Medium (DMEM) with l-glutamine (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS) (v/v) (Sigma-Aldrich) and 1% antibiotic-antimycotic (Thermo Fisher Scientific) in a standard tissue culture incubator (37°C, 5% CO2).
E. coli culture for protein expression
Competent E. coli strain BL21(DE3) (NEB) was used to produce recombinant proteins. Strains were propagated and chemically competent cells were generated. For protein expression, strains were transformed with the plasmid of interest and cultured in LB media under antibiotic selection (plasmid dependent). In all instances, protein expression was induced with isopropyl β-D-1-thiogalactopyranoside (IPTG) (concentration was protein dependent, described in purification methods). Growth conditions were optimized individually for each protein including temperature and induction period.
Method details
T. gondii RNA extraction and cDNA synthesis
Tachyzoite cultures were maintained as described above. Following a 72 h growth period, parasites were mechanically released from Vero host cell monolayers by scraping and passage through a 20-gauge needle and filtered through a 3μm polycarbonate membrane (ThermoFisher Scientific) to remove host cell debris. Parasites were pelleted at 4000xg for 10 min, after which the media supernatant was discarded. mRNA was extracted from tachyzoites pellets using a Quick-RNA Miniprep kit (Zymo Research) according to the manufacture’s protocol. Briefly, pellets were resuspended in RNA lysis buffer, precipitated with 100% EtOH and treated with DNase I to remove gDNA. Samples were then applied to filtration columns, washed, and eluted in DNase/RNase free water. RNA content was measured using a NanoDrop spectrophotometer (ThermoFisher Scientific). cDNA was synthesized with the following protocol: 500 ng of mRNA was mixed with 1 μL of random primer solution (5 μL total) (ThermoFisher Scientific) heated to 70°C for 5 min and then incubated for 5 min on ice. All enzymes and buffers used in cDNA synthesis were purchased from Promega. Each reaction mixture was then converted to cDNA based on the manufacture’s protocol (Promega).
Sequencing of TgPKS2 acyltransferase assembly gaps
The annotated genome sequence of the strain RH of T. gondii contains several sequencing gaps (GenBank assembly accession: GCA_001593265.1). Therefore, the genome of the closely related T. gondii strain GT1 (GenBank assembly accession: GCA_000149715.2) was used as a reference for our sequencing analysis. Based on the eukaryotic biosynthetic gene cluster prediction software fungiSMASH (Blin et al., 2019), PKS2 within T. gondii GT1 had multiple assembly gaps. To resolve these gaps, regions surrounding and overlapping the assembly gaps were PCR amplified from the RH strain of T. gondii cDNA. mRNA from tachyzoite T. gondii cultures was extracted and cDNA synthesized according to the manufacture’s protocols as described above. cDNA was used as a template for PCR amplification of assembly gap regions surrounding the AT domains of TgPKS2 using Q5 High-Fidelity DNA Polymerase (NEB). Reactions were run on 1% Tris Acetate-EDTA (TAE) agarose gels containing ethidium bromide, after which amplicons were subsequently extracted and purified using the NEB Monarch® DNA Gel Extraction Kit according to the manufactures protocol. Purified samples underwent Sanger sequencing. All PCR reagents and PCR cleanup kits were purchased from NEB. All primers and sequencing analyses were ordered from Eton Bioscience.
Plasmid construction
All protein sequences were codon optimized for E. coli or S. cerevisiae expression as noted using the IDT Codon optimization tool and purchased from Twist Biosciences. Plasmids were assembled via Gibson Assembly or restriction digest cloning as indicated. All enzymes and reagents were purchased from NEB. Codon optimized genes assembled by restriction digestion were amplified with corresponding restriction cut sites and assembled into appropriate vectors using restriction protein digestion and subsequent ligation with T4 ligase. Gibson assembly was performed using the NEBuilder® HiFi DNA Assembly kit (NEB) according to the manufactures protocol. All enzymes and buffers used for construct assembly were purchased from NEB. Proper gene insertion was verified by Sanger sequencing (Eton Bioscience).
Protein expression and purification
All purified proteins were stored in 50 mM triethanolamine, pH 7.5, 150 mM NaCl, 5 mM DTT in 25% glycerol at −80°C. Expression and purification conditions for each protein are described below. Protein concentrations were determined by Bradford assays.
TgAT1:E. coli codon optimized plasmid DNA encoding AT1 was transformed into E. coli BL21(DE3) cells and plated on LB medium supplemented with 100 μg/mL ampicillin. A 1 mL preculture was used to inoculate 8 L of LB medium in baffled Fernbach flasks supplemented with 100 μg/mL ampicillin. The culture was grown to mid-log phase (OD600 ∼ 0.6) at 37°C and induced by addition of 0.5 mM IPTG. Immediately after induction, the temperature was lowered to 18°C and the cells were allowed to express overnight. The cells were harvested by centrifugation (4,300xg, 20 min) and the cell pellet was resuspended in lysis buffer (50 mM KH2PO4, pH 8.0, 200 mM NaCl, 1 mM benzamidine, 5% glycerol (v/v%), 5 mM β-mercaptoethanol, 10 mM imidazole) supplemented with 1 cOmplete protease inhibitor tablet (Roche). Resuspended cells were disrupted using an FB120 Sonic Dismembrator (Fisher Scientific) and cellular debris was removed by centrifugation (4,300xg, 2 h). The histidine-tagged proteins were purified from the supernatant by using Ni-nitrilotriacetic acid agarose (QIAGEN). The bound protein was eluted with a stepwise imidazole gradient (25–150 mM) in lysis buffer at a flow rate of 1.50 mL/min. Pure fractions, as determined by SDS-PAGE were pooled and subjected to anion exchange chromatography using an HQ/10 column (Applied Biosciences). Protein yield was ∼0.00125 mg/L. Pure protein was concentrated using a 10-kDa cut-off centrifugal filter device (Vivaspin) and stored in 25% glycerol at −80°C.
TgAT1-E: E. coli codon optimized plasmid DNA encoding AT1 was transformed into E. coli BL21(DE3) cells and plated on LB medium supplemented with 100 μg/mL ampicillin. A 1 mL preculture was used to inoculate 4 L of LB medium in Ferbach flasks supplemented with 100 μg/mL ampicillin. The culture was grown to mid-log phase (OD600 ∼ 0.6) at 37°C and induced by addition of 0.5 mM IPTG. Immediately after induction, the temperature was lowered to 18°C and the cells were allowed to express overnight. The cells were harvested by centrifugation (4,300xg, 20 min) and the cell pellet was resuspended in lysis buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 1 mg/mL lysozyme, 10 mM imidazole) supplemented with 1 cOmplete protease inhibitor tablet (Roche). Resuspended cells were disrupted using an FB120 Sonic Dismembrator (Fisher Scientific) and cellular debris was removed by centrifugation (4,300xg, 2 h). The histidine-tagged proteins were purified from the supernatant using Ni-nitrilotriacetic acid agarose (QIAGEN). The bound protein was eluted with a stepwise imidazole gradient (25–150 mM) in lysis buffer at a flow rate of 1.50 mL/min. Pure fractions, as determined by SDS-PAGE were pooled and subjected to size exclusion chromatography using a HiLoad 16/600 superdex 200 pg column (Cytiva). Protein yield was ∼0.25 mg/L. Pure protein was concentrated using a 10-kDa cut-off centrifugal filter device (Vivaspin) and stored in 25% glycerol at −80°C.
TgAT2, TgAT2-E WT, and TgAT2-E-Q72H mutant: Constructs of TgPKS2 AT2 (AT2, AT2-E, and AT2-E-Q72H) were expressed and purified as C-terminally hexahistidine-tagged fusion proteins in E. coli BL21(DE3). A 1 mL preculture was used to inoculate 4 L of LB medium in Ferbach flasks supplemented with kanamycin (50 μg/mL). The cultures were grown to an OD600 of ∼0.6 at 37°C with shaking at 250 rpm. Expression was induced by the addition of 500 μM (AT2) or 1 mM (AT2-E and AT2-E Q72H) IPTG, and cells were incubated for 3 (AT2 and AT2-E Q72H) or 18 (AT2-E) hrs at 18°C with shaking at 250 rpm. Cells were then harvested, resuspended (50 mM KH2PO4, pH 8.0, 200 mM NaCl, 5% glycerol, 2 mM imidazole, 1 mg/mL lysozyme, 1 mM Pefabloc®, 1 mM benzamidine, 5 mM β-mercaptoethanol, supplemented with 1 cOmplete protease inhibitor tablet (Roche)), lysed using an FB120 Sonic Dismembrator (Fisher Scientific), and cellular debris was removed by centrifugation (4,300xg, 2 h). The soluble fraction was applied to Ni-nitrilotriacetic acid agarose (QIAGEN), washed with 10 column volumes, and eluted with buffer (50 mM KH2PO4, pH 8.0, 200 mM NaCl) with a stepwise gradient from 2–150 mM imidazole. Fractions were assessed by Western blot analysis with HisProbe (Thermo-Fisher) and SDS-PAGE before combining and concentrating. Yields were ∼0.5 mg/L (AT2), ∼10 mg/L (AT2-E), and ∼9 mg/L (AT2-E-Q72H) and all proteins were purified to >99% homogeneity. Pure protein was concentrated using a 10-kDa cut-off centrifugal filter device (Vivaspin) and stored in 50 mM triethanolamine, pH 8.0, 150 mM NaCl, 5 mM dithiothreitol, 25% glycerol at −80°C.
TgAT3, TgAT3-E WT, and TgAT3-E-H72Q mutant: E. coli codon optimized plasmid DNA encoding AT3 was transformed into E. coli BL21(DE3) cells and plated on LB medium supplemented with 100 μg/mL ampicillin. A 1 mL preculture was used to inoculate 4 L of LB medium in Fernbach flasks supplemented with 100 μg/mL ampicillin. The culture was grown to mid-log phase (OD600 ∼ 0.6) at 37°C and induced by addition of 1.0 mM IPTG. Immediately after induction, the temperature was lowered to 18°C and the cells were allowed to express overnight. The cells were harvested by centrifugation (4,300xg, 20 min) and the cell pellet was resuspended in lysis buffer (50 mM KH2PO4, pH 8.0, 200 mM NaCl, 1 mM benzamidine, 5% glycerol (v/v%), 5 mM β-mercaptoethanol, 10 mM imidazole) supplemented with 1 cOmplete protease inhibitor tablet (Roche). Resuspended cells were disrupted using an FB120 Sonic Dismembrator (Fisher Scientific) and cellular debris was removed by centrifugation (4,300xg, 2 h). The histidine-tagged proteins were purified from the supernatant using Ni-nitrilotriacetic acid agarose (QIAGEN). The bound protein was eluted with a stepwise imidazole gradient (25–150 mM) in lysis buffer at a flow rate of 1.50 mL/min. Pure fractions, as determined by SDS-PAGE were pooled and subjected to subjected to size exclusion chromatography using a HiLoad 16/600 superdex 200 pg column (Cytiva). Yields were ∼3 mg/L (AT3), ∼0.2 mg/L (AT3-E), and ∼0.65 mg/L (AT3-E-H72Q). Pure protein was concentrated using a 10-kDa cut-off centrifugal filter device (Vivaspin) and stored in 25% glycerol at −80°C.
TgAT substrate mediated hydrolysis via an α-ketoglutarate dehydrogenase (αKGDH) coupled assay
Hydrolytic activity of TgAT domains was assessed using an αKGDH coupled assay. Reactions were run with 0.1–0.8 μM purified recombinant TgAT domain with varying malonyl-CoA and methylmalonyl-CoA concentrations (0–150 μM), with AT-less control reactions run in parallel. Assay reaction mixtures were prepared in three different solutions as previously described. All solutions were prepared in 50 mM sodium phosphate buffer, pH 7.6, 10% glycerol, 1 mM TCEP, and 1 mM EDTA. Solution 1: αKGDH, NAD+, TPP, and α-ketoglutaric acid at four times their final concentration; solution 2: the acyl-CoA substrate (malonyl-CoA or methylmalonyl-CoA) prepared at four times its final concentration; and solution 3: TgAT domain prepared at twice its final concentration and 0.1 mg/mL BSA. To each well of a black polystyrene costar 96 well plate (Corning), 25 μL of solution 1, 25 μL of solution 2, and 50 μL were added for a final volume of 100 μL and final concentrations of 50 mM sodium phosphate, pH 7.6, 10% glycerol, 1 mM TCEP, 1 mM EDTA, 0.4 mU/μL αKGDH, 0.4 mM NAD+, 0.4 mM TPP, 2 mM α-ketoglutaric acid, 0.05 mg/mL BSA, and varying concentrations of TgAT acyl-CoA concentrations. Samples were illuminated with a 360 nm filter, while fluorescence was monitored using a 400 nm dichroic mirror with a 460 nm filter on an Envision® Multimode Plate Reader (PerkinElmer). Measurements were taken over a period of 15–20 min. Data analysis and determination of kinetic values were performed using Prism GraphPad software. Raw fluorescence values were trimmed to only include those in the linear range.
Circular dichroism spectroscopy
All CD spectra were collected with an Aviv Biomedical, Inc. Model 435 Circular Dichroism Spectrometer. Spectra were collected over a wavelength range of 350 to 190 nm at 25°C in 10 mM sodium phosphate buffer, pH 7, 50 mM NaCl. Protein concentration was 3 mg/mL (AT2 WT) and 5 mg/mL (AT2-E Q72H). All spectra were subtracted from a background buffer blank.
MALDI-TOF mass spectrometry
Proteins were excised from tris-glycine SDS gels and destained. Remaining gel pieces were reduced with DTT (10 mM), alkylated with iodoacetamide (55 mM), and digested with trypsin (20 μg/mL), as previously described (Shevchenko et al., 2007). Peptides were analyzed in positive mode on Bruker Autoflex Speed LRF MALDI-TOF System.
Site directed mutagenesis (SDM) of TgPKS2 AT2-E-Q72H and AT3-E-H72Q
Point mutations at the Q/H72 positions of AT2 and AT3 were introduced into the pET-30b-AT2-E and pET21a-AT3-E constructs (cloned as previously described) via the Q5® Site-Directed Mutagenesis Kit (NEB) based on the manufactures protocol. Briefly, each construct was amplified with primer sets annealing back-to-back at the 5′ ends containing the single nucleotide change in the forward direction by PCR with Q5-high fidelity polymerase (NEB). Following amplification, the PCR product was directly added to a Kinase-Ligase-DpnI (KLD) enzyme mix for 5 min at room temperature, and subsequently transformed into DH5α chemically competent E. coli cells and plated on LB-agar medium supplemented with 50 μg/mL kanamycin 100 μg/mL (pET-30b-AT2-E) or ampicillin (pET21a-AT3-E). Individual colonies were grown in overnight cultures, after which successful mutagenesis was verified by sequencing (Eton Bioscience).
TgAT domain protein modeling
Homology models of the TgAT domains were created using Deepmind’s AlphaFold2 with MMseqs2 (Jumper et al., 2021). Each extended AT (AT-E) was run separately through the Google Colab notebook, Colab-Fold: AlphaFold2 w/MMseqs2, with templates and a homooligomeric state of 1. All models had at least 1,000 sequences per position. For each, the internal AT structure (residues 59–335) had predicted local Distance Difference Test (lDTT) model confidence scores of greater than 75/100. The N-termini (1–58) and C-termini (336–367) had lower confidence scores ranging from 35–90/100. Models were visualized and aligned in PyMOL(Schrödinger). The AT3-E homology model highlighting the active site region was additionally built using Deepmind’s Alpha-Fold and the generated relaxed model was aligned atom-to-atom with the crystal structure of an acyltransferase crystallized with malonate (PDB ID: 4AMP), the reference structure was then removed excluding the depicted malonate moiety. All molecular representations were generated using PyMOL (Schrödinger).
Phylogenetic tree construction
To generate a maximum likelihood phylogenetic tree of AT protein sequences from various organisms were compiled, including 10 AT sequences from bacteria, 2 from chromerids, 3 from apicomplexans, 6 from fungi, 1 bacterial trans PKS AT, 1 bacterial iterative PKS AT, 2 mammalian FAS AT, and the E. coli FabD FASII sequence as an outgroup. The AT regions were determined by antiSMASH or fungiSMASH. Sequences were aligned using Clustal Omega to construct a maximum likelihood tree and visualized in iTol (Letunic and Bork, 2021; Sievers et al., 2011).
Molecular dynamic simulations
Homology models used for molecular dynamic (MD) simulations of AT3-E were built from residues 62–366 using Alphafold2 (Jumper et al., 2021). MD simulations were all performed with AMBER20 (Götz et al., 2012; Le Grand et al., 2013). The ff14SB force field was applied to each protein which was immersed into a periodic TIP3P water box containing 150 mM NaCl, extended 10 Å from any solute atom (Maier et al., 2015). A three-step minimization was performed for each protein and the Particle Mesh Ewald (PME) method was applied to handle long-range electrostatics (Darden et al., 1993). First, 500 cycles of steepest descent and 500 cycles of conjugate gradient with 50 kcal/mol/Å2 restraint on backbone carbons totaling 1,000 cycles of minimization were performed. Next, the same 1,000 minimization cycles were performed with a 10 kcal/mol/Å2 restraint on backbone carbons. Then 5,000 cycles of minimization were performed without restraint, including 1,000 cycles of steepest descent and 4,000 cycles of conjugate gradient. Afterward, the temperature of each system was elevated from 0 to 300 K in 50 ps with 2.0 kcal/mol/Å2 restraint on backbone carbons, and finally 100 ns NPT (T = 300 K and p = 1 atm) MD simulation was performed with a 2 fs time step. The SHAKE algorithm was used to handle all hydrogen bonded atoms (Ryckaert et al., 1977). Each simulation was repeated with a different random seed to support observations. RMSD for the loop region of AT3 (residues 67–78) was calculated using CPPTRAJ. Briefly, best-fit RMSD was calculated for residues 67–78, meaning coordinates were rotated/translated (to minimize the RMSD to the reference structure) using all atoms in residues 67–78 of frame 1 as a reference, calculated over the entire simulation. RMSF for the loop region of AT3 (residues 67–78) was calculated using CPPTRAJ. Briefly, a best-fit RMSD was calculated for residues 67–78 using all atoms in residues 67–78 of frame 1 as a reference, subsequently RMSF was calculated for 67–78 and output as the mass-weighted average by residue over the simulation (Roe and Cheatham, 2013).
Quantification and statistical analysis
NADH calibration curve generation and concentration quantification
All NADH fluorescence values obtained from the α-KGDH coupled kinetic assay were normalized to an NADH standard curve generate in triplicate measurements over a concentration of 0–10 μM.
Protein concentration quantification
The concentration of all recombinant protein was determined by incubation of samples with Pierce Coomassie Plus Bradford Assay Reagent (ThermoFisher Scientific), followed by measurement at 595 nm (EnVision, Perkin Elmer).
Statistical information
GraphPad Prism 7 software was used for data analysis and visualization. All results are represented as ± the standard error of the means (SEM). Measurements were obtained in 3 or 4 biological replicates as indicated. Statistical tests were performed using an unpaired Student’s t-test as indicated: ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
Acknowledgments
We thank the American Association for the Advancement of Science (AAAS Marion Milligan Mason Award to E.R.D.) and the Camille Dreyfus-Teacher Scholar Award (to E.R.D.). We also thank the Burroughs-Wellcome (to H.K.D.) and the IBIEM graduate training program (to A.M.K.) for fellowship support. We are thankful to Dr. Terrance Oas for his helpful discussions and assistance with circular dichroism spectroscopy. We additionally thank Dr. Peter Silinski for MALDI-TOF assistance.
Author contributions
H.K.D. performed T. gondii culturing, cDNA extractions, and TgPKS2 sequencing analyses. J.G.G. and A.M.K. performed molecular modeling, A.M.K performed molecular dynamics experiments. J.G.G. performed phylogenetic analyses. H.K.D, J.G.G., and A.M.K performed cloning, expression, and purification of proteins. H.K.D., J.G.G., and A.M.K. performed kinetic assays. H.K.D., J.G.G., and E.R.D. designed the experiments, all authors analyzed the data. H.K.D. and E.R.D. wrote the manuscript. All authors commented on the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: June 17, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104443.
Supplemental information
Data and code availability
-
•
Datasets of the sequenced ORFs from TgPKS2 are available as supplementary materials (See Table S1). Peptide list for MALDI-TOF determination of AT2-E from KS-AT2 truncation is available as a supplementary table (Table S2). Amino acid sequence and accession number of AT domains used in phylogenetic tree construction are available as a supplementary table (Table S3). Comparative AT sequences used in this study are additionally available as a supplementary table (Table S4).
-
•
Software used herein are available for either download or online usage.
-
•
This study did not generate any unique code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Bisang C., Long P.F., Cortés J., Westcott J., Crosby J., Matharu A.L., Cox R.J., Simpson T.J., Staunton J., Leadlay P.F. A chain initiation factor common to both modular and aromatic polyketide synthases. Nature. 1999;401:502–505. doi: 10.1038/46829. [DOI] [PubMed] [Google Scholar]
- Blin K., Shaw S., Steinke K., Villebro R., Ziemert N., Lee S.Y., Medema M.H., Weber T. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019;47:W81–W87. doi: 10.1093/nar/gkz310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brückner A., Kaltenpoth M., Heethoff M. De novo biosynthesis of simple aromatic compounds by an arthropod (Archegozetes longisetosus) Proc. Biol. Sci. 2020;287:20201429. doi: 10.1098/rspb.2020.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushkin G.G., Motari E., Carpentieri A., Dubey J.P., Costello C.E., Robbins P.W., Samuelson J. Evidence for a structural role for acid-fast lipids in oocyst walls of Cryptosporidium, Toxoplasma, and Eimeria. mBio. 2013;4 doi: 10.1128/mbio.00387-13. e00387-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castonguay R., Valenzano C.R., Chen A.Y., Keatinge-Clay A., Khosla C., Cane D.E. Stereospecificity of ketoreductase domains 1 and 2 of the tylactone modular polyketide synthase. J. Am. Chem. Soc. 2008;130:11598–11599. doi: 10.1021/ja804453p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan Y.A., Boyne M.T., Podevels A.M., Klimowicz A.K., Handelsman J., Kelleher N.L., Thomas M.G. Hydroxymalonyl-acyl carrier protein (ACP) and aminomalonyl-ACP are two additional type I polyketide synthase extender units. Proc. Natl. Acad. Sci. U S A. 2006;103:14349–14354. doi: 10.1073/pnas.0603748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekan J.R., Fallon T.R., Moore B.S. Biosynthesis of marine toxins. Curr. Opin. Chem. Biol. 2020;59:119–129. doi: 10.1016/j.cbpa.2020.06.009. [DOI] [PubMed] [Google Scholar]
- Chen A.Y., Cane D.E., Khosla C. Structure-based dissociation of a type I polyketide synthase module. Chem. Biol. 2007;14:784–792. doi: 10.1016/j.chembiol.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke T.F., Fischer C.R., Wu P., Jiang T.X., Xie K.T., Kuo J., Doctorov E., Zehnder A., Khosla C., Chuong C.M., Bustamante C.D. Genetic mapping and biochemical basis of yellow feather pigmentation in budgerigars. Cell. 2017;171:427–439.e21. doi: 10.1016/j.cell.2017.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T., York D., Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- Del Vecchio F., Petkovic H., Kendrew S.G., Low L., Wilkinson B., Lill R., Cortés J., Rudd B.A.M., Staunton J., Leadlay P.F. Active-site residue, domain and module swaps in modular polyketide synthases. J. Ind. Microbiol. Biotechnol. 2003;30:489–494. doi: 10.1007/s10295-003-0062-0. [DOI] [PubMed] [Google Scholar]
- Dunn B.J., Cane D.E., Khosla C. Mechanism and specificity of an acyltransferase domain from a modular polyketide synthase. Biochemistry. 2013;52:1839–1841. doi: 10.1021/bi400185v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta S., Whicher J.R., Hansen D.A., Hale W.A., Chemler J.A., Congdon G.R., Narayan A.R.H., Håkansson K., Sherman D.H., Smith J.L., Skiniotis G. Structure of a modular polyketide synthase. Nature. 2014;510:512–517. doi: 10.1038/nature13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmert E.A.B., Klimowicz A.K., Thomas M.G., Handelsman J. Genetics of zwittermicin A production by Bacillus cereus. Appl. Environ. Microbiol. 2004;70:104–113. doi: 10.1128/aem.70.1.104-113.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L., Gordon M.T., Liu Y., Basso K.B., Butcher R.A. Mapping the biosynthetic pathway of a hybrid polyketide-nonribosomal peptide in a metazoan. Nat. Commun. 2021;12:4912–4914. doi: 10.1038/s41467-021-24682-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach M.A., Walsh C.T. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic machinery, and mechanisms. Chem. Rev. 2006;37:3468–3496. doi: 10.1002/chin.200644265. [DOI] [PubMed] [Google Scholar]
- Ganley J.G., Derbyshire E.R. Linking genes to molecules in eukaryotic sources: an endeavor to expand our biosynthetic repertoire. Molecules. 2020;25:625–718. doi: 10.3390/molecules25030625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley J.G., Toro-Moreno M., Derbyshire E.R. Exploring the untapped biosynthetic potential of apicomplexan parasites. Biochemistry. 2017;57:365–375. doi: 10.1021/acs.biochem.7b00877. [DOI] [PubMed] [Google Scholar]
- Garg A., Xie X., Keatinge-Clay A., Khosla C., Cane D.E. Elucidation of the cryptic epimerase activity of redox-inactive ketoreductase domains from modular polyketide synthases by tandem equilibrium isotope exchange. J. Am. Chem. Soc. 2014;136:10190–10193. doi: 10.1021/ja5056998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale R.S., Lau J., Cane D.E., Khosla C. Functional orientation of the acyltransferase domain in a module of the erythromycin polyketide synthase. Biochemistry. 1998;37:2524–2528. doi: 10.1021/bi971887n. [DOI] [PubMed] [Google Scholar]
- Goto T., Toya Y., Ohgi T., Kondo T. Structure of amipurimycin, a nucleoside antibiotic having a novel branched sugar moiety. Tetrahedron Lett. 1982;23:1271–1274. doi: 10.1016/s0040-4039(00)87080-x. [DOI] [Google Scholar]
- Götz A.W., Williamson M.J., Xu D., Poole D., Le Grand S., Walker R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. generalized born. J. Chem. Theor. Comput. 2012;8:1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T., Kozone I., Hashimoto J., Suenaga H., Fujie M., Satoh N., Ikeda H., Shin-ya K. Identification, cloning and heterologous expression of biosynthetic gene cluster for desertomycin. J. Antibiot. 2020;73:650–654. doi: 10.1038/s41429-020-0319-0. [DOI] [PubMed] [Google Scholar]
- Herbst D.A., Jakob R.P., Zähringer F., Maier T. Mycocerosic acid synthase exemplifies the architecture of reducing polyketide synthases. Nature. 2016;531:533–537. doi: 10.1038/nature16993. [DOI] [PubMed] [Google Scholar]
- Hertweck C. The biosynthetic logic of polyketide diversity. Angew. Chem. Int. Ed. 2009;48:4688–4716. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- Irschik H., Kopp M., Weissman K.J., Buntin K., Piel J., Müller R. Analysis of the sorangicin gene cluster reinforces the utility of a combined phylogenetic/retrobiosynthetic analysis for deciphering natural product assembly by trans-AT PKS. Chembiochem. 2010;11:1840–1849. doi: 10.1002/cbic.201000313. [DOI] [PubMed] [Google Scholar]
- Ji H., Shi T., Liu L., Zhang F., Tao W., Min Q., Deng Z., Bai L., Zhao Y., Zheng J. Computational studies on the substrate specificity of an acyltransferase domain from salinomycin polyketide synthase. Catal. Sci. Technol. 2021;11:6782–6792. doi: 10.1039/d1cy00284h. [DOI] [Google Scholar]
- John U., Beszteri B., Derelle E., Van de Peer Y., Read B., Moreau H., Cembella A. Novel insights into evolution of protistan polyketide synthases through phylogenomic analysis. Protist. 2008;159:21–30. doi: 10.1016/j.protis.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkreuter E., Bingham K.S., Keeler A.M., Lowell A.N., Schmidt J.J., Sherman D.H., Williams G.J. Computationally-guided exchange of substrate selectivity motifs in a modular polyketide synthase acyltransferase. Nat. Commun. 2021;12:2193. doi: 10.1038/s41467-021-22497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kautsar S.A., Blin K., Shaw S., Navarro-Muñoz J.C., Terlouw B.R., Van Der Hooft J.J.J., Van Santen J.A., Tracanna V., Suarez Duran H.G., Pascal Andreu V., et al. MIBiG 2.0: a repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020;48:D454–D458. doi: 10.1093/nar/gkz882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keatinge-Clay A.T. The structures of type I polyketide synthases. Nat. Prod. Rep. 2012;29:1050–1073. doi: 10.1039/c2np20019h. [DOI] [PubMed] [Google Scholar]
- Keatinge-Clay A.T. The uncommon enzymology of cis-acyltransferase assembly lines. Chem. Rev. 2017;117:5334–5366. doi: 10.1021/acs.chemrev.6b00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keatinge-Clay A.T., Shelat A.A., Savage D.F., Tsai S.C., Miercke L.J.W., O’Connell J.D., Khosla C., Stroud R.M. Catalysis, specificity, and ACP docking site of Streptomyces coelicolor malonyl-CoA:ACP Transacylase. Structure. 2003;11:147–154. doi: 10.1016/s0969-2126(03)00004-2. [DOI] [PubMed] [Google Scholar]
- Khosla C., Tang Y., Chen A.Y., Schnarr N.A., Cane D.E. Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu. Rev. Biochem. 2007;76:195–221. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- Kimura K., Okuda S., Nakayama K., Shikata T., Takahashi F., Yamaguchi H., Skamoto S., Yamaguchi M., Tomaru Y. RNA sequencing revealed numerous polyketide synthase genes in the harmful dinoflagellate Karenia mikimotoi. PLoS One. 2015;10 doi: 10.1371/journal.pone.0142731. e0142731–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau Y.L., Lee W.C., Gudimella R., Zhang G.P., Ching X.T., Razali R., Aziz F., Anwar A., Fong M.Y. Deciphering the draft genome of Toxoplasma gondii RH strain. PLoS One. 2016;11:e0157901. doi: 10.1371/journal.pone.0157901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand S., Götz A.W., Walker R.C. SPFP: Speed without compromise - a mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013;184:374–380. doi: 10.1016/j.cpc.2012.09.022. [DOI] [Google Scholar]
- Letunic I., Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–W296. doi: 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew C.W., Nilsson M., Chen M.W., Sun H., Cornvik T., Liang Z.X., Lescar J. Crystal structure of the acyltransferase domain of the iterative polyketide synthase in enediyne biosynthesis. J. Biol. Chem. 2012;287:23203–23215. doi: 10.1074/jbc.m112.362210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little R.F., Hertweck C. Chain release mechanisms in polyketide and non-ribosomal peptide biosynthesis. Nat. Prod. Rep. 2021;39:163–205. doi: 10.1039/d1np00035g. [DOI] [PubMed] [Google Scholar]
- Lowell A.N., Demars M.D., Slocum S.T., Yu F., Anand K., Chemler J.A., Korakavi N., Priessnitz J.K., Park S.R., Koch A.A., et al. Chemoenzymatic total synthesis and structural diversification of tylactone-based macrolide antibiotics through late-stage polyketide assembly, tailoring, and C-H functionalization. J. Am. Chem. Soc. 2017;139:7913–7920. doi: 10.1021/jacs.7b02875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theor. Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema M.H., Blin K., Cimermancic P., De Jager V., Zakrzewski P., Fischbach M.A., Weber T., Takano E., Breitling R. AntiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39:339–346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirdita M., Ovchinnikov S., Steinegger M. ColabFold - making protein folding accessible to all. BioRxiv. 2021 doi: 10.1038/s41592-022-01488-1. Preprint at. 08.15.456425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretto L., Heylen R., Holroyd N., Vance S., Broadhurst R.W. Modular type I polyketide synthase acyl carrier protein domains share a common N-terminally extended fold. Sci. Rep. 2019;9:2325–2416. doi: 10.1038/s41598-019-38747-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K. The chemistry of brevetoxins: a review. Toxicon. 1985;23:473–479. doi: 10.1016/0041-0101(85)90031-5. [DOI] [PubMed] [Google Scholar]
- Nivina A., Yuet K.P., Hsu J., Khosla C. Evolution and diversity of assembly-line polyketide synthases. Chem. Rev. 2019;119:12524–12547. doi: 10.1021/acs.chemrev.9b00525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankewitz F., Hilker M. Polyketides in insects: ecological role of these widespread chemicals and evolutionary aspects of their biogenesis. Biol. Rev. 2008;83:209–226. doi: 10.1111/j.1469-185x.2008.00040.x. [DOI] [PubMed] [Google Scholar]
- Park H., Kevany B.M., Dyer D.H., Thomas M.G., Forest K.T. A polyketide synthase acyltransferase domain structure suggests a recognition mechanism for its hydroxymalonyl-acyl carrier protein substrate. PLoS One. 2014;9:e110965. doi: 10.1371/journal.pone.0110965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins T., Kapilivsky J., Cane D.E., Khosla C. Roles of conserved active site residues in the ketosynthase domain of an assembly line polyketide synthase. Biochemistry. 2016;55:4476–4484. doi: 10.1021/acs.biochem.6b00639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe D.R., Cheatham T.E. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theor. Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Romo A.J., Shiraishi T., Ikeuchi H., Lin G.M., Geng Y., Lee Y.H., Liem P.H., Ma T., Ogasawara Y., Shin-Ya K., et al. The amipurimycin and miharamycin biosynthetic gene clusters: unraveling the origins of 2-aminopurinyl peptidyl nucleoside antibiotics. J. Am. Chem. Soc. 2019;141:14152–14159. doi: 10.1021/jacs.9b03021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Sasso S., Pohnert G., Lohr M., Mittag M., Hertweck C. Microalgae in the postgenomic era: a blooming reservoir for new natural products. FEMS Microbiol. Rev. 2012;36:761–785. doi: 10.1111/j.1574-6976.2011.00304.x. [DOI] [PubMed] [Google Scholar]
- Scherlach K., Hertweck C. Mining and unearthing hidden biosynthetic potential. Nat. Commun. 2021;12:3864–3912. doi: 10.1038/s41467-021-24133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A., Tomas H., Havli J., Olsen J.V., Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2007;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Shou Q., Feng L., Long Y., Han J., Nunnery J.K., Powell D.H., Butcher R.A. A hybrid polyketide-nonribosomal peptide in nematodes that promotes larval survival. Nat. Chem. Biol. 2016;12:770–772. doi: 10.1038/nchembio.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegemann F., Grininger M. Transacylation kinetics in fatty acid and polyketide synthases and its sensitivity to point mutations. ChemCatChem. 2021;13:2771–2782. doi: 10.1002/cctc.202002077. [DOI] [Google Scholar]
- Tandel J., English E.D., Sateriale A., Gullicksrud J.A., Beiting D.P., Sullivan M.C., Pinkston B., Striepen B. Life cycle progression and sexual development of the apicomplexan parasite Cryptosporidium parvum. Nat. Microbiol. 2019;4:2226–2236. doi: 10.1038/s41564-019-0539-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y., Kim C.Y., Mathews I.I., Cane D.E., Khosla C. The 2.7-Å crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc. Natl. Acad. Sci. U S A. 2006;103:11124–11129. doi: 10.1073/pnas.0601924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y., Chen A.Y., Kim C.Y., Cane D.E., Khosla C. Structural and mechanistic analysis of protein interactions in module 3 of the 6-deoxyerythronolide B synthase. Chem. Biol. 2007;14:931–943. doi: 10.1016/j.chembiol.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres J.P., Lin Z., Winter J.M., Krug P.J., Schmidt E.W. Animal biosynthesis of complex polyketides in a photosynthetic partnership. Nat. Commun. 2020;11:2882. doi: 10.1038/s41467-020-16376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wagoner R.M., Satake M., Wright J.L.C. Polyketide biosynthesis in dinoflagellates: what makes it different? Nat. Prod. Rep. 2014;31:1101–1137. doi: 10.1039/c4np00016a. [DOI] [PubMed] [Google Scholar]
- Waldman B.S., Schwarz D., Wadsworth M.H., Saeij J.P., Shalek A.K., Lourido S. Identification of a master regulator of differentiation in Toxoplasma. Cell. 2020;180:359–372.e16. doi: 10.1016/j.cell.2019.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C.T. The chemical versatility of natural-product assembly lines. Acc. Chem. Res. 2008;41:4–10. doi: 10.1021/ar7000414. [DOI] [PubMed] [Google Scholar]
- Wenzel S.C., Hoffmann H., Zhang J., Debussche L., Haag-Richter S., Kurz M., Nardi F., Lukat P., Kochems I., Tietgen H., et al. Production of the bengamide class of marine natural products in myxobacteria: biosynthesis and structure-activity relationships. Angew. Chem. Int. Ed. 2015;54:15560–15564. doi: 10.1002/anie.201508277. [DOI] [PubMed] [Google Scholar]
- Wu N., Cane D.E., Khosla C. Quantitative analysis of the relative contributions of donor acyl carrier proteins, acceptor ketosynthases, and linker regions to intermodular transfer of intermediates in hybrid polyketide synthases. Biochemistry. 2002;41:5056–5066. doi: 10.1021/bi012086u. [DOI] [PubMed] [Google Scholar]
- Young J., Stevens D.C., Carmichael R., Tan J., Rachid S., Boddy C.N., Müller R., Taylor R.E. Elucidation of gephyronic acid biosynthetic pathway revealed unexpected sam-dependent methylations. J. Nat. Prod. 2013;76:2269–2276. doi: 10.1021/np400629v. [DOI] [PubMed] [Google Scholar]
- Yuzawa S., Kapur S., Cane D.E., Khosla C. Role of a conserved arginine residue in linkers between the ketosynthase and acyltransferase domains of multimodular polyketide synthases. Biochemistry. 2012;51:3708–3710. doi: 10.1021/bi300399u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Ji H., Ali I., Deng Z., Bai L., Zheng J. Structural and biochemical insight into the recruitment of acyl carrier protein-linked extender units in ansamitocin biosynthesis. ChemBioChem. 2020;21:1309–1314. doi: 10.1002/cbic.201900628. [DOI] [PubMed] [Google Scholar]
- Zhu G., LaGier M.J., Stejskal F., Millership J.J., Cai X., Keithly J.S. Cryptosporidium parvum: the first protist known to encode a putative polyketide synthase. Gene. 2002;298:79–89. doi: 10.1016/s0378-1119(02)00931-9. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Murphy A.C., Samborskyy M., Prediger P., Dias L.C., Leadlay P.F. Iterative mechanism of macrodiolide formation in the anticancer compound conglobatin. Chem. Biol. 2015;22:745–754. doi: 10.1016/j.chembiol.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.Z., Zhu H.J., Lin L.P., Zhang X., Ge H.M., Jiao R.H., Tan R.X. Dalmanol biosyntheses require coupling of two separate polyketide gene clusters. Chem. Sci. 2019;10:73–82. doi: 10.1039/c8sc03697g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Datasets of the sequenced ORFs from TgPKS2 are available as supplementary materials (See Table S1). Peptide list for MALDI-TOF determination of AT2-E from KS-AT2 truncation is available as a supplementary table (Table S2). Amino acid sequence and accession number of AT domains used in phylogenetic tree construction are available as a supplementary table (Table S3). Comparative AT sequences used in this study are additionally available as a supplementary table (Table S4).
-
•
Software used herein are available for either download or online usage.
-
•
This study did not generate any unique code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.