

Abstract
Aims
To measure and evaluate clinical response to nasal naloxone in opioid overdoses in the pre‐hospital environment.
Design
Randomised, controlled, double‐dummy, blinded, non‐inferiority trial, and conducted at two centres.
Setting
Participants were included by ambulance staff in Oslo and Trondheim, Norway, and treated at the place where the overdose occurred.
Participants
Men and women age above 18 years with miosis, rate of respiration ≤8/min, and Glasgow Coma Score <12/15 were included. Informed consent was obtained through a deferred‐consent procedure.
Intervention and comparator
A commercially available 1.4 mg/0.1 mL intranasal naloxone was compared with 0.8 mg/2 mL naloxone administered intramuscularly.
Measurements
The primary end‐point was restoration of spontaneous respiration of ≥10 breaths/min within 10 minutes. Secondary outcomes included time to restoration of spontaneous respiration, recurrence of overdose within 12 hours and adverse events.
Findings
In total, 201 participants were analysed in the per‐protocol population. Heroin was suspected in 196 cases. With 82% of the participants being men, 105 (97.2%) in the intramuscular group and 74 (79.6%) in the intranasal group returned to adequate spontaneous respiration within 10 minutes after one dose. The estimated risk difference was 17.5% (95% CI, 8.9%–26.1%) in favour of the intramuscular group. The risk of receiving additional naloxone was 19.4% (95% CI, 9.0%–29.7%) higher in the intranasal group. Adverse reactions were evenly distributed, except for drug withdrawal reactions, where the estimated risk difference was 6.8% (95% CI, 0.2%–13%) in favour of the intranasal group in a post hoc analysis.
Conclusion
Intranasal naloxone (1.4 mg/0.1 mL) was less efficient than 0.8 mg intramuscular naloxone for return to spontaneous breathing within 10 minutes in overdose patients in the pre‐hospital environment when compared head‐to‐head. Intranasal naloxone at 1.4 mg/0.1 mL restored breathing in 80% of participants after one dose and had few mild adverse reactions.
Keywords: Administration, drug overdose, injections, intramuscular, intranasal, naloxone, narcotic antagonists, physiological effects of drugs, substance‐related disorders
INTRODUCTION
Opioid overdose remains a global epidemic, with an annual death toll of more than 100 000 [1]. As a response, the main opioid antagonist naloxone has been made available to lay people in Take Home Naloxone (THN) Programmes from the late 1990s. THN was never meant to replace callout to and treatment by emergency medical services. It is a head start at the scene to shorten the time to the administration of the antidote while awaiting the emergency medical services for professional management and post‐overdose follow‐up.
The route of administration and dosing of naloxone in opioid overdoses in the community are debated, not least in the fentanyl era in North America [2]. Recommendations range from 0.04 to 2.0 mg via the intravenous or intramuscular route and titration to desired effect [3, 4]. The World Health Organisation (WHO) recommends starting at the lower end of that spectrum to avoid eliciting withdrawal [5]. Off‐label, unapproved, dilute nasal sprays have been used in THN programs [2, 6]. Nasal administration is preferred by lay people owing to its ease of use [7]. Since 2015, several nasal naloxone products with single doses ranging from 0.9 mg to 8.0 mg have entered the market. These formulations were approved based on phase I pharmacokinetic studies in healthy volunteers alone [8, 9, 10, 11, 12]. The lack of clinical trials of these high concentration/low volume sprays and the lack of trials comparing different naloxone regimens, leave an important knowledge gap in best practice for management of opioid overdoses in the community. Previous trials of intranasal (IN) naloxone have shown promise, but were limited in that the formulations investigated were neither specifically designed for IN use nor commercially available. They also lacked systematic information on adverse events and the risk of rebound overdose after initial naloxone revival [13, 14, 15, 16].
The nasal spray with 1.4 mg of naloxone hydrochloride dihydrate, equivalent to 1.26 mg naloxone (dne pharma as, Oslo, Norway), has been developed by the Norwegian University of Science and Technology (NTNU). The 1.4 mg/0.1 mL formulation was shown to provide adequate systemic concentrations compared to intramuscular 0.8 mg injection [10], and its absolute bioavailability was ~50% in healthy volunteers [17, 18]. However, exposure to the opioid remifentanil gives a relative bioavailability as high as 75% [19]. This highlights the need for clinical studies in the target population. Clinicians, lay people responders and policy makers should know precisely how a nasal naloxone spray performs in the field, compared to injectable antidotes. This requires studies that investigate both the effect and harm in the target population, allowing for evidence‐based decision‐making. The population of interest in this trial corresponds to patients suffering from severe opioid overdose who were treated by ambulance personnel outside the hospital. The intervention was the administration of a single dose of the 1.4 mg/0.1 mL dose naloxone nasal spray compared to 0.8 mg naloxone injected intramuscularly. The primary outcome was the return of spontaneous respiration within 10 minutes of drug administration. The main hypothesis was that, in a head‐to‐head comparison, the nasal spray would be non‐inferior to the injection.
METHODS
Study design
The NTNU Intranasal Naloxone Trial (NINA‐1) was a two‐centre, randomised, double‐dummy blinded, phase III, non‐inferiority trial [20]. Participants were recruited through ambulance services at Oslo University Hospital and St. Olav's University Hospital Trondheim, both in Norway. Extensive trial documentation, including information letters for consent and the protocol, is available at the NTNU Open Research Data repository [21].
Participants
Participants treated by ambulance services for suspected opioid overdose, recognised by reduced or absent spontaneous respiration (≤8 breaths/min), Glasgow Coma Score <12/15 and miosis, were included. However, those who had cardiac arrest, suspected pregnancy, age below 18 years or had received naloxone before the arrival of ambulance staff were excluded. A complete list of the inclusion and exclusion criteria and a flowchart of the consent procedure are provided in Supporting information Table S1, Fig. S1.
Naloxone formulation and dosing
The investigational medicinal product (IMP) was a 1.4 mg/0.1 mL naloxone hydrochloride dihydrate (equivalent to 1.26 mg naloxone) nasal spray produced by Sanivo Pharma, Oslo, Norway. The drug was administered as 1.4 mg/0.1 mL nasal spray using an unidose device (Aptar Pharma). The active comparator was a 2 mL intramuscular (IM) injection of 0.4 mg/mL naloxone hydrochloride (naloxone hydrochloride injection USP 4 mg/10 mL; Mylan Institutional). The IN placebo was similar to the IMP, except that it did not contain naloxone. The IM placebo was a 2 mL injection of sterile 9 mg/mL sodium chloride. The vials for injection were similar, blinded, and labelled for clinical trial use.
Randomisation and masking
To ensure blinding, a double‐dummy design was used. Active and placebo drugs were kept in a sealed box—a study kit that also contained case report forms, written information for consent and needles and syringes for IM injection. Study drugs were labelled, and kits were randomised, assembled and sealed by the Hospital Pharmacy, St. Olav's Hospital, Trondheim, Norway. Each ambulance only held one kit at a time, the drug contents of which were randomised to the nasal spray or vial for injection contained naloxone or a placebo. Staff were not randomised, but used the kit available in their vehicle. Participants were randomly assigned in a 1:1 ratio to receive either IN or IM naloxone. Randomisation was stratified by study centre, and random block sizes were used. Stratification was done both for practical reasons and to ensure balance of the treatment groups within each centre, because Trondheim does not have a safe injection facility that was a priory considered to be a possible prognostic factor. Computer generated randomisation lists were produced by The Clinical Trial Unit at Oslo University Hospital. The blinding was kept for all until after the database was locked, and only then did we perform the primary analysis. The whole study team, including the statistician, was blinded to the interventions. A procedure for emergency unblinding was in place, but never used.
Procedures
All participants were treated with airway control and ventilation using the bag‐mask technique before treatment with the study drug. Participants were treated in situ where the overdose occurred, not evacuated to an ambulance car or an emergency room before the administration of the study drug. Nasal spray and IM injection were administered simultaneously, or within 30 seconds of each other, with nasal spray always given first. Ambulance staff noted the time from the administration of the study drug to when a spontaneous respiration rate of ≥10 breaths/min was observed. The number of breaths per minute was counted manually. If the participant did not respond adequately or did not wake up after 10 minutes, additional intramuscular naloxone (0.4 mg/mL from either Naloxone B, Braun, Melsungen, or from Naloxon Hameln, Hameln, both in Germany) or other treatments were provided as clinically indicated. A 10‐minute cut‐off for the primary end‐point was similar to other trials in the field [14, 16]. After treatment and observation, participants were either left at the scene or transported to other health care sites following the local protocol at each site. Participants with a known national identity number were identified through an ambulance dispatch system for repeated naloxone treatment and for recurrence of opioid overdose within 12 hours after inclusion. A flowchart of the study treatment and a description of the dummy design kit are provided in Supporting information Figs. S2 and S3. To ensure fidelity to the study protocol, each ambulance worker underwent rigorous training that consisted of electronic learning and live scenarios. Re‐training and refresher courses were administered at all sites during the study period.
Outcomes
The primary outcome was the return of spontaneous respiration (≥10 breaths/min) within 10 minutes of administering the study drug. Secondary outcomes included the time from administration of naloxone to respiration of ≥10 breaths/min, receiving additional naloxone, and recurrence of opioid overdose within 12 hours of inclusion. Adverse reactions to naloxone formulation were assessed and coded according to the Medical Dictionary for Regulatory Activities (MedDRA). Symptoms of agitation or aggression, or statements from participants that they were in withdrawal, were coded as drug withdrawal syndrome, whereas nausea and vomiting were coded separately. A full list of pre‐specified outcomes and subgroups is provided in Supporting information (Table S2, Figure S4) and Study Protocol.
Statistical analysis
We assumed a probability of 88% for return to spontaneous respiration within 10 minutes in both groups and calculated that 200 cases were required to determine with 90% (power) confidence that the upper limit of the two‐sided 95% CI would exclude a difference of >15% in favour of the IM group. The non‐inferiority margin of 15% and the non‐inferiority of IN to IM administration were claimed if the 95% CI of the treatment difference for the primary end‐point lay fully within the margin. The 15% margin was not a mathematical calculation, but was based on clinical judgement and experience with naloxone. A similar range has been used to compare efficacy and safety in a biosimilar medication [22]. The primary efficacy analyses were conducted in the per‐protocol population, which comprised participants fully compliant with the pre‐specified treatment strategy. In non‐inferiority trials, analysis of the per‐protocol set is regarded as the primary analysis. This is a conservative approach, because a full analysis set (FAS)/intention to treat analysis is generally considered to be biased toward smaller differences between groups [23]. Protocol deviations that led to exclusion from the per‐protocol population are presented in Supporting information Table S3. Sensitivity analyses were performed in the FAS, which included all participants who received the study drug and did not withdraw consent. Safety analyses were conducted in all participants who received any study drugs, including those in the FAS as well as those who withdrew consent (safety set).
The primary and secondary dichotomous end‐points were analysed using logistic regression, wherein the treatment variable was adjusted for the study centre. To account for clustering in the data (the same individuals may have had several overdose events), generalised estimating equations with an exchangeable working correlation were used to estimate the parameters. The risk difference was calculated from the estimated model using average marginal means and corresponding 95% CIs using the delta method. The time‐to‐event end‐point of time to spontaneous respiration was analysed by calculating the difference in restricted mean survival time between the two treatment groups at each minute of follow‐up, adjusted for study centre. The time‐to‐event data were censored at 10 minutes. The jack‐knife technique was used to calculate the 95% CI, where one individual rather than one overdose event was left out in each jack‐knife sample, to account for clustering in the data. A complete overview of all pre‐specified end‐points and a detailed description of the statistical methods used are given in the Supplementary Statistical Analysis Plan.
Ethics and consent
The study was approved by the Norwegian Medicines Agency (EudraCT number: 2016–004072‐22) and Regional Committees for Medical and Health Research Ethics (REC 2016/2000). The trial was performed in accordance with the principles of the Declaration of Helsinki and adhered to the Good Clinical Practice guidelines of the International Council for Harmonisation of Technical Requirements. Participants were insured through the Drug Liability Association, Norway.
Informed consent was obtained through a deferred‐consent procedure. That is, participants were informed after regaining consciousness and the ability to consent, and two ambulance workers documented an orally given consent. The information stated that they were included in a clinical drugs trial, describing the intervention and information regarding the withdrawal procedure. Participants who did not respond to naloxone or were unable to give informed consent at the scene were provided written information and an option to withdraw later online or by telephone. In participants who withdrew, data on adverse events and safety end‐points were anonymised and retained. For more information, please consult Supporting information Figure S1 and S2.
Public consultation and involvement
A board of drug user representatives and family representatives of participants advised investigators in the study design, protocol, information letter writing and in applying the study for ethics committee approval. This work included assessing the burden of the deferred‐consent model for participants, compared to the burden in other consent models such as proxy consent or prior consent. The board actively informed the community throughout the inclusion period about the ongoing trial and will be part of disseminating the results. For details regarding the members, please consult the Supporting information.
RESULTS
Characteristics of the participants
From June 12, 2018, to August 4, 2020, a total of 147 cases of opioid overdose were randomised to IM naloxone treatment and 139 cases to IN (Figure 1). The per‐protocol sample was 108 for IM and 93 for IN. Overall, the groups were balanced in terms of baseline characteristics (Table 1). The overall allocation of treatment is balanced (Table 1). However, there is an unbalance among those individuals included several times in the study toward more often IM treatment (Table 1). Because of the apparently successful blinding procedure, we have no indication that this is anything but a chance occurrence. Characteristics of excluded participants and those in the FAS are available in Supporting information Table S4. Participants were included in both public places and private homes in n = 121/201, (60%), and in the Oslo Safe Injection Facility in n = 80/201 (40%). The dispatch time was 5.5 (SD, 3.5) minutes. Participants left at the scene were treated for 50.4 (SD, 18.0) minutes, whereas participants transferred elsewhere for further care were treated by ambulance staff for 40.0 (SD, 15.9) min. Heroin was suspected in n = 196/201 (98%) cases and concomitant drugs in n = 35/201 (17%) cases. Respiratory arrest was present in n = 56/201 (30%) of cases, they had no spontaneous breaths within 10 seconds despite a free airway. Another n = 82/201 (40%) had a respiratory rate of 4/min or less. The median respiratory rate was 3/min, and n = 157/201 (78%) had a Glasgow Coma Score of 3/15, which was also the median score (Figure 1).
FIGURE 1.
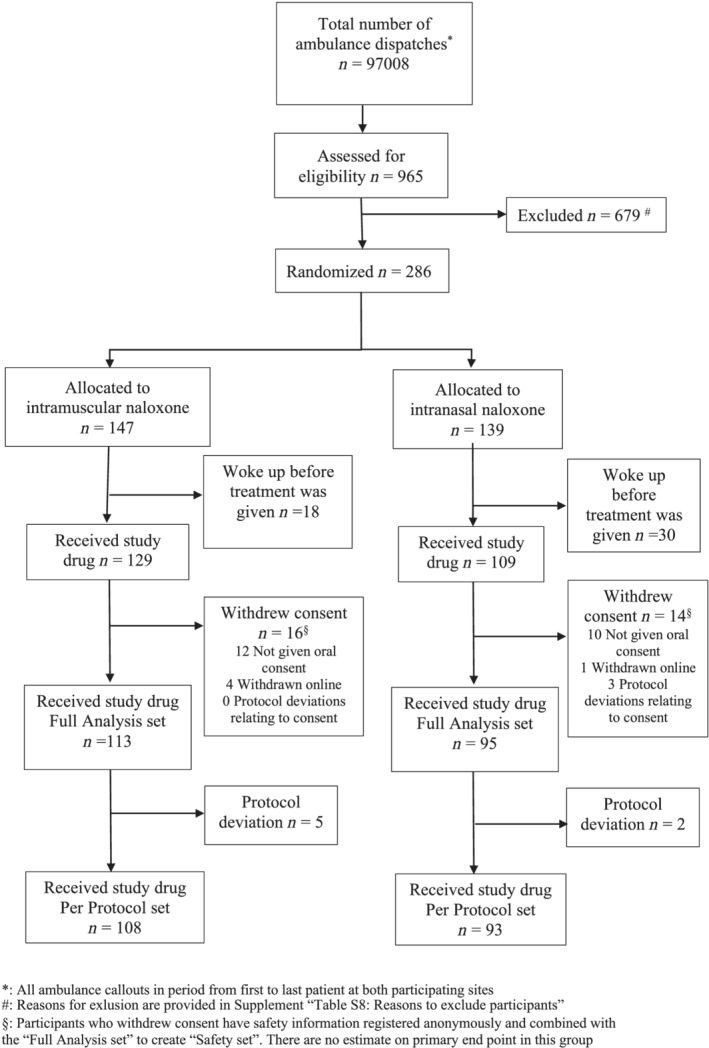
Flowchart of participants of the trial
TABLE 1.
Baseline overdose event characteristics of the per‐protocol population
| No. of overdose events with data | Intramuscular (n = 108) | Intranasal (n = 93) | Overall (n = 201) | ||
|---|---|---|---|---|---|
| Centre (%) | 201 | Oslo University Hospital | 101 (93.5) | 86 (92.5) | 187 (93.0) |
| St. Olav's Hospital, Trondheim | 7 (6.5) | 7 (7.5) | 14 (7.0) | ||
| Sex (%) | 201 | Female | 19 (17.6) | 17 (18.3) | 36 (17.9) |
| Male | 88 (81.5) | 75 (80.6) | 163 (81.1) | ||
| Unknown | 1 (0.9) | 1 (1.1) | 2 (1.0) | ||
| Age (mean [SD]) | 183 | 37.3 (10.2) | 38.5 (10.8) | 38.9 (10.5) | |
| Identity known (%) | 201 | Yes | 100 (92.6) | 83 (89.2) | 183 (91.0) |
| No | 8 (7.4) | 10 (10.8) | 18 (9.0) | ||
| Baseline respiratory rate in breaths/min (%) | 201 | 0 | 30 (27.8) | 26 (28.0) | 56 (27.9) |
| 1–4 | 46 (42.6) | 36 (38.7) | 82 (40.8) | ||
| 5–8 | 32 (29.6) | 31 (33.3) | 63 (31.3) | ||
| Baseline Glasgow Coma Score (%) | 201 | 3/15 | 86 (79.6) | 71 (76.3) | 157 (78.1) |
| 4–11/15 | 22 (20.4) | 22 (23.7) | 44 (21.9) | ||
| Primary suspected drug (%) | 201 | Heroin | 106 (98.1) | 90 (96.8) | 196 (97.5) |
| Methadone | 0 (0.0.) | 1 (1.1) | 1 (0.5) | ||
| Other opioids | 2 (1.9) | 2 (2.2) | 4 (2.0) | ||
| Benzodiazepines, alcohol, gamma hydroxybutyrate, or other drugs suspected (%) | 201 | Yes | 19 (17.6) | 16 (17.2) | 35 (17.4) |
| No | 89 (82.4) | 77 (82.8) | 166 (82.6) | ||
| Location of overdose (%) | 201 | Oslo Safe injection facility | 51 (47.2) | 29 (31.2) | 80 (39.8) |
| Private or public | 57 (52.8) | 64 (68.8) | 121 (60.2) | ||
| No. of times included (per protocol set) | 201 | 1 | 68 | 63 | 131 |
| 2 | 18 | 12 | 30 | ||
| 3 | 9 | 9 | 18 | ||
| 4 | 3 | 1 | 4 | ||
| 5 | 8 | 2 | 10 | ||
| 8 | 7 | 1 | 8 |
Primary outcome
There were 105 participants (97.2%) in the IM and 74 (79.6%) in the IN group with overdose events who achieved spontaneous breathing within 10 minutes after one dose of the study drug. The estimated risk difference between IM and IN naloxone was 17.5% (95% CI, 9.0%–26.1%) (Table 2, Figure 2). An unadjusted (for centre) post hoc robustness analysis gave a risk difference of 17.7% (95% CI, 9.0%, 26.3%). The primary analysis population in this non‐inferiority trial was the per‐protocol population. These results are consistent in an analysis of the FAS (Table 2). The FAS was the closest to a theoretical ITT population that is possible to get. The FAS did not contain patients that did not receive any treatment or patients that have withdrawn consent (see Figure 1). The results were also consistent across several pre‐specified subgroup analyses of possible prognostic factors (Supporting information Figure S4). For the Oslo centre, the estimate and 95% CI was 15.6% (6.9%, 24.4%). For the much smaller Trondheim centre, the estimate and 95% CI was 42.9% (7.1%, 78.6%) Furthermore, results are also consistent in post hoc analyses adjusting the treatment variable for each of the baseline variables given in Table 1 (Supporting information Table S5).
TABLE 2.
Primary outcome results in both the per‐protocol analysis and the full analysis set analysis
| Effect estimate | Analysis population | n_IM | n_IN | Estimate (95% CI) |
|---|---|---|---|---|
| Risk difference | Per‐protocol population | 105/108 | 74/93 | 17.5% (9.0%, 26.1%) |
| Risk difference | Full analysis set | 110/113 | 76/95 | 17.3% (8.9%, 25.7%) |
IM = intramuscular; IN = intranasal.
FIGURE 2.
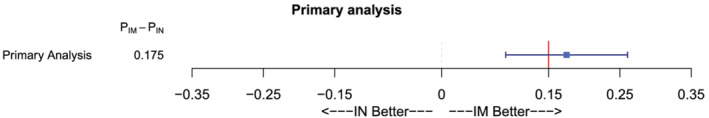
Results of primary analysis of the primary end‐point in the per‐protocol population. The risk difference with 95% CI is displayed. The red vertical line represents the non‐inferiority margin of 15%. IN, intranasal; IM, intramuscular
Figure 3(a) displays the probability of not breathing 10 spontaneous breaths per minute over time. The IN curve retained its linear shape in the 10‐minute observation period. Figure 3(b) displays the average delay in the time to spontaneous breathing in the IN group compared to the IM group quantified by the restricted mean survival time. After 4 minutes, a difference existed between the groups according to the upper 95% CI limit. Within the total follow‐up of 10 minutes, participants in the IM group returned to spontaneous respiration at an average of 2.3 (95% CI, 1.6–3.0) minutes earlier than in the IN group.
FIGURE 3.
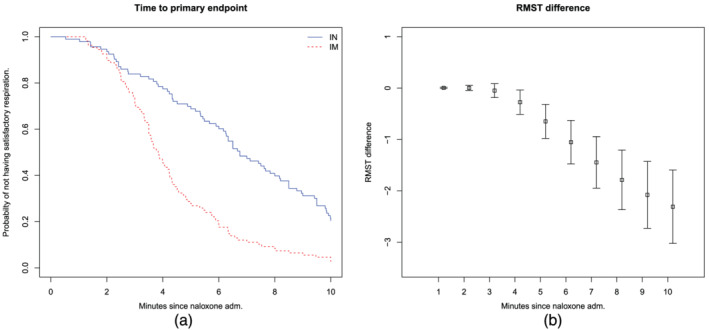
Probability of unsatisfactory respiration and average delay in spontaneous breathing. (a) Kaplan–Meier plot (unadjusted for study centre) showing the probability of not having reached satisfactory respiration (10 breaths/minute). (b) Restricted mean survival time (RMST) difference in minutes (intramuscular minus intranasal) at each minute of follow‐up time, from 1 to 10 minutes. IM, intramuscular; IN, intranasal
Secondary outcomes
In the per‐protocol population, additional naloxone was administered in 10 (9.3%) cases in the IM group and 27 (29.0%) in the IN group. The estimated risk difference was −19.4% (95% CI, −29.7% to −9.0%). Similar results were found when repeating the analysis in the FAS and safety set. The mean dose of additional naloxone administered was 0.6 (SD, 0.35) mg.
In the 201 overdose events in the per‐protocol population, four (3.7%) in the IM group and four (4.3%) in the IN group received treatment with naloxone by the ambulance service at another callout within 12 hours of inclusion. The estimated risk difference was −0.2% (95% CI, −6.7%, 6.3%). However, only 183 cases had known identities and could be followed up for recurrence.
In the per‐protocol population, there were 14 (13.0%) and 14 (15.1%) adverse reactions in the IM and IN groups, respectively. The estimated risk difference was −2.2% (95% CI, −11.5%–7.1%). Table 3 shows an overview of the adverse reactions in the safety set. One serious adverse event (self‐limiting bradycardia) was reported in the intranasal group. All participants survived during the treatment period. There were no reports of suspected unexpected serious adverse reactions. In the per‐protocol population, there were eight (7.5%) and five (5.4%) occurrences of drug withdrawal syndrome in the IM and IN groups, respectively. The estimated risk difference was 2.0% (95% CI, −4.6%–8.5%). However, in the safety set, a post hoc analysis revealed a borderline significant estimated risk difference of 6.8% (95% CI, 0.2%–13%), with a lower risk of withdrawal in the IN group. Among participants in the IM group with adverse events who refused or withdrew consent, six of the eight cases suffered withdrawal syndrome.
TABLE 3.
Number and proportion of cases from the safety set population with adverse reactions classified according to MedDRA
| System organ class | Preferred term | Treatment group | Overall (n = 238) | |
|---|---|---|---|---|
| Intramuscular (n = 129) | Intranasal (n = 109) | |||
| Cardiac disorders | Bradycardia (%) | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| Gastrointestinal disorders | Nausea (%) | 5 (3.9) | 7 (6.4) | 12 (5.0) |
| Vomiting (%) | 0 (0.0) | 2 (1.8) | 2 (0.8) | |
| General disorders and administration site conditions | Drug withdrawal syndrome (%) | 15 (11.6) | 5 (4.6) | 20 (8.4) |
| Nervous system disorders | Dizziness (%) | 1 (0.8) | 0 (0.0) | 1 (0.4) |
| Headache (%) | 5 (3.9) | 4 (3.7) | 9 (3.8) | |
DISCUSSION
A single dose of 1.4 mg/0.1 mL IN naloxone was inferior to 0.8 mg IM naloxone in terms of return to spontaneous breathing at 10 minutes after administration. In the IM naloxone group, 97% of cases achieved the primary end‐point, which outperformed our expectation of 88%. After a single 1.4 mg/0.1 mL spray, 80% achieved satisfactory respiration within 10 minutes. This likely resulted from an average slower uptake of naloxone in the IN group. After 3 minutes the stronger effect of IM became evident (Figure 3) and, within the follow‐up of 10 minutes, the effect of naloxone was 2.3 minutes slower in the IN group than in the IM group. The nasal effect curve was linear from about 3 minutes until censoring at 10 minutes, where non‐responders were administered additional IM naloxone according to protocol. Previous pharmacokinetic studies have shown that IN serum concentration continues to rise after 10 minutes, and measurement beyond 10 minutes would likely show an overall similar potency between IM and IN [10]. Both 0.8 mg IM and 1.4 mg/0.1 mL IN naloxone showed few and mostly mild, adverse reactions. There was no difference in the overall risk of adverse reactions, overdose complications, follow‐up after treatment or notable opioid overdose recurrence. However, more drug withdrawal reactions occurred in the IM group in the safety set. This is not a trivial matter, because over‐antagonism is associated with physical reactions, aggression, refusal of treatment and premature self‐discharge [3, 24].
To avoid over‐antagonism and triggering opioid withdrawal, naloxone should be titrated. Our findings that 0.8 mg IM was sufficient for reversal in almost 100% of cases indicate that it was too high as a starting dose and lower doses should be tested. This has also been seen previously in Australia [16]. Pharmacokinetic trials show that dose‐corrected concentrations of intravenous naloxone are many times as high as those achieved with IM naloxone [2]. This forms a strong argument for the efficacy and safety of the intramuscular route of administration in contrast to intravenous, which has a high probability of triggering withdrawal.
Role of 1.4 mg/0.1 mL in THN programs
Because the spray is primarily meant for THN distribution, it seems pertinent to discuss our findings in this context. THN aims to provide a head start in opioid reversal and the chain of overdose survival, to restore respiration, to regain consciousness and then to facilitate post‐overdose follow‐up, including addiction management and prevention of future overdoses. In this perspective, the slower onset of action of the 1.4 mg/0.1 mL IN dose, with an 80% probability of achieving spontaneous breathing within 10 minutes, seems a reasonable starting point for overdose treatment in THN. THN based on dose titration has worked in the past [25].
However, discussion on THN dosing of naloxone should also embrace fentanyl intoxications. Evidence indicating that large naloxone doses are required for fentanyl overdoses is limited and contradictory [2, 26]. The presence of fentanyl overdose deaths in Massachusetts has increased continuously, but the overdose rate has been stable since 2016 [27, 28]. A moderate increase in multiple naloxone dosing in the preceding years in the United States (US) has been reported, whereas the rate of additional nasal naloxone has not changed [29, 30, 31]. The amount of naloxone used for reversal has not increased either [32, 33]. However, the introduction of Narcan in 2016 [34] resulted in a dramatic rise of dose levels approaching those associated with serious pulmonary complications [35]. Ultimately, the major challenge with THN in preventing overdose deaths may not be the dose of naloxone, but whether there are bystanders present that carry naloxone [27].
Comparisons to other trials
Four previous trials of nasal naloxone used dilute IN formulations with unknown pharmacokinetic characteristics, making pharmacological assessment of the comparator impossible [13, 14, 15, 16]. However, all trials agreed that intranasal naloxone is a feasible and safe alternative to naloxone by the needle in opioid overdose. IM had a faster effect in all with less need for repeat doses. Therefore, the superiority of IM to IN in a bioequivalent head‐to‐head comparison in opioid exposed participants seemed not to completely overcome the slower action of IN, despite similar pharmacokinetics in healthy volunteers [8, 9, 10].
Advantages and limitations
The major advantage of this study was that the performance of a properly characterised and approved nasal naloxone spray was studied in the target population, strengthening the basis of evidence in the field. The inclusion criteria ensured that the overdoses studied were severe, and that the participants were in deep coma with inadequate spontaneous respiration. Compared to those in a non‐selected sample in Oslo, the participants had lower median respiratory rates (3 vs 7/min) and Glasgow Coma Score (3 vs 4/15) [36]. The nasal dose was chosen based on several pharmacokinetic studies of volunteers, including a study in which volunteers were exposed to an opioid [10, 17, 19]. The comparator dose exceeded the 0.4 mg IM dose required for regulatory purposes and was chosen based on a field study and recommendations of the WHO [5, 36]. The trial conformed to contemporary standards of clinical trial study design and conductance according to the Good Clinical Practice guidelines, including the registration, classification and publication of adverse events, such as recurrence of overdose in the 12 hours post‐inclusion. Our main results were consistent in all the trial populations.
The study is limited in that it only compares two single administrations of naloxone head‐to‐head and not regimens of titration, which would have been more relevant to the THN scenario. Administering up to two 1.4 sprays in one study arm to incremental doses of 0.4 mg IM naloxone in the other would have increased the value of this trial. The main end‐point number of breaths per minute was manually counted, which allowed for mistakes. The study drugs were administered simultaneously when possible and always within 30 seconds of each other, with IN first. Although we selected cases with severe overdoses, the low rate of fentanyl intoxications in this study is also a limitation. Future clinical studies should focus on overdose management, first aid response, the timely administration and titration of naloxone and follow‐up beyond the initial treatment. Studies should be conducted in areas with suspected fentanyl as overdose culprits. Policy and practitioners must recognise that opioid overdoses are a medical emergency that needs urgent first aid and antidote, but also follow‐up and prevention of new overdoses. The concept of ‘a chain of survival’ as seen in cardiac arrest may guide future practise [37]. For this to work, over‐antagonism with naloxone must be reduced and post‐overdose care must be expanded.
CONCLUSION
In conclusion, this study showed that 1.4 mg/0.1 mL IN naloxone was less efficient, owing to a slower onset, than 0.8 mg IM naloxone in terms of return of spontaneous breathing within 10 minutes in participants with serious opioid overdoses, and that 0.8 mg IM naloxone had an almost 100% success rate. However, notably, 1.4 mg/0.1 mL IN naloxone restored breathing in 80% of participants after one dose and was associated with few and mild adverse reactions, allowing for titration.
DECLARATION OF INTERESTS
A.K.S. spoke at a seminar arranged by dne pharma in Lisbon on October 2019 without an honorarium or other compensation. I.T. has nothing to disclose. M.V. has nothing to disclose. A.C.B. has nothing to disclose. J.D. has nothing to disclose. F.H. reports grants from Norwegian Air Ambulance Foundation during the conduct of the study. T.S. has nothing to disclose. J.B. has nothing to disclose. S.M. has nothing to disclose. O.D. reports grants from Regional Health Trust, Central Norway, grants from St. Olav's University Hospital, Trondheim, NO, grants from The Laerdal Foundation, during the conduct of the study; and NTNU; the Norwegian University of Science and Technology has signed a collaboration and licensing agreement with dne pharma to commercialise the nasal spray. Dne pharma has received marketing authorisation for a naloxone nasal spray (Ventizolve/Respinal) based on this collaboration. The formulation was invented by O.D., and the agreements ensure potential royalties for him through NTNU and NTNU's subsidiary Technology Transfer (TTO). The agreements do not restrict NTNU's opportunity to publish results from its own research on the product. O.D. was principal investigator (no personal honorarium) for dne pharma on a study of naloxone spray in volunteers and has been reimbursed for project related journeys between Oslo and Trondheim. He has received honoraria and travel support from dne pharma for presentations on marketing events.
AUTHOR CONTRIBUTIONS
A.K.S. and I.T. share first authorship of this manuscript. Together with M.V. and O.D., they prepared the first draft of the manuscript. A.C.B., J.D., J.B., F.H., S.M. and T.S. all revised that draft into its final form. No other writing assistance apart from language editing from Editage has been used on this manuscript. All authors have seen and accepted the version being submitted.
CLINICAL TRIAL REGISTRATION
ClinicalTrials.gov identifier: NCT03518021. EudraCT Number: 2016–004072‐22.
DATA SHARING
Trial documents and metadata are openly published in the Norwegian University of Science and Technology Open Repository (doi.org/10.18710/ABRUWW). Summaries of clinical study results will be posted in European Clinical Trials Database with 1 year of completion of the trial. Deidentified individual participant data will be made available on reasonable demand to recipients' conditional of data processor agreement being entered with NTNU.
Supporting information
Table S1: Full list of inclusion and exclusion criteria
Table S2: Primary and secondary endpoints
Table S3: Reasons for participants to be excluded from the Full Analysis Set
Table S4: Baseline characteristics of Full analysis set (FAS) and Excluded participants
Table S5: Post hoc adjusted estimates of primary endpoint
Table S6: Results prespecified secondary endpoints
Table S7: Baseline characteristics of included individuals
Table S8: Reasons to exclude participants
Figure S1: Flow chart of procedure for informed consent
Figure S2: Flow chart trial procedure during study visit
Figure S3: Photo double dummy kit
Figure S4: Subgroup analysis
Figure S5: Dichotomous secondary endpoints
Figure S6: Primary end point in Full Analysis Set
ACKNOWLEDGEMENTS
The participating patients and drug‐user representatives deserve great thanks. We also thank the 318 trained ambulance staff members who enthusiastically helped with patient enrolment and often did so even in challenging settings. They have all contributed well beyond what was expected. We are grateful to Tale Aurstad of the Hospital Pharmacy, St. Olav's Hospital, for assembling the study kits and for being supportive of our work. Oslo University Hospital Clinical Trials Unit have been invaluable in their work on the database, monitoring and support. We thank Per Farup, Jørgen Dahlberg, Øyvind Thomassen and Marissa E. LeBlanc for serving on the Data Monitoring and Safety Committee, Inge Christoffer Olsen for giving valuable statistical advice, Sanivo Pharma and dne pharma for supporting the NTNU unconditionally through the study kits and Kåre Stadskleiv for producing the foam casings for the study kits. We also acknowledge the support of the Central Norway Regional Health Authority, NTNU, St. Olav's Hospital, The Laerdal Foundation for Acute Medicine and the Norwegian Air Ambulance Foundation. This trial has been funded by The Joint Research Committee between St. Olavs Hospital and the Faculty of Medicine and Health Sciences, NTNU (FFU), The Central Norway Regional Health Authority, Oslo University Hospital, St Olavs Trondheim University Hospital, The Laerdal Foundation for Acute Medicine, the Norwegian Air Ambulance Foundation, and dne pharma as. The funders of the study had no role in study design, data collection, data analysis, data interpretation or writing of the report.
Skulberg AK, Tylleskär I, Valberg M, Braarud A‐C, Dale J, Heyerdahl F, et al. Comparison of intranasal and intramuscular naloxone in opioid overdoses managed by ambulance staff: a double‐dummy, randomised, controlled trial. Addiction. 2022;117:1658–1667. 10.1111/add.15806
Funding information dne pharma; Norwegian Air Ambulance Foundation; St Olavs Trondheim University Hospital; Oslo University Hospital; The Central Norway Regional Health Authority; The Joint Research Committee between St. Olavs Hospital and the Faculty of Medicine and Health Sciences, NTNU (FFU); The Laerdal Foundation for Acute Medicine
REFERENCES
- 1. Degenhardt L, Grebely J, Stone J, Hickman M, Vickerman P, Marshall BDL, et al. Global patterns of opioid use and dependence: Harms to populations, interventions, and future action. Lancet. 2019;394(10208):1560–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Strang J, McDonald R, Campbell G, Degenhardt L, Nielsen S, Ritter A, et al. Take‐home naloxone for the emergency interim Management of Opioid Overdose: The public health application of an emergency medicine. Drugs. 2019;79(13):1395–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim HK, Nelson LS. Reducing the harm of opioid overdose with the safe use of naloxone: A pharmacologic review. Expert Opin Drug Saf. 2015;14(7):1137–46. [DOI] [PubMed] [Google Scholar]
- 4. Clarke SF, Dargan PI, Jones AL. Naloxone in opioid poisoning: Walking the tightrope. Emerg Med J. 2005;22(9):612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. World Health Organization . Community management of opioid overdose. Geneva: World Health Organization; 2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK264311/ [PubMed] [Google Scholar]
- 6. Strang J, Darke S, Hall W, Farrell M, Ali R. Heroin overdose: The case for take‐home naloxone. BMJ. 1996;312(7044):1435–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mcdonald R, Breidahl S, Abel‐Ollo K, Akhtar S, Clausen T, Day E, et al. Take‐home naloxone kits: Attitudes and likelihood‐of‐use outcomes from a European survey of potential overdose witnesses. Eur Addict Res. 2021. [DOI] [PubMed] [Google Scholar]
- 8. Krieter P, Chiang N, Gyaw S, Skolnick P, Crystal R, Keegan F, et al. Pharmacokinetic properties and human use characteristics of an FDA‐approved intranasal naloxone product for the treatment of opioid overdose. J Clin Pharmacol. 2016;56(10):1243–53. [DOI] [PubMed] [Google Scholar]
- 9. McDonald R, Lorch U, Woodward J, Bosse B, Dooner H, Mundin G, et al. Pharmacokinetics of concentrated naloxone nasal spray for opioid overdose reversal: Phase I healthy volunteer study. Addiction. 2018;113(3):484–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Skulberg AK, Asberg A, Khiabani HZ, Rostad H, Tylleskar I, Dale O. Pharmacokinetics of a novel, approved, 1.4‐mg intranasal naloxone formulation for reversal of opioid overdose‐a randomized controlled trial. Addiction. 2019;114(5):859–67. [DOI] [PubMed] [Google Scholar]
- 11. Base de Données Publique des Médicaments . NALSCUE 0,9 mg/0,1 ml, Résumé des caractéristiques du produit, 2020 [cited 2020 5. october]. Available from: http://base-donnees-publique.medicaments.gouv.fr/affichageDoc.php?specid=60855566&typedoc=R [Google Scholar]
- 12. Knopf A. High‐dose naloxone nasal spray approved. Alcohol Drug Abuse Weekly. 2021;33(20):6. [Google Scholar]
- 13. Kelly AM, Kerr D, Dietze P, Patrick I, Walker T, Koutsogiannis Z. Randomised trial of intranasal versus intramuscular naloxone in prehospital treatment for suspected opioid overdose. Med J Aust. 2005;182(1):24–7. [DOI] [PubMed] [Google Scholar]
- 14. Kerr D, Kelly AM, Dietze P, Jolley D, Barger B. Randomized controlled trial comparing the effectiveness and safety of intranasal and intramuscular naloxone for the treatment of suspected heroin overdose. Addiction. 2009;104(12):2067–74. [DOI] [PubMed] [Google Scholar]
- 15. Sabzghabaee AM, Eizadi‐Mood N, Yaraghi A, Zandifar S. Naloxone therapy in opioid overdose patients: Intranasal or intravenous? A randomized clinical trial. Arch Med Sci. 2014;10(2):309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dietze P, Jauncey M, Salmon A, Mohebbi M, Latimer J, van Beek I, et al. Effect of intranasal vs intramuscular naloxone on opioid overdose: A randomized clinical trial. JAMA Netw Open. 2019;2(11):e1914977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tylleskar I, Skulberg AK, Nilsen T, Skarra S, Jansook P, Dale O. Pharmacokinetics of a new, nasal formulation of naloxone. Eur J Clin Pharmacol. 2017;73(5):555–62. [DOI] [PubMed] [Google Scholar]
- 18. Tylleskar I, Skulberg AK, Nilsen T, Skarra S, Dale O. Naloxone nasal spray ‐ bioavailability and absorption pattern in a phase 1 study. Tidsskr nor Laegeforen. 2019;139(13). [DOI] [PubMed] [Google Scholar]
- 19. Skulberg AK, Tylleskar I, Nilsen T, Skarra S, Salvesen O, Sand T, et al. Pharmacokinetics and ‐dynamics of intramuscular and intranasal naloxone: An explorative study in healthy volunteers. Eur J Clin Pharmacol. 2018;74(7):873–83. [DOI] [PubMed] [Google Scholar]
- 20. Skulberg AK, Tylleskär I, Braarud A‐C, Dale J, Heyerdahl F, Mellesmo S, et al. NTNU intranasal naloxone trial (NINA‐1) study protocol for a double‐blind, double‐dummy, non‐inferiority randomised controlled trial comparing intranasal 1.4 mg to intramuscular 0.8 mg naloxone for prehospital use. BMJ Open. 2020;10(11):e041556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Skulberg AK, Dale O. NTNU Intranasal Naloxone Trial (NINA‐1) Study documents. DataverseNO; 2020. Available from: https://doi.org/10.18710/ABRUWW [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jørgensen . K ea. ‘LB15 ‐ Biosimilar infliximab (CT‐P13) is not inferior to originator infliximab: results from the 52‐week randomized NOR‐SWITCH trial. In: United European Gastroenterology (UEG) Week meeting 2016, 15–19 October; Vienna, Austria. [Google Scholar]
- 23. Mauri L, D'Agostino RB Sr. Challenges in the design and interpretation of noninferiority trials. N Engl J Med. 2017;377(14):1357–67. [DOI] [PubMed] [Google Scholar]
- 24. Neale J, Strang J. Naloxone‐‐does over‐antagonism matter? Evidence of iatrogenic harm after emergency treatment of heroin/opioid overdose. Addiction. 2015;110(10):1644–52. [DOI] [PubMed] [Google Scholar]
- 25. Madah‐Amiri D, Clausen T, Lobmaier P. Rapid widespread distribution of intranasal naloxone for overdose prevention. Drug Alcohol Depend. 2017;173:17–23. [DOI] [PubMed] [Google Scholar]
- 26. Kim HK, Connors NJ, Mazer‐Amirshahi ME. The role of take‐home naloxone in the epidemic of opioid overdose involving illicitly manufactured fentanyl and its analogs. Expert Opin Drug Saf. 2019;18(6):465–75. [DOI] [PubMed] [Google Scholar]
- 27. Somerville NJ, O'Donnell J, Gladden RM, Zibbell JE, Green TC, Younkin M, et al. Characteristics of fentanyl overdose ‐ Massachusetts, 2014‐2016. MMWR Morb Mortal Wkly Rep. 2017;66(14):382–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Massachusetts Department of Public Health . Data Brief: Opioid‐Related Overdose Deaths among Massachusetts Residents 2020 Available from: https://www.mass.gov/doc/opioid-related-overdose-deaths-among-ma-residents-june-2020/download [Google Scholar]
- 29. Faul M, Lurie P, Kinsman JM, Dailey MW, Crabaugh C, Sasser SM. Multiple naloxone administrations among emergency medical service providers is increasing. Prehosp Emerg Care. 2017;21(4):411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weiner SG, Mitchell PM, Temin ES, Langlois BK, Dyer KS. Use of intranasal naloxone by basic life support providers. Prehosp Emerg Care. 2017;21(3):322–6. [DOI] [PubMed] [Google Scholar]
- 31. Klebacher R, Harris MI, Ariyaprakai N, Tagore A, Robbins V, Dudley LS, et al. Incidence of naloxone redosing in the age of the new opioid epidemic. Prehosp Emerg Care. 2017;1–6. [DOI] [PubMed] [Google Scholar]
- 32. Bell A, Bennett AS, Jones TS, Doe‐Simkins M, Williams LD. Amount of naloxone used to reverse opioid overdoses outside of medical practice in a city with increasing illicitly manufactured fentanyl in illicit drug supply. Subst Abus. 2019;40(1):52–5. [DOI] [PubMed] [Google Scholar]
- 33. Carpenter J, Murray BP, Atti S, Moran TP, Yancey A, Morgan B. Naloxone dosing after opioid overdose in the era of illicitly manufactured fentanyl. J Med Toxicol. 2020;16(1):41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mahonski SG, Leonard JB, Gatz JD, Seung H, Haas EE, Kim HK. Prepacked naloxone administration for suspected opioid overdose in the era of illicitly manufactured fentanyl: A retrospective study of regional poison center data. Clin Toxicol (Phila). 2020;58(2):117–23. [DOI] [PubMed] [Google Scholar]
- 35. Farkas A, Lynch MJ, Westover R, Giles J, Siripong N, Nalatwad A, et al. Pulmonary complications of opioid overdose treated with naloxone. Ann Emerg Med. 2020;75(1):39–48. [DOI] [PubMed] [Google Scholar]
- 36. Tylleskar I, Gjersing L, Bjornsen LP, Braarud AC, Heyerdahl F, Dale O, et al. Prehospital naloxone administration ‐ what influences choice of dose and route of administration? BMC Emerg Med. 2020;20(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tagami T, Hirata K, Takeshige T, Matsui J, Takinami M, Satake M, et al. Implementation of the fifth link of the chain of survival concept for out‐of‐hospital cardiac arrest. Circulation. 2012;126(5):589–97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Full list of inclusion and exclusion criteria
Table S2: Primary and secondary endpoints
Table S3: Reasons for participants to be excluded from the Full Analysis Set
Table S4: Baseline characteristics of Full analysis set (FAS) and Excluded participants
Table S5: Post hoc adjusted estimates of primary endpoint
Table S6: Results prespecified secondary endpoints
Table S7: Baseline characteristics of included individuals
Table S8: Reasons to exclude participants
Figure S1: Flow chart of procedure for informed consent
Figure S2: Flow chart trial procedure during study visit
Figure S3: Photo double dummy kit
Figure S4: Subgroup analysis
Figure S5: Dichotomous secondary endpoints
Figure S6: Primary end point in Full Analysis Set