

Abstract
Background and objectives
Vascular risk factors and elevated β-amyloid (Aβ) are commonly observed together among older adults. Here, we examined the interactive vs independent effects of systemic vascular risk and Aβ burden on longitudinal gray matter atrophy and how their co-occurrence may be related to cognitive decline in a cohort of clinically normal adults. A secondary goal was to examine whether vascular risk influences gray matter atrophy independently from markers of white matter injury.
Methods
Participants were 196 adults (age 73.8 ± 6.1 years) from the Harvard Aging Brain Study. Baseline Aβ burden was quantified with Pittsburgh compound B PET. Baseline vascular risk was measured with the Framingham Heart Study cardiovascular disease risk score. Brain atrophy was quantified longitudinally with structural MRI over a median of 4.50 (±1.26) years. Cognition was assessed yearly with the Preclinical Alzheimer Cognitive Composite over a median of 6.25 (±1.40) years. Linear mixed-effects models examined vascular risk and Aβ burden as interactive vs independent predictors of gray matter atrophy, with adjustment for age, sex, years of education, APOE ε4 status, intracranial volume (when appropriate), and their interactions with time. In subsequent models, we adjusted for markers of white matter injury to determine whether vascular risk accelerated brain atrophy independently from diffusion- and fluid-attenuated inversion recovery (FLAIR)–based markers. Mediation analyses examined whether brain atrophy mediated the interactive association of vascular risk and Aβ burden on cognitive decline.
Results
Higher vascular risk and elevated Aβ burden interacted to predict more severe atrophy in frontal and temporal lobes, thalamus, and striatum. Higher Aβ burden, but not vascular risk, was associated with more severe atrophy in parietal and occipital lobes, as well as the hippocampus. Adjusting for diffusion- and FLAIR-based markers of white matter injury had little impact on the above associations. Gray matter atrophy mediated the association between vascular risk and cognitive decline at higher levels of Aβ burden.
Discussion
We observed an interaction between elevated vascular risk and higher Aβ burden with longitudinal brain atrophy, which in turn influenced cognitive decline. These results support vascular risk factor management as a potential intervention to slow neurodegeneration and cognitive decline in preclinical Alzheimer disease.
Accumulating data suggest that vascular risk factors such as obesity, hypertension, high cholesterol, and diabetes are not only risk factors for cardiovascular disease but also are risk factors for Alzheimer disease (AD) dementia.1-3 Recent data from our group support the idea that vascular risk factors might act both independently and via canonical AD pathways to promote progression to AD dementia.4,5 Specifically, prior work demonstrated that combined elevated systemic vascular risk and higher β-amyloid (Aβ) burden was associated with greater cortical tau deposition5 and faster cognitive decline in clinically unimpaired adults.4 Elevated tau deposition and cognitive impairment have been closely linked to neurodegeneration6-8; therefore, neurodegeneration may be a final common pathway by which vascular risk and AD pathology lead to dementia.
Studies examining vascular risk and Aβ deposition as separate predictors of brain atrophy suggest that the 2 processes are associated with both overlapping and distinct patterns of brain atrophy as quantified by MRI.9-14 Vascular risk factors have been linked to atrophy primarily in frontal and temporal regions, whereas Aβ deposition has most commonly been associated with preferential cortical thinning in medial parietal and temporal regions.9-14 Few studies have examined the combined impact of vascular risk and Aβ burden on brain atrophy and how this in turn influences cognitive decline.15,16 In addition, it remains unclear whether vascular risk factors exacerbate brain atrophy via white matter changes or whether this occurs through independent pathways.17 Longitudinal studies are needed to advance our understanding of the interplay between vascular risk and Aβ burden on patterns of brain atrophy. Such studies may offer increased insight into shared pathways of disease and increase prognostic accuracy.
In the present study, we investigated the interactive vs independent associations of systemic vascular risk and Aβ burden on longitudinal patterns of brain atrophy in clinically unimpaired individuals from the Harvard Aging Brain Study (HABS). Secondarily, we examined whether systemic vascular risk is associated with brain atrophy independently from imaging markers of white matter injury, including diffusion-derived microstructural abnormalities and white matter hyperintensities (WMH). Last, building on prior work demonstrating that elevated vascular risk interacts with Aβ burden to accelerate cognitive decline,4,18,19 we examined whether this association is mediated by brain atrophy.
Methods
Participants
Participants were clinically unimpaired adults recruited from HABS.20 HABS began in 2010 and is ongoing. At study entry, participants were deemed clinically normal if they had a global Clinical Dementia Rating21 of 0, a Mini-Mental State Examination22 score ≥27 with educational adjustment, and intact performance on Logical Memory delayed recall.23 All participants were screened for major neurologic, psychiatric, and unstable medical illnesses and were required to have a Geriatric Depression Scale24 <11. Participants were excluded if they had unstable hypertension, uncontrolled diabetes, a Hachinski score of ≥5, a history of stroke with residual deficits, or a history of intracranial hemorrhage. For the present study, participants were required to have baseline Aβ PET data, baseline demographic and medical information to calculate a summary measure of vascular risk, baseline diffusion data, baseline WMH data, APOE data, and at least 2 MRI data points.
Standard Protocol Approvals, Registrations, and Patient Consents
The Partners Institutional Review Board approved the protocol for HABS. All study participants provided written informed consent before the performance of any study procedures.
Baseline Aβ PET
Baseline Aβ burden was quantified with 11C-Pittsburgh compound B (PiB) PET, as previously described.25 In brief, we calculated a summary distribution volume ratio for each participant by averaging the median PiB uptake value across frontal, lateral parietal and temporal, and retrosplenial cortices. Cerebellar gray matter served as the reference region. PET data were corrected for partial volume effects with the geometric transfer matrix method.26 Aβ burden was used as a continuous variable in all analyses. As expected, a bimodal distribution was observed for Aβ PET values. When we used log- or square root–transformed values of Aβ in the main models, the findings remained unchanged. We opted to report all results using the nontransformed Aβ values to enhance interpretability and to be consistent with prior studies from our group.
Baseline Cardiovascular Disease Risk
Consistent with previous studies from our group,4,5 baseline systemic vascular risk was quantified with the office-based Framingham Heart Study cardiovascular disease risk score (FHS-CVD).27 The FHS-CVD takes into account age, sex, antihypertensive treatment (yes or no), systolic blood pressure, body mass index (calculated as weight in kilograms divided by height in meters squared), history of diabetes (yes or no), and current cigarette smoking status (smoker or nonsmoker). The FHS-CVD provides a 10-year probability of future cardiovascular events. In our sample, the FHS-CVD ranged from 3.7% to 81.7% (mean 32.5%), with higher scores representing greater vascular risk.
Baseline Diffusion Tensor Imaging
MRI scanning was performed on a Siemens (Erlangen, Germany) TIM Trio 3T system with a 12-channel head coil at Massachusetts General Hospital (Athinoula A. Martinos Center for Biomedical Imaging). As previously reported,28 diffusion-weighted imaging was performed at baseline and collected with the following parameters: repetition time (TR) 8,040 milliseconds, echo time (TE) 84 milliseconds, time to inversion (TI) 2,100 milliseconds, 2 × 2 × 2–mm voxels, 64 transverse slices, b = 700 s/mm2, 30 diffusion directions, and 2× acceleration. Diffusion-weighted imaging data were processed in FSL version 5.0.9 (The Oxford Centre for Functional MRI of the Brain Software Library). We corrected for eddy current and motion distortions using the Eddy tool in FSL (FMRIB Software Library).29 We performed tract-based spatial statistics procedures30 and skeletonized the diffusion data to account for misregistration effects. After alignment to standard space, the average fractional anisotropy (FA) value was extracted from the full mask of the standard FSL FMRIB58 white matter skeleton. We refer to this metric as global FA.
Baseline WMH
WMH were assessed using fluid-attenuated inversion recovery images performed at baseline (TR 6,000 milliseconds, TE 454 milliseconds, TI 2,100 milliseconds, 1 × 1 × 1.5–mm voxels, 2× acceleration). Total WMH volume (cubic millimeters) was quantified with an automated algorithm31 as previously described.32,33 The distribution of WMH volumes was positively skewed and was log-transformed prior to analysis.
Longitudinal Structural Imaging
High-resolution 3D T1-weighted multiecho magnetization prepared rapid acquisition gradient-echo anatomic images were collected with the following parameters: TR 2,300 milliseconds, TE 2.95 milliseconds, TI 900 milliseconds, flip angle 9°, and 1.05 × 1.05 ×1.2–mm resolution. A subset of anatomic images were collected with a different set of parameters: TR 2,300 milliseconds, TE 2.98 milliseconds, TI 900 milliseconds, flip angle 9°, and 1.00 × 1.00 × 1.2–mm resolution. These 2 sequences have been deemed equivalent.
MRI structural data in HABS are acquired at years 1 (baseline), 4, and 6. At the time of the present analyses, MRI data were available for 196 participants at year 1, 190 participants at year 4, and 108 participants at year 6. Some of these participants received an additional scan at year 1 (n = 1), year 1.5 (n = 32), and/or year 4 (n = 54). All available data points were used in the present analyses to improve reliability. The median number of MRI scans per participant was 2.96 (±0.70) over a median of 4.50 (±1.26) years. Estimation of cortical thickness and subcortical volumetric segmentation was performed with FreeSurfer version 6.0.34,35 As previously described,20 MRI scans were grouped by participant and processed together with the FreeSurfer longitudinal processing stream.36 To reduce the number of analyses, we examined changes within bilateral frontal, temporal, parietal, and occipital cortices using a weighted average of FreeSurfer regions for each lobe (eTable 1, links.lww.com/WNL/B990, gives the regions included in each lobe). In addition to cortical regions, we examined subcortical and allocortical regions, including bilateral striatum, thalamus, and hippocampus. The striatum consisted of a volume-weighted average of bilateral putamen and bilateral caudate regions. The thalamus and hippocampus were each averaged across left and right hemispheres.
Longitudinal Cognition
Cognition was assessed annually with the Preclinical Alzheimer Cognitive Composite (PACC).4,37 As previously described,38 the composite includes tests of memory (Logical Memory delayed recall and the Free and Cued Selective Reminding Test), a timed measure of executive function (Digit Symbol Substitution Test), a measure of global cognition (Mini-Mental State Examination), and a category fluency task (animals, vegetables, and fruits).39 To calculate the PACC, raw scores were z transformed on the basis of the mean and SD from the baseline data and averaged together. At the time of the present analyses, cognitive data were available for 196 participants at years 1 through 4, 188 at year 5, 165 at year 6, 106 at year 7, 74 at year 8, and 8 at year 9. The median number of visits was 7.0 (±1.26) over a median of 6.25 (±1.40) years.
Data Availability
Data from HABS may be requested at: https://nmr.mgh.harvard.edu/lab/harvard-aging-brain-study/public-data-releases.
Statistical Analyses
Linear mixed-effects models implemented in R (version 3.5.1) were used to examine the associations between vascular risk and Aβ burden on longitudinal brain atrophy (nlme package). The initial predictor of interest was the 3-way interaction of vascular risk, Aβ burden, and time. If this term was not significant, it was removed from the model, and 2-way interactions of vascular risk with time and Aβ with time were examined. We ran separate models for each of the 4 lobes and for each subcortical/allocortical region (e.g., striatum, thalamus, and hippocampus). All models were adjusted for baseline age, sex, and APOE status (ε4 carrier vs noncarrier), as well as each variable interacted with time. When the outcome variable was an allocortical or subcortical (volumetric) region, we additionally adjusted for intracranial volume and its interaction with time. In analyses in which the outcome variable was cognition, we additionally included terms for education and its interaction with time. In all longitudinal analyses, a random intercept and slope were included for each participant. Time was represented as years from baseline for each participant. We inspected diagnostic plots of the residuals to ensure that the assumptions of linear models were met. To correct for multiple comparisons, we applied the Benjamini-Hochberg procedure40 with a false discovery rate (FDR) of 0.05.
A post hoc exploratory analysis examined the specific FreeSurfer cortical regions associated with the interaction of vascular risk and Aβ burden. To reduce the number of comparisons, FreeSurfer regions were averaged across the left and right hemispheres. Models were adjusted for the covariates described above, and FDR correction was applied.
To address whether the foregoing analyses may be driven by the age or sex components of the FHS-CVD, we repeated our analyses using age and sex in place of the FHS-CVD in separate models (i.e., vascular risk × Aβ × time was replaced with age × Aβ × time or sex × Aβ × time). The rationale here is that if the age or sex components are driving the associations, this should be reflected in significant associations when age or sex is replaced with the FHS-CVD in our analyses. These models were adjusted for the covariates described above, and FDR correction was applied.
In secondary analyses, we examined whether the interactive associations of vascular risk with Aβ burden on brain atrophy remained after adjustment for imaging markers of white matter injury, including diffusion-derived global FA and WMH. This allowed us to determine whether the impact of vascular risk on brain atrophy operates independently from imaging markers of white matter injury. To address this question, we repeated the primary set of analyses predicting lobar and allocortical/subcortical atrophy and included baseline global FA and WMH and their interactions with time in the models. Models were adjusted for the covariates described above, and FDR correction was applied.
A final goal of the study was to determine whether the interactive association of vascular risk with Aβ burden on cognitive decline was mediated by gray matter atrophy. To do so, we performed a moderated mediation analysis using the mediation package in R.41 Because the analyses were carried out in a linear regression framework, we extracted slopes of change for each FreeSurfer cortical region and for the PACC from separate linear mixed-effects models. To reduce the number of mediation analyses performed, we created a cortical atrophy composite that was based on atrophied regions associated with both the vascular risk–Aβ interaction (described above) and PACC decline. Significant associations between the FreeSurfer cortical slopes and PACC slopes were assessed in linear regression models, with adjustment for age, sex, education, and APOE ε4 status. FDR correction was applied to these analyses.
For the mediation analysis, we built 2 models: (1) a linear regression model in which the outcome was the cortical atrophy composite and the predictor was the vascular risk–Aβ interaction (mediator model) and (2) a linear regression model in which the outcome was PACC slope and the predictors were the vascular risk–Aβ interaction and the cortical atrophy composite (outcome model). Both the mediator model and the outcome model included the main effects of vascular risk and Aβ burden, as well as the following covariates: age, sex, education, and APOE ε4 status. To probe the moderating effect of Aβ burden on the association of vascular risk with PACC decline, we performed 2 mediation models: 1 at a low level of Aβ (Aβ value at the 15th percentile of the sample) and another at high level of Aβ (Aβ value at the 85th percentile of the sample). We used the test.modmed function to formally test the difference between the mediation effects at the 2 levels of Aβ. The mediated (average causal medication effect [ACME]) and direct (average direct effect [ADE]) effects were estimated from nonparametric bootstrapping (1,000 simulations, p < 0.05).
Results
There were 196 participants included in the present study. Participants were excluded if they did not have the following data: baseline Aβ PET data (n = 1 excluded), the necessary demographic or medical information to compute a summary measure of systemic vascular risk (n = 4 excluded), baseline diffusion data (n = 9 excluded), baseline WMH data (n = 0 excluded), APOE data (n = 6 excluded), and at least 2 MRI data points (n = 48 excluded). Table 1 summarizes the baseline characteristics of the study sample. We used partial Pearson correlations to examine the cross-sectional relationships of systemic vascular risk (as measured by FHS-CVD) with Aβ burden, WMH, and global FA, adjusting for age and sex. There was no association between vascular risk and Aβ burden (rpartial = −0.06, p = 0.43). As expected, there was a significant positive association between vascular risk and WMH (rpartial = 0.19, p = 0.009), a negative association between vascular risk and global FA (rpartial = −0.25, p < 0.001), and a negative association between WMH and global FA (rpartial = −0.25, p < 0.001).
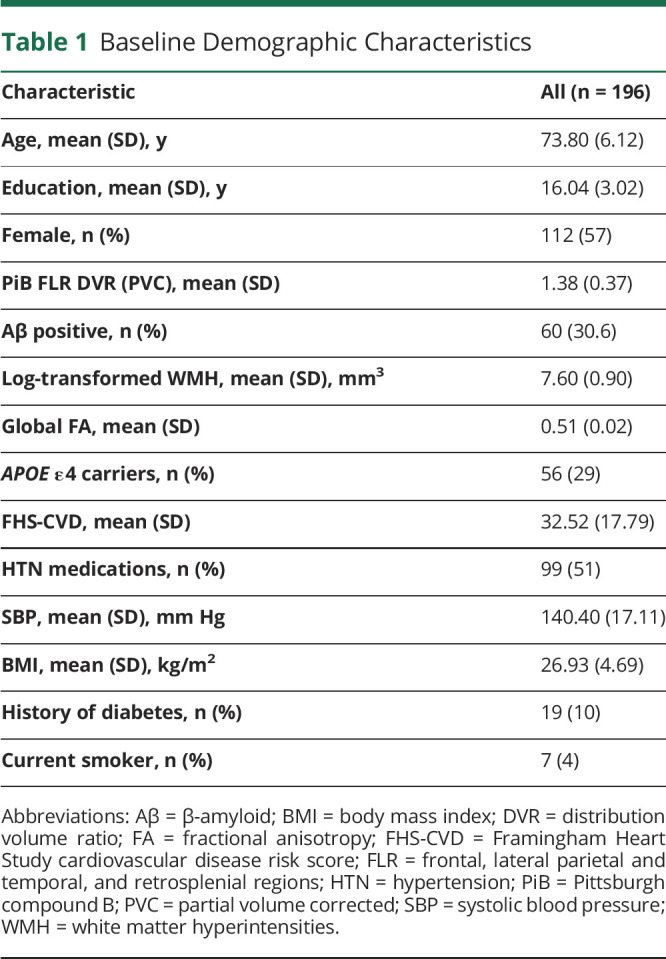
Table 1.
Baseline Demographic Characteristics
Vascular Risk and Aβ Are Interactively Associated With Longitudinal Gray Matter Loss
The combination of elevated vascular risk and higher Aβ burden was associated with faster cortical thinning in frontal and temporal lobes, as shown by significant 3-way interactions between vascular risk, Aβ burden, and time (Figure 1 and Table 2). In contrast, this same 3-way interaction was not significantly associated with cortical thinning in parietal or occipital regions (Table 2). Because of this, we next examined 2-way interactions of Aβ burden × time and vascular risk × time as predictors of parietal and occipital atrophy. We observed that higher Aβ burden, but not vascular risk, was associated with faster atrophy in parietal (Aβ × time: β = −0.067, SE = 0.016 [95% CI −0.098, −0.035], t = −4.14, p < 0.001; vascular risk × time: β = −0.036, SE = 0.024 [95% CI −0.082, 0.010], t = −1.54, p = 0.124) and occipital cortices (Aβ × time: β = −0.052, SE = 0.018 [95% CI −0.086, −0.018], t = −2.95, p = 0.003; vascular risk × time: β = −0.016, SE = 0.026 [95% CI −0.066, 0.034], t = −0.64, p = 0.526). With respect to subcortical and allocortical regions, higher vascular risk and elevated Aβ burden interacted to predict greater atrophy within the thalamus and striatum, although the latter did not survive FDR correction (Table 2). The 3-way interaction of vascular risk, Aβ burden, and time was not associated with hippocampal atrophy (Table 2). Instead, the 2-way interactions demonstrated that hippocampal atrophy was associated with higher Aβ burden but not vascular risk (Aβ × time: β = −0.025, SE = 0.009 [95% CI −0.043, −0.007], t = −2.72, p = 0.007; vascular risk × time: β = 0.003, SE = 0.013 [95% CI −0.022, 0.028], t = 0.207, p = 0.836, eTable 2, links.lww.com/WNL/B990, provides the full model output).
Figure 1. Spaghetti Plots Depicting Cortical Thinning Over Time Among Participants With High/Low Vascular Risk and High/Low Aβ Burden.
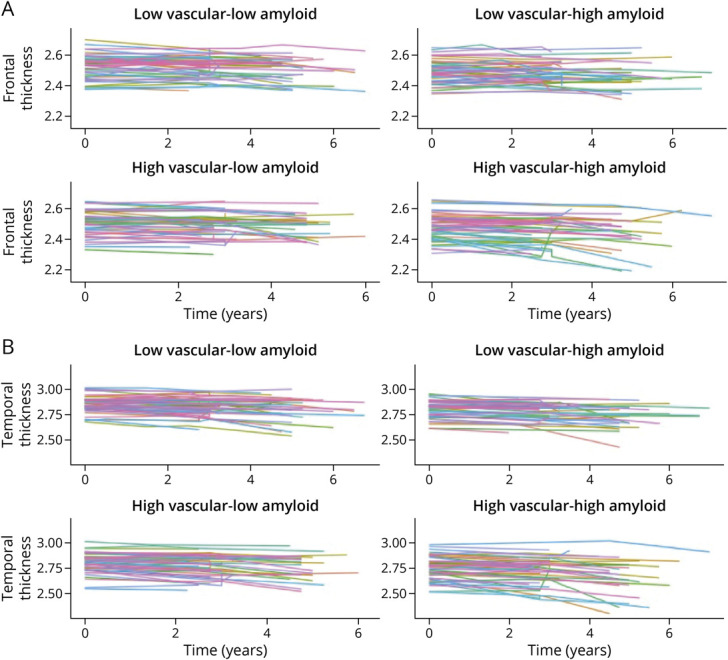
(A) Atrophy within frontal regions over time. (B) Atrophy within temporal regions over time. Participants were classified as high/low vascular risk and high/low β-amyloid (Aβ) burden on the basis of the median values of vascular risk and Aβ burden. The fastest rates of atrophy were observed in the high vascular risk–high Aβ group.
Table 2.
Interactive Associations of Aβ Burden and Vascular Risk on Longitudinal Brain Atrophy

In post hoc regional analyses, we investigated the specific frontal and temporal regions that were associated with the 3-way interaction of vascular risk, Aβ burden, and time. After FDR correction and adjustment for covariates (i.e., age, sex, APOE ε4 status, and their interactions with time), we found that higher vascular risk interacted with greater Aβ burden to predict more severe atrophy in several frontal regions, including bilateral rostral middle frontal, pars opercularis, pars triangularis, pars orbitalis, and lateral orbital frontal regions. The only temporal region to reach significance was the bilateral temporal pole (Figure 2).
Figure 2. FreeSurfer-Defined Cortical Regions in Which Longitudinal Atrophy Was Associated With the 3-Way Interaction of Vascular Risk, Aβ Burden, and Time.

Greater vascular risk interacted with higher β-amyloid (Aβ) burden to predict faster longitudinal atrophy in frontal and temporal regions and subcortically in the thalamus and striatum (subcortical regions not depicted). Regions were averaged across the left and right hemispheres. Models were adjusted for age, sex, APOE ε4 status, and their interactions with time. Color bars indicate the t statistic for the association of the 3-way interaction with brain atrophy. Regions shown are p < 0.05 after false discovery rate correction for multiple comparisons.
To assess whether the age or sex components of the FHS-CVD were driving the observed effects, we reran our primary analysis using age and sex in place of the FHS-CVD (i.e., age × Aβ × time or sex × Aβ × time) in separate models. These interaction terms were not significant in any of the models predicting cortical or subcortical/allocortical regions (all p > 0.05 after FDR correction), suggesting that the vascular risk factors that make up the FHS-CVD are driving the effects.
Associations of Vascular Risk and Aβ Burden on Brain Atrophy Remained After Adjustment for Markers of White Matter Injury
Next, we examined whether the associations of vascular risk with Aβ burden on brain atrophy remained after adjustment for imaging markers of white matter injury such as diffusion-derived global FA and WMH. These analyses allowed us to determine whether the impact of vascular risk on gray matter atrophy operates independently from imaging markers of white matter injury. As summarized in Table 3, adjusting for markers of white matter injury had little impact on the associations between vascular risk and Aβ-related gray matter atrophy. Specifically, we observed that the interaction of elevated vascular risk and higher Aβ burden remained a significant predictor of atrophy within frontal regions, temporal regions, thalamus, and striatum. In these models, lower global FA was independently but weakly associated with greater atrophy in frontal regions (this association did not survive FDR correction) and greater WMH burden was significantly associated with greater atrophy in temporal regions. There was also a weak counterintuitive association between lower WMH and more severe striatal atrophy; however, this association did not survive FDR correction.
Table 3.
Associations of Vascular Risk and Aβ Burden on Longitudinal Brain Atrophy, After Adjustment for Markers of White Matter Injury

Gray Matter Atrophy Mediated the Interactive Association of Vascular Risk and Aβ Burden on Cognitive Decline
A final goal of this study was to determine whether brain atrophy mediates the interactive association of vascular risk with Aβ burden on cognitive decline. In these models, we used a cortical atrophy composite that was based on atrophied regions associated with both the vascular risk–Aβ interaction and PACC decline. As depicted in Figure 3, PACC decline was associated with widespread atrophy across frontal, temporal, parietal, and occipital cortices, as well as the hippocampus and thalamus. The cortical regions associated with both the vascular risk–Aβ interaction and PACC decline included bilateral rostral middle frontal, pars opercularis, pars triangularis, pars orbitalis, and lateral orbital frontal, as well as bilateral temporal pole. These regions were used to create a cortical atrophy composite for the mediation analysis. Before performing the mediation analysis, we examined the pairwise associations between the vascular risk–Aβ interaction, cortical atrophy composite (slope), and PACC decline (slope). All associations were significant and are presented in eTable 3, links.lww.com/WNL/B990.
Figure 3. FreeSurfer-Defined Cortical Regions in Which Longitudinal Atrophy Was Associated With PACC Decline.

Preclinical Alzheimer Cognitive Composite (PACC) decline was associated with faster longitudinal atrophy across a wide set of cortical regions, as well as the hippocampus and thalamus (allocortical and subcortical regions not depicted). Regions were averaged across the left and right hemispheres. Models were adjusted for age, sex, education, APOE ε4 status, and their interactions with time. Color bars indicate the t statistic for the association between PACC decline and brain atrophy. Regions are shown at p < 0.05 after FDR correction for multiple comparisons.
As depicted in Figure 4, causal mediation models showed that the cortical atrophy composite mediated the effect of vascular risk on PACC decline at high levels of Aβ burden (ACME −0.08, p = 0.046) but not at low levels of Aβ burden (ACME 0.02, p = 0.24). There was a significant direct effect of vascular risk on PACC decline (i.e., an effect that was not mediated by atrophy) at both high (ADE −0.56, p < 0.001) and low (ADE −0.25, p < 0.001) levels of Aβ burden. In addition, we found that ACME significantly differed at high vs low levels of Aβ burden (estimate 0.10, p = 0.016). There was no significant group difference for ADE (estimate 0.31, p = 0.142).
Figure 4. Mediation Models of the Association of Vascular Risk, Brain Atrophy, and Cognitive Decline at High and Low Levels of Aβ Burden.

Mediation analyses were performed to determine whether brain atrophy mediates the association between vascular risk and cognitive decline at high vs low levels of β-amyloid (Aβ) burden. Because the analyses were carried out in a linear regression framework, we used slopes to represent gray matter atrophy and cognitive decline. At high levels of Aβ, there was a significant direct and atrophy-mediated effect of vascular risk on cognitive decline (left). At low levels of Aβ, there was a significant direct effect of vascular risk on cognitive decline but no significant indirect (mediated) effect as shown by the dotted line (right). ACME = average causal mediation effect; ADE = average direct effect. *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
The present study investigated the interactive vs independent associations of systemic vascular risk and Aβ burden with prospective patterns of brain atrophy among a group of well-characterized clinically unimpaired adults. We found that elevated vascular risk was associated with faster Aβ-related atrophy in frontal and anterior temporal regions and subcortically in the striatum and thalamus. Adjusting for markers of white matter injury (i.e., global FA and WMH) had little impact on these associations, indicating that the effects of systemic vascular risk on gray matter atrophy are not merely consequences of white matter changes. We additionally found that gray matter atrophy mediated the effect of vascular risk on cognitive decline at higher levels of Aβ burden.
A major finding of the study was that higher vascular risk interacted with higher Aβ burden to predict more severe atrophy in frontal and anterior temporal cortices, as well as in subcortical regions such as the thalamus and striatum. It was interesting to observe atrophy in subcortical regions given that we used a cortical composite of Aβ burden. However, this may be due to the high correlation between the cortical Aβ composite and Aβ in the striatum and thalamus in this sample. Furthermore, it was notable that the set of brain regions associated with the interaction of vascular risk and Aβ burden did not overlap with the canonical pattern of AD neurodegeneration, which shows preferential atrophy within temporoparietal regions. Our findings therefore suggest that heightened vascular risk does not merely accelerate Aβ-related atrophy in AD vulnerable regions.
The finding that vascular risk and Aβ burden interact to promote atrophy in frontal and anterior temporal regions diverges from those reported in a previous cross-sectional study.15 In that study, the authors observed an interactive effect of vascular risk and Aβ burden on cortical thinning in posterior parietal regions and an independent effect of vascular risk on cortical thinning in frontotemporal regions.15 The discrepant findings between the 2 studies may relate to methodologic differences such as sample characteristics (≈20% of participants in the prior study were mildy impaired15) or study design (cross-sectional vs longitudinal). The benefit of a longitudinal design is that it captures progressive brain atrophy, whereas cross-sectional designs might capture both progressive and nonprogressive processes. Additional longitudinal studies are needed to further clarify the independent vs interactive effects of vascular risk and Aβ burden on patterns of neurodegeneration.
In the present study, we had the opportunity to compare the effects of vascular risk with imaging markers of white matter injury on subsequent patterns of atrophy. After adjustment for markers of white matter injury, elevated vascular risk remained a predictor of Aβ-related atrophy in frontal and temporal regions, as well as subcortically in the thalamus and striatum. In contrast, atrophy patterns related to WMH and global FA were much more limited. Specifically, greater WMH burden independently predicted more severe atrophy in temporal regions. We also found that lower global FA was associated with greater atrophy in frontal regions, but this association did not survive FDR correction. These findings suggest that increased systemic vascular risk accelerates Aβ-related brain atrophy through pathways that are independent of those that cause white matter injury. Possible pathways to consider include reduced cerebral blood flow, increased systemic inflammation, tau deposition, and disrupted blood-brain barrier integrity.5,42-45
Given previous work showing that vascular risk accelerates Aβ-related cognitive decline,4 we were particularly interested in whether this association was mediated by gray matter atrophy. Our mediation analyses supported this hypothesis but also suggested that vascular risk influences cognitive trajectories independently of atrophy. We found that at higher levels of Aβ burden, gray matter atrophy mediated the association between vascular risk and cognitive decline. These data suggest that one way by which combined vascular risk and Aβ burden leads to cognitive impairment is via gray matter loss. There was also a direct effect of vascular risk on cognitive decline at both high and low levels of Aβ, suggesting that the detrimental effects of vascular risk on cognitive decline are not fully captured by gray matter atrophy. Additional mediators linking vascular risk to cognitive decline should be investigated in future work. Possible candidates include hypoperfusion, atherosclerosis, arteriolosclerosis, microinfarcts, inflammation, tau deposition, and blood-brain barrier breakdown.5,42-45
The present results build on accumulating evidence that aggressively targeting vascular risk factors may be an effective clinical strategy to slow progression to AD dementia. We previously showed that clinically unimpaired participants who had higher Aβ burden but lower vascular risk had less tau deposition in cortical regions.5 In another study, Gottesman and colleagues1 found that participants with fewer vascular risk factors in midlife were less likely to have elevated brain Aβ burden later in life. In the Systolic Blood Pressure Intervention Trial (SPRINT) Memory and Cognition in Decreased Hypertension (MIND) study, intensive blood pressure control (goal systolic blood pressure <120 mm Hg) relative to standard blood pressure control (goal <140 mm Hg) was associated with reduced incidence of mild cognitive impairment or dementia.46 Furthermore, recent data indicate a decline in the age-specific incidence and prevalence of dementia in some Western countries, potentially as a result of improved treatment of cardiovascular disease and vascular risk.47,48 Intervention studies targeting vascular risk factors are currently underway49 and will provide a clearer perspective on whether managing these risk factors can prevent or slow progression to AD dementia.
The strengths of the present study include the well-characterized sample and the prospective study design with longitudinal structural MRI. The latter is particularly important because cross-sectional studies may additionally capture nonprogressive processes of brain atrophy. Furthermore, the breadth of imaging data available at baseline allowed us to examine the effects of vascular risk on brain atrophy independently of imaging markers of white matter injury. However, the results here need to be considered in the context of the study sample and the measurements available in this cohort. First, the highest range of systemic vascular risk is likely underrepresented in our sample because HABS excludes individuals with symptomatic stroke, uncontrolled diabetes, and unstable hypertension. This leaves open the possibility that the interactive effects of vascular risk with Aβ burden on brain atrophy may be of even greater consequence in persons with higher vascular risk. Second, individuals with both high Aβ burden and high vascular risk are likely not well represented in the study given that these individuals are more likely to be cognitively impaired and therefore not eligible for HABS. Third, the participants in HABS are generally well educated, of relatively high socioeconomic status, and primarily White. These considerations may affect the generalizability of the results.
We observed that heightened vascular risk interacted with elevated Aβ to promote more severe brain atrophy in frontal and anterior temporal regions and subcortically in the thalamus and striatum. Adjusting for WMH and diffusion metrics had little impact on these associations, suggesting that white matter injury does not explain the effect of systemic vascular risk on neurodegeneration. Furthermore, we found that gray matter atrophy in frontal and anterior temporal regions partially mediated the association between elevated vascular risk and Aβ-related cognitive decline. Together, these results provide another line of support for managing vascular risk factors to reduce the emergence of cognitive impairment, especially in individuals with elevated Aβ burden.
Acknowledgment
The authors are grateful for the dedication of the HABS participants.
Glossary
- Aβ
β-amyloid
- ACME
average causal mediation effect
- AD
Alzheimer disease
- ADE
average direct effect
- FA
fractional anisotropy
- FDR
false discovery rate
- FHS-CVD
Framingham Heart Study cardiovascular disease risk score
- HABS
Harvard Aging Brain Study
- PACC
Preclinical Alzheimer Cognitive Composite
- PiB
11C-Pittsburgh compound B
- SPRINT MIND
Systolic Blood Pressure Intervention Trial Memory and Cognition in Decreased Hypertension
- TE
echo time
- TI
time to inversion
- TR
repetition time
- WMH
white matter hyperintensities
Appendix. Authors

Study Funding
This work was supported by the NIH (grant P01 AG036694 [Johnson, Sperling, Chhatwal]; R01 AG062667 [Chhatwal]; K24 AG035007 [Sperling]; R01AG054110, R01AG053509, P30AG066514 ([Hedden]; K23AG062750 [Yang]; K99AG061238 [Buckley]), Canadian Institutes of Health Research (postdoctoral fellowship [Rabin]), and Alzheimer's Association (Clinical Scientist Fellowship AACSF-20-685828 [Pruzin]; Research Fellowship AARF-20-675646 [R.F.B.]). This research was carried out in part at the Athinoula A. Martinos Center for Biomedical Imaging at the Massachusetts General Hospital, using resources provided by the Center for Functional Neuroimaging Technologies, P41EB015896, a P41 Biotechnology Resource Grant supported by the National Institute of Biomedical Imaging and Bioengineering, NIH. This work also involved the use of instrumentation supported by the NIH Shared Instrumentation Grant Program and/or High-End Instrumentation Grant Program, specifically grants S10RR021110, S10RR023401, and S10RR023043.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Gottesman RF, Schneider ALC, Zhou Y, et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA. 2017;317:1443-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:2672-2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnes D, Yaffe K. The pojected impact of risk factor reduction on Alzheimer's disease prevalence. Lancet Neurol. 2011;10:819-828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabin JS, Schultz AP, Hedden T, et al. Interactive associations of vascular risk and β-amyloid burden with cognitive decline in clinically normal elderly individuals: findings from the Harvard Aging Brain Study. JAMA Neurol. 2018;75:1124-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rabin JS, Yang H, Schultz AP, et al. Vascular risk and β-amyloid are synergistically associated with cortical tau. Ann Neurol. 2019;85:272-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bejanin A, Schonhaut DR, La Joie R, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer's disease. Brain. 2017;140:3286-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.La Joie R, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer's disease correlates with the intensity and topography of baseline tau-PET. Sci Transl Med. 2020;12:524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61:378-384. [DOI] [PubMed] [Google Scholar]
- 9.Chetelat G, Villemagne VL, Villain N, et al. Accelerated cortical atrophy in cognitively normal elderly with high β-amyloid deposition. Neurology. 2012;78:477-484. [DOI] [PubMed] [Google Scholar]
- 10.Andrews KA, Modat M, Macdonald KE, et al. Atrophy Rates in asymptomatic amyloidosis: implications for Alzheimer prevention trials. PLoS One. 2013;8:e58816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jagust W. Is amyloid-β harmful to the brain? Insights from human imaging studies. Brain. 2016;139:23-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cox SR, Lyall DM, Ritchie SJ, et al. Associations between vascular risk factors and brain MRI indices in UK Biobank. Eur Heart J. 2019;40:2290-2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye BS, Seo SW, Kim GH, et al. Amyloid burden, cerebrovascular disease, brain atrophy, and cognition in cognitively impaired patients. Alzheimer’s Dement. 2015;11:494-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamar M, Boots E, Arfanakis K, Barnes L, Schneider JA. Common brain structural alterations associated with cardiovascular disease risk factors and Alzheimer's dementia: future directions and implications. Neuropsychol Rev. 2020;30:546-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villeneuve S, Reed BR, Madison CM, et al. Vascular risk and Aβ interact to reduce cortical thickness in AD vulnerable brain regions. Neurology. 2014;83:40-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hohman TJ, Samuels LR, Liu D, et al. Stroke risk interacts with Alzheimer's disease biomarkers on brain aging outcomes. Neurobiol Aging. 2015;36:2501-2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villeneuve S, Jagust WJ. Imaging vascular disease and amyloid in the aging brain: implications for treatment. J Prev Alzheimers Dis. 2015;2:64-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider JA, Boyle PA, Arvanitakis Z, Bienias JL, Bennett DA. Subcortical infarcts, Alzheimer's disease pathology, and memory function in older persons. Ann Neurol. 2007;62:59-66. [DOI] [PubMed] [Google Scholar]
- 19.Clark LR, Koscik RL, Allison SL, et al. Hypertension and obesity moderate the relationship between β-amyloid and cognitive decline in midlife. Alzheimer's Dement. 2019;15:418-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dagley A, LaPoint M, Huijbers W, et al. Harvard Aging Brain Study: dataset and accessibility. Neuroimage. 2017;144:255-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412-2414. [DOI] [PubMed] [Google Scholar]
- 22.Folstein MF, Folstein SE, McHugh PR. Mini-Mental State: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189-198. [DOI] [PubMed] [Google Scholar]
- 23.Wechsler D. WMS-R: Wechsler Memory Scale-Revised. Psychological Corp; 1987. [Google Scholar]
- 24.Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a Geriatric Depression Screening Scale: a preliminary report. J Psychiatr Res. 1982;17:37-49. [DOI] [PubMed] [Google Scholar]
- 25.Johnson KA, Schultz A, Betensky RA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016;79:110-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rousset OG, Ma Y, Evans AC. Correction for partial volume effects in PET: principle and validation. J Nucl Med. 1998;39:904-911. [PubMed] [Google Scholar]
- 27.D'Agostino RB, Vasan RS, Pencina MJ, et al. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117:743-753. [DOI] [PubMed] [Google Scholar]
- 28.Rabin JS, Perea RD, Buckley RF, Johnson KA, Sperling RA, Hedden T. Synergism between fornix microstructure and beta amyloid accelerates memory decline in clinically normal older adults. Neurobiol Aging. 2019;81:38-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andersson JLR, Sotiropoulos SN. An integrated approach to correction for off-resonance effects and subject movement in diffusion MR imaging. Neuroimage. 2016;125:1063-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith SM, Jenkinson M, Johansen-Berg H, et al. Tract-based spatial statistics: voxelwise analysis of multi-subject diffusion data. Neuroimage. 2006;31:1487-1505. [DOI] [PubMed] [Google Scholar]
- 31.Wu M, Rosano C, Butters M, et al. A fully automated method for quantifying and localizing white matter hyperintensities on MR images. Psychiatry Res Neuroimaging. 2006;148:133-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rabin JS, Neal TE, Nierle HE, et al. Multiple markers contribute to risk of progression from normal to mild cognitive impairment. Neuroimage Clin. 2020;28:102400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wakana S, Jiang H, Nagae-Poetscher LM, van Zijl PC, Mori S. Fiber tract-based atlas of human white matter anatomy. Radiology. 2004;230:77-87. [DOI] [PubMed] [Google Scholar]
- 34.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341-355. [DOI] [PubMed] [Google Scholar]
- 35.Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968-980. [DOI] [PubMed] [Google Scholar]
- 36.Reuter M, Schmansky NJ, Rosas HD, Fischl B. Within-subject template estimation for unbiased longitudinal image analysis. Neuroimage. 2012;61:1402-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Donohue MC, Sperling RA, Salmon DP, et al. The Preclinical Alzheimer Cognitive Composite. JAMA Neurol. 2014;71:961-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rabin JS, Shirzadi Z, Swardfager W, et al. Amyloid-beta burden predicts prospective decline in body mass index in clinically normal adults. Neurobiol Aging. 2020;93:124-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papp KV, Rentz DM, Orlovsky I, Sperling RA, Mormino EC. Optimizing the preclinical Alzheimer's cognitive composite with semantic processing: the PACC5. Alzheimers Dement Transl Res Clin Interv. 2017;3:668-677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289-300. [Google Scholar]
- 41.Tingley D, Yamamoto T, Hirose K, Keele L, Imai K. Mediation: R package for Causal Mediation Analysis. J Stat Softw. 2014;59(5):1-38.26917999 [Google Scholar]
- 42.Snyder HM, Corriveau RA, Craft S, et al. Vascular contributions to cognitive impairment and dementia including Alzheimer's disease. Alzheimers Dement. 2015;11:710-717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wardlaw JM, Smith C, Dichgans M. Small vessel disease: mechanisms and clinical implications. Lancet Neurol. 2019;18:684-696. [DOI] [PubMed] [Google Scholar]
- 44.Smith EE, Schneider JA, Wardlaw JM, Greenberg SM. Cerebral microinfarcts: the invisible lesions. Lancet Neurol. 2012;11:272-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zlokovic BV, Seshadri S, Yaffe K, et al. Vascular contributions to cognitive impairment and dementia (VCID ): a report from the 2018 National Heart, Lung, and Blood Institute and National Institute of Neurological Disorders and Stroke workshop. Alzheimers Dement. 2020;16:1714-1733. [DOI] [PubMed] [Google Scholar]
- 46.Williamson J, Pajewski N, Auchus A. Effect of intensive vs standard blood pressure control on probable dementia: a randomized clinical trial. JAMA. 2019;321:553-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Satizabal CL, Beiser AS, Chouraki V, Chêne G, Dufouil C, Seshadri S. Incidence of dementia over three decades in the Framingham Heart Study. N Engl J Med. 2016;374:523-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langa KM, Larson EB, Crimmins EM, et al. A comparison of the prevalence of dementia in the United States in 2000 and 2012. JAMA Intern Med. 2017;177:51-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosenberg A, Mangialasche F, Ngandu T, Solomon A, Kivipelto M. Multidomain interventions to prevent cognitive impairment, Alzheimer's disease, and dementia: from FINGER to World-Wide FINGERS. J Prev Alzheimers Dis. 2020;7:29-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data from HABS may be requested at: https://nmr.mgh.harvard.edu/lab/harvard-aging-brain-study/public-data-releases.