

Abstract
Bimodal molecular probes combining nuclear magnetic resonance (NMR) and fluorescence have been widely studied in basic science, as well as clinical research. The investigation of spin phenomena holds promise to broaden the scope of available probes allowing deeper insights into physiological processes. Herein, a class of molecules with a bimodal character with respect to fluorescence and nuclear spin singlet states is introduced. Singlet states are NMR silent but can be probed indirectly. Symmetric, perdeuterated molecules, in which the singlet states can be populated by vanishingly small electron‐mediated couplings (below 1 Hz) are reported. The lifetimes of these states are an order of magnitude longer than the longitudinal relaxation times and up to four minutes at 7 T. Moreover, these molecules show either aggregation induced emission (AIE) or aggregation caused quenching (ACQ) with respect to their fluorescence. In the latter case, the existence of excited dimers, which are proposed to use in a switchable manner in combination with the quenching of nuclear spin singlet states, is observed
Keywords: aggregation induced emission, aggregation caused quenching, bimodal contrast agents, fluorescence, NMR spectroscopy
Long‐lived nuclear singlet states have been shown to be a viable tool in many different areas of research, from material sciences over the investigation of catalytic cycles to applications in biochemistry. The synthesis of four compounds, in which a nuclear singlet state in pairs of 13C and 15N nuclei can be populated is shown. Furthermore, bimodal fluorescence/singlet‐state NMR application for the determination of water concentration has been investigated, which can be useful in a variety of fields.
Multimodal molecular methods have been emerging as a powerful imaging tools in the scientific community as well as in the clinical practice. [1] Nuclear magnetic resonance (NMR) and fluorescence represent a particularly attractive combination for bimodal molecular probes because they can realize the complementary abilities of each modality – high spatial resolution with fluorescence and the detection of atomic‐scale changes even in deep tissue via magnetic resonance – and therefore achieve greater effects.[ 2 , 3 ]
Phenomena related to extended nuclear spin order lifetimes by using long‐lived nuclear singlet states (LLSS) are of interest for an emerging field of research aiming to discover novel molecular probes for magnetic resonance imaging.[ 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 ] Nuclear spin singlet states consist of two spin‐1/2 nuclei coupling to a total spin 0 state.[ 4 , 14 ] This property endows the nuclear singlet state immunity to direct dipole‐dipole interactions, a main contribution to longitudinal magnetization relaxation (T 1). Although the nuclear singlet state is NMR invisible and cannot be detected directly, Levitt and colleagues[ 15 , 16 ] revealed in 2004 that long‐lived states can be probed. To date, several methods have been developed to populate the singlet spin order and evaluate the singlet equilibration time (T S) between the singlet and triplet state.[ 4 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 ] The difference in chemical shift and J‐couplings have been explored to populate singlet states, which was recently shown to be even possible via localized spectroscopy in vivo..[ 15 , 16 , 17 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 ] One prerequisite for a molecular system to support long‐lived singlet lifetime is to incorporate an isolated spin‐1/2 homonuclear pair in almost exact chemical equivalence. Chemically equivalent systems have the potential of supporting long‐lived singlet lifetime because its symmetrical properties provide a protection against common relaxation mechanisms and thus do not allow for singlet‐triplet leakage.[ 33 , 43 ] However, the chemically equivalent spin pair has no chemical shift difference and J‐coupling difference is the only way allowing to populate singlet spin order since a transition between the two states requires a break in symmetry.[ 26 , 27 ] In such symmetric molecules, large (>5 Hz) J‐coupling have mainly been investigated indicating close proximity of the additional spins to the singlet spin‐pair. When using deuterons instead of protons, not only the J‐couplings are significantly reduced but also dipolar effects in proximity to the singlet spin pair are significantly reduced. This is the main point here. Additionally to further remove dipolar effects, one would like to move all spins that do not form the singlet as far away as possible in the molecule. This will automatically lead to a reduction in J‐coupling. Being able to access singlet states with very small J‐couplings or perturbations of the symmetry hence means that we can work towards optimized singlet molecules with long‐lifetime. In one example, a small J‐coupling difference (<1 Hz) has been used to read‐out long‐lived spin order when the singlet spin order originated from para‐hydrogen.[ 28 , 34 ] However the population of the singlet state in these cases was obtained by a reaction of a precursor with para‐hydrogen and subsequent transfer of the magnetization by means of field cycling [34] The particular long proton singlet states found in the latter studies underlines that an optimal design may be possible with minimized J‐couplings in symmetric molecules. Therefore, it is of great interest to develop new molecular systems with long‐lived singlet order created by the small J‐coupling difference.
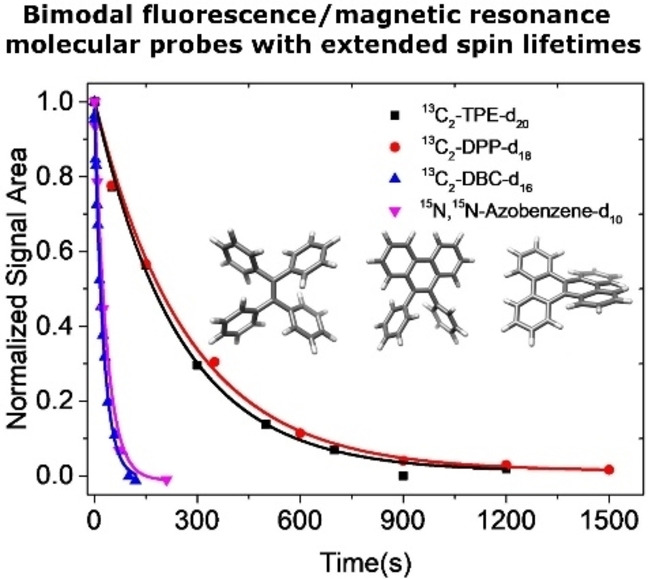
Herein, we introduce molecules that show fluorescent properties and display long‐lived nuclear spin states. The latter are accessed via very small J‐couplings (<1 Hz) between 13C or 15N and 2H spins to populate singlet states. The deuterium spins break the magnetic symmetry and significantly enhance the singlet lifetime compared to protonated counterparts. We have designed, synthesized and investigated perdeuterated 13C2‐TPE (tetraphenylethylene), DPP (diphenylphenanthrene) and DBC (dibenzochrysene) two of its derivatives, namely (figure 1a) and one fully deuterated 15N‐15N’‐diazobenzene compound (Figure 1b), which is a photo‐switchable molecule with long LLSS in its protonated form.[ 44 , 45 ] The synthesis of the molecules is described in detail in the Supporting Information. The singlet states were investigated using the SLIC (spin‐lock induced crossing) sequence with optimized spin‐lock durations and nutation frequencies (Supporting Information). [46] The singlet decay curves for the TPE derivatives at B0=7 T and for the azobenzene at B0=16.5 T are depicted in figure 1c with all relaxation and singlet equilibration times reported in table 1. From the analysis of the signal build‐up and 1D spectra, the 13C‐13C J‐coupling values of 13C2‐TPE‐d20 and its derivatives were respectively determined as 1 J cc=71 Hz, 3 J CD=0.82 Hz and 4 J CD<0.1 Hz for 13C2‐TPE‐d20, 1 J cc=62 Hz, 3 J CD=1.55 Hz and 4 J CD <0.1 Hz for 13C2‐DPP‐d18, and 1 J cc=57 Hz, 3 J CD=1.76 Hz and 4 J CD <0.1 Hz for 13C2‐DBC‐d16, whereby the 4 J coupling represents an estimate as this coupling could not be resolved.[ 26 , 27 ] These observations demonstrate that singlet spin order can still be populated by harnessing such small J‐couplings.
Figure 1.
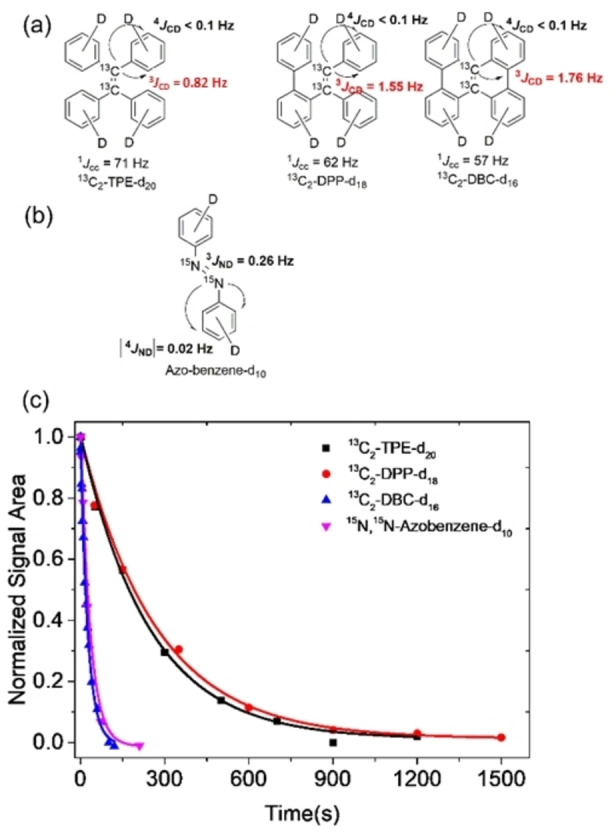
a) Molecular structure of fully deuterated 13C2‐TPE derivatives with J coupling values. b) The J couplings of trans‐15N, 15N’‐azobenzene‐d10. c) Experimental decay curves of long‐lived singlet state for 13C2‐TPE‐d20, 13C2‐DPP‐d18, 13C2‐DBC‐d16 and trans‐15 N,15 N‐Azobenzene‐d10. All the samples were degassed by three times freeze‐pump‐thaw cycling. Experimental data points of fully deuterated 13C2‐TPE derivatives and trans‐15N, 15N’‐azobenzene‐d10 were observed at the magnetic fields of 7.05 T and 16.5 T, respectively. The pulse sequence SLIC [46] was employed and the data points have been normalized to the value of the first point. Solid lines are fitted to the exponential decays.
Table 1.
Experimental value of T 1 and T s of 13C2‐TPE‐d20, 13C2‐DPP‐d18, 13C2‐DBC‐d16, 15N, 15N’‐azobenzene‐d10, 13C2‐TPE, 13C2‐DPP and 13C2‐DBC.
|
Compounds |
T s (s) |
T 1 (s) |
|---|---|---|
|
13C2‐TPE‐d20 |
243.5±20.9 |
22.7±0.1 |
|
13C2‐DDP‐d18 |
281.1±17.0 |
22.6±0.1 |
|
13C2‐DBC‐d16 |
26.8±1.8 |
15.2±0.0 |
|
15N, 15N‐Azobenzene‐d10 |
15.2±4.3 |
3.66±0.1 |
|
13C2‐TPE |
47.8±7.1 |
16.1±0.1 |
|
13C2‐DDP |
40.6±1.9 |
16.5±0.2 |
|
13C2‐DBC |
20.3±4.0 |
12.3±0.3 |
All samples were degassed by three freeze‐pump‐thaw cycles.
Another chemically equivalent system with even smaller J‐couplings that can be used to investigate singlet states is isotopically labeled 15N,15N’‐azobenzene‐d10. The heteronuclear J‐couplings found in the in trans‐form were determined as 1 J NN=16 Hz, 3 J ND=0.26 Hz, |4 J ND| <0.1 Hz. Here, none of the nitrogen‐deuterium couplings could be resolved and the couplings are estimated from the reported 15N‐1H values, scaled by the difference in gyromagnetic ratio between protons and deuterium.[ 44 , 45 ] The T 1 value of 13C2‐TPE‐d20 is about 22.7 s while the T s of 13C2‐TPE‐d20 dramatically increased to 243.5 s. The T 1 of 13C2‐DPP‐d18 and 13C2‐TPE‐d20 are similar and its T s is slightly increased to 281.1 s. Unlike 13C2‐TPE‐d20 and 13C2‐DPP‐d18, the T 1 and T s value of 13C2‐DBC‐d16 are significantly shortened to 15.2 s and 26.8 s. The determined T s of the respective compounds illustrate, that, apart from chemical shift difference and J‐couplings, the molecular symmetry plays a non‐negligible role in the stability of the singlet state. Due to a steric hindrance DBC cannot maintain a planar form but is twisted. This effect contributes to the decrease in T 1 and T s. The singlet lifetime of deuterated trans‐15N, 15N’‐azobenzene‐d10 is only 15.1 s, which is unexpectedly shorter than the protonated counterpart as reported by Ivanov et al.[ 44 , 45 ] We however note that in the deuterated molecule we observe a monoexponential decay only whereas in the reported protonated from, a much longer second exponential decay is observable. For comparison, we also synthesized the corresponding protonated molecules of the TPE derivatives and their T 1 and T s value were also evaluated under the same conditions (Table 1). The T 1 value of 13C2‐TPE and 13C2‐DPP are comparable with their corresponding counterparts that is 16.1 s and 16.5 s, respectively. However, the T s value of 13C2‐TPE and 13C2‐DPP shorten to 47.8 s and 40.6 s, respectively. 13C2‐DBC also has the similar tendency with its counterpart 13C2‐DBC‐d16 that both of its T 1 and T s shorten to 12.3 s and 20.3 s, respectively. The above results clearly show that the singlet state lifetime of 13C2‐TPE‐d20 and 13C2‐DPP‐d18 were extended more than 4‐folds due to the deuteration and are still accessible via very small J‐couplings adding another dimension to be considered when designing molecular probes with long lived singlet‐states. These findings illustrate the impact of the J‐coupling on the stability of the singlet‐states and thus the significance of the deuteration of the compounds.
To get insight into the singlet lifetime changes between the deuterated 13C molecules, their structure geometry were computed by the density functional theory (DFT) method B3LYP 6–31+G(d, p). The optimized molecular geometry is shown in Figure 2, which are in accordance with the reported literatures.[ 47 , 48 , 49 ] From the results, 13C2‐TPE‐d20 is a D2 symmetrical molecule, while 13C2‐DPP‐d18 is C2 and 13C2‐DBC‐d16 is D2. Since the molecular geometry is significantly different, their optimized geometry parameters (Table s1) accordingly changed. The results showed that distances between the atoms, such as d (13C‐13C), d (13C, 2H), d (2H, 2H) become shorter after the phenyl rings were locked. Thereafter, the chemical shift anisotropy CSA (13C) also become larger after locking the rings. Those results could draw a finding that the conformational change significantly affects the singlet state lifetime.
Figure 2.
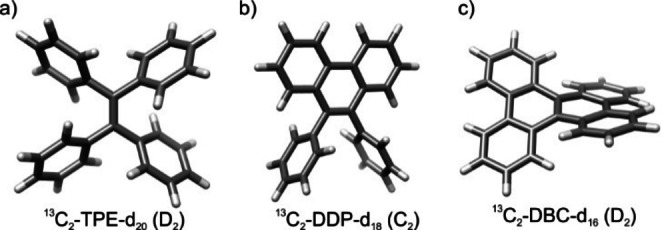
Structural models of 13C2‐TPE‐d20 (a), 13C2‐DPP‐d20 (b) and 13C2‐DBC‐d20 (c) optimized at B3LYP 6–31+G(d, p) density functional theory (DFT) level, using a polarizable continuum model for THF (for further information, see Supporting Information). The point groups resulting are given in parentheses.
Fluorescence performance of 13C2‐TPE‐d20, 13C2‐DPP‐d18 and 13C2‐DBC‐d16 were subsequently investigated. As shown from fluorometry in Figure 3, 13C2‐TPE‐d20 displays the same Aggregation Induced Emission (AIE) effect observed in protonated 13C2‐TPE. [31] The molecule of 13C2‐TPE‐d20 is weakly fluorescent in the dissolved state in THF solution, while it switched to emit strong fluorescence centered at 472 nm in the presence of 95 % water, where the aggregated state is formed. [50] In this aggregated state, TPE exhibits a fluorescence quantum yield of Φ F=0.13. [51] The extinction coefficient has been determined to be ϵ 319=2550 L ⋅ mol−1cm−1 leading to a brightness of 331 L ⋅ mol−1cm−1, which is in the same order of magnitude of other fluorescence probes with AIE properties.[ 52 , 53 ] In sharp contrast, dissolved 13C2‐DPP‐d18 emit strong fluorescence at the peak of 374 nm along with a shoulder at 358 nm, while the fluorescence intensity at the given wavelengths is quenched upon addition of 95 % water. At a fluorescence quantum yield of Φ F=0.053 in DPP [51] this leads to a brightness of 151 L ⋅ mol−1cm−1 at an extinction coefficient of ϵ 307=2550 L ⋅ mol‐1cm−1. At the same time a red shifted band appears at 480 nm which can be attributed to excited dimers. Similarly, dissolved 13C2‐DBC‐d16 emit strong fluorescence at the wavelength of 395 nm along with a shoulder at 408 nm, the fluorescence is quenched upon addition of 95 % water and a new fluorescent peak emerges at the wavelength of 482 nm. In this case, for the non‐aggregated compound, the extinction coefficient was determined to be ϵ 303=33700 L ⋅ mol−1cm−1. 13C2‐DPP‐d18 [51] and 13C2‐DBC‐d16 [49] display an opposite phenomenon of AIE, which is termed aggregation‐caused quenching (ACQ). The significant change in fluorescence behavior between 13C2‐TPE‐d20 and 13C2‐DPP‐d18, 13C2‐DBC‐d16 is related to their structural modification. Since the phenyl rings of 13C2‐DPP‐d18, 13C2‐DBC‐d16 are covalently locked to form a conjugated structure, their intramolecular motion is restricted and therefore their fluorescence behavior is affected. For both the AIE or ACQ effect, a bimodal switching process is observed effecting the fluorescence and nuclear spins at the same time: Upon the aggregation the nuclear spin cannot be detected in the liquid anymore due to dipolar broadening of the lines, however the fluorescence serves as an indicative read‐out. The change in fluorescence properties started at rather specific THF/H2O ratios of 30 : 70 for 13C2‐TPE‐d20, and 20 : 80 for 13C2‐DPP and 13C2‐DBC‐d16 respectively. In the same manner, the singlet state cannot be detected anymore at specific THF/D2O ratios of 50 : 50 for 13C2‐TPE‐d20, 80 : 20 for 13C2‐DPP, and 65 : 35 for 13C2‐DBC‐d16.The spectra proving this behavior are displayed in the Supporting Information (page 7). This process is reversible once the solvent system is changed towards organic solvents and the water removed.
Figure 3.
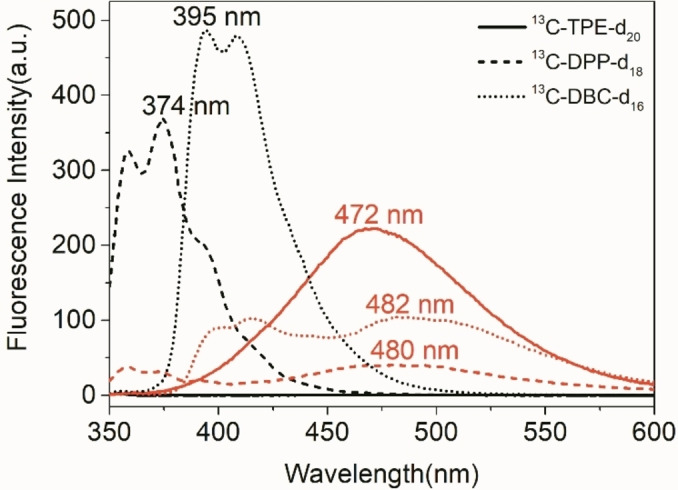
Fluorescence spectra of 13C2‐TPE‐d20 (solid line), 13C2‐DPP‐d18 (short dash line) and 13C2‐DBC‐d16 (short dot line) at the concentration of 10 uM in THF‐d8 with the fraction of 0 % (black) and 95 % water (red).
In conclusion, we have introduced a concept of ‘on‐off’ switchable singlet NMR/fluorescence bimodal probe, whereby its singlet spin order can be accessed with small J‐couplings (<1 Hz). We have analyzed the influence of conformational changes on the singlet relaxation lifetime and we have demonstrated that singlet lifetimes up to 4 min are achievable at high fields. Moreover, addition of water significantly quenches singlet lifetimes and effects their fluorescent properties. Although this has been mainly investigated here in an organic solvent system and the addition of water, tuning the molecules to for example sense ions in a biological context will be challenges for the future. In addition, the combination with techniques of magnetic resonance signal enhancement poses opportunities to develop the here proposed approaches into molecular probes for biomedicine.[ 54 , 55 , 56 , 57 , 58 ] Hence we envision the application potential as contrast agents or to map the bio‐distribution of photo‐pharmaceuticals. [59]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We gratefully thank the Max‐Planck‐Society and the Max‐Planck‐ Institute for Biophysical Chemistry for generous funding. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant agreement No. 949180). Open Access funding enabled and organized by Projekt DEAL.
S. Yang, P. Saul, S. Mamone, L. Kaltschnee, S. Glöggler, Chem. Eur. J. 2022, 28, e202104158.
References
- 1.
- 1a. Louie A., Chem. Rev. 2010, 110, 3146–3195; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Shin T.-H., Choi Y., Kim S., Cheon J., Chem. Soc. Rev. 2015, 44, 4501–4516; [DOI] [PubMed] [Google Scholar]
- 1c. Jennings L. E., Long N. J., Chem. Commun. 2009, 3511–3524; [DOI] [PubMed] [Google Scholar]
- 1d. Maldiney T., Doan B.-T., Alloyeau D., Bessodes M., Scherman D., Richard C., Adv. Funct. Mater. 2015, 25, 331–338; [Google Scholar]
- 1e. Angelovski G., Angew Chem. Int. Ed. 2016, 55, 7038–7046. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Verwilst P., Park S., Yoona B., Kim J. S., Chem. Soc. Rev. 2015, 44, 1791–1806; [DOI] [PubMed] [Google Scholar]
- 2b. Harrison V. S. R., Carney C. E., Macrenaris K. W., Meade T. J., Chem. Commun. 2014, 50, 11469–11471; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Yang S., Yuan Y., Jiang W., Ren L., Deng H., Bouchard L.-S., Liu M., Zhou X., Chem. Eur. J. 2017, 23, 7648–7652; [DOI] [PubMed] [Google Scholar]
- 2d. Harrison V. S. R., Carney C. E., MacRenaris K. W., Waters E. A., Meade T. J., J. Am. Chem. Soc. 2015, 137, 9108–9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Xu Z., Liu C., Zhao S., Chen S., Zhao Y., Chem. Rev. 2019, 119, 195–230; [DOI] [PubMed] [Google Scholar]
- 3b. Xie D., Yu M., Kadakia R. T., Que E. L., Acc. Chem. Res. 2020, 53, 2–10. [DOI] [PubMed] [Google Scholar]
- 4. Levitt M. H., Annu. Rev. Phys. Chem. 2012, 63, 89–105. [DOI] [PubMed] [Google Scholar]
- 5. Feng Y., Theis T., Wu T.-L., Claytor K., Warren W. S., J. Chem. Phys. 2014, 141, 134307. [DOI] [PubMed] [Google Scholar]
- 6. Tayler M. C. D., Levitt M. H., Phys. Chem. Chem. Phys. 2011, 13, 9128–9130. [DOI] [PubMed] [Google Scholar]
- 7. Ahuja P., Sarkar R., Vasos P. R., Bodenhausen G., J. Chem. Phys. 2007, 127, 134112. [DOI] [PubMed] [Google Scholar]
- 8. Levitt M. H., J. Magn. Reson. 2019, 306, 69–74. [DOI] [PubMed] [Google Scholar]
- 9. Zhang Y., Duan X., Soon P. C., Sychrowsky V., Canary J. W., Jerschow A., ChemPhysChem 2016, 17, 2967–2971. [DOI] [PubMed] [Google Scholar]
- 10. DeVience S. J., Walsworth R., Rosen M. S., NMR Biomed. 2013, 26, 1204–1212. [DOI] [PubMed] [Google Scholar]
- 11. Nikolaou P., Goodson B. M., Chekmenev E. Y., Chem. Eur. J. 2015, 21, 3156–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Glöggler S., Elliott S. J., Stevanato G., Brown R. C. D., Levitt M. H., RSC Adv. 2017, 55, 34574–34578. [Google Scholar]
- 13. Mamone S., Glöggler S., Phys. Chem. Chem. Phys. 2018, 20, 22463–22467. [DOI] [PubMed] [Google Scholar]
- 14. Pileio G., Prog. Nucl. Magn. Res. Spectr. 2010, 56, 217–231. [DOI] [PubMed] [Google Scholar]
- 15. Carravetta M., Johannessen O. G., Levitt M. H., Phys. Rev. Lett. 2004, 92, 153003. [DOI] [PubMed] [Google Scholar]
- 16. Carravetta M., Levitt M. H., J. Am. Chem. Soc. 2004, 126, 6228–6229. [DOI] [PubMed] [Google Scholar]
- 17. Pileio G., Carravetta M., Levitt M. H., Proc. Natl. Acad. Sci. USA 2011, 107, 17135–17139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mamone S., Rezaei-Ghaleh N., Opazo F., Griesinger C., Glöggler S., Sci. Adv. 2020, 6, eaaz1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sarkar R., Vasos P. R., Bodenhausen G., J. Am. Chem. Soc. 2007, 129, 328–334. [DOI] [PubMed] [Google Scholar]
- 20. Kiryutin A. S., Pravditsev A. N., Yurkovskaya A. V., Vieth H.-M., Ivanov K. L., J. Phys. Chem. B 2016, 120, 11978–11986. [DOI] [PubMed] [Google Scholar]
- 21. Pravdivtsev A. N., Kiryutin A. S., Yurkovskaya A. V., Vieth H. M., Ivanov K. L., J. Magn. Reson. 2016, 273, 56–64. [DOI] [PubMed] [Google Scholar]
- 22. Kharkov B., Duan X., Tovar E. S., Canary J. W., Jerschow A., Phys. Chem. Chem. Phys. 2019, 21, 2595–2600. [DOI] [PubMed] [Google Scholar]
- 23. Elliott S. J., Stevanato G., J. Magn. Reson. 2019, 301, 49–55. [DOI] [PubMed] [Google Scholar]
- 24. Tayler M. C. D., Levitt M. H., Phys. Chem. Chem. Phys. 2011, 13, 5556–5560. [DOI] [PubMed] [Google Scholar]
- 25. Pileio G., Carravetta M., Hughes E., Levitt M. H., J. Am. Chem. Soc. 2008, 130, 12582–12583. [DOI] [PubMed] [Google Scholar]
- 26. Feng Y., Davis R. M., Warren W. S., Nat. Phys. 2012, 8, 831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feng Y., Theis T., Liang X., Wang Q., Warren W. S., J. Am. Chem. Soc. 2013, 135, 9632–9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Franzoni M. B., Buljubasich L., Spiess H. W., Münnemann K., J. Am. Chem. Soc. 2012, 134, 10393–10396. [DOI] [PubMed] [Google Scholar]
- 29. Claytor K., Theis T., Feng Y., Yu J., Gooden D., Warren W. S., J. Am. Chem. Soc. 2014, 136, 15118–15121. [DOI] [PubMed] [Google Scholar]
- 30. Claytor K., Theis T., Feng Y., Warren W., J. Magn. Reson. 2014, 239, 81–86. [DOI] [PubMed] [Google Scholar]
- 31. Yang S., McCormick J., Mamone S., Bouchard L.-S., Glöggler S., Angew. Chem. Int. Ed. 2019, 58, 2879–2883. [DOI] [PubMed] [Google Scholar]
- 32. Warren W. S., Jensita E., Branca R. T., Chen X., Science 2009, 323, 1711–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stevanato G., Hill-Cousins J. T., Håkansson P., Roy S. S., Brown L. J., Brown R. C. D., Pileio G., Levitt M. H., Angew. Chem. Int. Ed. 2015, 54, 3740–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y., Soon P. C., Jerschow A., Canary J. W., Angew. Chem. Int. Ed. 2014, 53, 3396–3399. [DOI] [PubMed] [Google Scholar]
- 35. Procacci B., Roy S. S., Norcott P., Turner N., Duckett S. B., J. Am. Chem. Soc. 2018, 140, 16855–16864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saul P., Mamone S., Glöggler S., Chem. Sci. 2019, 10, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carravetta M., Levitt M. H., J. Chem. Phys. 2005, 122, 214505. [DOI] [PubMed] [Google Scholar]
- 38. Pileio G., Levitt M. H., J. Chem. Phys. 2009, 130, 214501. [DOI] [PubMed] [Google Scholar]
- 39. Pileio G., Bowen S., Laustsen C., Tayler M. C. D., Hill-Cousins J. T., Brown L. J., Brown R. C. D., Ardenskjær-Larsen J. H., Levitt M. H., J. Am. Chem. Soc. 2013, 135, 5084–5088. [DOI] [PubMed] [Google Scholar]
- 40. Theis T., G. X. Ortiz Jr , Logan A. W. J., Claytor K. E., Feng Y., Huhn W. P., Blum V., Malcolmson S. J., Chekmenev E. Y., Wang Q., Warren W. S., Sci. Adv. 2016, 2, e1501438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stavarache C., Hanganu A., Paun A., Paraschivescu C., Matache M., Vasos P. R., J. Magn. Reson. 2017, 284, 15–19. [DOI] [PubMed] [Google Scholar]
- 42.S. Mamone, A. B. Schmidt, N. Schwaderlapp, T. Lange, D. v. Elverfeldt, J. Hennig, J. Hennig, S. Glöggler, NMR Biomed. 10.1002/nbm.4400. [DOI] [PubMed]
- 43. Stevanato G., Singha Roy S., Hill-Cousins J., Kuprov I., Brown L. J., Brown R. C. D., Pileio G., Levitt M. H., Phys. Chem. Chem. Phys. 2015, 17, 5913–5922. [DOI] [PubMed] [Google Scholar]
- 44. Sheberstov K. F., Vieth H., Zimmermann H., Rodin B. A., Ivanov K. L., Kiryutin A. S., Yurkovskaya A. V., Sci. Rep. 2019, 9, 20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sheberstov K. F., Vieth H.-M., Zimmermann H., Ivanov K. L., Kiryutin A. S., Yurkovskaya A. V., Appl. Magn. Reson. 2018, 49, 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. DeVience S. J., Walsworth R. L., Rosen M. S., Phys. Rev. Lett. 2013, 111, 173002. [DOI] [PubMed] [Google Scholar]
- 47. Shustova N. B., Ong T.-C., Cozzolino A. F., Michaelis V. K., Griffin R. G., Dincă M., J. Am. Chem. Soc. 2012, 134, 15061–15070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cai Y., Du L., Samedov K., Gu X., Qi F., Sung H. H. Y., Patrick B. O., Yan Z., Jiang X., Zhang H., Lam J. W. Y., Williams I. D., Phillips D. L., Qin A., Tang B., Chem. Sci. 2018, 9, 4662–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.
- 49a. Hatakeyama T., Hashimoto S., Seki S., Nakamura M., J. Am. Chem. Soc. 2011, 133, 18614–18617; [DOI] [PubMed] [Google Scholar]
- 49b. Huang D.-C., Kuo C.-H., Ho M.-T., Lin B.-C., Peng W.-T., Chao I., Hsu C.-P., Tao Y.-T., J. Mater. Chem. C 2017, 5, 7935–7943; [Google Scholar]
- 49c. Ueda Y., Tsuji H., Tanaka H., Nakamura E., Chem. Asian J. 2014, 9, 1623–1628; [DOI] [PubMed] [Google Scholar]
- 49d. Hashimoto S., Ikuta T., Shiren K., Nakatsuka S., Ni J., Nakamura M., Hatakeyama T., Chem. Mater. 2014, 26, 6265–6271. [Google Scholar]
- 50. Mei J., Leung N. L. C., Kwok R. T. K., Lam J. W. Y., Tang B. Z., Chem. Rev. 2015, 115, 11718–11940. [DOI] [PubMed] [Google Scholar]
- 51. Aldred M. P., Li C., Zhu M.-Q., Chem. Eur. J. 2012, 18, 16037–16045. [DOI] [PubMed] [Google Scholar]
- 52. Mei J., Hong Y., Lam J. W. Y., Qin A., Tang Y., Tang B. Z., Adv. Mater. 2014, 26, 5429–5479. [DOI] [PubMed] [Google Scholar]
- 53. He Z., Ke C., Tang B. Z., ACS Omega 2018, 3, 3267–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Albers M. J., Bok R., Chen A. P., Cunningham C. H., Zierhut M. L., Zhang V. Y., Kohler S. J., Tropp J., Hurd R. E., Yen Y.-F., Nelson S. J., Vigneron D. B., Kurhanewicz J., Cancer Res. 2008, 68, 8607–8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rodrigues T. B., Serrao E. M., Kennedy B. W. C., Hu D.-E., Kettunen M. I., Brindle K. M., Nat. Med. 2014, 20, 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cabella C., Karlsson M., Canapè C., Catanzaro G., Serra S. C., Miragoli L., Poggi L., Uggeri F., Venturi L., Jensen P. R., Lerche M. H., Tedoldi F., J. Magn. Reson. 2013, 232, 45–52. [DOI] [PubMed] [Google Scholar]
- 57. Jensen P. R., Karlsson M., Meier S., Duus J. Ø., Lerche M. H., Chemistry 2009, 15, 10010–10012. [DOI] [PubMed] [Google Scholar]
- 58. Jiang W., Lumata L., Chen W., Zhang S., Kovacs Z., Sherry A. D., Khemtong C., Sci. Rep. 2015, 5, 9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lerche M. H., Meier S., Jensen P. R., Hustvedt S.-O., Karlsson M., Duus J. Ø., Ardenkjær-Larsen J. H., NMR Biomed. 2011, 24, 96–103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information