

Abstract
Aberrant glucocorticoid signaling via glucocorticoid receptors (GR) plays a critical role in alcohol use disorder (AUD). Acute alcohol withdrawal and protracted abstinence in dependent rats are associated with increased GR signaling and changes in GR-mediated transcriptional activity in the rat central nucleus of the amygdala (CeA). The GR antagonist mifepristone decreases alcohol consumption in dependent rats during acute withdrawal and protracted abstinence. Regulation of CeA synaptic activity by GR is currently unknown. Here, we utilized mifepristone and the selective GR antagonist CORT118335 (both at 10 μM) as pharmacological tools to dissect the role of GR on GABA transmission in male, adult Sprague-Dawley rats using slice electrophysiology. We subjected rats to chronic intermittent alcohol vapor exposure for 5–7 weeks to induce alcohol dependence. A subset of dependent rats subsequently underwent protracted alcohol withdrawal for 2 weeks, and air-exposed rats served as controls. Mifepristone reduced the frequency of pharmacologically-isolated spontaneous inhibitory postsynaptic currents (sIPSC) in the CeA (medial subdivision) without affecting postsynaptic measures in all groups, suggesting decreased GABA release with the largest effect in dependent rats. CORT118335 did not significantly alter GABA transmission in naive, but decreased sIPSC frequency in dependent rats. Similarly, mifepristone decreased amplitudes of evoked inhibitory postsynaptic potentials only in dependent rats and during protracted withdrawal. Collectively, our study provides insight into regulation of CeA GABAergic synapses by GR. Chronic ethanol enhances the efficiency of mifepristone and CORT118335, thus highlighting the potential of drugs targeting GR as a promising pharmacological avenue for the treatment of AUD.
Keywords: Mifepristone, Glucocorticoid receptors, Central amygdala, Alcohol use disorder, GABA, Synaptic transmission, Electrophysiology, sIPSC, eIPSP
1. Introduction
Alcohol use disorder (AUD) is a chronic relapsing disease characterized by compulsive and excessive alcohol intake accompanied by a loss of control over consumption and negative emotional state during abstinence (Koob and Mason, 2016). The pathogenesis of AUD is complex and multi-faceted involving hyperactive stress systems (Koob and Schulkin, 2019; Tunstall et al., 2017). Indeed, activation of the hypothalamic-pituitary-adrenal (HPA) axis is a critical mechanism mediating responses to both stress and alcohol (Stephens and Wand, 2012). Acute alcohol activates the HPA axis in alcohol-naïve subjects elevating cortisol (human) and corticosterone (rodents) levels (Richardson et al., 2008; Wemm and Sinha, 2019). Importantly, chronic alcohol exposure induces long-lasting alterations of HPA reactivity (Richardson et al., 2008), glucocorticoid release as well as glucocorticoid receptor (GR) function – mechanisms which likely contribute to continued alcohol consumption and increased vulnerability to relapse during abstinence (Koob, 2021). Basal cortisol levels are elevated in binge drinkers, and the expected cortisol rise in the HPA response to acute alcohol is blunted in heavy drinkers compared to light/moderate social drinkers. Alcohol stimulates cortisol levels in both dependent animals and humans and alcohol withdrawal is associated with increased cortisol levels, which remain elevated even during sustained abstinence compared to healthy subjects, suggesting persistent HPA neuroadaptations (Blaine and Sinha, 2017; Wemm and Sinha, 2019).
Glucocorticoid receptors (GR) are broadly expressed in reward and stress circuits (Cintra et al., 1994) and are involved in regulating physiological and pathological processes including learning and memory, and neuroendocrine negative feedback regulation (Garabedian et al., 2017; Lu et al., 2006). GR display low affinity for glucocorticoids, are activated by high circulating glucocorticoid levels as during stress or alcohol withdrawal, and exert their activity via both genomic and non-genomic mechanisms (Joëls, 2018).
The GR antagonist mifepristone is approved by the FDA for the treatment of hyperglycemia in Cushing’s syndrome (Fleseriu and Petersenn, 2015) and there is accumulating preclinical and clinical evidence for its therapeutic potential in the treatment of AUD (Donoghue et al., 2016; Higley et al., 2012; Howland, 2013; Vendruscolo et al., 2015). In preclinical models of alcohol dependence across different species, mifepristone reduced alcohol consumption (Repunte-Canonigo et al., 2015; Vendruscolo et al., 2015), prevented the development of alcohol dependence-induced escalation of alcohol drinking (Somkuwar et al., 2017; Vendruscolo et al., 2012), reduced escalated alcohol drinking during protracted alcohol abstinence – all in male rats- (Vendruscolo et al., 2012) and also reduced heavy alcohol drinking in non-human primates (Jimenez et al., 2020). Notably, mifepristone treatment also substantially reduced alcohol-cued craving, reduced alcohol consumption, and ameliorated liver enzymes in humans with AUD (Vendruscolo et al., 2015).
The central nucleus of the amygdala (CeA) is critically involved in mediating negative emotional behaviors associated with alcohol dependence (Koob, 2021; Roberto et al., 2020). The CeA is a predominantly GABAergic nucleus, which is highly sensitive to both acute and chronic effects of alcohol across species in that alcohol heightens CeA GABA transmission (Augier et al., 2018; Herman et al., 2016; Jimenez et al., 2019; Kirson et al., 2021; Roberto et al., 2004; Roberto et al., 2003). Importantly, increased GR activity in the CeA has been suggested to play a key role in alcohol-dependence associated behaviors (Repunte-Canonigo et al., 2015; Vendruscolo et al., 2015; Vendruscolo et al., 2012). Mifepristone injected directly in the CeA reduced yohimbine-induced reinstatement of ethanol-seeking in non-dependent rats (Simms et al., 2012) and alcohol drinking in dependent rats (Vendruscolo et al., 2015). Acute effects of mifepristone on CeA synaptic transmission representing a potential mechanism of action for its therapeutic efficacy in the treatment of AUD have not yet been explored. Here, we used mifepristone as a pharmacological tool to dissect GR regulation of CeA activity on the cellular level under basal alcohol-naïve conditions and to examine how GR-recruitment by alcohol dependence enhances CeA GABA transmission. We utilized chronic, intermittent ethanol vapor exposure over 5–7 weeks to induce alcohol dependence in adult, male Sprague Dawley rats and ex vivo slice electrophysiology to assess the effects of mifepristone on both spontaneous and locally evoked GABAA-receptor mediated synaptic transmission in the CeA and its interaction with acute and chronic ethanol. Specifically, we examined mifepristone and ethanol effects on CeA GABA transmission in rats that were either alcohol-naïve, alcohol-dependent, or undergoing protracted (2 weeks) alcohol withdrawal, thus providing detailed insights into chronic alcohol-induced neuroadaptions at CeA GABAergic synapses and how chronic alcohol and protracted withdrawal affect their regulation by GR. Lastly, mifepristone has been shown to bind also to molecular targets other than GR (e.g., progesterone or androgen receptors), thus the effects observed after mifepristone administration may result from interaction with multiple target sites. We thus also detemined the effects of a selective GR antagonist lacking activity at progesterone receptors (CORT118335; McGinn et al., 2021) on CeA GABAergic synapses in naïve and dependent rats enabling a more selective assessment of the role of GR in regulating CeA activity in a rat model of AUD.
2. Materials and methods
2.1. Animals
All procedures were approved by the Scripps Research Institutional Animal Care and Use Committee (IACUC) and are in line with the ARRIVE guidelines and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. For this study, we used 67 adult, male Sprague-Dawley rats (Charles River, Raleigh, NC) weighing 225–250 g upon arrival. Rats were group-housed (2–3 per cage) in standard plastic cages in a temperature- and humidity-controlled room under a reverse 12 h/12 h light/dark cycle and were given ad libitum access to food and water.
2.2. Chronic intermittent alcohol exposure
Alcohol dependence was modeled as previously described (Khom et al., 2020a, 2020b; Tunstall et al., 2019; Varodayan et al., 2017a; Vendruscolo and Roberts, 2014). Briefly, we exposed 37 rats to 14 h/day alcohol vapor (10 h air) using the standard chronic intermittent alcohol inhalation method in their home cages and determined blood alcohol levels (BAL) 1–2 times/week from tail-blood samples (average BAL: 158 ± 4 mg/dl). We subjected subsequently 14 dependent rats to protracted alcohol withdrawal for 2 weeks. Naïve controls were treated similarly, except that they were exposed to air only.
2.3. Ex vivo slice electrophysiology
Preparation of acute brain slices and electrophysiological recordings were performed as previously described (Khom et al., 2020a; Roberto et al., 2010; Roberto et al., 2004; Tunstall et al., 2019; Varodayan et al., 2018). In brief, we decapitated deeply anesthetized rats (3–5% isoflurane anesthesia), rapidly isolated their brains in ice-cold oxygenated high-sucrose cutting solution (composition in mM: 206 sucrose, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 1.2 NaH2PO4, 26 NaHCO3, 5 glucose, and 5 HEPES and cut 300 μm thick coronal slices containing the medial subdivision of CeA using a Leica VT 1000S. Dependent rats were euthanized during the last hour of their daily alcohol vapor exposure and trunk blood was collected to determine terminal BALs. Thus, animals were intoxicated at the time of euthanasia. However, as brain slices were incubated in ethanol-free solutions, the slices underwent acute in vitro withdrawal (1–10h).
2.4. Intracellular recording of evoked responses
We incubated CeA slices in an interface configuration for 15–20 min, and then completely submerged and continuously superfused them (flow rate of 2–4 ml/min) with 95% O2/5% CO2 equilibrated artificial cerebrospinal fluid (aCSF) of the following composition (all in mM): 130 NaCl, 3.5 KCl, 1.25 NaH2PO4, 1.5mMMgSO4•7H2O, 2.0 CaCl2; 24 NaHCO3, and 10 glucose. We recorded from 57 CeA (medial subdivision) neurons using sharp micropipettes filled with 3 M KCl in discontinuous current-clamp mode (Kirson et al., 2020; Tunstall et al., 2019) holding them near their resting membrane potential (−80.2 ± 0.7 mV). We acquired data with an Axoclamp-2A amplifier (Axon Instruments, Foster City, CA) and stored them for off-line analysis with pClamp software (Axon Instruments). Hyperpolarizing and depolarizing current steps (200 pA increments, 750 msec duration) were applied to generate I-V curves to monitor cell health and allow electrophysiological cell-typing (Beyeler and Dabrowska, 2020; Chieng et al., 2006). Recordings were obtained randomly from all neuronal cell-types commonly found in the CeA (Beyeler and Dabrowska, 2020; Chieng et al., 2006). We evoked GABAA-receptor mediated inhibitory postsynaptic potentials (eIPSPs) by stimulating locally within the CeA through a bipolar stimulating electrode and superfusing the slices with aCSF containing blockers of glutamate-mediated synaptic transmission [20 μM 6,7-dinitroquinoxaline-2,3-dione (DNQX) and 30 μM DL-2-amino-5-phosphonovalerate (AP-5)] and GABAB-receptors (1 μM CGP55845A). To determine the synaptic response parameters for each cell, we performed an input-output (I-O) protocol (Roberto et al., 2010; Roberto et al., 2004; Roberto et al., 2003; Tunstall et al., 2019) consisting of a range of five current stimulations (50–250 mA; 0.125 Hz), starting at the minimum current required to elicit an EPSP up to the strength required to elicit the maximum subthreshold amplitude. These stimulus strengths were maintained throughout the entire duration of the experiment. We examined paired-pulse facilitation (PPR) in each neuron using paired stimuli at 50 and 100 ms inter-stimulus interval (Andreasen and Hablitz, 1994; Logrip et al., 2017; Roberto et al., 2004). The stimulus strength was adjusted such that the amplitude of the first eIPSP was ~50% of the maximal amplitude determined by the I/O protocol. We calculated PPR as the amplitude of the second eIPSP over that of the first eIPSP and drug-induced changes in PPR generally reflect presynaptic effects such that an increase in PPR suggests a decrease in neurotransmitter (GABA) release (Roberto et al., 2004). All measures were taken before drug superfusion (control) and during drug superfusion. We performed washout-experiments (30–40 min) for a subset of CeA neurons to exclude potential run-down of neuronal responses in the presence of mifepristone, however, due to the lipophilic nature of mifepristone only partial recovery could be achieved.
2.5. Whole-cell patch clamp of spontaneous action-potential dependent and action-potential independent GABAA-receptor mediated synaptic transmission
We recorded from 114 neurons located in the medial subdivision of the CeA in the whole-cell voltage clamp configuration as previously described (Khom et al., 2020a; Kirson et al., 2021; Steinman et al., 2020). Neurons were visualized with infrared differential interference contrast optics using a 40× water-immersion objective (Olympus BX51WI), and a CCD camera (EXi Aqua, QImaging). All recordings were performed in gap-free acquisition mode with a 20 kHz sampling rate and 10 kHz low-pass filtering using a MultiClamp700B amplifier, Digidata 1440A, and pClamp 10 software (MolecularDevices).
We pulled patch pipettes from borosilicate glass (3–5 mΩ, King Precision) and filled them with a KCl-based internal solution composed of (all in mM): 135 KCl, 5 EGTA, 5 MgCl2, 10 HEPES, 2 Mg-ATP, and 0.2 Na-GTP (pH = 7.2–7.4 adjusted with 1 M KOH, 290–300 mOsm). We pharmacologically isolated action-potential dependent GABAA-receptor mediated spontaneous postsynaptic inhibitory currents (sIPSCs) by adding antagonists of glutamate-mediated transmission and GABAB-receptors to the bath solution as described above. To assess action-potential independent GABAA-receptor mediated transmission (mIPSCs), we added 0.5 μM tetrodotoxin (TTX) to block TTX-sensitive Na + channels and thus generation and propagation of action potentials. All neurons were held at −60 mV. Data are derived from neurons with access resistance (Ra) ≤15 MΩ and a maximum change of Ra of ≥20% during the recording as monitored by 10 mV pulses applied approximately every minute.
2.6. Drugs
We purchased mifepristone from Cayman Chemical (Ann Arbor, MI), ethanol from Remet (La Mirada, CA) and tetrodotoxin from Biotium (Hayward, CA). Chemicals other than these were purchased from Sigma Aldrich (St. Louis, MO). CORT118335 was provided by Corcept Therapeutics (Menlo Park, CA). We prepared stock solutions of AP-5, CGP55845A and tetrodotoxin in distilled water, while DNQX, mifepristone and CORT118335 were dissolved in 100% DMSO. All drugs including ethanol were applied to the bath solution to achieve the final desired concentrations. The final DMSO concentration did not exceed 0.15% in any of the recordings given DMSO effects at higher concentrations on neuronal viability or interference with synaptic transmission (Nakahiro et al., 1992; Tsvyetlynska et al., 2005; Zhang et al., 2017).
2.7. Data and statistical analysis
We analyzed data obtained from intracellular recordings using Clampfit 10.2 (Molecular Devices) and frequencies, amplitudes, rise and decay times of s/mIPSCs from whole-cell recordings in a semi-automatical mode utilizing Mini Analysis 5.1 software (Synaptosoft, Leonia, NJ). For each event (s/mIPSCs) the minimum amplitude was set to >5pA which was visually confirmed. For further analysis, we averaged s/mIPSC characteristics in 3 min bins. We used GraphPad Prism 9.0 software (GraphPad Software, San Diego, CA) for plotting of results and statistical analysis. Data are presented as means ± standard error of the mean (SEM) of either normalized data or raw values. Data sets for each experimental condition are derived from ≥5 rats, and n denotes the total number of recorded cells and N the number of used animals (exact values are indicated for each experiment). Data were pooled per experimental condition. To avoid pseudo-replication due to collecting multiple samples from individual animals, we set a cut-off ≤3 data points for a specific data set from single animals. The criterion for statistical significance for all experiments was set to P < 0.05. We used Kolmogorov-Smirnov tests to probe for normal data distribution (Gaussian), non-parametric Wilcoxon signed-rank tests to assess per se drug effects, and either Kruskcal-Wallis tests or - given Gaussian distributions- one-way ANOVAs or repeated measures two-way ANOVAs with appropriate post hoc mean comparisons to detect significant differences between treatments.
3. Results
3.1. Mifepristone decreases spontaneous, network-dependent GABA transmission in the CeA
We induced alcohol dependence in male Sprague Dawley rats by exposing them to chronic, intermittent ethanol vapor exposure (CIE) for 5–7 weeks. A subgroup of alcohol-dependent rats (Dep) was further subjected to 2 weeks of protracted withdrawal (WD). Air-exposed age-matched rats were used as alcohol-naïve controls (naïve). We utilized ex vivo slice electrophysiology to examine the effects of mifepristone on GABA transmission in the medial subdivision of the CeA given that elevated CeA GABA signaling is a critical hallmark of alcohol dependence across species (Roberto et al., 2020). Mifepristone levels in the CeA following systemic drug application are not known; thus, we used 10 μM mifepristone based on previous in vitro/ex vivo studies (Dalm et al., 2019; Paul et al., 2021; Suzuki et al., 2019).
First, we recorded pharmacologically isolated GABAA-receptor mediated, spontaneous action-potential dependent postsynaptic inhibitory currents (sIPSCs) in CeA neurons from all experimental groups and found that mifepristone (10 μM) significantly decreased sIPSC frequency irrespective of experimental group as illustrated in Fig. 1A–C (naïve: 86.3 ± 4.4%, P = 0.0166, n = 14 vs. Dep: 67.3 ± 5.8%, P = 0.0010, n = 12 vs. WD: 78.4 ± 4.8%, P = 0.0009, n = 14 Wilcoxon signed-rank tests) suggesting that mifepristone decreases presynaptic CeA GABA release.
Fig. 1.

Mifepristone decreases spontaneous synaptic transmission in the CeA. (A) Representative sIPSC recordings onto CeA neurons from the indicated groups (upper panel) and during superfusion with mifepristone (10 μM, lower panel) are shown. (B) Cumulative probability histograms comparing sIPSC frequency during baseline control (grey line) and during superfusion with mifepristone (red line) from individual CeA neurons from naïve (left), dependent (middle) and withdrawn rats, respectively are shown. Differences between conditions were calculated using the Kolmogorov–Smirnov test. Bars represent normalized means ± SEM of sIPSC (C) frequencies, (D) amplitudes, and (E) current kinetics in the presence of 10 μM mifepristone. Asterisks indicate significant differences from baseline assessed by Wilcoxon signed-rank tests (*) = P < 0.05, (***) = P < 0.001. Differences between groups were assessed with a Kruskal-Wallis test with ($) = P < 0.05 and a post hoc Dunn’s correction for multiple comparisons (#) = P < 0.05. Naïve: n = 14 (N = 10), Dep: n = 12 (N = 9), WD: n = 14 (N = 6). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Mifepristone did not significantly alter sIPSC amplitudes (naïve: 96.2 ± 4.6%, P = 0.3910; Dep: 92.4 ± 4.9%, P = 0.2661; WD: 95.2 ± 7.0%, P = 0.2958, Fig. 1D), rise (naïve: 101.3 ± 1.9%, P = 0.4631; Dep: 102.9 ± 1.9%, P = 0.1763; WD: 100.1 ± 3.1%, P = 0.9515, Fig. 1E) or decay times (naïve: 100.6 ± 4.2%, P > 0.9999; Dep: 98.6 ± 5.8%, P = 0.7334; WD: 99.3 ± 7.9%, P = 0.5016, all Wilcoxon signed-rank tests compared to baseline, Fig. 1E) in any experimental group indicating that postsynaptic GABAA-receptor function is not affected by mifepristone. Notably, mifepristone effects on CeA sIPSC frequency occurred rapidly (within <15 min) suggesting the involvement of non-genomic GR signaling (see Fig. S1 for time courses).
Chronic intermittent alcohol vapor exposure increased CeA sIPSC frequency indicative of heighthened CeA GABA release (one-way ANOVA, F(2,103) = 3.116, P = 0.0485, see Table 1 for details on sIPSC characteristics) lasting into protracted withdrawal and also significantly enhanced the inhibitory efficiency of mifepristone (Kruskal-Wallis test, P = 0.0414). Specifically, the effects of mifepristone on CeA sIPSC frequency were significantly larger in dependent rats than in naïve rats (P = 0.0356) and importantly, mifepristone effects in rats undergoing protracted withdrawal did not significantly differ from those in dependent rats (P = 0.3840, Dunn’s multiple comparison, Fig. 1C), indicative of more pronounced regulation of CeA synaptic activity by glucocorticoids resulting from chronic alcohol exposure.
Table 1.
Summary of baseline s/mIPSC characteristics and PPR from all recorded CeA neurons in this study.
| Frequency (HZ) | Amplitude (pA) | Rise time (ms) | Decay time (ms) | n | |
|---|---|---|---|---|---|
| sIPSCs | |||||
| Naïve | 1.41 ± 0.14 | 73.6 ± 4.4 | 2.68 ± 0.05 | 8.95 ± 0.59 | 41 |
| Dep. | 1.73 ± 0.15 | 63.7 ± 3.2 | 2.81 ± 0.06 | 9.13 ± 0.60 | 42 |
| WD | 2.12 ± 0.33 | 72.7 ± 5.8 | 2.53 ± 0.06 | 7.36 ± 0.51 | 23 |
| mIPSCs | |||||
| Naïve | 1.13 ± 0.37 | 71.3 ± 5.6 | 2.5 ± 0.15 | 7.0 ± 0.9 | 8 |
| PPR | |||||
| Naïve | 1.08 ± 0.2 | 8 | |||
| Dep | 0.82 ± 0.1 | 10 | |||
| WD | 0.71 ± 0.1 | 12 | |||
A one-way ANOVA reavealed a main effect of chronic alcohol exposure on sIPSC frequencies (F (2,103); P = 0.0485) and rise times (F (2, 103) = 5.015; P = 0.0083).
Data for each condition were derived from ≥5 animals and n denotes the number of recorded cells. Note that these recordings have been performed with 0.05% DMSO in the bath solution.
To gain mechanistic insight into how mifepristone decreases CeA GABA transmission, we further examined its effect on action-potential-independent (in the presence of tetrodotoxin [TTX]) miniature inhibitory postsynaptic currents (mIPSC) in naïve rats, and found that mifepristone also significantly decreased mIPSC frequency (76.5 ± 8.9% compared to baseline with Wilcoxon signed-rank tests, P = 0.0313, n = 7 see also Fig. S2) without altering postsynaptic measures including mIPSC amplitude (93.6 ± 7.4%, P = 0.6875) or kinetics (mIPSC rise time: 105.1 ± 7.0%, P = 0.5781 or mIPSC decay time: 90.2 ± 7.8%, P = 0.3750), indicating reduced vesicular CeA GABA release from presynaptic terminals as mechanism of action of mifepristone in naïve rats. Baseline characteristics of mIPSCs from naïve rats are summarized in Table 1.
3.2. Mifepristone blunts the effects of acute ethanol on CeA network activity after chronic intermittent alcohol exposure
We have reported that acute ethanol increases the frequency of CeA sIPSCs in naïve, male rats and this effect is unaffected by alcohol dependence or protracted alcohol withdrawal indicative of a lack of tolerance to acute ethanol, as previously reported (Roberto et al., 2003, 2004; Varodayan et al., 2017b; Khom et al., 2020a, 2020b; Kirson et al., 2021). Here, we recapitulated this experiment by demonstrating that CeA GABA transmission in the presence of 44 mM ethanol is similarly elevated in male, naïve and alcohol-dependent rats as well as during protracted withdrawal (sIPSC frequency in naïve: 141.8 ± 11.6%, P = 0.0010, n = 11; Dep.: 138.9 ± 10.9%, P = 0.0049, n = 11; WD: 149.1 ± 18.7 P = 0.0391, n = 9, Wilcoxon signed-rank tests compared to baseline control; Kruskal-Wallis test between treatments: P > 0.05, data not shown).
Next, we studied whether and how mifepristone interferes with this acute ethanol-induced GABA release. Thus, we applied 44 mM ethanol in the continued presence of mifepristone to a subset of neurons from all groups (see Fig. 2A) and found that mifepristone blunts the effects of acute ethanol only in alcohol-dependent rats and rats during protracted withdrawal (Fig. 2).
Fig. 2.

Mifepristone blunts effects of acute ethanol (44 mM) on action-potential dependent CeA GABA release after chronic ethanol exposure (A) Representative sIPSC recordings from CeA neurons from the indicated treatment groups before (upper panel), and during superfusion with 10 μM mifepristone (middle panel) and 10 μM mifepristone +44 mM ethanol (lower panel) are illustrated. Scatter dot diagrams depict averaged effects (means ± S.E.M.) of mifepristone and subsequent co-application of mifepristone and ethanol on sIPSC (B) frequency, (C) amplitude, (D) rise and (E) decay time. Significant differences from baseline (indicated as dashed line) were calculated using Wilcoxon signed-rank tests with (*) = P < 0.05, (**) = P < 0.01, (***) = P < 0.001. Differences between mifepristone vs. mifepristone plus ethanol effects in the respective treatment groups were calculated using two-way repeated-measures ANOVA with šidák multiple comparison testing ($ = P < 0.01). naïve: n = 12 (N = 8), Dep: n = 9 (N = 9), WD: n = 13 (N = 6).
Specifically, we found that despite a per se effect of mifepristone (sIPSC frequency: 81.0 ± 5.0%, P = 0.0093, n = 12) in naïve rats, ethanol significantly increased sIPSC frequency in the continued presence of mifepristone (mifepristone + ethanol: 99.74 ± 9.8%, P = 0.8501, n = 12 both compared to baseline with Wilcoxon signed-rank tests; Fig. 2B), suggesting that mifepristone does not alter the acute effects of ethanol.
Mifepristone also displayed a marked per se effect on CeA sIPSC frequency in alcohol-dependent rats (71.9 ± 8.7%, P = 0.0273, n = 9) as well as rats during protracted withdrawal (77.7 ± 5.1%; P = 0.0009, n = 13), and blunted the acute effects of ethanol (applied in the continued presence of mifepristone) (Dep: mifepristone + ethanol: 77.3 ± 12.9%, P = 0.1289, n = 9); WD: mifepristone + ethanol: 90.7 ± 7.1%, P = 0.2439, n = 13 compared to baseline with Wilcoxon signed-rank tests).
A repeated-measures two-way ANOVA revealed a significant main effect of acute drug treatment with no interaction effect (i.e., mifepristone vs. mifepristone + ethanol; F (1,31) = 8.420, P = 0.0068, šidák post hoc multiple comparison tests, naïve: P = 0.0383, Dep: P = 0.8848, WD: P = 0.1839), suggesting that mifepristone does not only blunt ethanol-induced CeA GABA release after chronic alcohol exposure but also provides evidence that alcohol dependence and withdrawal recruit brain stress systems such as the glucocorticoid/GR system to mediate both acute and chronic effects of alcohol.
Lastly, neither mifepristone nor mifepristone + ethanol significantly altered postsynaptic parameters (shown in Fig. 2C for sIPSC amplitudes, in Fig. 2D for sIPSC rise time and in Fig. 2E for sIPSC decay times; detailed data are summarized in Table 2) indicating that postsynaptic GABA-receptor function is not altered by mifepristone + ethanol.
Table 2.
Summary of postsynaptic sIPSC characteristics in presence of mifepristone for all treatment groups compared to baseline.
| sIPSC amplitude | sIPSC rise time | sIPSC decay time | n | ||
|---|---|---|---|---|---|
| Naïve | Mifepristone | 91.5 ± 4.1% P = 0.0923 | 102.9 ± 1.7% P = 0.2036 | 98.2 ± 4.6% P = 0.4697 | 12 |
| + Ethanol | 104.7 ± 10.0% P = 0.7910 | 102.1 ± 2.7% P = 0.3804 | 110.0 ± 10.3% P = 0.4697 | 12 | |
| Dep. | Mifepristone | 93.9 ± 7.7% P = 0.3594 | 102.2 ± 2.9%, P = 0.7344 | 103.9 ± 8.5% P = 0.6523 | 9 |
| + Ethanol | 97.0 ± 8.0% P = 0.7344 | 105.9 ± 2.5% P = 0.1289 | 131.2 ± 19.9% P = 0.1641 | 9 | |
| WD | Mifepristone | 95.7 ± 7.5% P = 0.3757 | 98.9 ± 3.1% P = 0.8394 | 100.1 ± 8.5% P = 0.6355 | 13 |
| + Ethanol | 101.8 ± 11.9%, P = 0.7354 | 105.7 ± 3.7% P = 0.1099 | 110.8 ± 13.2% P > 0.9999 | 13 |
Differences from baseline control conditions were calculated with Wilcoxon signed-rank tests (naïve: n = 12 (N = 8), Dep: n = 9 (N = 9), WD: n = 13 (N = 6)).
3.3. Mifepristone decreases electrically evoked GABA release in alcohol-dependent rats and rats undergoing protracted withdrawal and blunts effects of acute ethanol
Next, we recorded from 57 (resting membrane potential = −80.2 ± 0.7 mV; membrane resistance = 140 ± 5 MΩ) CeA neurons intracellularly with sharp pipettes to assess the effects of mifepristone on locally evoked, pharmacologically isolated inhibitory GABAA-receptor mediated postsynaptic potentials (eIPSP) by electrical stimulation in the CeA. Based on the I/O protocol, the stimulus strength was adjusted to evoke IPSPs at ~50% of the maximal amplitude and was maintained through the entire recording protocol. As shown in Fig. 3A and B, we found that mifepristone did not significantly alter eIPSP amplitudes in the CeA in naïve rats (93.3 ± 4.5%, P = 0.1934, n = 10), but decreased it in dependent rats (Dep: 84.1 ± 3.9%, P = 0.0002, n = 13) as well as during protracted withdrawal (WD: 88.7 ± 2.6%, P = 0.0015, n = 12, all Wilcoxon signed-rank tests compared to baseline), suggesting that mifepristone (10 μM) decreases evoked GABA transmission only after chronic alcohol exposure.
Fig. 3.
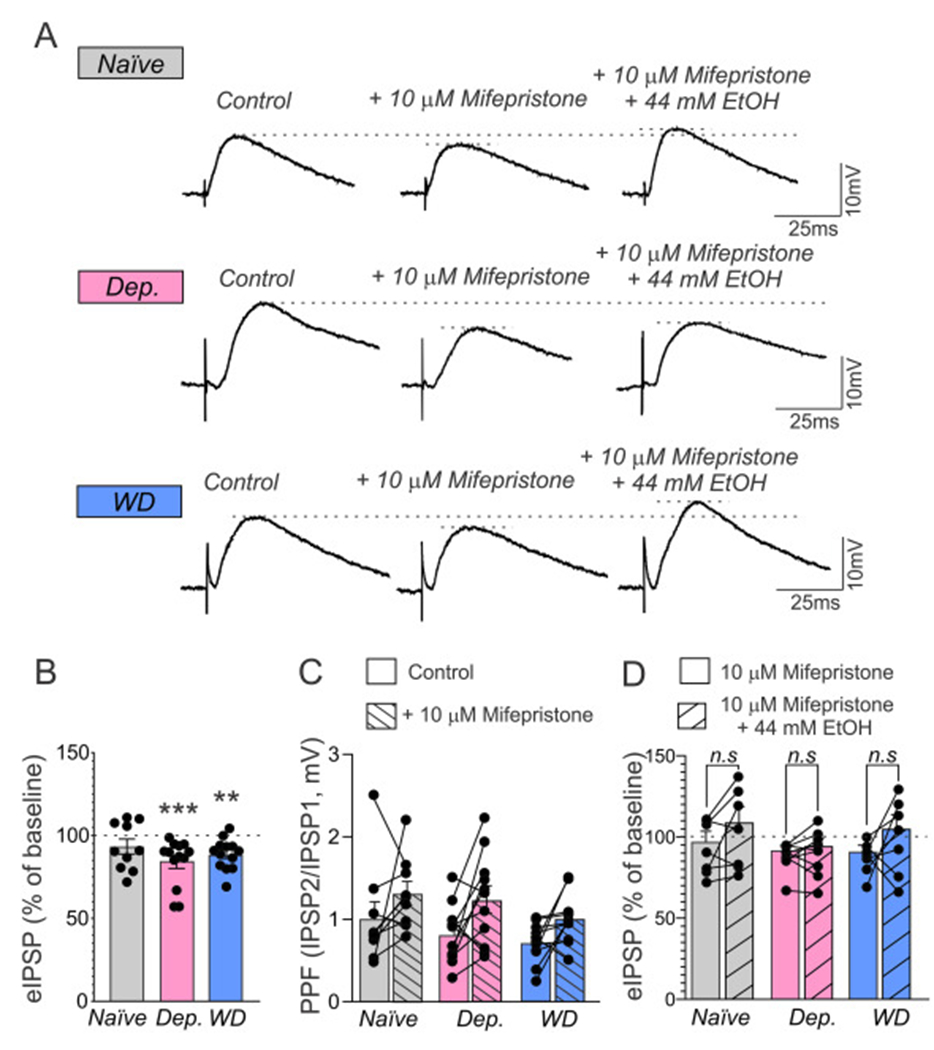
Mifepristone decreases evoked CeA GABA release only after chronic ethanol exposure, but blunts the effects of acute EtOH-induced GABA release in all groups. (A) Representative CeA eIPSPs before (left), and during superfusion with 10 μM mifepristone (middle), and 10 μM mifepristone and 44 mM ethanol are depicted. Bars in (B) represent means ±SEM of normalized effects of mifepristone from baseline (indicated as dashed line) were calculated using Wilcoxon signed-rank tests with (**) = P < 0.01, (***) = P < 0.001. (C) Bars depict paired-pulse facilitation (PPR) in absence (unfilled bar) and presence (dashed bar) of mifepristone. (D) Effects of mifepristone (unfilled bars) and mifepristone + ethanol (dashed bars) on eIPSP amplitudes are depicted as means ± SEM. Differences between groups in (B) was calculated by a Kruskal-Wallis test (naïve: n = 10 (N = 8), Dep: n = 13 (N = 9), WD: n = 12 (N = 8) and differences and (C) and (D) were assessed by a repeated measures-two way ANOVA and a post hoc mean comparison (šidák; n.s. = non-significant = P > 0.05; naïve: n = 7 (N = 6), Dep: n = 9 (N = 8), WD: n = 7 (N = 6).
A Kruskal-Wallis-test, however, revealed that mifepristone effects on eIPSP did not significantly differ between groups (P = 0.5242). Importantly, mifepristone did not alter input resistance, resting membrane potential or I-V-relationships of CeA neurons in either group (not shown) indicating that its effects on eIPSP amplitudes are not caused by changes in excitability but reflect indeed decreased GABAA-receptor mediated synaptic transmission in the presence of mifepristone.
Mifepristone significantly increased paired pulse ratio (PPR) of eIPSPs in the CeA of dependent rats (Dep: baseline PPR: 0.82 ± 0.11 vs. mifepristone PPR: 1.24 ± 0.17, P = 0.0481, n = 8), but did not affect PPR in neither naïve rats (naïve: baseline PPR: 1.02 ± 0.20, n = 10 vs. mifepristone PPR: 1.31 ± 0.15, P = 0.2857) nor rats undergoing protracted withdrawal (WD: baseline PPR: 0.71 ± 0.07 to PPR mifepristone: 1.0 ± 0.09, n = 12, P = 0.2414; repeated-measures-two-way ANOVA, F (1,28) = 11.81, P = 0.0019 for acute drug treatment, šidák post hoc mean comparison, Fig. 3C), suggesting distinct effects of mifepristone on evoked presynaptic GABA release in dependent rats. Next, we studied whether mifepristone would also interfere with the effects of acute ethanol on evoked GABA transmission. Acute ethanol (44 mM) increased eIPSP amplitudes similarly in all groups (naïve: 135.7 ± 3.4%, n = 6, P = 0.0313; Dep: 129.4 ± 3.9%, n = 7, P = 0.0156; WD: 137 ± 3.6%, n = 8, P = 0.0078, Wilcoxon signed-rank tests compared to baseline; Kruskal-Wallis test between groups: P = 0.2628) indicative of the lack of tolerance of CeA GABAergic synapses to acute effects of ethanol as reported (Gilpin et al., 2014; Roberto et al., 2004; Roberto et al., 2003; Tunstall et al., 2019). We then applied 44 mM ethanol (for 10–15 min) in the continued presence of mifepristone to a subset of neurons from all groups that had already received mifepristone for 15 min. As shown in Fig. 3D, both mifepristone and ethanol in the continued presence of mifepristone did not significantly alter evoked CeA GABA transmission in naïve rats (mifepristone: 92.8 ± 6.3%, P = 0.3750 vs. mifepristone + ethanol: 104.3 ± 9.3%, n = 7, P = 0.5781, Wilcoxon signed-rank tests compared to baseline control). In contrast, mifepristone significantly decreased CeA eIPSPs after chronic ethanol exposure in both dependent rats and during protracted withdrawal (mifepristone only: Dep: 87.8 ± 2.9%, n = 9, P = 0.0039; WD: 87.0 ± 3.8%, n = 7, P = 0.0156), while eIPSP amplitudes in presence of mifepristone plus ethanol did not significantly differ from baseline (mifepristone + EtOH: Dep: 90.2 ± 4.6%, n = 9, P = 0.0977, WD: 100.1 ± 8.8%, n = 7, P > 0.9999, Wilcoxon signed-rank tests compared to baseline). A repeated-measures two-way ANOVA revealed a significant main effect of acute drug treatment (i.e. mifepristone vs. mifepristone + EtOH; F(1,20) = 5.065, P = 0.0358) but no interaction; post hoc analyses (šidák) revealed that amplitudes of eIPSPs in presence of mifepristone and mifepristone plus ethanol, respectively, did not significantly differ in either naïve (P = 0.3321), Dep (P = 0.9762) or WD rats (P = 0.2292), suggesting that mifepristone blunts acute ethanol-induced CeA GABA release.
3.4. Selective GR antagonist confirms glucocorticoid regulation of CeA GABA transmission in alcohol-dependent rats
Lastly, given that mifepristone does not only interact with GR, but also binds to other molecular targets including progesterone (PR) and androgen receptors (AR), we examined the effects of the selective GR antagonist CORT118335 (10 μM) on CeA GABA transmission in naïve and dependent rats (Fig. 4A). Importantly, CORT118335 does not modulate PR or AR, but it also acts as mineralocortiocoid receptor antagonist. CORT118335 did not significantly alter sIPSC frequency in naïve rats (89.1 ± 6.7%, n = 10, P = 0.3223), but decreased it in dependent rats (80.9 ± 5.6%, n = 13, P = 0.0081, Fig. 4B) without affecting any postsynaptic measures, including sIPSC amplitude (naïve: 101.4 ± 3.3%, P = 0.6250; Dep: 108.1 ± 4.6%, P = 0.1099, Fig. 4C), rise time (naïve: 104.1 ± 2.8%, P = 0.2754; Dep: 103.4 ± 2.8%, P = 0.2163, Fig. 4D) or decay time (naïve: 98.4 ± 9.0%, P = 0.7695, Dep: 104.0 ± 10.2%, P = 0.7354, Fig. 4E; all Wilcoxon signed-rank tests compared to baseline).
Fig. 4.

CORT188335 decreases CeA GABA transmission only after chronic ethanol exposure. (A) Representative sIPSCs onto CeA neurons from the indicated groups before (upper panel) and during superfusion (lower panel) with CORT118335 (10 μM) are depicted. Bars represent means ± SEM of sIPSC (B) frequencies, (C) amplitudes, (D) rise time and (E) decay times in the presence of 10 μM CORT188335. Asterisks indicate significant difference from baseline assessed by Wilcoxon signed-rank tests; (**) = P < 0.01 (naïve: n 10 (N 6) and Dep: n = 13 (N = 8).)
A subsequent two-way ANOVA comparing the effects of CORT118335 and mifepristone on sIPSC frequency in naïve and dependent rats revealed a significant main effect of alcohol history (F(1, 44) = 4.763, P = 0.0345; dependent < naive), but no interaction (F(1, 44) = 1.349, P = 0.2518) and no main effect of acute drug (F(1, 44) = 1.535, P = 0.2219). These data show that targeting GR signaling in the CeA decreases GABA transmission in dependent rats and provides further evidence for CeA regulation by glucocorticoid signaling associated with alcohol dependence.
4. Discussion
Heightened CeA GABAergic signaling is a hallmark of excessive drinking and a key mechanism of alcohol-dependence associated behaviors across species (Augier et al., 2018; Herman et al., 2016; Jimenez et al., 2019; Roberto et al., 2004). Thus, drugs normalizing this aberrant CeA GABA activity hold promise to have therapeutic potential in the treatment of AUD (Falk et al., 2019; Mason and Heyser, 2021).
Increased CeA GABA transmission originates from dysregulation of multiple signaling systems including the CRF/CRF1 system (Ciccocioppo et al., 2009; Koob, 2021; Roberto et al., 2020; Roberto et al., 2010). In the CeA, the majority of CRF+ neurons also express GR, suggesting that glucocorticoids might be directly involved in CRF synthesis and release (Honkaniemi et al., 1992; Koob and Schulkin, 2019). Notably, although alcohol dependence is not associated with altered GR expression levels in the amygdala, recently increased fractions of phosphorylated CeA GR indicative of enhanced receptor activity have been reported in alcohol-dependent rats (Vendruscolo et al., 2015). In contrast, protracted alcohol abstinence or withdrawal have been shown to enhance CeA GR expression (Vendruscolo et al., 2012) similary suggesting elevated GR signaling. Importantly, the GR antagonist mifepristone decreases alcohol consumption in alcohol-dependent rats and alcohol-cue induced craving and alcohol consumption in human subjects suffering from AUD and mifepristone treatment was found to be safe and well tolerated in humans (Vendruscolo et al., 2015). However, the effects of GR antagonism on CeA synaptic GABA activity representing a potential mechanism of action for mifepristone remained to be determined. Likewise, regulation of CeA synaptic activity by GR under basal conditions but also in alcohol dependence and protracted withdrawal has not yet been explored. Thus, here we utilized mifepristone (10 μM) and the selective GR antagonist CORT118335 as pharmacological tools to unveil a potential GR regulation of CeA action-potential dependent, spontaneous synaptic network-driven (sIPSC) and electrically evoked GABA transmission (eIPSP) in ex vivo brain slices from male, adult rats. To induce alcohol dependence, we used the chronic intermittent vapor exposure (CIE) paradigm for 5–7 weeks. From each CIE cohort a subset of animals went through 2 weeks of protracted withdrawal, enabling assessment of CeA GR regulation at two different stages of alcohol dependence.
We found that mifepristone reduced spontaneous synaptic network-driven sIPSC GABA transmission in all treatment groups presumably via decreasing presynaptic neurotransmitter release from CeA GABAergic terminals given significantly decreased mIPSC frequency shown in naïve rats. Importantly, the decrease of sIPSC frequency was most pronounced in alcohol-dependent rats. This greater efficacy of mifepristone in dependent rats is in line with previous studies likely reflecting increased CeA GR function in alcohol dependence and protracted withdrawal (Vendruscolo et al., 2015). Indeed, mifepristone has been shown to be most efficacious in reducing excessive alcohol drinking in animal models of binge-like drinking, heavy drinking, and alcohol dependence all of which are strong stressors per se while mixed effects of mifepristone on alcohol consumption or anxiety-related behaviors in non-dependent animals as well as in strains genetically-selected for innate high anxiety levels such as the Marchigian Sardinian rats have been reported (Benvenuti et al., 2021; Calvo and Volosin, 2001; Fahlke et al., 1995; Holtyn and Weerts, 2019; Jacquot et al., 2008; Koenig and Olive, 2004; Newman et al., 2018; O’Callaghan et al., 2005; Ostroumov et al., 2016; Repunte-Canonigo et al., 2015; Savarese et al., 2020; Simms et al., 2012; Vendruscolo et al., 2015; Vendruscolo et al., 2012; Vozella et al., 2021; Yang et al., 2008).
Our data also suggest a basal regulation of CeA synaptic GABA activity by glucocorticoids even under naïve conditions as well as constitutive GR activity, which is highly sensitive to chronic ethanol exposure. Interestingly, mifepristone effects on CeA GABA signaling in rats during protracted withdrawal did not significantly differ from that in dependent rats indicative of further neuroadaptations of the GR system occuring during protracted withdrawal consistent with the previously reported increase in CeA GR expression (Vendruscolo et al., 2012). However, it also did not differ significantly from the effect of mifepristone in naïve rats which might also indicate a potential reduction of mifepristone efficacy in protracted withdrawal. In line, Jimenez et al. reported that mifepristone reduced heavy alcohol intake in non-human primates, but it did not eliminate baseline ethanol consumption. Upon cessation of mifepristone treatment animals rapidly returned to baseline alcohol consumption, and mifepristone did not prevent a relapse during early abstinence (Jimenez et al., 2020).
Interestingly, we found distinct effects of mifepristone on evoked CeA GABA transmission. Specifically, mifepristone decreased evoked CeA GABA transmission in rats only after chronic ethanol exposure but did not alter it in naïve rats. This apparent discrepancy of mifepristone effects on two distinct forms of isolated CeA GABA transmission further reflects the complex nature of glucocorticoid regulation of different transmitter vesicle pools at CeA GABAergic synapses. Moreover, these results provide further evidence that alcohol dependence engages the GR system to regulate synaptic activity. Similarly, a previous study found that application of the GR-agonist corticosterone alone does not alter evoked CeA glutamate transmission but when applied in combination with ethanol markedly reduced evoked glutamate transmission (Logrip et al., 2017), suggesting that prior ethanol exposure may prime GR at CeA synapses to enhance glucocorticoid regulation of evoked neurotransmitter release.
We also found distinct effects of mifepristone on the acute effects of alcohol on spontaneous action-potential dependent GABA transmission vs. evoked CeA GABA transmission. Specifically, our study revealed that mifepristone blocks the acute effects of alcohol on spontaneous, action-potential driven GABA transmission (i.e., spontaneous CeA network activity) in alcohol-dependent rats but did not affect alcohol effects in naïve rats. Similarly, we found that mifepristone blunted acute alcohol effects on evoked GABA transmission in dependent rats, but it also blunted the effects of alcohol in naïve rats. These results emphasize the suggested differential regulation of distinct vesicle pools by GR associated with chronic alcohol exposure. Roberto et al. (2010) demonstrated that the effect of acute alcohol on electrically evoked GABA release in both naïve and dependent rats involves a CRF1-mediated mechanism (Roberto et al., 2010), but the acute alcohol-induced augmentation of spontaneous, action-potential driven GABA transmission in naïve rats – but not in dependent–rats - was mediated by L-type calcium channels (Varodayan et al., 2017b). Thus, given that activated GR recruits CRF signaling in the CeA (Koob and Schulkin, 2019; Tunstall et al., 2017), the diverging effects of mifepristone on acute alcohol effects in naïve rats are indeed consistent with our previous studies and are likely to reflect the distinct pathways mediating elevated GABA signaling in response to acute alcohol.
Moreover, comparing the effects of the selective GR antagonist CORT118335 on CeA GABA transmission to those of mifepristone indicated indeed very similar pharmacological profiles of CORT118335 and mifepristone such as markedly stronger reduction of GABA release in alcohol-dependent rats further highlighting the critical role of GR in regulating CeA activity in alcohol dependence. Of note, these effects of CORT118335 occurred in the absence of antagonist activity at progesterone receptors associated with mifepristone, underscoring the specificity of GR activity of these drugs in alcohol dependence. CORT118335 has been recently shown to significantly reduce alcohol self-administration in non-dependent and dependent rats (McGinn et al., 2021). Importantly, although pure GR antagonism may be of therapeutic benefit, inhibiting other GR-mediated effects such as the negative feedback on the HPA axis could lead to increased cortisol levels, which could in turn, activate mineralocorticoid receptors (Zalachoras et al., 2013) and modulate alcohol dependence-related behaviors (Aoun et al., 2018).
Lastly, it is important to highlight that the effects of mifepristone and CORT118335 on CeA GABA synapses occurred fast (within <15 min). Thus, it is rather unlikely that they are due to changes in gene transcription but rather mediated by non-genomic GR activity. Indeed, non-genomic GR signaling mediated by either cytosolic GR or membrane-bound GR has recently gained recognition given that side effects of glucocorticoid therapy often stem from genomic GR effects. Furthermore, rapid effects of cortisol in increasing intracellular calcium levels (Panettieri et al., 2019) as well as on NMDA-evoked currents in hippocampal neurons (Zhang et al., 2012) and dexamethasone on inhibitory synaptic transmission in the prefrontal cortex have been recently reported (Teng et al., 2013). Importantly, most non-genomic glucocorticoid effects are sensitive to mifepristone antagonism (Panettieri et al., 2019), suggesting that also the effects on synaptic transmission in the CeA are mediated via non-genomic GR signaling. However, further studies are required to fully unravel the intracellular signaling pathways that mediate mifepristone’s rapid effects.
5. Conclusions
Collectively, our study sheds light on GR-mediated regulation of CeA GABAergic synapses – a system which is highly sensitive to chronic alcohol exposure. Specifically, our study revealed that the GR-antagonist mifepristone diminishes elevated GABA release in the CeA and also blocks acute effects of alcohol on CeA synaptic activity. Our data thus provide a strong mechanistic basis for mifepristone’s efficiency in reducing alcohol consumption in models of alcohol dependence but also in AUD patients and further support the potential of drugs targeting GR as a promising pharmacological avenue for the treatment of AUD.
Supplementary Material
Acknowledgments
This is manuscript number 30114 from the Scripps Research Institute. We thank Dr. Hazel Hunt and Corcept Therapeutics for providing CORT118335. This research was financially supported by NIH/NIAAA grants AA013498, AA021491, AA006420, AA017447, AA007456, AA027700 (to M.R), AA026638 (to D.K), AA023152 (B.J.M), NIH/NIDA IRP (L.F.V), the Austrian Science Fund FWF J-3942 (to S.K) and the Pearson Center for Alcoholism and Addiction Research.
Footnotes
Appendix A.: Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2022.105610.
References
- Andreasen M, Hablitz JJ, 1994. Paired-pulse facilitation in the dentate gyrus: a patch-clamp study in rat hippocampus in vitro. J. Neurophysiol 72, 326–336. 10.1152/jn.1994.72.1.326. [DOI] [PubMed] [Google Scholar]
- Aoun EG, Jimenez VA, Vendruscolo LF, Walter NAR, Barbier E, Ferrulli A, Haass-Koffler CL, Darakjian P, Lee MR, Addolorato G, Heilig M,Hitzemann R, Koob GF, Grant KA, Leggio L, 2018. A relationship between the aldosterone-mineralocorticoid receptor pathway and alcohol drinking: preliminary translational findings across rats, monkeys and humans. Mol. Psychiatry 23, 1466–1473. 10.1038/mp.2017.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augier E, Barbier E, Dulman RS, Licheri V, Augier G, Domi E, Barchiesi R, Farris S, Nätt D, Mayfield RD, Adermark L, Heilig M, 2018. A molecular mechanism for choosing alcohol over an alternative reward. Science 360, 1321–1326. 10.1126/science.aao1157. [DOI] [PubMed] [Google Scholar]
- Benvenuti F, Cannella N, Stopponi S, Soverchia L, Ubaldi M, Lunerti V,Vozella V, Cruz B, Roberto M, Ciccocioppo R, 2021. Effect of glucocorticoid receptor antagonism on alcohol self-administration in genetically-selected marchigian Sardinian alcohol-preferring and non-preferring wistar rats. Int. J. Mol. Sci 22 10.3390/ijms22084184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyeler A, Dabrowska J, 2020. Neuronal diversity of the amygdala and the bed nucleus of the stria terminalis. Handb. Behav. Neurosci 26, 63–100. 10.1016/b978-0-12-815134-1.00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaine SK, Sinha R, 2017. Alcohol, stress, and glucocorticoids: from risk to dependence and relapse in alcohol use disorders. Neuropharmacology 122, 136–147. 10.1016/j.neuropharm.2017.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo N, Volosin M, 2001. Glucocorticoid and mineralocorticoid receptors are involved in the facilitation of anxiety-like response induced by restraint. Neuroendocrinology 73, 261–271. 10.1159/000054643. [DOI] [PubMed] [Google Scholar]
- Chieng BCH, Christie MJ, Osborne PB, 2006. Characterization of neurons in the rat central nucleus of the amygdala: cellular physiology, morphology, and opioid sensitivity. J. Comp. Neurol 497, 910–927. 10.1002/cne.21025. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Gehlert DR, Ryabinin A, Kaur S, Cippitelli A, Thorsell A, Lê AD, Hipskind PA, Hamdouchi C, Lu J, Hembre EJ, Cramer J, Song M,McKinzie D, Morin M, Economidou D, Stopponi S, Cannella N, Braconi S, Kallupi M, de Guglielmo G, Massi M, George DT, Gilman J, Hersh J, Tauscher JT, Hunt SP, Hommer D, Heilig M, 2009. Stress-related neuropeptides and alcoholism: CRH, NPY, and beyond. Alcohol 43, 491–498. 10.1016/j.alcohol.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cintra A, Zoli M, Rosén L, Agnati LF, Okret S, Wikström AC, Gustaffsson JA, Fuxe K, 1994. Mapping and computer assisted morphometry and microdensitometry of glucocorticoid receptor immunoreactive neurons and glial cells in the rat central nervous system. Neuroscience 62, 843–897. 10.1016/0306-4522(94)90481-2. [DOI] [PubMed] [Google Scholar]
- Dalm S, Karssen AM, Meijer OC, Belanoff JK, de Kloet ER, 2019. Resetting the stress system with a mifepristone challenge. Cell. Mol. Neurobiol 39, 503–522. 10.1007/s10571-018-0614-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue K, Rose A, Coulton S, Milward J, Reed K, Drummond C, Little H, 2016. Double-blind, 12 month follow-up, placebo-controlled trial of mifepristone on cognition in alcoholics: the MIFCOG trial protocol. BMC Psychiatry 16, 40. 10.1186/s12888-016-0757-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlke C, Hård E, Eriksson CJ, Engel JA, Hansen S, 1995. Consequence of long-term exposure to corticosterone or dexamethasone on ethanol consumption in the adrenalectomized rat, and the effect of type I and type II corticosteroid receptor antagonists. Psychopharmacology 117, 216–224. 10.1007/BF02245190. [DOI] [PubMed] [Google Scholar]
- Falk DE, Ryan ML, Fertig JB, Devine EG, Cruz R, Brown ES, Burns H, Salloum IM, Newport DJ, Mendelson J, Galloway G, Kampman K, Brooks C, Green AI, Brunette MF, Rosenthal RN, Dunn KE, Strain EC, Ray L, Shoptaw S, Ait-Daoud Tiouririne N, Gunderson EW, Ransom J, Scott C, Leggio L, Caras S, Mason BJ, Litten RZ, National Institute on Alcohol Abuse and Alcoholism Clinical Investigations Group (NCIG) Study Group, 2019. Gabapentin enacarbil extended-release for alcohol use disorder: a randomized, double-blind, placebo-controlled, multisite trial assessing efficacy and safety. Alcohol. Clin. Exp. Res 43, 158–169. 10.1111/acer.13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleseriu M, Petersenn S, 2015. Medical therapy for Cushing’s disease: adrenal steroidogenesis inhibitors and glucocorticoid receptor blockers. Pituitary 18, 245–252. 10.1007/s11102-014-0627-0. [DOI] [PubMed] [Google Scholar]
- Garabedian MJ, Harris CA, Jeanneteau F, 2017. Glucocorticoid receptor action in metabolic and neuronal function. F1000Res 6, 1208. 10.12688/f1000research.11375.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Roberto M, Koob GF, Schweitzer P, 2014. Kappa opioid receptor activation decreases inhibitory transmission and antagonizes alcohol effects in rat central amygdala. Neuropharmacology 77, 294–302. 10.1016/j.neuropharm.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, Contet C, Roberto M, 2016. A functional switch in tonic GABA currents alters the output of central amygdala corticotropin releasing factor receptor-1 neurons following chronic ethanol exposure. J. Neurosci 36, 10729–10741. 10.1523/JNEUROSCI.1267-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley AE, Koob GF, Mason BJ, 2012. Treatment of alcohol dependence with drug antagonists of the stress response. Alcohol Res. 34, 516–521. [PMC free article] [PubMed] [Google Scholar]
- Holtyn AF, Weerts EM, 2019. Evaluation of mifepristone effects on alcohol-seeking and self-administration in baboons. Exp. Clin. Psychopharmacol 27, 227–235. 10.1037/pha0000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkaniemi J, Pelto-Huikko M, Rechardt L, Isola J, Lammi A, Fuxe K,Gustafsson JA, Wikström AC, Hökfelt T, 1992. Colocalization of peptide and glucocorticoid receptor immunoreactivities in rat central amygdaloid nucleus. Neuroendocrinology 55, 451–459. 10.1159/000126156. [DOI] [PubMed] [Google Scholar]
- Howland RH, 2013. Mifepristone as a therapeutic agent in psychiatry. J. Psychosoc. Nurs. Ment. Health Serv 51, 11–14. 10.3928/02793695-20130513-01. [DOI] [PubMed] [Google Scholar]
- Jacquot C, Croft AP, Prendergast MA, Mulholland P, Shaw SG, Little HJ, 2008. Effects of the glucocorticoid antagonist, mifepristone, on the consequences of withdrawal from long term alcohol consumption. Alcohol. Clin. Exp. Res 32, 2107–2116. 10.1111/j.1530-0277.2008.00799.x. [DOI] [PubMed] [Google Scholar]
- Jimenez VA, Herman MA, Cuzon Carlson VC, Walter NA, Grant KA,Roberto M, 2019. Synaptic adaptations in the central amygdala and hypothalamic paraventricular nucleus associated with protracted ethanol abstinence in male rhesus monkeys. Neuropsychopharmacology 44, 982–993. 10.1038/s41386-018-0290-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez VA, Walter NAR, Shnitko TA, Newman N, Diem K, Vanderhooft L, Hunt H, Grant KA, 2020. Mifepristone decreases chronic voluntary ethanol consumption in Rhesus macaques. J. Pharmacol. Exp. Ther 375, 258–267. 10.1124/jpet.120.000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M, 2018. Corticosteroids and the brain. J. Endocrinol 238, R121–R130. 10.1530/JOE-18-0226. [DOI] [PubMed] [Google Scholar]
- Khom S, Steinkellner T, Hnasko TS, Roberto M, 2020a. Alcohol dependence potentiates substance P/neurokinin-1 receptor signaling in the rat central nucleus of amygdala. Sci. Adv 6, eaaz1050. 10.1126/sciadv.aaz1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khom S, Wolfe SA, Patel RR, Kirson D, Hedges DM, Varodayan FP, Bajo M, Roberto M, 2020b. Alcohol dependence and withdrawal impair serotonergic regulation of GABA transmission in the rat central nucleus of the amygdala. J. Neurosci 10.1523/JNEUROSCI.0733-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirson D, Oleata CS, Roberto M, 2020. Taurine suppression of central amygdala GABAergic inhibitory signaling via glycine receptors is disrupted in alcohol dependence. Alcohol. Clin. Exp. Res 44, 445–454. 10.1111/acer.14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirson D, Khom S, Rodriguez L, Wolfe SA, Varodayan FP, Gandhi PJ, Patel RR, Vlkolinsky R, Bajo M, Roberto M, 2021. Sex differences in acute alcohol sensitivity of Naïve and alcohol dependent central amygdala GABA synapses. Alcohol Alcohol, 10.1093/alcalc/agab034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig HN, Olive MF, 2004. The glucocorticoid receptor antagonist mifepristone reduces ethanol intake in rats under limited access conditions. Psychoneuroendocrinology 29, 999–1003. 10.1016/j.psyneuen.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Koob GF, 2021. Drug addiction: Hyperkatifeia/negative reinforcement as a framework for medications development. Pharmacol. Rev 73, 163–201. 10.1124/pharmrev.120.000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Mason BJ, 2016. Existing and future drugs for the treatment of the dark side of addiction. Annu. Rev. Pharmacol. Toxicol 56, 299–322. 10.1146/annurev-pharmtox-010715-103143. [DOI] [PubMed] [Google Scholar]
- Koob GF, Schulkin J, 2019. Addiction and stress: an allostatic view. Neurosci. Biobehav. Rev 106, 245–262. 10.1016/j.neubiorev.2018.09.008. [DOI] [PubMed] [Google Scholar]
- Logrip ML, Oleata C, Roberto M, 2017. Sex differences in responses of the basolateral-central amygdala circuit to alcohol, corticosterone and their interaction. Neuropharmacology 114, 123–134. 10.10l6/j.neuropharm.2016.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu NZ, Wardell SE, Burnstein KL, Defranco D, Fuller PJ, Giguere V,Hochberg RB, McKay L, Renoir J-M, Weigel NL, Wilson EM, McDonnell DP, Cidlowski JA, 2006. International Union of Pharmacology. LXV. The pharmacology and classification of the nuclear receptor superfamily: glucocorticoid, mineralocorticoid, progesterone, and androgen receptors. Pharmacol. Rev 58, 782–797. 10.1124/pr.58.4.9. [DOI] [PubMed] [Google Scholar]
- Mason BJ, Heyser CJ, 2021. Alcohol use disorder: The role of medication in recovery. Alcohol Res. 41, 07. 10.35946/arcr.v41.1.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinn MA, Tunstall BJ, Schlosburg JE, Gregory-Flores A, George O, de Guglielmo G, Mason BJ, Hunt HJ, Koob GF, Vendruscolo LF, 2021. Glucocorticoid receptor modulators decrease alcohol self-administration in male rats. Neuropharmacology 188, 108510. 10.1016/j.neuropharm.2021.108510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahiro M, Arakawa O, Narahashi T, Ukai S, Kato Y, Nishinuma K,Nishimura T, 1992. Dimethyl sulfoxide (DMSO) blocks GABA-induced current in rat dorsal root ganglion neurons. Neurosci. Lett 138, 5–8. 10.1016/0304-3940(92)90459-k. [DOI] [PubMed] [Google Scholar]
- Newman EL, Albrechet-Souza L, Andrew PM, Auld JG, Burk KC, Hwa LS, Zhang EY, DeBold JF, Miczek KA, 2018. Persistent escalation of alcohol consumption by mice exposed to brief episodes of social defeat stress: suppression by CRF-R1 antagonism. Psychopharmacology 235, 1807–1820. 10.1007/s00213-018-4905-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan MJ, Croft AP, Jacquot C, Little HJ, 2005. The hypothalamopituitary-adrenal axis and alcohol preference. Brain Res. Bull 68, 171–178. 10.1016/j.brainresbull.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Ostroumov A, Thomas AM, Kimmey BA, Karsch JS, Doyon WM, Dani JA, 2016. Stress increases ethanol self-administration via a shift toward excitatory GABA signaling in the ventral tegmental area. Neuron 92, 493–504. 10.1016/j.neuron.2016.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panettieri RA, Schaafsma D, Amrani Y, Koziol-White C, Ostrom R, Tliba O, 2019. Non-genomic effects of glucocorticoids: an updated view. Trends Pharmacol. Sci 40, 38–49. 10.1016/j.tips.2Ol8.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul N, Raymond J, Lumbreras S, Bartsch D, Weber T, Lau T, 2021. Activation of the glucocorticoid receptor rapidly triggers calcium-dependent serotonin release in vitro. CNS Neurosci. Ther 27, 753–764. 10.1111/cns.13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repunte-Canonigo V, Shin W, Vendruscolo LF, Lefebvre C, van der Stap L, Kawamura T, Schlosburg JE, Alvarez M, Koob GF, Califano A, Sanna PP, 2015. Identifying candidate drivers of alcohol dependence-induced excessive drinking by assembly and interrogation of brain-specific regulatory networks. Genome Biol. 16, 68. 10.1186/s13059-015-0593-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson HN, Lee SY, O’Dell LE, Koob GF, Rivier CL, 2008. Alcohol self-administration acutely stimulates the hypothalamic-pituitary-adrenal axis, but alcohol dependence leads to a dampened neuroendocrine state. Eur. J. Neurosci 28, 1641–1653. 10.1111/j.1460-9568.2008.06455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR, 2003. Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc. Natl. Acad. Sci. U. S. A 100, 2053–2058. 10.1073/pnas.0437926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Stouffer DG, Parsons LH, Siggins GR, 2004. Increased GABA release in the central amygdala of ethanol-dependent rats. J. Neurosci 24, 10159–10166. 10.1523/JNEUROSCI.3004-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, Cottone P, Madamba SG, Stouffer DG, Zorrilla EP, Koob GF, Siggins GR, Parsons LH, 2010. Corticotropin releasing factor-induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence. Biol. Psychiatry 67, 831–839. 10.1016/j.biopsych.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Kirson D, Khom S, 2020. The role of the central amygdala in alcohol dependence. Cold Spring Harb. Perspect. Med 10.1101/cshperspect.a039339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarese AM, Ozburn AR, Metten P, Schlumbohm JP, Hack WR, LeMoine K, Hunt H, Hausch F, Bauder M, Crabbe JC, 2020. Targeting the glucocorticoid receptor reduces binge-like drinking in high drinking in the dark (HDID-1) mice. Alcohol. Clin. Exp. Res 44, 1025–1036. 10.1111/acer.14318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms JA, Haass-Koffler CL, Bito-Onon J, Li R, Bartlett SE, 2012. Mifepristone in the central nucleus of the amygdala reduces yohimbine stress-induced reinstatement of ethanol-seeking. Neuropsychopharmacology 37, 906–918. 10.1038/npp.2011.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somkuwar SS, Vendruscolo LF, Fannon MJ, Schmeichel BE, Nguyen TB,Guevara J, Sidhu H, Contet C, Zorrilla EP, Mandyam CD, 2017. Abstinence from prolonged ethanol exposure affects plasma corticosterone, glucocorticoid receptor signaling and stress-related behaviors. Psychoneuroendocrinology 84, 17–31. 10.1016/j.psyneuen.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman MQ, Kirson D, Wolfe SA, Khom S, D’Ambrosio SR, Spierling Bagsic SR, Bajo M, Vlkolinský R, Hoang NK, Singhal A, Sureshchandra S, Oleata CS, Messaoudi I, Zorrilla EP, Roberto M, 2020. Importance of sex and trauma context on circulating cytokines and amygdalar GABAergic signaling in a comorbid model of posttraumatic stress and alcohol use disorders. Mol. Psychiatry 10.1038/s41380-020-00920-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens MAC, Wand G, 2012. Stress and the HPA axis: role of glucocorticoids in alcohol dependence. Alcohol Res. 34, 468–483. [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Sato Y, Tamura K, Tamano H, Takeda A, 2019. Rapid intracellular Zn2+ dysregulation via membrane corticosteroid receptor activation affects in vivo CA1 LTP. Mol. Neurobiol 56, 1356–1365. 10.1007/s12035-018-1159-9. [DOI] [PubMed] [Google Scholar]
- Teng Z, Zhang M, Zhao M, Zhang W, 2013. Glucocorticoid exerts its non-genomic effect on IPSC by activation of a phospholipase C-dependent pathway in prefrontal cortex of rats. J. Physiol 591, 3341–3353. 10.1113/jphysiol.2013.254961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvyetlynska NA, Hill RH, Grillner S, 2005. Role of AMPA receptor desensitization and the side effects of a DMSO vehicle on reticulospinal EPSPs and locomotor activity. J. Neurophysiol 94, 3951–3960. 10.1152/jn.00201.2005. [DOI] [PubMed] [Google Scholar]
- Tunstall BJ, Carmack SA, Koob GF, Vendruscolo LF, 2017. Dysregulation of brain stress systems mediates compulsive alcohol drinking. Curr. Opin. Behav. Sci 13, 85–90. 10.1016/j.cobeha.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunstall BJ, Kirson D, Zallar LJ, McConnell SA, Vendruscolo JCM, Ho CP, Oleata CS, Khom S, Manning M, Lee MR, Leggio L, Koob GF, Roberto M, Vendruscolo LF, 2019. Oxytocin blocks enhanced motivation for alcohol in alcohol dependence and blocks alcohol effects on GABAergic transmission in the central amygdala. PLoS Biol. 17, e2006421 10.1371/journal.pbio.2006421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varodayan FP, Correia D, Kirson D, Khom S, Oleata CS, Luu G, Schweitzer P, Roberto M, 2017a. CRF modulates glutamate transmission in the central amygdala of naïve and ethanol-dependent rats. Neuropharmacology. 10.1016/j.neuropharm.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varodayan FP, de Guglielmo G, Logrip ML, George O, Roberto M, 2017b. Alcohol dependence disrupts Amygdalar L-type voltage-gated calcium channel mechanisms. J. Neurosci 37, 4593–4603. 10.1523/JNEUROSCI.3721-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varodayan FP, Khom S, Patel RR, Steinman MQ, Hedges DM, Oleata CS, Homanics GE, Roberto M, Bajo M, 2018. Role of TLR4 in the modulation of central amygdala GABA transmission by CRF following restraint stress. Alcohol Alcohol. 10.1093/alcalc/agx114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendruscolo LF, Roberts AJ, 2014. Operant alcohol self-administration in dependent rats: focus on the vapor model. Alcohol 48, 277–286. 10.1016/j.alcohol.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendruscolo LF, Barbier E, Schlosburg JE, Misra KK, Whitfield TW, Logrip ML, Rivier C, Repunte-Canonigo V, Zorrilla EP, Sanna PP, Heilig M, Koob GF, 2012. Corticosteroid-dependent plasticity mediates compulsive alcohol drinking in rats. J. Neurosci 32, 7563–7571. 10.1523/JNEUROSCI.0069-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendruscolo LF, Estey D, Goodell V, Macshane LG, Logrip ML, Schlosburg JE, McGinn MA, Zamora-Martinez ER, Belanoff JK, Hunt HJ, Sanna PP, George O, Koob GF, Edwards S, Mason BJ, 2015. Glucocorticoid receptor antagonism decreases alcohol seeking in alcohol-dependent individuals. J. Clin. Invest 125, 3193–3197. 10.1172/JCI79828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vozella V, Cruz B, Natividad LA, Benvenuti F, Cannella N, Edwards S, Zorrilla EP, Ciccocioppo R, Roberto M, 2021. Glucocorticoid receptor antagonist mifepristone does not Alter innate anxiety-like behavior in genetically-selected Marchigian Sardinian (msP) rats. Int. J. Mol. Sci 22,3095. 10.3390/ijms22063095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemm SE, Sinha R, 2019. Drug-induced stress responses and addiction risk and relapse. Neurobiol. Stress 10, 100148. 10.1016/j.ynstr.2019.100148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Wang S, Rice KC, Munro CA, Wand GS, 2008. Restraint stress and ethanol consumption in two mouse strains. Alcohol. Clin. Exp. Res 32, 840–852. 10.1111/j.1530-0277.2008.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalachoras I, Houtman R, Atucha E, Devos R, Tijssen AMI, Hu P, Lockey PM, Datson NA, Belanoff JK, Lucassen PJ, Joëls M, de Kloet ER, Roozendaal B, Hunt H, Meijer OC, 2013. Differential targeting of brain stress circuits with a selective glucocorticoid receptor modulator. Proc. Natl. Acad. Sci. U. S. A 110, 7910–7915. 10.1073/pnas.1219411110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sheng H, Qi J, Ma B, Sun J, Li S, Ni X, 2012. Glucocorticoid acts on a putative G protein-coupled receptor to rapidly regulate the activity of NMDA receptors in hippocampal neurons. Am. J. Physiol. Endocrinol. Metab 302, E747–E758. 10.1152/ajpendo.00302.2011. [DOI] [PubMed] [Google Scholar]
- Zhang C, Deng Y, Dai H, Zhou W, Tian J, Bing G, Zhao L, 2017. Effects of dimethyl sulfoxide on the morphology and viability of primary cultured neurons and astrocytes. Brain Res. Bull 128, 34–39. 10.1016/j.brainresbull.2016.11.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.