

Abstract
The melanocortin‐4 receptor (MC4R) has been critically investigated for the past two decades, and novel findings regarding MC4R signalling and its potential exploitation in weight loss therapy have lately been emphasized. An association between MC4R and obesity is well established, with disease‐causing mutations affecting 1% to 6% of obese patients. More than 200 MC4R variants have been reported, although conflicting results as to their effects have been found in different cohorts. Most notably, some MC4R gain‐of‐function variants seem to rescue obesity and related complications via specific pathways such as beta‐arrestin (ß‐arrestin) recruitment. Broadly speaking, however, dysfunctional MC4R dysregulates satiety and induces hyperphagia. The picture at the mechanistic level is complicated as, in addition to the canonical G stimulatory pathway, the ß‐arrestin signalling pathway and ions (particularly calcium) seem to interact with MC4R signalling to contribute to or alleviate obesity pathogenesis. Thus, the overall complexity of the MC4R signalling spectra has broadened considerably, indicating there is great potential for the development of new drugs to manage obesity and its related complications. Alpha‐melanocyte‐stimulating hormone is the major endogenous MC4R agonist, but structure‐based ligand discovery studies have identified possible superior and selective agonists that can improve MC4R function. However, some of these agonists characterized in vitro and in vivo confer adverse effects in patients, as demonstrated in clinical trials. In this review, we provide a comprehensive insight into the genetics, function and regulation of MC4R and its contribution to obesity. We also outline new approaches in drug development and emerging drug candidates to treat obesity.
Keywords: Ca2+ , drug design, Gs , MC4R, obesity, ß‐arrestin
1. INTRODUCTION
Obesity is characterized by excess fat mass, which affects physical health and increases the complexity of many associated diseases and conditions, including type 2 diabetes, cardiovascular complications and cancer. 1 The associated healthcare costs are huge, and obesity is accompanied by significant morbidity and mortality. Obesity is defined in terms of body mass index (BMI), that is, weight (kg)/height (m2); people with a BMI of more than 30 kg/m2 are considered obese, while those with a BMI of 35 to 39.9 30 kg/m2 are severely obese. 2 , 3 , 4 , 5 The intake of high‐calorie food and a lack of sufficient physical activity leads to an increased positive energy balance and subsequent weight gain. In addition, studies have suggested there is a strong genetic influence on obesity, involving a complex neurochemical system that regulates appetite, energy expenditure and weight loss. There is an overall synergistic relationship between genes, the environment and lifestyle. 6
The identification of genes involved in regulating body weight and maintaining energy homeostasis is crucial. Major genes associated with obesity include leptin, leptin receptor, proopiomelanocortin (POMC), proprotein convertase subtilin/kexin (PCSK1), adenylate cyclase 3, single‐minded 1 (SIM‐1), tyrosine kinase receptor tropomycin‐related kinase B (TRKB), brain‐derived neurotropic factor (BDNF), and melanocortin‐4 receptor (MC4R). 7 , 8 PCSK1 and POMC are critical in children with obesity, while MC4R is the most significant gene for obesity overall and the most widely investigated so far. 9 , 10 , 11 , 12 , 13 MC4R is localized at chromosome 18q22 and primarily expressed in the hypothalamus, encoding a 332‐amino acid transmembrane protein. 14 MC4R is a member of the heterotrimeric G‐protein‐coupled receptor (GPCR) family, and its primary functions are in regulating the intake of food, energy homeostasis and body weight. 15 , 16
The genetic basis of obesity was first discovered when disruption of the mouse MC4R was found to cause the accumulation of excess weight. 17 Several reports of frameshift mutations in human MC4Rs and their association with obesity were subsequently confirmed, 18 , 19 , 20 , 21 , 22 with 17, 58 and more than 200 human MC4R mutations identified by the years 2000, 23 , 24 2006 25 and 2021, 26 , 27 , 28 respectively. These mutations can cause partial or complete loss of function depending on the nature of the mutation and function of the mutated receptor. 25 Heterozygous MC4R variants alone are found in 1% to 6% of the obese population, and are particularly common in early‐onset or childhood obesity, with variable penetrance and expressivity resulting in mild to severe manifestations of obesity and associated complications. 7 , 16 , 29 , 30 Homozygous variants are reported less frequently but result in more severe manifestations of obesity. 31 , 32 In recent studies, MC4R signalling has been implicated as a viable target for antiobesity treatment. 10 , 13 , 33 , 34 , 35 , 36 For example, certain human MC4R variants are protective against obesity, and drugs targeting the receptor have shown efficacy for weight loss.
We discuss in the subsequent sections how MC4R, as a GPCR, relates to obesity. We begin with a brief introduction to GPCRs to promote a general understanding of the receptors. Then, MC4R‐signalling pathways, including the broader, centrally regulated melanocortinergic system, the canonical G‐stimulatory (Gs)‐signalling pathway, and the newly proposed roles of the ß‐arrestin and calcium (Ca2+) pathways, are detailed. We also discuss the loss of function/gain of function (GoF) caused by mutations of the MC4R and how this affects or alleviates disease pathology. The crystal structure of MC4R, the importance of ion‐binding sites, and the differences between MC4R and other GPCRs are discussed while focusing on the unique features of the receptor. Finally, we discuss in detail the development of MC4R agonist drugs and the current status of existing obesity drugs.
2. MELANOCORTIN‐4 RECEPTOR AS A GPCR
G‐protein‐coupled receptors are involved in most physiological functions in the body and in the manifestation of numerous diseases. 37 They represent the largest family of cell surface and transmembrane receptors, with more than 825 genes (~2% of the human genome) and corresponding gene products. 38 , 39 The structure of GPCRs, which is largely conserved, comprises seven transmembrane glycoproteins spanning the plasma membrane, 40 with additional extracellular N‐terminal and intracellular C‐terminal domains. The overall structure of GPCRs enables them to receive extracellular stimuli (eg, ions or peptides), to communicate via the otherwise impermeable plasma membrane, and to transfer stimuli to the interior of cells to induce functional changes. These processes involve a series of protein interactions and changes in the expression levels of biochemical mediators to ultimately effect physiological or even behavioural changes. GPCRs are considered very clinically significant protein targets; of the 1500 drugs approved by the US Food and Drug Administration (FDA) by 2020, 460 targeted GPCRs. 39 , 41 Among them, Class A GPCRs (rhodopsin‐like receptors) are major drug targets (94%), followed by Class B (secretin family, 4%), Class C (metabotropic glutamate receptors, 2%), and Class F (frizzled and smoothened receptors, 2%). 39
The MC4R is a rhodopsin‐like Class A GPCR, expressed in the paraventricular nucleus of the hypothalamus, and is a key component of the leptin‐melanocortin pathway. 17 MC4R is activated by POMC‐derived polypeptides obtained by the posttranslational processing of POMC that yields alpha (α)‐, beta (ß)‐ and gamma (γ)‐melanocyte‐stimulating hormone (MSH) and adrenocorticotropic hormone (ACTH). α‐MSH is very effective in regulating eating behaviour and energy homeostasis, and in addition to its primary function in melanogenesis, it also activates all (MC5R, MC4R, MC3R and MC1R) but one (MC2R) melanocortin receptors. Of these, MC4R is the most crucial, as mutations in this receptor cause different forms of obesity in humans. 15 , 42 The role of MC3R in energy homeostasis and in regulating satiety is also known and is under active investigation. 6 , 43 , 44 , 45 It is a key component of the central melanocortin pathway. 46 MC5R is involved in fatty acid and lipid metabolism as well as exocrine secretion. 6
3. MELANOCORTIN‐4 RECEPTOR SIGNALLING PATHWAYS
The next sections comprise an overview of the MC4R‐signalling pathways, including the leptin‐melanocortin pathway, followed by a more focused discussion of the specific details of G‐protein‐signalling pathways and, finally, a review of the latest proposed changes, particularly regarding the mechanisms of energy regulation via the ß‐arrestin and Ca2+‐regulated pathways. The leptin‐melanocortin pathway includes the overall, centrally (central nervous system) regulated pathway responses to anorectic/orectic signals. The canonical Gs pathway and the generation of cyclic adenosine monophosphate (cAMP) have been widely investigated for obesity‐related/energy balance issues, and the mechanisms have been fairly understood. 16 , 47 , 48 , 49 However, the recently proposed involvement of ß‐arrestin in the regulation of MC4R, often referred to as ß‐arrestin bias, is a bias or shift toward recruiting ß‐arrestin as the mode of action, 10 , 13 rather than canonical cAMP production via the Gs pathway. The role of Ca2+, another new discovery, 35 is also detailed below.
3.1. LEPTIN‐MELANOCORTIN PATHWAY
Under a fed state, adipose tissues secrete the hormone leptin. Binding of leptin to the leptin receptor stimulates POMC neurons to secrete α‐MSH, which binds to the MC4R, resulting in a satiety signal and hence reduced appetite and a signal to increase energy expenditure to achieve a reduced energy balance. 50 , 51 , 52 MC4R, however, can be blocked by the inhibitor, agouti‐related peptide (AgRP), expressed by neuropeptide Y (NPY) neurons in the arcuate nucleus. 5 Lack of food induces the increased expression of NYP/AgRP, resulting in hunger signals. A balance between these two hormones is, therefore, critical in regulating food intake and energy metabolism. A few other molecules included in this pathway that are not yet fully elucidated include SIM‐1, TRKB and BDNF. In brief, SIM‐1 is a transcription factor that causes hyperphagia and a reduction of the periventricular nucleus, resulting in severe obesity. 53 Mutations in BDNF, which is involved in downstream MC4R signalling, and its receptor, TRKB, also contribute to hyperphagia and obesity. 54
3.2. G‐PROTEIN SIGNALLING PATHWAY
G‐proteins are complex heterotrimeric guanine‐binding proteins with G‐alpha (Gα), G‐beta (Gß), and G‐gamma (Gγ) subunits. G‐proteins are also classified into four distinct groups according to the Gα subunits, Gαs, Gαi/o, Gαq/11 and Gα12/13. 34 , 53 In G‐protein‐signalling pathways (Figure 1), an exchange of guanosine triphosphate (GTP) for guanosine diphosphate (GDP) induces the dissociation of the α subunit from the ßγ dimer following communication with effectors. 55
FIGURE 1.
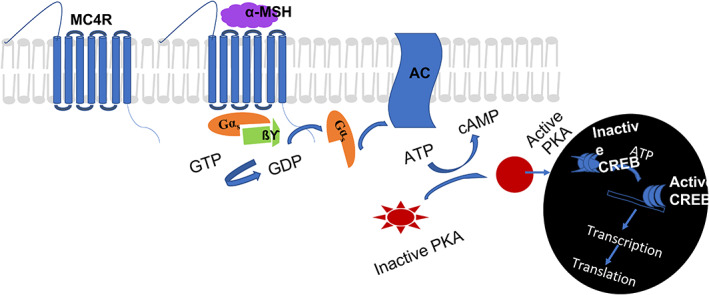
G‐protein signalling pathway. Schematic showing the canonical stimulatory G‐protein (Gs) pathway for melanocortin 4‐receptor (MC4R) signalling and gene expression. Binding of α‐melanocyte‐stimulating hormone (MSH) to the MC4R causes the activation of the G‐protein, with αßγ‐subunits dissociating into α‐/ßγ‐subunits. The dissociated Gαs causes the activation of adenylyl cyclase (AC), leading to the conversion of ATP to cyclic adenosine monophosphate (cAMP). cAMP activates inactive protein kinase A (PKA) which is translocated into the nucleus, activating the transcription factor cAMP response element binding protein (CREB) via phosphorylation of CREB, which regulates transcription. GTP, guanosine triphosphate
Alpha melanocyte stimulating hormone activates MC4R and catalyses the exchange of GDP for GTP on the stimulatory G‐protein (Gαs), resulting in the activation of adenylyl cyclase (AC) and the generation of intracellular cAMP. 56 cAMP, which may also be triggered by Gßγ subunits to increase AC activity via certain isozymes, 57 , 58 binds to protein kinase A (PKA) regulatory subunits, causing its dissociation and distribution to different cellular compartments. Activated PKA further affects numerous physiological processes by activating subsequent effector proteins (mostly via phosphorylation), including kinases, ion channels, and other signalling proteins/enzymes. 13 , 49 cAMP‐mediated transcriptional regulation is achieved via the binding of active PKA to the cAMP response element binding protein, causing PKA phosphorylation and the downstream transcription and translation of target genes and proteins. Coupling to Gαi/o deactivates or reduces AC activity, lowering cAMP levels. 56 Gαq/11 and Gα12/13 are mostly associated with functions other than obesity/energy homeostasis, such as the activation of phospholipase C. 59
In addition, MC4R also activates mitogen‐activated protein kinases (MAPK) and extracellular signal‐regulated kinases 1 and 2 (ERK1/2). Mo et al 60 reported various MC4R ligands, including AgRP and Ipsen 5i inverse agonists, at the Gs‐cAMP signalling pathway, to regulate ERK activation in wild‐type and six naturally occurring constitutively active mutant (CAM) MC4R. A significant increase in the phosphorylation of ERK was reported in some of them, suggesting that these MC4R inverse agonists could act as agonists in the MAPK pathway. 60 This study proposed the prevalence of multiple activation states of MC4R with ligand as well as mutant‐specific conformations that could couple differentially to the MC4R, giving rise to distinct signalling pathways or its constitutive activity. 60 This suggests abundant potential for future investigations into new novel mechanisms.
3.3. ß‐ARRESTIN PATHWAY
The knowledge of constitutive MC4R activity is not new, as described above and also detailed in the GoF section. This constitutive activity leads to a GoF effect, which can alleviate obesity pathology. However, the underlying molecular mechanism has only recently been postulated. 10 , 13 Mutations causing GoF may employ ß‐arrestin signalling, particularly those that are associated with a reduced risk of obesity‐associated diseases such as type 2 diabetes and cardiac ailments. There is strong evidence that mutations, particularly Valine 103 Isoleucine (V103I) and Isoleucine 251 Leucine (I251L), help to effectively recruit ß‐arrestin. 10 , 13 The binding of agonists, for example, α‐MSH and [Nle4, DPhe7]‐α‐MSH (NDP‐MSH; a synthetic analogue of α‐MSH), to these mutant MC4Rs may induce changes in the conformation of the receptor, affecting ligand receptor‐binding and either making the interaction strong enough to prevent internalization, resulting in longer retention of the MC4Rs on the plasma membrane, or allowing very rapid or much improved recycling so they are available in increased numbers on the plasma membrane compared with the wild type (Figure 2). This effect has yet to be investigated in detail, but research has been initiated by some groups 10 , 13 and will be discussed later in this review.
FIGURE 2.
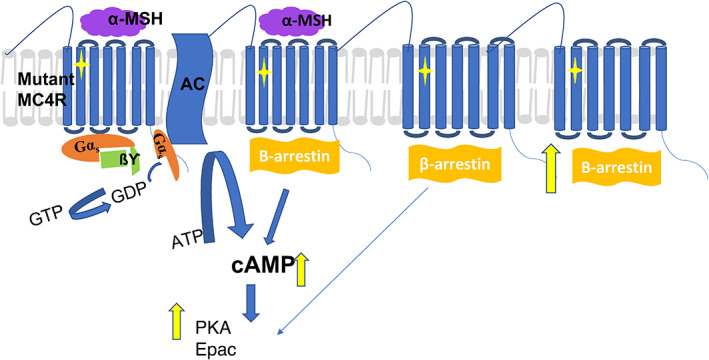
β‐arrestin signalling pathway. Schematic elucidating the mechanism of gain‐of‐function induced by a mutation (yellow diamond) in the melanocortin 4‐receptor (MC4R). The mutation causes an increase in the cell‐surface expression of MC4R, possibly via reduced internalization or rapid recycling, causing increased cyclic adenosine monophosphate (cAMP) production, as well as increased production of protein kinase A (PKA) and cAMP‐regulated guanine nucleotide exchange factors (Epac). AC, adenylyl cyclase; GDP, guanosine diphosphate; GTP, guanosine triphosphate; MSH, melanocyte‐stimulating hormone;
3.4. CA2+ ‐REGULATED PATHWAYS
Ca2+ is recognized to be a cofactor for ligand‐MC4R binding. The recent elucidation of the crystal structure of MC4R complexed with SHU9119, a potent cyclic peptide agonist, highlighted the role of Ca2+‐binding in the receptor's downstream signal regulation. 35 , 61 Briefly, both the ligand agonist and Ca2+ ions complementarily activate MC4R, which induces closure of the inwardly rectifying potassium channel (KIR7.1) to retain intracellular potassium levels (Figure 3). This leads to an overall anorexigenic effect with a negative energy balance and increased heart rate. 62 However, the antagonist AgRP has inhibitory effects that open KIR7.1, causing K+ to be pumped out of the cell. This promotes orexigenic effects by dysregulating energy homeostasis and ultimately creating a positive energy balance (Figure 3).
FIGURE 3.
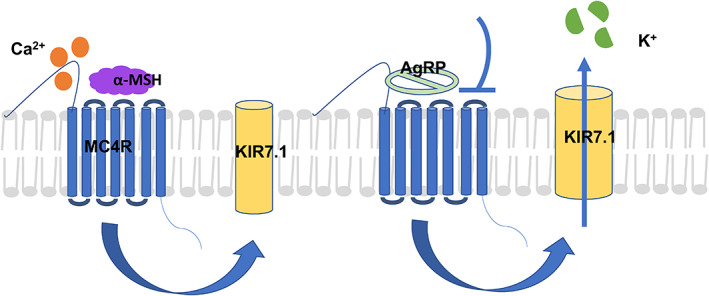
Ca2+ regulated pathway. The endogenous melanocortin 4‐receptor (MC4R) pathway is regulated by agonist α‐melanocyte‐stimulating hormone (MSH) and antagonist agouti‐related peptide (AgRP). Ca2+ ions are also important in the regulation of the MC4R pathway. α‐MSH, along with Ca2+, induces satiety and reduces food intake. This is regulated partly by closure of the potassium inward rectifying channel KIR7.1. Conversely, AgRP regulates orexigenic signals, also via the opening of the KIR7.1, in addition to other possible mechanisms
4. LOSS‐OF‐FUNCTION MUTATIONS, DISEASE PATHOLOGY AND RESCUE
Evidence has shown that mutations in MC4R are largely associated with severe obesity. These mutations cause either partial or complete loss of function, depending on the nature and function of the mutation, 16 , 27 , 48 , 63 , 64 , 65 and appear throughout the coding sequence. 25 A mutation‐based classification scheme was proposed as early as 2003. 25 , 66
Most loss‐of‐function mutations are heterozygous, exhibiting a phenotype intermediate between wild‐type and homozygous MC4R mutations. 15 , 63 The extent of the functional defect is sometimes conflicting, owing to the complexity of gene‐environment interactions and the varying expression of the dominant gene. Homozygous mutations, however, show pronounced effects on obesity, and patients carrying these mutations are characterized by high BMI, hyperphagia, 67 linear growth, 31 increased bone mineral density, 68 and hyperinsulinaemia. 32
In the functional characterization of most MC4R loss‐of‐function mutants, their intracellular retention suggests they undergo impaired receptor trafficking to the plasma membrane. This reduces the number of MC4Rs available on the cell surface, ultimately diminishing cAMP generation and all effector responses and downstream signalling, which leads to enhanced manifestation of disease. Changes in amino acid residue(s) as a result of mutation may also weaken ligand‐binding interactions, possibly because of changes in the protein conformation or reduced binding affinity, thus impairing or reducing downstream agonist‐stimulated signalling.
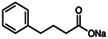
The intracellular retention of MC4R, which is a major cause of its functional defectiveness, results when it remains in the endoplasmic reticulum as a misfolded/ubiquitinated protein. Therapeutic rescue mechanisms, such as using pharmacological or chemical chaperones, could aid proper folding and enhance receptor expression at the cell surface or prevent ubiquitination and degradation of the protein by proteasomes. 69 Granell et al 70 , 71 first reported the rescuing potential of the chemical chaperone sodium 4‐phenylbutyrate and the ubiquitin‐activating enzyme inhibitor on MC4R in increasing the cell‐surface expression of mutant MC4R associated with severe obesity. The clinical utility of chemical chaperones has, however, been challenging to elucidate. Various pharmacological chaperones have been developed as antagonists of MC4R. 69 , 72 These return several misfolded MC4R mutants to the surface of the plasma membrane. 69 NBP (1‐(1‐(4‐fluorophenyl)‐ 2‐(4‐(4‐[naphthalene‐1‐yl] butyl) piperazin‐1‐yl)ethyl)‐4‐methylpiperazine) showed potential to rescue many MC4R mutants but failed to restore their NDP‐MSH binding responses, possibly due to the presence of a protracted binding, high‐affinity antagonist of inhibition constant (Ki) 2.4 nM. 72 ML00253764 (2‐[2‐[2‐[5‐Bromo‐2‐methoxyphenyl]ethyl]‐3‐fluorophenyl]‐4,5‐dihydro‐2‐1H‐imidazole) and DCPMP (N‐((2R)‐3(2,4‐dichlorophenyl)‐1‐(4‐(2‐([1‐methoxypropan2 ylamino]methyl)phenyl)piperazin‐1‐yl)‐1‐oxopropan2‐yl) propionamide) are efficient rescuers but have relatively low binding affinities, at Ki 0.17 μM and 0.02 μM, respectively, necessitating a high effective concentration (EC50 10 μM). 69 , 72 Encouragingly, Ipsen 5i and Ipsen 17 have wider rescue spectra and lower potency, facilitating rapid dissociation, thus they can rescue the mutant receptor at the plasma membrane at a low concentration yet still allow binding of endogenous ligands. 73 , 74 THIQ (N‐[(3R)‐1,2,3,4‐Tetrahydroisoquinolinium‐3‐ylcarbonyl]‐(1R)‐1‐(4‐chlorobenzyl)‐2‐[4‐cyclohexyl‐4‐(1H‐1,2,4‐triazol‐1‐ylmethyl) piperidin‐1‐yl]‐2‐oxoethylamine) was reported to rescue seven of the 10 mutants investigated in neuronal cell lines but only three in human embyonic kidney 293 cells, suggesting it has effective chaperone activity in neuronal cells. 75 The structures and Ki, half maximum inhibitory concentration (IC50) or effective concentration at a stable state inducing half of the maximum effect (EC50) values of important chemical/pharmacological chaperones are shown in Table 1.
TABLE 1.
Chemical structures of potent melanocortin‐4 receptor (MC4R) drug candidate examples with MC4R‐specific Ki/IC50/EC50 values
| Name | Type | Structure | Ki/IC50/EC50 value for MC4R | Reference |
|---|---|---|---|---|
| Sodium 4‐phenylbutyrate (4‐PBA) | Chemical chaperone |
|
— | 70 , 71 |
| THIQ |
Pharmacological chaperone agonist |
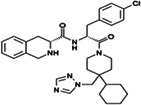
|
IC50 1.2 nM | 75 |
| NPB | Pharmacological chaperone antagonist |
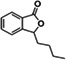
|
Ki 2.4 nM | 72 |
| Ipsen 17 | Pharmacological chaperone antagonist |
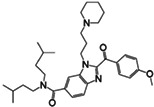
|
Ki 0.96 nM | 73 |
| RO‐273225 (Butyr‐His‐D‐Phe‐Arg‐Trp‐Sar‐NH2) | Linear peptide |
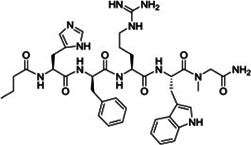
|
EC50 1 ± 0.3 | 113 |
| PL‐8905 | Cyclic peptide |
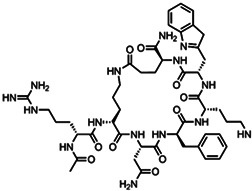
|
High affinity | 49 |
| Setmelanotide | Cyclic peptide |
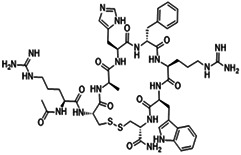
|
EC50 0.27 nM |
36 |
| 2Me‐2H tetrazole derivative | Nonpeptide agonists |
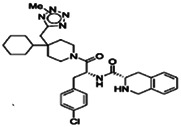
|
High affinity | 101 |
| Piperazine benzenes | Nonpeptide agonists |
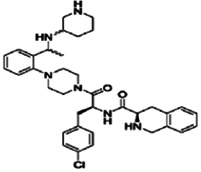
|
Ki 11 nM | 103 |
| 1,3,4‐trisubstituted‐2‐oxopiperazine | Nonpeptide agonists |
|
Ki 5.7 nM | 104 |
Abbreviations: EC50, half maximum effective concentration; IC50, half maximum inhibitory concentration; Ki, inhibition constant; MC4R, melanocortin‐4 receptor; THIQ, N‐[(3R)‐1,2,3,4‐Tetrahydroisoquinolinium‐3‐ylcarbonyl]‐(1R)‐1‐(4‐chlorobenzyl)‐2‐[4‐cyclohexyl‐4‐(1H‐1,2,4‐triazol‐1‐ylmethyl) piperidin‐1‐yl]‐2‐oxoethylamine.
5. GAIN‐OF‐FUNCTION MUTATIONS AND CONSTITUTIVE MC4R ACTIVITY
Interestingly, not all MC4R variants are associated with an increase in obesity pathology. A subset of variants has been reported to provide GoF, offering protection from obesity and associated complications. 24 , 64 , 76 In vitro assays developed to determine ligand binding, cell‐surface expression, and cAMP measurement as a function of Gs activation in wild‐type and variant MC4Rs have been successfully developed over the past two decades. The mutations S127L, 64 P230L 64 and L250Q 24 augment the constitutive activity of MC4R in heterologous expression systems. A new addition to this knowledge has been the quantification of ß‐arrestin recruitment, which was proposed as the mechanism of action of GoF variants associated with a considerable decrease in the risk of obesity and associated disorders. Lotta et al 13 screened UK Biobank data of 0.5 million people and characterized the variants to study their function as well as their association with BMI, type 2 diabetes and cardiometabolic diseases. Twelve of the 61 mutations identified in the UK population were nonsense/frameshift mutations, while 49 variants were functionally characterized and shown to be involved in the quantification of Gs‐mediated cAMP production and the recruitment of ß‐arrestin to MC4R. Of the 49 variants, 11 exhibited GoF: T11S, T101N, F201L, G231S, R236C, V103I, I251L, I289L, I317V, L304F and Y332C. 13 The first five exhibited bias toward cAMP production, the next four for ß‐arrestin recruitment, and the last two did not exhibit any bias for signalling. The frequency of reported heterozygous ß‐arrestin‐biased GoF alleles in the UK Biobank was 6.1% and that of homozygous alleles was 0.1%. Compared with noncarriers, heterozygous carriers had an intermediate risk of obesity, type 2 diabetes, and coronary artery disease, while homozygous carriers had a 50% lower risk of these conditions. 13 No change in protection from obesity was observed in carriers of the GoF variants with a preference toward cAMP production. 13
A detailed examination of the molecular mechanisms may explain the observed effects. The GoF variants that showed bias for ß‐arrestin alone (V103I, I251L, I289L and I317V) exhibited enhanced signalling via the MAPK pathway, 13 as confirmed by the overexpression of phosphorylated ERK 1/2, whereas no increase in the expression of this protein was observed in cAMP GoF variants. As expected, wild‐type MC4Rs translocated from the membrane to the cytoplasm upon agonist stimulation reduced the surface expression of MC4R, by 23%. The most frequently observed GoF variant, V103I, remained at the cell surface and showed no change in cell‐surface expression, 13 which could be because of impaired internalization or improved recycling.
6. MELANOCORTIN‐4 RECEPTOR CRYSTAL STRUCTURE FOCUSING ON THE ROLE OF Ca 2+: A COMPARISON WITH OTHER GPCRS AND THEIR IONIC BINDING
An in‐depth understanding of the function and pharmacological roles of ions and ion‐binding sites in GPCRs has also become possible in the past decade, with advances in their biophysical, structural and functional characterization, marking the beginning of new avenues for the discovery of potentially safer and more efficient drugs. Briefly, monovalent and divalent cations can act selectively and nonselectively at various sites of different GPCRs. Physiological concentrations of these ions can act as allosteric modulators in some cases. 77 Some GPCRs are selectively modulated by inorganic ions that facilitate the receptor's physiological function. 78 , 79 Some unique or highly conserved sites for specific ions have also been reported; for example, Class A GPCRs have a Na+‐binding site that functions as a near‐universal allosteric modulator for GPCR structure and function, and the origin of this site can be traced as far back as prokaryotic rhodopsin channels. 80 , 81 Fifteen residues in this highly conserved sodium pocket are conserved in 45 diverse receptors, with minor variants occurring in the majority of Class A GPCR members. Receptors lacking this Na+ pocket exhibit extremely compromised ligand‐induced signalling. 82 Also, structural changes that effect a shift in the position of the sodium pocket might result in changing the overall coordination and conservation pattern. 83 Crucial zinc (Zn2+)‐binding sites have also been reported, for example, in the Class A GPCR, platelet‐activating factor receptor. 84
With MC4R being an important drug target for obesity, gaining knowledge of its crystal structure is extremely beneficial. Yu et al 35 first reported the structure of human MC4R with the antagonist SHU9119 (a cyclic peptide) at 2.8 Å resolution. Analysis of the MC4R‐SHU9119 complex revealed the classic seven‐transmembrane helical structure in addition to details of the interactions involving the transmembrane and loop domains. Calcium was identified as a cofactor for ligand binding 35 and was complexed with amino acid residues of the receptor as well as the ligand (SHU9119). Intriguingly, extracellular calcium increased the affinity of endogenous α‐MSH by 37 times and the potency of α‐MSH more than 600‐fold, while showing no selective effect on AgRP binding or the antagonist SUH9119. 35 The Ca2+‐binding site in MC4R is distinct from the Na+‐ and Zn2+‐binding sites in other GPCRs. Although MC4R is a member of Class A, it has been reported to differ from other Class A GPCRs in many respects, including its ion/ion‐binding site, which confers its different functionality. MC4R shows more structural divergence as a GPCR, exhibiting a greater likeness to lipidic GPCRs than homologous peptidic GPCRs. 35 The structure of MC4R is in fact different from all other reported GPCRs. A comparison of the MC4R structure with other Class A GPCRs was carried out using the root‐mean‐square deviation of Cα atoms in the inactive state in the transmembrane regions. MC4R exceeded 2.2 Å, 35 which is closer to that of lysophosphatidic acid receptor 1 and is more than the 2.0 Å calculated for other GPCRs. The reason for the higher Cα value in MC4R could include the following: (a) the very short extracellular loop (ECL) 2; (b) the absence of the conserved disulphide bond that connects ECL2 to helix III in other Class A GPCRs 85 ; (c) the distinct outward position of the helix V; or (d) the presence of nonconserved residues, such as H5.5, D3.25 and G2.58. 35 It is interesting that Ca2+ binding only affects α‐MSH and has no effect on AgRP binding. The relatively unexplored selectivity of the ionic cofactors should be investigated further with respect to transducer coupling and downstream signalling, to boost drug discovery prospects. 86
7. ANTIOBESITY DRUG DEVELOPMENT WITH MC4R AS A TARGET
Melanocortin‐4 receptor, the key monogenic cause of obesity, is befitting as a strategic target for antiobesity drugs. The ligands of this receptor, including ACTH and α‐, ß‐ and γ‐MSH, are derived from the precursor POMC peptide. 87 ACTH [SYS MEHFRWGKPV GKKRRPVKVY PNGAEDESAE AFPLEF] is further processed to yield α‐MSH [SYS MEHFRWGKPV]. 87 , 88 α‐MSH adopts a ß‐turn conformation that presents histidine‐phenylalanine‐arginine‐tryptophan (HFRW) for receptor binding, interacting with the ionic and aromatic amino acids in the upper second and third transmembrane domains. 89 MSHs lack selectivity in humans, and they have additional roles in pigmentation, hormone regulation and antiinflammation, which limit their use in drug development. The hunt for a safe, potentially active, and highly specific drug continues.
The criteria for a good agonist include safety, selectivity, efficiency and bioavailability 49 ; the agonist must be harmless and incapable of causing any untoward effects in the body. The potency/ efficiency relates to its capacity to induce the desired response at the minimum possible concentration. Its selectivity refers to its ability to activate a single desired pathway, while an agonist's bioavailability depends on its degree of solubility in body fluid and ease of assimilation in the body. Most synthetic MC4R agonists fall into one of three categories: linear peptides, cyclic peptides and nonpeptides. The structure and Ki/IC50/EC50 values of important peptides/nonpeptides are shown in Table 1.
7.1. Linear peptides
Linear peptides are commonly between five and seven amino acid residues long, but may vary from four to 16 residues, and are held together by simple amide bonds. Most synthesis procedures are based on the substitution of amino acid residues in α‐MSH [S1Y2S3ME HFRW GKPV13], 49 particularly those in the core motif HFRW and/or two or three flanking sequences on either side. The core aim behind inducing and screening the various substitutions is to find novel potent ligand moieties with good selectivity and overall efficiency. A suitable agonist should have high potency (EC50 < 10 nm), high selectivity (>50 times EC50 for MC4R), 49 and better stability and safety compared with the unsubstituted parent peptide. The first step is the synthesis, which includes designing and inducing changes that might be useful, followed by a series of in vitro validation steps, including quantification of the functional activity of the proposed new peptide. For example, D‐Phe (synthetic dextro isomer of Phenylalanine)/D‐Phe analogues, the first substitutions reported, considerably increased the agonist's activity and ligand stability. 90 Haslach et al 91 reported that the use of a D‐Phe analogue with a halogen at the para position provided higher agonist activity and better ligand stability compared with that of D‐Phe. Histidine has been substituted with Tyrosine, Atc (2‐aminotetralin‐2‐carboxylic acid), Apc (1‐amino‐4‐phenylcyclohexane‐1‐carboxylic acid), and other residues to achieve more potent and selective peptides. Later, arginine was also proposed as a replacement for histidine. 91 Trptophan provides a better substitute than the Phe analogue, as it has an electron‐withdrawing group at its para position that enhances ligand‐receptor interactions. In addition to glutamine/glycine at the fifth position (first left of H), butyl and pentyl groups also act as effective ligands. Active ligands are formed by replacing glycine, first right of W, with acidic or neutral amino acids. However, in general, obtaining a potent/selective linear ligand for MC4R has been a challenge that has met with limited success.
Interestingly, the constitutive activity of MC4R is induced by its N‐terminal domain [HLWNRSS] and its transmembrane domain, which undergo spontaneous conformational transformations to change inactive MC4R to active MC4R. 92 The amino acid residues that take part in binding in case of constitutive activity differ from those involved in regular ligand binding, suggesting there is room for additional positive allosteric modulation and the potential to develop alternative therapeutic candidates. 93 , 94 , 95
7.2. Cyclic peptides
These are typically amino acids or amino acid analogues that are cyclized by disulphide bonds. The established core motif (HFRW) remains the same as in linear peptides; however, the potential substitutions and analogue designs differ to best fit the receptor, with the aim of conferring maximum efficiency and potency. Examples of the most effective substitutions include Phe to D‐Phe and/or D‐2‐naphthyalanine and His to either polar or nonpolar moieties. Acidic amino acids (glutamate, aspartate, or D‐alanine) are preferable at position 5, while alanine, lysine and cystine are optimum at position 10. In general, the replacement of Met at position 4 with lipophilic residues, such as norleucine or acidic residues, is favoured. Neutral or acidic compounds with short side chains at position 5 improve the potency of the ligand, while a change in chirality results in higher potency at position 7.
Multivalency can increase ligand‐receptor affinity, and introducing bivalent agonists reportedly increases potency. 96 The melanocortin bivalent agonist CJL‐1‐87, with two repeats in the structure that are linked by an oligomer, shows approximately seven times the potency of the monovalent structure. 97 Fernandes et al 98 examined the effect of homo/hetero bivalency, combining a linear, truncated NDP‐MSH with cyclic SHU9119 separated by a series of linkers of varying flexibility, for example, PEGO (19‐amino‐5‐oxo‐3,10,13,16‐tetraoxa‐6‐azanonadecan‐1‐oic acid) linkers. The heterobivalent ligand was five times more active against MC4R compared with the monovalent equivalents, indicating a cooperative effect upon binding, promoted by the flexible linker. 98
An example of an extremely efficient and safe cyclic peptide is setmelanotide. In addition to the tremendous (100‐fold) increase in downstream signalling on MC4R activation, it also reduces the undesirable side effects. This is suggested to be the result of biased signalling of setmelanotide at the MC4R. Compared with other tested drugs, setmelanotide is unique in that it is reported to activate nuclear factor of activated T cell (NFAT) signalling and restore the function of many MC4R variants. 26 , 99 Setmelanotide is more effective in stimulating cAMP accrual in the presence of AgRP compared with α‐MSH and LY2112688 (a first generation MC4R agonist). AgRP competes with α‐MSH and LY2112688 in the MC4R binding pocket but fails to displace setmelanotide owing to the superior binding affinity of setmelanotide. 26 , 99
7.3. Nonpeptide ligands
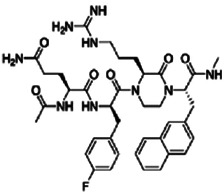
Substitutions within nonpeptide ligands are generally more effective than those in peptide ligands because the former have a compact, rigid structure. 49 Additionally, as nonpeptide ligands are resistant to proteolysis, they tend to be more stable compared with peptide ligands. Various nonpeptide agonists based on the ß‐turn motif were investigated by Haskell‐Luevano et al 100 using cyclic lactam templates of the, then leading, structures melanotan‐II and SHU9119. Sebhat et al 101 used a piperidine core and introduced triazoles/tetrazoles to develop the first potent and selective nonpeptide MC4R agonist. Fotsch et al 102 introduced tryptamine conjugated with cyclohexane 1,4‐diamine or butyl guanidine to mimic tryptophan and arginine, respectively. The resulting agonist was potent yet lacked specificity for MC4R; however, introducing piperazine as the principal scaffold yielded the required selectivity. 103 Following this, Tian et al 104 synthesized various 1,3,4‐trisubstituted 2‐oxopiperazines and further capped the ligand with a tetrapeptide core. The designed dipeptide and tripeptide analogues showed excellent binding affinity (nanomole scale), potency and selectivity for MC4R compared with MC1R. 104
8. DRUGS AT THE APPROVAL OR CLINICAL TRIAL STAGE
Structure‐based ligand discovery has provided superior and selective agonists to promote MC4R function. Some of the drugs in clinical trials include LY2112688, melanotan‐II, bremelanotide, PL‐8905 and setmelanotide. Although well characterized in vitro and in vivo, undesirable side effects have been reported for many of these drugs in clinical trials. LY2112688 caused increased blood pressure. 26 , 105 Melenotan‐II, a super‐potent cyclic MC4R agonist, caused penile erection in males and darkening of the skin. 49 , 106 Bremelanotide was more closely linked to sexual dysfunction than weight reduction in both men and women and failed as an antiobesity drug in clinical trials. 49 , 107 PL‐8905 is in clinical trials after exhibiting minimal side effects, such as changes in blood pressure, in preclinical studies. 49 , 108 Setmelanotide showed considerable promise with no observed side effects in phase III clinical trials 36 and has now been approved by the FDA. 108 Liraglutide, a glucagon‐like peptide‐1 receptor agonist (GLP‐1RA), also causes weight loss by reducing appetite. It has been reported to be effective in many cases of monogenic obesity. 109 Liraglutide treatment is reported to increase bone mass in common obesity, however, no change in bone metabolism was seen in obesity caused due to mutations in MC4R. 110 A combination of liraglutide therapy and exercise improves maintenance of weight loss (as weight regain after a weight loss is a common problem) compared to either exercise or drug treatment alone. 111 Sun et al 112 have proposed a gut‐intrinsic melanocortin signalling complex involving α‐MSH release and MC4R activation on L cells secretion in humans. This could directly target mucosal MC4R to treat human metabolic disorders including obesity. A comprehensive summary of all drugs developed thus far as agents to treat obesity, including MC4R agonists, and their targets, mode of action, potential side effects, and status with regards to FDA approval, is shown in Table 2.
TABLE 2.
List of antiobesity drugs
| Drug | Target | Mechanism of action | Usage | Side‐effects | Clinical status | Reference |
|---|---|---|---|---|---|---|
| Section I | ||||||
| Setmelanotide | MC4R | Decreased food intake and increased energy expenditure via MC4R binding | LT | Reported safe | Approved | 36, 108 |
| PL‐8905 | MC4R | ‐do‐* | — | Reported safe | Clinical studies | 49 |
| LY2112688 | MC4R | ‐do‐ | — | Increased systolic blood pressure | Failed in clinical studies | 26, 105 |
| Melanotan‐II | MC4R | ‐do‐ | — | Spontaneous penile erection; skin darkening | Failed for obesity | 49, 106 |
| Bremelanotide | MC4R | ‐do‐ | — | Increase blood pressure and sexual activity | Failed for obesity | 49, 107 |
| 4‐PBA | MC4R | Acts as chemical chaperone and helps rescue intracellular retention of variant MC4Rs | — | Lacks specificity | Preclinical | 69, 70, 71 |
| UBE‐41 | MC4R | ‐do‐ | — | Lacks specificity | Preclinical | 70, 71 |
| THIQ | MC4R | Acts as pharmacological chaperone and helps rescuing intracellular retention of variant MC4Rs | — | Prolonged exposure decreases cell surface expression and signalling | Preclinical | 75 |
| NBP | MC4R | ‐do‐ | — | ‐do‐ | Preclinical | 72 |
| ML00253764 | MC4R | ‐do‐ | — | High EC50 | Preclinical | 69, 72 |
| DCPMP | MC4R | ‐do‐ | — | High EC50 | Preclinical | 69, 72 |
| Ipsen 5i | MC4R | ‐do‐ | — | Reported efficient | Preclinical | 75 |
| Ipsen 17 | MC4R | ‐do‐ | — | Reported efficient | Preclinical | 73 |
| Section II | ||||||
| Orlistat | Pancreatic/stomach lipases | Decreases fat absorption | LT | Abdominal pain, diarrhea | Approved | 114 |
| Liraglutide | GLP‐1R | Centrally (CNS) mediated | LT | Adverse GI effects | Approved | 115, 116 |
| Semaglutide | GLP‐1R | ‐do‐ | LT | ‐do‐ | Approved | 117 |
| Naltrexone‐ Buproprion | α‐MSH/ß‐endorphin | Possible modulation of melanocortin system | LT | Adverse GI effects; dizziness/insomnia | Approved | 118 |
| Lorcaserin | Serotonin/5HT receptor | Modulates melanocortin system | LT | Headache, weakness, bradycardia, cognitive impairment | Approved | 119, 120 |
| Leptin | POMC/NPY neurons | Modulates the melanocortin system | LT | Exogenous administration largely ineffective | Approved as combinatorial therapy | 121, 122 |
| Section III | ||||||
| Amphetamine compounds | POMC/NPY neurons | High metabolic rate; stimulation of anorectic/inhibition of orectic signals | Short‐term | Addictive in nature | Approved (less addictive analogues now available) | 122, 123, 124 |
| Methamphetamine desoxyephedrine | ‐do‐ | ‐do‐ | ‐do‐ | ‐do‐ | Approved | 123, 125 |
| Deoxyphedrine | ‐do‐ | ‐do‐ | ‐do‐ | ‐do‐ | Approved | 126 |
| Amphetamine congeners (AC) | ‐do‐ | ‐do‐ | ‐do‐ | Additive in general | Approved | 127 |
| Diethylpropion (AC) | ‐do‐ | ‐do‐ | ‐do‐ | Limited drug efficiency | Approved | 128 |
| Phendimetrazine (AC) | ‐do‐ | ‐do‐ | ‐do‐ | Insomnia, dry mouth, constipation | Approved | 129 |
| Benzphetamine (AC) | ‐do‐ | ‐do‐ | ‐do‐ | Insomnia, dry mouth, mood swings | Approved | 130 |
| Phentermine | ‐do‐ | Increased energy consumption and anorexia | ‐do‐ | Insomnia, dry mouth, mood swings | Approved | 131, 132 |
| Phentermine/topiramate (Qsymia) | Glutamate and GABA receptors | Weight loss and decrease in CNS neuronal activity via Ca2+ channels | ‐do‐ | Insomnia, dry mouth, dizziness | Approved | 108, 120, 133 |
| Section IV | ||||||
| MEDI0382 | GLP‐1R/GCGR | Bi‐agonist targeting | — | — | Phase II | 134 |
| NNC0090‐2746 (RG7697) | GLP‐1R/GIPR | Bi‐agonist targeting | — | — | Phase IIa | 135 |
| LY3298176 | GLP‐1R/GIPR | Bi‐agonist targeting | — | — | Phase II complete | 136 |
| HM15211 | GLP‐1R/GCGR/GIPR | Tri‐agonist targeting | — | — | Preclinical | 137 |
| NN9423/NNC9204‐1706 | GLP‐1R/GCGR/GIPR | Tri‐agonist targeting | — | — | Phase I | 138 |
| Section V | ||||||
|
GLP‐1 delivering Estrogen |
Peptide mediated hormone delivery | Peripheral/central regulation by modulation of energy sensors | Long‐term | Risk of breast cancer, heart ailments, stroke, dementia | Preclinical | 139 |
| 17ß‐estradiol (E2) | ‐do‐ | ‐do‐ | Long‐term | ‐do‐ | Preclinical | 140 |
| Glucagon/T3 | ‐do‐ | Modulation of energy expenditure via BAT thermogenesis | — | — | Preclinical | 141 |
| GLP‐1 delivering dexamethasone | ‐do‐ | Energy balance and weight loss via hypothalamic neurocircuits | — | — | Preclinical | 142 |
| Section VI | ||||||
| Dinitrophenol | Mitochondrial uncoupling | High metabolic rate | — | Hyperthermia, tachycardia, nausea, vomiting | Withdrawn | 143 |
| Serotonergics | Seratonin/5HT | Seratonergic/Melanocortinergic system | — | Pulmonary hypertension; valvular heart disease | Withdrawn | 128, 131 |
| Fenfluramine | ‐do‐ | ‐do‐ | — | ‐do‐ | Withdrawn | 144, 145 |
| Dexfenfluramine | ‐do‐ | ‐do‐ | — | ‐do‐ | Withdrawn | 144, 145 |
| Sibutramine | Serotonin/norepinephrine inhibitor | ‐do‐ | — | High BP, cardiac arrhythmia | Withdrawn | 146 |
| Rimonabant | Type I CB1R | Weight loss by modulating hemostatic and hedonic feeding circuits | — | Adverse psychiatric effects | Withdrawn | 147 |
Section I: Antiobesity drugs targeting MC4R; Section II: General antiobesity drugs; Section III: Drugs approved for short‐term use only because of potential side effects and addictive nature. Section IV: Bi‐ and tri‐agonist drug targets (in developmental stage); Section V: Peptide‐hormone based drugs (in developmental stage); Section VI: Drugs that have been withdrawn as a result of extreme side effects.
Note: *‐do‐ Refers to repeat the exact words/content of the row above, in that specified column, to avoid writing the same information multiple times in the table.
Abbreviations: 4‐PBA, sodium 4‐phenylbutyrate; AC, adenylyl cyclase; ACTH, adrenocorticotropic hormone; BP, blood pressure; DCPMP, N‐((2R)‐3(2,4‐dichlorophenyl)‐1‐(4‐(2‐([1‐methoxypropan2‐ylamino] methyl) phenyl) piperazin‐1‐yl)‐1‐oxopropan2‐yl) propionamide; ECL, extracellular loop; GABA, gamma‐aminobutyric acid; GCGR, glucagon receptor; GI, gastrointestinal; GIPR, glucose‐dependent insulinotropic polypeptide; GLP‐1R, glucagon‐like peptide‐1 receptor; MC4R, melanocortin 4‐receptor; NBP, 1‐(1‐(4‐fluorophenyl)‐ 2‐(4‐(4‐[naphthalene‐1‐yl] butyl) piperazin‐1‐yl) ethyl)‐4‐ methylpiperazine; NPY, neuropeptide Y; POMC, proopiomelanocortin; THIQ, N‐[(3R)‐1,2,3,4‐Tetrahydroisoquinolinium‐3‐ylcarbonyl]‐(1R)‐1‐(4‐chlorobenzyl)‐2‐[4‐cyclohexyl‐4‐(1H‐1,2,4‐triazol‐1‐ylmethyl) piperidin‐1‐yl]‐2‐oxoethylamine; UBE‐41, ubiquitin activating enzyme inhibitor.
9. CONCLUDING REMARKS
The dramatic increase in obesity, its associated disorders, and related mortality is alarming. Most drugs approved so far to treat obesity cause considerable side effects, especially to the nervous and gastrointestinal systems. Some have been withdrawn or are only prescribed for short‐term use as a part of combination therapies. Overall, the development of effective drugs to treat obesity has been challenging. Encouragingly, however, our understanding of the genetics, molecular mechanisms, and structure of MC4R and other GPCRs, some of which are likely to contribute to the pathology of obesity, has increased tremendously. Setmelanotide, the latest FDA‐approved drug for use against obesity, reportedly induced no side effects in clinical trials, which gives us hope for its sustained and efficient use in the future. The biased NFAT signalling of setmelanotide, in addition to its efficient ligand binding, is probably the reason behind its success as a drug candidate, emphasizing the importance of designing effective ligand substitutes and investigating novel molecular pathways. ß‐arrestin‐biased signalling in the case of GoF variants, which provide protection against obesity and associated disorders, is another crucial example suggesting potential therapeutic approaches in the future would benefit from smart drug design and investigation of unconventional pathways as well as canonical ones. Thus, designing specific drugs that can selectively activate or block specific targets such as arrestin as well as improve ligand‐receptor interactions may be a promising therapeutic direction. All these advancements that elucidate the finer details of obesity pathology will undoubtedly provide useful insights into how to effectively target specific receptors, leading to the design of safe and efficient drugs with which to treat obesity and other diseases.
CONFLICT OF INTEREST
The author(s) declare(s) that they have no competing interests
AUTHOR CONTRIBUTIONS
Original draft preparation, Munazza Tamkeen Fatima and Ammira Al‐Shabeeb Akil; review and editing, Ammira Al‐Shabeeb Akil and Khalid Adnan Fakhro; visualization, Munazza Tamkeen Fatima; supervision, Ammira Al‐Shabeeb Akil; Ikhlak Ahmed; project administration, Ammira Al‐Shabeeb Akil, funding acquisition, Ammira Al‐Shabeeb Akil. All authors have read and agreed to the published version of the manuscript.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/dom.14618.
ACKNOWLEDGMENTS
This research was funded by Sidra Medicine through its Precision Medicine Program Grant—SDR#400149, Doha, Qatar.
Fatima MT, Ahmed I, Fakhro KA, Akil A‐S. Melanocortin‐4 receptor complexity in energy homeostasis,obesity and drug development strategies. Diabetes Obes Metab. 2022;24(4):583-598. doi: 10.1111/dom.14618
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Scully T, Ettela A, LeRoith D, Gallagher EJ. Obesity, type 2 diabetes, and cancer risk. Front Oncologia. 2021;10. doi: 10.3389/fonc.2020.615375 615375–615375. https://www.frontiersin.org/articles/10.3389/fonc.2020.615375/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. WHO (World Health Organization) . WHO obesity and overweight fact sheet no 311. Obes Oveweight Fact Sheet 2013.
- 3. WHO . Obesity and overweight. Fact sheet no 311 January 2015. Factsheeds 2015.
- 4. Corbin LJ, Timpson NJ. Body mass index: has epidemiology started to break down causal contributions to health and disease? Obesity (Silver Spring). 2016;24(8):1630‐1638. doi: 10.1002/oby.21554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huvenne H, Dubern B, Clément K, Poitou C. Rare genetic forms of obesity: clinical approach and current treatments in 2016. Obes Facts. 2016;9(3):158‐173. doi: 10.1159/000445061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singh RK, Kumar P, Mahalingam K. Molecular genetics of human obesity: a comprehensive review. C R Biol. 2017;340(2):87‐108. doi: 10.1016/j.crvi.2016.11.007 [DOI] [PubMed] [Google Scholar]
- 7. Fairbrother U, Kidd E, Malagamuwa T, Walley A. Genetics of severe obesity. Curr Diab Rep. 2018;18(10):85. doi: 10.1007/s11892-018-1053-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Flores‐Dorantes MT, Díaz‐López YE, Gutiérrez‐Aguilar R. Environment and Gene Association with obesity and their impact on neurodegenerative and neurodevelopmental diseases. Front Neurosci. 2020;14(863). doi: 10.3389/fnins.2020.00863. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7483585/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marenne G, Hendricks AE, Perdikari A, et al. Exome sequencing identifies genes and gene sets contributing to severe childhood obesity, linking PHIP variants to repressed POMC transcription. Cell Metab. 2020;31(6):1107‐1119.e12. doi: 10.1016/j.cmet.2020.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brouwers B, de Oliveira EM, Marti‐Solano M, et al. Human MC4R variants affect endocytosis, trafficking and dimerization revealing multiple cellular mechanisms involved in weight regulation. Cell Rep. 2021;34(12):108862. doi: 10.1016/j.celrep.2021.108862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anderson EJP, Çakir I, Carrington SJ, et al. 60 YEARS OF POMC: regulation of feeding and energy homeostasis by α‐MSH. J Mol Endocrinol. 2016;56(4):T157‐T174. doi: 10.1530/JME-16-0014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ramos‐Molina B, Martin MG, Lindberg I. PCSK1 variants and human obesity. Progress in Molecular Biology and Translational Science. Vol 140; 2016:47‐74. doi: 10.1016/bs.pmbts.2015.12.001. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6082390/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lotta LA, Mokrosiński J, Mendes de Oliveira E, et al. Human gain‐of‐function MC4R variants show signaling bias and protect against obesity. Cell. 2019;177(3):597‐607.e9. doi: 10.1016/j.cell.2019.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Magenis RE, Smith L, Nadeau JH, Johnson KR, Mountjoy KG, Cone RD. Mapping of the ACTH, MSH, and neural (MC3 and MC4) melanocortin receptors in the mouse and human. Mamm Genome. 1994;5(8):503‐508. doi: 10.1007/BF00369320 [DOI] [PubMed] [Google Scholar]
- 15. Tao Y‐X. The melanocortin‐4 receptor: physiology, pharmacology, and pathophysiology. Endocr Rev. 2010;31(4):506‐543. doi: 10.1210/er.2009-0037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Delhanty PJD, Bouw E, Huisman M, et al. Functional characterization of a new human melanocortin‐4 receptor homozygous mutation (N72K) that is associated with early‐onset obesity. Mol Biol Rep. 2014;41(12):7967‐7972. doi: 10.1007/s11033-014-3691-7 [DOI] [PubMed] [Google Scholar]
- 17. Huszar D, Lynch CA, Fairchild‐Huntress V, et al. Targeted disruption of the melanocortin‐4 receptor results in obesity in mice. Cell. 1997;88(1):131‐141. doi: 10.1016/S0092-8674(00)81865-6 [DOI] [PubMed] [Google Scholar]
- 18. Vaisse C, Clement K, Guy‐Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20(2):113‐114. doi: 10.1038/2407 [DOI] [PubMed] [Google Scholar]
- 19. Tao Y‐X. Molecular mechanisms of the neural melanocortin receptor dysfunction in severe early onset obesity. Mol Cell Endocrinol. 2005;239(1‐2):1‐14. doi: 10.1016/j.mce.2005.04.012 [DOI] [PubMed] [Google Scholar]
- 20. Hinney A, Volckmar A‐L, Knoll N. Melanocortin‐4 receptor in energy homeostasis and obesity pathogenesis. Progress in Molecular Biology and Translational Science. Vol 114; 2013:147‐191. doi: 10.1016/B978-0-12-386933-3.00005-4. http://www.ncbi.nlm.nih.gov/pubmed/23317785 [DOI] [PubMed] [Google Scholar]
- 21. Yeo GSH, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O'Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20(2):111‐112. doi: 10.1038/2404 [DOI] [PubMed] [Google Scholar]
- 22. Tao Y‐X. Chapter 6 mutations in melanocortin‐4 receptor and human obesity. Progress in Molecular Biology and Translational Science. Vol 88; 2009:173‐204. doi: 10.1016/S1877-1173(09)88006-X. http://www.ncbi.nlm.nih.gov/pubmed/20374728 [DOI] [PubMed] [Google Scholar]
- 23. Farooqi IS, Yeo GSH, Keogh JM, et al. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest. 2000;106(2):271‐279. doi: 10.1172/JCI9397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vaisse C, Clement K, Durand E, Hercberg S, Guy‐Grand B, Froguel P. Melanocortin‐4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest. 2000;106(2):253‐262. doi: 10.1172/JCI9238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. MacKenzie RG. Obesity‐associated mutations in the human melanocortin‐4 receptor gene. Peptides. 2006;27(2):395‐403. doi: 10.1016/j.peptides.2005.03.064 [DOI] [PubMed] [Google Scholar]
- 26. Kühnen P, Krude H, Biebermann H. Melanocortin‐4 receptor signalling: importance for weight regulation and obesity treatment. Trends Mol Med. 2019;25(2):136‐148. doi: 10.1016/j.molmed.2018.12.002 [DOI] [PubMed] [Google Scholar]
- 27. Trevellin E, Granzotto M, Host C, et al. A novel loss of function melanocortin‐4‐receptor mutation (MC4R‐F313Sfs*29) in morbid obesity. J Clin Endocrinol Metab. 2021;106(3):736‐749. doi: 10.1210/clinem/dgaa885 [DOI] [PubMed] [Google Scholar]
- 28. Namjou B, Stanaway IB, Lingren T, et al. Evaluation of the MC4R gene across eMERGE network identifies many unreported obesity‐associated variants. Int J Obes (Lond). 2021;45(1):155‐169. doi: 10.1038/s41366-020-00675-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aykut A, Özen S, Gökşen D, et al. Melanocortin 4 receptor (MC4R) gene variants in children and adolescents having familial early‐onset obesity: genetic and clinical characteristics. Eur J Pediatr. 2020;179(9):1445‐1452. doi: 10.1007/s00431-020-03630-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paolini B, Maltese PE, Del Ciondolo I, et al. Prevalence of mutations in LEP, LEPR, and MC4R genes in individuals with severe obesity. Genet Mol Res. 2016;15(3). doi: 10.4238/gmr.15038718. http://www.ncbi.nlm.nih.gov/pubmed/27706562 [DOI] [PubMed] [Google Scholar]
- 31. Farooqi IS, Keogh JM, Yeo GSH, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348(12):1085‐1095. doi: 10.1056/NEJMoa022050 [DOI] [PubMed] [Google Scholar]
- 32. Martinelli CE, Keogh JM, Greenfield JR, et al. Obesity due to melanocortin 4 receptor (MC4R) deficiency is associated with increased linear growth and final height, fasting hyperinsulinemia, and incompletely suppressed growth hormone secretion. J Clin Endocrinol Metab. 2011;96(1):E181‐E188. doi: 10.1210/jc.2010-1369 [DOI] [PubMed] [Google Scholar]
- 33. Nourbakhsh M, Sharifi R, Ghorbanhosseini SS, et al. Evaluation of plasma TRB3 and Sestrin 2 levels in obese and normal‐weight children. Child Obes. 2017;13(5):409‐414. doi: 10.1089/chi.2017.0082 [DOI] [PubMed] [Google Scholar]
- 34. Latorraca NR, Wang JK, Bauer B, et al. Molecular mechanism of GPCR‐mediated arrestin activation. Nature. 2018;557(7705):452‐456. doi: 10.1038/s41586-018-0077-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu J, Gimenez LE, Hernandez CC, et al. Determination of the melanocortin‐4 receptor structure identifies Ca2+ as a cofactor for ligand binding. Science. 2020;368(6489):428‐433. doi: 10.1126/science.aaz8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Clément K, Van Den AE, Argente J, et al. Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency:single‐arm, open‐label, multicentre, phase 3 trials. Lancet Diabetes Endocrinol. 2020;8:960‐970. [DOI] [PubMed] [Google Scholar]
- 37. Heng BC, Aubel D, Fussenegger M. An overview of the diverse roles of G‐protein coupled receptors (GPCRs) in the pathophysiology of various human diseases. Biotechnol Adv. 2013;31(8):1676‐1694. doi: 10.1016/j.biotechadv.2013.08.017 [DOI] [PubMed] [Google Scholar]
- 38. Sommer ME, Selent J, Carlsson J, et al. The European Research Network On Signal Transduction (ERNEST): toward a multidimensional holistic understanding of G protein‐coupled receptor signaling. ACS Pharmacol Transl Sci. 2020;3(2):361‐370. doi: 10.1021/acsptsci.0c00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alhosaini K, Azhar A, Alonazi A, Al‐Zoghaibi F. GPCRs: the most promiscuous druggable receptor of the mankind. Saudi Pharm J. 2021;29(6):539‐551. doi: 10.1016/j.jsps.2021.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pierce KL, Premont RT, Lefkowitz RJ. Seven‐transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3(9):639‐650. doi: 10.1038/nrm908 [DOI] [PubMed] [Google Scholar]
- 41. Rankovic Z, Brust TF, Bohn LM. Biased agonism: an emerging paradigm in GPCR drug discovery. Bioorg Med Chem Lett. 2016;26(2):241‐250. doi: 10.1016/j.bmcl.2015.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hill JO, Wyatt HR, Peters JC. Energy balance and obesity. Circulation. 2012;126(1):126‐132. doi: 10.1161/CIRCULATIONAHA.111.087213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. You P, Hu H, Chen Y, et al. Effects of melanocortin 3 and 4 receptor deficiency on energy homeostasis in rats. Sci Rep. 2016;6(1):34938. doi: 10.1038/srep34938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pei H, Patterson CM, Sutton AK, Burnett KH, Myers MG, Olson DP. Lateral hypothalamic Mc3R‐expressing neurons modulate locomotor activity, energy expenditure, and adiposity in male mice. Endocrinology. 2019;160(2):343‐358. doi: 10.1210/en.2018-00747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lensing CJ, Adank DN, Doering SR, et al. Ac‐Trp‐DPhe(p‐I)‐Arg‐Trp‐NH2, a 250‐fold selective melanocortin‐4 receptor (MC4R) antagonist over the melanocortin‐3 receptor (MC3R), affects energy homeostasis in male and female mice differently. ACS Chem Nerosci. 2016;7:1283‐1291. doi: 10.1021/acschemneuro.6b00156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baldini G, Phelan KD. The melanocortin pathway and control of appetite‐progress and therapeutic implications. J Endocrinol. 2019;241(1):R1‐R33. doi: 10.1530/JOE-18-0596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Glas E, Mückter H, Gudermann T, Breit A. Exchange factors directly activated by cAMP mediate melanocortin 4 receptor‐induced gene expression. Sci Rep. 2016;6(1):32776. doi: 10.1038/srep32776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Donohoue PA, Tao Y‐X, Collins M, Yeo GSH, O'Rahilly S, Segaloff DL. Deletion of codons 88‐92 of the melanocortin‐4 receptor gene: a novel deleterious mutation in an obese female. J Clin Endocrinol Metab. 2003;88(12):5841‐5845. doi: 10.1210/jc.2003-030903 [DOI] [PubMed] [Google Scholar]
- 49. Gonçalves JPL, Palmer D, Meldal M. MC4R agonists: structural overview on antiobesity therapeutics. Trends Pharmacol Sci. 2018;39(4):402‐423. doi: 10.1016/j.tips.2018.01.004 [DOI] [PubMed] [Google Scholar]
- 50. Gale SM, Castracane VD, Mantzoros CS. Energy homeostasis, obesity and eating disorders: recent advances in endocrinology. J Nutr. 2004;134(2):295‐298. doi: 10.1093/jn/134.2.295 [DOI] [PubMed] [Google Scholar]
- 51. Garfield AS, Lam DD, Marston OJ, Przydzial MJ, Heisler LK. Role of central melanocortin pathways in energy homeostasis. Trends Endocrinol Metab. 2009;20(5):203‐215. doi: 10.1016/j.tem.2009.02.002 [DOI] [PubMed] [Google Scholar]
- 52. Leibel RL. The molecular genetics of the melanocortin pathway and energy homeostasis. Cell Metab. 2006;3(2):79‐81. doi: 10.1016/j.cmet.2006.01.010 [DOI] [PubMed] [Google Scholar]
- 53. Ramachandrappa S, Raimondo A, Cali AMG, et al. Rare variants in single‐minded 1 (SIM1) are associated with severe obesity. J Clin Invest. 2013;123(7):3042‐3050. doi: 10.1172/JCI68016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Blanco AM, Bertucci JI, Hatef A, Unniappan S. Feeding and food availability modulate brain‐derived neurotrophic factor, an orexigen with metabolic roles in zebrafish. Sci Rep. 2020;10(1):10727. doi: 10.1038/s41598-020-67535-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cabrera‐Vera TM, Vanhauwe J, Thomas TO, et al. Insights into G protein structure, function, and regulation. Endocr Rev. 2003;24(6):765‐781. doi: 10.1210/er.2000-0026 [DOI] [PubMed] [Google Scholar]
- 56. Landry Y, Niederhoffer N, Sick E, Gies JP. Heptahelical and other G‐protein‐coupled receptors (GPCRs) signaling. Curr Med Chem. 2006;13(1):51‐63. doi: 10.2174/092986706775197953 [DOI] [PubMed] [Google Scholar]
- 57. Sunahara RK. Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Interv. 2002;2(3):168‐184. doi: 10.1124/mi.2.3.168 [DOI] [PubMed] [Google Scholar]
- 58. Clapham DE, Neer EJ. G protein beta gamma subunits. Annu Rev Pharmacol Toxicol. 1997;37:167‐203. doi: 10.1146/annurev.pharmtox.37.1.167 [DOI] [PubMed] [Google Scholar]
- 59. Gresset A, Sondek J, Harden TK. The phospholipase C isozymes and their regulation. Subcell Biochem. 2012;58:61‐94. doi: 10.1007/978-94-007-3012-0_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mo X‐L, Tao Y‐X. Activation of MAPK by inverse agonists in six naturally occurring constitutively active mutant human melanocortin‐4 receptors. Biochim Biophys Acta. 2013;1832(12):1939‐1948. doi: 10.1016/j.bbadis.2013.06.006 [DOI] [PubMed] [Google Scholar]
- 61. Chaturvedi M, Shukla AK. Calcium as a biased cofactor. Science. 2020;368(6489):369‐370. doi: 10.1126/science.abb4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ghamari‐Langroudi M, Digby GJ, Sebag JA, et al. G‐protein‐independent coupling of MC4R to Kir7.1 in hypothalamic neurons. Nature. 2015;520(7545):94‐98. doi: 10.1038/nature14051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Larsen LH, Echwald SM, Sørensen TIA, Andersen T, Wulff BS, Pedersen O. Prevalence of mutations and functional analyses of melanocortin 4 receptor variants identified among 750 men with juvenile‐onset obesity. J Clin Endocrinol Metab. 2005;90(1):219‐224. doi: 10.1210/jc.2004-0497 [DOI] [PubMed] [Google Scholar]
- 64. Hinney A, Schmidt A, Nottebom K, et al. Several mutations in the melanocortin‐4 receptor gene including a nonsense and a frameshift mutation associated with dominantly inherited obesity in humans. J Clin Endocrinol Metab. 1999;84(4):1483‐1486. doi: 10.1210/jcem.84.4.5728 [DOI] [PubMed] [Google Scholar]
- 65. Yeo GSH, Lank EJ, Farooqi IS, Keogh J, Challis BG, O'Rahilly S. Mutations in the human melanocortin‐4 receptor gene associated with severe familial obesity disrupts receptor function through multiple molecular mechanisms. Hum Mol Genet. 2003;12(5):561‐574. doi: 10.1093/hmg/ddg057 [DOI] [PubMed] [Google Scholar]
- 66. Tao Y‐X, Segaloff DL. Functional characterization of melanocortin‐4 receptor mutations associated with childhood obesity. Endocrinology. 2003;144(10):4544‐4551. doi: 10.1210/en.2003-0524 [DOI] [PubMed] [Google Scholar]
- 67. Branson R, Potoczna N, Kral JG, Lentes K‐U, Hoehe MR, Horber FF. Binge eating as a major phenotype of melanocortin 4 receptor gene mutations. N Engl J Med. 2003;348(12):1096‐1103. doi: 10.1056/NEJMoa021971 [DOI] [PubMed] [Google Scholar]
- 68. Di Monaco M, Vallero F, Di Monaco R, Mautino F, Cavanna A. Fat body mass, leptin and femur bone mineral density in hip‐fractured women. J Endocrinol Invest. 2003;26(12):1180‐1185. doi: 10.1007/BF03349154 [DOI] [PubMed] [Google Scholar]
- 69. Huang H, Wang W, Tao Y‐X. Pharmacological chaperones for the misfolded melanocortin‐4 receptor associated with human obesity. Biochim Biophys Acta. 2017;1863(10):2496‐2507. doi: 10.1016/j.bbadis.2017.03.001 [DOI] [PubMed] [Google Scholar]
- 70. Granell S, Mohammad S, Ramanagoudr‐Bhojappa R, Baldini G. Obesity‐linked variants of melanocortin‐4 receptor are misfolded in the endoplasmic reticulum and can be rescued to the cell surface by a chemical chaperone. Mol Endocrinol. 2010;24(9):1805‐1821. doi: 10.1210/me.2010-0071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Granell S, Serra‐Juhé C, Martos‐Moreno GÁ, et al. A novel melanocortin‐4 receptor mutation MC4R‐P272L associated with severe obesity has increased propensity to be ubiquitinated in the ER in the face of correct folding. PLoS One. 2012;7(12):e50894. doi: 10.1371/journal.pone.0050894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. René P, Le Gouill C, Pogozheva ID, et al. Pharmacological chaperones restore function to MC4R mutants responsible for severe early‐onset obesity. J Pharmacol Exp Ther. 2010;335(3):520‐532. doi: 10.1124/jpet.110.172098 [DOI] [PubMed] [Google Scholar]
- 73. Wang X‐H, Wang H‐M, Zhao B‐L, Yu P, Fan Z‐C. Rescue of defective MC4R cell‐surface expression and signaling by a novel pharmacoperone Ipsen 17. J Mol Endocrinol. 2014;53(1):17‐29. doi: 10.1530/JME-14-0005 [DOI] [PubMed] [Google Scholar]
- 74. Tao Y‐X, Huang H. Ipsen 5i is a novel potent pharmacoperone for intracellularly retained melanocortin‐4 receptor mutants. Front Endocrinol (Lausanne). 2014;5:131–131. doi: 10.3389/fendo.2014.00131. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4120685/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Huang H, Tao Y‐X. A small molecule agonist THIQ as a novel pharmacoperone for intracellularly retained melanocortin‐4 receptor mutants. Int J Biol Sci. 2014;10(8):817‐824. doi: 10.7150/ijbs.9625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Srinivasan S, Lubrano‐Berthelier C, Govaerts C, et al. Constitutive activity of the melanocortin‐4 receptor is maintained by its N‐terminal domain and plays a role in energy homeostasis in humans. J Clin Invest. 2004;114(8):1158‐1164. doi: 10.1172/JCI21927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Huang X‐P, Karpiak J, Kroeze WK, et al. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature. 2015;527(7579):477‐483. doi: 10.1038/nature15699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shiroishi M, Kobayashi T. Structural analysis of the histamine H1 receptor. Handb Exp Pharmacol. 2017;241:21‐30. doi: 10.1007/164_2016_10 [DOI] [PubMed] [Google Scholar]
- 79. Miller‐Gallacher JL, Nehmé R, Warne T, et al. The 2.1 Å resolution structure of cyanopindolol‐bound β1‐adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand‐free receptor. PLoS One. 2014;9(3):e92727. doi: 10.1371/journal.pone.0092727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC. Allosteric sodium in class A GPCR signaling. Trends Biochem Sci. 2014;39(5):233‐244. doi: 10.1016/j.tibs.2014.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shalaeva DN, Galperin MY, Mulkidjanian AY. Eukaryotic G protein‐coupled receptors as descendants of prokaryotic sodium‐translocating rhodopsins. Biol Direct. 2015;10(1):63. doi: 10.1186/s13062-015-0091-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. White KL, Eddy MT, Gao Z‐G, et al. Structural connection between activation microswitch and allosteric sodium site in GPCR signaling. Structure. 2018;26(2):259‐269.e5. doi: 10.1016/j.str.2017.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Isberg V, de Graaf C, Bortolato A, Cherezov V, Katritch V, Marshall FH, Mordalski S, Pin J‐P, Stevens RC, Vriend G, Gloriam DE. Generic GPCR residue numbers – aligning topology maps while minding the gaps. Trends in Pharmacological Sciences. 2015;36(1):22–31. 10.1016/j.tips.2014.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cao C, Tan Q, Xu C, et al. Structural basis for signal recognition and transduction by platelet‐activating‐factor receptor. Nat Struct Mol Biol. 2018;25(6):488‐495. doi: 10.1038/s41594-018-0068-y [DOI] [PubMed] [Google Scholar]
- 85. Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G‐protein‐coupled receptors. Nature. 2013;494(7436):185‐194. doi: 10.1038/nature11896 [DOI] [PubMed] [Google Scholar]
- 86. Wootten D, Christopoulos A, Marti‐Solano M, Babu MM, Sexton PM. Mechanisms of signalling and biased agonism in G protein‐coupled receptors. Nat Rev Mol Cell Biol. 2018;19(10):638‐653. doi: 10.1038/s41580-018-0049-3 [DOI] [PubMed] [Google Scholar]
- 87. Millington GWM. The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr Metab (Lond). 2007;4(1):18. doi: 10.1186/1743-7075-4-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Poggioli R, Vergoni AV, Bertolini A. ACTH‐(1–24) and α‐MSH antagonize feeding behavior stimulated by kappa opiate agonists. Peptides. 1986;7(5):843–848. 10.1016/0196-9781(86)90104-x [DOI] [PubMed] [Google Scholar]
- 89. Holder JR, Xiang Z, Bauzo RM, Haskell‐Luevano C. Structure‐activity relationships of the melanocortin tetrapeptide Ac‐His‐DPhe‐Arg‐Trp‐NH2 at the mouse melanocortin receptors. Part 3: modifications at the Arg position. Peptides. 2003;24(1):73‐82. doi: 10.1016/s0196-9781(02)00278-4 [DOI] [PubMed] [Google Scholar]
- 90. Sawyer TK, Hruby VJ, Wilkes BC, Draelos MT, Hadley MEBM. Comparative biological activities of highly potent active‐site analogues of alpha‐melanotropin. J Med Chem. 1982;25(9):1022‐1027. doi: 10.1021/jm00351a004 [DOI] [PubMed] [Google Scholar]
- 91. Haslach EM, Huang H, Dirain M, et al. Identification of tetrapeptides from a mixture based positional scanning library that can restore nM full agonist function of the L106P, I69T, I102S, A219V, C271Y, and C271R human melanocortin‐4 polymorphic receptors (hMC4Rs). J Med Chem. 2014;57(11):4615‐4628. doi: 10.1021/jm500064t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ersoy BA, Pardo L, Zhang S, et al. Mechanism of N‐terminal modulation of activity at the melanocortin‐4 receptor GPCR. Nat Chem Biol. 2012;8(8):725‐730. doi: 10.1038/nchembio.1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yuan X‐C, Tao Y‐X. Fenoprofen‐an old drug rediscovered as a biased allosteric enhancer for melanocortin receptors. ACS Chem Nerosci. 2019;10(3):1066‐1074. doi: 10.1021/acschemneuro.8b00347 [DOI] [PubMed] [Google Scholar]
- 94. Montero‐Melendez T, Forfar RAE, Cook JM, Jerman JC, Taylor DL, Perretti M. Old drugs with new skills: fenoprofen as an allosteric enhancer at melanocortin receptor 3. Cell Mol Life Sci. 2017;74(7):1335‐1345. doi: 10.1007/s00018-016-2419-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Pantel J, Williams SY, Mi D, et al. Development of a high throughput screen for allosteric modulators of melanocortin‐4 receptor signaling using a real time cAMP assay. Eur J Pharmacol. 2011;660(1):139‐147. doi: 10.1016/j.ejphar.2011.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Le Naour M, Akgün E, Yekkirala A, et al. Bivalent ligands that target μ opioid (MOP) and Cannabinoid1 (CB 1) receptors are potent analgesics devoid of tolerance. J Med Chem. 2013;56(13):5505‐5513. doi: 10.1021/jm4005219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lensing CJ, Adank DN, Wilber SL, et al. A direct in vivo comparison of the melanocortin monovalent agonist ac‐his‐DPhe‐Arg‐Trp‐NH2 versus the bivalent agonist Ac‐His‐DPhe‐Arg‐Trp‐PEDG20‐His‐DPhe‐Arg‐Trp‐NH2: a bivalent advantage. ACS Chem Nerosci. 2017;8(6):1262‐1278. doi: 10.1021/acschemneuro.6b00399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Fernandes SM, Lee YS, Gillies RJ, Hruby VJ. Synthesis and evaluation of bivalent ligands for binding to the human melanocortin‐4 receptor. Bioorg Med Chem. 2014;22(22):6360‐6365. doi: 10.1016/j.bmc.2014.09.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Clément K, Biebermann H, Farooqi IS, et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat Med. 2018;24(5):551‐555. doi: 10.1038/s41591-018-0015-9 [DOI] [PubMed] [Google Scholar]
- 100. Haskell‐Luevano C, Lim S, Yuan W, Cone RD, Hruby VJ. Structure activity studies of the melanocortin antagonist SHU9119 modified at the 6, 7, 8, and 9 positions. Peptides. 2000;21(1):49‐57. doi: 10.1016/s0196-9781(99)00167-9 [DOI] [PubMed] [Google Scholar]
- 101. Sebhat IK, Martin WJ, Ye Z, Barakat K, Mosley RT, Johnston DBR, Bakshi R, Palucki B, Weinberg DH, MacNeil T, Kalyani RN, Tang R, Stearns RA, Miller RR, Tamvakopoulos C, Strack AM, McGowan E, Cashen DE, Drisko JE, Hom GJ, Howard AD, MacIntyre D, van der Ploeg LHT, Patchett AA, Nargund RP. Design and Pharmacology of N‐[(3R)‐1,2,3,4‐Tetrahydroisoquinolinium‐ 3‐ylcarbonyl]‐(1R)‐1‐(4‐chlorobenzyl)‐ 2‐[4‐cyclohexyl‐4‐(1H‐1,2,4‐triazol‐ 1‐ylmethyl)piperidin‐1‐yl]‐2‐oxoethylamine (1), a Potent, Selective, Melanocortin Subtype‐4 Receptor Agonist. Journal of Medicinal Chemistry. 2002;45(21):4589–4593. 10.1021/jm025539h [DOI] [PubMed] [Google Scholar]
- 102. Fotsch C, Smith DM, Adams JA, et al. Design of a new peptidomimetic agonist for the melanocortin receptors based on the solution structure of the peptide ligand, Ac‐Nle‐cyclo[Asp‐Pro‐DPhe‐Arg‐Trp‐Lys]‐NH(2). Bioorg Med Chem Lett. 2003;13(14):2337‐2340. doi: 10.1016/s0960-894x(03)00412-8 [DOI] [PubMed] [Google Scholar]
- 103. Pontillo J, Tran JA, Arellano M, et al. Structure‐activity relationships of piperazinebenzylamines as potent and selective agonists of the human melanocortin‐4 receptor. Bioorg Med Chem Lett. 2004;14(17):4417‐4423. doi: 10.1016/j.bmcl.2004.06.059 [DOI] [PubMed] [Google Scholar]
- 104. Tian X, Mishra RK, Switzer AG, et al. Design and synthesis of potent and selective 1,3,4‐trisubstituted‐2‐oxopiperazine based melanocortin‐4 receptor agonists. Bioorg Med Chem Lett. 2006;16(17):4668‐4673. doi: 10.1016/j.bmcl.2006.05.087 [DOI] [PubMed] [Google Scholar]
- 105. Greenfield JR, Miller JW, Keogh JM, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360(1):44‐52. doi: 10.1056/NEJMoa0803085 [DOI] [PubMed] [Google Scholar]
- 106. Dorr RT, Lines R, Levine N, et al. Evaluation of melanotan‐II, a superpotent cyclic melanotropic peptide in a pilot phase‐I clinical study. Life Sci. 1996;58(20):1777‐1784. doi: 10.1016/0024-3205(96)00160-9 [DOI] [PubMed] [Google Scholar]
- 107. Clayton AH, Althof SE, Kingsberg S, et al. Bremelanotide for female sexual dysfunctions in premenopausal women: a randomized, placebo‐controlled dose‐finding trial. Womens Health (Lond Engl). 2016;12(3):325‐337. doi: 10.2217/whe-2016-0018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Jimenez‐Munoz CM, López M, Albericio F, Makowski K. Targeting energy expenditure—drugs for obesity treatment. Pharmaceuticals (Basel). 2021;14(5):435. doi: 10.3390/ph14050435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Iepsen EW, Zhang J, Thomsen HS, et al. Patients with obesity caused by melanocortin‐4 receptor mutations can be treated with a glucagon‐like peptide‐1 receptor agonist. Cell Metab. 2018;28:23‐32.e3. doi: 10.1016/j.cmet.2018.05.008 [DOI] [PubMed] [Google Scholar]
- 110. Iepsen EW, Zhang J, Hollensted M, et al. Adults with pathogenic MC4R mutations have increased final height and thereby increased bone mass. J Bone Miner Metab. 2020;38(1):117‐125. doi: 10.1007/s00774-019-01034-8 [DOI] [PubMed] [Google Scholar]
- 111. Lundgren JR, Janus C, Jensen SBK, et al. Healthy weight loss maintenance with exercise, liraglutide, or both combined. N Engl J Med. 2021;384(18):1719‐1730. doi: 10.1056/NEJMoa2028198 [DOI] [PubMed] [Google Scholar]
- 112. Sun EW, Iepsen EW, Pezos N, et al. A gut‐intrinsic melanocortin signaling complex augments L‐cell secretion in humans. Gastroenterology. 2021;161:536‐547.e2. doi: 10.1053/j.gastro.2021.04.014 [DOI] [PubMed] [Google Scholar]
- 113. Benoit SC, Schwartz MW, Lachey JL, et al. A novel selective melanocortin‐4 receptor agonist reduces food intake in rats and mice without producing aversive consequences. J Neurosci. 2000;20(9):3442‐3448. doi: 10.1523/jneurosci.20-09-03442.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Dombrowski SU, Knittle K, Avenell A, Araujo‐Soares V, Sniehotta FF. Long term maintenance of weight loss with non‐surgical interventions in obese adults: systematic review and meta‐analyses of randomised controlled trials. BMJ. 2014;348(may14 6):g2646‐g2646. doi: 10.1136/bmj.g2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Sadry SA, Drucker DJ. Emerging combinatorial hormone therapies for the treatment of obesity and T2DM. Nat Rev Endocrinol. 2013;9(7):425‐433. doi: 10.1038/nrendo.2013.47 [DOI] [PubMed] [Google Scholar]
- 116. Danne T, Biester T, Kapitzke K, et al. Liraglutide in an adolescent population with obesity: a randomized, double‐blind, placebo‐controlled 5‐week trial to assess safety, tolerability, and pharmacokinetics of liraglutide in adolescents aged 12‐17 years. J Pediatr. 2017;181:146‐153.e3. doi: 10.1016/j.jpeds.2016.10.076 [DOI] [PubMed] [Google Scholar]
- 117. Christou GA, Katsiki N, Blundell J, Fruhbeck G, Kiortsis DN. Semaglutide as a promising antiobesity drug. Obes Rev. 2019;20(6):805‐815. doi: 10.1111/obr.12839 [DOI] [PubMed] [Google Scholar]
- 118. Ornellas T, Chavez B. Naltrexone SR/bupropion SR (Contrave): a new approach to weight loss in obese adults. P T. 2011;36(5):255‐262. http://www.ncbi.nlm.nih.gov/pubmed/21785538 [PMC free article] [PubMed] [Google Scholar]
- 119. Thomsen WJ, Grottick AJ, Menzaghi F, et al. Lorcaserin, a novel selective human 5‐hydroxytryptamine 2C agonist: in vitro and in vivo pharmacological characterization. J Pharmacol Exp Ther. 2008;325(2):577‐587. doi: 10.1124/jpet.107.133348 [DOI] [PubMed] [Google Scholar]
- 120. Fleming JW, McClendon KS, Riche DM. New obesity agents: lorcaserin and phentermine/topiramate. Ann Pharmacother. 2013;47(7‐8):1007‐1016. doi: 10.1345/aph.1R779 [DOI] [PubMed] [Google Scholar]
- 121. Clemmensen C, Chabenne J, Finan B, et al. GLP‐1/glucagon coagonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes. 2014;63(4):1422‐1427. doi: 10.2337/db13-1609 [DOI] [PubMed] [Google Scholar]
- 122. Müller TD, Sullivan LM, Habegger K, et al. Restoration of leptin responsiveness in diet‐induced obese mice using an optimized leptin analog in combination with exendin‐4 or FGF21. J Pept Sci. 2012;18(6):383‐393. doi: 10.1002/psc.2408 [DOI] [PubMed] [Google Scholar]
- 123. Colman E. Anorectics on trial: a half century of federal regulation of prescription appetite suppressants. Ann Intern Med. 2005;143(5):380‐385. doi: 10.7326/0003-4819-143-5-200509060-00013 [DOI] [PubMed] [Google Scholar]
- 124. Dragano NRV, Fernø J, Diéguez C, López M, Milbank E. Recent updates on obesity treatments: available drugs and future directions. Neuroscience. 2020;437:215‐239. doi: 10.1016/j.neuroscience.2020.04.034 [DOI] [PubMed] [Google Scholar]
- 125. Jones BJ, Bloom SR. The new era of drug therapy for obesity: the evidence and the expectations. Drugs. 2015;75(9):935‐945. doi: 10.1007/s40265-015-0410-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Heisler LK, Jobst EE, Sutton GM, et al. Serotonin reciprocally regulates melanocortin neurons to modulate food intake. Neuron. 2006;51(2):239‐249. doi: 10.1016/j.neuron.2006.06.004 [DOI] [PubMed] [Google Scholar]
- 127. Müller TD, Clemmensen C, Finan B, DiMarchi RD, Tschöp MH. Anti‐obesity therapy: from rainbow pills to polyagonists. Pharmacol Rev. 2018;70(4):712‐746. doi: 10.1124/pr.117.014803 [DOI] [PubMed] [Google Scholar]
- 128. Luccheta R, Riveros B, Pontarola R, et al. Systematic review and meta‐analysis of the efficacy and safety of amfepramone and mazindol as a monotherapy for the treatment of obese or overweight patients. Clinics. 2017;72(5):317‐324. doi: 10.6061/clinics/2017(05)10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kwiker D, Godkar D, Lokhandwala N, Yakoby M. Rare case of rhabdomyolysis with therapeutic doses of phendimetrazine tartrate. Am J Ther. 2006;13(2):175‐176. doi: 10.1097/00045391-200603000-00015 [DOI] [PubMed] [Google Scholar]
- 130. Patel N, Mock DC, Hagans JA. Comparison of benzphetamine, phenmetrazine, d‐amphetamine, and placebo. Clin Pharmacol Ther. 1963;4:330‐333. doi: 10.1002/cpt196343330 [DOI] [PubMed] [Google Scholar]
- 131. Rothman RB, Baumann MH, Dersch CM, et al. Amphetamine‐type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39(1):32‐41. doi:10.1002/1098‐2396(20010101)39:1<32::AID‐SYN5>3.0.CO;2‐3 [DOI] [PubMed] [Google Scholar]
- 132. Apovian CM, Aronne LJ, Bessesen DH, et al. Pharmacological management of obesity: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2015;100(2):342‐362. doi: 10.1210/jc.2014-3415 [DOI] [PubMed] [Google Scholar]
- 133. Hsia DS, Gosselin NH, Williams J, et al. A randomized, double‐blind, placebo‐controlled, pharmacokinetic and pharmacodynamic study of a fixed‐dose combination of phentermine/topiramate in adolescents with obesity. Diabetes Obes Metab. 2020;22(4):480‐491. doi: 10.1111/dom.13910 [DOI] [PubMed] [Google Scholar]
- 134. Ambery PD, Klammt S, Posch MG, et al. MEDI0382, a GLP‐1/glucagon receptor dual agonist, meets safety and tolerability endpoints in a single‐dose, healthy‐subject, randomized, phase 1 study. Br J Clin Pharmacol. 2018;84(10):2325‐2335. doi: 10.1111/bcp.13688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Frias JP, Bastyr EJ, Vignati L, et al. The sustained effects of a dual GIP/GLP‐1 receptor agonist, NNC0090‐2746, in patients with type 2 diabetes. Cell Metab. 2017;26(2):343‐352.e2. doi: 10.1016/j.cmet.2017.07.011 [DOI] [PubMed] [Google Scholar]
- 136. Frias JP, Nauck MA, Van J, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP‐1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo‐controlled and active comparator‐controlled phase 2 trial. Lancet (London, England). 2018;392(10160):2180‐2193. doi: 10.1016/S0140-6736(18)32260-8 [DOI] [PubMed] [Google Scholar]
- 137. IY CHOI, JK KIM, JS LEE, et al. Effect of a novel long‐acting GLP‐1/GIP/glucagon triple agonist (HM15211) in a NASH and fibrosis animal model. Diabetes. 2018;67(1):1106‐110P. doi: 10.2337/db18-1106-P [DOI] [Google Scholar]
- 138. Brandt SJ, Müller TD, DiMarchi RD, Tschöp MH, Stemmer K. Peptide‐based multi‐agonists: a new paradigm in metabolic pharmacology. J Intern Med. 2018;284(6):581‐602. doi: 10.1111/joim.12837 [DOI] [PubMed] [Google Scholar]
- 139. López M, Tena‐Sempere M. Estradiol effects on hypothalamic AMPK and BAT thermogenesis: a gateway for obesity treatment? Pharmacol Ther. 2017;178:109‐122. doi: 10.1016/j.pharmthera.2017.03.014 [DOI] [PubMed] [Google Scholar]
- 140. González‐García I, Contreras C, Estévez‐Salguero Á, et al. Estradiol regulates energy balance by ameliorating hypothalamic ceramide‐induced ER stress. Cell Rep. 2018;25(2):413‐423.e5. doi: 10.1016/j.celrep.2018.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Finan B, Clemmensen C, Zhu Z, et al. Chemical hybridization of glucagon and thyroid hormone optimizes therapeutic impact for metabolic disease. Cell. 2016;167(3):843‐857.e14. doi: 10.1016/j.cell.2016.09.014 [DOI] [PubMed] [Google Scholar]
- 142. Quarta C, Clemmensen C, Zhu Z, et al. Molecular integration of incretin and glucocorticoid action reverses Immunometabolic dysfunction and obesity. Cell Metab. 2017;26(4):620‐632.e6. doi: 10.1016/j.cmet.2017.08.023 [DOI] [PubMed] [Google Scholar]
- 143. Grundlingh J, Dargan PI, El‐Zanfaly M, Wood DM. 2,4‐Dinitrophenol (DNP): a weight loss agent with significant acute toxicity and risk of death. J Med Toxicol. 2011;7(3):205‐212. doi: 10.1007/s13181-011-0162-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Cannistra LB, Davis SM, Bauman AG. Valvular heart disease associated with dexfenfluramine. N Engl J Med. 1997;337(9):636. doi: 10.1056/nejm199708283370912 [DOI] [PubMed] [Google Scholar]
- 145. Weissman NJ, Tighe JF, Gottdiener JS, Gwynne JT. An assessment of heart‐valve abnormalities in obese patients taking dexfenfluramine, sustained‐release dexfenfluramine, or placebo. Sustained‐Release Dexfenfluramine Study Group. N Engl J Med. 1998;339(11):725‐732. doi: 10.1056/NEJM199809103391103 [DOI] [PubMed] [Google Scholar]
- 146. James WPT, Caterson ID, Coutinho W, et al. Effect of Sibutramine on cardiovascular outcomes in overweight and obese subjects. N Engl J Med. 2010;363(10):905‐917. doi: 10.1056/NEJMoa1003114 [DOI] [PubMed] [Google Scholar]
- 147. Simon V, Cota D. MECHANISMS IN ENDOCRINOLOGY: endocannabinoids and metabolism: past, present and future. Eur J Endocrinol. 2017;176(6):R309‐R324. doi: 10.1530/EJE-16-1044 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.