

Abstract
Oligodendrocytes and their myelin sheaths play an intricate role in axonal health and function. The role of white matter pathology in a wide variety of central nervous system disorders has gained attention in recent years. Remyelination has therefore become a major target of therapeutic research, with the aim of protecting axons from further damage. The axon-myelin unit is elaborate, and demyelination causes profound changes in axonal molecular domains, signal transmission and metabolism. Remyelination is known to restore some of these changes, but many of its outcomes remain unknown. Understanding how different aspects of the axon-myelin unit are restored by remyelination is important for making effective, targeted therapeutics for white matter dysfunction. Additionally, understanding how subtle deficits relate to axonal function during demyelination and remyelination may provide clues into the impact of myelin on neuronal circuits. In this review, we discuss the current knowledge of the neuroprotective effects of remyelination, as well as gaps in our knowledge. Finally, we propose systems with unique myelin profiles that may serve as useful models for investigating remyelination efficacy.
Keywords: Oligodendrocytes, axon, demyelination
Introduction
Myelin is a specialized membrane produced by oligodendrocytes in the central nervous system (CNS), which ensheaths neuronal axons. Dr. Jean de Vellis’ group has studied numerous aspects of glial biology over more than 40 years, but one of the most impactful studies done by this group was the development of tissue culture protocols that allow us to study oligodendrocyte development and myelin membrane production. For many years, much of what we learned about CNS myelination came from in vitro studies focused on understanding how oligodendrocytes develop in isolation. The McCarthy and de Vellis protocol (1980) for culturing rat oligodendrocytes shaken and purified initially from mixed glial cultures has been a standard in hundreds of research laboratories since the early 1980s. It allowed the field to learn about some of the unique regulators of oligodendrocyte proliferation and differentiation, both extracellular regulators and intracellular mediators (Hardy and Reynolds 1991, Baron et al. 2000, Vela et al. 2002). Recently, such protocols have provided a platform for screening for molecules that can enhance differentiation of oligodendrocytes, hopefully for new therapeutic approaches for remyelination following demyelination (Zhang et al. 2011, Yuen et al. 2013, Rittchen et al. 2015).
The evolutionary advantages of having glial cells produce an ensheathing membrane for axons have been substantial. By establishing tightly organized molecular domains along the axon and spatially restricting ion exchange with the extracellular space, myelin sheaths dramatically increase the rate of long-range information flow in the brain. In addition to facilitating saltatory conduction, oligodendrocytes have been implicated in other crucial CNS functions, such as maintaining ion homeostasis (Menichella et al. 2006) and providing trophic/metabolic support to axons (reviewed in Morris et al 2013).
Myelin loss is a hallmark of developmental disorders such as leukodystrophies and of immune-mediated disorders such as multiple sclerosis (MS) (Kutzelnigg et al. 2005, Lassmann et al. 2012), but white matter damage has been implicated in a number of neurological conditions, including stroke (Dewar et al. 2003), Parkinson’s (Bohnen and Albin 2011, Hattori et al. 2012), schizophrenia (Whitford et al. 2012, Kubicki et al. 2009), and normal and pathological aging (Guttmann et al. 1998, Peters and Sethares 2002, reviewed in Bartzokis 2004). The link between myelin and axon health has driven interest in remyelination as a therapeutic target for neuroprotection, and a number of studies have investigated ways to promote de novo myelin generation in injury and disease models (reviewed in Dubois-Dalcq et al. 2005, Kremer et al. 2016). Despite this focus, we still know little about the degree to which remyelination restores axonal health and functional signal transduction. While remyelination has been shown to restore several measures of function, it does not fully recapitulate developmental myelination. Understanding the benefits and limitations of remyelination is crucial for quantifying the efficacy of potential remyelination therapeutics, and for integrating them into treatment of neurological disorders featuring myelin loss.
This review focuses on the current knowledge of outcomes following remyelination. Strategies for facilitating remyelination in different pathological contexts have been reviewed elsewhere (Gallo and Armstrong 2008, Rodgers et al. 2013, Franklin and Goldman 2015). Instead, we will discuss axon pathology in demyelination, as well as the known benefits and shortcomings of remyelination. Finally, we propose systems that lend themselves well to investigating the effectiveness of signal transduction during demyelination and remyelination.
Benefits of myelin
A primary function of oligodendrocytes is to facilitate fast, reliable electrical conduction in projection axons. The impact of myelin on axon electrical properties is two-fold. By increasing resistance along internodes, myelin prevents ion leakage, facilitating reliable spikes. Myelin also funnels ion exchange to a very small surface area at the node of Ranvier, decreasing the current, and thus the time, required to depolarize the membrane and generate the action potential. These properties have a profound impact on conduction speed, which is proportional to axon diameter for myelinated axons, but proportional to its square root for unmyelinated axons (Waxman and Bennett 1972, Rushton 1951). Myelin enables effective axonal transmission by establishing and maintaining the intricate molecular architecture of both the myelin sheath itself and the axon. The development of these membrane domains relies on bidirectional communication between myelinating oligodendrocytes and axons (reviewed in Rasband and Peles 2016), and it generates an interacting axo-myelin complex with several specialized myelin and axon domains, consisting of nodes of Ranvier, paranodes and paranodal junctions, juxtaparanodes, and internodes.
Axonal domains
Nodes of Ranvier (Figure 1) are ~1-μm wide gaps between adjacent myelin sheaths, which have a high density of voltage-activated Na+ (Nav) channels necessary for action potential generation (Lodish et al. 2000). Prior to myelination, axons express Nav 1.2 and 1.6 along their length. During myelination, both the expression and redistribution of Nav channels change: Nav1.2 expression is reduced as Nav1.6 localizes to nodes of Ranvier, restricting sites of ion exchange (Kaplan 2001). Each instance of axolemmal ion exchange takes time and requires re-establishment of ionic gradients by ATP-dependent ion pumps. Nodal organization thus speeds action potential conduction and reduces its associated energy costs.
Figure 1: Nodal alterations following demyelination and remyelination.
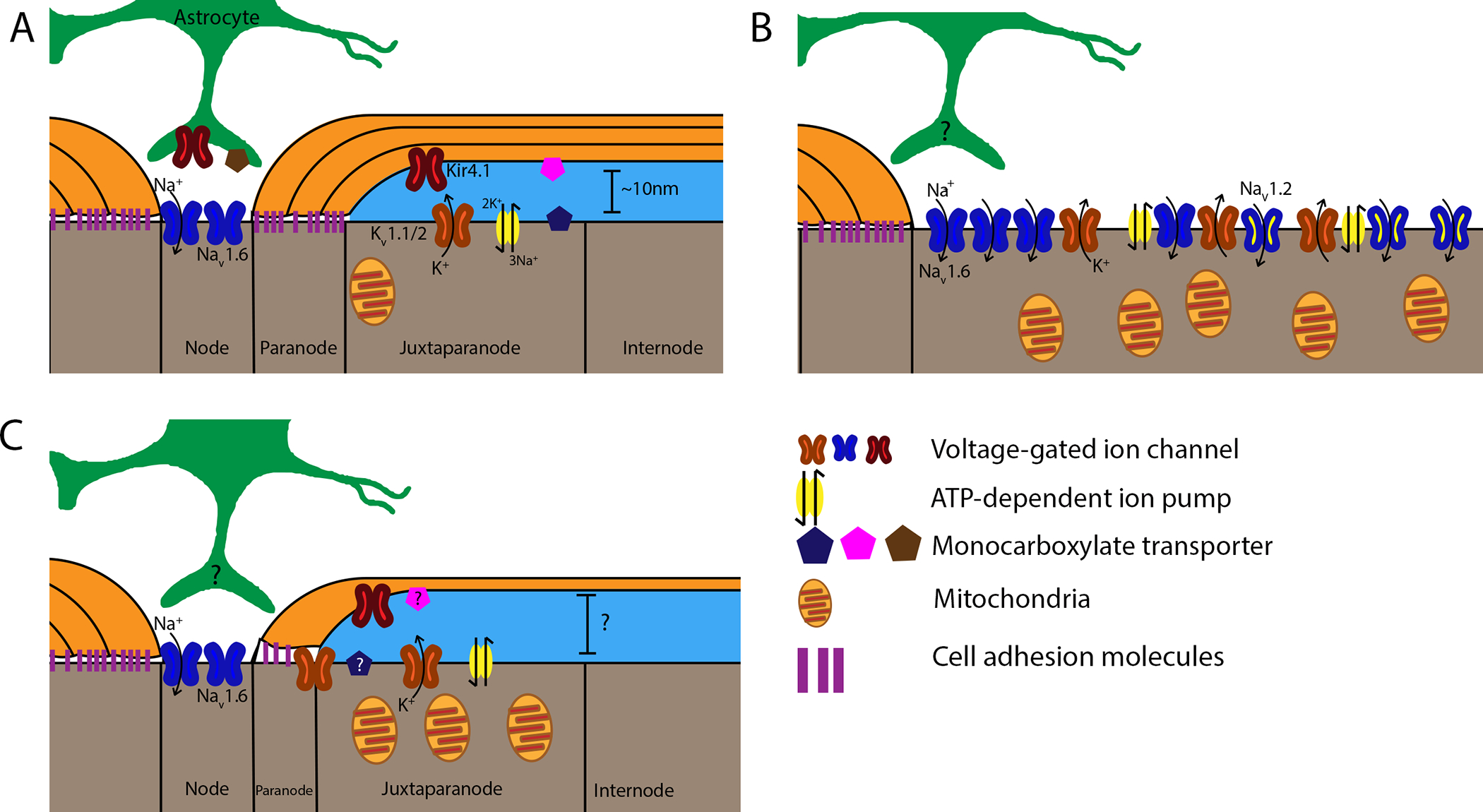
Healthy axo-myelin units are displayed on the left of each panel for comparison. A: In healthy myelinated axons, voltage-gated sodium channels (Nav1.6, blue) are located at the node of Ranvier, while voltage-gated potassium channels (Kv1.1,2, orange), ATP-dependent Na+/K+ pumps (yellow), and mitochondria localize to the juxtaparanode. Inward-rectifying K+ channels (Kir4.1, red) are expressed by both oligodendrocytes and astrocytes, which extend perinodal processes that contribute to node function. Different monocarboxylate transporters (MCTs) are expressed by oligodendrocytes (MCT1, pink), axons (MCT2, blue), and astrocytes (MCT4, brown), indicating an important role for lactate metabolism. B: Loss of paranodal junctions (purple) due to demyelination results in Kv channel diffusion along the axolemma. Nav clusters also diffuse, and axons begin expressing Nav1.2 along the former internode. This increases the area across which ion gradients must be maintained, causing broad expression of Na+/K+ pumps and increasing mitochondrial density to meet axonal energy demands. Axonal MCT expression is reduced, potentially to compensate for high extracellular lactate levels. C: Following remyelination, ion channel and pump localization is restored, with some Kv channels persisting in the paranodal region. Mitochondrial density remains elevated, while the expression of MCTs during this period is unknown. Notably, the presence or absence of perinodal astrocyte processes during demyelination and remyelination has yet to be elucidated. Other morphological factors of remyelinated axons, such as the size of the periaxonal space, also remain unknown.
Nodes are bordered by the paranodal loops of the myelin sheath, which form septate-like junctions with the paranodal axolemma. The paranode separates nodal Nav channels from juxtaparanodal shaker K+ (Kv) channels (reviewed in Girault et al. 2002). The paranode not only serves as a barrier to lateral channel diffusion, but also limits diffusion of K+ into nodes of Ranvier. This focal distribution facilitates K+ buffering by oligodendrocytes (Menichella 2006) and astrocytes (Neusch et al. 2006), both of which express inward rectifying K+ channels and may cooperate in K+ homeostasis.
Oligodendrocyte support of axons
Oligodendrocytes have also been implicated in providing trophic support unrelated to myelin structure. Studies of several myelin protein mutants provide support for this concept. For example, mouse deletion mutants of the myelin proteolipid protein gene (Plp−/−, Griffiths et al. 1998) or the 2’,3’-cyclic nucleotide 3’ phosphodiesterase gene (Cnp−/−, Lappe-Siefke et al. 2003) develop axonal pathology despite the absence of gross myelin abnormalities. New techniques may provide evidence of subtle structural deficits in these mice, which may not have been apparent in previous studies. For example, Cnp was recently shown to play a role in preventing premature myelin compaction (Snaidero et al. 2014), suggesting that CNP-deficient mutants may have myelin deficits that occur during development and contribute to long-term axonal pathology as the animal ages. No similar role has been found for PLP, but the loss of a quantitatively major component of the myelin sheath likely impacts myelin membrane structure and function.
Action potential propagation is an energy intensive process that requires restoration of the electrochemical gradient following depolarization at nodes of Ranvier. These energy demands must be met at sites as much as several centimeters or more from the neuronal cell body. Local glia have been proposed to play a role in meeting axonal metabolic needs. Early studies suggested that aerobic glycolysis in astrocytes produces lactate as a byproduct (Pellerin and Magistretti 1994), which can then be released through the monocarboxylate transporter 4 (MCT4) and metabolized by neurons expressing the high-affinity MCT2 (Suzuki et al. 2012). More recent findings suggest similar metabolic coupling between neurons and neighboring oligodendrocytes (Funfschilling et al. 2012). Oligodendrocytes strongly express MCT1, and both genetic and shRNA-mediated MCT1 loss induce axonal pathology without obvious oligodendrocyte damage (Lee et al. 2012). However, further research of lactate dynamics between neurons, astrocytes, and oligodendrocytes is necessary to understand its role in axonal function during different levels of activity.
Impact of Demyelination
Myelin loss is a common feature of a number of disorders and brain injuries. Demyelination results in a breakdown of tightly controlled axonal molecular domains and cellular interactions (Coman et al. 2006), which can profoundly compromise communication between distributed brain regions. Furthermore, prolonged demyelination has been associated with axonal damage and eventual degeneration, which is thought to underlie long-term functional deficits in disorders such as MS (De Stefano et al. 1998, Trapp et al. 1998, Kornek et al. 2000). Demyelination and axonal dysfunction go hand-in-hand, but it can be difficult to prove a causal relationship, as primary axonal damage must be definitively ruled out. This is especially complicated when demyelination occurs in a context of oxidative stress or inflammation, which likely induce both oligodendrocyte and axon responses. Nevertheless, dysmyelinating mutants, toxin-induced myelin damage, and genetic oligodendrocyte ablation have given us insights into the effects of myelin dysfunction on axonal function in a number of pathological contexts. Perhaps the most immediate consequence of demyelination is a loss of the molecular domains at and surrounding the node of Ranvier, which has profound consequences for ion channel distribution and saltatory conduction (Volman and Ng 2014, reviewed in Susuki 2013).
Axonal conduction changes after demyelination
Axonal signal propagation relies on a complex intersection of morphological, electrical, and chemical factors (reviewed in Bucher and Goaillard 2011), and demyelination can have a number of important effects on action potential propagation. Spike failure is the most obvious symptom of demyelination, and is associated with many of the negative symptoms in MS (Smith 1994). Increased capacitance causes action potentials entering the demyelinated region to more quickly deplete the local ionic gradients necessary for conduction, and action potentials slow, which can induce failure of subsequent spike propagation during bursting activity. Demyelination can also induce aberrant electrical activity, which manifest either as spikes that occur without stimulation, or as bursts of activity that outlast stimulation. Na+ can mediate small membrane potential oscillations that contribute to ectopic spikes (Kapoor et al. 1997), potentially due to increased expression of Nav1.2 channels (Majumdar and Sikdar 2005). On the other hand, spike reflection can occur at the transition from myelinated to demyelinated regions. In myelinated axons, backward spike propagation is prevented by Nav channel inactivation in the previous node. Demyelinated regions depolarize much more slowly, allowing propagation to previous nodes whose refractory period has ended. Reflected spikes can shunt forward propagating spikes, but may also induce persistent ectopic spiking (Zlochiver 2010).
In addition to the intrinsic axonal conduction abnormalities above, demyelination can cause pathological intercellular signaling between naked axons, known as ephaptic signaling. In myelinated axons, the electrical environment is tightly controlled, with Kv channels sequestered beneath the myelin sheath, and the node surrounded by adjacent sheaths and astrocytic processes (Figure 1A). Demyelination exposes these ionic currents to the extracellular space, allowing activity in one axon to induce failed, ectopic, and reflected spikes in another (Nielsen 2004). Ephaptic signaling may underlie the seizures and diverse paroxysmal attacks suffered by individuals with MS (Osterman and Westerberg 1975, Sokić et al. 2008).
Ion channel redistribution in demyelinated axons
Many of these changes result from specific redistribution of ion channels (Figure 1B). As the initiators of action potential generation, Nav channels play an important role in axonal responses to demyelination. Nav channel clustering is influenced by the presence of oligodendrocytes but does not rely on compact myelin (Ishibashi et al. 2003, Dupree et al. 2004), and clusters can persist to a lesser degree in demyelinated regions (Coman et al., 2006). During early stages of demyelination, nodal Nav clusters elongate (Doppler et al. 2013), which can dramatically impact conduction parameters (Babbs and Shi 2013). Meanwhile, Nav channels spread to axonal domains of former internodes, resulting in a distribution similar to unmyelinated axons. During the spontaneous demyelination that occurs in adult mice overexpressing PLP, Nav1.2 expression increases (Rasband et al. 2003). This may be a compensatory change, as Nav1.6 can reopen during prolonged activity, causing persistent currents (Chatelier et al. 2010). Because demyelination results in larger ion flux during activity, these persistent currents could potentially deplete the ionic gradients and even reverse Na+/Ca2+ pumps, causing Ca2+ influx and secondary axonal damage (Stys et al. 1992, reviewed in Waxman 2006).
Unlike Nav channels, Kv channel localization appears to depend directly on the presence of compact myelin (Baba et al. 1999). Normally isolated from the extracellular space by the myelin sheath, Kv channels laterally diffuse into nodes and internodes following demyelination. Kv channel expression also changes with demyelination, and enrichment of Kv1.1 subunits contribute to a lower voltage activation of hyperpolarizing K+ currents (Bagchi et al. 2014). Increased capacitance results in increased K+ efflux, and may contribute to extracellular accumulation in conjunction with impaired glial K+ buffering. These changes in the ionic environment can make axons silent or hyperexcitable, depending on the pathological context.
Metabolic changes in demyelinated axons
The redistribution of ion channels during demyelination dramatically alters the metabolic demands of signal propagation. Increased movement of Na+ and K+ during action potentials requires increased activity of ATP-dependent Na+/K+ pumps to maintain the electrochemical gradients necessary for depolarization. This change in energy demand is associated with mitochondrial accumulation that matches that of healthy unmyelinated axons (Mutsaers and Carroll 1998). Importantly, a number of changes that affect lactate dynamics occur in MS, including an increase in CSF lactate concentrations (Albanese et al. 2016), which may be mediated by infiltrating macrophages (López-Villegas et al. 1995). Demyelinated lesions exhibit changes in astrocytic and neuronal expression of lactate-permeable monocarboxylate transporters (Nijland et al. 2014), which as noted above could impact energy status in axons. These changes may be due to impaired mitochondrial function in axons, or be an adaptive response to elevated lactate by which astrocytes increase and axons decrease uptake, but these mechanisms and their consequences remain unclear.
Response of demyelinated axons to secondary injury
Demyelination seldom occurs in isolation, and often occurs in the context of inflammation and oxidative stress. The ability of demyelinated axons to cope with these factors is therefore an important component of axonal pathology. The axonal dysfunction of PLP−/− and CNP−/− mice in the absence of other insults suggests that demyelinated axons would be a vulnerable population, but so far few injury or disease models have been applied to these mutants. As expected, focal demyelination increases axonal susceptibility to some stressors: Nitric oxide, which plays a role in inflammatory immune response and is elevated in MS, preferentially blocks conduction in demyelinated CNS axons (Redford et al. 1997). However, both amyelinated (Waxman et al. 1990) and demyelinated (Imaizumi et al. 1998) axons appear more resistant to action potential failure following in vitro anoxia and reperfusion. These electrophysiological studies did not measure the long-term health of myelinated and demyelinated axons under these different conditions. However, this differential susceptibility of demyelinated axons underlines the complex changes that occur during demyelination, whose impact likely depends heavily on the surrounding pathological context. Understanding these dynamics may inform windows of opportunity for neuroprotective therapies following demyelination.
Benefits of Remyelination
The dramatic impact of demyelination on long-range neuronal signaling, and its prevalence in a wide variety of injury and disease models, has inspired extensive research into facilitating remyelination as a neuroprotective therapy. While some of the benefits of remyelination have been clearly demonstrated, there are still many unanswered questions regarding axo-myelin interactions, and their functional consequences, during this period.
The efficacy of remyelination in restoring axonal health and function likely depends on the amount of time axons spend in an unmyelinated state. Notably, remyelination cannot proceed in the presence of myelin debris. Oligodendrocytes produce massive amounts of myelin: A single oligodendrocyte is estimated to produce five hundred to several thousand times the surface area of its soma in myelin membrane (Pfeiffer et al. 2003). Myelin decompaction thus results in large amounts of myelin debris, which inhibits differentiation of recruited oligodendrocyte precursors (Robinson et al. 1999, Kotter et al. 2006). Remyelination relies on debris clearance by recruitment of microglia (Lampron et al. 2015) and macrophages (Kotter et al. 2001).
Restoration of nodal architecture
Extensive pathological analysis of MS lesions as early as the 1960s identified remyelinated axons, which have shorter, thinner internodes than healthy individuals (Périer and Grégoire 1965, Prineas et al 1984). Perhaps the most obvious readout of functional remyelination is the degree to which axonal molecular domains and associated saltatory conduction are restored. Functionally, remyelination in animal models is associated with mitigation of behavioral deficits following demyelination (Mayer 1971, Jeffery and Blakemore 1997, Duncan et al. 2009), which has been linked to increased conduction velocity (Mayer 1971) and restoration of nodal structure (Sasaki 2006) (Figure 1C). This has been corroborated in human MS brains: demyelinated lesions are characterized by diffusion of Nav and Kv channels, while remyelinated shadow plaques exhibit a restoration of paranodal channel separation (Coman et al. 2006). These changes alone indicate a neuroprotective role for remyelination due to the greatly reduced metabolic cost of saltatory conduction. However, it is important to note that we know little about the time needed to re-establish proper channel domains following remyelination in the human CNS. Observations in the rodent PNS after lysolethicin injections indicate that Kv channels still remain in nodes of Ranvier a month after remyelination initiates (Rasband et al. 1998). Such slow changes in ion channel distribution raise questions regarding the stability of signal transmission during active remyelination, which has been proposed as a period of increased axonal vulnerability (Smith et al. 2006). Axonal adaptations may be particularly relevant when demyelination and remyelination are recurrent or chronic occurrences that induce continuous flux of ion channels.
Axonal metabolism
Can remyelination impact axonal metabolism by mechanisms other than node restoration? Notably, the increased mitochondrial density and distribution following demyelination appear to persist in remyelinated axons (Mutsaers and Carroll 1998), but the functional implications of this persistence are unknown. No studies to date have directly investigated lactate metabolism following remyelination, but the steep metabolic demands of myelin biogenesis may shift oligodendrocytes toward high lactate utilization, which has been reported in vitro (Sánchez-Abarca 2001). Thus, oligodendrocytes may not be able to support axonal lactate metabolism until they have switched from active remyelination to myelin maintenance. Astrocytic lactate shuttling during or after remyelination is also unclear, as there is little information about the presence of perinodal astrocyte processes at newly formed nodes following injuries. The metabolic environment of remyelination is further complicated by changes in lactate transport by monocarboxylate transporters (MCTs) that occur during demyelination, where MCT2 normally expressed by axons is downregulated in inactive lesions (Nijland et al. 2014). The distribution of MCTs has not been studied in remyelination, which presents a major gap in our knowledge of the impact of remyelination on axonal health.
Neuroprotection by remyelination
A crucial question is whether axonal degeneration following demyelination can be mitigated by remyelination. Irvine et al. (2008) investigated this question by exposing the corpus callosum of mice to X-irradiation in the third week of cuprizone treatment. X-irradiation killed local cells while sparing axons, thus compromising local remyelinating capacity. Axonal degeneration and spheroids were more severe in X-irradiated mice compared to controls that were only fed cuprizone. Notably, this damage was diminished by grafting neural stem cells, which differentiated into oligodendrocytes and remyelinated callosal axons (Irvine et al. 2008). This was one of the first studies to establish sufficiency of remyelination in preventing axonal pathology following demyelination, but the mechanisms are still unknown. Possibilities include restoration of channel distribution, physical separation of the axolemma from the extracellular environment, and restoration of metabolic support. The close relationships among these factors makes it particularly hard to parse out their specific contributions to axonal protection. Moreover, delayed axonal pathology can occur despite remyelination after cuprizone-induced demyelination, which may reflect subtle alterations in the ability of newly formed myelin to support axonal function (Manrique-Hoyos et al. 2012). Nevertheless, the methods employed by Irving and coworkers may provide a valuable system for examining the impact of remyelination on several aspects of axonal function such as axonal transport and mitochondrial distribution.
The ability of remyelination to prevent axonal damage is an exciting prospect for therapeutics, but what about axons that are already damaged? Progressive axonal damage can follow or precede demyelination and likely underlies many of the long-term behavioral deficits seen in disorders that injure white matter. Axonal swelling can remain stable over time and has been shown to reverse spontaneously in the experimental autoimmune encephalomyelitis (EAE) model of MS (Nikić et al. 2011), but it is not known whether oligodendrocytes can contribute to recovery. Early observations in human MS and mouse EAE tissue suggest that axonal damage is prevalent in demyelinated lesions and absent in remyelinated shadow plaques (Kornek et al. 2000). This may indicate axon protection by remyelinating oligodendrocytes, but it is also possible that remyelination can only take place at sites of minimal axonal damage. Further studies are necessary to dissociate these possibilities, which will be important for determining therapeutic targets.
Thin myelin sheaths were one of the first recorded characteristics of remyelination, which may have important consequences for signal transduction and metabolic support. Exciting findings from the Bansal and Fyffe-Maricich laboratories indicate a potential target for increasing myelin thickness. Constitutive activation of Mek1 and the consequent increased ERK1/2 MAP activity in the oligodendrocyte lineage results in faster remyelination and thicker sheaths following lysolethicin-induced demyelination (Ishii 2012; Fyffe-Maricich 2013). This provides a fascinating therapeutic target, but must be approached carefully. Mice that overexpress constitutively active Akt in oligodendrocytes (Akt-DD) are characterized by hypermyelination. At young ages, these mice exhibit faster-latency visually evoked potentials than WT controls (Yu et al. 2011). As they age, however, Akt-DD mice exhibit delayed latencies correlated with myelin abnormalities. Furthermore, myelin continues to accumulate in these mice, eventually becoming pathologic, as they die around one year of age (Flores et al., 2008). Clearly modulating signaling pathways to regulate myelin thickness would need to be paired with electrophysiological and behavioral measures to characterize the effects of myelin thickness on remyelinated axons, and the regulation of myelin thickness would need to be tightly controlled.
Myelin plasticity: implications for remyelination and circuit function
The role of myelination in higher brain function is still poorly understood. However, as we gain a greater appreciation for the role of temporally precise oscillatory and synchronous neuronal activity in complex cognitive functions, it becomes more important for us to understand the axo-myelin unit as a controller of signal transmission timing in health and disease. In recent years it has become clear that myelination is not an all-or-none, uniform process, and myelin plasticity and heterogeneity may have important functional consequences for signal propagation and circuit function.
Even in the adult, new myelin can be generated, and recent studies suggest that axonal activity can drive that production of new myelin in the adult (Gibson et al. 2014). Additionally, some cortical axons are discontinuously myelinated, with large unmyelinated sections interspersed between myelinated segments in a layer-specific pattern (Tomassy et al. 2014). Remarkably, these unmyelinated regions appear to contain synaptic connections, suggesting a functional role for discontinuous myelin. The composition of these unmyelinated axon segments in discontinuously myelinated axons is unknown, and it will be fascinating to discover how they facilitate signal transmission, and whether they represent a site of myelin remodeling. Discontinuous myelination also presents an excellent opportunity to explore another dimension of remyelination efficacy. Cortical remyelination is known to occur in MS patients (Albert et al. 2007, Chang et al 2012), but the degree to which the channel composition of the unmyelinated regions and the relative laminar myelin patterns change during myelin injury and recovery is currently unknown.
Return to near-normal conduction velocity is an important measure of remyelination success. In order to better understand the functional impact of remyelination on neuronal communication, we must integrate the recovery of circuits with specific myelin profiles, transmission requirements, and behavioral correlates. To some extent, such measures are most detectable for sensory signaling to the CNS. Clearly synchrony of neuronal firing is essential for olfactory detection (reviewed in Mori et al. 2013), and altered conduction latencies can contribute to visual deficits (Wist et al. 1978, reviewed in Balcer et al. 2014). A particularly important readout of circuit restoration is the ability of remyelinated axons to mediate temporally specific neuronal signaling. Problems in temporal synchrony of neuronal firing are particularly clear with MS patients, who often have trouble with sound localization, even in the absence of gross auditory deficits (Cranford et al. 1990, reviewed in Furst and Levine 2015). Localization of low-frequency sounds is mediated by interaural time differences (ITD), which are computed by microsecond resolution coincident detection in the auditory brainstem, and thus rely on tight control of conduction timing. Signaling from the ear is mediated by neurons with bifurcating axons that project to both sides of the medial superior olive. Contralateral action potentials must therefore travel over twice as far as ipsilateral projections to reach their target, clearly a challenge to coincident detection. Seidl and Rubel (2016) recently reported that contralateral and ipsilateral projections are differentially myelinated in gerbils. Contralateral branches established longer internodes than ipsilateral branches prior to hearing onset, which was followed by an increase in axonal diameter. This heterogeneous myelin profile for individual axons is likely an important factor in ITD detection, and could be a powerful system for investigating circuit function in relation to demyelination and remyelination. An important question is whether after demyelination the axo-myelin unit can adjust to restore the behaviorally relevant interaural circuit function. Myelination by engrafted OPCs can restore gross auditory latencies in the hypomyelinated shiverer mice (Abiraman 2015), but ITD-mediated sound localization has not been studied in that system. The auditory brainstem offers a unique opportunity to pursue these questions at the level of molecular architecture, electrical signal propagation, and behavioral performance in sound localization. These approaches would provide information not only about the advantages and limitations of remyelination, but also the role of myelin as an active player in neuronal networks.
Conclusion
Myelin produced by oligodendrocytes is a crucial component of proper CNS function. In addition to the classic demyelinating diseases, myelin dysfunction is becoming recognized as a component of many CNS disorders traditionally associated with neuronal function. Remyelination provides an exciting avenue of therapeutic research. However, understanding the mechanisms of remyelination, in particular whether remyelination results from essentially the recapitulation of developmental myelination is important. How axons have changed after myelination, demyelination and remyelination is still an area of important investigation. Thus, the neuroprotective capacity of remyelination is still not well understood, and its ability to restore the circuit deficits underlying symptoms in neurological disorders is even more unclear, although as a goal, increasing remyelination after damage is clearly important. Given the wide variety of pathological and injury contexts in which demyelination occurs, it is crucial to learn more about how remyelination impacts axonal health and neuronal function.
Significance:
Loss of oligodendrocytes and the myelin they produce is implicated in a wide range of central nervous system pathologies, with profound effects on axonal health and function. Facilitating remyelination has become a major therapeutic target. Thus, understanding its neuroprotective impact is crucial for using remyelination in treatment. This review integrates the current literature on remyelination as a neuroprotective strategy, as well as gaps in our knowledge and how these gaps can be addressed in different model systems and circuits.
Acknowledgements:
This work was supported by a grant from the American Heart Association Bugher Foundation grant.
Footnotes
Conflict of Interest: The authors declare no known conflict of interest.
Associate Editor: Kazuhiro Ikenaka
References
- Abiraman K, Pol SU, O ‘bara MA, Chen G-D, Khaku ZM, Wang J, Thorn DA, Vedia BH, Ekwegbalu EC, Li J-X, Salvi RJ, Sim FJ. 2015. Anti-Muscarinic Adjunct Therapy Accelerates Functional Human Oligodendrocyte Repair. J Neurosci 25:3676–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanese M, Zagaglia S, Landi D, Boffa L, Nicoletti CG, Marciani MG, Mandolesi G, Marfia GA, Buttari F, Mori F, Centonze D. 2016. Cerebrospinal fluid lactate is associated with multiple sclerosis disease progression. J Neuroinflammation 13:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert M, Antel J, Brück W, Stadelmann C. 2007. Extensive cortical remyelination in patients with chronic multiple sclerosis. Brain Pathol 17:129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba H, Akita H, Ishibashi T, Inoue Y, Nakahira K, Ikenaka K. 1999. Completion of myelin compaction, but not the attachment of oligodendroglial processes triggers K(+) channel clustering. J Neurosci Res 58:752–764. [PubMed] [Google Scholar]
- Babbs CF, Shi R. 2013. Subtle Paranodal Injury Slows Impulse Conduction in a Mathematical Model of Myelinated Axons. PLoS One 8:e67767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi B, Al-Sabi A, Kaza S, Scholz D, O’Leary VB, Dolly JO, Ovsepian SV. 2014. Disruption of myelin leads to ectopic expression of KV1.1 channels with abnormal conductivity of optic nerve axons in a cuprizone-induced model of demyelination. de Castro F, ed. PLoS One 9:e87736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcer LJ, Miller DH, Reingold SC, Cohen JA. 2015. Vision and vision-related outcome measures in multiple sclerosis. Brain 138:11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron W, Metz B, Bansal R, Hoekstra D, de Vries H. 2000. PDGF and FGF-2 signaling in oligodendrocyte progenitor cells: regulation of proliferation and differentiation by multiple intracellular signaling pathways. Mol Cell Neurosci 15:314–329. [DOI] [PubMed] [Google Scholar]
- Bartzokis G 2004. Age-related myelin breakdown: A developmental model of cognitive decline and Alzheimer’s disease. Neurobiol Aging 25:5–18. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Albin RL. 2011. White matter lesions in Parkinson disease. Nat Rev Neurol 7:229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher D, Goaillard JM. 2011. Beyond faithful conduction: Short-term dynamics, neuromodulation, and long-term regulation of spike propagation in the axon. Prog Neurobiol 94:307–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Staugaitis SM, Dutta R, Batt CE, Easley KE, Chomyk AM, Yong VW, Fox RJ, Kidd GJ, Trapp BD. 2012. Cortical remyelination: A new target for repair therapies in multiple sclerosis. Ann Neurol 72:918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatelier A, Zhao J, Bois P, Chahine M. 2010. Biophysical characterisation of the persistent sodium current of the Nav1.6 neuronal sodium channel: A single-channel analysis. Pflugers Arch Eur J Physiol 460:77–86. [DOI] [PubMed] [Google Scholar]
- Coman I, Aigrot MS, Seilhean D, Reynolds R, Girault JA, Zalc B, Lubetzki C. 2006. Nodal, paranodal and juxtaparanodal axonal proteins during demyelination and remyelination in multiple sclerosis. Brain 129:3186–3195. [DOI] [PubMed] [Google Scholar]
- Cranford JL, Boose M, Moore CA. 1990. Tests of the precedence effect in sound localization reveal abnormalities in multiple sclerosis. Ear Hear 11:282–288. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Matthews PM, Antel JP, Preul M, Francis G, Arnold DL. 1995. Chemical pathology of acute demyelinating lesions and its correlation with disability. Ann Neurol 38:901–909. [DOI] [PubMed] [Google Scholar]
- Dewar D, Underhill SM, Goldberg MP. 2003. Oligodendrocytes and ischemic brain injury. J Cereb Blood Flow Metab 23:263–274. [DOI] [PubMed] [Google Scholar]
- Doppler K, Werner C, Sommer C. 2013. Disruption of nodal architecture in skin biopsies of patients with demyelinating neuropathies. J Peripher Nerv Syst 18:168–176. [DOI] [PubMed] [Google Scholar]
- Dubois-Dalcq M, Ffrench-Constant C, Franklin RJM. 2005. Enhancing central nervous system remyelination in multiple sclerosis. Neuron 48:9–12. [DOI] [PubMed] [Google Scholar]
- Duncan ID, Brower A, Kondo Y, Curlee JF, Schultz RD. 2009. Extensive remyelination of the CNS leads to functional recovery. Proc Natl Acad Sci U S A 106:6832–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Mason JL, Marcus JR, Stull M, Levinson R, Matsushima GK, Popko B. 2004. Oligodendrocytes assist in the maintenance of sodium channel clusters independent of the myelin sheath. Neuron Glia Biol 1:179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJM, Goldman SA. 2015. Glia disease and repair - Remyelination. Cold Spring Harb Perspect Biol 7:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Möbius W, Diaz F, Meijer D, Suter U, Hamprecht B, Sereda MW, Moraes CT, Frahm J, Goebbels S, Nave K-A. 2012. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485:517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furst M, Levine RA. 2015. Hearing disorders in multiple sclerosis. Handb Clin Neurol 129:649–665. [DOI] [PubMed] [Google Scholar]
- Fyffe-Maricich SL, Schott A, Karl M, Krasno J, Miller RH. 2013. Signaling through ERK1/2 controls myelin thickness during myelin repair in the adult central nervous system. J Neurosci 33:18402–18408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Armstrong RC. 2008. Myelin repair strategies: a cellular view. Curr Opin Neurol 21:278–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, Barres BA, Woo PJ, Vogel H, Monje M. 2014. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344:1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girault JA, Peles E. 2002. Development of nodes of Ranvier. Curr Opin Neurobiol 12:476–485. [DOI] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, McCulloch M, Nadon N, Nave KA. 1998. Axonal Swellings and Degeneration in Mice Lacking the Major Proteolipid of Myelin. Science 280:1610–1613. [DOI] [PubMed] [Google Scholar]
- Guttmann CR, Jolesz FA, Kikinis R, Killiany RJ, Moss MB, Sandor T, Albert MS. 1998. White matter changes with normal aging. Neurology 50:972–978. [DOI] [PubMed] [Google Scholar]
- Hardy R, Reynolds R. 1991. Proliferation and differentiation of rat forebrain oligodendroglial progenitors both in vitro and in vivo. Development 111:1061–1080. [DOI] [PubMed] [Google Scholar]
- Hattori T, Orimo S, Aoki S, Ito K, Abe O, Amano A, Sato R, Sakai K, Mizusawa H. 2012. Cognitive status correlates with white matter alteration in Parkinson’s disease. Hum Brain Mapp 33:727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi T, Kocsis JD, Waxman SG. 1998. Resistance to anoxic injury in the dorsal columns of adult rat spinal cord following demyelination. Brain Res 779:292–296. [DOI] [PubMed] [Google Scholar]
- Irvine KA, Blakemore WF. 2008. Remyelination protects axons from demyelination-associated axon degeneration. Brain 131:1464–1477. [DOI] [PubMed] [Google Scholar]
- Ishibashi T 2004. Tetraspanin Protein CD9 Is a Novel Paranodal Component Regulating Paranodal Junctional Formation. J Neurosci 24:96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Fyffe-Maricich SL, Furusho M, Miller RH, Bansal R. 2012. ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. J Neurosci 32:8855–8864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery ND, Blakemore WF. 1997. Locomotor deficits induced by experimental spinal cord demyelination are abolished by spontaneous remyelination. Brain 120:27–37. [DOI] [PubMed] [Google Scholar]
- Kaplan MR, Cho MH, Ullian EM, Isom LL, Levinson SR, Barres BA. 2001. Differential control of clustering of the sodium channels Nav1.2 and Nav1.6 at developing CNS nodes of Ranvier. Neuron 30:105–119. [DOI] [PubMed] [Google Scholar]
- Kapoor R, Li YG, Smith KJ. 1997. Slow sodium-dependent potential oscillations contribute to ectopic firing in mammalian demyelinated axons. Brain 120:647–652. [DOI] [PubMed] [Google Scholar]
- Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, Linington C, Schmidbauer M, Lassmann H. 2000. Multiple Sclerosis and Chronic Autoimmune Encephalomyelitis. Am J Pathol 157:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotter MR. 2006. Myelin Impairs CNS Remyelination by Inhibiting Oligodendrocyte Precursor Cell Differentiation. J Neurosci 26:328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotter M 2012. CNS Remyelination – Can We Wrap It Up? – Regulators of Neural Stem/Precursor Cells in the Process of Myelin Regeneration. QScience Proc 2012:39. [Google Scholar]
- Kremer D, Göttle P, Hartung HP, Küry P. 2016. Pushing Forward: Remyelination as the New Frontier in CNS Diseases. Trends Neurosci 39:246–263. [DOI] [PubMed] [Google Scholar]
- Kubicki M, Niznikiewicz M, Connor E, Ungar L, Nestor PG, Bouix S, Dreusicke M, Kikinis R, McCarley RW, Shenton ME. 2009. Relationship between white matter integrity, attention, and memory in schizophrenia: A diffusion tensor imaging study. Brain Imaging Behav 3:191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H. 2005. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 128:2705–2712. [DOI] [PubMed] [Google Scholar]
- Lampron A, Larochelle A, Laflamme N, Préfontaine P, Plante M-M, Sánchez MG, Yong VW, Stys PK, Tremblay M-È, Rivest S. 2015. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med 212:481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave K-A. 2003. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33:366–374. [DOI] [PubMed] [Google Scholar]
- Lassmann H, van Horssen J, Mahad D. 2012. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 8:647–656. [DOI] [PubMed] [Google Scholar]
- Lassmann H 2010. Axonal and neuronal pathology in multiple sclerosis: What have we learnt from animal models. Exp Neurol 225:2–8. [DOI] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang P-W, Pellerin L, Magistretti PJ, Rothstein JD. 2012. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487:443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. 2000. The Action Potential and Conduction of Electric Impulses. Mol Cell Biol 4th Ed 2000. [Google Scholar]
- López-Villegas D, Lenkinski RE, Wehrli SL, Ho WZ, Douglas SD. 1995. Lactate production by human monocytes/macrophages determined by proton MR spectroscopy. Magn Reson Med 34:32–38. [DOI] [PubMed] [Google Scholar]
- Majumdar S, Sikdar SK. 2005. Fast pseudo-periodic oscillation in the rat brain voltage-gated sodium channel alpha subunit. J Membr Biol 208:1–14. [DOI] [PubMed] [Google Scholar]
- Manrique-Hoyos N, Jürgens T, Grønborg M, Kreutzfeldt M, Schedensack M, Kuhlmann T, Schrick C, Brück W, Urlaub H, Simons M, Merkler D. 2012. Late motor decline after accomplished remyelination: Impact for progressive multiple sclerosis. Ann Neurol 71:227–244. [DOI] [PubMed] [Google Scholar]
- Mason JL, Langaman C, Morell P, Suzuki K, Matsushima GK. 2001. Episodic demyelination and subsequent remyelination within the murine central nervous system: Changes in axonal calibre. Neuropathol Appl Neurobiol 27:50–58. [DOI] [PubMed] [Google Scholar]
- Mayer RF. 1971. Conduction velocity in the central nervous system of the cat during experimental demyelination and remyelination. Int J Neurosci 1:287–308. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, De Vellis J. 1980. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85:890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella DM, Majdan M, Awatramani R, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. 2006. Genetic and Physiological Evidence That Oligodendrocyte Gap Junctions Contribute to Spatial Buffering of Potassium Released during Neuronal Activity. J Neurosci 26:10984–10991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Manabe H, Narikiyo K, Onisawa N. 2013. Olfactory consciousness and gamma oscillation couplings across the olfactory bulb, olfactory cortex, and orbitofrontal cortex. Front Psychol 4:743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BM, Lee Y, Rothstein JD. 2013. Oligodendroglia: Metabolic supporters of axons. Trends Cell Biol 23:644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutsaers SE, Carroll WM. 1998. Focal accumulation of intra-axonal mitochondria in demyelination of the cat optic nerve. Acta Neuropathol 96:139–143. [DOI] [PubMed] [Google Scholar]
- Neusch C 2006. Lack of the Kir4.1 Channel Subunit Abolishes K+ Buffering Properties of Astrocytes in the Ventral Respiratory Group: Impact on Extracellular K+ Regulation. J Neurophysiol 95:1843–1852. 10.1152/jn.00996.2005. Accessed June 12, 2016. [DOI] [PubMed] [Google Scholar]
- Nijland PG, Michailidou I, Witte ME, Mizee MR, Van Der Pol SMA, Van Het Hof B, Reijerkerk A, Pellerin L, van der Valk P, de Vries HE, van Horssen J. 2014. Cellular distribution of glucose and monocarboxylate transporters in human brain white matter and multiple sclerosis lesions. Glia 62:1125–1141. [DOI] [PubMed] [Google Scholar]
- Nikić I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM, Brück W, Bishop D, Misgeld T, Kerschensteiner M. 2011. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 17:495–499. [DOI] [PubMed] [Google Scholar]
- Osterman PO, Westerberg CE. 1975. Paroxysmal Attacks in Multiple Sclerosis. Brain 98:189–202. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. 1994. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A 91:10625–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Périer O, Grégoire A. 1965. Electron microscopic features of multiple sclerosis lesions. Brain 88:937–952. [DOI] [PubMed] [Google Scholar]
- Peters A, Sethares C. 2002. Aging and the myelinated fibers in prefrontal cortex and corpus callosum of the monkey. J Comp Neurol 442:277–291. [DOI] [PubMed] [Google Scholar]
- Pfeiffer S, Warrington A, Bansal R. 1993. The oligodendrocyte and its many cellular processes. Trends Cell Biol 3:191–197. [DOI] [PubMed] [Google Scholar]
- Prineas JW, Connell F. 1979. Remyelination in multiple sclerosis. Ann Neurol 5:22–31. [DOI] [PubMed] [Google Scholar]
- Prineas JW, Kwon EE, Cho ES, Sharer LR. 1984. Continual breakdown and regeneration of myelin in progressive multiple sclerosis plaques. Ann N Y Acad Sci 436:11–32. [DOI] [PubMed] [Google Scholar]
- Rasband MN, Trimmer JS, Schwarz TL, Levinson SR, Ellisman MH, Schachner M, Shrager P. 1998. Potassium channel distribution, clustering, and function in remyelinating rat axons. J Neurosci 18:36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Kagawa T, Park EW, Ikenaka K, Trimmer JS. 2003. Dysregulation of axonal sodium channel isoforms after adult-onset chronic demyelination. J Neurosci Res 73:465–470. [DOI] [PubMed] [Google Scholar]
- Rasband MN, Peles E. 2016. The nodes of Ranvier: Molecular assembly and maintenance. Cold Spring Harb Perspect Biol 8:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redford EJ, Kapoor R, Smith KJ. 1997. Nitric oxide donors reversibly block axonal conduction: Demyelinated axons are especially susceptible. Brain 120:2149–2157. [DOI] [PubMed] [Google Scholar]
- Rittchen S, Boyd A, Burns A, Park J, Fahmy TM, Metcalfe S, Williams A. 2015. Myelin repair in vivo is increased by targeting oligodendrocyte precursor cells with nanoparticles encapsulating leukaemia inhibitory factor (LIF). Biomaterials 56:78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson S, Miller RH. 1999. Contact with central nervous system myelin inhibits oligodendrocyte progenitor maturation. Dev Biol 216:359–368. [DOI] [PubMed] [Google Scholar]
- Rodgers JM, Robinson AP, Miller SD. 2013. Strategies for protecting oligodendrocytes and enhancing remyelination in multiple sclerosis. Discov Med 16:53–63. [PMC free article] [PubMed] [Google Scholar]
- Roy T 2006. Seizures in multiple sclerosis in Eastern India. Neurol Asia 42:123–127. [Google Scholar]
- Rushton WAH. 1951. A Theory of the Effects of Fibre Size in Medullated Nerve. J Physiol 5:101–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Abarca LI, Tabernero A, Medina JM. 2001. Oligodendrocytes use lactate as a source of energy and as a precursor of lipids. Glia 36:321–329. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Black JA, Lankford KL, Tokuno HA, Waxman SG, Kocsis JD. 2006. Molecular Reconstruction of Nodes of Ranvier after Remyelination by Transplanted Olfactory Ensheathing Cells in the Demyelinated Spinal Cord. J Neurosci 26:1803–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl AH, Rubel EW. 2016. Systematic and differential myelination of axon collaterals in the mammalian auditory brainstem. Glia 64:487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ. 1994. Conduction properties of central demyelinated and remyelinated axons, and their relation to symptom production in demyelinating disorders. Eye (Lond) 8 (Pt 2):224–237. [DOI] [PubMed] [Google Scholar]
- Smith KJ. 2006. Axonal protection in multiple sclerosis - A particular need during remyelination? Brain 129:3147–3149. [DOI] [PubMed] [Google Scholar]
- Snaidero N, Möbius W, Czopka T, Hekking LHP, Mathisen C, Verkleij D, Goebbels S, Edgar J, Merkler D, Lyons DA, Nave KA, Simons M. 2014. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell 156:277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK. 2005. General mechanisms of axonal damage and its prevention. In: Journal of the Neurological Sciences 233(2005):3–13. [DOI] [PubMed] [Google Scholar]
- Susuki K 2013. Node of Ranvier disruption as a cause of neurological diseases. ASN Neuro 5:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. 2011. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144:810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomassy GS, Berger DR, Chen H-H, Kasthuri N, Hayworth KJ, Vercelli A, Seung HS, Lichtman JW, Arlotta P. 2014. Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science 344:319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Peterson JW, Ransohoff RM, Rudick RA, Mork S, Bo L, Mörk S, Bö L. 1998. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338:278–285. [DOI] [PubMed] [Google Scholar]
- Vela J 2002. Interleukin-1 Regulates Proliferation and Differentiation of Oligodendrocyte Progenitor Cells. Mol Cell Neurosci 20:489–502. [DOI] [PubMed] [Google Scholar]
- Volman V, Ng LJ. 2014. Primary paranode demyelination modulates slowly developing axonal depolarization in a model of axonal injury. J Comput Neurosci 37:439–457. [DOI] [PubMed] [Google Scholar]
- von Büdingen H-C, Mei F, Greenfield A, Jahn S, Shen Y-AA, Reid HH, McKemy DD, Chan JR. 2015. The myelin oligodendrocyte glycoprotein directly binds nerve growth factor to modulate central axon circuitry. J Cell Biol 210:891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Bennet MVL. 1972. © 1972 Nature Publishing Group. Nature 235:36. [Google Scholar]
- Waxman SG, Davis PK, Black JA, Ransom BR. 1990. Anoxic injury of mammalian central white matter: decreased susceptibility in myelin-deficient optic nerve. Ann Neurol 28:335–340. [DOI] [PubMed] [Google Scholar]
- Waxman SG. 2006. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci 7:932–941. [DOI] [PubMed] [Google Scholar]
- Whitford TJ, Ford JM, Mathalon DH, Kubicki M, Shenton ME. 2012. Schizophrenia, myelination, and delayed corollary discharges: A hypothesis. Schizophr Bull 38:486–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wist ER, Hennerici M, Dichgans J. 1978. The Pulfrich spatial frequency phenomenon: a psychophysical method competitive to visual evoked potentials in the diagnosis of multiple sclerosis. J Neurol Neurosurg Psychiatry 41:1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Narayanan SP, Wang F, Morse E, MacKlin WB, Peachey NS. 2011. Visual abnormalities associated with enhanced optic nerve myelination. Brain Res 1374:36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Johnson KR, Miron VE, Zhao C, Quandt J, Harrisingh MC, Swire M, Williams A, McFarland HF, Franklin RJM, Ffrench-Constant C. 2013. Identification of endothelin 2 as an inflammatory factor that promotes central nervous system remyelination. Brain 136:1035–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Jarjour AA, Boyd A, Williams A. 2011. Central nervous system remyelination in culture - A tool for multiple sclerosis research. Exp Neurol 230:138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlochiver S 2010. Persistent reflection underlies ectopic activity in multiple sclerosis: A numerical study. Biol Cybern 102:181–196. [DOI] [PubMed] [Google Scholar]