

Abstract
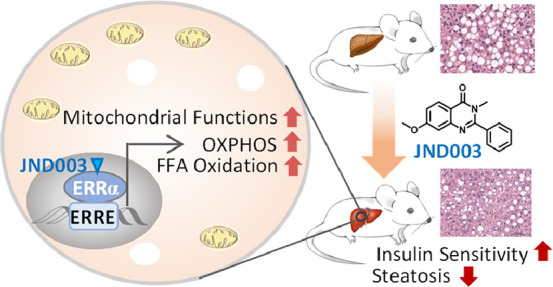
Nonalcoholic fatty liver disease (NAFLD) is one of the most prevalent forms of chronic liver diseases and is causally linked to hepatic insulin resistance and reduced fatty acid oxidation. Therapeutic treatments targeting both hepatic insulin resistance and lipid oxidative metabolism are considered as feasible strategies to alleviate this disease. Emerging evidence suggests estrogen-related receptor alpha (ERRα), the first orphan nuclear receptor identified, as a master regulator in energy homeostasis by controlling glucose and lipid metabolism. Small molecules improving the functions of ERRα may provide a new option for management of NAFLD. In the present study, using liver-specific Errα knockout mouse (Errα-LKO), we showed that liver-specific deletion of ERRα exacerbated diet-evoked fatty liver hepatic and systemic insulin resistance in mice. A potent and selective ERRα agonist JND003 (7) was also discovered. In vitro and in vivo investigation demonstrated that the compound enhanced the transactivation of ERRα downstream target genes, which was accompanied by improved insulin sensitivity and fatty liver symptoms. Furthermore, the therapeutic effects were completely abolished in Errα-LKO mice, indicative of its on-target efficacy. Our study thus suggests that hepatic ERRα is a viable target for NAFLD and that the ERRα agonist may serve as an intriguing pharmacological option for management of metabolic diseases.
Keywords: estrogen-related receptor alpha, nonalcoholic fatty liver disease, liver-specific Errα knockout mouse, ERRα agonist, fatty acid oxidation
Introduction
Nonalcoholic fatty liver disease (NAFLD) is one of the most prevalent forms of chronic liver disease.1 NAFLD increases the susceptibility of the liver to acute liver injury and is therefore a key etiological event preceding nonalcoholic steatohepatitis (NASH), cirrhosis, and hepatocellular cancer.2,3 Accumulating evidence also indicates that NAFLD is an independent risk factor for subclinical and clinical cardiovascular diseases.4,5 Although a number of compounds with various mechanisms have been advanced into different stages of clinical investigation, to date, no therapeutic drug for NAFLD is approved by the US FDA.6,7 The first-line treatment for NAFLD is still weight loss through a combination of exercise and a controlled diet. Hepatic steatosis is the first stage of fatty liver disease with lipid accumulation observed in the liver. Although this condition is reversible and benign, it is commonly associated with hepatic insulin resistance characterized by increased gluconeogenesis, lipogenesis, and reduced fatty acid oxidation. These events subsequently induce the formation of reactive oxygen species, Kupffer cell activation, and hepatocyte apoptosis.8 Therapeutic treatments targeting both insulin resistance and glucose/lipid metabolism might be an efficient strategy for prevention and treatment of NAFLD.
Estrogen-related receptors (ERRs) belong to an orphan nuclear receptor superfamily. Three isoforms of ERRs (i.e., ERRα, β, and γ) have been identified.9 ERRα is the first-discovered and most-studied member of this subgroup. It was cloned in 1988 based on its sequence similarity in the DNA-binding domain to that of estrogen receptor alpha (ERα).10 ERRα binds on an extended estrogen response element half-site (5′-TCAAGGTCA-3′) which was later named ERRα response element (ERRE).3 Despite this, ERRα does not bind natural estrogens nor does it directly participate in classic estrogen signaling pathways or biological processes.11 ERRα was originally designated as an orphan nuclear receptor because of the lack of the natural ligand, but recent study suggested that cholesterol (2) is possibly an endogenous ligand of ERRα and mediates its biological functions in bone, macrophage, and muscle cells.43
Most of the early studies focused on the roles of ERRα in cancer, bone differentiation, and development.4,12 Several small-molecule ERRα inverse agonists have been identified with antitumor potential.13 During the past decade, emerging evidence suggests that ERRα plays a central role in energy metabolism by serving as a major transcriptional regulator of many metabolism-related genes.14 Genome-wide identification of ERRα-binding sites reveals that it is preferentially associated with genes participating in glucose metabolism, lipid oxidation, mitochondrial biogenesis, and energy sensing.15 ERRα had been identified as a primary effector of the peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) that is a key coactivator in energy homeostasis through analyses of transcriptional profiles using a computational strategy.16,17 A master regulatory role of ERRα in metabolism was further supported by reduced expression of ERRα-regulated genes in insulin-resistant subjects, while overexpression of a chimeric, constitutively active form of ERRα was sufficient to induce mitochondrial gene expression and biogenesis.11 Studies have also confirmed that ERRα participates in transcriptional regulation of specific genes required for fatty acid oxidation.18 ERRα is abundantly expressed in tissues with high rates of fatty acid oxidation.19 Specifically, two gatekeepers controlling mitochondrial fatty acid oxidation, that is, medium-chain acyl CoA dehydrogenase (Mcad and Acadm) and malonyl CoA decarboxylase (Mlycd), are ERRα target genes, providing a robust link between lipid metabolism and ERRα function.20 In addition to lipid utilization, ERRα plays a key role in glucose metabolism as it regulates genes involved both in glycolysis and glycolysis-associated reactions as well as genes encoding the glucose transporter family.10,21,22 The expression of pyruvate dehydrogenase kinase 4 (Pdk4) is also driven by ERRα, thus facilitating the entry of glucose-derived pyruvate into the tricarboxylic acid cycle (TCA) in mitochondria.23,24 Interestingly, ERRα also negatively regulates hepatic gluconeogenesis by serving as a repressor of phosphoenolpyruvate carboxykinase (Pepck) gene transcription.25
It is therefore conceivable that within the liver, one of the organs with the highest rates of glucose and lipid metabolism, ERRα may act as a key player in energy homeostasis. Indeed, genetic and pharmacological inhibition of ERRα activity has been reported to exacerbate rapamycin-induced hepatic hyperlipidemia in mice.26 However, whole-body ERRα-deficient mice were interestingly found to protect themselves from high-fat diet-induced NAFLD and glucose intolerance,13,27 which might at least partially attribute to the deficiency of an ERRα direct downstream target, apolipoprotein B48, to prevent lipid absorption in the intestine.28 Systemic administration of a small-molecule inverse agonist or a polyamine-based ERRE-binding inhibitor of ERRα was also reported to normalize serum triglyceride (TG), improve glucose tolerance, and block liver steatosis and steatohepatitis in various animal models.29−31 These inconsistent results suggest the complexity of regulatory functions of ERRα in metabolic homeostasis. Additionally, in contrast to the significant progress on ERRα antagonist discovery, rare ERRα agonists have been characterized, possibly due to the tiny binding pocket of the ERRα ligand-binding domain (LBD) which is as small as 100 Å3.25,44 Although several dietary products (3) and statins (compounds 4–6) were reported to upregulate the transcriptional functions of ERRα in preliminary screening assays32,33 (Figure 1), their pharmacological efficacies and action modes were not properly investigated. It remains an open question whether positively or negatively targeting ERRα will achieve therapeutic benefit for human metabolic disorders.
Figure 1.

Chemical structures of the representatives of potential ERRα agonists.
In the present study, we first generated a liver-specific Errα knockout mouse model (Errα-LKO) to validate the critical functions of ERRα in hepatic energy homeostasis. Different from the previous observation in whole-body Errα-knockout animals, liver-specific deletion of Errα exacerbated diet-evoked fatty liver and systemic insulin resistance in mice. A small molecule, JND003, was also discovered as a new ERRα agonist through a scaffold-hopping strategy from our previously reported molecules. Further in vitro and in vivo investigation demonstrated that pharmacological agonism of ERRα enhanced transcription of ERRα downstream target genes, improved insulin sensitivity, and alleviated fatty liver symptoms. Our study provides a “proof-of-concept” investigation to support the hypothesis that agonism of ERRα may be a new therapeutic strategy against obesity-associated NAFLD.
Results and Discussion
Liver-Restricted Deletion of Errα Exacerbates Glucose/Lipid Dysregulation and NAFLD in Mice
In order to validate the physiological functions of ERRα in the liver and its pathogenic contribution to fatty liver symptoms, a liver-restricted Errα knockout mouse model (Errα-LKO) was generated as described in experiments and Figure 2A. The presence of albumin-driven Cre recombinase leads to hepatocyte-selective deletion of exon 2 that includes the start codon and part of the DNA-binding domain (DBD) of ERRα. The selective deletion of ERRα in the liver of mice was confirmed by western blot analysis (Figure 2B, J).
Figure 2.

Reduced expression of ERRα and fatty acid metabolic related genes in liver tissue of Errα- LKO mice. (A) ERRα-loxP strategy; (B) ERRα proteins were analyzed in the liver, adipose, heart, and kidney of wild-type and Errα- LKO mice by western blot analysis; (C) animals’ body weight change upon high-fat diet feeding; (D) IGTT showing plasma glucose responses determined after intraperitoneal injection of glucose after 12 h fasting; (E) intraperitoneal ITT showing plasma glucose responses determined after intraperitoneal injection of insulin after 6 h fasting; (F) glucose uptake of liver tissue; (G) FFA oxidation; (H) CS activity; (I) relative RNA expression of Errα, Pdk4, Mcad, ATP5β, Cpt1a, Cs, and Sdh; and (J) western blot analysis on the liver with ERRα, SDH, MCAD, and ATP5β. *p < 0.05, compared with WT, n = 5.
To examine the role of ERRα in obesity-associated NAFLD and metabolic syndromes, male Errα-LKO mice were fed high-fat diet for 8 weeks and the phenotypes were monitored accordingly. Male littermates without the Cre recombinase transgene were used as the wild-type (WT) control. Compared to the WT mice, Errα-LKO mice gained more weight upon high-fat diet feeding (Figure 2C), while the food intake between WT and LKO mice was similar (Supporting Information Figure S1A). Intraperitoneal glucose tolerance test (IGTT) results revealed that Errα-LKO mice were more susceptible to diet-induced glucose intolerance (Figure 2D). Moreover, insulin resistance evoked by the high-fat diet was more prominent in Errα-LKO mice as determined by the intraperitoneal insulin tolerance test (ITT) (Figure 2E). The impaired glucose disposal at least partially attributed to a dampened glucose uptake in liver tissue under both basal and insulin-stimulated conditions (Figure 2F).
The liver tissues of Errα-LKO mice also exhibited a significant reduction in free fatty acid (FFA) oxidation rate (Figure 2G) and citrate synthase (CS) activity (Figure 2H), indicating a lower mitochondrial capacity. The reduced glucose and lipid metabolism in liver tissues of Errα-LKO mice were also corroborated by reduced mRNA levels of Pdk4, Atp5β, Mcad, Cpt1a, CS, and Sdh (Figure 2I). Protein levels of SDH, MCAD, and ATP5β were also obviously decreased in the Errα-LKO mice (Figure 2J).
In accordance with impaired lipid catabolism in the Errα-LKO liver, higher plasma levels of TG, total cholesterol (TC), and FFA were observed in these mice (Figure 3A–C). Additionally, liver-restricted deletion of Errα caused more pronounced hepatosteatosis, as determined by oil red O and hematoxylin–eosin (HE) staining (Figure 3D). Quantification of total lipids and TG within liver tissues confirmed the pronounced lipid deposition in livers of Errα-LKO mice (Figure 3E,F). In addition to simple steatosis, Errα-LKO mice also exhibited more prominent liver injury, as exemplified by elevated serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels (Figure 3G,H). These data collectively supported the fact that deletion of Errα in the liver led to diet-induced hepatic insulin resistance and hepatosteatosis.
Figure 3.

Enhanced liver steatosis and serum lipid disorders in Errα-LKO mice. (A) Plasma TG; (B) TC; (C) FFA; (D) HE staining and oil red O staining of the liver; (E) relative lipid accumulation and (F) TG contents in the liver; and (G) ALT and (H) AST levels. *p < 0.05, compared with WT, n = 5.
It is noteworthy that the results obtained here are in contrast to the previous observation that whole-body ERRα-deficient mice were protective from high-fat diet-induced NAFLD and glucose intolerance.26,34,35 This should not be surprising because ERRα demonstrates multifaceted roles in different tissues through modulating a panel of genes, and its functions are likely location- and context-dependent. For instance, ERRα facilitates adipocyte differentiation and adipogenesis in adipose tissue such that mice with a global deficiency of ERRα exhibit reduced adiposity.36,37 This may explain why global Errα KO mice are more insulin-sensitive and display improved fatty liver symptoms. ERRα was also demonstrated as a key transcriptional regulator of apolipoprotein B48 in the intestine. Whole-body Errα KO significantly reduced apolipoprotein B48 to suppress lipid absorption in the intestine, which explains why these knockout mice are lean and resistant to a high-fat diet.28,35 Further tissue-specific deletion of Erra, for example, adipocyte-, skeletal muscle-, and kidney-restricted knockout, is warranted to elucidate the functions and the pathophysiological roles of ERRα.
Discovery of JND003 (7) as a New ERRα-Selective Agonist
The Errα-LKO mouse investigation showed that hepatic ERRα played an essential role in maintaining hepatic and systemic metabolic homeostasis and that deficiency of ERRα in the liver accelerated NAFLD. Pharmacological agonism of ERRα function was thus hypothesized to be protective against diet-induced NAFLD and insulin resistance. We previously discovered a pyrido[1,2-α]pyrimidin-4-one-based molecule (1, JND045) which potently improved the transcriptional functions of ERRα.29 Based on the structural geometry of compound 1, 7-methoxy-3-methyl-2-phenyl quinazolin-4(3H)-one (compound 7, JND003) was designed and synthesized as a new potential ERRα agonist by utilizing a scaffold-hopping strategy (Figure 4A). The designed compound 7 was readily synthesized using 2-amino-4-methoxybenzoic acid (8) as the starting material. Briefly, compound 8 was reacted with benzoyl chloride under basic conditions to afford intermediate 9, which was further condensed with methylamine hydrochloride to obtain the target compound (Figure 4A). Preliminary biological evaluation revealed that compound 7 effectively improved the ERRα-driven luciferase activity at 5.0 μM after 24 h of incubation in a heterologous cell-based reporter assay, suggesting its potential agonism on the transcriptional functions of ERRα (Supporting Information Figure S2).21,22 The ERRα transcriptional function enhanced by compound 7 was further validated in AAB-Luc cells stably expressing ERRα luciferase reporter33 to exhibit an EC50 value of 2.7 μΜ (Figure 4B). Binding of the compound with ERRα-LBD was also confirmed using an AlphaScreen assay to display an EC50 value of 86.0 nM enhancing the interaction between ERRα and PGC1α peptide (Figure 4C). The in situ binding of the molecule with ERRα was further validated using a thermal stability assay (Figure 4D,E). It was found that the compound 7 obviously inhibited thermal degradation of ERRα in HepG2 cells at a concentration as low as 1.0 μM.
Figure 4.

Compound 7 potently and selectively agonizes the transcriptional function of ERRα. (A) Design strategy and chemical synthesis of compound 7 (JND003); (B) ERRα expression stimulated by 7 in AAB-Luc cells. (C) Compound 7 enhances interaction of ERRα with the PGC1α peptide determined by an AlphaScreen assay. (D,E) Compound 7 improves thermal stability of ERRα in HepG2 cells determined using a CETSA. (F) Compound 7 exhibits selective transcriptional improvement of ERRα over ERRβ and ERRγ. (G) Compound 7 elevates mRNA levels of ERRα-targeted genes, that is, Mcad, Atp5β, and Pdk4. *p < 0.05 vs DMSO.
Real-time polymerase chain reaction (PCR) analysis also confirmed that compound 7 selectively elevated the transcriptional level of ERRα by approximately 2.5-fold at 5.0 μM after 24 h of incubation, whereas it did not display an obvious effect on ERRβ and ERRγ, suggesting its favorable isoform selectivity (Figure 4F). Likewise, the expression of other nuclear hormone receptors, such as ERα, ERβ, ERγ, and PPARγ, was also not obviously affected by the treatment of compound 7 (Supporting Information Figure S3). The agonistic efficacy of compound 7 was further evaluated by investigating its impact on the expression of ERRα-targeted genes, for example, MCAD, PDK4, and ATP5β. It was shown that the compound dose-dependently elevated mRNA levels of the downstream genes in a quantitative real-time PCR assay (Figure 4G).
Compound 7 Reduces Lipid Accumulation and Enhances Fatty Acid Oxidization in Hepatocytes
To examine the role of ERRα on glucose metabolism and lipid oxidation, insulin-resistant cell models were established by treating HepG2 cells with high glucose/high insulin or palmitic acid (HepG2-IR), respectively.10,38 Oil red O staining indicated that treatment of compound 7 significantly alleviated lipid accumulation in both of the two insulin-resistant hepatocyte models at 10.0 μM after 48 h of incubation (Figure 5A–D). Meanwhile, transient transfection (overexpression) of Errα exerted similar effects to JND003-treatment, while shRNA-mediated knockdown of Errα aggravated lipid accumulation in hepatocytes (Figure 5A–D). Further investigation demonstrated that compound 7 also significantly enhanced glucose uptake and fatty acid oxidation in HepG2-IR cells (Figure 5E,F). Expression analysis demonstrated that ERRα target genes, including Errα itself, were increased by the molecule in a dose-dependent manner (Supporting Information Figure S2), which was similar to the trend observed in Errα-overexpressing cells (Supporting Information Figure S2). On the contrary, expression levels of the ERRα target genes were reduced in HepG2 cells with knocked down Errα (Supporting Information Figure S2).
Figure 5.

Compound 7 reduces lipid accumulation and enhances FFA oxidation in HepG2-IR cells. (A) Oil red O staining of glucose/insulin-induced HepG2-IR cells. The glucose/insulin-induced HepG2-IR cell model was established with high glucose and insulin. The HepG2-IR cells were treated with DMSO or compound 7 at 10 μM. The ERRα overexpressing or knockdown groups were utilized as the positive and negative controls, respectively. (B) Quantitation of lipid accumulation in glucose/insulin-induced HepG2-IR cells from A. (C) Oil red O staining of FFA-induced HepG2-IR cells. The FFA-induced HepG2-IR cell model was established with FFA incubation. FFA-induced HepG2-IR cells were treated with DMSO or compound 7, respectively. The ERRα overexpressing or knockdown groups were utilized as the positive and negative controls, respectively. (D) Quantitation of lipid accumulation in FFA-induced HepG2-IR cells of C. (E) Glucose uptake in HepG2-IR cells after being treated with DMSO or compound 7, respectively. (F) FFA oxidation in HepG2-IR cells after being treated with DMSO or compound 7. *p < 0.05, vs DMSO.
Compound 7 Is Orally Bioavailable and Exhibits High Grade of Distribution in the Liver and Abdominal Adipose Tissues
A preliminary pharmacokinetic (PK) investigation of compound 7 was also conducted in SD rats (Supporting Information, Table S1). The results showed that compound 7 was rapidly absorbed to reach a maximum plasma concentration of 531.03 ± 120.31 ng/mL (2.0 ± 0.45 μM) after oral administration of the compound at a dose of 10 mg/kg in 15 min. However, compound 7 in plasma could also be quickly eliminated with a t1/2 value of 0.73 h. The short t1/2 value also resulted in a relatively low plasma exposure with an area under curve (AUC) value of 382.57 ± 110.38 h·ng/mL, whereas the corresponding AUC value for IV (intravenous) injection was approximately 857.18 ± 68.62 h·ng/mL at a dose of 2.0 mg/kg. Thus, the compound exhibited a relatively low oral bioavailability of 9.0% (Supporting Information, Table S1).
The distribution of compound 7 was also investigated by monitoring the drug concentrations in different rat tissues after oral drug administration (Figure 6 and Supporting Information, Table S2). It was found that compound 7 exhibited significantly higher grade of distribution in the liver and abdominal adipose tissues compared with that in plasma. For instance, compound 7 exhibited 58.9-, 39.8-, 28.6-, and 47.4-fold higher concentrations in rat liver tissues than that in plasma, at 0.25, 0.5, 2.0, and 4.0 h time points after drug administration, respectively. The compound also demonstrated higher concentrations in abdominal adipose tissues than that in plasma with factors of 7.4-, 30.1-, 22.8-, and 234.8-fold, respectively, at the corresponding time points. Similar to many other drugs, compound 7 also demonstrated a significantly high grade of distribution in the major absorption tissues such as the stomach, large intestine, and small intestine after oral administration. However, its distributions in other tissues, for example, muscle, heart, lung, spleen, kidney, testis, and brain, were significantly lower. It is noteworthy that ERRα is predominately expressed in metabolically active tissues such as muscle, liver, adipose, and so forth. The preferred distribution of compound 7 in the liver of healthy rats inspired us to hypothesize that it could selectively agonize the functions of hepatocyte ERRα to achieve protection from diet-induced NAFLD and improve the insulin sensitivity after oral administration. However, it is intriguing to examine whether the diseased state would potentially affect the biodistribution of the drug and warrant future study.
Figure 6.

Relative levels of compound 7 in different tissues compared with that in plasma after oral administration (30 mg/kg).
Compound 7 Improves Insulin Sensitivity and Serum Lipid Disorders In Vivo
HFD-induced obese mice were orally treated with a vehicle or compound 7 (10 and 30 mg/kg/day body weight) for 4 weeks during administration of high-fat diet. Body weight and fasting serum glucose of the mice were monitored during the treatment period. It was found that although compound 7 did not obviously affect body weight of the mice, animals treated with compound 7 displayed decreased fasting glucose levels compared to those in vehicle control groups (Figure 7A,B), while food intake between compound 7-treated and PBS-fed mice was similar (Supporting Information, Figure 1B). Administration of compound 7 also exhibited improved glucose tolerance and insulin resistance (Figure 7C–F) as determined by IGTT and ITT, respectively. Further investigation revealed that compound 7 significantly sensitized Akt signaling pathways in the liver (Figure 7G,H).
Figure 7.

Antidiabetic effects of compound 7 in HFD-fed C57 mice. (A) Body weight; (B) blood fasting glucose; (C) glucose tolerance test showing plasma glucose responses determined after intraperitoneal injection of glucose after 12 h fasting; (D) AUC analysis of (C); (E) ITT showing plasma glucose responses after intraperitoneal injection of insulin after 6 h fasting; (F) AUC analysis of (E); (G) western blot analyses on liver tissues; and (H) densitometric quantifications of the western blot showing the ratio of p-Akt to total Akt in Figure 7G. *p < 0.05, compound 7 treatment compared with mice with a PBS vehicle, #p < 0.05, HFD group compared with LFD (low-fat-diet) mice, n = 5.
Obesity-associated outcomes including insulin resistance and steatosis coincide with changes in plasma markers for glucose and lipid homeostasis.39 Upon high-fat diet feeding, mice showed significantly higher plasma levels of TG, TC, FFA, and insulin, all of which were attenuated by supplementation of compound 7 (Figure 8A–D), supporting that pharmacological activation of ERRα exerts a beneficial impact on systemic insulin sensitivity and lipidemia independent of body weight and adiposity.
Figure 8.

Compound 7 alleviates liver steatosis and serum lipid disorders in HFD-fed C57 mice. Plasma (A) TGs; (B) TC; (C) FFA; (D) insulin; (E) ALT and (F) AST levels in response to compound 7; and (G) representative HE staining and oil red O staining of liver sections with the corresponding quantitation of (H) relative lipid accumulation and (I) TG contents in response to compound 7. *p < 0.05, compound 7-treated mice compared with PBS-fed mice, #p < 0.05, HFD group compared with LFD mice, n = 5.
Compound 7 Protects Obese Mice from HFD-Induced NAFLD and Enhances FFA Oxidation and Mitochondrial Functions in Livers of HFD-Induced C57 Mice
In addition to glucose disposal, the effect of compound 7 on the pathogenesis of NAFLD was also evaluated. As determined by Oil red O and HE staining, daily oral administration of compound 7 potently mitigated hepatic steatosis in obese mice at both doses of 10 and 30 mg/kg body weight (Figure 8H,I). Moreover, serum ALT and AST levels were also dampened by compound 7 (Figure 8E,F). Notably, the effects of compound 7 on systemic and liver insulin resistance were almost completely abolished in Errα-LKO mice at a dose of 30 mg/kg body weight (Figure 9A–I). These data collectively suggested that the favorable effect of compound 7 was, at least in part, mediated through liver ERRα, although it could also modulate the functions of ERRα in other tissues because of the broad tissue distributions.
Figure 9.

Beneficial effects of compound 7 on liver steatosis are almost completely abolished in Errα-LKO mice. Plasma (A) TGs; (B) TC; (C) FFAs; (D) HE staining and oil red O staining of the liver with the corresponding quantification of (E) relative lipid accumulation and (F) TG contents in the liver in response to compound 7; (G) ALT and (H) AST levels in response to compound 7; and (I) glucose uptake of liver tissue.
Preliminary pharmacodynamic investigation was also conducted to show that compound 7 significantly elevated transcription of Errα and its target genes in the mouse liver, that is, Pdk4, Atp5β, and Pgc1α, at both doses of 10 and 30 mg/kg body weight (Figure 10A). Two important genes involved in FFA oxidation, that is, Mcad and Cpt1a, were also obviously enhanced in the compound-treated group (Figure 10A). Western blot assays further validated that the compound significantly increased protein levels of ERRα, PDK4, MCAD, and ATP5β (Figure 10B).
Figure 10.

Compound 7 enhances mitochondrial function and FFA oxidation. (A) Relative RNA expression of Errα, Pdk4, Atp5β, Pgc1α, Mcad, and cpt1a in response to compound 7; (B) western blot analysis on the liver with ERRα, PKD4, MCAD, and ATP5β; (C) relative RNA expression of Cs, TfII, CoxII, Sdh, Acat2, Cyp4a12b, and Aldh1b1 in response to compound 7; (D) compound 7 enhances the expression of mitochondria DNA; (E) compound 7 enhances the activity of CS and FFA oxidation (F); and (G) western blot analysis on the liver with AOX1, Cytochrome C, SDH, and Cpt1A. (H) Electron microscopic analysis and (I) O2 consumption. *p < 0.05, compound 7 compared with PBS mice during a 12 h light/dark cycle, #p < 0.05, HFD group compared with LFD mice, n = 5.
Reduced mitochondrial biogenesis and mitochondrial dysfunction are characteristic features in NAFLD. The effect of compound 7 on mitochondrial function was also evaluated by determining the expression of several mitochondria-related genes including Cs, TfII, Sdh, Acat2, Cyp4a12b, and Aldhlb1. It was found that the molecule significantly increased the mRNA levels of these genes (Figure 10C). The copy number of mitochondrial DNA was also elevated after treatment of the compound and accompanied by higher CS activity and fatty acid oxidation (Figure 10D–F). Moreover, protein levels of important mitochondrial function proteins, that is, AOX1, Cytochrome C, SDH, and CPT1A, were also elevated (Figure 10G). JND003-induced mitochondrial biogenesis was further supported by an electron microscopic analysis (Figure 10H). It is also noteworthy that mice treated with compound 7 exhibited elevated oxygen consumption, coinciding with the increased lipid oxidation and energy expenditure of these mice with a metabolic cage (Figure 10I).
In summary, we have demonstrated the crucial beneficial functions of liver ERRα in the development of NAFLD and insulin resistance using liver-specific Errα KO mice. Hepatocyte-specific deletion of Errα exacerbates diet-induced hepatosteatosis and hepatic/systemic insulin resistance. Moreover, JND003 (compound 7) was discovered as a new selective small-molecule ERRα agonist. Administration of the molecule significantly improved the transcriptional functions of ERRα and evoked beneficial effects on type 2 diabetes (T2DM) and liver steatosis through improved insulin sensitivity, enhanced fatty acid oxidation, and mitochondrial function both in vitro and in vivo. However, the therapeutic benefits of the ERRα agonist were largely abrogated in the liver-specific Errα KO mice, supporting its on-target effect. To the best of our knowledge, this is the first proof-of-concept in vivo investigation of a selective ERRα agonist, and the results agree with the previous observation that treatment of mice with mTOR inhibitors in combination with ERRα inhibiting agents aggravated the development of NAFLD.26 Although the detailed action mechanism of JND003 remains elusive, our study may provide some important knowledge and a research tool for further investigation of ERRα agonists as new potential therapeutic agents for T2DM and liver steatosis. Currently, it remains unknown whether or not compound 7 induces a conformational change in ERRα that engages the activation of its activity. Further studies are warranted to address this question which will facilitate the application and further optimization of the lead molecule.
Experiments
Chemistry
All reagents and solvents were obtained from commercial sources and were used without further purification. Flash chromatography was performed using silica gel (200–300 mesh). The reactions were monitored by TLC, using silica gel plates with fluorescence GF254 and UV light visualization. Melting points were determined on OptiMelt. 1H and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer at 400 and 100 MHz, respectively. Coupling constants (J) are expressed in hertz (Hz). Chemical shifts (δ) of NMR are reported in parts per million (ppm) units relative to the internal control (TMS). High-resolution ESI-MS spectra were recorded on an AB Sciex X500r QTOF mass spectrometer. The purity of JND003 was determined by reverse-phase high-performance liquid chromatography (HPLC) analysis to be 99.70%. HPLC instrument: Dionex Summit HPLC (Column: Diamonsil C18, 5.0 μm, 4.6 × 250 mm (Dikma Technologies); detector: PDA-100 photodiode array; inJector: ASI-100 autoinJector; pump: p-680A). A flow rate of 1.0 mL/min was used with the mobile phase of MeOH in H2O.
7-Methoxy-2-phenyl-4H-benzo[d][1,3]oxazin-4-one (9)
A solution of 2-amino-4-methoxybenzoic acid (1.67 g, 10 mmol) in pyridine (10 mL) was added drop-wise to the solution of benzoyl chloride (1.4 g, 10 mmol) in pyridine (5 mL) at room temperature. The reaction was stirred for 6 h at room temperature. Then, the mixture was poured into ice water (50 g), extracted with ethyl acetate, dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the residue was purified by flashing chromatography on silica gel (ethyl acetate: petroleum ether = 1: 4) to obtain 1.96g of intermediate 9 as a white solid (yield: 77.5%). 1H NMR (400 MHz, DMSO-d6): δ 8.20 (d, J = 7.5 Hz, 2H), 8.07 (d, J = 8.5 Hz, 1H), 7.68 (t, J = 7.3 Hz, 1H), 7.61 (t, J = 7.5 Hz, 2H), 7.25–7.14 (m, 2H), 3.96 (s, 3H). ESI-MS m/z: = 254.0 [M + H]+.
7-Methoxy-3-methyl-2-phenyl Quinazolin-4(3H)-one (7, JND003)
Intermediate 9 (0.253 g, 1.0 mmol) and methylamine hydrochloride (0.675 g, 10 mmol) were dissolved in DMF (10 mL). The reaction mixture was heated to reflux for 6 h. Then, the mixture was poured into ice water (50 g), extracted with ethyl acetate, dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the residue was purified by flashing chromatography on silica gel (ethyl acetate/petroleum ether = 1:1) to afford the desired compound 7 as a white solid (0.14 g, 51.0%). mp: 179.7–179.9 °C. 1H NMR (400 MHz, CD3OD): δ 8.15 (d, J = 8.9 Hz, 1H), 7.72–7.49 (m, 5H), 7.24–6.97 (m, 2H), 3.92 (s, 3H), 3.44 (s, 3H). 13C NMR (101 MHz, CD3OD): δ 165.04, 162.32, 157.73, 149.14, 135.15, 130.00, 128.50, 127.85, 127.70, 116.92, 113.51, 106.93, 54.90, 33.14. HPLC analysis: MeOH: H2O (80:20), 5.95 min, 99.70% purity. HRMS (ESI): calcd for C16H15N2O2 [M + H]+, 267.1128; found, 267.1130.
Generation of Errα Liver-Specific Knockout Mice
A 3.4 and a 3.6 kb genomic fragment upstream and downstream of mouse ERRα exon 2 were used as recombination arms, respectively, in constructing the targeting vector. The targeting vector was performed according to the following protocol: C57BL/6J embryonic stem (ES) cells were electroporated with the linearized targeting construct. After being selected in the presence of G418, clones with targeted alleles were identified through PCR analysis. ES cells with targeted alleles were injected into the blastocysts of C57BL6/J mice. The chimeric males were mated with females of the same strain to obtain heterozygous mutant mice. Then, the Errα-floxed mice were crossed with albumin-Cre mice, and the homozygous Errα KO mice were subsequently generated.
Animal Study
Male mice (8 weeks old) in the C57BL/6J background were housed in an SPF mouse room and maintained in a 12 h light–dark cycle at 23 °C and fed with a high-fat diet (21.9 kJ/g, 60% of energy as fat, 20% of energy as protein, 20% of energy as carbohydrate; D12492; Research Diet, New Brunswick, NJ, USA) or low-fat diet (3.25 kJ/g, 10% of energy as fat, 20% of energy as protein, 70% of energy as carbohydrate; D12450B; Research Diet, New Brunswick, NJ, USA) for 8 weeks, followed by daily oral administration of JND003 (10 or 30 mg/kg body weight) or vehicle [phosphate-buffered saline (PBS)] by gavage and diluting the solution with 0.5% carboxymethylcellulose sodium. The animals were fasted overnight and sacrificed 4 weeks later and tissues were collected for analysis. All experimental procedures were carried out in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals. Animal experiments were approved by the Animal Care and Use Committee of Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences.
IGTT and ITT
IGTT and ITT were performed at the end of the experiment. For IGTT, mice were fasted overnight and i.p. injected with 10% glucose at a dose of 1.0 g/kg body weight. For ITT, mice were fasted for 6 h and i.p. injected with 0.5 U/kg body weight of recombinant human insulin (Sigma). Blood glucose was monitored from the tail vein blood using a glucometer (ACCU-CHEK Advantage; Roche Diagnostics China, Shanghai, China) at regular time points.
Transient Transfection Mammalian Two-Hybrid Reporter Gene Assay
HEK293T cells (Invitrogen) were seeded in 96-well plates at a density of 10,000 cells/well in DMEM containing 10% fetal bovine serum (FBS). After plating for 18–24 h, transient transfections were performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Total DNA for transfections included GAL4-DBD-ERRα-LBD vector (0.5 ng), pcDNA-PGC-1α vector (0.5 ng), pFR vector (25 ng), and internal control vector pRLTKrenilla (3.0 ng). After cells were transfected for 6 h, 5.0 μM compounds were then added for an additional 24 h. Luciferase activity was measured using a dual luciferase reporter assay system (Promega) according to the manufacturer’s instruction on a Veritas Microplate Luminometer. Relative luciferase light units were ratios of the absolute activity of firefly luciferase to that of renilla luciferase. The experiment was conducted in triplicate, and results are representatives of at least three independent experiments.
Establishment of an AAB-GFP-LUC Stably Expressed Cell Line
The sequence of natural ERRα pleiotropic nuclear receptor enhancer multiple hormone response element designated as AAB was synthesized and cloned into a pCDH-CMV-MCS-EF1-copGFP-Luc-Puro lenti-reporter vector that contains both green fluorescent protein (GFP) and Luc marker reporters and designated as AAB-GFP-Luc.33 The construct was verified by sequencing and packaged into lentivirus that were used to infect the cells. All lentivirus were packed in HEK293T cells according to the published protocol.40 Briefly, HEK293T cells were transiently transfected with pMD2G, psPAX2, and transfer vector containing the desired gene using Lipofectamine 2000 (Invitrogen). The supernatant was collected 48 h post-transfection and cleared from debris before storage. A multiplicity of infection (MOI) of 7 was used for the transduction of the aforementioned cell line to create an HEK293T strain that stably expresses AAB-GFP-Luc. Using optimized ERRα luciferase reporter gene assays multiplexed with a cell viability assay, the compounds were screened in three independent experiments.
AlphaScreen Assay
The binding affinity of JND003 to ERRα-LBD was determined by AlphaScreen assays using a hexahistidine detection kit from PerkinElmer. The experiments were conducted with 200 nM receptor LBD and 50 nM biotinylated cofactor peptides in the presence of 10 mg/mL donor and acceptor beads in a buffer containing 50 mM MOPS, 50 mM_NaF, 0.05 mM_CHAPS, and 0.1 mg/mL bovine serum albumin (BSA), all adjusted to pH 7.4. The multiple proportion dilution of the abovementioned compounds were selected and used in this experiment from 10 mM to 2 nM. The tested EC50 of JND003 is 86.0 nM.
Cellular Thermal Shift Assay
To validate the interaction of JND003 and ERRα protein, a cellular thermal shift assay (CETSA) was performed.41 HepG2 cells were seeded in 10 cm cell culture dishes and grown until ∼90% confluence. The cells were incubated with DMSO or JND003 (1.0 or 5.0 μM in fresh growth medium) in a 37 °C incubator with 5% CO2. After 4 h of incubation, the medium was aspirated. The cells were washed with PBS, harvested with trypsin, and collected by centrifugation. The cell pellets were further washed with PBS twice, resuspended in 700 μL of PBS, and distributed into seven different 1.5 mL tubes with 100 μL in each tube for both DMSO control and JND003-treated cells. Then, the tubes were heated at designated temperature end points (48–60 °C) for 3 min on a heating block. After heating, the tubes were removed and incubated at room temperature for 10 min. Subsequently, the cells were freeze–thawed thrice using liquid nitrogen. About 80 μL of the supernatant was collected by centrifugation (Eppendorf centrifuge 5415R, 13000 rpm × 30 min) from each resulting cell lysate at 4 °C. Finally, the residue was mixed with 20 μL of 5× standard sodium dodecyl sulfate (SDS)-loading buffer, heated at 95 °C for 10 min, separated by SDS-polyacrylamide gel electrophoresis (PAGE), followed by transferring to a poly(vinylidene difluoride) (PVDF) membrane for WB analysis.
Induction and Evaluation of Lipid Accumulation in HepG2 Cells
Cellular lipid accumulation was induced by a slight modification of the previously described protocol.10 HepG2 cell (Invitrogen) cultures were incubated with DMEM containing 10% FBS (Gibco, Thermo, Australia) and 1% BSA, supplemented with FFA (oleic and palmitic acid in association) in the following final concentrations: mixtures of palmitic acid (0.33 mM) and oleic acid 0.66 mM (final fatty acid concentration 1.0 mM). Control cell cultures were incubated with plain medium or with medium added with the vehicle in which fatty acids were dissolved. After 24 h of incubation with FFA, the extent of steatosis and gene expression were evaluated. After fixation with formaldehyde, neutral lipids were stained using 0.5% oil red O (Sigma-Aldrich) in isopropanol for 30 min and nuclei were stained with hematoxylin. Another model of insulin-resistant cells was established in HepG2 by treatment with high glucose and insulin,38 and the extent of lipid accumulation was evaluated as detailed above.
Pharmacokinetics and Tissue Distribution Assays
Pharmacokinetics (PK) and tissue distribution studies were performed at Medicilon Inc. (Shanghai, China). Male Sprague–Dawley rats were dosed with compound 7 intravenously (i.v.) at 2.0 mg/kg and by oral gavage (p.o.) at 10 mg/kg. The formulation was 5% DMSO/95% hydroxypropyl β-cyclodextrin (20% w/v) at a concentration of 1 mg/mL. Blood samples (0.2 mL) were then obtained via orbital sinus puncture at 5, 15, and 30 min and 1, 2, 4, 6, 8, and 24 h time points and collected into heparinized tubes. Samples were centrifuged for cell removal, and the plasma supernatant was then transferred to a clean vial and subsequently stored at −80 °C prior to analysis. The sample concentrations were determined by LC/MS, and PK parameters were calculated using Phoenix Winnolin R7.0 software.
Compound 7 was given to male SD mice by oral gavage as a single dose of 30 mg/kg. The compound was formulated with 0.5% CMC-Na. At 0.25, 0.5, 2, and 4 h after oral administration (3 mice at each time point), 0.5 mL of whole blood was collected from each animal, placed in heparinized tubes, and centrifuged at 11,000 rpm for 5 min, and plasma was separated and stored frozen in a refrigerator at −80 °C. After the animals were sacrificed, the brain, heart, lung, kidney, liver, spleen, bladder, abdominal fat, stomach, small intestine, large intestine, skeletal muscle, and testis were dissected, and the residual blood was washed with icy saline. After blotting, the label was sealed and stored in a refrigerator at −80 °C. The concentration of each compound in plasma and tissue was determined by LC/MS. The data were analyzed using Graphpad Prism5 (Graphpad Software, Inc.).
Enzymatic Assays
Specific activities for CS enzymes were assessed using dedicated enzyme activity assays from Abcam, according to manufacturer’s instructions (CS activity assay kit, ab119692).
Glucose Uptake Assay
Cells were starved in serum-free medium for 2 h and washed twice with glucose-free KRPH buffer [140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1.2 mM KH2 PO4, 2.5 mM MgSO4, 5 mM NaHCO3, 25 mM HEPES, pH 7.4, and 0.2% fatty acid free bovine serum albumin]. Then, the cells were incubated with KRPH buffer containing 5.0 μM JND003, 5 mM d-glucose, and 0.5 mCi/well of 2-deoxy-D [3H]-glucose (PerkinElmer, Boston, MA) for 30 min. Cells were then washed three times with ice-cold PBS and lysed with 0.5 M NaOH and 0.1% SDS. Cell lysates were neutralized with HCl. Radioactivity was measured by liquid scintillation counting (Tri-Carb 2800; PerkinElmer Co.).29
Fatty Acid Oxidation Assay
Oxidation of uniformly (U) 14C-labeled palmitate to CO2 and acid-soluble products in cultured cells was measured as previously described.42 Briefly, HepG2 cells were cultivated in 12-well plates with JND003 or vehicle for 24 h, and then, the medium was removed and cells were further incubated at 37 °C for 1.5 h in fresh medium containing 0.2 mM l-carnitine (Sigma, St. Louis, MO) and 200 μM [14C(U)] palmitate (0.1 μCi/mL, from PerkinElmer, Boston, MA), in the continuous presence of JND003 or vehicle. Prior to the 1.5 h incubation, each well was covered with a piece of Whatman paper and the multiwell plate sealed with parafilm. Following 1.5 h incubation, the Whatman paper was soaked with 0.1 mL of methylbenzylamine/methanol (1:1) to trap the CO2 produced, and 0.2 mL of 6 M HCl was injected into the wells to release the CO2 present in the liquid phase to the gaseous phase. After 1 h of CO2 capturing at room temperature, the pieces of Whatman paper were removed and transferred to scintillation vials for radioactivity counting. Acid-soluble products in 1 mL of the culture medium were extracted from intact [14C(U)] palmitate present in the media by the addition of 0.5 mL cold 1.5 M HClO4. After centrifugation (15 min, 1800g), radioactivity in the supernatant was measured by scintillation counting. Protein concentration in cell lysates of extra parallel culture wells was measured using the BCA protein assay kit (Pierce, Rockford, IL). For liver tissues, fresh isolated liver tissues (50 mg) were cut into 1 mm × 1 mm × 1 mm granules with scissors for the determination of fatty acid oxidation in liver tissues as described above.
Indirect Calorimetry and Calculated Energy Expenditure
Whole-body oxygen consumption was measured with an open-circuit indirect calorimetry system with automatic temperature and light controls (Comprehensive Lab Animal Monitoring System, Columbus Instruments, OH). Mice had ad libitum access to chow and water in respiration chambers, and data were recorded for 48 h, including 24 h of acclimatization. Energy expenditure was calculated as recommended by the manufacturer.
RNA Extraction, Reverse Transcription, and Quantitative PCR
Total RNA was isolated from liver tissue or cultured cell using TRIzol Reagent (Invitrogen). First-strand cDNA synthesis was performed with SuperScript III Reverse Transcriptase (Invitrogen). Quantification of mRNA levels was performed using SYBR Premix Ex Taq (TaKaRa) under optimized conditions following the manufacturer’s protocol. Primer sequences are shown in Table S2. Gapdh was used as the reference gene to normalize the expression level.
Western Blot
Liver tissues were washed twice with ice-cold PBS and lysed with RIPA buffer (Beyotime) for 30 min on ice. Liver tissue lysates were centrifuged at 12,000g for 15 min at 4 °C, and supernatants were collected. About 40 μg of tissue proteins was resolved by 12% SDS-PAGE gel and transferred to the PVDF membrane (Millipore). Membranes were probed overnight with specific antibodies at 4 °C, washed three times with Tris-buffered saline with 0.05% Tween 20 (TBST), blocked with 5% evaporated skimmed milk, and then incubated with rabbit-horseradish-peroxidase-conjugated secondary antibody for 4 h at 4 °C. Membranes were developed by applying ECL Plus developing agent (GE Healthcare). The primary antibodies used in the experiments were ERRα (1:1000; Abcam; Ab10983), SDH (1:4000; Epitomics; P31040), MCAD (1:5000; Epitomics; P11310), ATP5β (1:1000; Abcam; Ab150291), GSK3β (1:1000; Abcam; Ab93926), p-GSK3β (1:1000; Abcam; Ab75814), AKT (1:1000; Abcam; Ab8805), p-AKT (1:1000; Abcam; Ab38449), and β-actin (1:2000; Abcam; Ab115777).
Serum Analysis
Serum concentrations of TC, TG, ALT, AST, and FFA were measured using COD-PAP and GPO-PAP methods (ApplyGen) with an automatic analyzer BAYER ADVIA-2400. Insulin concentrations were determined by ELISA (R&D Systems).
Histochemistry
Liver tissues were fixed in 4% formaldehyde overnight at room temperature, paraffinized, and sectioned by microtome. The slides were stained with HE (Sigma) following the standard protocol.
Statistical Analysis
Data are expressed as means ± SEM. All comparisons were analyzed by one-way ANOVA, followed by the LSD method. A p value of less than 0.05 is considered significant.
Acknowledgments
The authors appreciate the financial support from the Natural Science Foundation of China (81820108029, 81874284, 22037003, and 81800774), Guangdong Province (2018B030337001), Guangzhou city (201805010007, 201806010166, and 83653513), Pearl River S&T Nova Program of Guangzhou (201806010166), Frontier Research Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (2018GZR110105019), the CSIRO-International Partnership Program (154144KYSB20180063), a grant from the Chinese Academy of Sciences (XDA12040325), and a Guangzhou International Collaborative grant (2019A050510027). The authors also thank Dr. Chiwai Wong, a former colleague in Guangzhou Institutes of Biomedicine and Health (CAS), for introduction of biological knowledge in ERR area.
Glossary
Abbreviations
- ERRα
estrogen-related receptor alpha
- ERRE
ERRα response element
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- Errα-LKO
liver-specific Errα knockout mouse model
- PGC-1α
peroxisome proliferator-activated receptor gamma coactivator 1 alpha
- Mcad
medium-chain acyl CoA dehydrogenase
- Mlycd
malonyl CoA decarboxylase
- Pdk4
pyruvate dehydrogenase kinase 4
- TCA
tricarboxylic acid cycle
- Pepck
phosphoenolpyruvate carboxykinase
- LBD
ligand-binding domain
- WT
wild-type
- IGTT
intraperitoneal glucose tolerance test
- ITT
intraperitoneal insulin tolerance test
- FFA
free fatty acid
- CS
citrate synthase
- HE
hematoxylin–eosin staining
- TG
triglyceride
- TC
total cholesterol
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- IR
insulin-resistant
- HFD
high-fat diet
- LFD
low-fat diet
- AUC
area under curve
- PCR
polymerase chain reaction
- GFP
green fluorescent protein
- CETSA
cellular thermal shift assay
- ATP5β
ATP synthase β subunit
- Cpt1a
carnitine palmitoyltransferase 1A
- SDH
succinate dehydrogenase
- GSK3β
glycogen synthase kinase 3 beta
- Akt
protein kinase B
- FDA
Food and Drug Administration
- LC
liquid chromatography
- Ms
mass spectrometry
- SD
Sprague–Dawley
- HP-β-CD
hydroxypropyl-β-cyclodextrin
- MOI
multiplicity of infection
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomedchemau.1c00050.
Food intake of mice, supporting information about the effects of JND003 on ERRα and related genes, selectivity of JND003, IGTT, and ITT in ERRα-LKO mice, effects of JND003 on the differentiation of adipocytes, pharmacokinetic results of JND003 in SD rat, primers used for quantitative PCR, and chemical data (PDF)
Author Contributions
‡‡ L.M., L.P., and X.R. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Xie C.; Yagai T.; Luo Y.; Liang X.; Chen T.; Wang Q.; Sun D.; Zhao J.; Ramakrishnan S. K.; Sun L.; Jiang C.; Xue X.; Tian Y.; Krausz K. W.; Patterson A. D.; Shah Y. M.; Wu Y.; Jiang C.; Gonzalez F. J. Activation of intestinal hypoxia-inducible factor 2α during obesity contributes to hepatic steatosis. Nat. Med. 2017, 23, 1298–1308. 10.1038/nm.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda K.; Uto H.; Mawatari S.; Ido A. Clinical features of hepatocellular carcinoma associated with nonalcoholic fatty liver disease: a review of human studies. J. Clin. Gastroenterol. 2015, 8, 1–9. 10.1007/s12328-014-0548-5. [DOI] [PubMed] [Google Scholar]
- Farrell G. C.; Larter C. Z. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- Francque S. M.; van der Graaff D.; Kwanten W. J. Non-alcoholic fatty liver disease and cardiovascular risk: Pathophysiological mechanisms and implications. J. Hepatol. 2016, 65, 425–443. 10.1016/j.jhep.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Hashiba M.; Ono M.; Hyogo H.; Ikeda Y.; Masuda K.; Yoshioka R.; Ishikawa Y.; Nagata Y.; Munekage K.; Ochi T.; Hirose A.; Nozaki-Fujimura Y.; Noguchi S.; Okamoto N.; Chayama K.; Suganuma N.; Saibara T. Glycemic variability is an independent predictive factor for development of hepatic fibrosis in nonalcoholic fatty liver disease. PLoS One 2013, 8, e76161 10.1371/journal.pone.0076161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi-Sales E.; Ebrahimi-Kalan A.; Alipour M. R. Preventive effect of trans-chalcone on non-alcoholic steatohepatitis: Improvement of hepatic lipid metabolism. Biomed. Pharmacother. 2019, 109, 1306–1312. 10.1016/j.biopha.2018.10.196. [DOI] [PubMed] [Google Scholar]
- Sporea I.; Popescu A.; Dumitrascu D.; Brisc C.; Nedelcu L.; Trifan A.; Gheorghe L.; Braticevici C. F. Nonalcoholic Fatty Liver Disease: Status Quo. J. Gastrointestin. Liver Dis. 2018, 27, 439–448. 10.15403/jgld.2014.1121.274.quo. [DOI] [PubMed] [Google Scholar]
- Sato N. Central role of mitochondria in metabolic regulation of liver pathophysiology. J. Gastroenterol. Hepatol. 2007, 22, S1–S6. 10.1111/j.1440-1746.2007.04963.x. [DOI] [PubMed] [Google Scholar]
- Eichner L. J.; Giguère V. Estrogen related receptors (ERRs): a new dawn in transcriptional control of mitochondrial gene networks. Mitochondrion 2011, 11, 544–552. 10.1016/j.mito.2011.03.121. [DOI] [PubMed] [Google Scholar]
- Ricchi M.; Odoardi M. R.; Carulli L.; Anzivino C.; Ballestri S.; Pinetti A.; Fantoni L. I.; Marra F.; Bertolotti M.; Banni S.; Lonardo A.; Carulli N.; Loria P. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. 10.1111/j.1440-1746.2008.05733.x. [DOI] [PubMed] [Google Scholar]
- Deblois G.; Giguère V. Functional and physiological genomics of estrogen-related receptors (ERRs) in health and disease. Biochim. Biophys. Acta 2011, 1812, 1032–1040. 10.1016/j.bbadis.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Sladek R.; Bader J. A.; Giguère V. The orphan nuclear receptor estrogen-related receptor alpha is a transcriptional regulator of the human medium-chain acyl coenzyme A dehydrogenase gene. Mol. Cell. Biol. 1997, 17, 5400–5409. 10.1128/mcb.17.9.5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y.; Song L.; Zhang L.; Ling H.; Zhang Y.; Chen H.; Qi H.; Shi X.; Li Q. The discovery of novel, potent ERR-alpha inverse agonists for the treatment of triple negative breast cancer. Eur. J. Med. Chem. 2017, 136, 457–467. 10.1016/j.ejmech.2017.04.050. [DOI] [PubMed] [Google Scholar]
- Ranhotra H. S. The estrogen-related receptor alpha: the oldest, yet an energetic orphan with robust biological functions. J. Recept. Signal Transduct. Res. 2010, 30, 193–205. 10.3109/10799893.2010.487493. [DOI] [PubMed] [Google Scholar]
- Audet-Walsh É.; Giguére V. The multiple universes of estrogen-related receptor alpha and gamma in metabolic control and related diseases. Acta Pharmacol. Sin. 2015, 36, 51–61. 10.1038/aps.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha V. K.; Handschin C.; Arlow D.; Xie X.; Pierre J.; Sihag S.; Yang W.; Altshuler D.; Puigserver P.; Patterson N.; Willy P. J.; Schulman I. G.; Heyman R. A.; Lander E. S.; Spiegelman B. M. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 6570–6575. 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell B. B.; Shulman G. I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- Ranhotra H. S. The orphan estrogen-related receptor alpha and metabolic regulation: new frontiers. J. Recept. Signal Transduct. Res. 2015, 35, 565–568. 10.3109/10799893.2015.1024853. [DOI] [PubMed] [Google Scholar]
- Bookout A. L.; Jeong Y.; Downes M.; Yu R. T.; Evans R. M.; Mangelsdorf D. J. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 2006, 126, 789–799. 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao M.; Parimoo B.; Tanaka K. Developmental, nutritional, and hormonal regulation of tissue-specific expression of the genes encoding various acyl-CoA dehydrogenases and alpha-subunit of electron transfer flavoprotein in rat. J. Biol. Chem. 1993, 268, 24114–24124. 10.1016/s0021-9258(20)80500-6. [DOI] [PubMed] [Google Scholar]
- Wang H.; Gao M.; Wang J. Kaempferol inhibits cancer cell growth by antagonizing estrogen-related receptor alpha and gamma activities. Cell Biol. Int. 2013, 37, 1190–1196. 10.1002/cbin.10152. [DOI] [PubMed] [Google Scholar]
- Wang J.; Fang F.; Huang Z.; Wang Y.; Wong C. Kaempferol is an estrogen-related receptor alpha and gamma inverse agonist. FEBS Lett. 2009, 583, 643–647. 10.1016/j.febslet.2009.01.030. [DOI] [PubMed] [Google Scholar]
- Wende A. R.; Huss J. M.; Schaeffer P. J.; Giguère V.; Kelly D. P. PGC-1alpha coactivates PDK4 gene expression via the orphan nuclear receptor ERRalpha: a mechanism for transcriptional control of muscle glucose metabolism. Mol. Cell. Biol. 2005, 25, 10684–10694. 10.1128/mcb.25.24.10684-10694.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Ma K.; Sadana P.; Chowdhury F.; Gaillard S.; Wang F.; McDonnell D. P.; Unterman T. G.; Elam M. B.; Park E. A. Estrogen-related receptors stimulate pyruvate dehydrogenase kinase isoform 4 gene expression. J. Biol. Chem. 2006, 281, 39897–39906. 10.1074/jbc.m608657200. [DOI] [PubMed] [Google Scholar]
- Herzog B.; Cardenas J.; Hall R. K.; Villena J. A.; Budge P. J.; Giguère V.; Granner D. K.; Kralli A. Estrogen-related receptor alpha is a repressor of phosphoenolpyruvate carboxykinase gene transcription. J. Biol. Chem. 2006, 281, 99–106. 10.1074/jbc.m509276200. [DOI] [PubMed] [Google Scholar]
- Chaveroux C.; Eichner L. J.; Dufour C. R.; Shatnawi A.; Khoutorsky A.; Bourque G.; Sonenberg N.; Giguère V. Molecular and genetic crosstalks between mTOR and ERRalpha are key determinants of rapamycin-induced nonalcoholic fatty liver. Cell Metab. 2013, 17, 586–598. 10.1016/j.cmet.2013.03.003. [DOI] [PubMed] [Google Scholar]
- B’Chir W.; Dufour C. R.; Ouellet C.; Yan M.; Tam I. S.; Andrzejewski S.; Xia H.; Nabata K.; St-Pierre J.; Giguère V. Divergent role of estrogen-related receptor alpha in lipid- and fasting-induced hepatic steatosis in mice. Endocrinology 2018, 159, 2153–2164. 10.1210/en.2018-00115. [DOI] [PubMed] [Google Scholar]
- Carrier J. C.; Deblois G.; Champigny C.; Levy E.; Giguère V. Estrogen-related receptor alpha (ERRalpha) is a transcriptional regulator of apolipoprotein A-IV and controls lipid handling in the intestine. J. Biol. Chem. 2004, 279, 52052–52058. 10.1074/jbc.m410337200. [DOI] [PubMed] [Google Scholar]
- Peng L.; Gao X.; Duan L.; Ren X.; Wu D.; Ding K. Identification of pyrido[1,2-α]pyrimidine-4-ones as new molecules improving the transcriptional functions of estrogen-related receptor alpha. J. Med. Chem. 2011, 54, 7729–7733. 10.1021/jm200976s. [DOI] [PubMed] [Google Scholar]
- Chen C.-y.; Li Y.; Zeng N.; He L.; Zhang X.; Tu T.; Tang Q.; Alba M.; Mir S.; Stiles E. X.; Hong H.; Cadenas E.; Stolz A. A.; Li G.; Stiles B. L. Inhibition of estrogen-related receptor alpha blocks liver steatosis and steatohepatitis and attenuates triglyceride biosynthesis. Am. J. Pathol. 2021, 191, 1240–1254. 10.1016/j.ajpath.2021.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patch R. J.; Searle L. L.; Kim A. J.; De D.; Zhu X.; Askari H. B.; O’Neill J. C.; Abad M. C.; Rentzeperis D.; Liu J.; Kemmerer M.; Lin L.; Kasturi J.; Geisler J. G.; Lenhard J. M.; Player M. R.; Gaul M. D. Identification of diaryl ether-based ligands for estrogen-related receptor alpha as potential antidiabetic agents. J. Med. Chem. 2011, 54, 788–808. 10.1021/jm101063h. [DOI] [PubMed] [Google Scholar]
- Teng C. T.; Beames B.; Merrick B. A.; Martin N.; Romeo C.; Jetten A. M. Development of a stable cell line with an intact PGC-1alpha/ERRalpha axis for screening environmental chemicals. Biochem. Biophys. Res. Commun. 2014, 444, 177–181. 10.1016/j.bbrc.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng C. T.; Hsieh J.-H.; Zhao J.; Huang R.; Xia M.; Martin N.; Gao X.; Dixon D.; Auerbach S. S.; Witt K. L.; Merrick B. A. Development of Novel Cell Lines for High-Throughput Screening to Detect Estrogen-Related Receptor Alpha Modulators. SLAS Discov. 2017, 22, 720–731. 10.1177/2472555216689772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong E.-J.; Levasseur M.-P.; Dufour C. R.; Perry M.-C.; Giguère V. Loss of estrogen-related receptor alpha promotes hepatocarcinogenesis development via metabolic and inflammatory disturbances. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 17975–17980. 10.1073/pnas.1315319110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J.; Sladek R.; Carrier J.; Bader J.-A.; Richard D.; Giguère V. Reduced fat mass in mice lacking orphan nuclear receptor estrogen-related receptor alpha. Mol. Cell. Biol. 2003, 23, 7947–7956. 10.1128/mcb.23.22.7947-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijichi N.; Ikeda K.; Horie-Inoue K.; Yagi K.; Okazaki Y.; Inoue S. Estrogen-related receptor alpha modulates the expression of adipogenesis-related genes during adipocyte differentiation. Biochem. Biophys. Res. Commun. 2007, 358, 813–818. 10.1016/j.bbrc.2007.04.209. [DOI] [PubMed] [Google Scholar]
- Ju D.; He J.; Zheng X.; Yang G. Cloning, expression of the porcine estrogen-related receptor alpha gene and its effect on lipid accumulation in mature adipocytes. Sheng Wu Gong Cheng Xue Bao 2009, 25, 1627–1632. [PubMed] [Google Scholar]
- Zheng X.; Ke Y.; Feng A.; Yuan P.; Zhou J.; Yu Y.; Wang X.; Feng W. The mechanism by which amentoflavone improves insulin resistance in HepG2 cells. Molecules 2016, 21, 624. 10.3390/molecules21050624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sookoian S.; Pirola C. J. Systematic review with meta-analysis: risk factors for non-alcoholic fatty liver disease suggest a shared altered metabolic and cardiovascular profile between lean and obese patients. Aliment. Pharmacol. Ther. 2017, 46, 85–95. 10.1111/apt.14112. [DOI] [PubMed] [Google Scholar]
- Barde I.; Salmon P.; Trono D. Production and titration of lentiviral vectors. Curr. Protoc. Neurosci. 2010, 53, 4.21.1–4.21.23. 10.1002/0471142301.ns0421s53. [DOI] [PubMed] [Google Scholar]
- Jafari R.; Almqvist H.; Axelsson H.; Ignatushchenko M.; Lundbäck T.; Nordlund P.; Molina D. M. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- Prinsen F. M. C.; Veerkamp H. J. Transfection of L6 myoblasts with adipocyte fatty acid-binding protein cDNA does not affect fatty acid uptake but disturbs lipid metabolism and fusion. Biochem. J. 1998, 329, 265–273. 10.1042/bj3290265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W.; Schwaid A. G.; Wang X.; Wang X.; Chen S.; Chu Q.; Saghatelian A.; Wan Y. Ligand Activation of ERRα by Cholesterol Mediates Statin and Bisphosphonate Effects. Cell Metab. 2016, 23, 479–491. 10.1016/j.cmet.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt S. M.; Lockamy E. L.; Stein R. A.; McDonnell D. P.; Miller A. B.; Orband-Miller L. A.; Willson T. M.; Zuercher W. J. On the Intractability of Estrogen-Related Receptor; as a Target for Activation by Small Molecules.. J. Med. Chem. 2007, 50, 6722–6724. 10.1021/jm7012387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.