

ABSTRACT
Staphylococcus aureus is a leading cause of skin and soft tissue infections (SSTIs). Studies examining the immune response to S. aureus have been conducted, yet our understanding of the kinetic response to S. aureus subcutaneous skin infection remains incomplete. In this study, we used C57BL/6J mice and USA300 S. aureus to examine the host-pathogen interface from 8 h postinfection to 15 days postinfection (dpi), with the following outcomes measured: lesion size, bacterial titers, local cytokine and chemokine levels, phenotype of the responding leukocytes, and histopathology and Gram staining of skin tissue. Lesions were largest at 1 dpi, with peak necrotic tissue areas at 3 dpi, and were largely resolved by 15 dpi. During early infection, bacterial titers were high, neutrophils were the most abundant immune cell type, there was a decrease in most leukocyte populations found in uninfected skin, and many different cytokines were produced. Histopathological analysis demonstrated swift and extensive keratinocyte death and robust and persistent neutrophil infiltration. Gram staining revealed subdermal S. aureus colonization and, later, limited migration into upper skin layers. Interleukin-17A/F (IL-17A/F) was detected only starting at 5 dpi and coincided with an immediate decrease in bacterial numbers in the following days. After 9 days, neutrophils were no longer the most abundant immune cell type present as most other leukocyte subsets returned, and surface wounds resolved coincident with declining bacterial titers. Collectively, these data illustrate a dynamic immune response to S. aureus skin infection and suggest a key role for precisely timed IL-17 production for infection clearance and healthy tissue formation.
KEYWORDS: skin infection, virulence, methicillin-resistant Staphylococcus aureus, MRSA, immune response
INTRODUCTION
The Gram-positive pathogen Staphylococcus aureus can cause a wide range of diseases, from mild, localized skin and soft tissue infections (SSTIs) to severe systemic infections with a high mortality rate. The most prevalent types of infection that S. aureus causes are SSTIs (1), and the majority of SSTIs are caused by S. aureus (2). Over the past few decades, the knowledge gained about S. aureus SSTIs has been invaluable in decreasing the rate of methicillin-resistant S. aureus (MRSA) infections. However, the rate of decline for MRSA infections decreased and the rate of methicillin-susceptible S. aureus (MSSA) infections increased from 2012 to 2017 (3). Considering the impact of S. aureus on human health, it is imperative that we work toward generating a comprehensive view of how this pathogen causes disease.
Mouse models of skin infections are commonly used to gain a better understanding of the complex relationship between S. aureus and the host immune system. Previous studies identified both bacterial and host factors that contribute to pathogenesis and immune defense. Upon infection, neutrophils are the first leukocytes recruited to the site of infection by cytokines and chemokines produced by damaged skin cells (reviewed in reference 4) and are crucial for sequestering and clearing S. aureus from the infection (5, 6). Recently, it has been shown that bacterial factors such as phenol-soluble modulins (PSMs) are directly involved in recruiting neutrophils to the site of infection (7). The skin has a variety of antigen-presenting cells (APCs) that help orchestrate the immune response, including dendritic cells (DCs), Langerhans cells (LCs), and macrophages. The skin contains several DC populations essential for the immune response. While not specifically studied in the skin, DCs are important during S. aureus infection models despite their poor ability to phagocytose and kill S. aureus (8, 9). Their importance likely lies in their ability to produce cytokines and activate T cells (9, 10). LCs are a subset of CD207 (langerin)-expressing APCs in the epidermis. While LCs have also not been extensively studied in S. aureus skin infections, they are key cells in skin immune homeostasis (11–13). In addition, mice expressing human langerin on LCs have an altered immune response to S. aureus in the skin (14). Dermal, monocyte-derived, and perivascular macrophages are key components of the immune response to S. aureus infection and help recruit neutrophils to the site of infection (15, 16). In C57BL/6 mice, interleukin-17 (IL-17) is critical for host defense against S. aureus during skin infection (17–19). γδ T cells produce IL-17, and the absence of these cells decreases neutrophil recruitment to the site of infection and increases bacterial titers at the site of infection (17, 20, 21). Other studies have also implicated a role for CD4+ T cells in S. aureus SSTIs (22–25). Thus, multiple leukocyte subsets respond to S. aureus SSTIs and are required for successful infection control and a return to skin homeostasis.
While studies performed to date do not necessarily disagree with each other, direct comparisons between studies to gain a complete picture of the immune response to S. aureus skin infections are challenging. This is due, in part, to the use of different mouse strains that exhibit notable immunological differences, C57BL/6 versus BALB/c, for example (22). In addition, studies have used a variety of S. aureus strains, each with different virulence factor expression potential. Furthermore, the immune response varies depending on the layer of skin infected (6). Finally, it is not clear what time point after infection is appropriate for any given parameter that one wishes to interrogate.
In previous studies, we (26–29) and others observed changes in skin pathology and local cytokine/chemokine levels during infection by mutant bacteria, demonstrating an altered immune response. The reason for these changes was not clear, and thus, the dynamics of the host-pathogen interaction during infection are of great interest. To fully decipher the complexity of this host-pathogen interface, we must first understand the kinetic immune response to S. aureus infection. To this end, we subcutaneously infected the genetically tractable C57BL/6J mouse strain with a derivative of the USA300 community-associated MRSA strain LAC. We then sacrificed mice at 8 h postinfection (hpi) and at 1, 3, 5, 7, 9, 11, 13, and 15 days postinfection (dpi) (Fig. 1a). At each time point, we quantified CFU; measured lesion size, necrosis area, and cytokine and chemokine production; immunophenotyped leukocyte populations; and collected skin samples for histopathological examination. The culmination of these data allowed us to assemble a timeline of the immune response and pathology of S. aureus subcutaneous skin infection. We observed dramatic changes in immune populations, with most cell types decreasing upon infection and a spike in IL-17 levels immediately preceding bacterial clearance and wound resolution.
FIG 1.

Kinetic macroscopic tissue damage and bacterial titer enumeration over the course of infection. (a) Schematic of the experimental design. (b) Mice were imaged, and the total lesion size (blanching and necrosis) and surface necrosis area were measured using ImageJ. Data represent the means with standard errors of the means (SEM) (n = 21 to 186). Errors bars are present and may be smaller than the symbols. (c) Bacterial titers were determined. Each symbol represents the value for a single animal (n = 10 to 13), and the line represents the median value. “#” indicates a P value of <0.05 by a Mann-Whitney test compared to 0.3 dpi.
RESULTS
S. aureus causes skin lesions in a subcutaneous infection model, and bacterial clearance begins at around day 9 postinfection.
Our first goal was to macroscopically determine the kinetics of lesion formation, a typical measure of pathology in mouse skin infection models. This model produces two readily observable skin manifestations: initial skin blanching, likely resulting from ischemia, followed by a hard necrotic scab (necrosis). No surface lesion was apparent at 8 h postinfection (Fig. 1b and Fig. 2). The total surface lesion, which includes the larger blanched area and necrosis within the blanched area, was largest at 1 day postinfection (dpi) (Fig. 1b); however, by 3 dpi, the surface lesion consisted of all necrotic tissue (Fig. 1b and Fig. 2). Thereafter, the necrotic area steadily declined until 15 dpi when the wound neared resolution.
FIG 2.

Tissue damage and bacterial localization. Representative photographs of lesions (macroscopic) throughout the infection, H&E staining of histology slides, and modified Gram staining of tissue are shown. Bars on macroscopic mouse pictures, 0.5 cm. Sections are 5 μm thick. Bars on tissue sections, 1 mm. Arrows indicate S. aureus.
Another key metric in skin infection models is the bacterial burden within the tissue surrounding and including the lesion. Bacterial titers were between 106 and 107 CFU per mg tissue from 8 hpi to 7 dpi, did not significantly decrease until 9 dpi, and steadily declined through 15 dpi (Fig. 1c). Based on these data, we defined 3 phases of the infection. First, there was an early phase from 8 hpi to 3 dpi when bacterial titers were high, surface lesions grew, and tissue necrosis occurred; next, there was a preresolution phase from 5 to 7 dpi when bacterial levels remained high but surface necrosis began to decline; and finally, there was a resolution phase from 9 dpi onward when bacterial titers declined and the surface wound continually resolved.
Histopathological analysis and Gram staining of S. aureus skin lesions.
While two-dimensional measurements of surface lesions are commonly used as an indicator of skin pathology during S. aureus skin infection, they neither allow the examination of changes in skin architecture below the surface nor provide details on cellular localization. In our studies, we sought to understand subsurface disease progression as well. Therefore, we performed histopathological analysis and Gram staining of S. aureus skin lesions to assess tissue destruction and recovery, bacterial localization, and leukocyte recruitment. At 8 hpi, there was scant infiltration of neutrophils and mild inflammation, and bacteria were aggregated underneath the skeletal muscle (Fig. 2; see also Fig. S1 in the supplemental material). Twenty-four hours after infection, there was more obvious neutrophil infiltration with foci of neutrophil abscesses; tissue overlaying the bacterial colonies exhibited signs of necrosis, as evidenced by sporadic nuclear dissolution (Fig. 2 and Fig. S2). Transitioning from 1 dpi to 3 dpi, there was an extensive neutrophilic abscess with neutrophilic karyorrhexis (nuclear fragmentation during cell death where chromatin is irregularly distributed in the cytoplasm) and a mild mononuclear cell infiltrate with necrosis of both the skin and subcutaneous tissue (Fig. 2 and Fig. S3). The bacteria were observed laterally in the tissue under the lesion. During the preresolution phase (5 to 7 dpi), there was well-developed, full-thickness tissue necrosis involving the epidermis, dermis, adipose tissue, and skeletal muscle, leading to the formation of a discrete ulcer; bacterial colonies were scattered within the necrotic tissue (Fig. 2 and Fig. S4 and S5). The viable tissue flanking the lesion showed reparative epidermal changes and early granulation tissue formation. At 9 dpi, reparative changes were more evident, with an ulcer flanked by reactive epidermis and granulation tissue (Fig. 2 and Fig. S6). Early reepithelization was also observed. At 11 dpi, there was significant epidermal reepithelialization, bacteria were rare and intermixed with the necrotic tissue, and granulation tissues were well formed beneath the scab/wound composed of mononuclear inflammatory cells, neovascularization, and fibroblasts (Fig. 2 and Fig. S7). By 13 and 15 dpi, there were areas of nearly complete or complete epidermal reepithelialization overlaying mature granulation tissue; wound remodeling had occurred with early scar formation (Fig. 2 and Fig. S8 and S9). Together, the histopathology results show dynamic changes in the skin during infection where at the beginning, there was brisk neutrophil recruitment, followed by major destruction of the skin tissue. Upon the resolution of the surface lesion, the subsurface skin architecture remained disrupted.
The cytokine and chemokine profiles were dynamic throughout the infection.
Cytokine and chemokine profiling is commonly used to gauge changes in the immune response to S. aureus infection. These mediators are important for the host response to infection and are necessary for immune cell differentiation, activation, proliferation, and recruitment. Thus, we measured cytokine and chemokine levels at the site of infection. All the cytokines that we measured were below the limit of detection in control mice that were only depilated (naive) or mock infected with phosphate-buffered saline (PBS), except for IL-1α (Fig. S10). This is consistent with previous reports demonstrating high intracellular IL-1α pools within keratinocytes (30–32).
Our kinetic study revealed the production of specific cytokines/chemokines during the three phases of the infection. Many key cytokines important for neutrophil, monocyte, and DC recruitment, differentiation, or activation were readily detectable by as early as 8 hpi and persisted within the lesion for several days (Fig. 3). These included IL-6, CXCL-1 (KC), CCL2 (monocyte chemoattractant protein 1 [MCP-1]), CCL3 (macrophage inflammatory protein 1α [MIP-1α]), granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-4, and IL-1β. Many cytokines and chemokines were at their highest abundance during the preresolution phase of the infection (days 5 to 7 postinfection [p.i.]). These included tumor necrosis factor (TNF), CCL4 (MIP-1β), and CCL5 (RANTES) (Fig. 3). CCL3 was observed during all phases and was absent only in uninfected tissue and at 15 dpi. We were particularly interested in IL-17 since it is critical for combating S. aureus infections (17, 18, 20, 33). The IL-17A level was highest at 5 to 9 dpi, while IL-17F was primarily observed on day 7 p.i., coinciding with initial wound healing and preceding the decrease in bacterial titers. As the infection resolved, most cytokines and chemokines declined and by 15 dpi were below the limit of detection, as observed in uninfected mice. Overall, we observed a dynamic temporal shift in the cytokine/chemokine profile throughout the infection. There was a robust response of a variety of proinflammatory cytokines and chemokines during the early phase of the infection, a separate response marked by IL-17A/F production during the preresolution phase, and, finally, a return to baseline levels during the resolution phase. We did not detect interferon gamma (IFN-γ) or IL-12/IL-23p40 at any time point.
FIG 3.

Cytokines and chemokines detected in the skin during S. aureus infection. The indicated cytokines and chemokines were quantified by cytometric bead arrays. Each symbol represents the value for an individual mouse (n = 9 to 13). Red symbols represent samples whose cytokine/chemokine values were below the limit of detection.
Neutrophil and antigen-presenting cell dynamics in the S. aureus lesion.
From our cytokine and chemokine data, it was clear that the immune environment was dynamic over the course of infection. One way to assess the overall immune response is to quantify and phenotype infected skin leukocytes over time. To do this, we first performed flow cytometry to quantify the number of total immune cells present using CD45, the common leukocyte antigen. As expected, there were detectable leukocytes present in the skin of both depilated (Nair-treated) and mock-infected (PBS-injected) mice. CD45+ cell numbers remained stable for the first week of infection (Fig. 4a). In contrast, there was an increase in the CD45+ cell number during the resolution phase (9 to 15 dpi). This was initially surprising considering the influx of neutrophils to the site of infection observed by histochemical analysis. Neutrophils were not observed in depilated (Nair) and mock-infected (PBS) mice but were readily detectable at 8 hpi as the major leukocyte population present (Fig. 4b). Neutrophils remained the most abundant leukocyte type through 9 dpi (Fig. 4c). After 9 dpi, neutrophil numbers declined as the infection was cleared.
FIG 4.
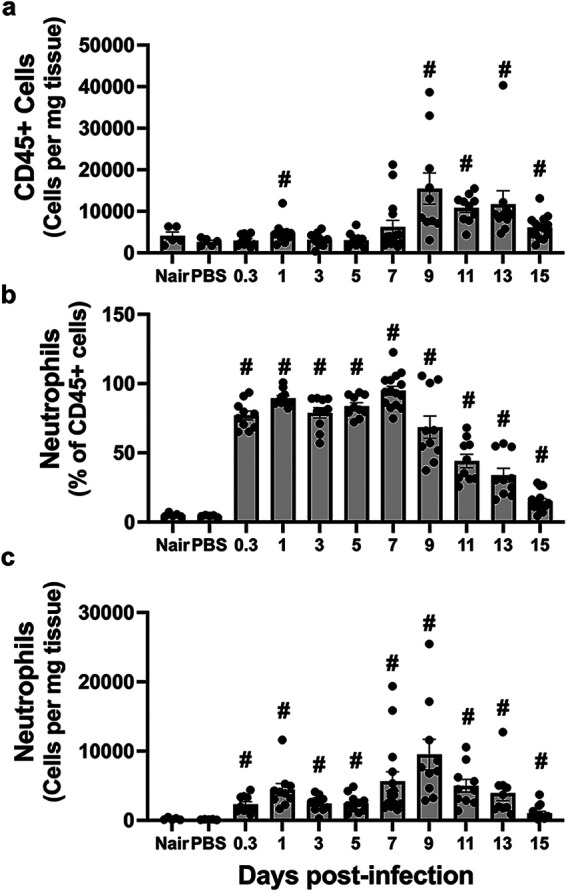
Total leukocytes and neutrophils at the site of infection. (a) Total CD45+ cells per milligram of tissue of either depilated (Nair)-only, mock-infected (PBS), or infected mice at the indicated time points. (b and c) Neutrophils (Ly6G+ CD11b+) were determined as the percentage of total CD45+ cells (b) or the number of neutrophils per milligram of tissue (c). Each symbol represents the value for one mouse. The bars represent the means with SEM (n = 5 to 15 mice/group). “#” indicates a P value of <0.05 by a Mann-Whitney test relative to PBS.
Antigen-presenting cells (APCs), such as macrophages and dendritic cells, process and present S. aureus antigens to T cells. There are resident APCs in the skin, but we found that upon infection, this population significantly decreased (Fig. 5). They remained at significantly reduced numbers until 9 dpi when they then became the major cell type in the skin during the resolution phase (Fig. 5). Given the diverse skin APC populations, we separated them into subsets based on CD11b, CD11c, CCR2, and CD207 expression. First, we found that inflammatory monocytes (CD11b+ Ly6C+ CCR2+) were present in uninfected skin but were largely absent during the early and preresolution phases of the infection. Their numbers returned at around 9 dpi, and they remained until 15 dpi (Fig. S11a). Next, we observed that langerin-positive DCs (CD11b− CD11c+ CD207+) were near homeostatic levels throughout the infection (Fig. S11b). LCs (F4/80+ CD207+ CD11b+ CD11c+) remained largely absent at days 1 through 7 p.i. and then returned to the numbers found in uninfected skin (Fig. S11c). F4/80+ macrophages (F4/80+ CD11b+) were found in uninfected skin, immediately decreased upon infection, and returned at 15 dpi (Fig. S11d). Additional CD11b+ and/or CD11c+ macrophage/DC populations together made up approximately 35% of the immune cells in uninfected skin (Fig. S12). All these cell populations decreased upon infection and remained low during the early and preresolution phases of the infection but returned to nearly homeostatic levels during the resolution phase.
FIG 5.
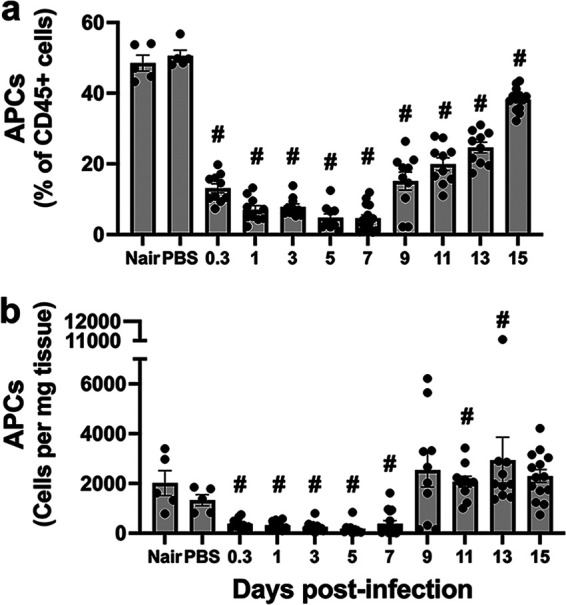
Antigen-presenting cells at the site of infection. Single-cell suspensions derived from skin homogenates from control mice (Nair and PBS) or infected mice isolated at each time point were analyzed to determine the percentage (a) and number (b) of antigen-presenting cells (APCs) (Ly6G− MHC-II+ [major histocompatibility complex class II positive] and CD11b+ and/or CD11c+). Each symbol represents the value for one mouse. The bars represent the means with SEM (n = 5 to 15 mice/group). “#” indicates a P value of <0.05 by a Mann-Whitney test relative to PBS.
Innate lymphocyte dynamics in the S. aureus lesion.
Non-natural killer (NK) innate lymphoid cells (ILCs) are resident skin cells involved in tissue homeostasis and contribute to inflammation during S. aureus epicutaneous infection (19). In noninfected skin, ILCs were prominent, constituting ~30% of all leukocytes (Fig. 6a). Following infection, the number of ILCs decreased rapidly and remained low through 7 dpi. The ILCs began repopulating the skin on day 9 p.i. and were present throughout the resolution phase of the infection.
FIG 6.

Dynamics of the innate lymphoid cell response at the site of infection. Single-cell suspensions derived from skin homogenates from control mice (Nair and PBS) or infected mice isolated at each time point were analyzed to determine the percentages and numbers of ILCs (B220− GR1− CD11b− CD11c− CD3− NK1.1− CD90+) (a) and NK cells (CD3− NK1.1+) (b). Each symbol represents the value for one mouse. The bars represent the means with SEM (n = 5 to 15 mice/group). “#” indicates a P value of <0.05 by a Mann-Whitney test relative to PBS.
NK cells are both tissue-resident and infiltrating cells that play a role in inflammation and immune homeostasis. While they are best known for combating viral infections, NK cells are also important for combating S. aureus lung infections (34). In addition, NK cells are involved in wound healing by delaying resolution to locally contain the pathogen and prevent its systemic spread during group A Streptococcus infection (35). We observed few NK cells in uninfected skin but readily identified them at 8 hpi (Fig. 6b). NK cells were maintained during infection and did not show the rapid decrease observed in the non-NK ILC population. Indeed, NK cells represented ~7% of the CD45+ cells from 8 hpi to 7 dpi, and their elevated number in the tissue was maintained through day 13 (Fig. 6b). NK cells decreased during the resolution phase, approaching baseline levels by 15 dpi.
γδ T cells are found in healthy skin and are key players in tissue homeostasis, responding rapidly to pathogens by producing key cytokines like IL-17 that recruit neutrophils (36, 37). In addition, previous studies have demonstrated their importance in S. aureus skin infections, including the production of IL-17 (17, 18, 20, 21). We observed that γδ T cells were present in the skin prior to infection and at 8 hpi. However, their numbers declined over the subsequent several days and then increased as the infection and tissue damage resolved during the resolution phase (Fig. 7a).
FIG 7.

Dynamics of the T cell response at the site of infection. Single-cell suspensions derived from skin homogenates from control mice (Nair and PBS) or infected mice isolated at each time point were analyzed to determine the percentages and numbers of γδ T cells (CD3+ γδTCR+ [γδ T cell receptor positive]) (a), NKT cells (CD3+ NK1.1+) (b), CD8+ T cells (CD3+ CD8+) (c), and CD4+ T cells (CD3+ CD4+) (d). Each symbol represents the value for one mouse. The bars represent the means and SEM (n = 5 to 15 mice/group). “#” indicates a P value of <0.05 by a Mann-Whitney test relative to PBS.
NKT cells detect and respond to microbial lipids and are not abundant in uninjured skin (38). As expected, we observed very low numbers of NKT cells in uninfected skin, constituting 0.3% of CD45+ cells (Fig. 7b). NKT cell numbers stayed low after infection until the resolution phase, where we observed an increase in a subset of mice.
Adaptive lymphocyte dynamics in the S. aureus lesion.
αβ CD8+ and CD4+ T cells have shown mixed importance in their contribution to combating S. aureus infection, and their contribution may depend on the location of the infection, the strain of S. aureus used, and whether it is a primary or secondary infection (20, 24, 25, 39, 40). We found that CD4+ and CD8+ T cells were present in uninfected skin (Fig. 7c and d). Both cell types were largely absent at 8 hpi and were not detected again until the resolution phase of the infection.
S. aureus produces virulence factors that target B cells, and antibodies against S. aureus are easily detected in blood, yet B cells are thought to not play a major role in combating S. aureus during systemic infection (40). As expected, we identified B cells in uninfected skin, representing ~15% of leukocytes (Fig. S13). Like other cell types, the numbers of B cells in the skin declined upon infection and remained low until 9 dpi, returning to the levels found in naive (Nair) and mock-infected (PBS) mice by 13 dpi.
DISCUSSION
The results of this study describe the kinetic response to S. aureus subcutaneous skin infection in a mouse model. Based on lesion development and bacterial titers, we categorized the infection into three phases: the early response (8 hpi to 3 dpi), the preresolution phase (5 to 7 dpi), and the resolution phase (9 to 15 dpi). Our data demonstrate a dynamic immune response during S. aureus skin infection; the leukocyte population summary is represented in Fig. 8. Prior to infection, ILCs and APCs were the predominant immune cells present in the skin, and there were few to no cytokines present. Upon infection, neutrophils entered the skin and remained the predominant cell type for the first 9 days, which we observed by both flow cytometry and histopathological analysis. Despite the high neutrophil abundance, bacterial titers remained consistently high during this time and spread laterally in the tissue under the lesion. During early infection, NK cells and neutrophils were present at higher numbers than before infection, but most other cell types decreased; bacterial titers remained consistent at around 106 CFU/mg tissue; and G-CSF, CXCL-1, IL-6, IL-4, MCP-1, and IL-1β were abundant. During the preresolution phase, neutrophils remained the predominant cell type; NK cells were still present at higher numbers; bacterial titers remained at around 106 CFU/mg tissue; and CCL4, IL-17A, and IL-17F levels increased. Necrosis was observed from the epidermis down to the skeletal muscle underneath the skin, and bacteria were seen throughout the necrotic tissue. During the resolution phase, neutrophils, while still present, were no longer the predominant cell type. Instead, ILCs and APCs returned to the infection site, CD4+ and CD8+ T cells arrived in the tissue, NK cells decreased, and most cytokines/chemokines were either at or below the limit of detection. Reepithelialization and tissue repair began at around day 9 and continued through day 15. Collectively, these data demonstrate the complex dynamics of the host response to S. aureus skin infection at both the immunological and histological levels.
FIG 8.
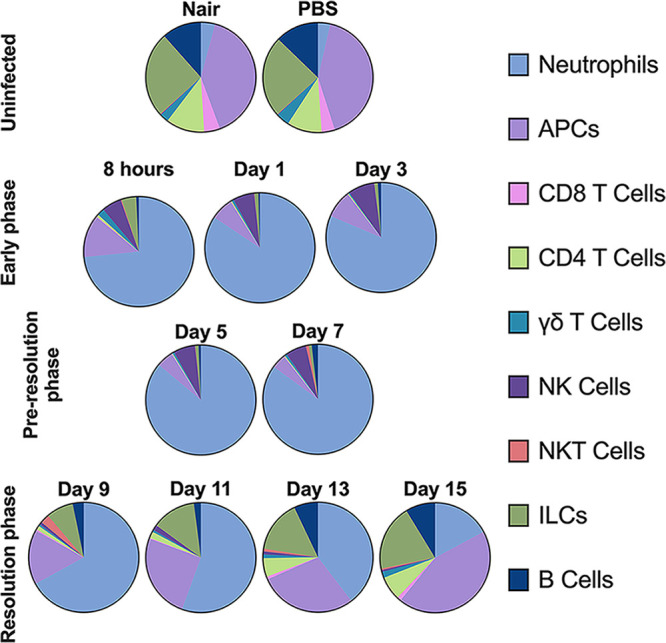
Summary of leukocyte changes during S. aureus skin infection compared to uninfected skin. Data are the average percentages of each cell population.
Upon tissue damage or infection, skin-resident cells, including leukocytes and keratinocytes, produce chemokines to recruit additional leukocyte populations. Initially, we were surprised to find that the number of CD45+ cells at the infection site did not change above those of uninfected skin for the first 7 days despite the recruitment of neutrophils; however, this large neutrophil influx was offset by the loss of other resident immune cell types, such as ILCs, APCs, and T cells. Indeed, every cell population examined, other than neutrophils, CD207+ DCs, and NK cells, decreased upon infection. This countereffect resulted in no net change in leukocyte numbers at the site of infection but a drastic change in the composition of the leukocytes found in the skin. This held true through the first 7 days of infection, after which there was a rebalance of the leukocyte populations as the infection was cleared and the wound was repaired during the resolution phase.
The decrease in the cell populations that we observed may be the result of leukocytes leaving the infection site or cell death. The numbers of both ILCs and APCs largely decreased after infection. Although ILCs can migrate, they are typically tissue-resident cells, and we hypothesize that the loss of ILCs in the skin after infection is the result of cell death during the infection. Similarly, APCs are also tissue resident but migrate out of tissues upon stimulation as a key component of their role in the immune response. Considering the amount of observable tissue damage, we hypothesize that the general loss of these cells was also due to extensive cell death. This is not surprising considering the number of cytolytic proteins produced by S. aureus, including the phenol-soluble modulins and alpha-hemolysin, which contribute to necrosis and can kill host cells, including various leukocyte subsets (7, 27, 29, 41–46). We did identify several cell types whose numbers did not decrease upon infection. For example, we found an immediate increase in NK cells, whose numbers persisted for 7 days. Either there was a continued influx of NK cells to the infection site or NK cells were resistant to killing during infection. Discerning between these possibilities is a focus of ongoing studies. In addition, the numbers of CD207+ DCs remained similar to those in control animals even after infection. Again, whether these cells are resistant to killing, proliferate, or are continually replenished at the site of infection will require additional investigation.
Considering studies that demonstrated an important role of IL-17 in combating S. aureus infections, we were particularly interested in the timing of IL-17 production. We originally chose the 8-h time point due to a previous report that IL-17A and IL-17F mRNA spiked then during intradermal infection (17); however, our study showed that IL-17F protein levels were either absent or below the level of detection at 8 hpi. Indeed, we observed IL-17F only in all animals at 7 dpi and in a subset of animals at 9 dpi. For IL-17A, we observed very low levels of protein, except between days 5 and 9, where IL-17A levels were elevated. While there are scant IL-17 kinetic data from other studies, IL-17 protein is readily detectable at 7 dpi in other studies (18, 19, 24). Of note, the peak IL-17 levels that we observed immediately preceded bacterial clearance and the start of the resolution phase of the infection. Given the striking correlation of IL-17A/F production just prior to the decrease in bacterial titers in our study, this supports the hypothesis that IL-17 production at this later time point is important for bacterial clearance.
IL-17A/F is critical for combating S. aureus skin infections (18). While many cells produce IL-17, its production in response to S. aureus skin infection is thought to be controlled largely by γδ T cells, although other cells, such as ILCs, Th17 cells, or CD8+ T cells, may be important as well (17–20, 23, 24, 47). Mice deficient in γδ T cells have higher skin bacterial titers early in an S. aureus intracutaneous infection model (21). During cutaneous infection, C57BL/6J mice deficient in either γδ T cells or IL-17 receptor (IL-17R) had both larger surface lesions and higher bacterial titers over 2 weeks of infection (17). A similar result was observed using a surgical site infection model with C57BL/6 mice lacking IL-17R, IL-1R, or γδ T cells (20); however, the relative importance depended on the strain of S. aureus used. IL-17 also contributes to altered inflammation and the ability to combat S. aureus skin infection using epicutaneous (19, 24) and intradermal (18) models. While most of these studies attribute IL-17 production to γδ T cells to recruit neutrophils, they also point to the potential roles of type 3 ILCs (ILC3s), CD4+ T cells, and Th17 cells as additional IL-17 producers.
The IL-17 spike that we observed immediately prior to bacterial clearance supports the body of evidence that IL-17 is a key mediator in immune defense against S. aureus. Therefore, we are intrigued as to which cell type(s) is responsible for the production of IL-17 in the preresolution phase of the infection. We observed the highest numbers of γδ T cells at 8 hpi and then again at 9 dpi and beyond. This timing did not align with the observed elevated IL-17A/F quantities in the skin. While we cannot rule out that γδ T cells are producing IL-17 early in infection (8 h), we did not observe a protein spike as seen in a previous mRNA study, which used an epicutaneous model of infection (17). A spike in IL-17 mRNA expression at the early time point in this model could be because the bacteria are in the epidermis, where γδ T cells reside, much earlier than in our studies using the subcutaneous infection model where it takes longer for the bacteria to access this site (Fig. 2). CD4+ Th17 cells also produce IL-17 and contribute to skin inflammation during epicutaneous S. aureus infection (24); however, we found that the CD4+ population returned after the observed production of IL-17, suggesting that they may not be the main producer of IL-17 during the transition to the resolution phase. Two other candidate cells for IL-17 production include ILC3s and NK cells. ILC3s produce IL-17 and contribute to skin inflammation during S. aureus infection (19). Again, ILC populations were small at 5 to 9 dpi when IL-17 was maximally detected. NK cells produce IL-17 and help combat S. aureus lung infections (34). The numbers of NK cells increased during infection and were maintained until 7 dpi. NK cells were present in elevated numbers when we observed the peak level of IL-17, but they were also abundant earlier when little IL-17 was detected. Finally, while CD8+ T cells are not considered major players in the IL-17 response to S. aureus skin infections, these cells produce IL-17A and impact wound repair in response to S. epidermidis (47). Identifying whether the late IL-17A/F spike is indeed critical for bacterial clearance, as well as the cells producing this IL-17, is a major focus of our ongoing studies.
The goal of this study was to provide a kinetic examination of the immune response to S. aureus subcutaneous infections. Identifying how the leukocyte populations change over time is key to understanding the immune response during these infections. We have demonstrated a striking and immediate loss of most skin leukocyte populations that recovers only after a critical event corresponding to reductions in recruited neutrophil and NK numbers and a precisely timed yet ephemeral IL-17A/F burst. Moreover, this study provides a foundation on which further studies can dissect the mechanisms behind S. aureus pathogenesis, the immune response to S. aureus infection, and the process of wound repair. This resource will be invaluable for investigations of how the inactivation of genes in either the host or the bacterium impacts the pathogenesis of S. aureus skin infections.
MATERIALS AND METHODS
Preparation of inocula.
For these experiments, we used the AH1263 strain (48), a derivative of the USA300 strain LAC lacking the LAC-p03 plasmid. Bacteria were retrieved from −80°C freezer stocks and inoculated into 3 mL of tryptic soy broth (TSB). The culture was incubated at 37°C for 16 h with shaking at 250 rpm with a 1-in. orbit. The culture was then diluted 1:100 in 10 mL TSB in a 50-mL conical tube with a loose cap and grown for ~4.5 h at 37°C with shaking at 250 rpm. Bacteria were pelleted at 4,000 × g for 10 min, washed with Dulbecco’s phosphate-buffered saline (DPBS) without calcium and magnesium, pelleted again, and resuspended to 3.85 × 107 to 4.71 × 107 CFU per 50-μL injection. The CFU concentrations were confirmed by dilution plating.
Subcutaneous infection model.
All animal studies were conducted in strict accordance with approved protocols (2018-2451 and 2021-2613) by the University of Kansas Medical Center Institutional Animal Care and Use Committee. Eight-week-old female C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) were infected as previously described (28, 29), with modifications. In brief, mice were anesthetized using isoflurane 5 days before infection for mechanical hair removal using a veterinary shaver. The following day, the mice were treated with Chapstick, and chemical depilation (Nair with cocoa butter; Church & Dwight, Ewing, NJ) was performed to destroy the remaining hair follicles and delay hair regrowth. The Nair was removed using an isopropanol wipe, and the mice were then treated with ointment (Aquaphor; Beiersdorf, Inc., Wilton, CT). For the following 2 days, Aquaphor was applied. On infection day, mice were again anesthetized using isoflurane and inoculated subcutaneously with ~4 × 107 CFU suspended in 50 μL DPBS into the back or flank of mice using a 27-gauge needle. Mice were weighed and their infection site was photographed every other day. A ruler was included in the frame to use for measurement reference. For each time point, mice were euthanized using CO2 asphyxiation. The lesion and ~3 mm of the surrounding healthy tissue were excised. Skin extracts were placed into either cassettes (for histology), Hanks’ balanced salt solution (HBSS) (no cations) with 0.2% (wt/vol) human serum albumin (HSA) (catalog number A5843; Sigma-Aldrich, St. Louis, MO) and 10 mM HEPES (for CFU and cytokines), or gentleMACS tissue storage solution (Miltenyi Biotec, Bergisch Gladbach, Germany) (for immune cell isolation). Control mice included naive mice (depilated, with no subcutaneous injection) or mice mock inoculated with DPBS after depilation and were then treated as infected mice; tissue was harvested from these two control mouse groups at a time correlating to 3 dpi. The lesion size was determined from photographs using ImageJ software (NIH).
Histological procedures.
Lesions were dissected from mouse skin, maintaining an ~3-mm border of healthy tissue around each lesion. Next, the lesions were bisected along the shortest dimension, and one-half was placed into a histology cassette with a tissue sponge (25 by 31 mm; Epredia, Kalamazoo, MI) to limit skin curling during fixation. The cassette was then placed into 10% neutral buffered formalin and placed onto a platform orbital shaker (New Brunswick Innova 2050; Eppendorf, Enfield, CT) at 115 rpm (1.9-cm radius) for 16 to 18 h at room temperature. After fixation, the fixative was replaced with 70% ethanol, and the tissues were again incubated on a platform orbital shaker as described above. The tissues were stored at 4°C until they were processed, embedded in paraffin wax, and sectioned using standard procedures (Kansas Intellectual and Developmental Disabilities Research Center Histology Core). Skin sections (5 μm) were placed onto glass slides and stained with hematoxylin and eosin (H&E), after which coverslips were attached to the slides using Permount (Electron Microscopy Sciences, Hatfield, PA). H&E-stained skin sections were scanned using an Aperio AT2 instrument (Leica Biosystems, Buffalo Grove, IL) at a ×200 magnification.
Alcoholic saffron-modified Gram stain.
Gram staining of tissue sections was performed as previously described (49). Skin sections (5 μm thick) were deparaffinized in CitriSolv (3 times for 3 min each), after which they were rehydrated in a series of ethanol baths (100% ethanol, twice for 1 min each; 95% ethanol, once for 1 min; 70% ethanol, once for 1 min; and 30% ethanol, once for 1 min) and then incubated in reverse-osmosis water (twice for 1 min each) with agitation on a platform orbital shaker (115 rpm, 1.9-cm radius). Next, the slides were incubated in crystal violet for 5 min, rinsed in cool tap water until the water ran clear, incubated in Gram iodine for 2 min, rinsed again, incubated in Gram decolorizer for 30 s, and rinsed again in cool tap water. Next, the slides were incubated in Gram safranin for 1 min 40 s and rinsed a final time in cool tap water. Thereafter, the sections were dehydrated in a series of ethanol baths (95% ethanol, once for 1 min, and 100% ethanol, once for 1 min) and then incubated in alcoholic saffron for 5 min. After a final ethanol incubation step (100%, once for 1 min), the slides were submerged in fresh CitriSolv for 5 min, and the coverslips were then attached using Cytoseal XYL (Richard Allan Scientific, Kalamazoo, MI). Alcoholic saffron-modified Gram-stained sections were imaged using an Aperio AT2 instrument at a ×200 magnification.
Histopathology analysis.
Microscopic evaluations of the H&E-stained sections and Gram-stained sections were performed by a board-certified dermatopathologist using light microscopy (BX46; Olympus, Tokyo, Japan). The histopathological changes at each time point were recorded.
Bacterial titer enumeration.
As described previously (29), dissected skin was bisected, minced, and placed into two lysing matrix H 2-mL tubes (MP Biomedicals, Irvine, CA) containing buffer (HBSS with 0.2% HSA and 10 mM HEPES). Skin samples were then homogenized according to the manufacturer’s protocol for human skin in a FastPrep-24 5G homogenizer (MP Biomedicals). The previously separated samples were combined and serially diluted in PBS for CFU enumeration on tryptic soy agar plates. The remaining homogenate was clarified by centrifugation at 10,000 × g for 10 min at 4°C. Complete protease inhibitor (Roche, Basel, Switzerland) and 0.8 mM EDTA (Fisher Chemical, Hampton, NH) were added to the supernatant, and the sample was stored at −80°C for future cytokine and chemokine analyses.
Isolating immune cells from the skin.
A protocol modified from the one for the Miltenyi Biotec whole-skin dissociation kit for human skin was used to generate single-cell suspensions. Briefly, skin extracts were washed in gentleMACS tissue storage solution, minced, and placed into a C tube with 435 μL RPMI 1640. Enzymes A (2.5 μL), D (50 μL), and R (12.5 μL) from multitissue dissociation kit 1 were added to each tube, and the samples were incubated in a 37°C water bath for 3 h. Once removed from the water bath, 500 μL of cold cell culture medium (RPMI 1640 plus 10% fetal bovine serum [FBS]) was added to the C tube. The tissue was dissociated using protocol h_skin_01 in a gentleMACS dissociator. C tubes were centrifuged at 200 × g briefly to collect tissue to the bottom of the tube. The tubes were rinsed with 4 mL cold cell culture medium, and samples were poured through a 40-μm strainer into a 50-mL tube. The 50-mL tubes were then centrifuged at 300 × g at 4°C for 10 min, and the cells were resuspended in buffer (PBS containing 2% FBS and 0.1% sodium azide) supplemented with brilliant stain buffer plus (BD Biosciences). Live cells were enumerated using trypan blue staining and a hemocytometer.
Flow cytometry.
Isolated cells were separated into 3 samples, treated with 0.5 μg mL−1 Fc block (BD Biosciences, Franklin Lakes, NJ) for 5 min at 4°C, and incubated with optimized antibody cocktails as outlined in Table 1. Panel 1 was used to immunophenotype neutrophils and antigen-presenting cells. Panel 2 was used to immunophenotype ILCs and B cells. Finally, panel 3 was used to immunophenotype T cells and NK cells. Antibodies were incubated with samples in the dark at 4°C for 20 min. The samples were treated with 4′,6-diamidino-2-phenylindole (DAPI) and analyzed using a BD LSRII instrument at the University of Kansas Flow Cytometry Core. Analysis was performed using FlowJo software, version 10. Gating strategies for identifying each cell population are shown in Fig. S14 to S16 in the supplemental material. Panel 1 did not contain a CD45 antibody; therefore, to determine the percentages and numbers of the cell populations, we used the average for CD45+ cells found in the other two panels.
TABLE 1.
Antibody panels
| Marker | Colorb | Manufacturer, catalog no.a |
|---|---|---|
| Panel 1 | ||
| CCR2 | BV786 | BD, 747966 |
| CD11b | BV510 | BD, 562950 |
| CD11c | PerCP/Cy5.5 | BL, 117328 |
| CD207 | APC | MB, 130-113-053 |
| F4/80 | BV650 | BD, 744338 |
| Ly6C | AF700 | BL, 128023 |
| Ly6G | FITC | BD, 551460 |
| MHC-II (I-Ab) | PE | BD, 553552 |
| Dead cells | DAPI | BD, 561907, or Invitrogen, D1306 |
| Panel 2 | ||
| CD11b | BV510 | BD, 562950 |
| CD11c | PerCP/Cy5.5 | BL, 117328 |
| CD45 | BV711 | BD, 563709 |
| CD3ε | BV605 | BL, 100351 |
| CD90.2 | APC | BD, 561974 |
| GR1 | FITC | BD, 553126 |
| NK1.1 | PE | BD, 553165 |
| B220 | BV786 | BD, 563894 |
| Dead cells | DAPI | BD, 561907, or Invitrogen, D1306 |
| Panel 3 | ||
| CD45 | PE-CF594 | BD, 562420 |
| CD3ε | BV605 | BL, 100351 |
| CD4 | PE-Cy5 | BL, 130312 |
| CD8α | BV711 | BL, 100759 |
| NK1.1 | APC | MB, 130-120-507 |
| γδTCR | FITC | eB, 11-5711-82 |
| Dead cells | DAPI | BD, 561907, or Invitrogen, D1306 |
BD, BD Biosciences; BL, BioLegend; MB, Miltenyi Biotec; eB, eBioscience.
PerCP, peridinin chlorophyll protein; APC, allophycocyanin; FITC, fluorescein isothiocyanate; PE, phycoerythrin; BV, brilliant violet; AF, alexa fluor.
Measuring cytokine production.
Frozen homogenates were thawed, and cytokines and chemokines were quantified using cytometric bead array assays (BD Biosciences) according to the manufacturer’s recommendations and protocols on a BD LSRII instrument. A custom Flex set was used to assess the following cytokines: KC (CXCL1), IL-6, MIP-1α (CCL3), TNF, MIP-1β (CCL4), IL-12/IL-23p40, IL-1β, GM-CSF, RANTES, IFN-γ, G-CSF, MCP-1 (CCL2), IL-17F, and IL-1α. The enhanced-sensitivity Flex set (BD Biosciences) was used for IL-4, IL-17A, and IL-10 quantification.
Statistical analysis.
Statistical analyses were performed using GraphPad Prism, version 9. Significance in the figures was determined using a Mann-Whitney test compared to mock-infected (PBS) control mice, with tissues harvested correlating to 3 dpi (or 8 hpi in Fig. 1c). Pairwise comparisons between time points and controls for all leukocyte populations in the text are provided in Table S1 in the supplemental material and were conducted by a nonparametric Kruskal-Wallis test comparing the mean ranks of each column (Nair, PBS, or time point) without correction for multiple comparisons (uncorrected Dunn’s test).
ACKNOWLEDGMENTS
We thank Jing Huang at the Kansas Intellectual and Developmental Disabilities Research Center Histology Core for her assistance.
This research was supported by funding from National Institute of Allergy and Infectious Diseases (NIAID) award R01AI121073, pilot project grants from K-INBRE (P20GM130418) and University of Kansas Chemical Biology of Infectious Disease COBRE grant P20GM113117 to J.L.B., as well as grant MF-2104-01575 from The Harold G. and Leila Y. Mathers Foundation to M.T.P. We acknowledge the Kansas Intellectual and Developmental Disabilities Research Center Histology Core, which is sponsored in part by the NIH/NICHD (U54 HD090216), and the Flow Cytometry Core Laboratory, which is sponsored in part by NIH/NIGMS COBRE grant P30 GM103326 and NIH/NCI Cancer Center grant P30 CA168524.
J.L.B. serves on the scientific advisory boards and is a consultant for Azitra, Inc., and Merck & Co, Inc. M.A.M. serves as a consultant for Design-Zyme. These activities did not financially support and are unrelated to the current study.
Footnotes
Supplemental material is available online only.
Contributor Information
Mary A. Markiewicz, Email: mmarkiewicz@kumc.edu.
Jeffrey L. Bose, Email: jbose@kumc.edu.
Nancy E. Freitag, University of Illinois at Chicago
REFERENCES
- 1.Tognetti L, Martinelli C, Berti S, Hercogova J, Lotti T, Leoncini F, Moretti S. 2012. Bacterial skin and soft tissue infections: review of the epidemiology, microbiology, aetiopathogenesis and treatment: a collaboration between dermatologists and infectivologists. J Eur Acad Dermatol Venereol 26:931–941. 10.1111/j.1468-3083.2011.04416.x. [DOI] [PubMed] [Google Scholar]
- 2.Moet GJ, Jones RN, Biedenbach DJ, Stilwell MG, Fritsche TR. 2007. Contemporary causes of skin and soft tissue infections in North America, Latin America, and Europe: report from the SENTRY Antimicrobial Surveillance Program (1998-2004). Diagn Microbiol Infect Dis 57:7–13. 10.1016/j.diagmicrobio.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Kourtis AP, Hatfield K, Baggs J, Mu Y, See I, Epson E, Nadle J, Kainer MA, Dumyati G, Petit S, Ray SM, Emerging Infections Program MRSA Author Group, Ham D, Capers C, Ewing H, Coffin N, McDonald LC, Jernigan J, Cardo D. 2019. Vital signs: epidemiology and recent trends in methicillin-resistant and in methicillin-susceptible Staphylococcus aureus bloodstream infections—United States. MMWR Morb Mortal Wkly Rep 68:214–219. 10.15585/mmwr.mm6809e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Oliveira S, Rosowski EE, Huttenlocher A. 2016. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol 16:378–391. 10.1038/nri.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mölne L, Verdrengh M, Tarkowski A. 2000. Role of neutrophil leukocytes in cutaneous infection caused by Staphylococcus aureus. Infect Immun 68:6162–6167. 10.1128/IAI.68.11.6162-6167.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsumoto M, Nakagawa S, Zhang L, Nakamura Y, Villaruz AE, Otto M, Wolz C, Inohara N, Núñez G. 2021. Interaction between Staphylococcus Agr virulence and neutrophils regulates pathogen expansion in the skin. Cell Host Microbe 29:930–940.e4. 10.1016/j.chom.2021.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen TH, Cheung GYC, Rigby KM, Kamenyeva O, Kabat J, Sturdevant DE, Villaruz AE, Liu R, Piewngam P, Porter AR, Firdous S, Chiou J, Park MD, Hunt RL, Almufarriji FMF, Tan VY, Asiamah TK, McCausland JW, Fisher EL, Yeh AJ, Bae JS, Kobayashi SD, Wang JM, Barber DL, DeLeo FR, Otto M. 2022. Rapid pathogen-specific recruitment of immune effector cells in the skin by secreted toxins. Nat Microbiol 7:62–72. 10.1038/s41564-021-01012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagl M, Kacani L, Müllauer B, Lemberger E-M, Stoiber H, Sprinzl GM, Schennach H, Dierich MP. 2002. Phagocytosis and killing of bacteria by professional phagocytes and dendritic cells. Clin Diagn Lab Immunol 9:1165–1168. 10.1128/cdli.9.6.1165-1168.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schindler D, Gutierrez MG, Beineke A, Rauter Y, Rohde M, Foster S, Goldmann O, Medina E. 2012. Dendritic cells are central coordinators of the host immune response to Staphylococcus aureus bloodstream infection. Am J Pathol 181:1327–1337. 10.1016/j.ajpath.2012.06.039. [DOI] [PubMed] [Google Scholar]
- 10.Mitsui H, Watanabe T, Saeki H, Mori K, Fujita H, Tada Y, Asahina A, Nakamura K, Tamaki K. 2004. Differential expression and function of Toll-like receptors in Langerhans cells: comparison with splenic dendritic cells. J Invest Dermatol 122:95–102. 10.1046/j.0022-202X.2003.22116.x. [DOI] [PubMed] [Google Scholar]
- 11.West HC, Bennett CL. 2017. Redefining the role of Langerhans cells as immune regulators within the skin. Front Immunol 8:1941. 10.3389/fimmu.2017.01941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seneschal J, Clark RA, Gehad A, Baecher-Allan CM, Kupper TS. 2012. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity 36:873–884. 10.1016/j.immuni.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deckers J, Hammad H, Hoste E. 2018. Langerhans cells: sensing the environment in health and disease. Front Immunol 9:93. 10.3389/fimmu.2018.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Dalen R, De La Cruz Diaz JS, Rumpret M, Fuchsberger FF, van Teijlingen NH, Hanske J, Rademacher C, Geijtenbeek TBH, van Strijp JAG, Weidenmaier C, Peschel A, Kaplan DH, van Sorge NM. 2019. Langerhans cells sense Staphylococcus aureus wall teichoic acid through langerin to induce inflammatory responses. mBio 10:e00330-19. 10.1128/mBio.00330-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feuerstein R, Seidl M, Prinz M, Henneke P. 2015. MyD88 in macrophages is critical for abscess resolution in staphylococcal skin infection. J Immunol 194:2735–2745. 10.4049/jimmunol.1402566. [DOI] [PubMed] [Google Scholar]
- 16.Abtin A, Jain R, Mitchell AJ, Roediger B, Brzoska AJ, Tikoo S, Cheng Q, Ng LG, Cavanagh LL, von Andrian UH, Hickey MJ, Firth N, Weninger W. 2014. Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat Immunol 15:45–53. 10.1038/ni.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, Magorien JE, Blauvelt A, Kolls JK, Cheung AL, Cheng G, Modlin RL, Miller LS. 2010. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest 120:1762–1773. 10.1172/JCI40891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marchitto MC, Dillen CA, Liu H, Miller RJ, Archer NK, Ortines RV, Alphonse MP, Marusina AI, Merleev AA, Wang Y, Pinsker BL, Byrd AS, Brown ID, Ravipati A, Zhang E, Cai SS, Limjunyawong N, Dong X, Yeaman MR, Simon SI, Shen W, Durum SK, O’Brien RL, Maverakis E, Miller LS. 2019. Clonal Vγ6+Vδ4+ T cells promote IL-17-mediated immunity against Staphylococcus aureus skin infection. Proc Natl Acad Sci USA 116:10917–10926. 10.1073/pnas.1818256116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakagawa S, Matsumoto M, Katayama Y, Oguma R, Wakabayashi S, Nygaard T, Saijo S, Inohara N, Otto M, Matsue H, Núñez G, Nakamura Y. 2017. Staphylococcus aureus virulent PSMalpha peptides induce keratinocyte alarmin release to orchestrate IL-17-dependent skin inflammation. Cell Host Microbe 22:667–677.e5. 10.1016/j.chom.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maher BM, Mulcahy ME, Murphy AG, Wilk M, O’Keeffe KM, Geoghegan JA, Lavelle EC, McLoughlin RM. 2013. Nlrp-3-driven interleukin 17 production by γδT cells controls infection outcomes during Staphylococcus aureus surgical site infection. Infect Immun 81:4478–4489. 10.1128/IAI.01026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molne L, Corthay A, Holmdahl R, Tarkowski A. 2003. Role of gamma/delta T cell receptor-expressing lymphocytes in cutaneous infection caused by Staphylococcus aureus. Clin Exp Immunol 132:209–215. 10.1046/j.1365-2249.2003.02151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nippe N, Varga G, Holzinger D, Löffler B, Medina E, Becker K, Roth J, Ehrchen JM, Sunderkötter C. 2011. Subcutaneous infection with S. aureus in mice reveals association of resistance with influx of neutrophils and Th2 response. J Invest Dermatol 131:125–132. 10.1038/jid.2010.282. [DOI] [PubMed] [Google Scholar]
- 23.Ferraro A, Buonocore SM, Auquier P, Nicolas I, Wallemacq H, Boutriau D, van der Most RG. 2019. Role and plasticity of Th1 and Th17 responses in immunity to Staphylococcus aureus. Hum Vaccin Immunother 15:2980–2992. 10.1080/21645515.2019.1613126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H, Archer NK, Dillen CA, Wang Y, Ashbaugh AG, Ortines RV, Kao T, Lee SK, Cai SS, Miller RJ, Marchitto MC, Zhang E, Riggins DP, Plaut RD, Stibitz S, Geha RS, Miller LS. 2017. Staphylococcus aureus epicutaneous exposure drives skin inflammation via IL-36-mediated T cell responses. Cell Host Microbe 22:653–666.e5. 10.1016/j.chom.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLoughlin RM, Solinga RM, Rich J, Zaleski KJ, Cocchiaro JL, Risley A, Tzianabos AO, Lee JC. 2006. CD4+ T cells and CXC chemokines modulate the pathogenesis of Staphylococcus aureus wound infections. Proc Natl Acad Sci USA 103:10408–10413. 10.1073/pnas.0508961103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zapf RL, Wiemels RE, Keogh RA, Holzschu DL, Howell KM, Trzeciak E, Caillet AR, King KA, Selhorst SA, Naldrett MJ, Bose JL, Carroll RK. 2019. The small RNA Teg41 regulates expression of the alpha phenol-soluble modulins and is required for virulence in Staphylococcus aureus. mBio 10:e02484-18. 10.1128/mBio.02484-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krute CN, Krausz KL, Markiewicz MA, Joyner JA, Pokhrel S, Hall PR, Bose JL. 2016. Generation of a stable plasmid for in vitro and in vivo studies of Staphylococcus species. Appl Environ Microbiol 82:6859–6869. 10.1128/AEM.02370-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bose JL, Daly SM, Hall PR, Bayles KW. 2014. Identification of the Staphylococcus aureus vfrAB operon, a novel virulence factor regulatory locus. Infect Immun 82:1813–1822. 10.1128/IAI.01655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ridder MJ, Daly SM, Triplett KD, Seawell NA, Hall PR, Bose JL. 2020. Staphylococcus aureus fatty acid kinase FakA modulates pathogenesis during skin infection via proteases. Infect Immun 88:e00163-20. 10.1128/IAI.00163-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wood LC, Elias PM, Calhoun C, Tsai JC, Grunfeld C, Feingold KR. 1996. Barrier disruption stimulates interleukin-1 alpha expression and release from a pre-formed pool in murine epidermis. J Invest Dermatol 106:397–403. 10.1111/1523-1747.ep12343392. [DOI] [PubMed] [Google Scholar]
- 31.Lee RT, Briggs WH, Cheng GC, Rossiter HB, Libby P, Kupper T. 1997. Mechanical deformation promotes secretion of IL-1 alpha and IL-1 receptor antagonist. J Immunol 159:5084–5088. [PubMed] [Google Scholar]
- 32.Freedberg IM, Tomic-Canic M, Komine M, Blumenberg M. 2001. Keratins and the keratinocyte activation cycle. J Invest Dermatol 116:633–640. 10.1046/j.1523-1747.2001.01327.x. [DOI] [PubMed] [Google Scholar]
- 33.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y. 2009. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30:108–119. 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 34.Small C-L, McCormick S, Gill N, Kugathasan K, Santosuosso M, Donaldson N, Heinrichs DE, Ashkar A, Xing Z. 2008. NK cells play a critical protective role in host defense against acute extracellular Staphylococcus aureus bacterial infection in the lung. J Immunol 180:5558–5568. 10.4049/jimmunol.180.8.5558. [DOI] [PubMed] [Google Scholar]
- 35.Sobecki M, Krzywinska E, Nagarajan S, Audigé A, Huỳnh K, Zacharjasz J, Debbache J, Kerdiles Y, Gotthardt D, Takeda N, Fandrey J, Sommer L, Sexl V, Stockmann C. 2021. NK cells in hypoxic skin mediate a trade-off between wound healing and antibacterial defence. Nat Commun 12:4700. 10.1038/s41467-021-25065-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ness-Schwickerath KJ, Morita CT. 2011. Regulation and function of IL-17A- and IL-22-producing γδ T cells. Cell Mol Life Sci 68:2371–2390. 10.1007/s00018-011-0700-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qi C, Wang Y, Li P, Zhao J. 2021. Gamma delta T cells and their pathogenic role in psoriasis. Front Immunol 12:627139. 10.3389/fimmu.2021.627139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wun KS, Cameron G, Patel O, Pang SS, Pellicci DG, Sullivan LC, Keshipeddy S, Young MH, Uldrich AP, Thakur MS, Richardson SK, Howell AR, Illarionov PA, Brooks AG, Besra GS, McCluskey J, Gapin L, Porcelli SA, Godfrey DI, Rossjohn J. 2011. A molecular basis for the exquisite CD1d-restricted antigen specificity and functional responses of natural killer T cells. Immunity 34:327–339. 10.1016/j.immuni.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown AF, Murphy AG, Lalor SJ, Leech JM, O’Keeffe KM, Mac Aogáin M, O’Halloran DP, Lacey KA, Tavakol M, Hearnden CH, Fitzgerald-Hughes D, Humphreys H, Fennell JP, van Wamel WJ, Foster TJ, Geoghegan JA, Lavelle EC, Rogers TR, McLoughlin RM. 2015. Memory Th1 cells are protective in invasive Staphylococcus aureus infection. PLoS Pathog 11:e1005226. 10.1371/journal.ppat.1005226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmaler M, Jann NJ, Ferracin F, Landmann R. 2011. T and B cells are not required for clearing Staphylococcus aureus in systemic infection despite a strong TLR2-MyD88-dependent T cell activation. J Immunol 186:443–452. 10.4049/jimmunol.1001407. [DOI] [PubMed] [Google Scholar]
- 41.Cheung GYC, Rigby K, Wang R, Queck SY, Braughton KR, Whitney AR, Teintze M, DeLeo FR, Otto M. 2010. Staphylococcus epidermidis strategies to avoid killing by human neutrophils. PLoS Pathog 6:e1001133. 10.1371/journal.ppat.1001133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forsman H, Christenson K, Bylund J, Dahlgren C. 2012. Receptor-dependent and -independent immunomodulatory effects of phenol-soluble modulin peptides from Staphylococcus aureus on human neutrophils are abrogated through peptide inactivation by reactive oxygen species. Infect Immun 80:1987–1995. 10.1128/IAI.05906-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berube BJ, Bubeck Wardenburg J. 2013. Staphylococcus aureus α-toxin: nearly a century of intrigue. Toxins (Basel) 5:1140–1166. 10.3390/toxins5061140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castleman MJ, Pokhrel S, Triplett KD, Kusewitt DF, Elmore BO, Joyner JA, Femling JK, Sharma G, Hathaway HJ, Prossnitz ER, Hall PR. 2018. Innate sex bias of Staphylococcus aureus skin infection is driven by alpha-hemolysin. J Immunol 200:657–668. 10.4049/jimmunol.1700810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tkaczyk C, Hamilton MM, Datta V, Yang XP, Hilliard JJ, Stephens GL, Sadowska A, Hua L, O’Day T, Suzich J, Stover CK, Sellman BR. 2013. Staphylococcus aureus alpha toxin suppresses effective innate and adaptive immune responses in a murine dermonecrosis model. PLoS One 8:e75103. 10.1371/journal.pone.0075103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kennedy AD, Bubeck Wardenburg J, Gardner DJ, Long D, Whitney AR, Braughton KR, Schneewind O, DeLeo FR. 2010. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J Infect Dis 202:1050–1058. 10.1086/656043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Linehan JL, Harrison OJ, Han S-J, Byrd AL, Vujkovic-Cvijin I, Villarino AV, Sen SK, Shaik J, Smelkinson M, Tamoutounour S, Collins N, Bouladoux N, Dzutsev A, Rosshart SP, Arbuckle JH, Wang C-R, Kristie TM, Rehermann B, Trinchieri G, Brenchley JM, O’Shea JJ, Belkaid Y. 2018. Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell 172:784–796.e18. 10.1016/j.cell.2017.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boles BR, Thoendel M, Roth AJ, Horswill AR. 2010. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5:e10146. 10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Becerra SC, Roy DC, Sanchez CJ, Christy RJ, Burmeister DM. 2016. An optimized staining technique for the detection of Gram positive and Gram negative bacteria within tissue. BMC Res Notes 9:216. 10.1186/s13104-016-1902-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1-16. Download iai.00065-22-s0001.pdf, PDF file, 2.8 MB (2.8MB, pdf)
Table S1. Download iai.00065-22-s0002.xlsx, XLSX file, 0.02 MB (17.9KB, xlsx)