

ABSTRACT
Non-small cell lung cancer (NSCLC) is the most common lung cancer with a highest mortality rate. MiR-497-5p has been reported as tumor suppressor in many cancers, but the role and mechanism of miR-497-5p in regulating NSCLC progression are still largely unknown in vitro and in vivo. Here, miR-497-5p was significantly downregulated in human NSCLC tissues and cell lines, compared with matched adjacent tissues and normal lung epithelial cell line. Then, miR-497-5p mimic and inhibitor were, respectively, transfected into human NSCLC cells A549 and H460, CCK-8 assay, transwell assay, and flow cytometry were used to detect the capacities of cell proliferation, invasion and apoptosis. MiR-497-5p negatively regulated proliferation and invasion of NSCLC cancer cells. MiR-497-5p was demonstrated to directly bound to 3’-UTR of CX3CL1 mRNA and post-transcriptionally suppressed its expression thus inactivating its downstream oncogenic pathway ERK/AKT. Moreover, transfection with short hairpin RNA (shRNA) against CX3CL1 decreased capacity of cell proliferation and invasion and promoted cell apoptosis in NSCLC cells. In addition, ERK inhibitor U0126 attenuated the promotion effect of miR-497-5p inhibitor on activation of ERK/AKT and cell proliferation and migration. Finally, overexpression of miR-497-5p substantially suppressed activation of the ERK/AKT pathway and tumor growth in tumor-bearing mice in vivo. Taken together, our findings showed that miR-497-5p is downregulated in human NSCLC tissues and cell lines, and it inhibited tumor growth and cell invasion by targeting CX3CL1 gene to inactivate the ERK/AKT pathway in NSCLC cells.
KEYWORDS: miR-497-5p, CX3CL1, NSCLC, cell proliferation, invasion, the ERK/AKT pathway
Introduction
Recent studies have shown that lung cancer as one of the most dangerous malignancies is the leading cause of cancer-related mortality in the world. [1,2]. There are two different types of lung cancer depending on the pathological characteristics: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) [3]. In the past few years, researches on NSCLC have not been thorough enough. However, it is clear that NSCLC is more common than SCLC, and the histologic subtypes of NSCLC include squamous-cell carcinoma (SCC), adenocarcinoma (ADC), and large-cell lung cancer (LCC) [4]. Although there has been substantive progress in the research of diagnosis and treatment for lung cancer, lung cancer mortality remains very unfavorable [5]. Efforts of new approaches should be made to develop therapeutic strategies in human lung cancer treatment.
MicroRNAs (miRNAs), a well-known class of non-coding RNAs with 19–25 nucleotides, which regulate gene expression at a posttranscriptional level through interacting with 3’ untranslated region (3’-UTR) of the target genes [6]. MiRNAs may act as oncogenes or tumor suppressors to make a key role in regulating tumor pathogenesis in virtually all forms of cancer, such as breast cancer [7], prostate cancer [8] and lung cancer [9]. Increasing attention is being paid to the role of miRNAs, which involved in many cellular processes, such as cell proliferation, invasion and apoptosis [10]. Growing evidence has demonstrated that miR-497-5p may act as a tumor suppressor in different cancers. For instance, expression of miR-497-5p was remarkably downregulated in clinical samples of colorectal cancer (CRC), and miR-497-5p inhibited CRC cell proliferation and invasion via targeting PTPN3 [11]. Overexpression of miR-497-5p also suppressed the proliferation and colony growth of gastric cancer cells [12]. In addition, it has been demonstrated that miR-497-5p may act as a tumor suppressor miRNA in lung cancer by targeting FGF2 [13].
CX3CL1, also known as fractalkine, is a unique chemokine and type I transmembrane protein with the CX3C chemokine domain [14]. It can bind to its specific receptor CX3CR1, which is abundantly expressed by monocytes/macrophages, natural killer cells and T cells [15]. The chemokine is a type of cytokine secreted by leukocytes or some implanted cells that binds to the specific receptor. The chemokine has chemotaxis and activation effects on certain immune cells, such as neutrophils, lymphocytes, and monocytes, through its specific receptor binding to exert biological effects [16,17].
In this study, we identified the expression of miR-497-5p was downregulated in human NSCLC tissue samples and cell lines. Then, we explored the role and underlying mechanisms of miR-497-5p in regulating the proliferation, invasion and apoptosis of NSCLC cells. Our data revealed the protective role of miR-497-5p in NSCLC progression by targeting CX3CL1 gene through the ERK/AKT pathway to inhibit cell proliferation and migration.
Materials and methods
Tissue samples and cell culture
The NSCLC tissue samples (20 patients) and adjacent non-tumor tissue samples (20 patients) were obtained from the Third People’s Hospital of Xinjiang Uygur Autonomous Region (Urumqi, China). This study received written informed consent from each patient and was approved by the Ethics Committee of the Xi’an Central Hospital. Human NSCLC cell lines A549, H460, H226 and H1299 and normal human lung epithelial cell lines BEAS-2B were purchased from ATCC (Manassas, VA, USA). Cells were routinely cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA). Moreover, cells were added 100 U/mL penicillin (HyClone, Logan, UT, USA) and 100 μg/mL streptomycin sulfate (HyClone, Logan, UT, USA) in the complex, which was then mixed. At last, all the dishes were placed at 37°C, under a 5% CO2 environment.
Cell transfection
MiR-497-5p mimic, miR-497-5p inhibitor and their corresponding negative controls were designed and purchased from GenePharama (Shanghai, China). pGPU6/GFP/Neo vector (GenePharama, Shanghai, China) was used to construct vector with short hairpin RNA (shRNA) against CX3CL1, and the non-targeting sequences (scramble shRNA). The full-length DNA coding sequence of CX3CL1 was inserted into pcDNA3.1 empty vector to construct pcDNA-CX3CL1 vector. For transfection, cells were maintained in 6-well plates with 70% confluence and transfected with oligonucleotides or plasmids using Lipofectamine 3000 Transfection Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. After 48 h transfection, cells were harvested for validation of transfection efficiency using RT-qPCR analysis.
RNA extraction and real-time quantitative polymerase chain reaction (RT-qPCR) analysis
Total RNA of tissue samples and human lung cancer cells were extracted using TRIzol Reagent (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). Then, total RNA was reverse transcribed into cDNA with High-Capacity complementary DNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA, USA). Next, mRNA level was quantified by RT-qPCR using TB Green™ Premix Ex Taq™ II (Takara Biomedical Technology, Beijing, China) on ABI 7300 Real‐time PCR system (Applied Biosystems, Waltham, MA, USA) under the following conditions: 95°C for 1 min followed by 35 cycles of 95°C for 20s, then 56°C for 10s and 72°C for 15s. The RT-qPCR reaction system was 20 µL: cDNA (1 μL), specific primers (1 μL), SYBR Green Mix (10 μL), and ddH2O (7 μL). U6 RNA was used as the endogenous control for miR-497-5p; GAPDH was used as the endogenous control for CX3CL1. The experimental operation was repeated at least in three times independently. Data were analyzed using the relative quantification 2−ΔΔCT method.
Cell proliferation assay
After 48 h transfection, Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) method was used to measure the cell proliferation efficiency. When the cells of each group reached a density of ~70%, the cells were washed twice with PBS. First, 1.0 × 104 cells were seeds in 24-well plates containing 10 μL CCK-8 reagent and 100 μL FBS-free medium. After cells were cultured in a humidified incubator containing 5% CO2 at 37°C for 48 h, then measured the cell proliferation efficiency at 0, 24, 48, 72 and 96 h after culture medium was removed. The absorbance OD450 was detected by a microplate reader. The experiment was independently repeated three times.
Cell invasion assay
For cell invasion assay, cells were suspended in 200 μL of serum-free medium and placed in the upper chamber pre-coated with Matrigel (50 μg/well, BD Biosciences, SanJose, CA, USA) of 24-well transwell plates (Corning, NY, USA) after transfection. The lower wells were added 600 μL of DMEM medium containing 10% FBS. Then, invading cells were stained with 0.5% crystal violet solution under a light microscope (Olympus, Tokyo, Japan) to count.
Luciferase reporter gene assay
Target gene analysis for miR-497-5p was performed using the online database of TargetScan biological prediction website (http://www.targetscan.org/vert_71/) to verify whether CX3CL1 was a direct target gene. The full length of the 3’ untranslated region (3’-UTR) of the CX3CL1 was cloned. Then CX3CL1 PCR product was cloned into the pmirGLO luciferase gene (Promega, Madison, WI, USA). The pRL-TK vector expressing Renilla luciferase (Takara Biotechnology Co., Ltd., Dalian, China) was used as the internal reference for transfection efficiency to adjust for the number of cells. Cells were transfected with miR-497-5p mimic and negative control in HEK293T cells (Shanghai, China). At 48 h post-transfection, cell lysate (20 µL per sample) was detected by a chemiluminescence detector (Modulus™; Turner BioSystems; Promega) at a wavelength of 560 nm. The relative luciferase activity was the ratio of firefly luciferase activity to Renilla luciferase activity. The experiment was independently repeated three times.
Cell apoptosis assay
Cells were treated differently for 24 h and then were collected at a centrifuge of 300 g at 4°C for 5 min. 5 μL of Annexin V‐FITC and 5 μL of propidium iodide (PI) were added at room temperature in dark and mix gently for 20 min. After washing with binding buffer, cells were examined using flow cytometry (BD Biosciences, San Jose, CA, USA).
Western blot analysis
The NSCLC tissue samples and adjacent non-tumor tissue samples were added with protein extraction reagent at a ratio of 1:10 (g/L). PBS was used to wash the NSCLC tissue samples twice and cell lyzed using RIPA lysis buffer (CW Biotech, Beijing, China). The extracted protein was homogenized and centrifuged and the supernatant was collected. The protein concentration was detected using a bicinchoninic acid quantitative kit (Thermo Fisher Scientific, Inc.) and adjusted to 3 μg/μL prior to conducting Western blotting. 10 μL boiled proteins per well with 5 × loading buffer (Beyotime Biotechnology, China) were separated in 12% SDS-PAGE at 70 V for 20 minutes and 100 V for 100 minutes. Samples were then transferred to PVDF membranes (Millipore, Bedford, MA, USA) at 350 mA for 105 minutes. Then the membranes were blocked with 5% nonfat milk 2 hours incubated with primary antibodies. The following antibodies were, respectively, used to incubate with the membrane at 4°C overnight: ERK antibody (1:500 dilution, ab176660, Abcam, Cambridge, UK), AKT antibody (1:400 dilution, ab142088, Abcam, Cambridge, UK), CX3CL1 antibody (1: 500 dilution, ab89229, Abcam, Cambridge, UK), MMP-2 antibody (1:600 dilution, ab97779, Abcam, Cambridge, UK), and MMP-9 antibody (1:400 dilution, ab38898, Abcam, Cambridge, UK), GAPDH antibody (1:300 dilution, ab181602, Abcam, Cambridge, UK). GAPDH was set as the endogenous control. After 3 times washing with TBS-T, the secondary antibody (1:1200 dilution, ab205718, Abcam) were, respectively, used to incubate with the membrane for 1 h at room temperature. The ECL system was used to show the banding of proteins and ImageJ 1.33 u software (National Institutes of Health, Bethesda, Md, USA) was used to analyze the relative light density of the blot bands. The relative expression level of the target gene was displayed as the fold change versus control group. The experiment was performed at least three times to obtain a mean value.
In vivo tumor growth assay in nude mice
0.2 mL of A549 lung carcinoma suspension (containing 107 cells) were subcutaneously into C57BL/6 nude mice (7 weeks old, weighing about 18 g) at right axilla to generate patient-derived xenografts (PDX). MiR-497-5p mimic (2 mg/Kg) and NC mimic (2 mg/Kg) were, respectively, locally injected into the mice every 2 days. Every 7 days, tumor volumes were detected. After 30 days, the mice were euthanized, and the tumors were obtained.
Statistical analysis
Data analysis was performed with SPSS version 22.0 software (IBM SPSS. Armonk, NY, USA). Each measurement was obtained from at least triple experiments and expressed as the mean ± standard error of mean (SEM). One-way analysis of variance was applied to perform comparisons among multiple groups followed by student’s t-test. P < 0.05 was considered to indicate a statistically significant difference.
Results
MiR-497-5p was downregulated in human NSCLC tissues and cell lines
RT-qPCR analysis was performed to explore the potential role of miR-497-5p in human NSCLC tissues and cell lines. In comparison with the adjacent normal tissues, the expression of miR-497-5p in NSCLC tissues was significantly downregulated (Figure 1a). The levels of miR-497-5p in cervical cancer cell lines A549, H460, H226 and H1299 markedly lower than those in human normal lung epithelial cells BEAS-2B (Figure 1b). The above results demonstrated that downregulation of miR-497-5p was a critical event in NSCLC and miR-497-5p might play an important role in progression of NSCLC.
Figure 1.
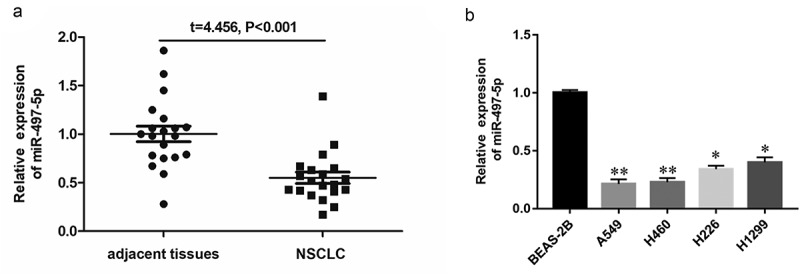
Expression of miR-497-5p in NSCLC tissues and different cell lines. (a) Expression of miR-497-5p in NSCLC tissues (n = 30) and adjacent normal tissues (n = 30) were measured by RT-qPCR. U6 was used as an internal reference. (b) Expression of miR-497-5p in different NSCLC cell lines. Statistical significance was determined using an independent sample t-test. Values are expressed as means ± SEM, n = 5. * P < 0.05 and ** P < 0.01 vs. normal or control.
MiR-497-5p overexpression inhibited cell proliferation and invasion of NSCLC cells and promoted apoptosis
To further explore the potential effect of miR-497-5p on the biological function of NSCLC, the miR-497-5p mimic, miR-497-5p inhibitor and their corresponding negative control oligonucleotides were, respectively, transfected into A549 and H460 cells. Expression of miR-497-5p was significantly upregulated when transfected with miR-497-5p mimic, while significantly downregulated by miR-497-5p inhibitor in A549 and H460 cells (Figure 2a). Transwell assay indicated that upregulation of miR-497-5p significantly decreased the invasive ability of NSCLC cells, while downregulation of miR-497-5p promoted the invasive ability (Figure 2b). The CCK-8 assay indicated that overexpression of miR-497-5p significantly inhibited the proliferation of A549 (Figure 2c) and H460 cells (Figure 2d), while downregulation of miR-497-5p promoted the cell proliferation ability (Figure 2(c,d)). As can be seen in Figure E, upregulation of miR-497-5p promoted the apoptotic ability of NSCLC cells, while low expression of miR-497-5p decreased the apoptotic ability of NSCLC cells (Fig. E). These results indicated that miR-497-5p overexpression significantly decreased the cell proliferation and invasion ability in A549 and H460 cells.
Figure 2.
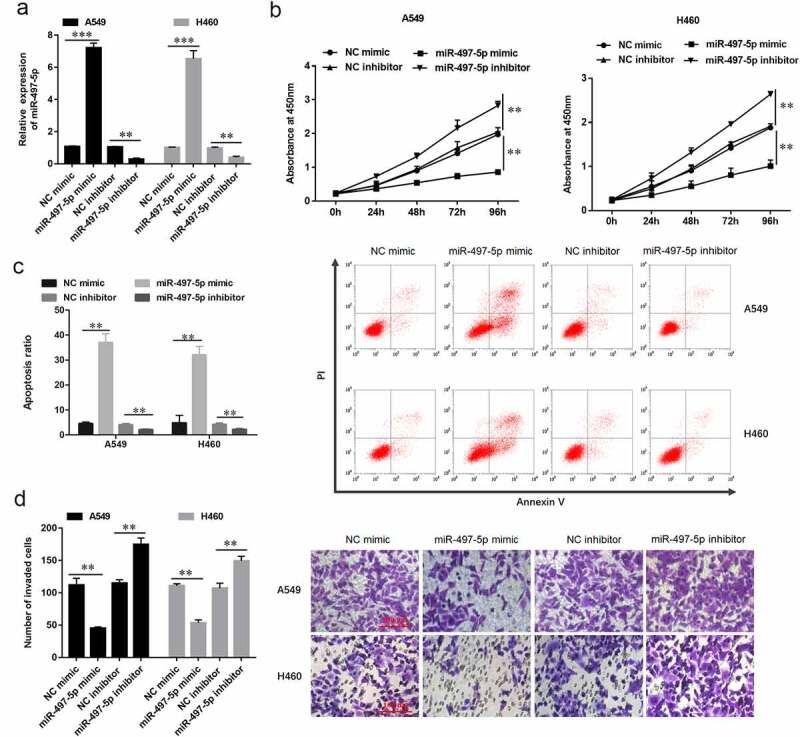
MiR-497-5p overexpression inhibited cell proliferation and invasion of NSCLC cells and promoted apoptosis. A549 or H460 cells were transfected with miR-497-5p mimic (40 nM) and miR-497-5p inhibitor (20 nM) for 48 h. (a) RT-qPCR was performed to determine expression of miR-497-5p in A549 or H460 cells transfected with miR-497-5p mimic, miR-497-5p inhibitor, or their negative control. (b) Cell invasion was examined by Transwell assay. (c) Cell proliferation was examined using the CCK-8 assay in A549 or (d) H460 cells. (e) Cell apoptosis was detected by flow cytometry. Statistical significance was determined using an independent sample t-test. Values are expressed as means ± SEM, n = 5. * P < 0.05 and ** P < 0.01.
CX3CL1 was a target gene of miR-497-5p and was upregulated in NSCLC cells
Bioinformatic analysis was used online database of TargetScan biological prediction website (http://www.targetscan.org/vert_71/) to verify whether CX3CL1 was a potential target gene for miR-497-5p. As showed in Figure 3a, the result suggested output of TargetScan showed that there is a CX3CL1 binding site (ACGACGA) in the sequence of miR-497-5p (Figure 3a). Further validation of the binding relationship between CX3CL1 and miR-497-5p using luciferase reporter assay. The results indicated miR-497-5p directly bind to CX3CL1 3’-UTR (Figure 3b). Moreover, it was found that CX3CL1 was significantly upregulated in human NSCLC tissues and cell lines using RT-qPCR analysis (Figure 3(c,d)) and western blotting analysis (Figure 3e). Furthermore, overexpression of miR-497-5p significantly downregulated CX3CL1 mRNA expression in A549 and H460 cells (figure 3f), and Western blot analysis was used to detect CX3CL1 protein levels (Figure 3g). These results demonstrated that miR-497-5p negatively modulated CX3CL1 expression by binding to its 3′-UTR.
Figure 3.
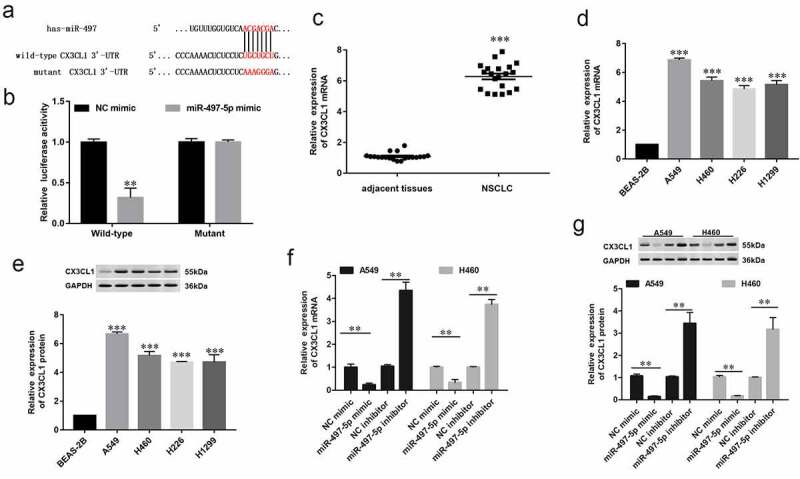
MiR-497-5p directly targeting CX3CL1 gene. (a) The sequence of miR-497-5p binding sites with WT/MUT CX3CL 3′-UTR. (b) Relative luciferase reporter activities of WT/MUT CX3CL1 in HEK293 cells. (c) Expression of CX3CL1 in NSCLC tissues (n = 30) and adjacent normal tissues (n = 30) was measured by RT-qPCR. (d) RT-qPCR and (e) Western blot analysis was performed to determine the expression of CX3CL1 in different cell lines, including A549, H460, H226 and H1299 and BEAS-2B. (f) RT-qPCR and (g) Western blotting analysis were performed to determine expression of CX3CL in A549 or H460 cells transfected with miR-497-5p mimic, miR-497-5p inhibitor, or their negative controls. Values are expressed as means ± SEM, n = 5. * P < 0.05 and ** P < 0.01.
CX3CL1 promoted cell proliferation and invasion in NSCLC cells
To investigate the underlying mechanism of CX3CL1 regulating NSCLC cell function, CX3CL1 was down-regulated in NSCLC cells. The results indicated that CX3CL1 was significantly downregulated when A549 and H460 cells were transfected with short hairpin RNA (shRNA) against CX3CL1 (Figure 4a). To study the effect of CX3CL1 on the biological function of lung cancer cells, we detected cell proliferation, invasion and apoptosis ability after transfected with CX3CL1 shRNA in lung cancer cells A549 and H460. CCK-8 assay indicated that transfection with CX3CL1 shRNA significantly decreased the cell proliferation ability and promoted cell apoptosis in A549 and H460 cells (Figure 4(b,c)). Moreover, downregulation of CX3CL1 decreased the cell invasion ability of A549 and H460 cells (Figure 4d). To further study the mechanism of CX3CL1 in regulating NSCLC cell functions, we investigated the phosphorylation of extracellular signal-regulated kinase (ERK) and protein kinase B (AKT), which are key kinases potentially activated by CX3CL1. It was observed that phospho-ERK and phospho-AKT expression levels were decrease following transfection with CX3CL1 shRNA, while the total ERK and AKT expression levels were unchanged in NSCLC cells (figure 4(f-h)). MMPs have been shown to play a key role in regulating cell invasion and apoptosis, so we detected expression of CX3CL1 after transfected with CX3CL1 shRNA. The mRNA expression levels of MMP2 and MMP9 were significantly downregulation after transfected with CX3CL1 shRNA (Figure 4(i,j)), and western blotting also showed the downgrade trend of protein expression levels of MMP2 and MMP9 (Figure 4(k-m)). The above results demonstrated that CX3CL1 promoted the cell proliferation and invasion and increased the phosphorylation of ERK and AKT in NSCLC cells.
Figure 4.
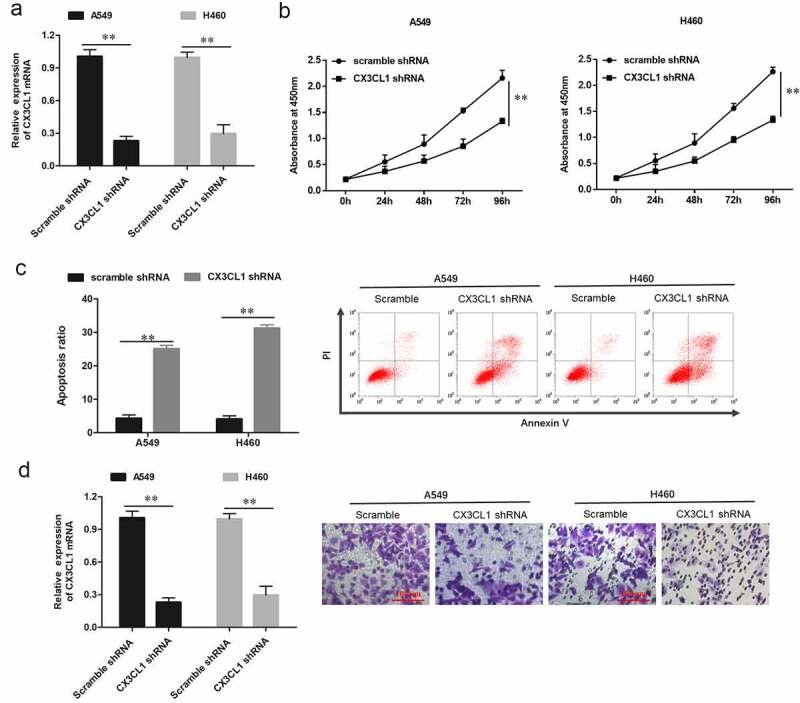
Knockdown of CX3CL1 suppressed the cell proliferation and invasion in A549 and H460 NSCLC cells. A549 and H460 cells were respectively transfected with 1 µg/mL lentiviral interference vector against CX3CL1 (CX3CL1 shRNA) and negative control vector (scrambled shRNA, scramble). Following transfection for 48 h, (a) Expression of CX3CL1 was measured by RT-qPCR in A549 and H460 cells. GAPDH was used as an internal reference. (b) Cell proliferation was examined using the CCK-8 assay in A549 and H460 cells. (c) Cell apoptosis was detected by flow cytometry. (d) Cell invasion was examined by Transwell assay. n = 5. **P < 0.01.
MiR-497-5p inhibited the phosphorylation of ERK and AKT through suppressing CX3CL1
To investigate whether ERK and AKT are involved in miR-497-5p/CX3CL1 mediated NSCLC cell functions, miR-497-5p mimic, pcDNA-CX3CL1 and their corresponding negative control oligonucleotides were transfected into A549 and H460 cells, respectively. It was observed that phospho-ERK and phospho-AKT levels were decreased following transfection with miR-497-5p mimic, while CX3CL1 overexpression reversed and increased phospho-ERK and phospho-AKT levels (Figure 5a). Furthermore, as important marker proteins of lung cancer invasion, the mRNA expression levels of MMP-2 and MMP-9 were significantly downregulated by miR-497-5p mimic, which could be reversed by upregulation of CX3CL1 reversed (Figure 5b). Western blotting also showed that overexpression of miR-497-5p could downregulate the expression of MMP-2 and MMP-9 protein, which could be also reversed by CX3CL1 overexpression (5C). The above results demonstrated that ERK/AKT were involved in miR-497-5p/CX3CL1 mediated NSCLC cell functions.
Figure 5.
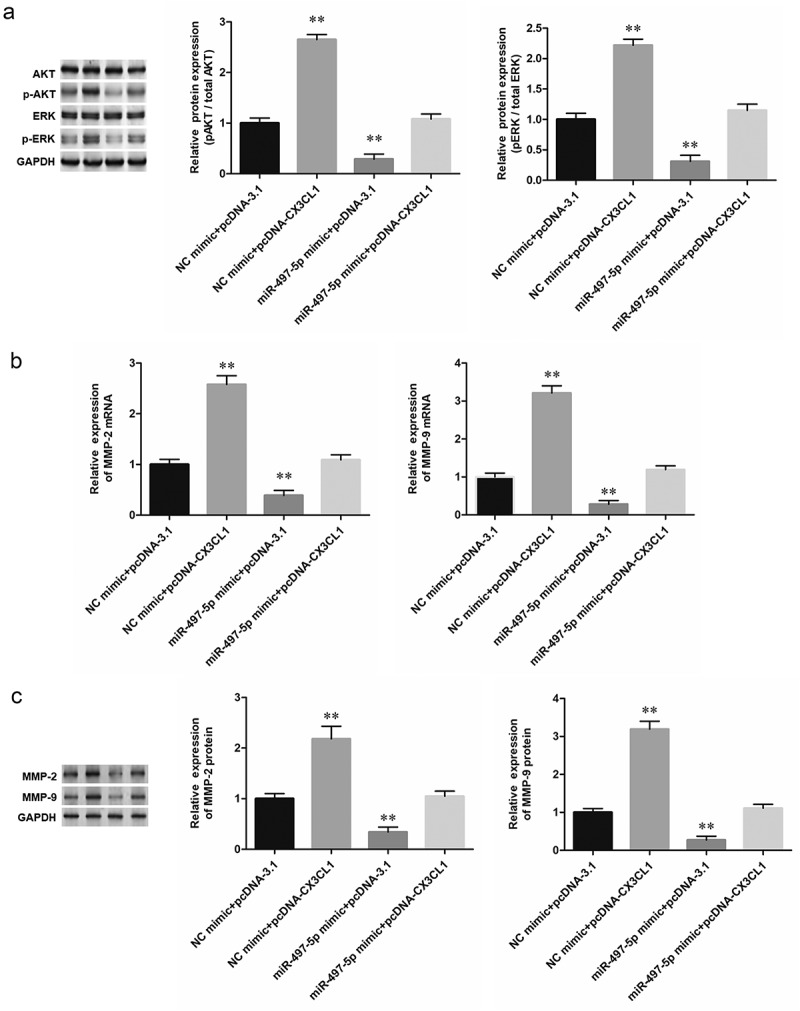
CX3CL1 rescued the suppression of miR-497-5p on phosphorylation of AKT and ERK in A549 cells. Human NSCLC A549 cells were respectively transfected with 40 nM miR-497-5p mimic alone or together with 1.2 µg/mL pcDNA vector expressing CX3CL1 (pcDNA-CX3CL1). Following transfection for 48 h, (a) The phosphorylation of AKT and ERK was observed by Western blotting analysis. (b) RT-qPCR was performed to determine expression of MMP-2 and MMP-9 mRNA in A549 NSCLC cells. (c) Western blotting was performed to determine expression of MMP-2 and MMP-9 protein in NSCLC cells. Quantification of band intensity was performed by ImageJ software, normalized to GAPDH. Values are expressed as means ± SEM, n = 5. * P < 0.05 and **P < 0.01 vs. negative control group (NC mimic + pcDNA3.1).
MiR-497-5p inhibited cell growth and invasion through and promoted cell apoptosis by inactivating the ERK/AKT pathway in NSCLC cells
To further investigate the regulatory mechanisms of miR-497-5p, miR-497-5p inhibitor was transfected into NSCLC cells alone or together with ERK inhibitor U0126 (15 μM). The expression levels of miR-497-5p were detected in A549 and H460 cells. Transfection with miR-497-5p inhibitor caused a significant decrease in miR-497-5p level, but ERK/AKT pathway inhibitor U0126 had no effect on the expression of upstream miR-497-5p and CX3CL1 in NSCLC cells (Figure 6(a,b)). As expected, miR-497-5p inhibitor enhanced phosphorylation of ERK and AKT, and ERK inhibitor U0126 reversed ERK and AKT phosphorylation induced by down-regulation of miR-497-5p in A549 and H460 cells (Figure 6(c-g)). Moreover, Transwell assay showed that ERK inhibitor U0126 reversed the promotion effect of miR-497-5p inhibitor on proliferation and invasion of A549 and H460 cells (Figure 7(a,b)). These results indicated that miR-497-5p negatively regulated the proliferation and invasion of NSCLC cells by suppressing CX3CL1-mediated activation of the ERK/AKT pathway.
Figure 6.
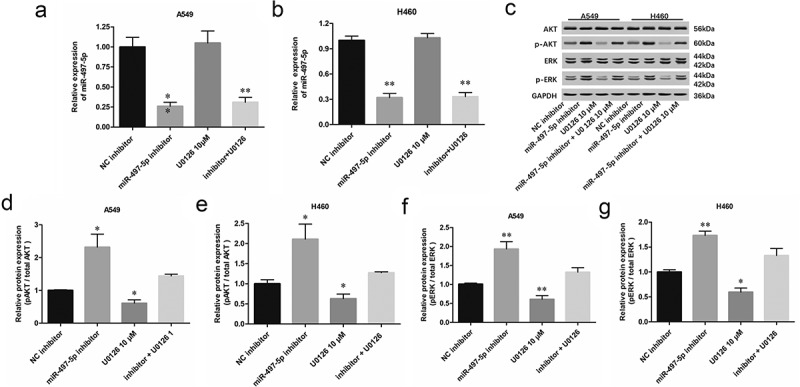
MiR-497-5p suppressed the phosphorylation of ERK and AKT. A549 or H460 cells were treated with miR-497-5p mimic (40 nM) or together with ERK inhibitor U0126 (10 µM) for 48 h. (a and b) Expression of miR-497-5p was detected with RT-qPCR. (c-g) The phosphorylation of AKT and ERK was observed by Western blotting analysis. Quantification of band intensity was quantified by ImageJ software, normalized to GAPDH. Statistical significance was determined with one-way ANOVA test. Values are expressed as means ± SEM, n = 5. * P < 0.05 and ** P < 0.01 vs. NC inhibitor.
Figure 7.
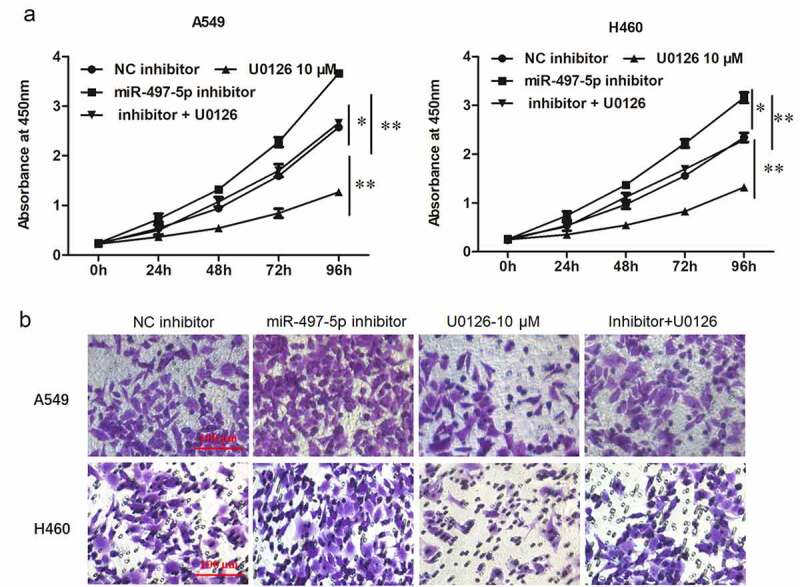
Inhibition of MiR-497-5p promoted cell growth and invasion in NSCLC cells, which could be abated by blockade of the ERK/AKT pathway. A549 or H460 cells were treated with miR-497-5p inhibitor (20 nM) alone or together with ERK inhibitor U0126 (10 µM) for 48 h. A. CCK-8 assay was used to detect cell viability. B. Transwell invasion assay was used to detect cell invasion. Statistical significance was determined with one-way ANOVA test. Values are expressed as means ± SEM, n = 5. * P < 0.05 and ** P < 0.01 vs. NC inhibitor.
Overexpression of miR-497-5p inhibited tumor growth in vivo
To confirm the regulatory role of miR-497-5p and ERK/AKT pathway in vivo, A549 cells were subcutaneously injected into the nude mice to generate patient-derived xenografts (PDX), and miR-497-5p mimic (2 mg/Kg) and NC mimic (2 mg/Kg) were, respectively, locally injected into the mice every 2 days. MiR-497-5p knockdown significantly suppressed tumor growth, evidenced by the diminished tumor volume (Figure 8(a,b)) and tumor weight (Figure 8c). Additionally, RT-qPCR assay revealed that miR-497-5p levels were lowered (Figure 8d). Consistent with the in vitro findings, down-regulation of miR-497-5p suppressed the levels of p-ERK, p-AKT, MMP-9 and MMP-2 in xenograft tumors (Figure 8(e,f)).
Figure 8.
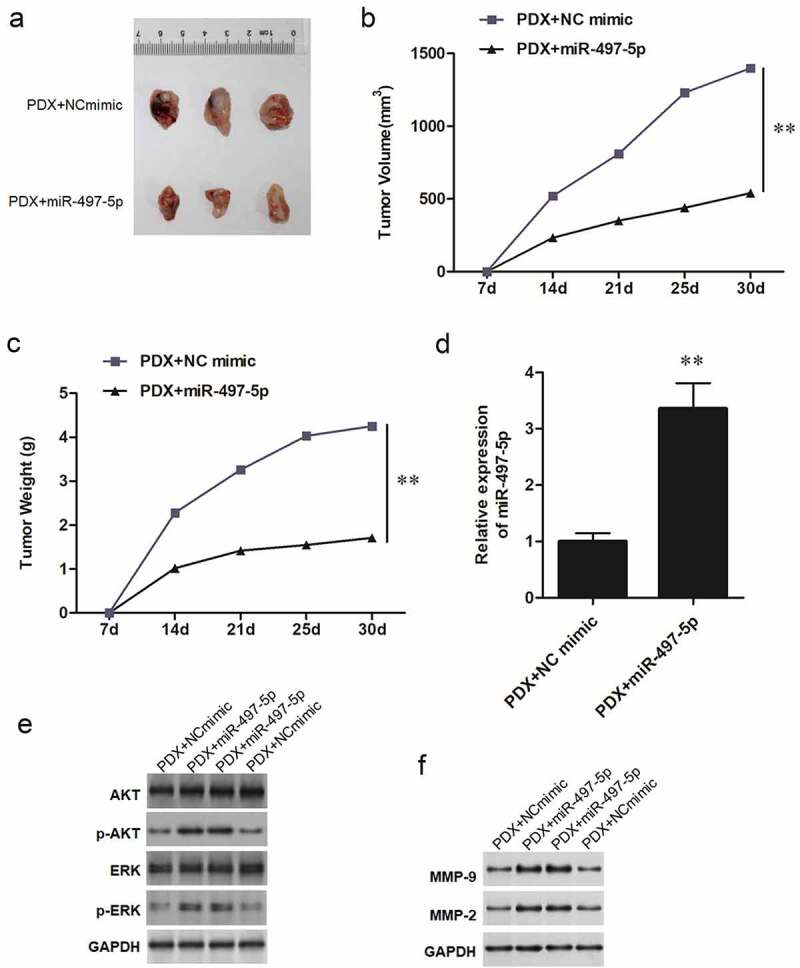
Overexpression of miR-497-5p inhibited tumor growth in vivo. A549 lung carcinoma suspension (containing 107 cells) was subcutaneously into C57BL/6 nude mice at right axilla to generate patient-derived xenografts (PDX). MiR-497-5p mimic (2 mg/Kg) and NC mimic (2 mg/Kg) were respectively locally injected into the mice every two days. After 30 days, the mice were euthanized, and the tumors were obtained. (a) Representative tumors from xenograft mice locally injected with miR-497-5p mimic or NC mimic respectively. (b) Average tumor volumes were measured in xenograft mice every 7 days. (c) Average tumor weight at the end of indicated treatment. (d) RT-qPCR was used to detect the expression of miR-497-5p in tumors. (e,f) expression of p-AKT, p-ERK, MMP-9 and MMP-2 proteins was measured by Western blot. Statistical significance was determined by using an independent sample t-test. Values are expressed as means ± SEM, n = 8. ** P < 0.01.
Discussion
MiRNAs play a pivotal role in many biologic processes, and are involved in the control of multiple molecular pathways leading to gene expression changes. During the past few years, a large body of evidence has shown that the dysregulation of miRNAs expression is associated with the carcinogenesis and development of malignant tumors and implicates the control of cell proliferation, migration and apoptosis [18,19]. Our studies focused on the involvement of miR-497-5p in NSCLC and its possible molecular mechanism. Our results showed that miR-497-5p was low expressed in NSCLC cells and regulated cell proliferation and invasion of NSCLC cells. Moreover, CX3CL1 was a target of miR-497-5p and was upregulated in lung cancer cells. Therefore, this study elucidated the important role of the miR-497-5p/CX3CL1 axis in NSCLC (Figure 7), and also provided a therapeutic target for NSCLC.
MiR-497-5p, an important cancer-associated miRNA, has been shown to be involved in numerous cellular processes including the control of cell proliferation, invasion and apoptosis in several cancers [10]. Growing evidence has demonstrated that miR-497-5p may act as a tumor suppressor. For instance, miR-497-5p inhibited colorectal cancer cell proliferation and invasion via targeting PTPN3 [11]. Additionally, it was also demonstrated that miR-497-5p may act as a tumor suppressor miRNA in lung cancer by targeting FGF2 [13]. In the last decade, miRNAs have emerged as biomarkers for diagnosis, prognosis, and prediction of response to treatment [20]. Researchers also found that miR-497-5p overexpression inhibited NSCLC cell proliferation, migration and invasion, and induced cell apoptosis through inhibiting SOX5 gene expression [21]. These findings corroborated with our study findings, which revealed that miR-497-5p might regulate cell proliferation and invasion of NSCLC. As our results showed, miR-497-5p was downregulated in NSCLC tissues and different cell lines, while upregulation of miR-497-5p inhibited the cell proliferation and invasion in NSCLC cells. To further understand the mechanism of miR-497-5p in NSCLC, we used bioinformatic tools to predict the downstream targets of miR-497-5p. As expected, luciferase reporter gene assays showed the combination of miR-497-5p and CX3CL1.
CX3CL1 can participate in a variety of physiological and pathological processes of diseases, such as renal diseases [22], allergic diseases [23] and chronic obstructive pulmonary disease [24]. For example, CX3CL1 exerted cytotoxic effects on the endothelium as well as anti-apoptotic and proliferative effects on vascular cells, affecting the context and stability of the atherosclerotic plaque [25]. It is observed that miR-125b directly regulated tumor cell-derived chemokine CSF1 and CX3CL1, which are known to control the recruitment of tumor-associated macrophages to tumor sites [26]. Similar to our experimental results, CX3CL1 can participate in chronic obstructive pulmonary disease [24]. For instance, CX3CL1 activates the proinflammatory pathway mediated by the transcription factor NF-κB as an early response and CX3CL1 induced several kinases such as MAPK’s in microglial cells [27]. Similarly, CX3CL1/CX3CR1-mediated microglial activation played a detrimental role in ischemic brain via p38/MAPK/PKC signaling [28]. CX3CL1 activates c-Raf, MEK, ERK, and NF-κB on the MMP-3 promoter through CX3CR1, thus contributing to cartilage destruction during Osteoarthritis [29]. And miR-195 exerted neuroprotective roles mainly through inhibiting CX3CR1-mediated neuroinflammation and subsequent neuronal cell apoptosis [30,31]. It is also indicated that histone deacetylases and NF-κB signaling coordinate epithelial expression of CX3CL1 to promote mucosal antimicrobial defense through suppression of the mir-424-5p and mir-503-5p 31. FKN stimulates angiogenesis by activating ERK/Akt signaling pathway through CX3CR1. CX3CL1 promotes MMP-3 production in osteoarthritic synovial fibroblasts through c-Raf, MEK and ERK signaling pathways. Finally, the relationship among miR-497-5p and CX3CL1 was explored, and it was found that miR-497-5p inhibited cell growth and invasion by targeting CX3CL1 through inactivating the ERK/AKT pathway in NSCLC cells. Furthermore, IL-8 production from lung cancer cells could be initiated by their own produced factors, leading to the recruitment of inflammatory cells in the cancer tissue, and the EGFR-PI3K-AKT-ERK pathway can be the potential target of therapies for inflammatory microenvironment in lung cancer [32]. Similarly, miR-7 was a common regulator of the PI3K/ATK and Raf/MEK/ERK pathways, both of which were launched by EGFR through its two direct targets, the transcription factors PI3K and Raf-1, respectively. Enforced expression of miR-7 markedly decreased expression of PI3K, phosphorylated AKT, Raf-1, phosphorylated MEK 1/2, and cyclin D1, as well as slightly reduced expression of EGFR [33].
In conclusion, miR-497-5p is downregulated in human NSCLC tissues and cell lines, and it inhibited tumor growth and cell invasion by targeting CX3CL1 gene to inactivate the ERK/AKT pathway in NSCLC cells. The miR-497-5p/CX3CL1 axis played a crucial role in the pathogenesis of NSCLC, and it also provided a potential therapeutic approach for NSCLC patients.
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Ethics approval
This study was approved by the Ethics Committee of Xi’an Central Hospital (Approval number: XACH-2019027).
References
- [1].Torre LA, Siegel RL, Jemal A.. Lung cancer statistics. Adv Exp Med Biol. 2016;893:1–19. [DOI] [PubMed] [Google Scholar]
- [2].Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. [DOI] [PubMed] [Google Scholar]
- [3].Zakaria N, Satar NA, Abu Halim NH, et al. Targeting lung cancer stem cells: research and clinical impacts. Front Oncol. 2017;7:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Travis WD, Brambilla E, Riely GJ. New pathologic classification of lung cancer: relevance for clinical practice and clinical trials. J Clin Oncol. 2013;31(8):992–1001. [DOI] [PubMed] [Google Scholar]
- [5].Alcala N, Leblay N, Gabriel AAG, et al. Integrative and comparative genomic analyses identify clinically relevant pulmonary carcinoid groups and unveil the supra-carcinoids. Nat Commun. 2019;10(1):3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Leclercq M, Diallo AB, Blanchette M. Computational prediction of the localization of microRNAs within their pre-miRNA. Nucleic Acids Res. 2013;41(15):7200–7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Majd SM, Salimi A, Ghasemi F. An ultrasensitive detection of miRNA-155 in breast cancer via direct hybridization assay using two-dimensional molybdenum disulfide field-effect transistor biosensor. Biosens Bioelectron. 2018;105:6–13. [DOI] [PubMed] [Google Scholar]
- [8].Das R, Gregory PA, Fernandes RC, et al. MicroRNA-194 promotes prostate cancer metastasis by inhibiting SOCS2. Cancer Res. 2017;77(4):1021–1034. [DOI] [PubMed] [Google Scholar]
- [9].Choi KH, Shin CH, Lee WJ, et al. Dual-strand tumor suppressor miR-193b-3p and −5p inhibit malignant phenotypes of lung cancer by suppressing their common targets. Biosci Rep. 2019;39(7). DOI: 10.1042/BSR20190634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hill M, Tran N. MicroRNAs regulating microRNAs in cancer. Trends Cancer. 2018;4(7):465–468. [DOI] [PubMed] [Google Scholar]
- [11].Hong S, Yan Z, Wang H, et al. Up-regulation of microRNA-497-5p inhibits colorectal cancer cell proliferation and invasion via targeting PTPN3. Biosci Rep. 2019;39(8). DOI: 10.1042/BSR20191123 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [12].Feng L, Cheng K, Zang R, et al. miR-497-5p inhibits gastric cancer cell proliferation and growth through targeting PDK3. Biosci Rep. 2019;39(9). DOI: 10.1042/BSR20190654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Huang X, Wang L, Liu W, et al. MicroRNA-497-5p inhibits proliferation and invasion of non-small cell lung cancer by regulating FGF2. Oncol Lett. 2019;17(3):3425–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Su YC, Chang H, Sun SJ, et al. Differential impact of CX3CL1 on lung cancer prognosis in smokers and non-smokers. Mol Carcinog. 2018;57(5):629–639. [DOI] [PubMed] [Google Scholar]
- [15].Ferretti E, Pistoia V, Corcione A. Role of fractalkine/CX3CL1 and its receptor in the pathogenesis of inflammatory and malignant diseases with emphasis on B cell malignancies. Mediators Inflamm. 2014;2014:480941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cardoso AL, Fernandes A, Aguilar-Pimentel JA, et al. Towards frailty biomarkers: candidates from genes and pathways regulated in aging and age-related diseases. Ageing Res Rev. 2018;47:214–277. [DOI] [PubMed] [Google Scholar]
- [17].Liu WM, Bian C, Liang Y, et al. CX3CL1: a potential chemokine widely involved in the process spinal metastases. Oncotarget. 2017;8:15213–15219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang Y, Yang Q, Wang S. MicroRNAs: a new key in lung cancer. Cancer Chemother Pharmacol. 2014;74(6):1105–1111. [DOI] [PubMed] [Google Scholar]
- [19].Kunz M, Pittroff A, Dandekar T. Systems biology analysis to understand regulatory miRNA networks in lung cancer. Methods Mol Biol. 2018;1819:235–247. [DOI] [PubMed] [Google Scholar]
- [20].Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29(4):452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li G, Wang K, Wang J, et al. miR-497-5p inhibits tumor cell growth and invasion by targeting SOX5 in non-small-cell lung cancer. J Cell Biochem. 2019;120(6):10587–10595. [DOI] [PubMed] [Google Scholar]
- [22].Zhuang Q, Cheng K, Ming Y. CX3CL1/CX3CR1 axis, as the therapeutic potential in renal diseases: friend or Foe? Curr Gene Ther. 2017;17(6):442–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Julia V. CX3CL1 in allergic diseases: not just a chemotactic molecule. Allergy. 2012;67(9):1106–1110. [DOI] [PubMed] [Google Scholar]
- [24].Hao W, Li M, Zhang C, et al. High Serum Fractalkine/CX3CL1 in patients with chronic obstructive pulmonary disease: relationship with emphysema severity and frequent exacerbation. Lung. 2019;197(1):29–35. [DOI] [PubMed] [Google Scholar]
- [25].Apostolakis S, Spandidos D. Chemokines and atherosclerosis: focus on the CX3CL1/CX3CR1 pathway. Acta Pharmacol Sin. 2013;34(10):1251–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Batool A, Wang YQ, Hao XX, et al. A miR-125b/CSF1-CX3CL1/tumor-associated macrophage recruitment axis controls testicular germ cell tumor growth. Cell Death Dis. 2018;9(10):962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Galan-Ganga M, Garcia-Yague AJ, Lastres-Becker I. Role of MSK1 in the Induction of NF-kappaB by the Chemokine CX3CL1 in Microglial Cells. Cell Mol Neurobiol. 2019;39(3):331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ma B, Jing R, Liu J, et al. Gremlin is a potential target for posterior capsular opacification. Cell cycle (Georgetown, Tex) 2019:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hou SM, Hou C-H, Liu J-F. CX3CL1 promotes MMP-3 production via the CX3CR1, c-Raf, MEK, ERK, and NF-κB signaling pathway in osteoarthritis synovial fibroblasts. Arthritis Res Ther. 2017;19(1):282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yang G, Liu Z, Wang L, et al. MicroRNA-195 protection against focal cerebral ischemia by targeting CX3CR1. J Neurosurg. 2018;1–10. 10.3171/2018.5.JNS173061. [DOI] [PubMed] [Google Scholar]
- [31].Zhou R, Gong AY, Chen D, et al. Histone deacetylases and NF-kB signaling coordinate expression of CX3CL1 in epithelial cells in response to microbial challenge by suppressing miR-424 and miR-503. PLoS One. 2013;8(5):e65153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang Y, Wang L, Zhang M, et al. Potential mechanism of interleukin-8 production from lung cancer cells: an involvement of EGF–EGFR–PI3K–Akt–Erk pathway. J Cell Physiol. 2012;227(1):35–43. [DOI] [PubMed] [Google Scholar]
- [33].Liu Z, Jiang Z, Huang J, et al. miR-7 inhibits glioblastoma growth by simultaneously interfering with the PI3K/ATK and Raf/MEK/ERK pathways. Int J Oncol. 2014;44(5):1571–1580. [DOI] [PubMed] [Google Scholar]