

ABSTRACT
5-Lipoxygenase (5-LO) is an enzyme required for the production of leukotrienes and lipoxins and interferes with parasitic infections. In vitro, Toxoplasma gondii inhibits leukotriene B4 (LTB4) production, and mice deficient in 5-LO are highly susceptible to infection. The aim of this study was to investigate the effects of the pharmacological inhibition of the 5-LO pathway and exogenous LTB4 supplementation during experimental toxoplasmosis. For this purpose, susceptible C57BL/6 mice were orally infected with T. gondii and treated with LTB4 or MK886 (a selective leukotriene inhibitor through inhibition of 5-LO-activating protein [FLAP]). The parasitism, histology, and immunological parameters were analyzed. The infection decreased 5-LO expression in the small intestine, and treatment with MK886 reinforced this reduction during infection; in addition, MK886-treated infected mice presented higher intestinal parasitism, which was associated with lower local interleukin-6 (IL-6), interferon gamma (IFN-γ), and tumor necrosis factor (TNF) production. In contrast, treatment with LTB4 controlled parasite replication in the small intestine, liver, and lung and decreased pulmonary pathology. Interestingly, treatment with LTB4 also preserved the number of Paneth cells and increased α-defensins expression and IgA levels in the small intestine of infected mice. Altogether, these data demonstrated that T. gondii infection is associated with a decrease in 5-LO expression, and on the other hand, treatment with the 5-LO pathway product LTB4 resulted in better control of parasite growth in the organs, adding to the knowledge about the pathogenesis of T. gondii infection.
KEYWORDS: Paneth cells, antimicrobial peptides, eicosanoids, experimental toxoplasmosis, intestinal immune response
INTRODUCTION
5-Lipoxygenase (5-LO) is an enzyme required for the biosynthesis of leukotrienes (LTs), which are lipid mediators derived from arachidonic acid (AA) (1). 5-LO is expressed in several cell types, including neutrophils, eosinophils, monocytes/macrophages, dendritic cells, mast cells, B lymphocytes, and human foam cells (reviewed in reference 2). To produce LTs, phospholipase A2 (PLA2) catalyzes the mobilization of AA from membrane phospholipids (reviewed in reference 3). The release of AA from membrane phospholipids can be triggered by a variety of stimuli, such as antigens, microbes, cytokines, complement, oxidants, immune complexes, and toxins (3). The formation of LTs is initiated by the oxidation of AA at C-5 by 5-LO to generate the epoxide intermediate leukotriene A4 (LTA4) (4), and this enzyme acts in concert with 5-LO-activating protein (FLAP) to generate LTA4 (4, 5). LTA4 can then be converted to LTB4 through the action of LTA4 hydrolase or LTC4 by the addition of glutathione. The LTC4 compound is metabolized to LTD4 and LTE4 by the successive elimination of a γ-glutamyl residue and glycine (reviewed in reference 6).
LTB4 has broad functions in the immune response, including increasing the phagocytosis of microorganisms by neutrophils and macrophages; enhancing phagocyte migration, chemotaxis, and activation; and producing inflammatory mediators (7). LTB4 signals through two G-protein-coupled receptors (GPCRs), BLT1 and BLT2. BLT2 is a low-affinity receptor expressed ubiquitously, whereas BLT1 is a high-affinity receptor broadly expressed in leukocytes (8), which mediates the chemoattractant and proinflammatory properties of LTB4 (reviewed in reference 9). In contrast to the proinflammatory effects of LTB4, the cooperation between 5-LO and 12- or 15-lipoxygenase results in the production of lipoxin A4 (LXA4), a lipid mediator with anti-inflammatory properties (reviewed in reference 10).
5-LO and LTB4 participate in the immune response during several inflammatory disorders, including airway diseases (11), inflammatory bowel disease (12), colitis (13), inflammatory dermatosis (14), inflammatory arthritis (15), tumor metastasis (16), Alzheimer’s disease (17), and diabetes (18).
Furthermore, the role of the 5-LO pathway has also been studied in the immune response to pathogens, and its effects on the control of infection are particular to each infectious disease. The absence of 5-LO activity or 5-LO inhibition associated with low levels of LTB4 enhances the parasitic burden during infection with Histoplasma capsulatum (19), Mycobacterium tuberculosis (20), or Strongyloides venezuelensis (21). Although there was increased parasitemia, genetically 5-LO-deficient (5-LO−/−) mice infected with Trypanosoma cruzi presented with higher survival rates than those of wild-type (WT) mice (22). In contrast, 5-LO−/− mice are more susceptible to Toxoplasma gondii infection (23, 24), which is associated with severe encephalitis and a lower parasitic burden in the brain during the chronic phase of the infection (23).
Although toxoplasmosis is usually asymptomatic in immunocompetent individuals, the infection is more severe in immunocompromised individuals and in cases of congenital infection (25). The infected hosts develop a Th1-type immune response (reviewed in reference 26) characterized by the recruitment of neutrophils, macrophages, and dendritic cells; the production of the proinflammatory cytokines tumor necrosis factor (TNF), interferon gamma (IFN-γ), and interleukin-12 (IL-12) (27); and the participation of CD8+ and CD4+ T cells in eliminating infected cells and producing IFN-γ, respectively (26). However, an uncontrolled and exacerbated immune response to T. gondii results in severe ileitis and early mortality in susceptible mice (28). Therefore, regulatory mechanisms, such as IL-10 production, are necessary to prevent the pathological consequences of the infection (29) and the overproduction of IL-12, IFN-γ, and TNF (30).
Previous studies demonstrated that T. gondii inhibits LTB4 production in infected human macrophages (31) and decreases LTB4 levels in HIV-1-seropositive patients with toxoplasmic encephalitis (32).
Although it is known that 5-LO deficiency leads to enhanced susceptibility to T. gondii and that the infection decreases LTB4 production, the role of 5-LO and LTB4 in the immunopathology and parasitism of the small intestine, the main route of parasite entry, is not yet elucidated. Here, we found that T. gondii decreases 5-LO expression in the small intestine and that treatment with MK886 (a FLAP inhibitor, known for its specific inhibition of LT production [33]) reinforces this reduction and increases the parasitic load. On the other hand, treatment with exogenous LTB4 decreased the parasitic burden in the lung, liver, and small intestine of infected mice. The control of parasitism in the small intestine was associated with decreased Paneth cell hypoplasia and increased antimicrobial peptide (AMP) expression and T. gondii-specific IgA production. Our results highlight the important role of the 5-LO pathway and LTB4 in the control of tissue parasitism during infection with T. gondii.
RESULTS
T. gondii infection decreases 5-LO expression in the small intestine.
It was previously shown that T. gondii infection decreases LTB4 production by human macrophages in vitro (31), and LTB4 was not detected in the cerebrospinal fluid of HIV-1-seropositive subjects with toxoplasmic encephalitis (32). Here, we first verified whether T. gondii could alter the mRNA expression of 5-LO or BLT1 in the small intestine during the acute phase of infection. For this purpose, C57BL/6 and BALB/c mice, which present susceptible and resistant major histocompatibility complex (MHC) alleles to toxoplasmosis, H2b and H2d, respectively (34), and their congenic strains CB10-H2 (H2b) and C57BKs/J (H2d) were infected, and the expression levels of 5-LO and BLT1 were measured in the small intestine at day 7 postinfection (Fig. 1A). Infection with T. gondii decreased the 5-LO mRNA expression levels in resistant and susceptible mice and their congenic mouse lineages in relation to uninfected mice (Fig. 1B). Similarly, infection with the parasite decreased the expression of BLT1 in C57BL/6, BALB/c, and CB10-H2 mouse lineages (Fig. 1C). These results indicate that regardless of the genetic background or MHC haplotype, T. gondii interferes with both the 5-LO and BLT1 mRNA expression levels in the small intestine of mice, except for BLT1 in the small intestine of C57BKs/J mice.
FIG 1.

Toxoplasma gondii impairs 5-lipoxygenase expression in the small intestine. (A) C57BL/6 and BALB/c mice and their congenic strains C57BKs/J and CB10-H2, respectively, were infected with 5 cysts of the ME49 strain of T. gondii. (B and C) On day 7 postinfection, the 5-LO (B) and BLT-1 (C) mRNA expression levels in the ilea were measured by real-time quantitative PCR (qPCR). (D) In the following experiments, C57BL/6 mice were infected with 10 cysts of the ME49 strain of T. gondii and treated daily with MK886 (5 mg/kg/day) or the vehicle (20% ethanolic solution) only. (E and F) On the 7th day after infection, levels of 5-LO mRNA (E) and BLT-1 (F) were measured in the ileum by qPCR. Gene expression was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Tg, T. gondii infected mice; DI, days of infection. *, P < 0.05 compared to noninfected (NI) mice; &, P < 0.05 compared to vehicle-treated mice (by a Mann-Whitney test [B and C] and one-way analysis of variance [ANOVA] with a Bonferroni posttest [E and F]). Data are presented as means ± standard errors of the means (SEM) and are representative of results from one of two independent experiments.
Additional experiments were conducted with C57BL/6 mice, which are highly susceptible to T. gondii infection. In the next step, C57BL/6 mice were treated with MK886 to inhibit 5-LO activity (Fig. 1D). It is noteworthy that treatment with MK886 inhibited the 5-LO mRNA expression levels in the small intestine compared with vehicle-treated infected mice (Fig. 1E). In relation to LTB4 receptors, T. gondii infection decreased BLT1 expression levels irrespective of treatment with MK886 (Fig. 1F), but it did not alter BLT2 expression levels in the small intestine (data not shown).
Treatment with MK886 or LTB4 altered neither the clinical parameters nor the survival rates of infected mice.
Since it was observed that T. gondii decreases 5-LO mRNA expression in infected mice, in the next step, animals were treated with LTB4, a known end product of the 5-LO pathway, or with MK886, a 5-LO inhibitor, and infected with the parasite (Fig. 2A).
FIG 2.

MK886 or LTB4 treatment did not increase C57BL/6 mouse survival during Toxoplasma gondii infection. (A) C57BL/6 mice were infected with 10 T. gondii cysts and treated with MK886 (5 mg/kg/day by the oral route), LTB4 (1 μg/kg/day by the intraperitoneal route), or the vehicle (20% ethanolic solution by the oral route) for 7 days consecutively (n = 5 per group). (B and C) Mice were observed daily for body weight changes (B) and mortality (C) during 60 days postinfection. *, P < 0.05 compared to LTB4- and vehicle-treated mice (by two-way ANOVA). Data are presented as means ± SEM from one of two independent experiments.
T. gondii-infected mice presented with a notable loss of body weight, which was not altered by treatment with MK886 or LTB4 (Fig. 2B). Moreover, the survival rates of T. gondii-infected mice were similar in the MK886-, LTB4-, and vehicle-treated groups (Fig. 2C). Together, these data suggest that the pharmacological inhibition of the 5-LO pathway or high systemic LTB4 levels altered neither the survival nor the loss of body weight in T. gondii-infected mice.
Treatment with LTB4 decreases the parasitic burden in the small intestine and enhances T. gondii-specific IgA levels in the feces of infected mice.
Previous studies demonstrated that C57BL/6 mice develop severe intestinal inflammation in response to oral infection with the ME49 strain of T. gondii (28). In this context, we investigated the effects of 5-LO pathway inhibition by treatment with MK886 or exogenous LTB4 supplementation on intestinal alterations when C57BL/6 mice were infected by the oral route (Fig. 3A).
FIG 3.

LTB4 controls Toxoplasma gondii replication in the small intestine and enhances T. gondii-specific fecal IgA levels in C57BL/6 mice. (A) C57BL/6 mice were infected with 10 T. gondii cysts and treated daily with MK886 (5 mg/kg/day by the oral route) or LTB4 (1 μg/kg/day by the intraperitoneal route). On day 7 postinfection, the small intestines of mice were collected and submitted to histological analysis. (B and C) Small intestine length (B) and parasite quantification (C) by immunohistochemistry of infected mice. (D to F) Representative photomicrographs from small intestine sections stained with hematoxylin and eosin (H&E) (D) and inflammatory scores (E) and goblet cell counts (F) in the small intestine. (G) T. gondii-specific IgA levels in mouse fecal samples were measured by an enzyme-linked immunosorbent assay (ELISA). OD492, optical density at 492 nm. (H) Representative photomicrographs from small intestine sections stained with alcian blue. Bars, 200 μm (D) and 100 μm (H). *, P < 0.05 compared to noninfected (NI) mice; &, P < 0.05 for comparison between groups of infected mice (by a Kruskal-Wallis test with Dunn’s posttest [B and C] and one-way ANOVA with a Bonferroni comparison posttest [E to G]). Data are presented as means ± SEM and are representative of results from one of two independent experiments.
First, macroscopic analysis showed that mice infected with T. gondii and treated with MK886 or LTB4 presented with a smaller intestinal length than that of noninfected mice (Fig. 3B).
Interestingly, the parasitism in the small intestine evaluated by immunohistochemistry showed that treatment with LTB4 decreased the parasitic burden in this organ, while the opposite effect was observed when mice were treated with MK886 (Fig. 3C).
The analysis of histological alterations in the small intestine showed that, as expected for oral infection with 10 T. gondii cysts, the small intestine of infected mice presented with intense inflammatory infiltration into the lamina propria (LP) and submucosa in some areas of the organ (Fig. 3D and E). C57BL/6 mice treated with MK886 or LTB4 presented with lesions similar to those of infected untreated animals (Fig. 3D and E). In addition, the quantification of goblet cells in the small intestine of infected mice showed that T. gondii infection decreased the number of goblet cells, irrespective of the treatment given (Fig. 3F and H).
Secretory IgA (SIgA) plays an important role in the mucosal immune barrier, preventing pathogen attachment or invasion of the mucosal surfaces (reviewed in reference 35). Therefore, we measured T. gondii-specific IgA levels in the fecal samples of orally infected mice treated with MK886 or LTB4 (Fig. 3G). T. gondii-specific IgA antibody levels in feces were similar in the vehicle- and MK886-treated infected mice (Fig. 3G). However, the group of infected mice that received treatment with LTB4 showed significantly higher anti-T. gondii (Fig. 3G)-specific IgA levels in their feces than those in the other infected groups.
Taken together, these results suggest that although treatment with MK886 and LTB4 altered neither the histological changes nor the numbers of goblet cells in T. gondii-infected mice, 5-LO activity is important for the control of parasitism in the small intestine during acute toxoplasmosis.
Exogenous LTB4 attenuates Paneth cell hypoplasia and the reduction of α-defensins levels triggered by T. gondii infection.
Paneth cells are crucial for the maintenance of intestinal homeostasis and produce AMPs, such as defensins and lysozyme, that participate in the immune response against intestinal pathogens (reviewed in reference 36). To evaluate the effects of the 5-LO pathway on the number of Paneth cells during T. gondii infection, we quantified these cells in the small intestine of infected mice treated with MK886 or LTB4 (Fig. 4A and B). It was observed that T. gondii infection decreased the number of Paneth cells in the small intestine of infected mice, even in those treated with MK886 or LTB4 (Fig. 4A and B). However, LTB4-treated infected mice presented with a higher number of Paneth cells than those detected in T. gondii-infected or T. gondii-infected MK886-treated mice (Fig. 4A and B), suggesting that treatment with LTB4 partially prevents the loss of Paneth cells caused by the infection.
FIG 4.

LTB4 treatment attenuates Paneth cell hypoplasia and increases α-defensin 1 expression in the small intestine of T. gondii-infected mice. C57BL/6 mice were infected with 10 T. gondii cysts and treated daily with MK886 (5 mg/kg/day by the oral route) or LTB4 (1 μg/kg/day by the intraperitoneal route). On day 7 postinfection, the small intestines of mice were collected and analyzed. (A and B) Quantification (A) and representative photomicrographs (B) of Paneth cells obtained from small intestine sections stained with H&E. (C to H) qPCR analysis of α-defensin 3 (C), α-defensin 5 (D), α-defensin 20 (E), α-defensin 21 (F), α-defensin 24 (G), and lysozyme (H) expression in the small intestine of T. gondii-infected mice treated or not with LTB4. Arrows indicate intestinal crypts with Paneth cells. Bars, 100 μm. log2FC, log2 fold change, normalized to GAPDH. *, P < 0.05 compared to noninfected (NI) vehicle-treated mice; #, P < 0.05 compared to noninfected LTB4-treated mice; &, P < 0.05 for comparison between groups of infected mice (by one-way ANOVA with a Bonferroni comparison posttest [A and C to G] and a Kruskal-Wallis test with Dunn’s posttest [H]). Data are presented as means ± SEM and are representative of results from one of two independent experiments.
Treatment with MK886 did not alter the lower number of Paneth cells caused by the infection, whereas treatment with LTB4 partially preserved the number of these cells. In the next step, the mRNA expression levels of α-defensins 3, 5, 20, 21, and 24 and lysozyme in the intestine of infected mice receiving treatment with LTB4 were evaluated.
It was verified that T. gondii infection decreased the mRNA expression levels of all of the α-defensins tested and lysozyme in the small intestine of infected mice (Fig. 4C to H). Interestingly, the expressions of α-defensins 3, 5, 21, and 24 were improved in the small intestine of noninfected mice treated with LTB4 (Fig. 4C, D, F, and G).
Moreover, treatment with LTB4 presented the ability to increase α-defensin expression levels in the small intestine, even during T. gondii infection, since α-defensin 3, 5, 21, and 24 mRNA levels were higher in LTB4-treated infected mice than in infected mice treated with the vehicle only (Fig. 4C, D, F, and G). These increases could reflect the high Paneth cell numbers in LTB4-treated infected mice compared to vehicle-treated mice.
Additional inhibition of the 5-LO pathway in T. gondii infection downregulates IL-6, IFN-γ, and TNF in the small intestine.
To evaluate whether 5-LO inhibition or treatment with LTB4 affects the production of cytokines involved in T. gondii control, the cytokine levels in serum samples and ileum homogenates of infected mice treated with MK886 or LTB4 were measured (Fig. 5).
FIG 5.
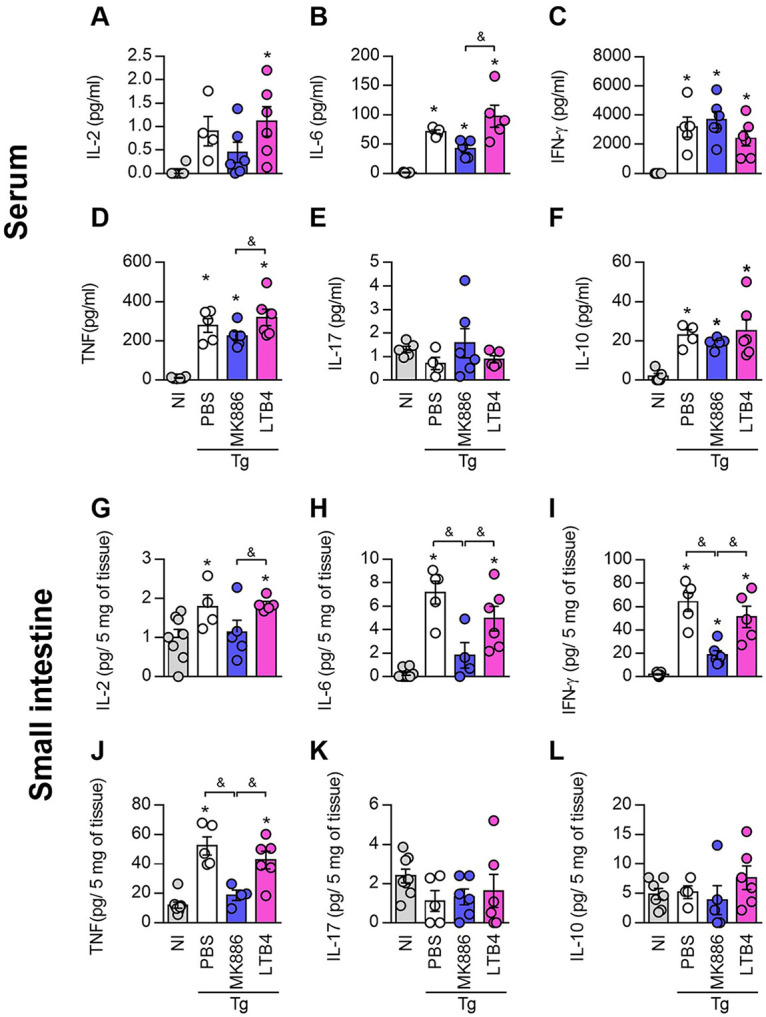
5-LO inhibition decreases cytokine production during Toxoplasma gondii infection. Cytokine levels were quantified in serum samples (A to F) or small intestine homogenates (G to L) of C57BL/6 mice infected with 10 T. gondii cysts and treated daily with MK886 (5 mg/kg/day by the oral route) or LTB4 (1 μg/kg/day by the intraperitoneal route). IL-2 (A and G), IL-6 (B and H), IFN-γ (C and I), TNF (D and J), IL-17 (E and K), and IL-10 (F and L) levels were measured using a cytometric bead array (CBA) cytokine kit. *, P < 0.05 compared to noninfected (NI) mice; &, P < 0.05 for comparison between groups of infected mice (by one-way ANOVA). Data are presented as means ± SEM and are representative of results from one of two independent experiments. PBS, phosphate-buffered saline.
When systemic cytokine production was measured in sera, it was observed that T. gondii infection increased the production of IL-2 (Fig. 5A), IL-6 (Fig. 5B), IFN-γ (Fig. 5C), TNF (Fig. 5D), and IL-10 (Fig. 5F), irrespective of the treatment, even though IL-6 (Fig. 5B) and TNF (Fig. 5D) levels were lower in MK886-treated mice than in LTB4-treated mice. Infection with the parasite did not alter systemic IL-17 production (Fig. 5E).
In relation to the cytokine profile in the small intestine, although T. gondii infection increased IL-2 (Fig. 5G), IL-6 (Fig. 5H), IFN-γ (Fig. 5I), and TNF (Fig. 5J) production, the IL-6, IFN-γ, and TNF levels were lower in MK886-treated infected mice than in vehicle- or LTB4-treated infected mice (Fig. 5H to J). Likewise, the levels of IL-2 production in MK886-treated infected mice were lower than those in infected LTB4-treated mice (Fig. 5G). The levels of IL-17 (Fig. 5K) were not altered in the small intestine of infected mice, and the levels of IL-10 (Fig. 5L) were below the limit of detection of the kit. These data suggest that 5-LO pathway inhibition was able to downregulate the production of IL-6, IFN-γ, and TNF in the small intestine with T. gondii infection.
LTB4 controls T. gondii replication in the lungs and livers of infected mice.
T. gondii can migrate across the intestinal epithelial barrier and infect various cell types, disseminating to several organs, such as the lungs, livers, and kidneys, and neural and muscular tissues (reviewed in reference 37). Therefore, we evaluated the effects of the inhibition of the 5-LO pathway and treatment with LTB4 on the lungs and livers of T. gondii-infected mice (Fig. 6).
FIG 6.
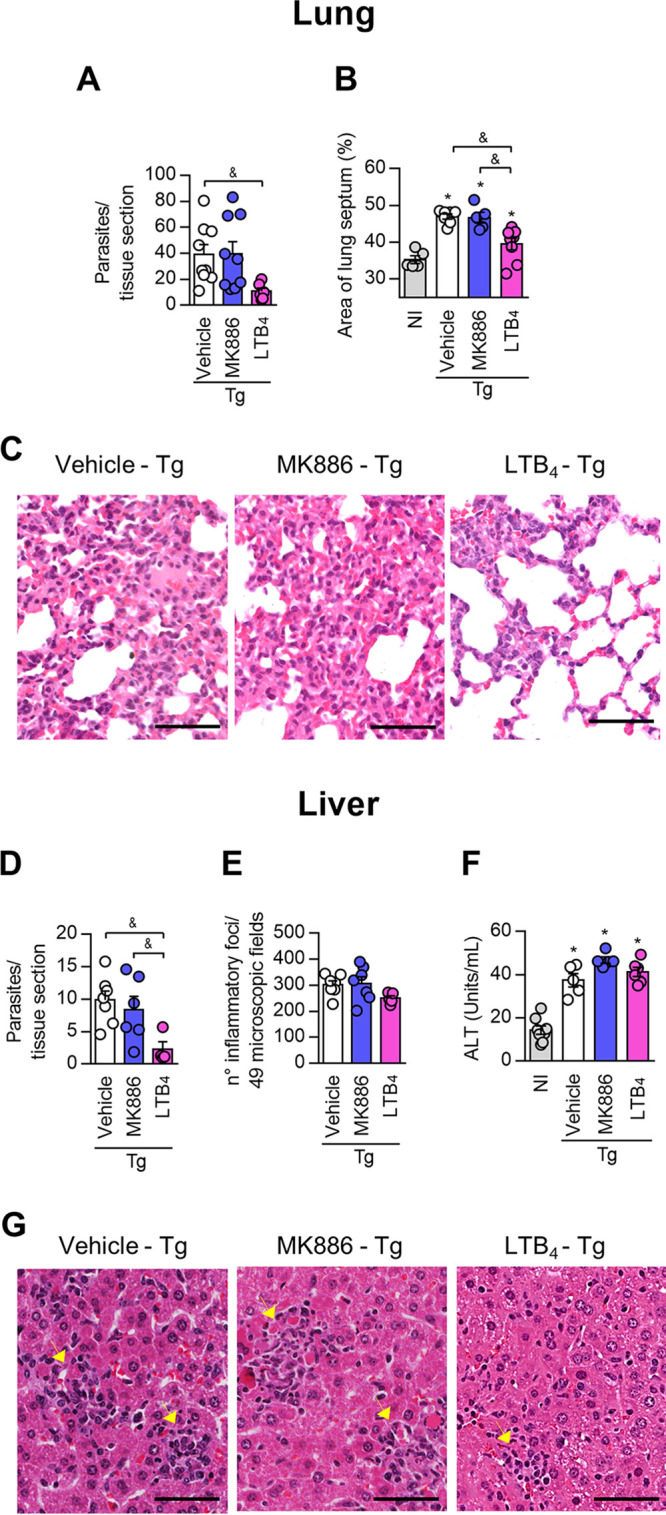
LTB4 controls Toxoplasma gondii replication in the lung and liver of C57BL/6 mice. C57BL/6 mice were infected with 10 T. gondii cysts and treated daily with MK886 (5 mg/kg/day by the oral route) or LTB4 (1 μg/kg/day by the intraperitoneal route). (A, B, D, and E) On day 7 postinfection, the lung and liver were collected, and tissue parasitism (A and D) and inflammatory scores (B and E) were quantified in tissue sections. (F) ALT levels were measured by an analytic kit in serum samples as an indication of liver damage. (C and G) Photomicrographs showing the histological alterations in the lung (C) and liver (G) of infected and treated mice. Arrowheads indicate inflammatory foci (G). Bars, 50 μm. *, P < 0.05 compared to noninfected (NI) mice; &, P < 0.05 for comparison between groups of infected mice (by a Kruskal-Wallis test with Dunn’s posttest [A, D, and F] or one-way ANOVA [B and E]). Data are presented as means ± SEM and are representative of results from one of two independent experiments.
The data obtained from lung (Fig. 6A to C) and liver (Fig. 6D to G) analyses revealed that treatment with MK886 altered neither the parasitic burden nor inflammatory changes in T. gondii-infected mice. Moreover, T. gondii infection increased serum alanine aminotransferase (ALT) concentrations, irrespective of the treatment given to the animals (Fig. 6F). In contrast, infected mice treated with LTB4 showed a decreased parasitic load in the lung (Fig. 6A) and reduced alveolar septum thickening caused by the infiltration of inflammatory cells (Fig. 6B and C). Similar to the results obtained in the lung analysis, treatment with LTB4 decreased the parasitism in the liver (Fig. 6D). However, treatment with LTB4 did not alter the number of inflammatory foci in this organ (Fig. 6E).
DISCUSSION
The enzyme 5-LO and its metabolites participate in the immune response during different inflammatory and pathogenic diseases. Previously, it was demonstrated that infection with Brucella abortus induces the upregulation of 5-LO mRNA and LTB4 production (38), while H. capsulatum (19) and Streptococcus pyogenes (39) infections induce LTB4 production in infected mice. Additionally, infection with Plasmodium berghei ANKA increases 5-LO expression in the brain of susceptible C56BL/6 mice, although no expression is detected in the brain of resistant BALB/c mice (40). In relation to T. gondii infection, in vitro studies have demonstrated that viable T. gondii inhibits calcium ionophore-induced LTB4 released by human macrophages (31), and HIV-seropositive patients with toxoplasmic encephalitis present with LTB4 levels below the detection limit in cerebrospinal fluid (32). Although the effects of 5-LO deficiency during chronic experimental toxoplasmosis were previously investigated (23), the participation of the 5-LO pathway during the acute phase of T. gondii infection has not yet been elucidated. Therefore, we first evaluated 5-LO and LTB4 receptor expression in the small intestine of susceptible C57BL/6 and resistant BALB/c mice (41) and their congenic counterparts on the seventh day of oral T. gondii infection. Our results demonstrated that T. gondii infection decreased 5-LO and BLT1 expression levels in the small intestine of infected C57BL/6 and BALB/c mice and their respective congenic strains, suggesting that T. gondii infection interferes with the 5-LO pathway regardless of the genetic background or MHC haplotype.
It was previously shown that 5-LO−/− mice are more susceptible to T. gondii infection than their WT counterparts (B6, 129S F2/J, or 129/SvEvTac) when infected with 20 or 100 T. gondii ME49 cysts by the intraperitoneal or oral route, respectively (23, 24). Moreover, chronically infected 5-LO−/− mice presented with a low parasitic burden and increased leukocyte infiltration in the brain compared to the control (B6 and 129J F2) mice (23). In the present experimental work, it was observed that treatment with MK886 enhanced the parasitic load in the small intestine, although the inflammatory changes in the organ and survival rates were similar to those observed in infected untreated C57BL/6 mice.
The different data observed in our study compared with those of Aliberti et al. (23) could be explained by the different experimental conditions. Although the ME49 strain of T. gondii was used in both studies, the routes of infection, the analyzed organs, and the phases of infection were different. Additionally, our previous study demonstrated that 129/SvEvTac mice are more resistant to oral T. gondii ME49 infection than 129/SvEvTac 5-LO−/− mice, which were orally inoculated with the parasite (24).
Similar to our findings with intestinal parasitism, the administration of MK886 enhanced the replication of other pathogens, such as M. tuberculosis (20, 42), H. capsulatum (43), and S. venezuelensis (21), in murine models. Likewise, 5-LO−/− mice presented with a higher parasitic burden during infection with T. cruzi (44), Paracoccidioides brasiliensis (45), or Leishmania infantum (46). Those previous studies also demonstrated that 5-LO deficiency or treatment with MK886 impairs LTB4 production (20–22, 43, 45).
The enhanced proliferation of parasites in the small intestine of MK886-treated mice could be related to the lower IL-6, IFN-γ, and TNF levels measured in this organ since these cytokines are involved in the control of T. gondii infection (47–51). Additionally, no difference in IL-10 production in the small intestine was observed, irrespective of the treatment received. A previous study has shown that treatment with MK886 also decreased TNF and IFN-γ levels and maintained IL-10 levels, followed by impaired mycobacterial clearance in the lungs of M. tuberculosis-infected mice (20).
The pharmacological inhibition or deletion of 5-LO downregulates proinflammatory cytokines, even in nonparasitic disease. The decreased production of IL-1β, TNF, and IL-17 after treatment of mice with a specific 5-LO inhibitor, zileuton, in a polyposis model was observed (52), and treatment with the lipoxygenase inhibitor nordihydroguaiaretic acid (NDGA) caused impaired TNF production in splenocytes in a murine model of asthma (53). Similarly, 5-LO−/− mice presented with low levels of TNF in skin wounds in comparison with their WT counterparts (54). Although IFN-γ and TNF are crucial cytokines for protection against toxoplasmosis, their exacerbated production contributes to the development of intestinal lesions (28). On the other hand, IL-10 is necessary for the prevention of intestinal pathology in T. gondii-infected mice (29). However, even though treatment with MK886 reduced IL-6, IFN-γ, and TNF production, the histological changes in the small intestine caused by T. gondii infection were not attenuated, suggesting that the decreased amounts of proinflammatory cytokines were not sufficient to control the inflammation in this organ.
Contrary to 5-LO inhibition, treatment with LTB4 enhanced monocyte chemoattractant protein 1 (MCP-1) (55), IL-8 (56), IL-1 (57), IL-6 (58), prime IL-2-dependent TNF (59), IL-2, and IFN-γ (60) production in vitro by human cells and increased IFN-β, IL-6, and TNF production in mice infected with influenza virus (61). So it was expected that treatment with LTB4 would enhance cytokine production during T. gondii infection. However, the cytokine profile induced by T. gondii infection was not altered by treatment with exogenous LTB4, suggesting that the protective effect of LTB4 was independent of cytokine production. Furthermore, although LTB4 presents with well-known chemoattractant proprieties (9), treatment with exogenous LTB4 did not enhance inflammatory cell infiltration provoked by T. gondii in the small intestine.
Treatment with LTB4 was able to enhance the expression of some AMPs, such as α-defensins 3, 5, 20, 21, and 24 and lysozyme, in the small intestine of noninfected mice. Other studies demonstrated the effect of LTB4 on the production of AMPs. In vivo treatment with LTB4 increased plasmatic α-defensin levels in monkeys and enhanced the in vitro release of α-defensins by human neutrophils (62) and α-defensins, cathepsin G, elastase, lysozyme C, and cathelicidin LL-37 by human polymorphonuclear leukocytes (PMNs) (63). Contrary to treatment with LTB4, T. gondii infection reduced the expression of α-defensins 3, 5, 20, 21, and 24 and lysozyme in the small intestine. In accordance, others have shown that T. gondii infection reduced the expression of lysozyme (62, 64), α-defensin 1 (64), and criptidin 2 (65, 66) in the small intestine of infected mice. Although the expression levels of the AMPs α-defensin 3, 5, 20, 21, and 24 and lysozyme in LTB4-treated mice infected with T. gondii are quite lower than those in noninfected mice, among the infected groups, treatment with LTB4 significantly increased the intestinal expression of α-defensins 3, 5, 21, and 24 in relation to vehicle-treated infected mice. In the small intestine, α-defensins, lysozyme, C-type lectins, and phospholipase A2 (PLA2) are produced by Paneth cells (reviewed in reference 67). We and others have previously demonstrated that T. gondii oral infection results in the elimination (64, 68) or hypoplasia (69, 70) of Paneth cells. Here, our results showed that treatment with LTB4 attenuated the loss of Paneth cells and improved the expression of α-defensins 3, 5, 21, and 24 in the small intestine.
Previously, other studies have demonstrated that treatment with LTB4 controls pathogen replication in other infectious models, such as in in vitro H. capsulatum (19), Candida albicans (71), Klebsiella pneumoniae (72), and Streptococcus pneumoniae (73) infection models as well as Leishmania species infection models (74–76). It is noteworthy that some investigations have demonstrated that the beneficial role of LTB4 is mediated by the activity of AMPs. Therefore, during Achromobacter xylosoxidans infection, the improvement of bactericidal activity induced by LTB4 in the lungs of mice is dependent on α-defensin 1 activity (77). Additionally, treatment of mice with LTB4 controlled influenza virus replication through the upregulated production of β-defensin 3 and a cathelicidin-related AMP (63), and the plasma obtained from monkeys treated with LTB4 presented ex vivo antimicrobial activity (62). Although it is known that exogenous LTB4 promotes the in vitro intracellular killing of T. gondii by the induction of cytotoxicity in tachyzoites (31), the toxoplasmacidal mechanisms trigged by LTB4 during in vivo infection were not elucidated. Additionally, our data suggest that one of these mechanisms, at least in the small intestine, could be related to the expression of both Paneth cells and α-defensins.
In addition to an increase in α-defensin expression levels in relation to those in nontreated mice, the findings presented here showed that treatment with LTB4 improved IgA production against T. gondii in the small intestine of infected mice. Others have shown that T. gondii infection induces the production of IgA specific for the parasite in the small intestine of mice (78, 79). Recently, it was demonstrated that the microbe-dependent proliferation of IgA-positive (IgA+) plasma cells and the production of antigen-specific IgA require BLT1 expression in the small intestine (80). Given that our data showed that T. gondii infection reduced the intestinal expression levels of both 5-LO and BLT1, treatment with LTB4 could be affecting the LTB4-BLT1 pathway and improving the production of T. gondii-specific IgA in the small intestine. Moreover, it was previously demonstrated that SIgA from human milk controls T. gondii replication in enterocytes in vitro (81).
T. gondii has the ability to disseminate to extraintestinal tissues such as the spleen, liver, lung, and brain (82) and triggers an inflammatory immune response in these organs (83) during the acute phase of infection. Indeed, our group had shown that T. gondii is detected in the lung and liver of mice together with histological alterations even after oral or intraperitoneal infection with a low number of cysts (69, 84). In the current study, 5-LO inhibition altered neither parasitism nor histopathology changes, whereas treatment with LTB4 reduced parasitism in the lung and liver and attenuated pulmonary inflammation triggered by T. gondii infection. Therefore, our data reinforce that treatment with LTB4 has a positive effect in controlling parasitism, and this beneficial role is not restricted to the small intestine.
In conclusion, our results demonstrate that T. gondii decreases 5-LO expression in the small intestine of infected mice, and on the other hand, LTB4 supplementation leads to a diminished parasitic burden in the small intestine, lung, and liver of T. gondii-infected mice. In the small intestine, the microbicidal effects seem to be associated with the preservation of Paneth cell and α-defensins expression and increased IgA production (Fig. 7). Taken together, these data demonstrate the importance of the 5-LO pathway during acute oral T. gondii infection in a susceptible mouse lineage, adding to the knowledge about the pathogenesis of T. gondii infection and suggesting LTB4 as a complementary therapeutic approach for the management of toxoplasmosis.
FIG 7.

Schematic representation of the effects of treatment with MK886 or LTB4 on T. gondii-infected mice. T. gondii infection reduces 5-LO expression in the small intestine of C57BL/6 mice. The inhibition of the 5-LO pathway (MK886 treatment) downregulates IL-6, IFN-γ, and TNF and increases the parasite burden in the small intestine. In contrast, treatment with LTB4 reduces parasitism in the small intestine, lung, and liver. In the small intestine, treatment with LTB4 preserved Paneth cell numbers and increased defensin expression and IgA levels. Therefore, treatment with LTB4 presented a beneficial effect during toxoplasmosis. (Y.C. drew the figure).
MATERIALS AND METHODS
Animals.
Eight-week-old female C57BL/6 major histocompatibility complex (MHC) haplotype H2b, C57BKs/J (H2d haplotype; more than 70% of its genome is derived from C57BL/6J mice), BALB/c (H2d haplotype), CB10-H2 (H2b haplotype; derivative congenic strain derived from BALB/c mice), and Swiss Webster mice were used in this experimental work. The animals were bred and maintained in the specific-pathogen-free animal facility (Rede de Biotérios de Roedores [REBIR/UFU]) at the Universidade Federal de Uberlândia, with 12-h-light/dark cycles and free access to food and filtered water. All animal experiments were performed in accordance with the Brazilian Government’s ethical guidelines and were approved by the Comitê de Ética na Utilização de Animais (CEUA) of the Universidade Federal de Uberlândia, under protocol no. 078/14.
T. gondii strain.
The ME49 T. gondii strain was used in this experimental work and was maintained by inoculating Swiss Webster mice by the intraperitoneal route with 10 T. gondii cysts. Thirty days after infection, ME49 cysts were harvested from Swiss brain mice and quantified under a light microscope using a 10× objective. For experimental infection, mice were orally infected with 5 or 10 cysts in 0.2 mL phosphate-buffered saline (PBS) (0.01 mol/L, pH 7.2).
Drugs.
MK886 (Cayman Chemical, Ann Arbor, MI, USA), an inhibitor of FLAP, was dissolved in absolute ethanol and then diluted in water, resulting in a 20% ethanolic solution (85). LTB4 (Cayman Chemical) was obtained as an ethanolic solution and prepared by the dilution of ethanolic LTB4 in 0.9% saline (61).
Treatment of mice and infection.
In order to analyze the influence of the MHC haplotype and genetic background of mice on 5-LO and BLT1 mRNA expression levels in the small intestine, resistant BALB/c (H2d) and susceptible C57BL/6 (H2b) mice and their congenic mice, C57BKs/J (H2d) and CB10-H2 (H2b), were orally infected with 5 cysts, and the organs were assayed on day 7 of infection.
In the following experiments, groups of C57BL/6 mice were orally treated with MK886 (5 mg/kg of body weight/day in 0.2 mL) 1 h before infection with 10 cysts and were treated for 7 additional days or intraperitoneally treated daily with LTB4 (1 μg/kg/day in 0.1 mL) from days 1 to 7 of infection. As all vehicle treatments presented similar results, only the data from 20% ethanolic solution-treated mice are presented as the vehicle control treatment.
At 7 days postinfection, groups of mice were anesthetized with ketamine (Syntec Brasil Ltd., Cotia, SP, Brazil) and xylazine (Schering-Plough Coopers, Cotia, SP, Brazil) by the intraperitoneal route and euthanized by cervical dislocation. Blood samples were collected by puncture of the retro-orbital plexus for serological assays, and tissue samples, such as the lung, liver, and small intestine, were collected, fixed in 10% buffered formalin, and processed routinely for paraffin embedding and sectioning or frozen immediately and stored at −80°C for PCR or cytometric bead array (CBA) analysis.
In parallel, groups of infected C57BL/6 mice treated with MK886 or LTB4 were observed daily for weight changes and mortality for 60 days postinfection, when mice that survived were euthanized. In order to prevent unnecessary suffering, mice that simultaneously showed starry stiff coat, reluctance to move, and 20% weight loss were euthanized as described previously (69).
Histological and biochemical analyses.
To analyze histological changes after T. gondii infection, deparaffinized tissue sections were stained with hematoxylin and eosin (H&E), and all analyses were performed in a blind manner.
In the small intestine, the inflammatory scores were analyzed using a 10× objective as previously described (70). The histological changes in the pulmonary tissue were estimated by measurement of the lung septal area assessed using the NIH ImageJ program (https://imagej.nih.gov/ij/) from 10 random images of each lung tissue sample.
Related to the liver, the number of inflammatory foci was counted in 40 microscopic fields using a 10× objective. In addition to histological analysis, alanine aminotransferase (ALT) levels were measured in serum samples of infected C57BL/6 mice by using an analytic kit (Labtest, Lagoa Santa, MG, Brazil) according to the manufacturer’s instructions. The absorbance was obtained at 505 nm, and ALT levels were expressed in units per milliliter.
Quantification of goblet and Paneth cells in the small intestine.
For goblet and Paneth cell quantification in the small intestine, alcian blue- or H&E-stained tissue sections were analyzed, respectively. The numbers of goblet cells were quantified in 200 microscopic fields and the numbers of Paneth cells were measured in 400 intestinal crypts using a 40× objective in a blind manner.
Quantification of tissue parasitism.
The parasite burden was evaluated in the organs by immunohistochemistry as previously described (69). Briefly, deparaffinized tissue sections were incubated with 3% hydrogen peroxide to block endogenous peroxidase activity, and antigenic unmasking was performed in a microwave oven. Tissue sections were incubated overnight at 4°C with polyclonal anti-T. gondii serum (obtained from Swiss Webster mice chronically infected with the ME49 strain) diluted in 0.01% saponin. After incubation with biotinylated goat anti-mouse antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA), the assay sensitivity was improved by adding an avidin-biotin-peroxidase complex (ABC kit, catalog no. PK-4000; Vector Laboratories, Inc., Burlingame, CA, USA). The reactions were developed with 0.03% H2O2 plus 3,3′-diaminobenzidine tetrahydrochloride (DAB; Sigma) for 5 min. The sections were counterstained with Harris hematoxylin and examined under a light microscope using a 40× objective. Tissue parasitism was scored by counting the stained parasites per tissue section.
Quantitative real-time PCR.
Fragments of the ileum (100 mg) were used for mRNA extraction using TRIzol reagent according to the manufacturer’s instructions (Life Technologies, Carlsbad, CA, USA). The RNA concentration was determined (GeneQuant 1300 spectrophotometer; GE Healthcare), and cDNA was synthesized using 1 μg RNA by a reverse transcription reaction according to the manufacturer’s instructions (Promega, Madison, WI, USA). Quantitative PCR (qPCR) assays were performed using SYBR green reagent (Roche, Basel, Switzerland) in an Applied Biosystems 7500 real-time PCR system (Life Technologies). The standard PCR conditions were 95°C for 10 min and 40 cycles at 95°C (15 s), 50°C (30 s), and 60°C (1 min), using the following specific primers: forward (F) primer 5′-GGAGAAACCTGCCAAGTATGATG-3′ and reverse (R) primer 5′-CAGTGTAGCCCAAGATGCCC-3′ for gapdh, F primer 5′-AGCTGCCTGCTGTGCATCCC-3′ and R primer 5′-CCCGGTGGCATTGGCCTTGT-3′ for alox5 (38), F primer 5′-TCCTCCACCATTCCTGAGTC-3′ and R primer 5′-GTCTCTCTGCCCTGACTTGC-3′ for blt1, F primer 5′-CAGGCTGTGTCTGTCTCTTTTG-3′ and R primer 5′-TCAGCGACAGCAGAGTGTGTA-3′ for defa3 (86), F primer 5′-TTGTCCTCCTCTCTGCCCTTGT-3′ and R primer 5′-ATGAAGAGCAGACCCTTCTTGG-3′ for defa5 (86), F primer 5′-GAGAGATCTGATATGCTATTG-3′ and R primer 5′-AGAACAAAAGTCGTCCTGAG-3′ for defa20 (87), F primer 5′-GAGAGATCTGATCTGCCTTTG-3′ and R primer 5′-CCTCTATTGCAGCGACGA-3′ for defa21 (87), F primer 5′-GATCTGGTATGCTATTGTAGAG-3′ and R primer 5′-GACAGCAGAGCATGTACAA-3′ for defa24 (87), and F primer 5′-GCCAAGGTCTACAATCGTTGTGAGTTG-3′ and R primer 5′-CAGTCAGCCAGCTTGACACCACG-3′ for lyz (64). The unreferenced primers were designed using Primer Express V3 software (Life Technologies).
The expression of each target gene was normalized to that of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene using the 2−ΔΔCT method (88).
Quantification of secretory IgA in the feces of infected mice.
The measurement of secretory IgA (SIgA) levels in fecal samples was performed by an enzyme-linked immunosorbent assay (ELISA) as previously described (69). First, mouse fecal samples were collected and diluted 1:5 with fecal dilution buffer (90 mL 0.01 M PBS [pH 7.2], 10 mL 0.5 M EDTA [pH 8], 10 mg aprotinin [Sigma], and 666 μL 100 mM phenylmethylsulfonyl fluoride [PMSF; Sigma]). Next, the samples were centrifuged at 10,000 × g for 5 min, and the supernatants were collected (fecal solutions).
For the ELISA, low-binding microtiter plates were coated with 10 μg/mL T. gondii soluble antigen (STAg) (69) extract in carbonate buffer for 16 h at 4°C. After washes with 0.05% Tween 20–PBS (PBS-T), plates were blocked with PBS–5% bovine serum albumin (BSA) for 1 h. In the next step, the fecal solution was diluted 1:7 and incubated for 16 h at 4°C. Subsequently, the plates were washed and incubated with peroxidase-labeled anti-mouse IgA (1:2,000) antibody (Santa Cruz Biotechnology, Dallas, TX, USA) for 1 h at 37°C. The reaction was developed with 1 mg/mL of o-phenylenediamine (OPD; Sigma) and 0.03% hydrogen peroxide. Optical density (OD) values were determined in a microplate reader (VersaMax; Molecular Devices, San Jose, CA, USA) at 492 nm.
Cytokine measurement.
For cytokine quantification in the small intestine, fragments of the ileum (200 mg) from C57BL/6 mice were dissected, washed in PBS, and homogenized in 1 mL extraction buffer diluted in PBS (5 mM EDTA [pH 8.0], 0.016 mM aprotinin, 1 mM benzamidine, 0.21 mM leupeptin, 1.6 mM PMSF). After centrifugation at 3,000 × g for 10 min at 4°C, the supernatants were collected and stored at −80°C until use.
Measurements of the IL-2, IL-6, IFN-γ, TNF, IL-17A, and IL-10 cytokines in serum samples and supernatants of ileum homogenates were performed using a CBA mouse cytokine kit (BD Biosciences-Pharmingen, San Diego, CA, USA) according to the manufacturer’s instructions, and the results were recorded with a FACSCanto-II flow cytometer (BD) and analyzed with FACSDiva software (BD). According to the instruction manual of the kit, the theoretical limits of detection are 0.1 pg/mL for IL-2, 1.4 pg/mL for IL-6, 0.5 pg/mL for IFN-γ, 0.9 pg/mL for TNF, 0.8 pg/mL for IL-17A, and 16.8 pg/mL for IL-10.
Statistical analysis.
Statistical analysis was carried out using GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA). The Kaplan-Meier method was applied to estimate the percentage of mice surviving at each time point after infection, and survival curves were compared using the log rank test. The data distribution was evaluated by a Kolmogorov-Smirnov normality test and the appropriate statistical test used for each experimental analysis. To compare more than two experimental groups, analysis of variance (ANOVA) with the Bonferroni comparison posttest or a Kruskal-Wallis test with Dunn’s comparison posttest was applied for parametric or nonparametric data, respectively. The Mann-Whitney test was used to compare two different conditions with a nonparametric data distribution. Differences were considered statistically significant when the P value was <0.05.
ACKNOWLEDGMENTS
This work was partially supported by the Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) and the Conselho Nacional de Pesquisa Científica e Tecnológica (CNPq). In addition, this study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brazil (CAPES), finance code 001. N.M.S. is a research fellow of the CNPq.
E.C.B.A. performed the majority of the experiments, infected and treated animals, collected tissue and serum samples, and performed cytokine measurement, data analysis, and manuscript preparation; M.P.P.B. contributed to the extraction of mRNA, the qPCR assay, and its data analysis; Y.C. contributed to histological and morphometric analysis; M.C.O. was involved in IgA measurement, extraction of mRNA, and T. gondii strain maintenance; M.P.O.A. was involved in infection and treatment of animals, IgA measurement, and T. gondii strain maintenance; N.C.D.M. contributed to daily observation of weight changes and survival rates; N.M.S. conceived the idea and contributed to the histological analysis, interpretation of results, and manuscript preparation and revision.
Contributor Information
Neide Maria Silva, Email: nmsilva@ufu.br.
Jeroen P. J. Saeij, UC Davis School of Veterinary Medicine
REFERENCES
- 1.Borgeat P, Samuelsson B. 1979. Metabolism of arachidonic acid in polymorphonuclear leukocytes. Structural analysis of novel hydroxylated compounds. J Biol Chem 254:7865–7869. 10.1016/S0021-9258(18)36026-5. [DOI] [PubMed] [Google Scholar]
- 2.Rådmark O, Samuelsson B. 2010. Regulation of the activity of 5-lipoxygenase, a key enzyme in leukotriene biosynthesis. Biochem Biophys Res Commun 396:105–110. 10.1016/j.bbrc.2010.02.173. [DOI] [PubMed] [Google Scholar]
- 3.Peters-Golden M, Brock TG. 2003. 5-Lipoxygenase and FLAP. Prostaglandins Leukot Essent Fatty Acids 69:99–109. 10.1016/S0952-3278(03)00070-X. [DOI] [PubMed] [Google Scholar]
- 4.Rouzer CA, Matsumoto T, Samuelsson B. 1986. Single protein from human leukocytes possesses 5-lipoxygenase and leukotriene A4 synthase activities. Proc Natl Acad Sci USA 83:857–861. 10.1073/pnas.83.4.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller DK, Gillard JW, Vickers PJ, Sadowski S, Léveillé C, Mancini JA, Charleson P, Dixon RAF, Ford-Hutchinson AW, Fortin R, Gauthier JY, Rodkey J, Rosen R, Rouzer C, Sigal IS, Strader CD, Evans JF. 1990. Identification and isolation of a membrane protein necessary for leukotriene production. Nature 343:278–281. 10.1038/343278a0. [DOI] [PubMed] [Google Scholar]
- 6.Samuelsson B. 1983. Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science 220:568–575. 10.1126/science.6301011. [DOI] [PubMed] [Google Scholar]
- 7.Brandt SL, Serezani CH. 2017. Too much of a good thing: how modulating LTB4 actions restore host defense in homeostasis or disease. Semin Immunol 33:37–43. 10.1016/j.smim.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yokomizo T. 2011. Leukotriene B4 receptors: novel roles in immunological regulations. Adv Enzyme Regul 51:59–64. 10.1016/j.advenzreg.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 9.He R, Chen Y, Cai Q. 2020. The role of the LTB4-BLT1 axis in health and disease. Pharmacol Res 158:104857. 10.1016/j.phrs.2020.104857. [DOI] [PubMed] [Google Scholar]
- 10.Romano M. 2010. Lipoxin and aspirin-triggered lipoxins. ScientificWorldJournal 10:1048–1064. 10.1100/tsw.2010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pal K, Feng X, Steinke JW, Burdick MD, Shim YM, Sung SS, Teague WG, Borish L. 2019. Leukotriene A4 hydrolase activation and leukotriene B4 production by eosinophils in severe asthma. Am J Respir Cell Mol Biol 60:413–419. 10.1165/rcmb.2018-0175OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh VP, Patil CS, Kulkarni SK. 2004. Effect of 5-lipoxygenase inhibition on events associated with inflammatory bowel disease in rats. Indian J Exp Biol 42:667–673. [PubMed] [Google Scholar]
- 13.Cuzzocrea S, Rossi A, Mazzon E, Di Paola R, Genovese T, Muià C, Caputi AP, Sautebin L. 2005. 5-Lipoxygenase modulates colitis through the regulation of adhesion molecule expression and neutrophil migration. Lab Invest 85:808–822. 10.1038/labinvest.3700276. [DOI] [PubMed] [Google Scholar]
- 14.Penno CA, Jäger P, Laguerre C, Hasler F, Hofmann A, Gass S, Wettstein-Ling B, Schaefer D, Avrameas A, Raulf F, Wieczorek G, Lehmann JCU, Loesche C, Roth L, Röhn TA. 2020. Lipidomics profiling of hidradenitis suppurativa skin lesions reveals lipoxygenase pathway dysregulation and accumulation of pro-inflammatory leukotriene B4. J Invest Dermatol 140:2421–2432.e10. 10.1016/j.jid.2020.04.011. [DOI] [PubMed] [Google Scholar]
- 15.Chen M, Lam BK, Luster AD, Zarini S, Murphy RC, Bair AM, Soberman RJ, Lee DM. 2010. Joint tissues amplify inflammation and alter their invasive behavior via leukotriene B4 in experimental inflammatory arthritis. J Immunol 185:5503–5511. 10.4049/jimmunol.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nosaka T, Baba T, Tanabe Y, Sasaki S, Nishimura T, Imamura Y, Yurino H, Hashimoto S, Arita M, Nakamoto Y, Mukaida N. 2018. Alveolar macrophages drive hepatocellular carcinoma lung metastasis by generating leukotriene B4. J Immunol 200:1839–1852. 10.4049/jimmunol.1700544. [DOI] [PubMed] [Google Scholar]
- 17.Chen F, Ghosh A, Lin J, Zhang C, Pan Y, Thakur A, Singh K, Hong H, Tang S. 2020. 5-Lipoxygenase pathway and its downstream cysteinyl leukotrienes as potential therapeutic targets for Alzheimer’s disease. Brain Behav Immun 88:844–855. 10.1016/j.bbi.2020.03.022. [DOI] [PubMed] [Google Scholar]
- 18.Ramalho T, Ramalingam L, Filgueiras L, Festuccia W, Jancar S, Moustaid-Moussa N. 2019. Leukotriene-B4 modulates macrophage metabolism and fat loss in type 1 diabetic mice. J Leukoc Biol 106:665–675. 10.1002/JLB.MA1218-477RR. [DOI] [PubMed] [Google Scholar]
- 19.Secatto A, Rodrigues LC, Serezani CH, Ramos SG, Dias-Baruffi M, Faccioli LH, Medeiros AI. 2012. 5-Lipoxygenase deficiency impairs innate and adaptive immune responses during fungal infection. PLoS One 7:e31701. 10.1371/journal.pone.0031701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peres CM, de Paula L, Medeiros AI, Sorgi CA, Soares EG, Carlos D, Peters-Golden M, Silva CL, Faccioli LH. 2007. Inhibition of leukotriene biosynthesis abrogates the host control of Mycobacterium tuberculosis. Microbes Infect 9:483–489. 10.1016/j.micinf.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Machado ER, Ueta MT, Lourenço EV, Anibal FF, Sorgi CA, Soares EG, Roque-Barreira MC, Medeiros AI, Faccioli LH. 2005. Leukotrienes play a role in the control of parasite burden in murine strongyloidiasis. J Immunol 175:3892–3899. 10.4049/jimmunol.175.6.3892. [DOI] [PubMed] [Google Scholar]
- 22.Pavanelli WR, Gutierrez FR, Mariano FS, Prado CM, Ferreira BR, Teixeira MM, Canetti C, Rossi MA, Cunha FQ, Silva JS. 2010. 5-Lipoxygenase is a key determinant of acute myocardial inflammation and mortality during Trypanosoma cruzi infection. Microbes Infect 12:587–597. 10.1016/j.micinf.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 23.Aliberti J, Serhan C, Sher A. 2002. Parasite-induced lipoxin A4 is an endogenous regulator of IL-12 production and immunopathology in Toxoplasma gondii infection. J Exp Med 196:1253–1262. 10.1084/jem.20021183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benevides L, Cardoso CR, Milanezi CM, Castro-Filice LS, Barenco PV, Sousa RO, Rodrigues RM, Mineo JR, Silva JS, Silva NM. 2013. Toxoplasma gondii soluble tachyzoite antigen triggers protective mechanisms against fatal intestinal pathology in oral infection of C57BL/6 mice. PLoS One 8:e75138. 10.1371/journal.pone.0075138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montoya JG, Liesenfeld O. 2004. Toxoplasmosis. Lancet 363:1965–1976. 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- 26.Sturge CR, Yarovinsky F. 2014. Complex immune cell interplay in the gamma interferon response during Toxoplasma gondii infection. Infect Immun 82:3090–3097. 10.1128/IAI.01722-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park J, Hunter CA. 2020. The role of macrophages in protective and pathological responses to Toxoplasma gondii. Parasite Immunol 42:e12712. 10.1111/pim.12712. [DOI] [PubMed] [Google Scholar]
- 28.Liesenfeld O, Kosek J, Remington JS, Suzuki Y. 1996. Association of CD4+ T cell-dependent, interferon-gamma-mediated necrosis of the small intestine with genetic susceptibility of mice to peroral infection with Toxoplasma gondii. J Exp Med 184:597–607. 10.1084/jem.184.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki Y, Sher A, Yap G, Park D, Neyer LE, Liesenfeld O, Fort M, Kang H, Gufwoli E. 2000. IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii. J Immunol 164:5375–5382. 10.4049/jimmunol.164.10.5375. [DOI] [PubMed] [Google Scholar]
- 30.Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kühn R, Müller W, Trinchieri G, Sher A. 1996. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J Immunol 157:798–805. [PubMed] [Google Scholar]
- 31.Yong EC, Chi EY, Henderson WR, Jr.. 1994. Toxoplasma gondii alters eicosanoid release by human mononuclear phagocytes: role of leukotrienes in interferon gamma-induced antitoxoplasma activity. J Exp Med 180:1637–1648. 10.1084/jem.180.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayatepek E, Flock B, Zelezny R, Kreutzer K, von Giesen HJ. 1999. LTB4 and LTC4 are absent in the cerebrospinal fluid of human immunodeficiency virus type 1-seropositive persons with toxoplasmic encephalitis: evidence for inhibition of 5-lipoxygenase by Toxoplasma gondii. J Infect Dis 179:714–716. 10.1086/314619. [DOI] [PubMed] [Google Scholar]
- 33.Gür ZT, Çalışkan B, Banoglu E. 2018. Drug discovery approaches targeting 5-lipoxygenase-activating protein (FLAP) for inhibition of cellular leukotriene biosynthesis. Eur J Med Chem 153:34–48. 10.1016/j.ejmech.2017.07.019. [DOI] [PubMed] [Google Scholar]
- 34.Brown CR, McLeod R. 1990. Class I MHC genes and CD8+ T cells determine cyst number in Toxoplasma gondii infection. J Immunol 145:3438–3441. [PubMed] [Google Scholar]
- 35.Lamm ME. 1997. Interaction of antigens and antibodies at mucosal surfaces. Annu Rev Microbiol 51:311–340. 10.1146/annurev.micro.51.1.311. [DOI] [PubMed] [Google Scholar]
- 36.Santaolalla R, Abreu MT. 2012. Innate immunity in the small intestine. Curr Opin Gastroenterol 28:124–129. 10.1097/MOG.0b013e3283506559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dubey JP, Lindsay DS, Speer CA. 1998. Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin Microbiol Rev 11:267–299. 10.1128/CMR.11.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fahel JS, de Souza MB, Gomes MT, Corsetti PP, Carvalho NB, Marinho FA, de Almeida LA, Caliari MV, Machado FS, Oliveira SC. 2015. 5-Lipoxygenase negatively regulates Th1 response during Brucella abortus infection in mice. Infect Immun 83:1210–1216. 10.1128/IAI.02592-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soares EM, Mason KL, Rogers LM, Serezani CH, Faccioli LH, Aronoff DM. 2013. Leukotriene B4 enhances innate immune defense against the puerperal sepsis agent Streptococcus pyogenes. J Immunol 190:1614–1622. 10.4049/jimmunol.1202932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borges TK, Alves ÉA, Vasconcelos HA, Carneiro FP, Nicola AM, Magalhães KG, Muniz-Junqueira MI. 2017. Differences in the modulation of reactive species, lipid bodies, cyclooxygenase-2,5-lipoxygenase and PPAR-γ in cerebral malaria-susceptible and resistant mice. Immunobiology 222:604–619. 10.1016/j.imbio.2016.11.010. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki Y, Yang Q, Remington JS. 1995. Genetic resistance against acute toxoplasmosis depends on the strain of Toxoplasma gondii. J Parasitol 81:1032–1034. 10.2307/3284069. [DOI] [PubMed] [Google Scholar]
- 42.Franco LH, Paula MO, Wowk PF, Fonseca DM, Sérgio CA, Fedatto PF, Gembre AF, Ramos SG, Silva CL, Medeiros AI, Faccioli LH, Bonato VL. 2010. Leukotrienes are not essential for the efficacy of a heterologous vaccine against Mycobacterium tuberculosis infection. Braz J Med Biol Res 43:645–650. 10.1590/s0100-879x2010007500053. [DOI] [PubMed] [Google Scholar]
- 43.Medeiros AI, Sá-Nunes A, Soares EG, Peres CM, Silva CL, Faccioli LH. 2004. Blockade of endogenous leukotrienes exacerbates pulmonary histoplasmosis. Infect Immun 72:1637–1644. 10.1128/IAI.72.3.1637-1644.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Panis C, Victorino VJ, Tatakihara VLH, Cecchini R, Rizzo LV, Yamauchi LM, Yamada-Ogatta SF, Martins-Pinge MC, Pinge-Filho P. 2019. Differences in cNOS/iNOS activity during resistance to Trypanosoma cruzi infection in 5-lipoxygenase knockout mice. Mediators Inflamm 2019:5091630. 10.1155/2019/5091630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santos PC, Santos DA, Ribeiro LS, Fagundes CT, de Paula TP, Avila TV, Baltazar LDM, Madeira MM, Cruz RDC, Dias ACF, Machado FS, Teixeira MM, Cisalpino PS, Souza DG. 2013. The pivotal role of 5-lipoxygenase-derived LTB4 in controlling pulmonary paracoccidioidomycosis. PLoS Negl Trop Dis 7:e2390. 10.1371/journal.pntd.0002390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sacramento LA, Cunha FQ, de Almeida RP, da Silva JS, Carregaro V. 2014. Protective role of 5-lipoxigenase during Leishmania infantum infection is associated with Th17 subset. Biomed Res Int 2014:264270. 10.1155/2014/264270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suzuki Y, Orellana MA, Schreiber RD, Remington JS. 1988. Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 240:516–518. 10.1126/science.3128869. [DOI] [PubMed] [Google Scholar]
- 48.Deckert-Schlüter M, Rang A, Weiner D, Huang S, Wiestler OD, Hof H, Schlüter D. 1996. Interferon-gamma receptor-deficiency renders mice highly susceptible to toxoplasmosis by decreased macrophage activation. Lab Invest 75:827–841. [PubMed] [Google Scholar]
- 49.Suzuki Y, Rani S, Liesenfeld O, Kojima T, Lim S, Nguyen TA, Dalrymple SA, Murray R, Remington JS. 1997. Impaired resistance to the development of toxoplasmic encephalitis in interleukin-6-deficient mice. Infect Immun 65:2339–2345. 10.1128/iai.65.6.2339-2345.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jebbari H, Roberts CW, Ferguson DJ, Bluethmann H, Alexander J. 1998. A protective role for IL-6 during early infection with Toxoplasma gondii. Parasite Immunol 20:231–239. 10.1046/j.1365-3024.1998.00152.x. [DOI] [PubMed] [Google Scholar]
- 51.Deckert-Schlüter M, Bluethmann H, Rang A, Hof H, Schlüter D. 1998. Crucial role of TNF receptor type 1 (p55), but not of TNF receptor type 2 (p75), in murine toxoplasmosis. J Immunol 160:3427–3436. [PubMed] [Google Scholar]
- 52.Gounaris E, Heiferman MJ, Heiferman JR, Shrivastav M, Vitello D, Blatner NR, Knab LM, Phillips JD, Cheon EC, Grippo PJ, Khazaie K, Munshi HG, Bentrem DJ. 2015. Zileuton, 5-lipoxygenase inhibitor, acts as a chemopreventive agent in intestinal polyposis, by modulating polyp and systemic inflammation. PLoS One 10:e0121402. 10.1371/journal.pone.0121402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim SY, Kim TB, Moon KA, Kim TJ, Shin D, Cho YS, Moon HB, Lee KY. 2008. Regulation of pro-inflammatory responses by lipoxygenases via intracellular reactive oxygen species in vitro and in vivo. Exp Mol Med 40:461–476. 10.3858/emm.2008.40.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guimarães FR, Sales-Campos H, Nardini V, da Costa TA, Fonseca MTC, Júnior VR, Sorgi CA, da Silva JS, Chica JEL, Faccioli LH, de Barros Cardoso CR. 2018. The inhibition of 5-lipoxygenase (5-LO) products leukotriene B4 (LTB4) and cysteinyl leukotrienes (cysLTs) modulates the inflammatory response and improves cutaneous wound healing. Clin Immunol 190:74–83. 10.1016/j.clim.2017.08.022. [DOI] [PubMed] [Google Scholar]
- 55.Huang L, Zhao A, Wong F, Ayala JM, Struthers M, Ujjainwalla F, Wright SD, Springer MS, Evans J, Cui J. 2004. Leukotriene B4 strongly increases monocyte chemoattractant protein-1 in human monocytes. Arterioscler Thromb Vasc Biol 24:1783–1788. 10.1161/01.ATV.0000140063.06341.09. [DOI] [PubMed] [Google Scholar]
- 56.Aoki Y, Qiu D, Zhao GH, Kao PN. 1998. Leukotriene B4 mediates histamine induction of NF-kappaB and IL-8 in human bronchial epithelial cells. Am J Physiol 274:L1030–L1039. 10.1152/ajplung.1998.274.6.L1030. [DOI] [PubMed] [Google Scholar]
- 57.Rola-Pleszczynski M, Lemaire I. 1985. Leukotrienes augment interleukin 1 production by human monocytes. J Immunol 135:3958–3961. [PubMed] [Google Scholar]
- 58.Rola-Pleszczynski M, Stanková J. 1992. Leukotriene B4 enhances interleukin-6 (IL-6) production and IL-6 messenger RNA accumulation in human monocytes in vitro: transcriptional and posttranscriptional mechanisms. Blood 80:1004–1011. 10.1182/blood.V80.4.1004.1004. [DOI] [PubMed] [Google Scholar]
- 59.Stanková J, Dupuis G, Gagnon N, Thivierge M, Turcotte S, Rola-Pleszczynski M. 1993. Priming of human monocytes with leukotriene B4 enhances their sensitivity in IL-2-driven tumor necrosis factor-alpha production. Transcriptional and post-transcriptional up-regulation of IL-2 receptors. J Immunol 150:4041–4051. [PubMed] [Google Scholar]
- 60.Rola-Pleszczynski M, Chavaillaz PA, Lemaire I. 1986. Stimulation of interleukin 2 and interferon gamma production by leukotriene B4 in human lymphocyte cultures. Prostaglandins Leukot Med 23:207–210. 10.1016/0262-1746(86)90187-3. [DOI] [PubMed] [Google Scholar]
- 61.Le Bel M, Gosselin J. 2015. Leukotriene B4 enhances NOD2-dependent innate response against influenza virus infection. PLoS One 10:e0139856. 10.1371/journal.pone.0139856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Flamand L, Tremblay MJ, Borgeat P. 2007. Leukotriene B4 triggers the in vitro and in vivo release of potent antimicrobial agents. J Immunol 178:8036–8045. 10.4049/jimmunol.178.12.8036. [DOI] [PubMed] [Google Scholar]
- 63.Gaudreault E, Gosselin J. 2008. Leukotriene B4 induces release of antimicrobial peptides in lungs of virally infected mice. J Immunol 180:6211–6221. 10.4049/jimmunol.180.9.6211. [DOI] [PubMed] [Google Scholar]
- 64.Raetz M, Hwang SH, Wilhelm CL, Kirkland D, Benson A, Sturge CR, Mirpuri J, Vaishnava S, Hou B, Defranco AL, Gilpin CJ, Hooper LV, Yarovinsky F. 2013. Parasite-induced TH1 cells and intestinal dysbiosis cooperate in IFN-γ-dependent elimination of Paneth cells. Nat Immunol 14:136–142. 10.1038/ni.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.López-Yglesias AH, Burger E, Araujo A, Martin AT, Yarovinsky F. 2018. T-bet-independent Th1 response induces intestinal immunopathology during Toxoplasma gondii infection. Mucosal Immunol 11:921–931. 10.1038/mi.2017.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burger E, Araujo A, López-Yglesias A, Rajala MW, Geng L, Levine B, Hooper LV, Burstein E, Yarovinsky F. 2018. Loss of Paneth cell autophagy causes acute susceptibility to Toxoplasma gondii-mediated inflammation. Cell Host Microbe 23:177–190.e4. 10.1016/j.chom.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kurashima Y, Kiyono H. 2017. Mucosal ecological network of epithelium and immune cells for gut homeostasis and tissue healing. Annu Rev Immunol 35:119–147. 10.1146/annurev-immunol-051116-052424. [DOI] [PubMed] [Google Scholar]
- 68.Villeret B, Brault L, Couturier-Maillard A, Robinet P, Vasseur V, Secher T, Dimier-Poisson I, Jacobs M, Zheng S-G, Quesniaux VF, Ryffel B. 2013. Blockade of IL-1R signaling diminishes Paneth cell depletion and Toxoplasma gondii induced ileitis in mice. Am J Clin Exp Immunol 2:107–116. [PMC free article] [PubMed] [Google Scholar]
- 69.Araujo ECB, Cariaco Y, Almeida MPO, Briceño MPP, de Sousa JEN, Lima WR, Costa-Cruz JM, Silva NM. 2021. Beneficial effects of Strongyloides venezuelensis antigen extract in acute experimental toxoplasmosis. Parasite Immunol 43:e12811. 10.1111/pim.12811. [DOI] [PubMed] [Google Scholar]
- 70.Oliveira MC, Coutinho LB, Almeida MPO, Briceño MP, Araujo ECB, Silva NM. 2020. The availability of iron is involved in the murine experimental Toxoplasma gondii infection outcome. Microorganisms 8:560. 10.3390/microorganisms8040560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morato-Marques M, Campos MR, Kane S, Rangel AP, Lewis C, Ballinger MN, Kim SH, Peters-Golden M, Jancar S, Serezani CH. 2011. Leukotrienes target F-actin/cofilin-1 to enhance alveolar macrophage anti-fungal activity. J Biol Chem 286:28902–28913. 10.1074/jbc.M111.235309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Serezani CH, Aronoff DM, Jancar S, Mancuso P, Peters-Golden M. 2005. Leukotrienes enhance the bactericidal activity of alveolar macrophages against Klebsiella pneumoniae through the activation of NADPH oxidase. Blood 106:1067–1075. 10.1182/blood-2004-08-3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mancuso P, Lewis C, Serezani CH, Goel D, Peters-Golden M. 2010. Intrapulmonary administration of leukotriene B4 enhances pulmonary host defense against pneumococcal pneumonia. Infect Immun 78:2264–2271. 10.1128/IAI.01323-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Serezani CH, Perrela JH, Russo M, Peters-Golden M, Jancar S. 2006. Leukotrienes are essential for the control of Leishmania amazonensis infection and contribute to strain variation in susceptibility. J Immunol 177:3201–3208. 10.4049/jimmunol.177.5.3201. [DOI] [PubMed] [Google Scholar]
- 75.Chaves MM, Sinflorio DA, Thorstenberg ML, Martins MDA, Moreira-Souza ACA, Rangel TP, Silva CLM, Bellio M, Canetti C, Coutinho-Silva R. 2019. Non-canonical NLRP3 inflammasome activation and IL-1β signaling are necessary to L. amazonensis control mediated by P2X7 receptor and leukotriene B4. PLoS Pathog 15:e1007887. 10.1371/journal.ppat.1007887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Saini S, Singh B, Prakash S, Kumari S, Kureel AK, Dube A, Sahasrabuddhe AA, Rai AK. 2020. Parasitic load determination by differential expressions of 5-lipoxygenase and PGE2 synthases in visceral leishmaniasis. Prostaglandins Other Lipid Mediat 147:106390. 10.1016/j.prostaglandins.2019.106390. [DOI] [PubMed] [Google Scholar]
- 77.Prado MKB, Locachevic GA, Zoccal KF, Paula-Silva FWG, Fontanari C, Ferreira JC, Pereira PAT, Gardinassi LG, Ramos SG, Sorgi CA, Darini ALC, Faccioli LH. 2017. Leukotriene B4 is essential for lung host defence and alpha-defensin-1 production during Achromobacter xylosoxidans infection. Sci Rep 7:17658. 10.1038/s41598-017-17993-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mineo JR, McLeod R, Mack D, Smith J, Khan IA, Ely KH, Kasper LH. 1993. Antibodies to Toxoplasma gondii major surface protein (SAG-1, P30) inhibit infection of host cells and are produced in murine intestine after peroral infection. J Immunol 150:3951–3964. [PubMed] [Google Scholar]
- 79.Zorgi NE, Costa A, Galisteo AJ, Jr, do Nascimento N, de Andrade HF, Jr.. 2011. Humoral responses and immune protection in mice immunized with irradiated T. gondii tachyzoites and challenged with three genetically distinct strains of T. gondii. Immunol Lett 138:187–196. 10.1016/j.imlet.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 80.Nagatake T, Hirata SI, Koga T, Kuroda E, Kobari S, Suzuki H, Hosomi K, Matsumoto N, Yanrismet Y, Shimojou M, Morimoto S, Sasaki F, Ishii KJ, Yokomizo T, Kunisawa J. 2019. BLT1 mediates commensal bacteria-dependent innate immune signals to enhance antigen-specific intestinal IgA responses. Mucosal Immunol 12:1082–1091. 10.1038/s41385-019-0175-z. [DOI] [PubMed] [Google Scholar]
- 81.Mack DG, McLeod R. 1992. Human Toxoplasma gondii-specific secretory immunoglobulin A reduces T. gondii infection of enterocytes in vitro. J Clin Invest 90:2585–2592. 10.1172/JCI116153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sumyuen MH, Garin YJ, Derouin F. 1995. Early kinetics of Toxoplasma gondii infection in mice infected orally with cysts of an avirulent strain. J Parasitol 81:327–329. 10.2307/3283948. [DOI] [PubMed] [Google Scholar]
- 83.Heimesaat MM, Dunay IR, Bereswill S. 2019. Comprehensive kinetic survey of intestinal, extra-intestinal and systemic sequelae of murine ileitis following peroral low-dose Toxoplasma gondii infection. Front Cell Infect Microbiol 9:98. 10.3389/fcimb.2019.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Silva NM, Vieira JC, Carneiro CM, Tafuri WL. 2009. Toxoplasma gondii: the role of IFN-gamma, TNFRp55 and iNOS in inflammatory changes during infection. Exp Parasitol 123:65–72. 10.1016/j.exppara.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 85.Sforcin JM, Nunes GA, Missima F, Sá-Nunes A, Faccioli LH. 2007. Effect of a leukotriene inhibitor (MK886) on nitric oxide and hydrogen peroxide production by macrophages of acutely and chronically stressed mice. J Pharm Pharmacol 59:1249–1254. 10.1211/jpp.59.9.0009. [DOI] [PubMed] [Google Scholar]
- 86.Foureau DM, Mielcarz DW, Menard LC, Schulthess J, Werts C, Vasseur V, Ryffel B, Kasper LH, Buzoni-Gatel D. 2010. TLR9-dependent induction of intestinal alpha-defensins by Toxoplasma gondii. J Immunol 184:7022–7029. 10.4049/jimmunol.0901642. [DOI] [PubMed] [Google Scholar]
- 87.Castillo PA, Nonnecke EB, Ossorio DT, Tran MTN, Goley SM, Lönnerdal B, Underwood MA, Bevins CL. 2019. An experimental approach to rigorously assess Paneth cell α-defensin (Defa) mRNA expression in C57BL/6 mice. Sci Rep 9:13115. 10.1038/s41598-019-49471-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]