

Abstract
Alzheimer's disease (AD) is histopathologically characterized by Aβ plaques and the accumulation of hyperphosphorylated Tau species, the latter also constituting key hallmarks of primary tauopathies. Whereas Aβ is produced by amyloidogenic APP processing, APP processing along the competing nonamyloidogenic pathway results in the secretion of neurotrophic and synaptotrophic APPsα. Recently, we demonstrated that APPsα has therapeutic effects in transgenic AD model mice and rescues Aβ-dependent impairments. Here, we examined the potential of APPsα to mitigate Tau-induced synaptic deficits in P301S mice (both sexes), a widely used mouse model of tauopathy. Analysis of synaptic plasticity revealed an aberrantly increased LTP in P301S mice that could be normalized by acute application of nanomolar amounts of APPsα to hippocampal slices, indicating a homeostatic function of APPsα on a rapid time scale. Further, AAV-mediated in vivo expression of APPsα restored normal spine density of CA1 neurons even at stages of advanced Tau pathology not only in P301S mice, but also in independent THY-Tau22 mice. Strikingly, when searching for the mechanism underlying aberrantly increased LTP in P301S mice, we identified an early and progressive loss of major GABAergic interneuron subtypes in the hippocampus of P301S mice, which may lead to reduced GABAergic inhibition of principal cells. Interneuron loss was paralleled by deficits in nest building, an innate behavior highly sensitive to hippocampal impairments. Together, our findings indicate that APPsα has therapeutic potential for Tau-mediated synaptic dysfunction and suggest that loss of interneurons leads to disturbed neuronal circuits that compromise synaptic plasticity as well as behavior.
SIGNIFICANCE STATEMENT Our findings indicate, for the first time, that APPsα has the potential to rescue Tau-induced spine loss and abnormal synaptic plasticity. Thus, APPsα might have therapeutic potential not only because of its synaptotrophic functions, but also its homeostatic capacity for neuronal network activity. Hence, APPsα is one of the few molecules which has proven therapeutic effects in mice, both for Aβ- and Tau-dependent synaptic impairments and might therefore have therapeutic potential for patients suffering from AD or primary tauopathies. Furthermore, we found in P301S mice a pronounced reduction of inhibitory interneurons as the earliest pathologic event preceding the accumulation of hyperphosphorylated Tau species. This loss of interneurons most likely disturbs neuronal circuits that are important for synaptic plasticity and behavior.
Keywords: Alzheimer's disease, APPsα, hippocampus, interneurons, synaptic plasticity, Tau
Introduction
Alzheimer's disease (AD) is the most prevalent neurodegenerative disease in the elderly, characterized by two major pathologic hallmarks: extracellular senile amyloid-β (Aβ) plaques, and intracellular neurofibrillary tangles that consist of hyperphosphorylated Tau protein (for review, see Ballatore et al., 2007; De Strooper and Karran, 2016). Under physiological conditions, Tau is enriched in axons where it binds to microtubules and regulates their dynamics (Y. Wang and Mandelkow, 2016). Under pathologic conditions, Tau becomes increasingly phosphorylated, which leads to its detachment from microtubules and promotes its mislocalization to the somatodendritic compartment. Eventually, Tau undergoes a transition from soluble to insoluble oligomers that fibrillize and are finally deposited as neurofibrillary tangles (Fitzpatrick et al., 2017; Wegmann et al., 2018).
Recent studies indicated that in addition to impairments induced by pathologic Tau and Aβ species, network abnormalities caused by a disturbed balance of excitation and inhibition (E/I) are frequent in AD patients (Nakamura et al., 2017) and may precede the formation of protein aggregates (Harris et al., 2020). Inhibitory interneurons are key regulators of network activity and impaired GABAergic neurotransmission can lead to network hyperexcitability (for review, see Vossel et al., 2017; Harris et al., 2020; Y. Xu et al., 2020). In AD mouse models, interneuron dysfunction and hyperexcitability have previously been observed in conjunction with Aβ-mediated pathology. Importantly, these Aβ-mediated impairments crucially involved Tau-dependent signaling cascades (Roberson et al., 2007, 2011; Verret et al., 2012; Ittner et al., 2016; Palop and Mucke, 2016).
Aβ is generated by sequential cleavage of APP by β- and γ-secretase. In the competing and physiologically predominant nonamyloidogenic pathway, APP is cleaved by α-secretase (Lammich et al., 1999; Lichtenthaler et al., 2011), which liberates the neuroprotective ectodomain APPsα and precludes the formation of Aβ. The levels of endogenous APPsα are reduced in neurologic disorders, including AD (Lannfelt et al., 1995; Kim et al., 2009) and low APPsα levels in the CSF correlate with poor memory performance in both aged humans and rats (Almkvist et al., 1997; Anderson et al., 1999). Shifting APP processing toward nonamyloidogenic processing has therefore been suggested as a therapeutic strategy for AD (Mockett et al., 2017; Müller et al., 2017). Indeed, APPsα protects neurons against different forms of stress (Müller et al., 2017), stimulates neurite outgrowth and branching (Young-Pearse et al., 2008; Weyer et al., 2014), increases spine density in APP-deficient mice (Weyer et al., 2011, 2014; Tyan et al., 2012; Hick et al., 2015) and in WT mice upon environmental enrichment (Zou et al., 2016). Moreover, APPsα facilitates LTP (Taylor et al., 2008; Hick et al., 2015; Richter et al., 2018; Mockett et al., 2019) and enhances memory (Ring et al., 2007; Xiong et al., 2017; Richter et al., 2018). Importantly, we recently demonstrated that APPsα has therapeutic potential in transgenic APP/PS1ΔE9 mice with plaque pathology, as viral vector-mediated APPsα expression in the hippocampus of these mice rescued deficits in synaptic plasticity, spine density, and spatial memory and also reduced Aβ levels (Fol et al., 2016). These results raised the question of whether the beneficial in vivo effects of APPsα may also be exploited for Aβ-independent impairments, in particular Tau-induced pathology.
Here, we used Tau transgenic mice, the hTau.P301S mouse line (further referred to as P301S mice) (Allen et al., 2002), and the THY-Tau22 mouse line (Schindowski et al., 2006; van der Jeugd et al., 2011) to examine whether APPsα has a more generalized therapeutic effect and could ameliorate Tau-induced synaptic deficits. As a baseline for these studies, we first characterized P301S mice in more detail. Electrophysiological recordings indicated an aberrantly increased LTP in hippocampal slices of P301S mice that could be restored to WT levels by recombinant (rec) APPsα. In vivo, AAV-mediated APPsα expression in the hippocampus rescued spine density deficits of pyramidal neurons both in P301S mice and in THY-Tau22 mice. Strikingly, the aberrant increase in LTP in P301S mice was paralleled by a so far unrecognized progressive loss of inhibitory GABAergic interneurons present already early postnatally and clearly preceding detectable Tau pathology.
Materials and Methods
Ethics statement
All animal experiments were performed in accordance with the guidelines and regulations set forth by the German Animal Welfare Act and the Regierungspräsidium Karlsruhe (Germany) to reduce the numbers of animals and prevent unnecessary suffering. All procedures performed were approved by the Regierungspräsidium Karlsruhe. (Aktenzeichen 35-9185.81/G-153/16, 35-9185.81/G-151/16, 35-9185.81/G-157/21).
Mice
Animals were housed in a 12 h light/dark cycle in Makrolon Type II cages with standard bedding and ad libitum access to water and food. Homozygous Tau transgenic mice overexpressing a human Tau (hTau) isoform harboring a mutation at position 301 were originally generated on a C57Bl/6J and CBA mixed background, backcrossed to C57Bl/6J and a kind gift from Michel Goedert. Generation and genotyping of hTau.P301S mice (further referred to as P301S) were described previously (Allen et al., 2002). C57Bl/6J mice were used as the age-matched control group (further referred to as WT). Because of the expression of the transgene in the motor neurons of the spinal cord, P301S mice show a progressing hindlimb paralysis. Hence, they are classified as having a burdened phenotype. The decline of their motor performance is monitored on a daily basis and a score is assigned: 0, no symptoms; 0.5, paralysis of the tip of the tail; 1, paralysis of the tail and beginning ataxia; 1.5, paresis of the tail, weakness of the hindlimbs; 2, severe one-sided paresis of the hindlimbs and ataxia. Animals had to be sacrificed when reaching a score of 2 to prevent unnecessary suffering.
Generation and genotyping of THY-Tau22 mice were described previously (Schindowski et al., 2006). Heterozygous THY-Tau22 mice and internal WT littermate controls of both sexes were used.
Protein extraction based on solubility
For Sarkosyl-soluble/insoluble protein extraction, mice were sacrificed at the age of 12-14, 16-18, or 20-22 weeks by cervical dislocation, and the brain was rapidly removed. Brain samples from animals of the same age were processed strictly in parallel. The cerebral hemispheres were separated, the hippocampi were dissected, snap-frozen in liquid nitrogen, and stored at −80°C upon handling. Samples were homogenized in 250 µl of buffer H (10 mm Tris, pH 7.4, 0.8 m NaCl, 1 mm EGTA, 10% sucrose, 1 mm PMSF, protease and phosphatase inhibitors) using a bead mill (Omni Bead Ruptor 24, Omni International; 2 × 20 s at 3.1 m/s). The homogenates were kept on ice for 20 min. Subsequently, the samples were centrifuged for 20 min at 21,200 × g at 4°C, and the supernatants (S1) were collected. The resulting pellets (P1) were homogenized a second time in the same volume of buffer H and centrifuged again. The supernatants (S2) were mixed with the corresponding supernatants from the first centrifugation step (S1) in a new tube and the beads were washed with 50 µl of buffer H, that was added to the supernatants afterward. The pellets (P2) were resuspended in 100 µl Tris-buffered saline, snap-frozen in liquid nitrogen, and stored at −80°C. The combined supernatants were adjusted to 1% Sarkosyl and incubated for 1 h at 37°C on an orbital shaker (350-400 rpm). Afterward, an ultracentrifugation step for 68 min at 127,900 × g and at 4°C was used to form the pellets (P3) and the supernatants (S3), containing Sarkosyl-insoluble and Sarkosyl-soluble Tau species, respectively. The pellets (Sarkosyl-insoluble fraction) were resuspended in 30 µl Tris-buffered saline. From the supernatants (S3), 5 µl was used for protein quantification (BCA assay). From the pellets (P3), 30 µl was used directly for sample preparation. Protein quantification and sample preparation were performed on the same day as the fractionation.
Western blot analysis
Sarkosyl-soluble (S) and insoluble (P) fractions (20 µg/20 µl) were used for SDS-PAGE. Proteins were separated using 4% stacking and 10% running Tris-glycine gels in Laemlli buffer (0.025 m Tris, 0.2 m glycine, 0.1% (w/v) SDS, add H2O). The gels contain TCE (Trichlorethanol, Merck Millipore) to allow for the detection of total protein. Before blotting, gels were placed on an UV transilluminator and activated for 1 min. Proteins were transferred to 0.45 µm PVDF membranes (GE Healthcare) using a tank blot (1 h, 450 mA). After the electro transfer, stain-free blot images were acquired using the ChemiDoc MP system without further activation to detect total protein amount on the membrane.
Membranes were blocked at room temperature (RT) for 1 h in PBS-T (0.05% Tween-20 in PBS) with 5% BSA. Primary antibodies were diluted in PBS-T with 5% BSA. The following antibodies were used: AT8 (mouse monoclonal, 1:500, #MN1020, Thermo Fisher Scientific), AT180 (mouse monoclonal, 1:1000, #MN1040, Thermo Fisher Scientific). After washing with PBS-T, membranes were incubated with an HRP-coupled secondary antibody against mouse primary antibodies (goat anti-mouse HRP, 1:10,000, #115-165-146, Dianova), followed by another washing step. The membranes were developed using Signal-Fire ECL reagent (#6883, Cell Signaling Technology) and signals were detected using the Bio-Rad Chemidoc MP imager (Bio-Rad, Hercules).
To re-probe the membranes with the HT7 antibody (mouse monoclonal, 1:1000, #MN1000, Thermo Fisher Scientific), they were incubated in stripping buffer (62.5 mm Tris, pH 6.7, 2% [w/v] SDS, 100 mm β-mercaptoethanol, add H2O) for 30 min at 65°C. After washing with PBS-T, the membranes were blocked again using 3% nonfat milk in PBS-T for 1 h at RT and then incubated with HT7 diluted in blocking buffer. Afterward, the membranes were incubated in secondary antibody and imaged as described above.
Immunohistochemistry (IHC)
Animals of the same age and treatment group were always sacrificed on the same day, and brain samples were stained in parallel. For IHC, mice were sacrificed with CO2 and transcardially perfused with ice-cold PBS, followed by 4% PFA in PBS. Brains were dissected from the skull and postfixed in 4% PFA in PBS for 24 h at 4°C. 40 µm coronal brain sections were cut on a vibratome (HM650V Vibratome, Thermo Fisher Scientific) and collected in PBS.
Standard IHC
Slices were blocked and permeabilized (5% BSA, 5% NGS, 0.4% Triton X-100 in PBS) for 2 h at RT. After washing in PBS, the slices were stained free-floating overnight at 4°C in a 24-well plate using the following primary antibodies in corresponding primary antibody solutions (5% NGS, 0.2% Triton X-100 in PBS): parvalbumin (PV, guinea pig, 1:1500, #195004, Synaptic Systems), calretinin (CR, rabbit, 1:1500, #214102, Synaptic Systems), Somatostatin (SST, rat, incubated for 48 h, #MAB354, Merck Milipore). Afterward, the slices were washed in PBS and incubated in the corresponding secondary antibody solutions (0.1% BSA, 0.05% Triton X-100 in PBS) for 2 h at RT. The following secondary antibodies were used: donkey anti-rabbit Cy3 (1:1000, #711-165-152, Jackson ImmunoResearch Laboratories), goat anti-rat Cy3 (1:1000, #112-165-167, Dianova), goat anti-guinea pig Alexa-488 (1:1000, #106-545-003, Jackson ImmunoResearch Laboratories), goat anti-guinea pig Alexa-568 (1:500, #A11075, Thermo Fisher Scientific). Following secondary antibody incubation, the slices were washed in PBS and nuclei were counterstained with DAPI. Slices were mounted in Mowiol on Superfrost microscope slides (Menzel). Images were taken with a Nikon C2 Plus confocal microscope.
Fluorescent ABC staining
Remaining PFA was neutralized using 50 nm NH4Cl in PBS for 15 min at RT. After washing, the slices were permeabilized (2% Triton X-100 in PBS) overnight at RT, followed by a blocking step (M.O.M. (Mouse on Mouse) Blocking Reagent, 1:100, # MKB-2213, Vector Laboratories) for 6 h at RT. After washing in PBS, the slices were stained free-floating overnight at RT in a 24-well plate using the following primary antibodies (5% NGS in PBS): HT7 (mouse, 1:200, #MN1000, Thermo Fisher Scientific), AT8 (mouse, 1:200, #MN1020, Thermo Fisher Scientific), and AT180 (mouse, 1:200, #MN1040, Thermo Fisher Scientific). Afterward, the slices were washed in PBS and afterward incubated in the corresponding biotinylated secondary antibody (1:250, goat anti-mouse IgG antibody, biotinylated, #BA-9200, Vector Laboratories) for 2 h at RT (3% NGS, 1% BSA in PBS). Brain sections were washed again and incubated in ABC solution for 2 h at RT (1:100, Vectastain ABC Kit, #PK-4000, Vector Laboratories). After another washing step in PBS, a streptavidin rhodamine-RedX conjugate was used to detect the biotinylated secondary antibody (8 µg/µl, S6366, Thermo Fisher Scientific). Following streptavidin incubation, the slices were washed in PBS and nuclei were counterstained with DAPI. Slices were mounted in Mowiol on Superfrost microscope slides (Menzel). Images were taken with a Nikon C2 Plus confocal microscope as described below.
Production of recAPPsα
Recombinant His-HA-tagged recAPPsα was purified by affinity chromatography from the supernatant of stably transfected HEK293T cells as described previously (Hick et al., 2015). Briefly, HEK293T cells were stably transfected with pIRES-His-TEV-HA-muAPPsα-Puro and cultivated in DMEM, supplemented with 10% FCS, 1% L-Glut, and 2.5 µg/µl puromycin, in T-1000 flasks. When reaching 70%-80% confluency, the medium was changed to serum-reduced OptiMEM. Medium was conditioned with secreted His-HA-APPsα for 24 at 37°C. The conditioned medium was applied to a diafiltration process using a 10 kDa cutoff diafiltration cartridge (Vivaflow 200, 10,000 MWCO, PES, # VF20P0, Sartorius) on a modified ÄKTA PrimePlus chromatography system (Cytiva). For the affinity purification of the His-tagged APPsα, either a cobalt resin (Amintra Cobalt IDA Resin, Expedeon) or prepacked nickel columns were used (HisTrap High Performance, Cytiva). Afterward, the bound protein was eluted from the column using 50 mm sodium phosphate buffer, pH 7.4, 300 mm NaCl, 300 mm imidazole, and 1 mm benzamidine. During the purification process, protein-containing fractions were collected and analyzed using stain-free SDS gels and Western Blot with the following antibodies: anti-HA (mouse, 1:1000, #2367, Cell Signaling Technology), anti-His (mouse, 1:3000, #27-410-01, Sigma Aldrich), anti-APP (m3.2, mouse, 1:1000, a kind gift from Paul Mathews), and anti-APP (clone 22c11, mouse, 1:200, #MAB348, MerckMillipore) to check for C- and N-terminal integrity of the protein. Fractions containing APPsα were pooled and concentrated using centrifugal concentrators (VivaSpin 20, 30 000 MWCO, #VS2021, Sartorius). Using a HiPrep Desalting column (GE Healthcare), the physiological conditions in the preparation were restored by exchanging the buffer to PBS. The protein concentration was determined by BCA assay and the integrity and purity of produced His-HA-APPsα was assessed using stain-free SDS gels and Western blot with aforementioned antibodies.
Electrophysiology
Slice preparation for extracellular field recordings
In vitro extracellular recordings were performed on acute hippocampal slices of WT controls and P301S mice aged 16-18 weeks.
Acute hippocampal transversal slices were prepared from isoflurane-anesthetized mice. Following decapitation, the brain was removed and quickly transferred into ice-cold carbogenated (95% O2, 5% CO2) ACSF containing 125.0 mm NaCl, 2.0 mm KCl, 1.25 mm NaH2PO4, 2.0 mm MgCl2, 26.0 mm NaHCO3, 2.0 mm CaCl2, and 25.0 mm glucose. The hippocampus was sectioned into 400-µm-thick transversal slices with a vibrating microtome (VT1200S, Leica) and maintained in carbogenated ACSF at RT for at least 1.5 h.
recAPPsα application
Acute hippocampal slices of P301S mice and WT controls were preincubated in 30 ml gently carbogenated ACSF containing recombinant APPsα (recAPPsα) at a concentration of 10 nm, in a custom-made incubation chamber for 1 h at RT. Afterwards, slices were transferred into the recording chamber where again 30 ml of ACSF with recAPPsα was circulating in a closed loop during the entire experiment at 32°C.
Muscimol application
The GABA-R agonist muscimol (Tocris Bioscience) solved in Aqua dest. was used at a final concentration of 0.2 µm and applied 10 min before Theta-Burst Stimulation delivery.
Extracellular field recordings
Slices were placed in a submerged recording chamber and perfused with carbogenated ACSF (32°C; 125.0 mm NaCl, 2.0 mm KCl, 1.25 mm NaH2PO4, 2.0 mm MgCl2, 26.0 mm NaHCO3, 2.0 mm CaCl2, 25.0 mm glucose) at a rate of 1.2-1.5 ml/min. fEPSPs were recorded in stratum radiatum of CA1 region with a borosilicate glass micropipette (resistance 1-4 mΩ) filled with 3 m NaCl at a depth of ∼150-200 µm. Monopolar tungsten electrodes were used for stimulating the Schaffer collaterals at a frequency of 0.1 Hz. Stimulation intensity was adjusted to ∼40% of maximum fEPSP slope for 20 min baseline recording. LTP was induced by applying Theta-Burst Stimulation, TBS: 10 trains of four pulses at 100 Hz in a 200 ms interval, repeated 3 times. To induce LTD, a low-frequency stimulus (1 Hz) was applied for 15 min after 20 min of baseline recording.
Basal synaptic transmission properties were analyzed via input-output (IO) measurements and short-term plasticity was examined via paired-pulse facilitation (PPF). The IO measurements were performed by application of defined current values (25-250 µA) or by adjusting the stimulus intensity to achieve specific fiber volley (FV) amplitudes (0.1–0.8 mV). Presynaptic function and short-term plasticity were assessed with the PPF paradigm by applying a pair of two closely spaced stimuli in interstimulus intervals (ISIs) ranging from 10–160 ms.
Whole-cell electrophysiological recordings
16-18-week-old WT and P301S mice were anesthetized with isoflurane and intracardially perfused with sucrose dissection solution containing 212 mm sucrose, 3.0 mm KCl, 1.25 mm NaH2PO4, 7.0 mm MgCl2, 26.0 mm NaHCO3, 0.02 mm CaCl2, and 10.0 mm glucose. Brains were quickly removed and cut into 250-µm-thick acute transverse slices with the help of a tissue slicer (Leica; razor blade: Personna). Slices were incubated at 37°C for 15 min in ACSF containing 125 mm NaCl, 2.5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 25 mm NaHCO3, 1.25 mm NaH2PO4, and 25 mm glucose (bubbled with 95% O2/5% CO2 to pH 7.4). The holding chamber was slowly cooled down to RT and slices were incubated for 45 min before recordings. mEPSCs and mIPSCs were recorded from the same cell using glass capillaries filled with an internal solution containing 120 mm Cs-methane sulfonate, 5 mm MgCl2, 0.4 mm GTP, 4 mm MgATP, 10 mm Na-phosphocreatine, 0.6 mm EGTA, and 30 mm HEPES, pH 7.3. Recordings were acquired in 30°C ACSF containing 50 µm APV (Biotrend) and 1 µm TTX (Biotrend) in voltage-clamp mode at a holding potential of −70 mV to record mEPSCs or 10 mV to record mIPSCs. Series resistance was monitored, but no compensation was performed.
AAV plasmid design and vector production
The codon-optimized murine APPsα coding sequence was cloned under control of the synapsin promoter into a single-stranded rAAV2-based shuttle vector, as described previously (Fol et al., 2016). To enable easy detection, an N-terminal double HA-tag was inserted downstream of the APP signal peptide. The bicistronic DNA construct harbors a T2A site that connects the cDNA of membrane-anchored lckVenus and muAPPsα. The monocistronic AAV-Venus vector serves as a control and encodes only the yellow fluorescent protein Venus, which contains a lymphocyte-specific protein tyrosine kinase (lck)-derived peptide motif which tethers it to the plasma membrane. To produce viral particles, HEK293 cells were transfected with the abovementioned expression cassettes and the helper plasmid pDP9rs as previously described (Richter et al., 2018). Briefly, cell lysate and supernatant were collected, and virus particles were purified by ultracentrifugation on an iodixanol density gradient followed by buffer exchange to 0.01% pluronic/PBS (1× DPBS) via a 100 kDa Amicon centrifugal filter unit (Merck Millipore). The concentration was determined by free inverted terminal repeat-specific quantitative TaqMan PCR and expressed as genomic copies per µl of concentrated stocks (gc/µl) as previously described (Richter et al., 2018).
Stereotactic injection of AAVs
Mice were anesthetized by intraperitoneal injection of sleep mix (medetomidine: 500 µg/kg, midazolam: 5 mg/kg, fentanyl: 50 µg/kg in isotonic NaCl solution) and positioned on a stereotactic frame (World Precision Instruments). For optimized vector spread, AAV-Venus or AAV-APPsα vectors were bilaterally injected into two spots of the hippocampus. At each injection spot, 1 µl AAV vector stock titer (5 × 108 to 1 × 109 gc/µl) was applied at a rate of 0.2 µl/min. To prevent efflux of viral vector preparations, the cannula was left to rest for 1 min. Stereotactic coordinates of injection sites are as follows (relative to bregma): anteroposterior −2 mm, mediolateral: ±1 mm, dorsoventral −2.25 mm and −1.75 mm.
Neuronal morphology and spine counts
Biocytin fillings of neurons
For morphologic analysis, CA1 pyramidal neurons of P301S or WT mice were filled with a solution containing 0.1%-0.5% biocytin (Sigma Aldrich) through the patch pipette while recording. Acute slices were fixed in 4% Histofix (Carl Roth) after recording. After 2-10 d, the slices were washed in 1× PBS for 3 × 10 min. Permeabilization was performed for 1 h in 0.2% PBST (0.2% Triton X-100 in 1× PBS). Slices were stained overnight with Alexa-594-conjugated Streptavidin directed against biocytin (Invitrogen). On the next day, the slices were washed again for 3 × 10 min in 1× PBS. After air-drying the slices at RT for 1 h, they were mounted with a coverslip in ProLong Gold Antifade (Invitrogen).
Golgi staining
Golgi staining was done using the Rapid Golgi Staining Kit according to the manufacturer's protocol (FD NeuroTechnologies). Briefly, AAV-injected mice were sacrificed by cervical dislocation either at the age of 16-18 or 20-22 weeks for the P301S mouse line, or at the age of 12 months for the THY-Tau22 mouse line. Brains were removed from the skull and parted into hemispheres, from which one was used for Western blot analysis while the other one was used for Golgi staining. All procedures were performed in the dark. The hemispheres were immersed according to the manufacturer's protocol. Then brains were rapidly frozen on dry ice, and 100 µm coronal sections were cut using a cryotome (HM550, Thermo Fisher Scientific). Sections were mounted with Solution C on adhesive microscope slides precoated with 0.5% gelatin/0.05% Chromalaun and let dry at RT. The sections were stained according to the manufacturer's protocol. Finally, slices were coverslipped with Permount (Thermo Fisher Scientific).
Image acquisition
Images were acquired at the inverted fluorescence microscope Axio Observer Z1 using Plan Apo 20×/0.8 DICII and Plan Apo 63×/1.4 Oil DICII objectives (Zeiss). Overview images of the whole neuron for reconstruction were taken with a 20× objective and a z-step size of 0.5 µm. Basal and apical dendrites were imaged individually with two overlapping stacks. More detailed images of basal and apical dendritic segments for spine density analysis were acquired with a 63× oil objective and a z-step size of 130 nm. Exposure time was individually set for each cell so that the complete range of the grayscale was used.
Sholl and spine density analysis
Biocytin-filled hippocampal CA1 neurons were manually reconstructed using the Neurolucida software (MicroBrightField) by an experimenter blind to genotype. Neurons were only included in Sholl analysis if they showed a completely filled apical or basal tree and well-defined dendritic endings. The morphometric Sholl analysis was done using the NeuroExplorer software (MicroBrightField). In short, a series of concentric spheres (centered on the soma) was drawn with an intersection interval of 30 µm and the number of dendrites crossing each sphere as well as the dendritic length in between each sphere was calculated. This analysis was done separately for basal and apical dendrites of CA1 pyramidal cells and was plotted against the distance from the soma. For evaluation of dendritic spine density, at least three different dendritic segments of the basal dendritic arbor were imaged. Spine density was determined as described (Richter et al., 2018; Steubler et al., 2021) following the criteria of Holtmaat et al. (2009) and was plotted as spines per µm of dendrite. Before statistical analysis and blind to genotype, neurons were excluded if the image quality (poor signal-to-noise ratio) was not sufficient for counting of spines. Data acquisition and analysis were performed blind to genotype.
Nesting behavior
Species-specific nesting behavior was assessed as described previously (Deacon, 2006). The nesting test was conducted in the Interdisciplinary Biomedical Facility at the University of Heidelberg. Thirty mice (15 WT and 15 P301S mice) of both sexes were housed in individual cages containing regular bedding. Mice were kept under a 12 h light/dark cycle (lights on at 6:00 A.M.). A nestlet of roughly 3 g of compressed cotton (Ancare, Bellmore) was placed into each cage at the beginning of the dark phase. The exact weight of the nestlet was measured and noted.
Experimental design and statistical analysis
All analyses were performed with the person analyzing the data blind to the genotype and experimental condition. For each experiment, the exact number of animals (N) as well as the number of slices analyzed (n) are given in the corresponding figure.
For all experiments, male and female mice were used at different ages as indicated in the main text and figure legends (age: 4, 6, 12-14, 16-18, 20-22 weeks).
Data analysis and statistics of Western blot experiments
For the analysis of Western blots, the Bio-Rad Image Lab Software was used (version 6.1.0, build 7). The signal intensity of the AT8/AT180 bands was normalized to the corresponding HT7 signal in the same fraction. Using GraphPad Prism (versions 8 and 9), the datasets were analyzed for Gaussian distribution applying the D'Agostino-Pearson omnibus test. Values that deviated more than 2 times the SD from the mean value were classified as outliers and were removed from the dataset. Data were analyzed by one-way ANOVA followed by Tukey's post hoc test. Values of p = 0.05 were considered significant and plotted as follows: *p < 0.05; **p < 0.01; ***p < 0.001. All data are indicated as mean ± SEM. Figures were created using the graphics program Affinity Designer (version 1.8.6).
Data analysis of Tau immunohistochemistry
For quantification of Tau immunohistochemistry (HT7, AT8, AT180), z-stack mosaic images of the hippocampus were taken using a 10× objective. Hippocampal sections from all groups were imaged with consistent settings (number of steps: 11; step size: 2.7 µm) and with the same laser intensities avoiding overexposure. For quantification, maximum intensity projections of the images were prepared and four hippocampal subregions were analyzed: stratum oriens (SO), pyramidal cell layer (Pyr), stratum radiatum (SR), and CA3. In each of these regions, 4-6 nonoverlapping rectangles were positioned and the mean intensity was measured using Fiji (version: 2.0.0-rc69&1.52r), with the experimenter blind to genotype and age of specimen. The mean intensities were analyzed relative to WT that was set to 100%. Using GraphPad Prism (version 8), the datasets were analyzed for Gaussian distribution with the D'Agostino-Pearson omnibus test. Values deviating from the mean by more than 2 times the SD were classified as outliers and removed. Datasets were analyzed for each subregion separately by one-way ANOVA, followed by Bonferroni's post hoc test. Non-Gaussian distributed data were analyzed by the nonparametric Kruskal–Wallis test, followed by Dunn's test for multiple comparisons. Values of p = 0.05 were considered significant and plotted as follows: *p < 0.05; **p < 0.01; ***p < 0.001. All data are indicated as mean ± SEM. Image analysis was performed on raw data, while images shown in the figures were subject to postprocessing to enhance contrast and brightness. Figures were created using the graphics program Affinity Designer (version 1.8.6).
Data analysis and statistics of interneuron subpopulations
For the analysis of interneuron subpopulations, z-stack mosaic images of the hippocampus using a 10× objective were taken. Slices from all groups were imaged with the same settings (number of steps: 11; step size: 2.7 µm) and with the same laser intensities. At minimum, 3 or 4 animals per group and at least three slices per animal were imaged. For quantification, maximum intensity projections of the images were prepared and the area of the hippocampus was measured using Fiji (version: 2.0.0-rc-69&1.52r). PV+, CR+, and SST+ cell profiles in all hippocampal subfields were counted manually using the multipoint tool in Fiji, with the experimenter blind to the genotype and treatment. The number of cell profiles identified in the z-stack projections was given as % of cell profiles/mm2 relative to the WT. Using GraphPad Prism (versions 8 and 9), the datasets were analyzed for Gaussian distribution with the D'Agostino-Pearson omnibus test and outliers were removed. Values that deviated >2 times the SD from the mean value were classified as outliers. Data were analyzed by either using a Student's t test (2 groups) or a one-way ANOVA (>2 groups), followed by Bonferroni's post hoc test using GraphPad Prism. Values of p = 0.05 were considered significant and plotted as follows: *p < 0.05; **p < 0.01; ***p < 0.001. All data are indicated as mean ± SEM. Image analysis was performed on raw data, while images shown in the figures were subject to postprocessing. In order to increase the clarity of the figures, contrast and brightness were adjusted and the same changes were applied to the entire image. All images in a figure were processed strictly in parallel to maintain comparability. Figures were created using the graphics program Affinity Designer (version 1.8.6).
Data analysis and statistics of extracellular field recordings
Data of electrophysiological field recordings were collected, stored and analyzed with Labview software (National Instruments). The initial slope of fEPSPs elicited by stimulation of the Schaffer collaterals was measured over time, normalized to baseline, and plotted as average ± SEM. Analysis of the PPF data was performed by calculating the ratio of the slope of the second fEPSP divided by the slope of the first one and multiplied by 100. The statistical analysis was performed using Microsoft Excel or GraphPad Prism. Data obtained between two genotypes or two different experimental conditions were compared using an unpaired two-tailed student's t test. Data including more than two different groups were analyzed using a one-way ANOVA followed by a Bonferroni's post hoc test. Values of p = 0.05 were considered significant and plotted as follows: *p < 0.05; **p < 0.01; ***p < 0.001. All data are indicated as mean ± SEM. Figures were created using the graphics program Affinity Designer (version 1.8.6).
Data analysis and statistics of patch-clamp recordings
Miniature events were detected with a template search using Clampfit software (Molecular Devices). Statistical analyses were performed with GraphPad Prism using t test for normally distributed data and Mann–Whitney tests for not-normally-distributed data. Data are represented as median ± SEM for normally distributed data and median ± interquartile range (IQR) for not normally distributed data.
Data analysis and statistics of spine counts
The number of spines was determined per micrometer of dendritic length at apical and basal segments using Neurolucida software (version 2019.1.2), Neurolucida Explorer (version 2019.2.1) (MicroBrightField Bioscience). At minimum, 5 mice per genotype (N) and 5 neurons per animal were imaged. In total, 100 µm per neuron from not more than three segments per picture (n) was analyzed, blinded to genotype, and injected viral vector. Before statistical analysis, the dataset was analyzed for Gaussian distribution with the D'Agostino-Pearson omnibus test and outliers were removed. Values that deviated >2 times the SD from the mean value were classified as outliers. If the dataset was normally distributed, a one-way ANOVA was conducted, followed by Bonferroni's post hoc test using GraphPad Prism (version 8). Values of p = 0.05 were considered significant and plotted as follows: *p < 0.05; **p < 0.01; ***p < 0.001. All data are indicated as mean ± SEM. Figures were created using the graphics program Affinity Designer (version 1.8.6).
Data analysis and statistics of Sholl analysis
Dendritic trees were reconstructed using Neurolucida software (Neurolucida (version 2019.1.2), and Sholl analysis was performed using Neurolucida Explorer (version 2019.2.1), MicroBrightField Bioscience). At minimum, 5 mice per genotype (N) and 5 neurons per animal were imaged. Before statistical analysis, the dataset was analyzed for Gaussian distribution with the D'Agostino-Pearson omnibus test and outliers were removed. Values that deviated >2 times the SD from the mean value were classified as outliers. If the dataset was normally distributed, a two-way repeated-measures ANOVA was conducted, followed by Sidak's post hoc multiple comparisons test using GraphPad Prism (version 8). Bar graphs were analyzed using either unpaired Student's t test or Mann–Whitney test, depending on the distribution of the data using GraphPad Prism (version 8). Values of p = 0.05 were considered significant and plotted as follows: *p < 0.05; **p < 0.01; ***p < 0.001. All data are indicated as mean ± SEM. Figures were created using the graphics program Affinity Designer (version 1.8.6).
Data analysis and statistics of the nest building test
Data analysis and statistics of the nest building test. After the dark phase, the nest was rated according to the scale published by Deacon (2006): 1, nestlet > 90% intact; 2, nestlet 50%-90% intact; 3, nestlet mostly shredded but no identifiable nest site; 4, identifiable but flat nest; 5, crater-shaped nest. Remaining intact parts of the nestlet were weighted. As the data did not follow a Gaussian distribution, a Mann-Whitney test was performed.
Results
Progressive Tau pathology in the hippocampus of P301S mice
Previous studies indicated an early onset of Tau pathology in transgenic P301S mice (Allen et al., 2002; Hampton et al., 2010; Scattoni et al., 2010) with functional impairments in spatial memory that were associated with reduced spine density from 10 weeks of age onwards (H. Xu et al., 2014). So far, however, a systematic immunohistochemical analysis of the temporal course of pathologic Tau species accumulation in the brain of P301S mice had not yet been performed. Here, we concentrated predominantly on the hippocampus because of its crucial role in synaptic plasticity, as well as cognition and being one of the brain regions affected early during AD pathogenesis. We choose three different time points for our analysis: 12-14 weeks of age (shortly after the onset of learning and memory impairments in P301S mice), 16-18 weeks as an intermediate time point, and 20-22 weeks as the latest possible time point. At this age, P301S mice need to be sacrificed because of paralysis of the hindlimbs, which occurs as a consequence of Tau accumulation in motor neurons and their subsequent degeneration (Allen et al., 2002). We performed stainings for total hTau and used two different anti-Tau antibodies to visualize pathologically phosphorylated Tau species. Total hTau, as indicated by HT7 staining, was found in all hippocampal subfields at the earliest time point of analysis (12–14 weeks) and was strongly increased at the latest time point (Fig. 1a–d, left). AT8 immunoreactivity, directed against hTau phosphorylated at Ser202, Thr205, and Ser208, was detected mainly in the mossy fibers at 12-14 and 16-18 weeks of age. With progressing age, AT8+ cells were prominently stained in the hilus of the dentate gyrus (DG) and the CA1 region of the hippocampus with immunoreactivity detected both in dendritic regions and also in somata (Fig. 1a–d, middle). AT180 immunoreactivity detecting paired helical filament hTau, phosphorylated at Thr231, was present at low levels in the CA1-CA3 region at the early and intermediate time points of analysis. At the age of 20–22 weeks, somata and dendrites of both CA1 and CA3 pyramidal neurons showed strong immunoreactivity for AT180, as well as the somata of neurons in the hilus of the DG (Fig. 1a–d, right). Together, P301S mice express hTau in all hippocampal subfields and exhibit progressive Tau pathology, indicated by increasingly hyperphosphorylated Tau species.
Figure 1.
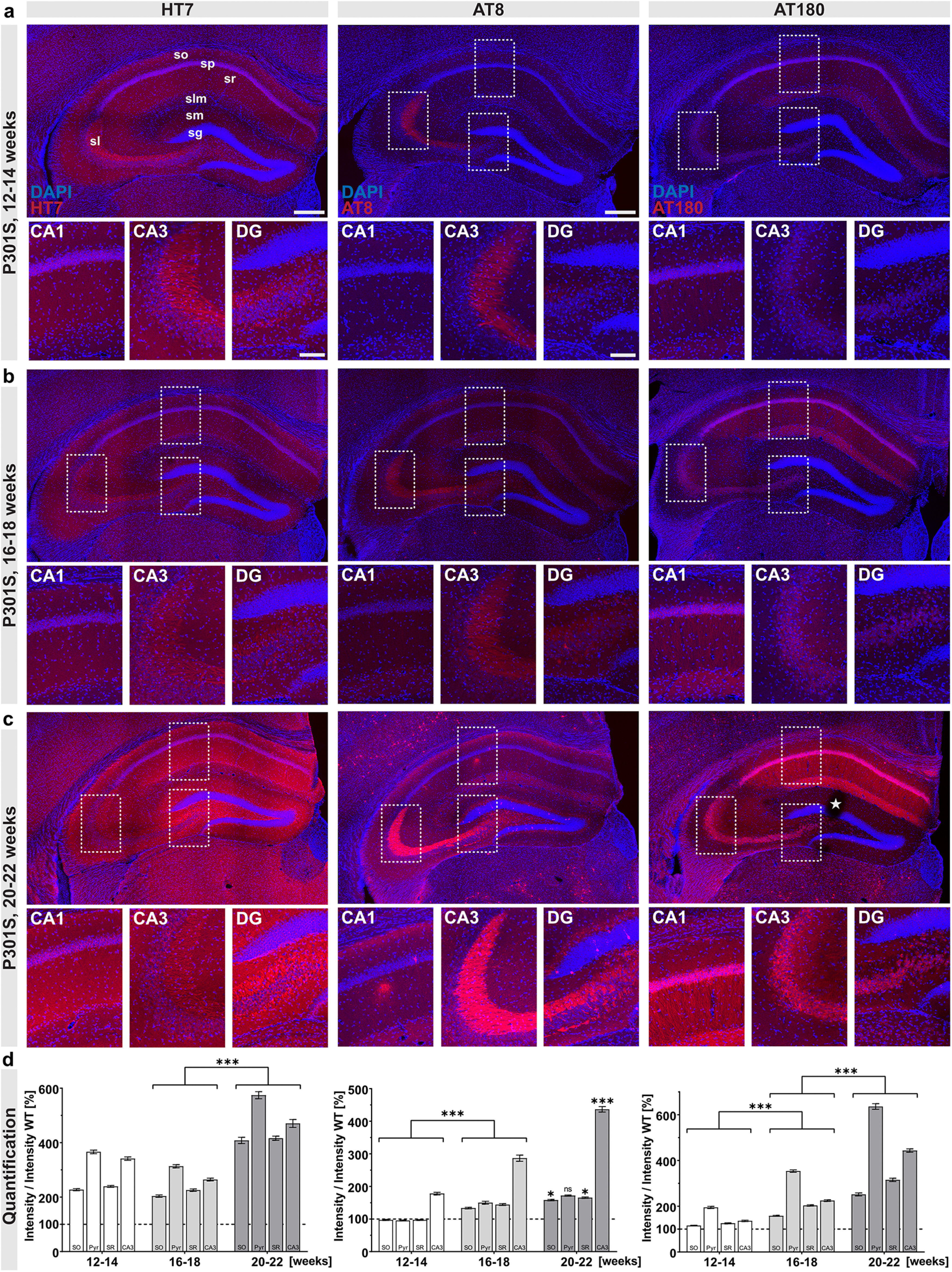
Progressive Tau pathology in P301S mice. Overview and zoom-in (CA1, CA3, DG) of the hippocampus of P301S mice. a, At 12-14 weeks of age, immunoreactivity with the HT7 antibody, detecting total hTau, was present in all hippocampal subfields. AT8 immunoreactivity was mainly observed in stratum lucidum (sl), while AT180 immunoreactivity was present at low levels in the stratum pyramidale (sp) of the CA1-CA3 region and to a lesser extent in the stratum oriens (so), stratum radiatum (sr), and lacunosum moleculare (slm). b, At the age of 16–18 weeks, all hippocampal subfields show HT7 immunoreactivity. A weak AT8 immunoreactivity was detected in all layers of the CA1 region as well as in the DG, where AT8+ cells were present in the hilus. Immunoreactivity for AT180+ Tau increased in all layers of the CA1-CA3 region compared with 12–14 weeks. c, At the latest time point analyzed (20–22 weeks), HT7 immunoreactivity massively increased in all hippocampal subfields. AT8+ cells were detected throughout the CA1 region, with a massive accumulation of AT8 immunoreactivity in the mossy fibers of sl. In the DG, AT8+ cells were present in the hilus and the stratum granulosum (sg), showing a somatodendritic staining pattern. Somata and dendrites of CA1 pyramidal neurons show strong immunoreactivity for AT180, also somata of CA3 neurons show AT180 staining. d, Quantification of HT7, AT8, and AT180 immunoreactivity in hippocampal regions over time. Staining intensity in the four subregions was quantified relative to the respective earlier time points. AT8 (center) and AT180 (right) immunoreactivity significantly increased in all subregions from 12–14 to 16–18 weeks (AT8: 12–14 vs 16–18 weeks, SO, Pyr, SR, CA3, ***p < 0.0001; AT180: 12–14 vs 16–18 weeks, SO, Pyr, SR, CA3, ***p < 0.0001), while HT7 immunoreactivity was not altered (not significant.) From 16-18 to 20-22 weeks, all markers (HT7, AT8, AT180) showed a significant increase, except for AT8 immunoreactivity in the pyramidal cell layer (HT7: 16–18 vs 20–22 weeks, PO, Pyr, SR, CA3, ***p < 0.0001; AT8: 16–18 vs 20–22 weeks, SO, SR, *p < 0.05, CA3, ***p < 0.0001, Pyr not significant; AT180: 16–18 vs 20–22 weeks, SO, Pyr, SR, CA3, ***p < 0.0001). Background staining from WT slices was set to 100% (dashed line). Data are mean ± SEM. Intensities for each subregion were analyzed using either one-way ANOVA with Bonferroni's post hoc test, or using Kruskal–Wallis test with Dunn's test: *p < 0.05; ***p < 0.0001. All sections were imaged with constant laser intensity to visualize progressing Tau pathology. N = 3 animals per genotype and age, maximum intensity projections of representative confocal images, 40 µm coronal sections. Scale bars: hippocampal overview, 300 µm, zoom-in CA1, CA3, and DG, 100 µm. *Slicing artifact.
In addition, we performed Western blot analysis at the same time points to further assess changes in the abundance of different Tau species over time. Figure 2a shows a representative Western blot of hippocampal fractions probed with the AT8 antibody, stripped, and re-probed with HT7 to detect total hTau. No signal was observed with HT7, AT8 or AT180 for nontransgenic WT controls, indicating that antibodies used are specific for hTau epitopes. While the amount of soluble and insoluble AT8+ Tau (normalized to the HT7 signal in the soluble or insoluble fraction, respectively), was comparable at all time points (Fig. 2b,c), the ratio of insoluble to soluble AT8+ Tau significantly increased by ∼3-fold at the latest time point analyzed (Fig. 2d).
Figure 2.

Progressive accumulation of insoluble Tau species in P301S mice. a, Representative Western blots of hippocampal extracts from WT and P301S mice at different ages probed with AT8 antibody, stripped, and re-probed with HT7 to detect total hTau. Note the absence of a detectable AT8 or HT7 signal in WT extracts (first two lanes). b-d, The amount of AT8+ Sarkosyl-soluble Tau normalized to total hTau in the supernatant and the amount of insoluble AT8+ Tau normalized to total hTau in the pellet is statistically indistinguishable between the investigated time points (not significant). The ratio of insoluble/soluble Tau increased with age (12–14 weeks vs 20–22 weeks, ***p = 0.0008; 16–18 weeks vs 20–22 weeks, **p = 0.0011). e, Representative Western blots of hippocampal extracts from WT and P301S mice at different ages probed with AT180, stripped, and re-probed with HT7 to detect total hTau. Note the absence of a detectable AT180 signal in WT extracts. f-h, The amount of AT180-immunoreactive Sarkosyl-soluble Tau normalized to total hTau (HT7) in the supernatant did not change over time (not significant), while the amount of insoluble AT180+ Tau normalized to total hTau in the pellet increased with age (12–14 weeks vs 20–22 weeks, *p = 0.0401). The ratio of insoluble/soluble Tau increased significantly with age (12–14 weeks vs 20-22 weeks, **p = 0.0026; 16–18 weeks vs 20–22 weeks, **p = 0.0073). Data are mean ± SEM. n = number of hippocampi as indicated in the bar graphs from N = 3 or 4 animals. Data were analyzed using one-way ANOVA with Tukey post hoc test: *p < 0.05; **p < 0.01; ***p < 0.001.
The amount of AT180-immunoreactive soluble Tau (normalized to the HT7 signal in the soluble fraction) was comparable at all time points (Fig. 2e,f), while the amount of insoluble AT180-positive (AT180+) Tau increased over time, reaching significance at the latest time point analyzed (Fig. 2g). Further, the ratio of insoluble to soluble Tau increased by ∼3-fold until 20-22 weeks of age (Fig. 2h). Together, our Western blot data indicate a progressive increase of insoluble Tau species over time but only mild, statistically not significant alterations until 16-18 weeks of age. Only at the latest time point of analysis (20-22 weeks), a significant increase in hTau aggregation, as expressed by the ratio of Sarkosyl-insoluble to soluble Tau, was detectable.
P301S mice exhibit reduced spine density but otherwise normal dendritic length and complexity
To study synaptic impairments of P301S mice, we first determined neuronal morphology and spine density of CA1 pyramidal cells at 16-18 weeks of age. For Sholl analysis, biocytin-filled neurons and their dendritic tree were imaged and reconstructed (Fig. 3b). The number of intersections was plotted against the distance from the soma (Fig. 3a, measured within circles centered on the soma). When comparing CA1 neurons from P301S and WT mice, we observed no significant difference in the overall morphology or complexity of basal or apical dendrites (Fig. 3c,f). Further, no significant difference in total dendritic length was detectable (Fig. 3d, apical: WT 3601.86 ± 124.42 vs P301S 3771.88 ± 173.09, not significant; Fig. 3g, basal: WT 1430.49 ± 153.67 vs P301S 1454.04 ± 120.51, not significant). Next, we assessed spine density as a correlate of excitatory synapses. Spine density was significantly reduced in apical dendrites of P301S neurons by ∼7% (Fig. 3e, WT 100.00 ± 2.41% vs P301S 92.58 ± 1.47%, *p = 0.0104), while basal dendrites were unaffected at this age (Fig. 3h, WT 100.00 ± 2.96% vs P301S 102.46 ± 2.60%, not significant; for the later time point at 20-22 weeks of age, see Fig. 7e,f).
Figure 3.
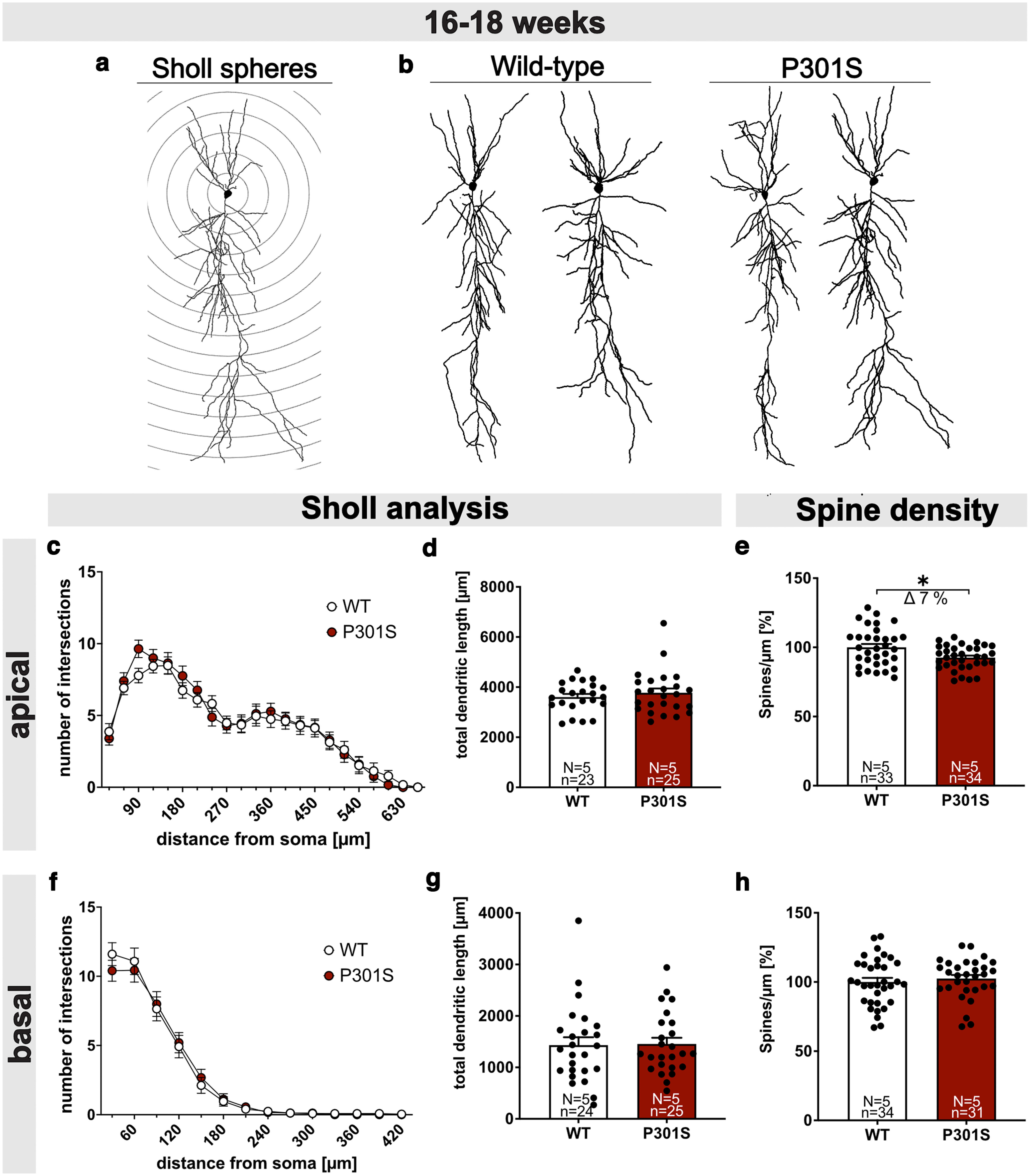
P301S mice show reduced spine density but normal neuronal morphology. a, Schematic representation of Sholl spheres around the soma (radius increment 30 µm). b, Representative 3D reconstructions of CA1 pyramidal neurons from WT (left) and P301S mice (right). c-e, Sholl and spine density analysis in apical dendrites. Sholl analysis of Biocytin-filled CA1 pyramidal cells revealed no differences in the number of intersections (c) and total dendritic length (d) of apical dendritic segments between WT and P301S mice. e, The spine density of apical dendrites was reduced by 7% in 16- to 18-week-old P301S mice compared with WT mice (WT vs P301S, *p = 0.0104). f-h, Sholl and spine density analysis in basal dendrites. Sholl analysis revealed no differences in the number of dendritic intersections (f), total dendritic length (g), and spine density (h) between WT and P301S mice at the age of 16-18 weeks. Data are mean ± SEM. n = number of neurons; N = number of animals. Data were analyzed by Student's t test: *p < 0.05.
Figure 7.

AAV-APPsα expression rescues spine density deficits in P301S and THY-Tau22 mice. a, Schematic representation of monocistronic and bicistronic AAV constructs enabling neuron-specific expression of (aI) Venus and (aII) HA-tagged APPsα linked via a T2A side with Venus. ITR, Inverted terminal repeat; Synapsin, neuron-specific promotor; T2A, Thosea asigna virus 2A site; SP, signal peptide; HA, influenza hemagglutinin tag used to visualize APPsα. b, Scheme of the hippocampus with coordinates of the two injection sites for intracranial AAV injection (black stars). c, d, The spine density deficit of Venus-injected P301S mice is rescued by APPsα to WT control levels at 16-18 weeks of age. c, Spine density in midapical CA1 dendrites was reduced by 6% in P301S-Venus mice compared with the WT-Venus group (**p = 0.0029) and rescued by the injection of AAV-APPsα (WT-Venus vs P301S-APPsα: not significant; P301S-Venus vs P301S-APPsα: Δ 10%, ***p < 0.0001). d, The spine density of basal CA1 dendrites was comparable in WT and P301S mice. APPsα led to a significant increase in basal spine density in P301S-APPsα-injected mice compared with P301S-Venus injected mice (WT-Venus vs P301S-Venus: not significant; P301S-Venus vs P301S-APPsα: Δ 6%, **p = 0.0078; WT-Venus vs P301S-APPsα: not significant). e, f, The spine density deficit of Venus-injected P301S mice is rescued by APPsα to WT control levels at 20-22 weeks of age. e, The spine density deficit in midapical CA1 dendrites of P301S-Venus mice amounted to ∼8% and was rescued to WT levels by the injection of AAV-APPsα (WT-Venus vs P301S-Venus: ***p < 0.0001, WT-Venus vs P301S-APPsα: not significant; P301S-Venus vs P301S-APPsα: Δ 8%, ***p < 0.0001). f, In basal CA1 dendrites, the spine density deficit of Venus-injected P301S mice was normalized to WT level by the expression of APPsα (WT-Venus vs P301S-Venus: Δ 14%, ***p < 0.0001, WT-Venus vs P301S-APPsα: not significant; P301S-Venus vs P301S-APPsα: Δ 11% ***p < 0.0001). g, h, The spine density deficit of Venus-injected THY-Tau22 mice is rescued by APPsα to WT control levels at 12 months of age. g, Spine density in midapical CA1 dendrites was reduced by 12% in THY-Tau22-Venus mice compared with the WT-Venus group (***p < 0.0001) and rescued by the injection of AAV-APPsα (WT-Venus vs THY-Tau22-APPsα: not significant; THY-Tau22-Venus vs THY-Tau22-APPsα: Δ 8%, **p = 0.0077). h, In basal CA1 dendrites, the spine density deficit of Venus-injected THY-Tau22 mice was normalized to WT level by the expression of APPsα (WT-Venus vs THY-Tau22-Venus: Δ 11%, ***p = 0.0001, WT-Venus vs THY-Tau22-APPsα: not significant; THY-Tau22-Venus vs THY-Tau22-APPsα: Δ 12% ***p < 0.0001). Data are mean ± SEM. n = number of analyzed segments; N = number of animals per condition. Data were analyzed by one-way ANOVA followed by Bonferroni's post hoc test: *p < 0.05; **p < 0.01; ***p < 0.001.
P301S mice exhibit impaired synaptic function with deficits in basal synaptic transmission
Despite being one of the earliest generated Tau-transgenic lines which dates back to 2002 (Allen et al., 2002), electrophysiological properties of neurons from P301S mice have not been investigated up to now. As a baseline for further studies, we therefore analyzed synaptic physiology by recording AMPA receptor-mediated mEPSCs and GABA receptor-mediated mIPSCs in CA1 pyramidal cells (Fig. 4a–k). We found that mEPSC frequency was significantly higher in CA1 neurons of P301S mice compared to neurons of WT mice (Fig. 4b, WT: 1 [0.84-1.6] Hz vs P301S: 2 [1.7-2.6] Hz; median [IQR], Mann–Whitney test: p < 0.0001). mEPSC peak amplitudes of neurons from P301S mice were also significantly larger than those of WT neurons (Fig. 4c, WT: 8.9 [7.6-9.7] pA vs P301S: 9.56 [9.2-11.1] pA; median [IQR], t test: p = 0.0028). Finally, charge transfer (area) of mEPSCs was increased in neurons from P301S mice (Fig. 4d, WT: 39.84 [29.01-53.67] fC per second vs P301S: 67.61 [50.53-101.5] fC per second; median [IQR]; Mann–Whitney test: p = 0.0001). While mIPSC frequencies of CA1 neurons were comparable between genotypes (Fig. 4f, WT: 7.3 [5.9-9.3] Hz vs P301S: 8 [6.6-10.1] Hz; median [IQR], t test: p = 0.2446), mIPSC peak amplitudes were slightly but significantly larger in neurons from P301S mice (Fig. 4g, WT: 20.74 [18.2-23.8] pA vs P301S 22.77 [21.22-44.22] pA; median [IQR], Mann–Whitney test: 0.0023). Charge transfer of mIPSCs was not different between genotypes (Fig. 4h, WT: 1752 [1100-2304] fC per second vs P301S: 1828 [1201-2571] fC per second; median [IQR]; t- test: p = 0.9818). To assess E/I balance, we calculated the ratio of mEPSCs divided by mIPSCs. Interestingly, the ratio of mean mEPSC/mIPSC frequency and charge transfer was significantly higher in P301S mice (Fig. 4i,k, Mann–Whitney test for frequency: p = 0.0002; Mann–Whitney test for charge transfer: p = 0.0027), while no change in mEPSC/mIPSC amplitude (Fig. 4j, t test: p = 0.1618) was observed. Overall, mEPSC and mIPSC recordings revealed rather mild changes of synaptic physiology in mice with a slight increase in E/I ratio.
Figure 4.
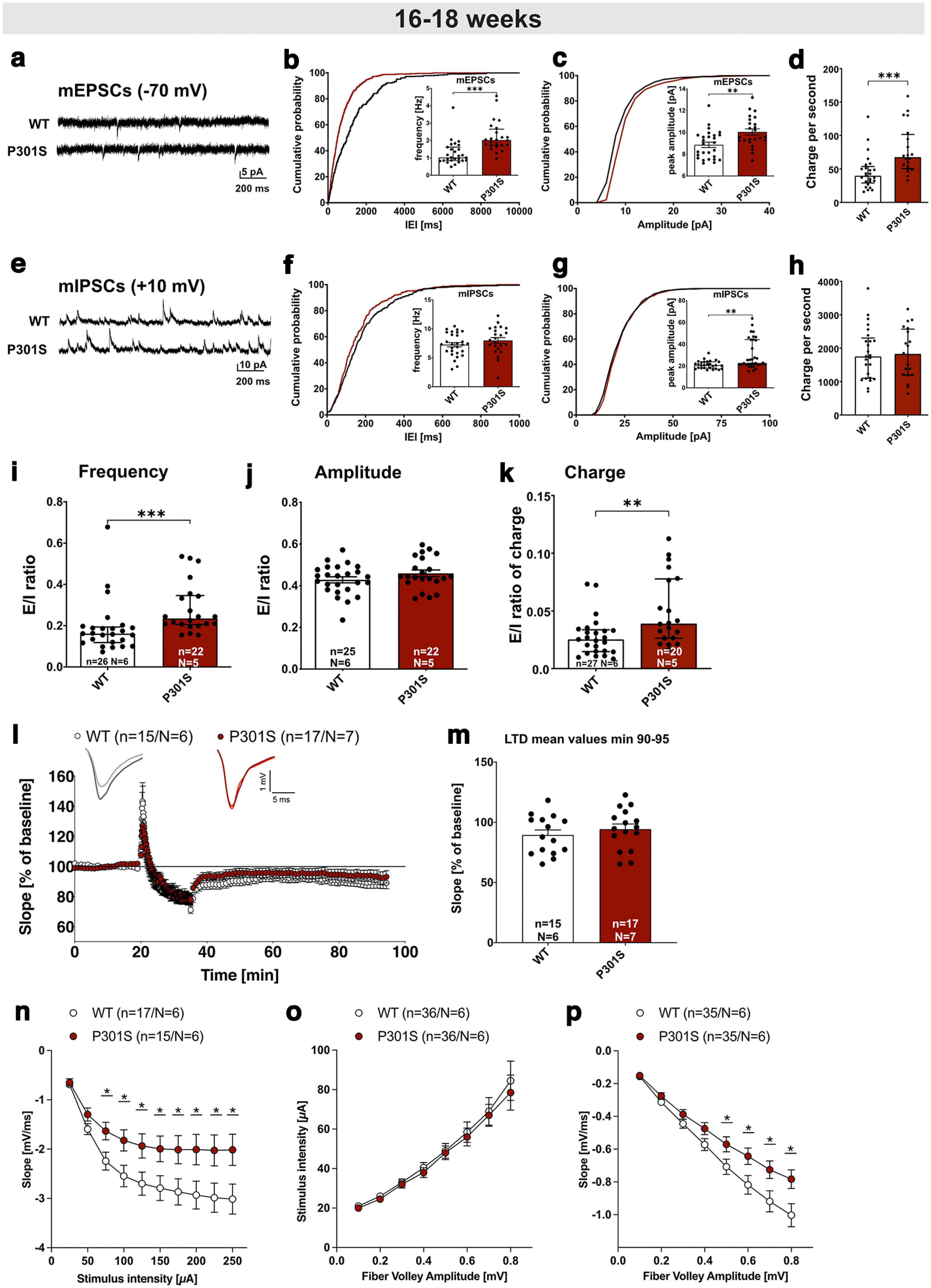
Electrophysiological analysis reveals impaired hippocampal network function in P301S mice. a, mEPSC sample traces of recordings from CA1 pyramidal cells, measured at −70 mV. b, Cumulative distribution plot of mEPSC interevent intervals (IEI) and bar graph of mEPSC frequencies. The cumulative distribution of IEIs is shifted to the left in P301S mice, consistent with the increased mEPSC frequency in CA1 neurons of P301S mice (n = 24, N = 5) compared with those of WT mice (***p < 0.0001, n = 27, N = 6). c, mEPSC amplitude was increased in CA1 pyramidal cells of P301S mice (n = 24, N = 5) compared with WT cells (**p = 0.0028, n = 27, N = 6). d, mEPSC charge transfer per second was increased in pyramidal cells of P301S mice (n = 20, N = 5) compared with WT cells (***p = 0.0001, n = 27, N = 6). e, mIPSC sample traces of recordings from CA1 pyramidal cells, measured at 10 mV. f, mIPSC frequency was not different in CA1 pyramidal neurons of P301S mice (n = 23, N = 5) vs WT cells (not significant, n = 28, N = 6). g, mIPSC amplitude was increased in CA1 pyramidal cells of P301S mice (n = 34, N = 5) compared with WT neurons (**p = 0.0023, n = 26, N = 6). h, mIPSC charge transfer per second was unchanged in pyramidal cells of P301S mice (n = 20, N = 5) compared with WT cells (p = 0.9818, n = 27, N = 6). i, j, E/I ratios of mEPSC/mIPSC frequencies were higher in neurons of P301S mice compared with WT neurons (***p = 0.0002), while the E/I ratio of mEPSC/mIPSC amplitudes was not altered (not significant). k, E/I ratio of charge transfer was increased in neurons of P301S mice compared with WT neurons (**p = 0.0027). Data are represented as median ± SD for normally distributed data and median ± IQR for not normally distributed data. n = number of recorded cells; N = number of animals. l, m, After 20 min baseline recording, LTD was induced by application of a low-frequency stimulus (LFS, 1 Hz) for 15 min. LTD was indistinguishable between P301S and WT mice (not significant, 55-60 min after LFS). n, fEPSP amplitudes were reduced in P301S mice compared with WT mice reaching significance at a stimulus intensity of 75 μA (*p = 0.028 for 75 µA). o, When the stimulus intensity was compared with the FV amplitude, there was no difference between the WT and the P301S group. p, The fEPSP slope at FV amplitudes between 0.5 and 0.8 V was reduced in P301S mice compared with WT mice (*p = 0.042 for 0.5 mV). Data are mean ± SEM. n = number of recorded slices; N = number of animals. Data were analyzed using either Student's t test (k-m) or one-way ANOVA with Bonferroni's post hoc test and Bartlett's test for equal variances: *p < 0.05; **p < 0.01; ***p < 0.001.
Previous studies had indicated impairments in synaptic plasticity in several Tau transgenic models that affected either LTP or LTD (Yoshiyama et al., 2007; Polydoro et al., 2009; Hoover et al., 2010; Lasagna-Reeves et al., 2011; Levenga et al., 2013; Fa et al., 2016). Here, we first assessed LTD that was induced by low-frequency stimulation of Schaffer collateral fibers. The slope of fEPSPs recorded after LTD induction was, however, not significantly different in P301S mice compared with WT mice (Fig. 4l,m). Next, we performed extracellular field recordings of the CA3/CA1 pathway to study basal synaptic transmission within the hippocampal network. Comparing the fEPSP size at increasing stimulus intensities revealed significantly reduced fEPSP responses in P301S slices at stimulus intensities from 100 to 250 µA (Fig. 4n). This finding could in principle be explained by a reduced number of CA3 axons firing action potentials at given stimulus intensities. We therefore recorded the fiber volley (FV), which represents the number of axons firing action potentials, at different stimulus intensities. There was no genotype difference in the dependency of FV on stimulus intensity (Fig. 4o). Thus, the reduced fEPSP size in slices of P301S mice does not result from a decreased excitability of axons but rather from decreased synaptic strength. Consistently, there was a reduction on fEPSP slope at fiber volleys between 0.5 and 0.8 mV in slices of P301S mice compared with WT (Fig. 4p). In conclusion, the electrophysiological recordings revealed an increased E/I balance of miniature postsynaptic currents and altered synaptic function that is attributable to CA1 neurons, as axonal firing and presynaptic vesicle release (see PPF paradigm, Fig. 5d) is not changed in P301S mice.
Figure 5.

Aberrantly increased LTP in P301S mice is rescued by application of recAPPsα. a, b, After 20 min baseline recording, LTP was induced by application of TBS (arrow). P301S mice at the age of 16-18 weeks displayed a significantly higher LTP than WT mice (min 75-80, ***p = 0.0005). The application of recombinant APPsα (10 nm) normalized the elevated LTP values of P301S mice to WT level (WT-Venus vs P301S+recAPPsα: not significant; P301S vs P301S+recAPPsα: **p = 0.0085). The LTP induction rate is shown as percentage (%) of mean baseline slope. c, fEPSP amplitudes were smaller in P301S mice compared with WT mice reaching significance at a stimulus intensity of 150 μA (*p = 0.0145 for 150 µA). When APPsα was applied, the neuronal excitability in P301S slices and WT slices was comparable at all stimulus intensities. d, PPF was not different between the WT and the P301S group. PPF was quantified by the ratio of the slopes of EPSPs evoked by two stimuli with different ISI. APPsα treatment led to the highest PPF curve within that paradigm, reaching significance at the 10 and 20 ms ISI (10 ms, WT vs P301S: not significant, WT vs P301S+recAPPsα: ###p = 0.0008, P301S vs P301S+recAPPsα: •p = 0.0201; 20 ms, WT vs P301S: not significant, WT vs P301S+recAPPsα: ##p = 0.0018, P301S vs P301S+recAPPsα: not significant). e, f, Application of recAPPsα (10 nm) does not significantly affect LTP in WT mice (age 16-18 weeks). LTP was induced by application of TBS (arrow). The LTP induction rate is shown as percentage (%) of mean baseline slope. Data are mean ± SEM. n = number of recorded slices; N = number of animals. Data were analyzed by Student's t test: *p < 0.05; **p < 0.01; ***p < 0.001. g, h, AAV-APPsα expression in the hippocampus of WT mice (age 16-18 weeks; also see Fig. 7a,b) does not alter spine density in midapical (g) and basal dendrites (h) of CA1 neurons. Data are mean ± SEM. n = number of analyzed dendritic segments; N = number of animals. Data were analyzed by Student's t test.
P301S mice show elevated LTP that is rescued by application of recAPPsα
P301S mice have been reported to show deficits in spatial learning and memory at 10 weeks of age (H. Xu et al., 2014) that may be related to altered synaptic plasticity. As LTD was normal in P301S mice, we now studied LTP. To this end, we used hippocampal slices of WT and P301S mice at the age of 16–18 weeks (Fig. 5a,b). After 20 min of stable baseline recording, that was indistinguishable when comparing P301S and WT mice, LTP was induced at the Schaffer collateral to the CA1 pathway by application of Theta-Burst Stimulation (TBS) (arrowhead; Fig. 5a). Surprisingly, P301S mice displayed considerably higher LTP values compared with WT mice. Sixty minutes after TBS induction (corresponding to 75–80 min after baseline recording), slices of P301S mice showed a potentiation of 153.29 ± 5.25% that was significantly increased compared with WT slices that exhibited a potentiation of 128.06 ± 2.48% (Fig. 5b, WT vs P301S: ***p = 0.0005, n = 16–22 slices from N = 6–8 mice; t75-t80 after start of baseline recording). In previous studies, we and others have shown that nanomolar amounts of recAPPsα can rescue impaired LTP (Hick et al., 2015; Mockett et al., 2019). Here, we asked whether APPsα may also normalize aberrantly increased LTP of P301S slices. Acute slices of P301S mice were preincubated for 60 min at RT with native 10 nm recAPPsα. After preincubation and 20 min of stable baseline recording, LTP was induced by TBS and recorded for 60 min. During the whole measurement, the recAPPsα peptide diluted in ACSF was circulating in a closed-loop system. Strikingly, and consistent with our hypothesis that APPsα can restore normal synaptic plasticity, we found that the application of recAPPsα normalized aberrantly increased LTP in P301S slices resulting in an LTP curve closely overlapping with that of WT mice (Fig. 5a). At 60 min after TBS induction, slices of P301S mice treated with recAPPsα showed a potentiation statistically indistinguishable from that of untreated WT slices (Fig. 5b, WT vs P301S+recAPPsα: 128.06 ± 2.48% vs 128.94 ± 3.59%, not significant; P301S vs P301S+recAPPsα: 153.29 ± 5.25% vs 128.94 ± 3.59%, **p = 0.0085). Next, we asked whether recAPPsα may also mitigate impairments in basal synaptic properties, as studied by field recordings (see also Fig. 4n,o). In a separate set of experiments, we compared the fEPSP size at increasing stimulus intensities, which again revealed significantly reduced fEPSP responses at stimulus intensities between 150 and 250 µA in P301S slices (Fig. 5c, WT vs P301S: *p = 0.0145 for 150 µA). When recAPPsα was applied, the fEPSP slopes were comparable to those of the WT group (Fig. 5c). Presynaptic function was assessed by PPF, which was statistically not different in P301S slices compared with slices from WT mice at all ISI tested (Fig. 5d). Interestingly, the application of recAPPsα led to a significantly increased PPF at 10 and 20 ms ISIs (Fig. 5d, 10 ms, WT vs P301S: not significant, WT vs P301S+recAPPsα: ###p = 0.0008, P301S vs P301S+recAPPsα: •p = 0.0201; 20 ms, WT vs P301S: not significant, WT vs P301S+recAPPsα: ##p = 0.0018, P301S vs P301S+recAPPsα: not significant).
Previously, we had shown that APPsα can rescue Aβ-related impairments of LTP in APP/PS1ΔE9 mice (Fol et al., 2016). Moreover, APPsα also had beneficial effects in an Aβ-independent pathology with synaptic impairments, namely, NexCre conditional double knockout mice lacking APP and APLP2. In these mice, APPsα rescued LTP deficits both in vitro (Hick et al., 2015) and in vivo (Richter et al., 2018), while heat inactivated APPsα (Hick et al., 2015) or scrambled peptides had no effects (Richter et al., 2018). Nevertheless, to exclude any potential adverse effects of APPsα, we now also recorded LTP in hippocampal slices of WT mice in the presence of recAPPsα (Fig. 5e,f). Upon recAPPsα treatment of WT slices, we detected a trend toward slightly increased fEPSP slopes shortly after LTP induction (WT vs WT+recAPPsα: 165.36 ± 6.52% vs 196.32 ± 12.92%, p = 0.051, Fig. 5e) and values not significantly different from untreated WT slices 60 min after TBS at the maintenance phase of LTP (WT vs WT+recAPPsα: 132.74 ± 5.01% vs 148.69 ± 8.80%, not significant, p = 0.144, Fig. 5f). In line with this, AAV-mediated expression of APPsα in the hippocampus of WT mice did not affect spine density of CA1 dendrites (Fig. 5g,h), thus excluding any adverse effect of APPsα. Together, these experiments revealed an aberrant increase in LTP in P301S mice that was rescued to normal WT level by recAPPsα application pointing toward a homeostatic function of APPsα under conditions of synaptic impairments including Tau pathology.
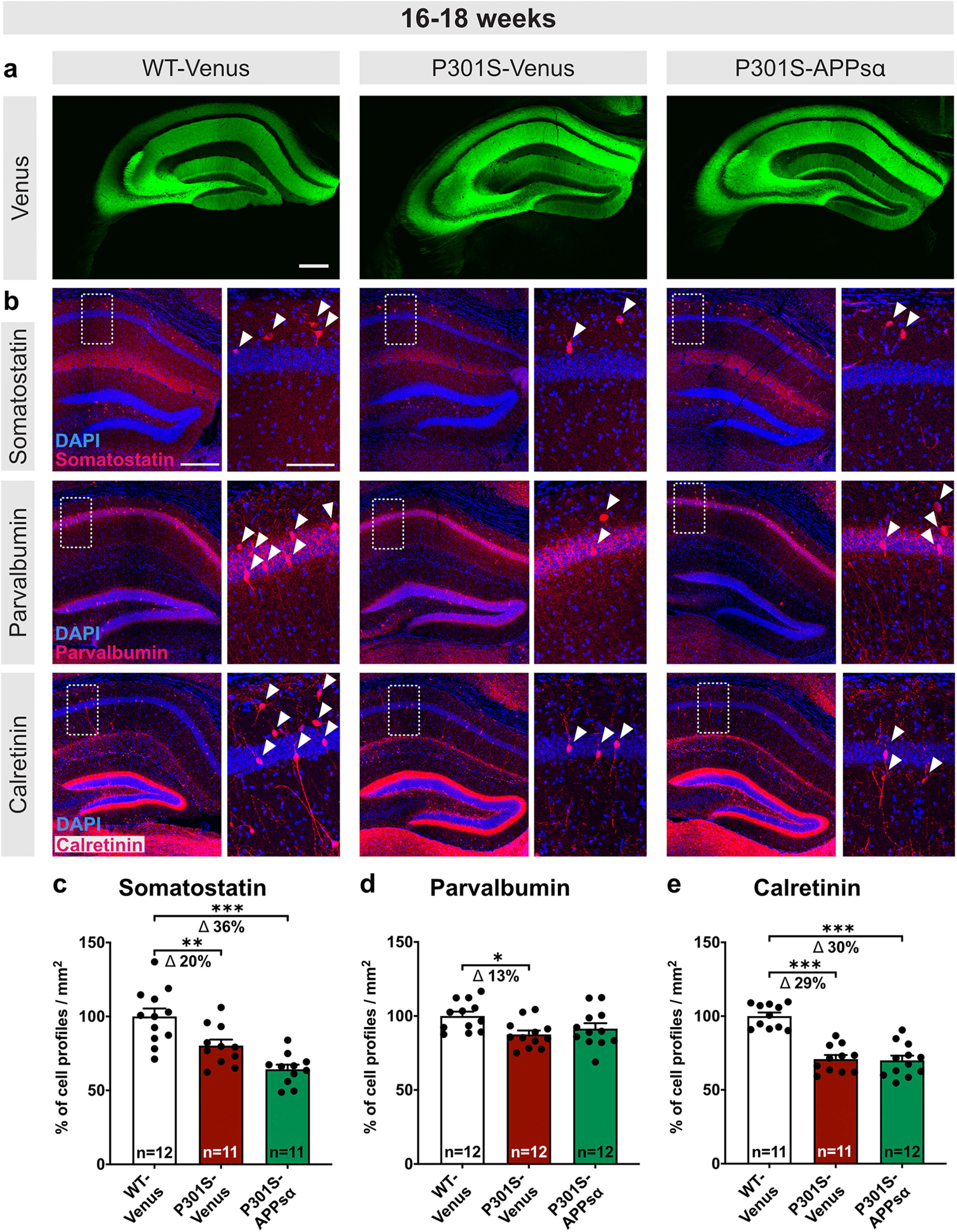
P301S mice show a pronounced loss of inhibitory interneurons
Having established this aberrant increase in LTP at a time point at which P301S mice exhibit cognitive deficits (H. Xu et al., 2014), we next aimed to elucidate the underlying mechanism. Accumulating evidence indicates that hippocampal dysfunction and deficits in cognition may be caused not only by deficits in excitatory principal cells, but also in inhibitory interneurons that can regulate LTP induction and hippocampal network activity (Chen et al., 2010; Levenga et al., 2013; Huh et al., 2016). As reduced GABAergic inhibition or reduced interneuron numbers may facilitate the induction of LTP and the degree of potentiation during LTP expression, this prompted us to assess whether P301S mice may exhibit differences in the abundance of interneurons. To this end, we determined the number of three different interneuron subtypes in the hippocampus of P301S mice using antibodies against SST, CR, and PV (Fig. 6a–d). Strikingly, the number of SST+, PV+, and CR+ profiles of interneurons was decreased by 15.01 ± 6.307% (Fig. 6b, *p = 0.0301), 21.63 ± 7.609% (Fig. 6c, *p = 0.0118), and 30.17 ± 11.5% (Fig. 6d, *p = 0.0156), respectively, in the hippocampus of 16- to 18-week-old P301S mice. Further, to investigate the functional relevance of reduced GABAergic inhibition and to test whether reduced GABAergic function contributes to aberrant LTP, we treated slices with the GABA-R agonist muscimol. Remarkably, treatment of P301S hippocampal slices 10 min before TBS with muscimol at a concentration of 0.2 µm significantly reduced LTP induction (Fig. 6e, min 31-35: P301S vs P301S+muscimol 187.14 ± 10.11% vs 154.78 ± 8.03%, p= 0.018), while it had no significant effect in WT slices (Fig. 6f). Hence, reduced GABAergic inhibition contributes to enhanced LTP in P301S mice. Together, our findings indicate that increased LTP in P301S mice is associated with a degeneration of interneurons that impairs GABAergic inhibition of the hippocampal network.
Figure 6.
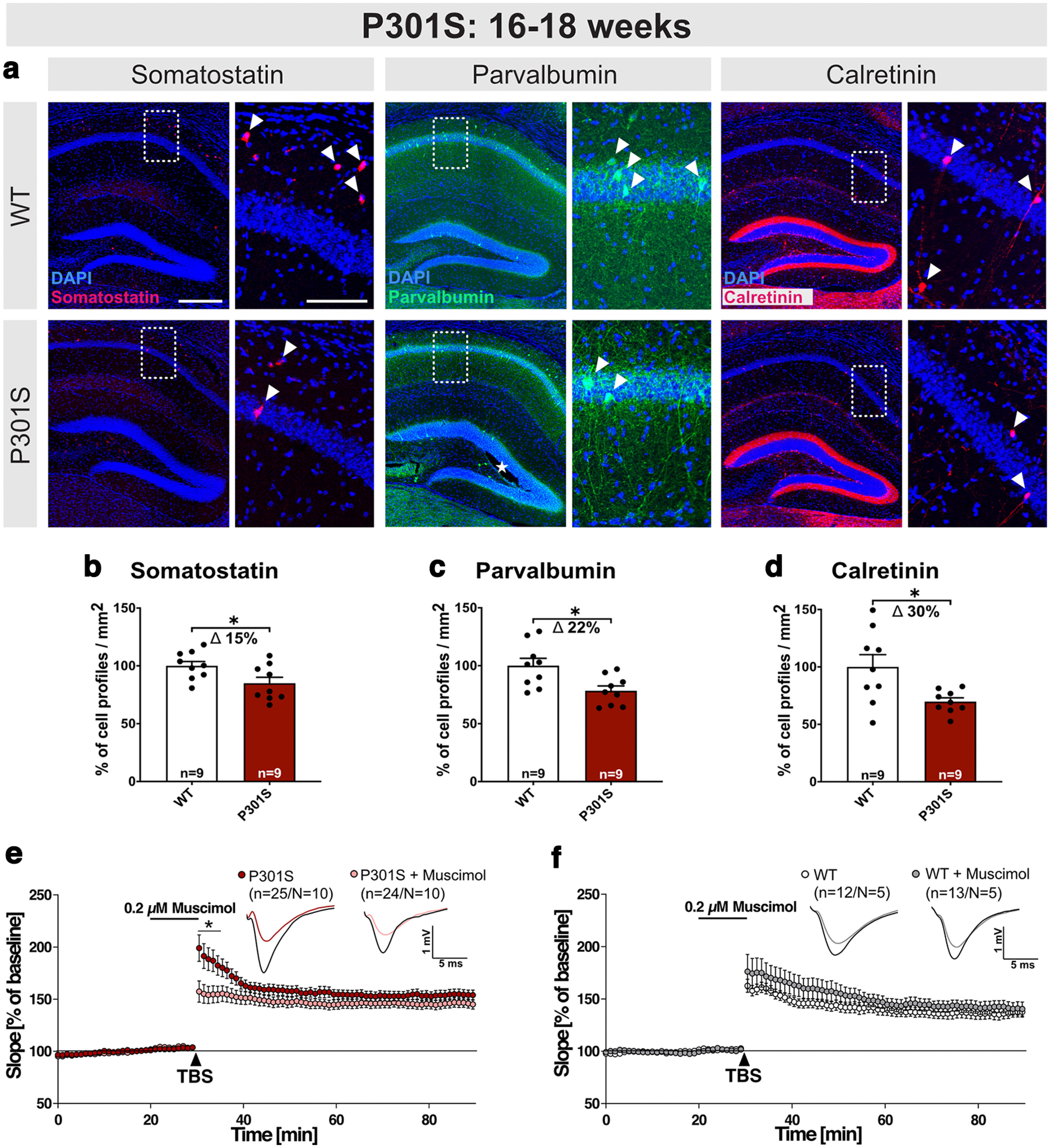
P301S mice exhibit a loss of interneurons at 16-18 weeks. a, Representative coronal brain sections (40 µm) depicting the hippocampus of WT and P301S mice at 16-18 weeks of age. Boxed areas of CA1 are displayed at higher magnification on the right. SST+, PV+, and CR+ inhibitory interneurons were identified using specific antibodies. Arrowheads indicate exemplary cells for each marker. b–d, Hippocampi from P301S mice show a loss of SST+, PV+, and CR+ cell profiles/mm2 (SST: *p = 0.0301; PV: *p = 0.0118; CR: *p = 0.0156) compared with WT controls. e, f, Application of muscimol reduced LTP induction in P301S slices, while LTP in WT slices was not significantly different. Age of mice: 16-18 weeks Scale bars (a): overview, 300 µm; zoom-in, 100 µm; 10× mosaic confocal images, maximum intensity projections, slicing artifact marked with an asterisk. b–d, Data are mean ± SEM. n = number of sections from N = 3 mice/genotype, as indicated in the bar graphs. e, f, n = number of recorded slices; N = number of animals. Data were analyzed by Student's t test: *p < 0.05; **p < 0.01; ***p < 0.001.
AAV-APPsα expression rescues spine density deficits in P301S mice
Encouraged by the effects of APPsα in vitro, we next aimed to further investigate its therapeutic potential in vivo. To this end, APPsα was expressed in the dorsal hippocampus by stereotactic injection of AAV vectors. To enable tracking of transduced cells, we used an AAV9-based bicistronic viral vector (AAV-APPsα; Fig. 7a) encoding for Venus and HA-tagged murine APPsα, which were fused by a T2A site (Richter et al., 2018). Venus- and HA-APPsα expression was driven by the neuron-specific Synapsin promotor (Fig. 7aII). As a control, a monocistronic AAV-Venus vector was used (Fig. 7aI). P301S mice were either injected with AAV-APPsα or AAV-Venus at the age of 6 weeks before the onset of Tau pathology. WT mice served as a control and received only AAV-Venus. For optimized vector spread, AAV-Venus or AAV-APPsα vectors were bilaterally injected into the stratum lacunosum-moleculare and the DG of the dorsal hippocampus. At each injection spot, 1 µl vector stock was applied (Fig. 7b). Animals were sacrificed either at the age of 16-18 weeks (10 weeks after injection, same age as used for LTP measurements) or at 20–22 weeks (late time point, 14 weeks after injection).
Next, we investigated spine density in the hippocampus as a correlate of excitatory synapses using Golgi/Cox staining. Spine density was analyzed in midapical and basal dendrites of CA1 pyramidal neurons using Golgi staining. Consistent with a study that reported reduced spine density in P301S mice at 10 weeks of age (H. Xu et al., 2014), we detected a significant reduction in spine density in AAV-Venus-injected P301S mice compared with AAV-Venus-injected WT mice at 16-18 weeks of age in midapical dendritic segments of CA1 pyramidal cells (Fig. 7c, WT-Venus 100.00 ± 1.56% vs P301S-Venus: 93.61 ± 1.13%, **p = 0.0029, Δ 6%). Interestingly, the expression of AAV-APPsα increased spine density in midapical dendrites by ∼10% to levels not significantly different from WT neurons (Fig. 7c, WT-Venus 100.00 ± 1.56% vs P301S-APPsα 103.54 ± 1.37%; not significant; P301S-Venus 93.61 ± 1.13% vs P301S-APPsα 103.54 ± 1.37%, ***p < 0.0001). In basal dendrites, no significant difference in spine density was detectable between 16- and 18-week-old transgenic P301S mice and WT mice injected with AAV-Venus (Fig. 7d, WT-Venus 100.00 ± 1.33% vs P301S-Venus 95.86 ± 1.11%, not significant). Nevertheless, AAV-APPsα significantly increased spine density of basal dendritic segments of P301S transgenic mice by 6% (Fig. 7d, P301S-Venus 95.86 ± 1.11% vs P301S-APPsα 101.48 ± 1.45%, **p = 0.0078). Strikingly, AAV-APPsα also rescued spine density deficits at advanced stages of severe Tau pathology at the age of 20-22 weeks. At this age, transgenic P301S mice injected with AAV-Venus exhibited more pronounced reductions in spine density compared with the earlier time point. We detected a significant reduction of spine density by ∼8% in midapical dendritic segments of P301S mice injected with AAV-Venus compared with AAV-Venus-injected WT mice (Fig. 7e, WT-Venus 100.00 ± 1.46% vs P301S-Venus 91.51 ± 1.48%, ***p < 0.0001). For basal dendrites, the spine density deficit amounted to 14% in AAV-Venus-injected P301S mice compared with the WT-Venus group (Fig. 7f, WT-Venus 100.00 ± 1.44% vs P301S-Venus 86.43 ± 1.13%, ***p < 0.0001). In contrast, P301S mice that received AAV-APPsα revealed spine densities not significantly different from AAV-Venus-injected WT mice in midapical (Fig. 7e, WT-Venus 100.00 ± 1.46% vs P301S-APPsα 100.03 ± 1.07%, not significant; P301S-Venus 91.51 ± 1.48% vs P301S-APPsα 100.03 ± 1.07%, ***p < 0.0001) as well as in basal dendritic segments (Fig. 7f, WT-Venus 100.00 ± 1.44% vs P301S-APPsα 96.33 ± 1.33%, not significant; P301S-Venus 86.43 ± 1.13% vs P301S-APPsα 96.33 ± 1.33%, ***p < 0.0001). Together, these findings indicate that APPsα not only restored LTP and basal synaptic transmission when applied in vitro, but also restored normal synaptic density in P301S mice in vivo.
AAV-APPsα expression rescues spine density in THY-Tau22 mice
Finally, we intended to test the more general potential of APPsα to rescue Tau-induced spine density deficits. To this end, we used transgenic THY-Tau22 mice, which express hTau (4R1N) mutated at sites G272V and P301S under control of the murine Thy1 promoter (Schindowski et al., 2006). THY-Tau22 mice show, compared with P301S mice, less rapid Tau accumulation and pathology that are restricted to cortex and hippocampus. Pathologic Tau phosphorylation starts at 3-6 months of age and becomes highly abundant in CA1 cells at 12 months of age. At this age, CA1 cells also exhibit prominent neurofibrillary tangle-like inclusions (Schindowski et al., 2006). Moreover, Tau pathology is associated with reduced spine density in CA1 neurons from 9 months onward (Burlot et al., 2015). We injected THY-Tau22 mice and WT littermates with either AAV-APPsα or AAV-Venus at 9 months of age and sacrificed them for spine density analysis at 12 months of age. We detected a significant reduction of spine density by ∼12% in midapical dendritic segments of transgenic THY-Tau22 mice injected with AAV-Venus compared with AAV-Venus-injected WT mice (Fig. 7g, WT-Venus 100.00 ± 1.72% vs THY-Tau22-Venus: 88.41 ± 1.84%, ***p < 0.0001, Δ 12%). In basal dendrites, the spine density deficit amounted to 11% in AAV-Venus-injected THY-Tau22 mice compared with the WT-Venus group (Fig. 7h, WT-Venus 100.00 ± 1.61% vs THY-Tau22-Venus: 89.34 ± 1.79%, ***p = 0.0001, Δ 11%). In sharp contrast, THY-Tau22 mice that received AAV-APPsα revealed spine densities not significantly different from AAV-Venus-injected WT mice in both midapical (Fig. 7g, WT-Venus 100.00 ± 1.72% vs THY-Tau22-APPsα 96.43 ± 2.06%, not significant; THY-Tau22-Venus 88.41 ± 1.84% vs THY-Tau22-APPsα 96.43 ± 2.06%, **p = 0.0077) and basal dendrites (Fig. 7h, WT-Venus 100.00 ± 1.61% vs THY-Tau22-APPsα 101.55 ± 1.68%, not significant; THY-Tau22-Venus 89.34 ± 1.79% vs THY-Tau22-APPsα 101.55 ± 1.68%, ***p < 0.0001). Together, this indicates that APPsα can normalize spine density deficits in two independent Tau transgenic mouse lines.
AAV-APPsα injected at 6 weeks of age does not rescue the interneuron deficit in adult P301S mice
Previous studies indicated that APPsα has potent neuroprotective effects in vitro against excitotoxicity, growth factor deprivation, proteasomal stress, or stress following traumatic or ischemic brain injury in vivo (for review, see Mockett et al., 2017; Müller et al., 2017). We therefore asked whether APPsα expression might also rescue the interneuron deficit of P301S mice. To this end, 6-week-old WT and P301S mice were intracranially injected with AAV-APPsα or AAV-Venus. Again, animals were sacrificed at the age of 16-18 and 20-22 weeks and SST+, PV+, and CR+ interneuron populations in the hippocampus were quantified by immunohistochemistry. Representative images show the vector spread in WT and P301S mice as well as exemplary images of all three interneuron markers at the age of 16-18 weeks (Fig. 8a,b). In line with our previous findings in noninjected mice, the number of SST+ cells as indicated by cell profiles/mm2 was significantly reduced in P301S-Venus injected mice (Fig. 8c, WT-Venus vs P301S-Venus, Δ 19.72 ± 6.15%, **p = 0.0085). Likewise, we confirmed a severe loss of both PV+ and CR+ interneurons in P301S mice, compared with WT mice injected with AAV-Venus (PV, Fig. 8d, WT-Venus vs P301S-Venus: Δ 12.48 ± 4.42%, *p = 0.0213; CR, Fig. 8e, WT-Venus vs P301S-Venus: Δ 29.15 ± 4.10%, ***p < 0.0001). When P301S mice received the AAV-APPsα vector, the number of SST+ and CR+ cell profiles/mm2 was, however, still reduced compared with the WT-Venus group (SST, Fig. 8c, WT-Venus vs P301S-APPsα: Δ 35.70 ± 6.15%, ***p < 0.0001; CR, Fig. 8e, WT-Venus vs P301S-APPsα: Δ 30.03 ± 4.01%, ***p < 0.0001). In summary, injection of AAV-APPsα at 6 weeks of age failed to rescue the loss of inhibitory interneurons.
Figure 8.
AAV-APPsα does not rescue the interneuron deficit in adult P301S mice. a, Representative coronal brain sections (40 µm) of WT-Venus, P301S-Venus, and P301S-APPsα-injected mice analyzed at the age of 16-18 weeks. The Venus signal indicates widespread expression of both vectors in all subregions of the hippocampus. b, Representative coronal sections (40 µm) of WT-Venus, P301S-Venus, and P301S-APPsα-injected mice analyzed at the age of 16-18 weeks. Magnifications display the boxed CA1 region. SST+, PV+, and CR+ inhibitory interneuron populations in the hippocampus were analyzed. Arrowheads indicate exemplary cells for each marker. c-e, At the age of 16-18 weeks, the number of SST+, PV+, and CR+ cell profiles/mm2 was significantly reduced in P301S-Venus and P301S-APPsα mice compared with WT-Venus controls (SST, WT-Venus vs P301S-Venus: **p = 0.0085; WT-Venus vs P301S-APPsα: ***p < 0.0001; PV, WT-Venus vs P301S-Venus: *p = 0.0213; WT-Venus vs P301S-APPsα: not significant; CR, WT-Venus vs P301S-Venus: ***p < 0.0001; WT-Venus vs P301S-APPsα: ***p < 0.0001). Data are mean ± SEM. n = number of sections from N = 4 mice/genotype, indicated in the bar graphs. Data were analyzed by one-way ANOVA followed by Bonferroni's post hoc test: *p < 0.05; **p < 0.01; ***p < 0.001. Scale bars: 300 µm; zoom-in, 100 µm; 10× mosaic confocal images, maximum intensity projections.
Early postnatal onset and temporal progression of interneuron loss in the hippocampus of P301S mice
Next, we explored the onset and temporal progression of interneuron loss in the hippocampus of P301S mice. Already at the age of 4 weeks, the number of SST+ interneurons as indicated by the number of cell profiles/mm2 was reduced by 25.08 ± 9.56% in P301S mice compared with WT mice (Fig. 9a, WT vs P301S: *p = 0.0159), while the number of PV+ and CR+ interneurons was not significantly altered (Fig. 9b,c). We also asked whether the hTau transgene may not only be expressed in excitatory principal cells of the hippocampus (Fig. 1, HT7 panels), but may also be expressed in interneurons, which had not been studied so far. Indeed, double staining using HT7 (detecting total hTau) and antibodies against interneuron marker proteins revealed prominent colocalization of HT7 with SST+, CR+, and PV+ cells, indicating hTau expression from 4 weeks of age (Fig. 9d–f).
Figure 9.
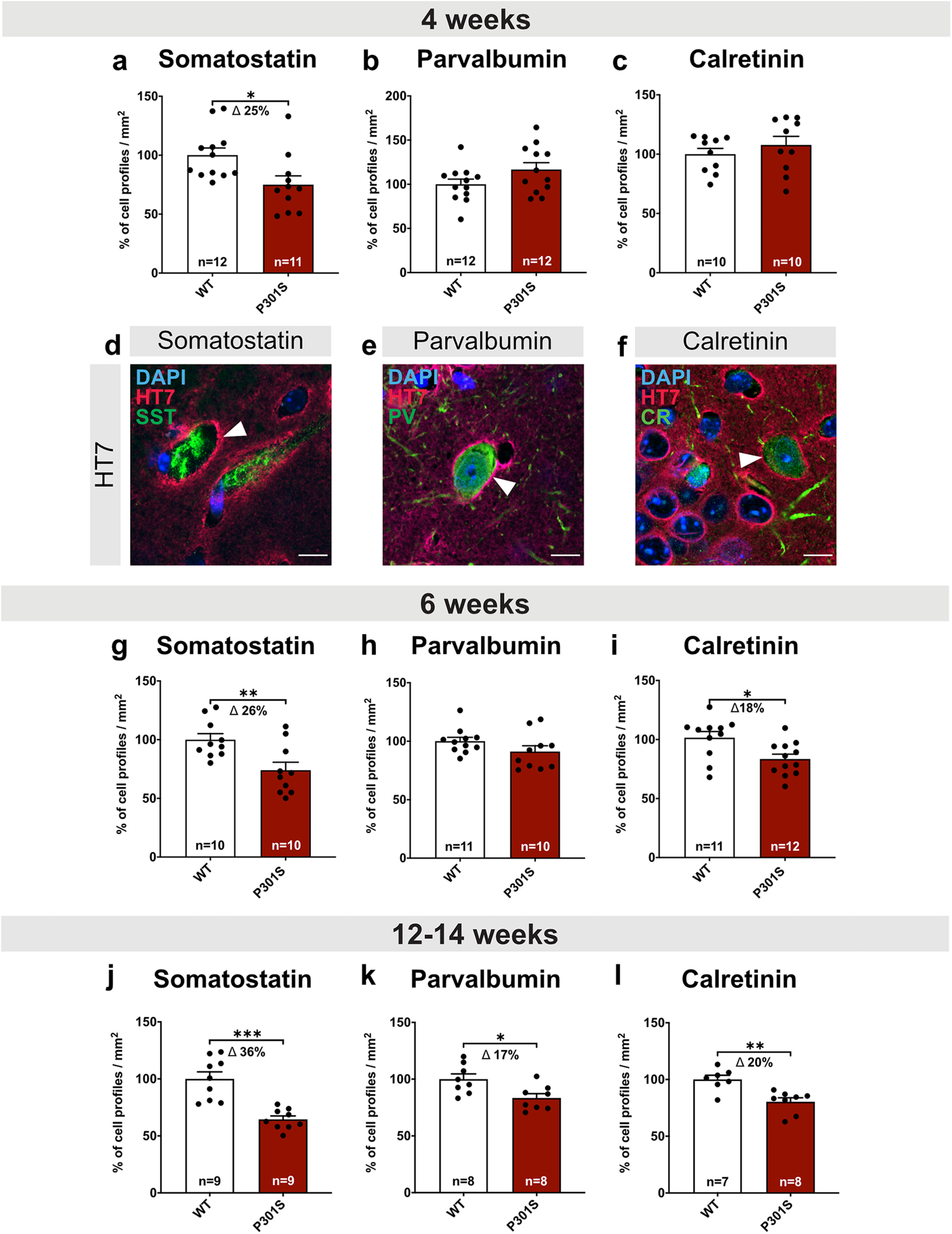
Early onset and temporal progression of interneuron loss in P301S mice. a-c, Already at the age of 4 weeks, the number of SST+ cell profiles/mm2 in the hippocampus was significantly reduced in P301S compared with WT mice (*p = 0.0159). When analyzing PV+ and CR+ interneurons, P301S mice were indistinguishable from the WT group. d-f, Double immunostaining of interneuron markers (SST, PV, and CR) with total hTau (HT7, arrowhead) indicates expression of hTau in hippocampus of P301S mice at 4 weeks of age. Arrowhead indicates HT7-positive somata. g, i, The number of SST+ and CR+ cell profiles/mm2 was significantly reduced in 6-week-old P301S mice compared with WT mice (SST: **p = 0.0067; CR: *p = 0.0121). h, At this age, the number of PV+ cell profiles/mm2 was not significantly different between groups. j-l, At the age of 12-14 weeks, the number of all three interneuron subtypes (SST+, PV+, and CR+) was significantly reduced as indicated by a reduction in cell profiles/mm2 (SST: ***p < 0.0001; PV: *p = 0.0148; CR: **p = 0.0020). Scale bars: 300 µm; zoom-in, 100 µm; 10× mosaic confocal images, maximum intensity projections. Data are mean ± SEM. n = number of sections from N = 3 or 4 mice/genotype, indicated in the bar graphs. Data were analyzed by Student's t test: *p < 0.05; **p < 0.01; ***p < 0.001.
Next, we studied interneuron numbers at 6 and 12-14 weeks of age. At the age of 6 weeks, not only SST+ interneurons (Fig. 9g, WT vs P301S, Δ 25.95 ± 8.47%, **p = 0.0067), but also CR+ interneurons were significantly reduced (Fig. 9i, WT vs P301S: Δ 18.03 ± 6.57%, *p = 0.0121). The number of PV+ cells was indistinguishable between WT and P301S mice (Fig. 9h). Further, at the age of 12-14 weeks, all three analyzed subclasses of GABAergic interneurons were drastically reduced (Fig. 9j–l). The number of SST+ cells in P301S mice was reduced by 35.50 ± 6.83% (Fig. 9j, WT vs P301S: ***p < 0.0001). Likewise, the number of PV+ and CR+ cells was significantly reduced by 16.64 ± 5.99% and 19.64 ± 5.10%, respectively (PV Fig. 9k, WT vs P301S: *p = 0.0148; CR Fig. 9l, WT vs P301S: **p = 0.0020). Together with the interneuron deficits observed at 16 weeks of age (see Fig. 6a–d), we saw a progressive interneuron loss with age. Overall, these novel findings indicate a pronounced and very early loss of several interneuron subtypes in P301S mice that may impair hippocampal circuitry and function from early postnatal stages onward.
P301S mice show early postnatal impairment in hippocampus-dependent nest building
The interneuron loss in 4-week-old P301S mice suggests that hippocampal function might be impaired already at this early time point in development. We therefore studied nest building, an innate species-specific behavior that is highly sensitive to hippocampal lesions and dysfunction (Deacon, 2006). Interestingly, P301S mice showed severe impairments in nesting at 4 weeks of age (Fig. 10). The nests were rated after 24 h on a scale ranging from 1 (nesting material unused) to 5 (perfect nest, >90% of the nesting material used up). While WT mice reached a mean score of 2.533 ± 0.1856, P301S mice had an average score of 1.267 ± 0.1182 (Fig. 10, ***p < 0.0001). These findings indicate that the onset of interneuron loss and the associated hippocampal dysfunction develop early postnatally in P301S mice, well before aggregated Tau species and pathology are detectable.
Figure 10.

P301S mice show early postnatal impairment in hippocampus-dependent nest building. P301S mice showed significantly lower scores of nest building at 4 weeks of age (***p < 0.0001). Data are mean ± SEM. n = number of animals, indicated in the bar graphs. Mann–Whitney test for not normally distributed data: *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
In this study, we examined the potential of neurotrophic, secreted APPsα to ameliorate Tau-induced synaptic and neuronal deficits in P301S mice. Our study revealed several novel findings: (1) We demonstrate that P301S mice exhibit an aberrantly increased LTP at 16-18 weeks of age, whereas LTD was unaffected. (2) We show that acute application of nanomolar amounts of recAPPsα to brain slices restores normal LTP, and basal synaptic transmission, indicating a homeostatic function of recAPPsα on a rapid time scale. (3) Further, in vivo expression of APPsα in the hippocampus restored normal spine density of CA1 pyramidal cells even at advanced stages of Tau pathology, both in P301S mice as well as in THY-Tau22 mice. Together, these data corroborate therapeutic effects of APPsα for Tau-induced synaptic pathology. (4) As a further key finding, we identified a pronounced loss of GABAergic interneurons in the hippocampus of P301S mice as the earliest pathologic event that clearly precedes the accumulation of pathologic Tau species. (5) Thus, interneuron loss and aberrant LTP may disturb neuronal circuits important for hippocampal function and cognitive behavior.
Previous work had indicated a crucial role of APPsα for neuroprotection against various forms of cellular stress, including glutamate and Aβ-induced toxicity in vitro (for review, see Mockett et al., 2017; Müller et al., 2017) and an essential in vivo role of endogenous APPsα for synaptic plasticity and cognition (Ring et al., 2007; Taylor et al., 2008; Hick et al., 2015; Moreno et al., 2015; Richter et al., 2018). Moreover, endogenously produced APPsα has been shown to protect against acute forms of brain injury in vivo (Smith-Swintosky et al., 1994; Corrigan et al., 2012; Hefter et al., 2016; Plummer et al., 2016). Together, these studies had indicated that APPsα is neuroprotective under injury conditions and suggested that these properties may be leveraged for therapeutic application in AD. To this end, we and others recently showed that AAV-mediated expression of APPsα in aged APP/PS1ΔE9 mice with established plaque pathology restored synaptic plasticity and rescued spine density deficits (Fol et al., 2016; Tan et al., 2018). Here, we set out to explore the role of APPsα for Tau-induced pathology as the second hallmark of AD and other primary tauopathies. Although P301S mice have been widely used as a model of human tauopathy (Allen et al., 2002), the temporal progression of pathologic Tau species had not been studied in depth within the hippocampus. The earliest time point of our IHC analysis (12-14 weeks of age) parallels the onset of memory impairments. Somewhat surprisingly, AT8 and AT180 staining was not widespread at this age but was restricted to the mossy fiber axons while being hardly detectable in somatodendritic regions. In older animals, we observed a pronounced and progressive increase in pathologic phospho-Tau species that now accumulated in the cell bodies of pyramidal CA1 and CA3 cells, in hilar cells of the DG, and in dendritic regions of all hippocampal subfields. Western blot analysis of hippocampal extracts also showed a progressive increase in Sarkosyl-insoluble phospho-Tau, that reached significance, however, only at 20 weeks of age. Thus, accumulation of insoluble and pathologic Tau species occurs later than memory impairments.
Electrophysiological recordings revealed that P301S mice exhibit a complex phenotype with subtle alterations in synaptic physiology of CA1 cells, including an increase in mEPSC amplitude and frequency. Extracellular field recordings indicated altered basal postsynaptic transmission in IO measurements, while presynaptic function and short-term plasticity were unchanged. The most striking finding was that LTP was significantly increased in P301S mice. At first glance, this increase in LTP may appear counterintuitive given the previously shown cognitive impairments of P301S mice (H. Xu et al., 2014). Importantly, however, increased LTP in conjunction with memory impairments is not uncommon and has also been reported for gene-targeted mice of several synaptic proteins, including PSD95, GluR2, components of the cannabinoid system, and others (Gerlai et al., 1998; Migaud et al., 1998; Uetani et al., 2000; Kaksonen et al., 2002; Monory et al., 2015; Zimmermann et al., 2019). Thus, not only reduced but also abnormally increased LTP can be regarded as indicative of an impaired hippocampal network (Korte and Schmitz, 2016). Further, using the GABA-R agonist muscimol, we demonstrate that impaired GABAergic inhibition of the CA3-CA1 network in P301S mice contributes to the increase in LTP.
Regarding the interneuron deficit, it was, however, surprising to see that CA1 cells revealed no decrease in mIPSCs, but rather an increase in mEPSC frequency, amplitude, and charge transfer leading to an overall disturbed E/I balance. We hypothesize that this might, at least in part, be related to compensatory mechanisms. Among these, the increase in mEPSC frequency might be because of the unsilencing of silent synapses as a consequence of altered interneuron input or increased LTP. Similarly, it is conceivable that there are changes in function (e.g., increased release probability) or anatomy (increased axonal arbor) of interneurons in P301S mice to compensate for the decrease in interneuron number. Such compensatory changes might explain normal mIPSC frequency despite reduced interneuron number. Irrespective of the primary cause of these alterations, P301S mice exhibit complex synaptic and network alterations, such as abnormal LTP, that may disturb hippocampal function.
Our previous studies in APP/PS1ΔE9 mice had indicated that APPsα can rescue impaired LTP. Here, we now demonstrate that APPsα can also normalize aberrantly increased LTP. This action occurred on a rapid time scale as incubating P301S slices with nanomolar amounts of recombinant APPsα was sufficient to restore normal LTP. Although the mechanism of how precisely APPsα counteracts this abnormal increase in LTP deserves further investigation, the known inhibitory action of APPsα on L-type calcium channels may play an important role. Recently, Hefter et al. (2016) demonstrated that APPsα can normalize hypoxia-induced increased intracellular Ca2+ levels and neuronal network function by inhibiting L-type calcium channels. Consistent with this, APP can biochemically interact with Cav1.2 L-type calcium channels to suppress voltage-dependent calcium influx into neurons (Yang et al., 2009).
In addition, it is conceivable that APPsα might limit excessive presynaptic glutamate release via binding to GABABR1a receptors (Rice et al., 2019). However, this interaction required much higher concentrations (in the low micromolar range) of recAPPsα (Rice et al., 2019), which is in contrast to the nanomolar concentration used in our study. Taken together, a dual function of APPsα for LTP is emerging: (1) under conditions of LTP impairments APPsα can increase postsynaptic responses and facilitate LTP by several mechanisms such as the modulation of α7-nicotinic acetylcholine receptors (α7nAChR) or glutamate receptors, by increasing local protein synthesis (Claasen et al., 2009; Richter et al., 2018; Mockett et al., 2019) and by regulating the intracellular calcium content of endoplasmic reticulum stores (Ludewig et al., 2021); (2) under conditions of abnormally increased LTP, however, as observed in P301S mice, APPsα can also reduce LTP to restore WT-like levels.
Importantly, APPsα also rescued Tau-induced spine density deficits of CA1 neurons in vivo, not only in P301S mice but also in independent Tau-transgenic THY-Tau22 mice. APPsα may thus have a more general potential for synaptic repair, regardless of whether Tau pathology is associated with interneuron deficits or not. Remarkably, synaptotrophic effects of APPsα were not restricted to early time points but were also found for advanced stages of pathology with pronounced Tau aggregation in both Tau transgenic models (at 20-22 weeks in P301S mice and at 12 months in THY-Tau22 mice). Together, our results indicate that APPsα has therapeutic potential for synaptic deficits induced by human mutant Tau.
Degeneration of excitatory pyramidal cells has previously been observed in multiple transgenic Tau lines (see, e.g., Santacruz et al., 2005; Yoshiyama et al., 2007; Maeda et al., 2016), including P301S mice investigated here, which show neuronal loss in cortical layers I-II at 3 months of age (Hampton et al., 2010). Upon aging, P301S mice exhibit massive Tau aggregation in the brainstem and spinal cord which ultimately leads to the loss of motor neurons (Allen et al., 2002). In contrast, the interneuron deficit in the hippocampus that emerges at 4 weeks of age clearly precedes the development of Tau aggregates and constitutes the by far earliest Tau-induced pathology of P301S mice described to date. From the onset and time course of interneuron loss, we conclude that Tau-induced interneuron degeneration in the P301S hippocampus may either be related to toxic soluble or oligomeric Tau species (Lasagna-Reeves et al., 2011; Fa et al., 2016) secreted by excitatory principal cells or to cell autonomous effects of hTau expression in interneurons themselves (Fig. 9d,e). Interneurons may be particularly sensitive to mutant hTau overexpression, due to their high metabolic demand (Kann, 2016). Our data are also consistent with and extend findings in a different transgenic hTau line (JNPL3) that showed increased late-phase L-LTP and impaired memory associated with a reduction in interneurons in aged mice (Levenga et al., 2013). In contrast to our findings, however, interneuron degeneration observed in JNPL3 mice occurred at advanced stages of Tau pathology, suggesting Tau aggregation-dependent neurotoxicity, whereas young JNPL3 mice were normal (Levenga et al., 2013).
Strikingly, nest building behavior, a species-typic innate behavior that is highly sensitive to hippocampal lesions (Deacon et al., 2002), was significantly impaired concomitant with the loss of SST+ and CR+ interneurons, suggesting already early impairments of hippocampal functions in P301S mice. SST+ interneurons have recently been shown to shape postnatally the development and maturation of PV-dependent circuits (C. Z. Wang et al., 2019). Interestingly, the reported onset of learning and memory impairments of P301S mice (H. Xu et al., 2014) coincides with the time point at which we identified reduced numbers of PV+ cells in the hippocampus. PV+ interneurons comprise ∼25% of GABAergic interneurons in the CA1 region (Pelkey et al., 2017) and provide the majority of inhibitory input onto hippocampal pyramidal cells to control the oscillatory activity and rhythmic output of the hippocampus that is crucial for learning and memory (Murray et al., 2011). Most importantly, our data indicate that mutant hTau on its own can drive early interneuron degeneration, which may contribute to Tau-induced hippocampal network dysfunction and cognitive impairments in AD and in other tauopathies. Together, our data corroborate that APPsα is one of the few molecules with therapeutic potential to rescue both Aβ- and Tau-induced synaptic pathology and support the concept of targeting Tau-dependent network abnormalities as a therapeutic strategy in neurodegenerative diseases.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant MU1457/14-1; Baden-Württemberg-Stiftung Grant BWST-ISF2020-46 to U.C.M. and Phospholipid Research Center Grant UMU-2019-077/1-1 to U.C.M. We thank Michel Goedert for providing P301S mice; Luc Buée for providing THY-Tau22 mice; Tobias Abel and Gundula Braun (Paul Ehrlich Institute, Langen) for the production of AAVs; Martina Hühn and Silvio Krasemann for taking care of animals; Julia Gobbert and Elisabeth Blumenschein for excellent technical assistance; and the Nikon Imaging center at the University of Heidelberg.
The authors declare no competing financial interests.
References
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M (2002) Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S Tau protein. J Neurosci 22:9340–9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almkvist O, Basun H, Wagner SL, Rowe BA, Wahlund LO, Lannfelt L (1997) Cerebrospinal fluid levels of alpha-secretase-cleaved soluble amyloid precursor protein mirror cognition in a Swedish family with Alzheimer disease and a gene mutation. Arch Neurol 54:641–644. 10.1001/archneur.1997.00550170111022 [DOI] [PubMed] [Google Scholar]
- Anderson JJ, Holtz G, Baskin PP, Wang R, Mazzarelli L, Wagner SL, Menzaghi F (1999) Reduced cerebrospinal fluid levels of alpha-secretase-cleaved amyloid precursor protein in aged rats: correlation with spatial memory deficits. Neuroscience 93:1409–1420. 10.1016/S0306-4522(99)00244-4 [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci 8:663–672. 10.1038/nrn2194 [DOI] [PubMed] [Google Scholar]
- Burlot MA, et al. (2015) Cholesterol 24-hydroxylase defect is implicated in memory impairments associated with Alzheimer-like Tau pathology. Hum Mol Genet 24:5965–5976. 10.1093/hmg/ddv268 [DOI] [PubMed] [Google Scholar]
- Chen YJ, Zhang M, Yin DM, Wen L, Ting A, Wang P, Lu YS, Zhu XH, Li SJ, Wu CY, Wang XM, Lai C, Xiong WC, Mei L, Gao TM (2010) ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proc Natl Acad Sci USA 107:21818–21823. 10.1073/pnas.1010669107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claasen AM, Guevremont D, Mason-Parker SE, Bourne K, Tate WP, Abraham WC, Williams JM (2009) Secreted amyloid precursor protein-alpha upregulates synaptic protein synthesis by a protein kinase G-dependent mechanism. Neurosci Lett 460:92–96. 10.1016/j.neulet.2009.05.040 [DOI] [PubMed] [Google Scholar]
- Corrigan F, Vink R, Blumbergs PC, Masters CL, Cappai R, van den Heuvel C (2012) sAPPalpha rescues deficits in amyloid precursor protein knockout mice following focal traumatic brain injury. J Neurochem 122:208–220. 10.1111/j.1471-4159.2012.07761.x [DOI] [PubMed] [Google Scholar]
- De Strooper B, Karran E (2016) The cellular phase of Alzheimer's disease. Cell 164:603–615. 10.1016/j.cell.2015.12.056 [DOI] [PubMed] [Google Scholar]
- Deacon RM (2006) Assessing nest building in mice. Nat Protoc 1:1117–1119. 10.1038/nprot.2006.170 [DOI] [PubMed] [Google Scholar]
- Deacon RM, Croucher A, Rawlins JN (2002) Hippocampal cytotoxic lesion effects on species-typical behaviours in mice. Behav Brain Res 132:203–213. 10.1016/S0166-4328(01)00401-6 [DOI] [PubMed] [Google Scholar]
- Fa M, et al. (2016) Extracellular tau oligomers produce an immediate impairment of LTP and memory. Sci Rep 6:19393. 10.1038/srep19393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick AW, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SH (2017) Cryo-EM structures of tau filaments from Alzheimer's disease. Nature 547:185–190. 10.1038/nature23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fol R, Braudeau J, Ludewig S, Abel T, Weyer SW, Roederer JP, Brod F, Audrain M, Bemelmans AP, Buchholz CJ, Korte M, Cartier N, Muller UC (2016) Viral gene transfer of APPsalpha rescues synaptic failure in an Alzheimer's disease mouse model. Acta Neuropathol 131:247–266. 10.1007/s00401-015-1498-9 [DOI] [PubMed] [Google Scholar]
- Gerlai R, Henderson JT, Roder JC, Jia Z (1998) Multiple behavioral anomalies in GluR2 mutant mice exhibiting enhanced LTP. Behav Brain Res 95:37–45. 10.1016/s0166-4328(98)00002-3 [DOI] [PubMed] [Google Scholar]
- Hampton DW, Webber DJ, Bilican B, Goedert M, Spillantini MG, Chandran S (2010) Cell-mediated neuroprotection in a mouse model of human tauopathy. J Neurosci 30:9973–9983. 10.1523/JNEUROSCI.0834-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SS, Wolf F, De Strooper B, Busche MA (2020) Tipping the scales: peptide-dependent dysregulation of neural circuit dynamics in Alzheimer's disease. Neuron 107:417–435. 10.1016/j.neuron.2020.06.005 [DOI] [PubMed] [Google Scholar]
- Hefter D, Kaiser M, Weyer SW, Papageorgiou IE, Both M, Kann O, Muller UC, Draguhn A (2016) Amyloid precursor protein protects neuronal network function after hypoxia via control of voltage-gated calcium channels. J Neurosci 36:8356–8371. 10.1523/JNEUROSCI.4130-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hick M, Herrmann U, Weyer SW, Mallm JP, Tschape JA, Borgers M, Mercken M, Roth FC, Draguhn A, Slomianka L, Wolfer DP, Korte M, Muller UC (2015) Acute function of secreted amyloid precursor protein fragment APPsalpha in synaptic plasticity. Acta Neuropathol 129:21–37. 10.1007/s00401-014-1368-x [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Knott G, Lee WC, Mostany R, Mrsic-Flogel TD, Nedivi E, Portera-Cailliau C, Svoboda K, Trachtenberg JT, Wilbrecht L (2009) Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc 4:1128–1144. 10.1038/nprot.2009.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68:1067–1081. 10.1016/j.neuron.2010.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh S, Baek SJ, Lee KH, Whitcomb DJ, Jo J, Choi SM, Kim DH, Park MS, Lee KH, Kim BC (2016) The reemergence of long-term potentiation in aged Alzheimer's disease mouse model. Sci Rep 6:29152. 10.1038/srep29152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner A, et al. (2016) Site-specific phosphorylation of tau inhibits amyloid-beta toxicity in Alzheimer's mice. Science 354:904–908. 10.1126/science.aah6205 [DOI] [PubMed] [Google Scholar]
- Kaksonen M, Pavlov I, Voikar V, Lauri SE, Hienola A, Riekki R, Lakso M, Taira T, Rauvala H (2002) Syndecan-3-deficient mice exhibit enhanced LTP and impaired hippocampus-dependent memory. Mol Cell Neurosci 21:158–172. 10.1006/mcne.2002.1167 [DOI] [PubMed] [Google Scholar]
- Kann O (2016) The interneuron energy hypothesis: implications for brain disease. Neurobiol Dis 90:75–85. 10.1016/j.nbd.2015.08.005 [DOI] [PubMed] [Google Scholar]
- Kim M, Suh J, Romano D, Truong MH, Mullin K, Hooli B, Norton D, Tesco G, Elliott K, Wagner SL, Moir RD, Becker KD, Tanzi RE (2009) Potential late-onset Alzheimer's disease-associated mutations in the ADAM10 gene attenuate alpha-secretase activity. Hum Mol Genet 18:3987–3996. 10.1093/hmg/ddp323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Schmitz D (2016) Cellular and system biology of memory: timing, molecules, and beyond. Physiol Rev 96:647–693. 10.1152/physrev.00010.2015 [DOI] [PubMed] [Google Scholar]
- Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F (1999) Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA 96:3922–3927. 10.1073/pnas.96.7.3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannfelt L, Basun H, Wahlund LO, Rowe BA, Wagner SL (1995) Decreased alpha-secretase-cleaved amyloid precursor protein as a diagnostic marker for Alzheimer's disease. Nat Med 1:829–832. 10.1038/nm0895-829 [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R (2011) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener 6:39. 10.1186/1750-1326-6-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenga J, Krishnamurthy P, Rajamohamedsait H, Wong H, Franke TF, Cain P, Sigurdsson EM, Hoeffer CA (2013) Tau pathology induces loss of GABAergic interneurons leading to altered synaptic plasticity and behavioral impairments. Acta Neuropathol Commun 1:34. 10.1186/2051-5960-1-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler SF, Haass C, Steiner H (2011) Regulated intramembrane proteolysis: lessons from amyloid precursor protein processing. J Neurochem 117:779–796. 10.1111/j.1471-4159.2011.07248.x [DOI] [PubMed] [Google Scholar]
- Ludewig S, Herrmann U, Michaelsen-Preusse K, Metzdorf K, Just J, Bold C, Müller UC, Korte M (2021) APPsα rescues impaired Ca2+ homeostasis in APP- and APLP2-deficient hippocampal neurons. Proc Natl Acad Sci USA 118:e2011506118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Djukic B, Taneja P, Yu GQ, Lo I, Davis A, Craft R, Guo W, Wang X, Kim D, Ponnusamy R, Gill TM, Masliah E, Mucke L (2016) Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep 17:530–551. 10.15252/embr.201541438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG (1998) Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396:433–439. 10.1038/24790 [DOI] [PubMed] [Google Scholar]
- Mockett BG, Richter M, Abraham WC, Muller UC (2017) Therapeutic potential of secreted amyloid precursor protein APPsalpha. Front Mol Neurosci 10:30. 10.3389/fnmol.2017.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockett BG, Guevremont D, Elder MK, Parfitt KD, Peppercorn K, Morrissey J, Singh A, Hintz TJ, Kochen L, Tom Dieck S, Schuman E, Tate WP, Williams JM, Abraham WC (2019) Glutamate receptor trafficking and protein synthesis mediate the facilitation of LTP by secreted amyloid precursor protein-alpha. J Neurosci 39:3188–3203. 10.1523/JNEUROSCI.1826-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monory K, Polack M, Remus A, Lutz B, Korte M (2015) Cannabinoid CB1 receptor calibrates excitatory synaptic balance in the mouse hippocampus. J Neurosci 35:3842–3850. 10.1523/JNEUROSCI.3167-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno L, Rose C, Mohanraj A, Allinquant B, Billard JM, Dutar P (2015) sAbetaPPalpha improves hippocampal NMDA-dependent functional alterations linked to healthy aging. J Alzheimers Dis 48:927–935. 10.3233/JAD-150297 [DOI] [PubMed] [Google Scholar]
- Müller UC, Deller T, Korte M (2017) Not just amyloid: physiological functions of the amyloid precursor protein family. Nat Rev Neurosci 18:281–298. 10.1038/nrn.2017.29 [DOI] [PubMed] [Google Scholar]
- Murray AJ, Sauer JF, Riedel G, McClure C, Ansel L, Cheyne L, Bartos M, Wisden W, Wulff P (2011) Parvalbumin-positive CA1 interneurons are required for spatial working but not for reference memory. Nat Neurosci 14:297–299. 10.1038/nn.2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Cuesta P, Kato T, Arahata Y, Iwata K, Yamagishi M, Kuratsubo I, Kato K, Bundo M, Diers K, Fernandez A, Maestu F, Ito K (2017) Early functional network alterations in asymptomatic elders at risk for Alzheimer's disease. Sci Rep 7:6517. 10.1038/s41598-017-06876-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L (2016) Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci 17:777–792. 10.1038/nrn.2016.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, McBain CJ (2017) Hippocampal GABAergic inhibitory interneurons. Physiol Rev 97:1619–1747. 10.1152/physrev.00007.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer S, Van den Heuvel C, Thornton E, Corrigan F, Cappai R (2016) The neuroprotective properties of the amyloid precursor protein following traumatic brain injury. Aging Dis 7:163–179. 10.14336/AD.2015.0907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polydoro M, Acker CM, Duff K, Castillo PE, Davies P (2009) Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci 29:10741–10749. 10.1523/JNEUROSCI.1065-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice HC, de Malmazet D, Schreurs A, Frere S, Van Molle I, Volkov AN, Creemers E, Vertkin I, Nys J, Ranaivoson FM, Comoletti D, Savas JN, Remaut H, Balschun D, Wierda KD, Slutsky I, Farrow K, De Strooper B, de Wit J (2019) Secreted amyloid-beta precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science 363:eaao4827. 10.1126/science.aao4827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter MC, Ludewig S, Winschel A, Abel T, Bold C, Salzburger LR, Klein S, Han K, Weyer SW, Fritz AK, Laube B, Wolfer DP, Buchholz CJ, Korte M, Muller UC (2018) Distinct in vivo roles of secreted APP ectodomain variants APPsalpha and APPsbeta in regulation of spine density, synaptic plasticity, and cognition. EMBO J 37:e9833. 10.15252/embj.201798335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, Herms J, Buchholz C, Eckman CB, Korte M, Wolfer DP, Muller UC (2007) The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci 27:7817–7826. 10.1523/JNEUROSCI.1026-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L (2007) Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science 316:750–754. 10.1126/science.1141736 [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L (2011) Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci 31:700–711. 10.1523/JNEUROSCI.4152-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309:476–481. 10.1126/science.1113694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scattoni ML, Gasparini L, Alleva E, Goedert M, Calamandrei G, Spillantini MG (2010) Early behavioural markers of disease in P301S tau transgenic mice. Behav Brain Res 208:250–257. 10.1016/j.bbr.2009.12.002 [DOI] [PubMed] [Google Scholar]
- Schindowski K, Bretteville A, Leroy K, Bégard S, Bion JP, Hamdane M, Buée L (2006) Alzheimer's disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol 169:599–616. 10.2353/ajpath.2006.060002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Swintosky VL, Pettigrew LC, Craddock SD, Culwell AR, Rydel RE, Mattson MP (1994) Secreted forms of beta-amyloid precursor protein protect against ischemic brain injury. J Neurochem 63:781–784. 10.1046/j.1471-4159.1994.63020781.x [DOI] [PubMed] [Google Scholar]
- Steubler V, Erdinger S, Back MK, Ludewig S, Fässler D, Richter M, Han K, Slomianka L, Amrein I, von Engelhardt J, Wolfer DP, Korte M, Müller UC (2021) Loss of all three APP family members during development impairs synaptic function and plasticity, disrupts learning, and causes an autism-like phenotype. EMBO J 40:e107471. 10.15252/embj.2020107471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan VT, Mockett BG, Ohline SM, Parfitt KD, Wicky HE, Peppercorn K, Schoderboeck L, Yahaya MF, Tate WP, Hughes SM, Abraham WC (2018) Lentivirus-mediated expression of human secreted amyloid precursor protein-alpha prevents development of memory and plasticity deficits in a mouse model of Alzheimer's disease. Mol Brain 11:7. 10.1186/s13041-018-0348-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CJ, Ireland DR, Ballagh I, Bourne K, Marechal NM, Turner PR, Bilkey DK, Tate WP, Abraham WC (2008) Endogenous secreted amyloid precursor protein-alpha regulates hippocampal NMDA receptor function, long-term potentiation and spatial memory. Neurobiol Dis 31:250–260. 10.1016/j.nbd.2008.04.011 [DOI] [PubMed] [Google Scholar]
- Tyan SH, Shih AY, Walsh JJ, Maruyama H, Sarsoza F, Ku L, Eggert S, Hof PR, Koo EH, Dickstein DL (2012) Amyloid precursor protein (APP) regulates synaptic structure and function. Mol Cell Neurosci 51:43–52. 10.1016/j.mcn.2012.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetani N, Kato K, Ogura H, Mizuno K, Kawano K, Mikoshiba K, Yakura H, Asano M, Iwakura Y (2000) Impaired learning with enhanced hippocampal long-term potentiation in PTPdelta-deficient mice. EMBO J 19:2775–2785. 10.1093/emboj/19.12.2775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Jeugd A, Ahmed T, Burnouf S, Belarbi K, Hamdame M, Grosjean ME, Humez S, Balschun D, Blum D, Buee L, D'Hooge R (2011) Hippocampal tauopathy in tau transgenic mice coincides with impaired hippocampus-dependent learning and memory, and attenuated late-phase long-term depression of synaptic transmission. Neurobiol Learn Mem 95:296–304. 10.1016/j.nlm.2010.12.005 [DOI] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ (2012) Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149:708–721. 10.1016/j.cell.2012.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, Miller BL (2017) Epileptic activity in Alzheimer's disease: causes and clinical relevance. Lancet Neurol 16:311–322. 10.1016/S1474-4422(17)30044-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CZ, Ma J, Xu YQ, Jiang SN, Chen TQ, Yuan ZL, Mao XY, Zhang SQ, Liu LY, Fu Y, Yu YC (2019) Early-generated interneurons regulate neuronal circuit formation during early postnatal development. Elife 8:e44649. 10.7554/eLife.44649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci 17:5–21. 10.1038/nrn.2015.1 [DOI] [PubMed] [Google Scholar]
- Wegmann S, Eftekharzadeh B, Tepper K, Zoltowska KM, Bennett RE, Dujardin S, Laskowski PR, MacKenzie D, Kamath T, Commins C, Vanderburg C, Roe AD, Fan Z, Molliex AM, Hernandez-Vega A, Muller D, Hyman AA, Mandelkow E, Taylor JP, Hyman BT (2018) Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J 37:e98049. 10.15252/embj.201798049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer SW, Klevanski M, Delekate A, Voikar V, Aydin D, Hick M, Filippov M, Drost N, Schaller KL, Saar M, Vogt MA, Gass P, Samanta A, Jaschke A, Korte M, Wolfer DP, Caldwell JH, Muller UC (2011) APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. EMBO J 30:2266–2280. 10.1038/emboj.2011.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer SW, Zagrebelsky M, Herrmann U, Hick M, Ganss L, Gobbert J, Gruber M, Altmann C, Korte M, Deller T, Muller UC (2014) Comparative analysis of single and combined APP/APLP knockouts reveals reduced spine density in APP-KO mice that is prevented by APPsalpha expression. Acta Neuropathol Commun 2:36. 10.1186/2051-5960-2-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong M, Jones OD, Peppercorn K, Ohline SM, Tate WP, Abraham WC (2017) Secreted amyloid precursor protein-alpha can restore novel object location memory and hippocampal LTP in aged rats. Neurobiol Learn Mem 138:291–299. 10.1016/j.nlm.2016.08.002 [DOI] [PubMed] [Google Scholar]
- Xu H, Rosler TW, Carlsson T, de Andrade A, Bruch J, Hollerhage M, Oertel WH, Hoglinger GU (2014) Memory deficits correlate with tau and spine pathology in P301S MAPT transgenic mice. Neuropathol Appl Neurobiol 40:833–843. 10.1111/nan.12160 [DOI] [PubMed] [Google Scholar]
- Xu Y, Zhao M, Han Y, Zhang H (2020) GABAergic inhibitory interneuron deficits in Alzheimer's disease: implications for treatment. Front Neurosci 14:660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Wang Z, Wang B, Justice NJ, Zheng H (2009) Amyloid precursor protein regulates Cav1.2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. J Neurosci 29:15660–15668. 10.1523/JNEUROSCI.4104-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53:337–351. 10.1016/j.neuron.2007.01.010 [DOI] [PubMed] [Google Scholar]
- Young-Pearse TL, Chen AC, Chang R, Marquez C, Selkoe DJ (2008) Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev 3:15. 10.1186/1749-8104-3-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann T, Bartsch JC, Beer A, Lomazzo E, Guggenhuber S, Lange MD, Bindila L, Pape HC, Lutz B (2019) Impaired anandamide/palmitoylethanolamide signaling in hippocampal glutamatergic neurons alters synaptic plasticity, learning, and emotional responses. Neuropsychopharmacology 44:1377–1388. 10.1038/s41386-018-0274-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C, Crux S, Marinesco S, Montagna E, Sgobio C, Shi Y, Shi S, Zhu K, Dorostkar MM, Muller UC, Herms J (2016) Amyloid precursor protein maintains constitutive and adaptive plasticity of dendritic spines in adult brain by regulating D-serine homeostasis. EMBO J 35:2213–2222. 10.15252/embj.201694085 [DOI] [PMC free article] [PubMed] [Google Scholar]